
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild and controlled lignin methylation with trimethyl phosphate: towards a precise control of lignin functionality†
Antoine
Duval
and
Luc
Avérous
 *
*
BioTeam/ICPEES-ECPM, UMR 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: luc.averous@unistra.fr; Fax: +333 68852716; Tel: +333 68852784
First published on 14th February 2020
Abstract
The methylation of lignin is a key reaction for many different purposes, such as increasing the thermal stability and controlling lignin functionality for polymer applications, and increasing the yield of aromatic hydrocarbons during lignin pyrolysis. Methylation most often requires the use of toxic reagents, such as dimethyl sulfate. We developed an alternative protocol based on trimethyl phosphate as a safer and milder methylating reagent. The reaction proceeds without a solvent for only one hour to ensure complete and selective methylation of phenolic hydroxyl groups. This specific protocol was successfully applied to several lignins from different botanical origins (softwood, hardwood, and annual plants) and fractionation processes (Kraft, soda, and organosolv). This approach further allows a precise control of methylation in order to prepare lignins with tunable contents of phenolic hydroxyl groups for a large range of potential applications.
Introduction
Lignins are renewable polyphenols abundantly found in all vascular plants and constitute the main feedstock of biobased aromatic structures. They are now more and more seen as a potential source of aromatic building blocks in chemistry and polymer science. New processes have been developed in the past decades to extract lignins via Kraft1,2 or sulfur-free processes. Lignocellulosic biorefineries have the potential to bring high amounts of lignins to the market with a variety of architectures depending on the resources and fractionation processes.The use of lignins in polymer or materials science mainly requires preliminary chemical modifications.3,4 Methylation is probably one of the oldest chemical reactions that was employed by lignin chemists. It was already described in the early stages of the study of the lignin chemical structure by Brauns in 1939.5 Methylation served then as a chemical tool to characterize the structure of lignin, mainly by comparing the samples before and after methylation, for instance to quantify phenolic hydroxyl (Ph-OH) and carboxylic (COOH) groups.6,7 It was also a part of the process of the characterization of lignins by the degradative permanganate oxidation method.8,9 Since then, these characterization techniques have been replaced by more modern techniques, such as NMR spectroscopy, but methylation reactions still find utility for analytical purposes, for instance to increase the solubility in solvents for size-exclusion chromatography (SEC)10 or gas-chromatography coupled with mass spectrometry (GC-MS).11
Following the evolution towards the use of lignins in materials and polymer science, methylation reactions later found renewed interest. Methylated lignins were shown to form miscible blends with aliphatic polyesters or polyethers,12–14 leading to improved mechanical properties. Recently, it has been shown that methylation can enhance the compatibility of lignin with natural rubber, making it a potential alternative to carbon black.15 The methylation of lignins has also been shown to increase their thermal stability by limiting condensation crosslinking reactions, thus potentially facilitating their thermomechanical processing for incorporation into polymer materials.16
In the context of depletion of some fractions from fossil oils, lignin appears to be a potential source of aromatic hydrocarbons such as benzene, toluene and xylene (BTX). However, during the pyrolysis of lignin catalyzed by zeolites, the phenolic OH groups can cause the deactivation of the catalyst, and contribute to increase coke formation by condensation reactions.17 The methylation of phenolic OH groups can prevent these phenomena and lead to a significant increase in the yields of BTX and polycyclic aromatic hydrocarbons.18 Similarly, methylation can increase the yield of aromatics obtained during reductive depolymerization of lignin,19 and increase the storage stability of bio-oils obtained by solvent liquefaction of lignin.20
Historically, the first protocol that described the methylation of the phenolic OH groups of lignin used diazomethane as a methylating agent. Diazomethane is a highly toxic and explosive gas which requires extreme caution during handling. It has progressively been replaced by other methylating agents, the most common being dimethyl sulfate (DMS), even though it is also highly toxic and carcinogenic.21 The importance of the methylation reaction has thus led researchers to search for safer and greener protocols. Recently, the use of dimethyl carbonate (DMC) has appeared as an attractive approach.22 However, DMC methylation requires long reaction times in pressurized reactors with DMSO as a solvent. The aliphatic OH groups of lignin are also masked, and the sulfur content of lignin increases as a result of a side reaction with DMSO,20 which can be a serious drawback for some applications.
Trialkyl phosphates have been recognized as potential alkylating agents for phenols since the 1930s.23 They have commonly been used to esterify COOH groups and to methylate aromatic amines or N-heterocyclic compounds,24 but only scarcely for the methylation of phenols. Nelson described in a 1984 patent the methylation of various phenolic compounds, such as vanillin and syringaldehyde, with trimethyl phosphate (TMP).25 Some naphthols and phenols were also reported to be successfully methylated by TMP using BF3 as a catalyst under microwave activation.26 More recently, TMP has been used to methylate aliphatic polyols in melt-phase reactions with iron triflate as a catalyst.27
In this paper, we describe for the first time the use of TMP to methylate various lignins in a precisely controlled manner, leaving the aliphatic OH (Al-OH) groups largely unaffected (Scheme 1). We first determined the best conditions to obtain fully methylated lignin in agreement with several green chemistry principles, i.e. via a solvent-free protocol, under ambient pressure and in a short reaction time. The applicability of the process to several industrial sulfur-based (Kraft) and sulfur-free lignins (soda and organosolv) was then assessed. A protocol was finally established to obtain lignins with a tunable content of phenolic OH (Ph-OH) groups by controlled partial methylations.
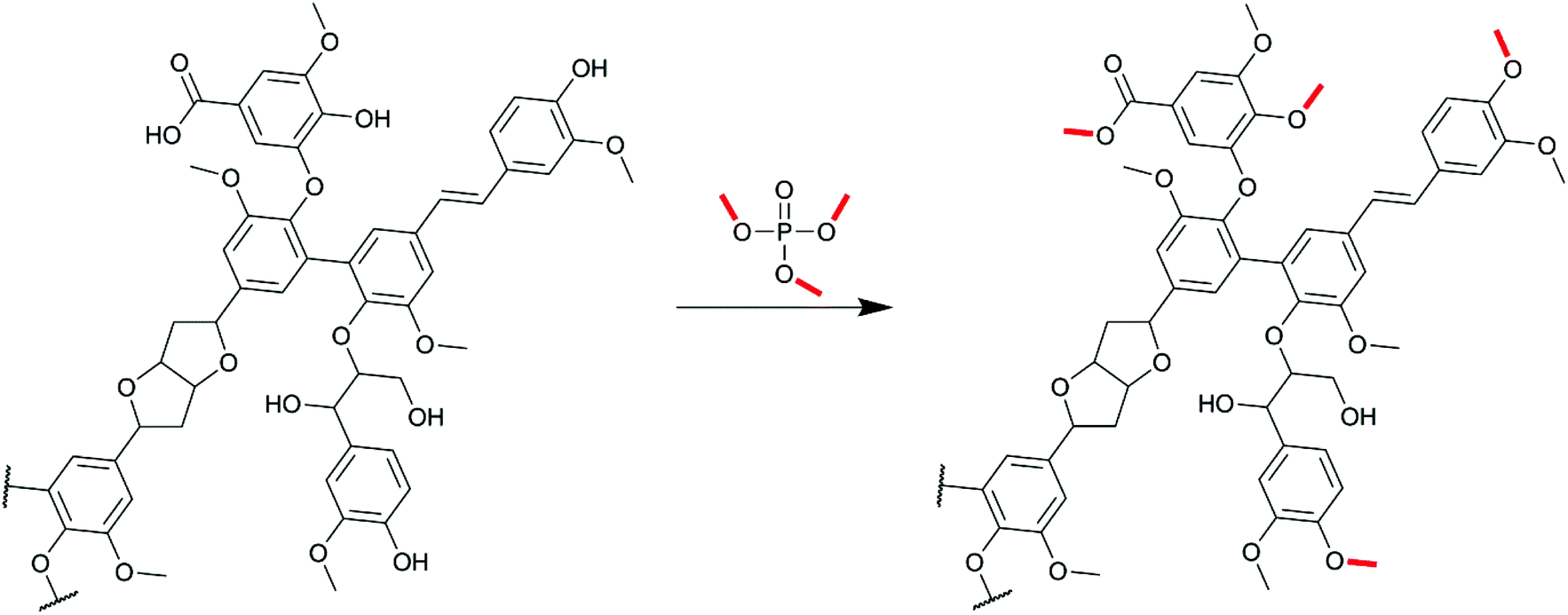 | ||
| Scheme 1 Methylation of Kraft lignin with trimethyl phosphate (TMP). | ||
Materials and methods
Materials
Softwood Kraft lignin (SW-KL) and soda lignin from mixed grasses (wheat straw and Sarkanda grass, WS-SL) were obtained from Mead Westvaco (Indulin AT) and Green Value SA (Protobind 1000), respectively. Hardwood organosolv lignins from beech (HW-OL1) and birch (HW-OL2) were produced at the Fraunhofer CBP pilot plant (Leuna, Germany). They were extracted by the acetone-based FABIOLA™ process,28–30 and recovered by precipitation of the lignin after dilution with water. Some characteristics of the different lignins are provided in Table 1.| Abbreviation | Extraction process | Supplier | Botanical source | Functional group contents (mmol g−1) | ||
|---|---|---|---|---|---|---|
| Phenolic OH (Ph-OH) | Aliphatic OH (Al-OH) | COOH | ||||
| SW-KL | Kraft | Mead Westvaco | Softwood | 4.18 | 2.42 | 0.36 |
| WS-SL | Soda | Green Value SA | Wheat straw and Sarkanda | 3.34 | 2.06 | 1.28 |
| HW-OL1 | Organosolv | Fraunhofer CBP | Hardwood (beech) | 3.58 | 1.89 | 0.06 |
| HW-OL2 | Organosolv | Fraunhofer CBP | Hardwood (birch) | 3.76 | 1.79 | 0.09 |
Trimethyl phosphate (TMP, 99%) was purchased from Acros Organics and used as received.
Methylation reactions
Lignin (0.25 g), K2CO3 (0 to 1 equivalent with respect to the reactive groups in lignin, i.e. the sum of Ph-OH and COOH groups) and TMP (10 equivalents) were introduced into a 25 mL round-bottom flask. The mixture was then flushed with argon and placed in an oil bath regulated at a given temperature (80, 100 or 120 °C) for various reaction times (from 30 minutes to 16 h). Then the mixture was directly poured into ca. 100 mL of water previously acidified to pH 2 by the addition of a 2 M HCl solution. The precipitate was then recovered by filtration using 0.45 μm PVDF membranes (Durapore®, Merck Millipore), further washed on the filter with 100 mL acidified water, and dried under vacuum at 40 °C overnight.The yields were calculated taking into account the increase in mass caused by grafting, as previously reported,31 according to eqn (1):
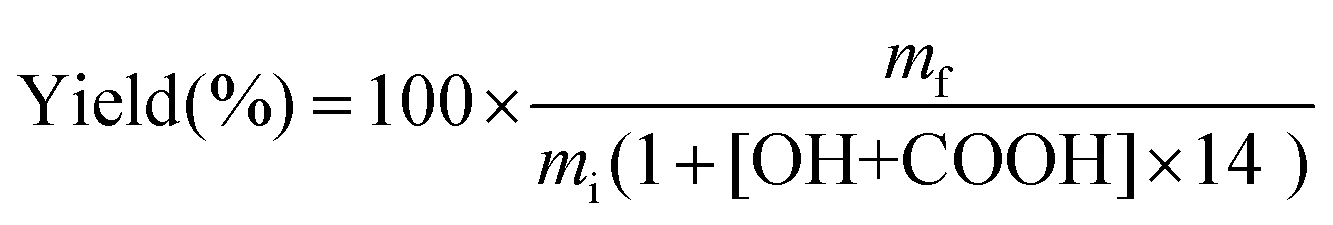 | (1) |
For kinetic studies, the amount of lignin was increased to 0.5 g. Aliquots of the reaction mixture (about 300 μL) were then taken at given reaction times and treated as previously described.
Acetylation
Acetylation of lignins was performed according to a protocol previously described.32 Briefly, 100 mg samples were dissolved in 2 mL pyridine/acetic anhydride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and stirred at room temperature for 24 h. The mixture was then precipitated in about 50 mL water previously acidified to pH 2 by the addition of a 2 M HCl solution, and filtered using 0.45 μm PVDF membranes (Durapore®, Merck Millipore).
:1 v/v) and stirred at room temperature for 24 h. The mixture was then precipitated in about 50 mL water previously acidified to pH 2 by the addition of a 2 M HCl solution, and filtered using 0.45 μm PVDF membranes (Durapore®, Merck Millipore).
Characterization
All NMR spectra were recorded on a Bruker 400 MHz spectrometer at 25 °C. For 1H NMR spectroscopy, about 20 mg of samples were dissolved in 550 μL DMSO-d6. 100 μL of a solution of 2,3,4,5,6-pentafluorobenzaldehyde was added as the internal standard. 16 scans were recorded. For standard 31P NMR spectroscopy, the samples were dissolved in DMSO-d6 and 16 scans were recorded with a standard proton-decoupling sequence. For quantitative 31P NMR spectroscopy, about 30 mg of the lignin samples were first derivatized with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane in pyridine/CDCl3 (1.6:1) in the presence of cholesterol as the internal standard, according to the standard literature protocol.33 128 scans were recorded using an inverse-gated decoupling and 15 s relaxation delay.
Fourier transform infrared (FTIR) spectra were recorded on a Nicolet iS10 spectrometer (Thermo Scientific) in the attenuated total reflectance (ATR) mode. 32 scans were recorded between 500 and 4000 cm−1 at 4 cm−1 resolution.
Dynamic scanning calorimetry (DSC) was performed using a TA Q200 calorimeter. The samples were first maintained at 105 °C for 15 min to erase the thermal history, then cooled to 0 °C at 10 °C min−1 and heated to 200 °C at 10 °C min−1. The glass transition temperature (Tg) was taken as the midpoint of the change in slope during the heating run.
Size-exclusion chromatography (SEC) was performed using a Waters Acquity Advanced Polymer Chromatography (APC) system equipped with three 150 mm APC XT columns (a 45 Å, 1.7 μm column; a 200 Å, 2.5 μm column; and a 450 Å, 2.5 μm column) thermostated at 40 °C. Tetrahydrofuran (THF, HPLC grade, Fisher Scientific) was used as the eluent at a flow rate of 0.6 mL min−1. Detection was performed with an Acquity refractive index (RI) detector and an Acquity tunable UV (TUV) detector operating at 280 nm. The samples were dissolved in THF at 5 mg mL−1 and filtered through 0.2 μm PTFE syringe filters prior to injection. When required to ensure complete solubility in THF, the samples were first acetylated as described above. The average molar masses and the dispersity were calculated from a calibration with polystyrene standards.
Results and discussion
Determination of optimum reaction conditions on SW-KL
Lignin chemistry is complicated because of the low solubility of lignins in common organic solvents. To solubilize lignin samples, polar aprotic solvents such as DMSO and DMF are usually needed. In order to avoid such solvents, we have recently developed several reactions with cyclic carbonates, which can act as both solvent and reagent to modify lignins.31,32,34 Under these conditions, it is possible to quantitatively derivatize lignins without the need for any organic solvents. Following these results, we applied a similar strategy to methylate lignins with TMP as a mild methylating agent. The reaction conditions were first tested on SW-KL.Using 10 eq. of TMP with respect to the acidic groups in lignins (i.e. the sum of Ph-OH and COOH groups), it is possible to obtain a homogeneous solution without the need for an additional solvent, confirming that TMP is a powerful polar aprotic solvent.24 K2CO3 was chosen as a catalyst, and preliminary trials were performed at 80 °C. The corresponding results are presented in Table 2.
| K2CO3 eq. | t (h) | Ph-OH conversion (%) | COOH conversion (%) |
|---|---|---|---|
| 0.1 | 5 | 22.3 | 100 |
| 0.1 | 16 | 23.6 | 100 |
| 0.5 | 5 | 73.1 | 100 |
| 0.5 | 16 | 82.2 | 100 |
| 1.0 | 5 | 77.5 | 100 |
| 1.0 | 16 | 87.7 | 100 |
Using 0.1 eq. K2CO3, the conversion of Ph-OH groups was limited to 22–23% and increasing the reaction time did not lead to any amelioration. Increasing the catalyst content to 0.5 eq. resulted in up to 82% conversion, whereas almost 90% conversion was obtained using a stoichiometric amount of K2CO3.
Using 1 eq. K2CO3 as a catalyst, methylation reactions were then performed at different temperatures (80, 100 and 120 °C), and the evolution of the conversion with the reaction time was followed (Fig. 1). At 80 °C, the maximum conversion achieved was 88%. Quantitative conversions could be achieved at 100 °C in 6 h and at 120 °C in only 1 h.
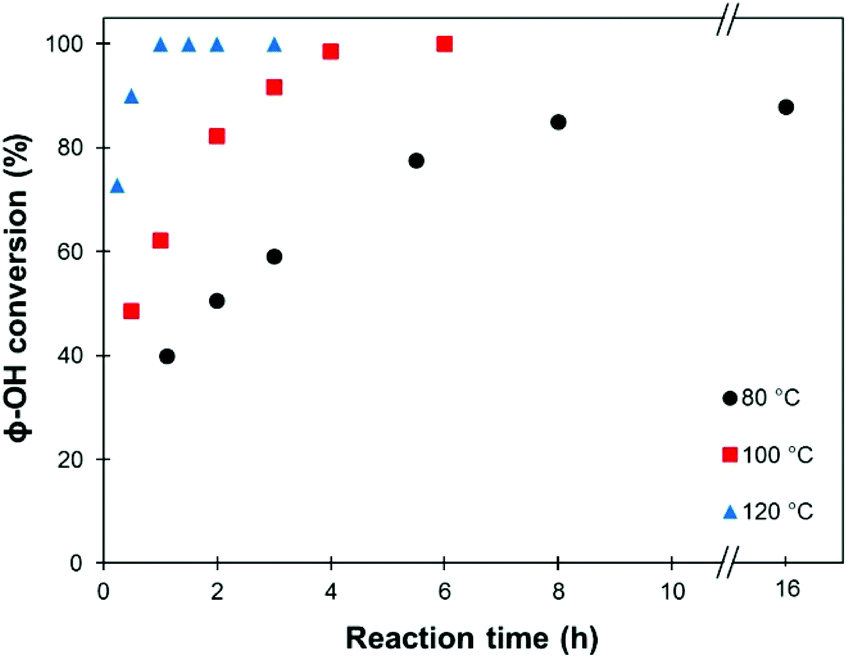 | ||
| Fig. 1 Evolution of the conversion of the phenolic OH groups in SW-KL with the reaction time (10 eq. TMP, 1 eq. K2CO3). | ||
Fully methylated SW-KLs were soluble in THF and could thus be directly analyzed by SEC. Fig. 2a shows their SEC distributions and acetylated SW-KL distributions as a reference. SW-KLs methylated for 6 h at 100 °C and for 1 h at 120 °C have purely identical SEC traces. Both reaction conditions can thus be used indifferently to methylate lignin samples. Slight differences are observed between the SEC distributions of methylated and acetylated SW-KLs, due to the differences between both derivatizations. Indeed, the groups grafted onto the lignins are different (methyl vs. acetyl), and the grafting does not affect the same functional groups in lignins. Acetylation converts Al-OH and Ph-OH groups, but leaves COOH groups unaffected, whereas the present methylation converts Ph-OH and COOH groups but leaves aliphatic OH groups unaffected. Nevertheless, the similarity of the SEC distributions confirms that the developed methylation protocol does not lead to significant changes in molar mass.
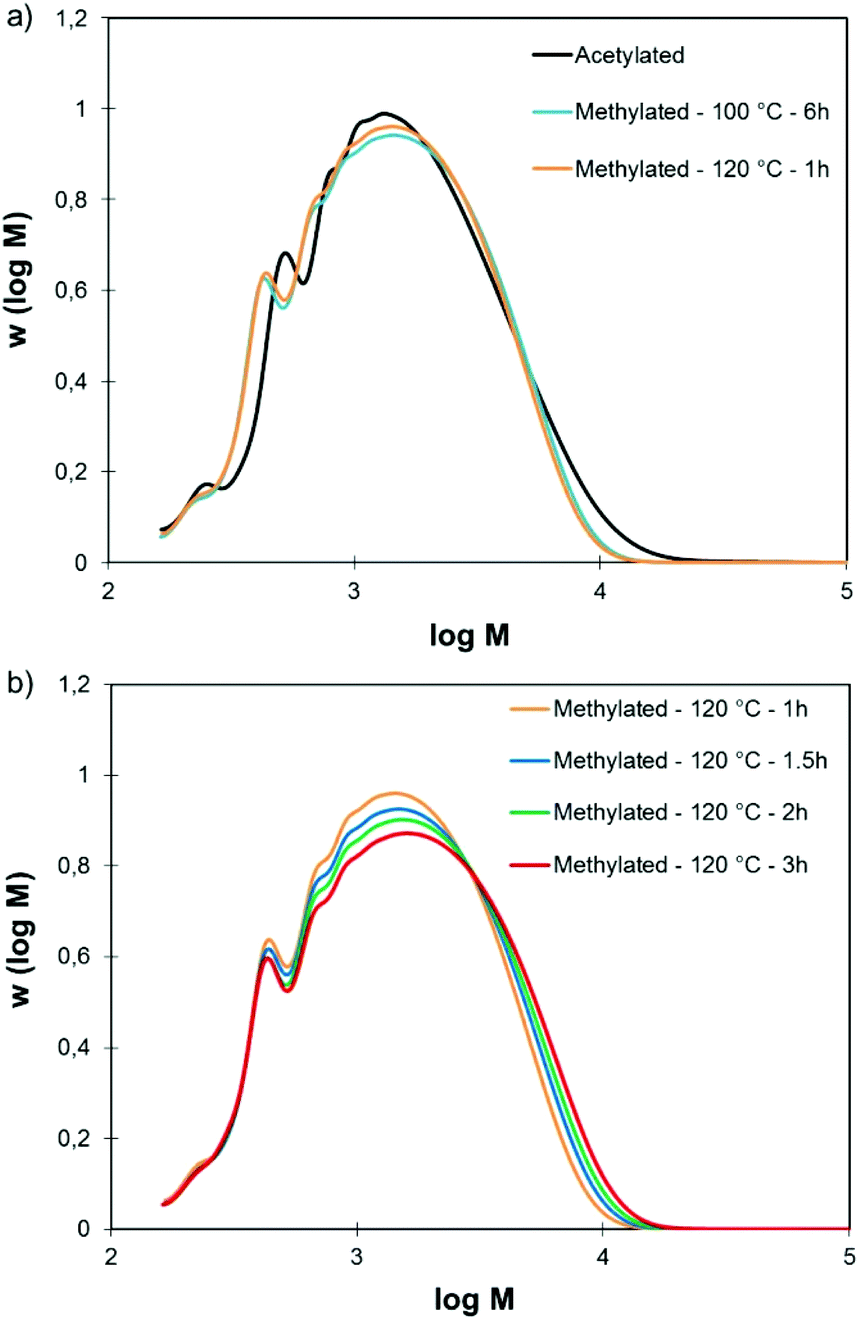 | ||
| Fig. 2 SEC traces of SW-KL acetylated, methylated at 100 and 120 °C (a), and methylated at 120 °C for various reaction times (b). Methylation conditions: 10 eq. TMP, 1 eq. K2CO3. | ||
Prolonged reaction times at 120 °C however caused gradual changes in the SEC distributions (Fig. 2b). There is a shift toward higher molar mass when the reaction time is increased (ESI, Fig. S1†). This calls for a strict control of the reaction time at 120 °C to avoid the occurrence of side reactions.
Methylation of lignins from diverse botanical origins and extraction processes
The conditions determined previously were then applied to the methylation of various lignins originating from different fractionation processes (Kraft, soda and organosolv) and botanical origins (softwood, hardwood and annual plants, Table 1). They present significant differences in terms of their chemical structure. SW-KL is almost entirely composed of guaiacyl units (G-type Ph-OH, Scheme 2), with a significant proportion of them engaged in linkages at position 5 (condensed G-type Ph-OH, Scheme 2). HW-OLs contain mainly syringyl units (S-type Ph-OH), with significant amounts of guaiacyl units that can also be engaged in linkages at position 5. 31P NMR spectra do not allow separate quantification of syringyl units and guaiacyl units with linkages at position 5, and both are thus grouped as 5-substituted Ph-OH.35 WS-SLs contain all types of phenol groups (H-type, G-type and 5-substituted PhOH, Scheme 2).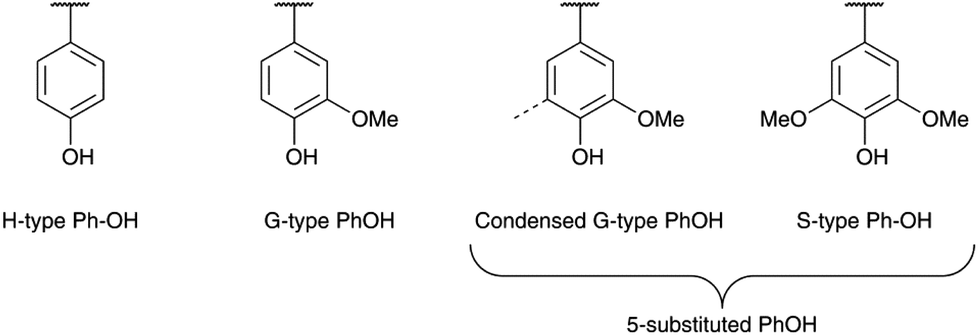 | ||
| Scheme 2 Chemical structures of the different types of phenolic groups of lignins. | ||
All lignins were successfully methylated under the predetermined conditions (Table 3). 31P spectra show the complete disappearance of signals for Ph-OH and COOH groups, which are converted into methoxyls and methyl esters, respectively (Fig. 3). It seems that there is no difference in reactivity between the various types of phenol groups present in the different lignin samples (Scheme 2). The content of Al-OH groups is also reduced, but the decrease was limited to about 22% for SW-KLs, and 32 to 36% for WS-SLs and HW-OLs, respectively. Part of this decrease is directly caused by the increase in mass related to the grafting of methyl groups onto the phenols. If we consider a sample of initial mass mi containing nAl-OH moles of Al-OH groups, its initial content of Al-OH groups as measured by 31P NMR is given by eqn (2):
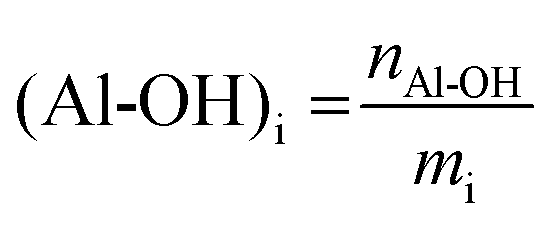 | (2) |
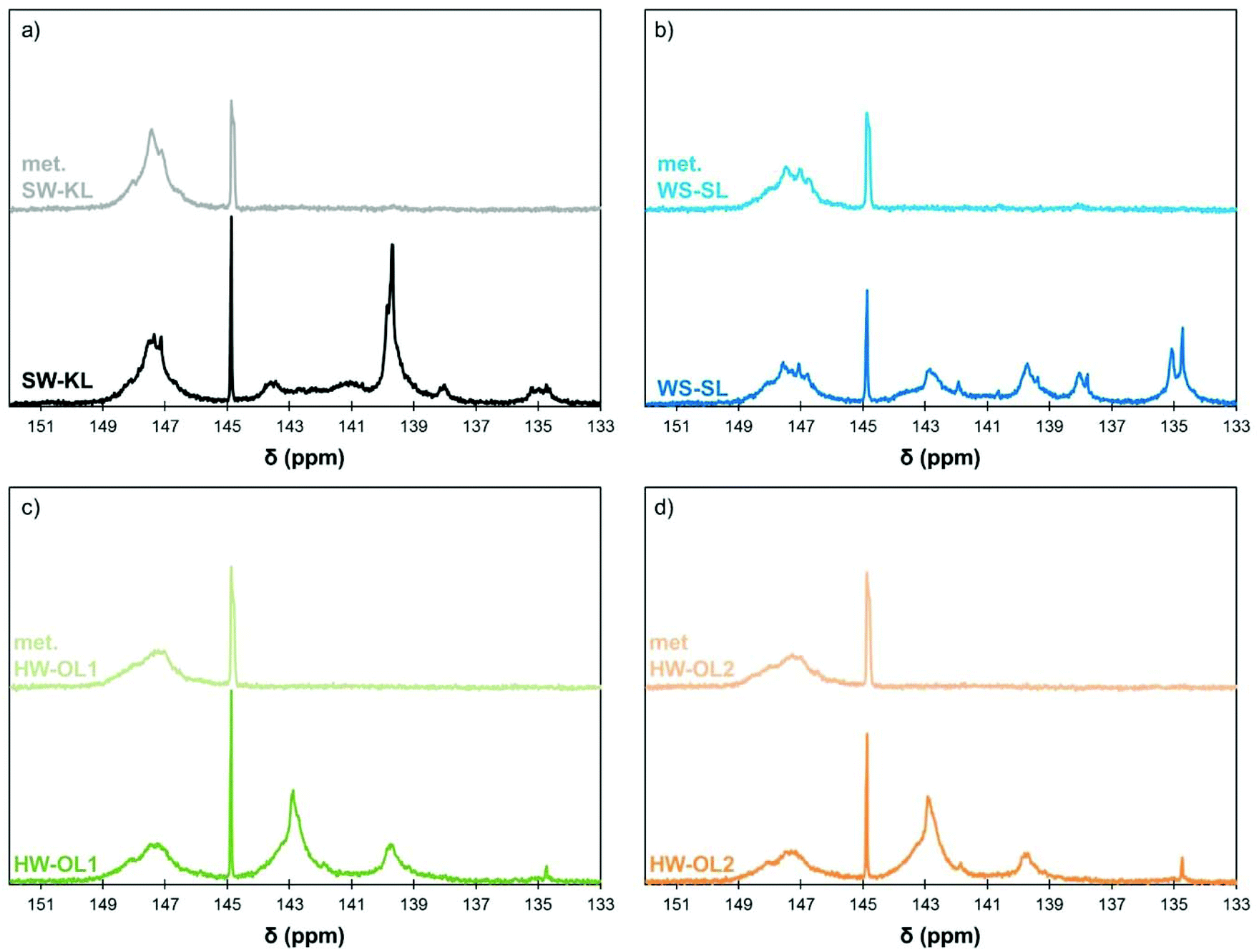 | ||
| Fig. 3 31P NMR spectra of initial and methylated lignins (10 eq. TMP, 1 eq. K2CO3, 120 °C, 1 h): SW-KL (a), WS-SL (b), HW-OL1 (c) and HW-OL2 (d). | ||
| Substrate | Reduction by 31P NMR (%) | Yield (%) | ||
|---|---|---|---|---|
| Ph-OH | Al-OH | COOH | ||
| a Three distinct experiments under similar reaction conditions. b n.a. = not available (aliquots of the reaction mixture were taken at regular intervals to follow the kinetics by NMR). | ||||
| SW-KL – 1a | 100 | 20.6 | 100 | n.a.b |
| SW-KL – 2a | 100 | 20.2 | 100 | 90.6 |
| SW-KL – 3a | 100 | 23.8 | 100 | n.a.b |
| SW-KL – average | 100 | 21.5 ± 1.6 | 100 | — |
| WS-SL | 100 | 32.4 | 100 | 90.2 |
| HW-OL1 | 100 | 32.1 | 100 | 94.1 |
| HW-OL2 | 100 | 35.9 | 100 | 87.1 |
After the methylation of all phenol and carboxylic groups, its final mass mf is increased (eqn (3)):
| mf = mi[1 + (nPh-OH + nCOOH)×14] | (3) |
The final content of Al-OH measured by 31P NMR is then obtained using eqn (4):
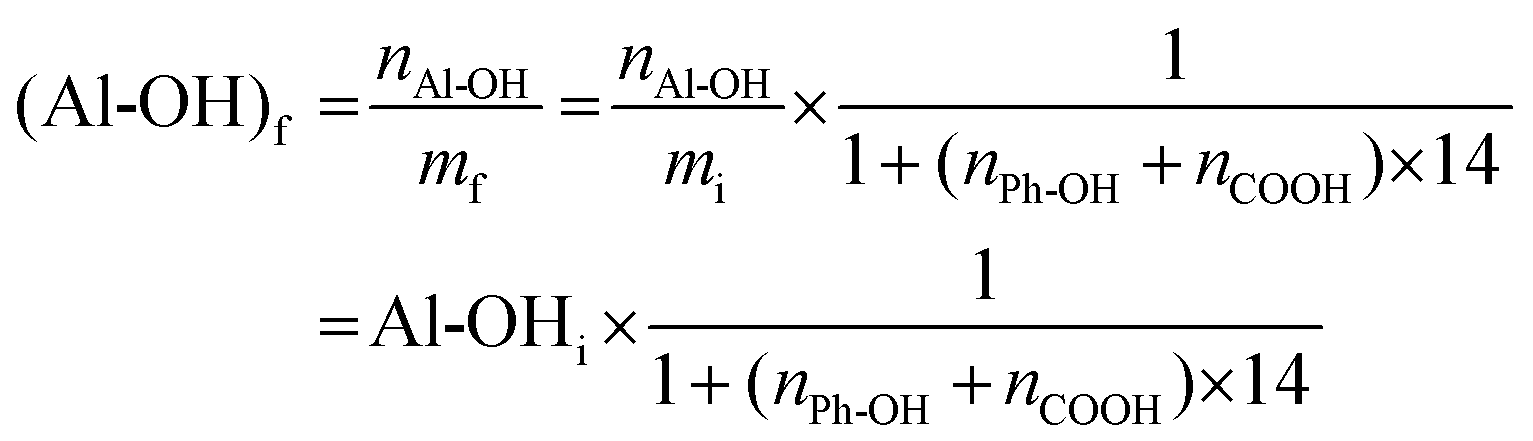 | (4) |
It thus appears that 7 to 9% of the decrease of Al-OH groups is in fact caused by the increase in mass, showing that only about 12% of Al-OH groups in SW-KLs and 23 to 29% in WS-SLs and HW-OLs, respectively, have been converted into methoxyls. The proposed methylation protocol thus leaves Al-OH groups largely unaffected, and produces purely aliphatic polyols from lignins, which are recovered in high yields (Table 3).
FTIR spectra also confirm the successful methylation (Fig. 4). The large OH band, initially centered between 3220 and 3360 cm−1 depending on the lignin, is shifted toward higher wavenumbers (3430–3500 cm−1) as a result of the masking of Ph-OH groups. Indeed, the signals in the 3220–3240 cm−1 region have been assigned to intermolecular hydrogen bonds in phenols.36 Similarly, the modification of polyphenols by oxypropylation, which leaves only Al-OH groups, has been shown to induce a similar shift of the OH absorption band in FTIR.34 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch band in COOH, initially located for SW-KLs and WS-SLs below 1700 cm−1, is also shifted to higher wavenumbers (1720–1730 cm−1), confirming the conversion of COOH into the corresponding methyl esters. 1H NMR spectra (available in the ESI, Fig. S2†) confirm the disappearance of phenolic protons, but are unable to provide further information, since the protons of the new methoxyl groups overlap with those initially present in the lignin. SEC of HW-OL1 and HW-OL2 after methylation gives results close to those of their acetylated counterparts (ESI, Fig. S3 and Table S1†), indicating the absence of significant side reactions. The solubility of WS-SL in THF was enhanced by methylation, but it was however not completely soluble and thus could not be analyzed by SEC.
O stretch band in COOH, initially located for SW-KLs and WS-SLs below 1700 cm−1, is also shifted to higher wavenumbers (1720–1730 cm−1), confirming the conversion of COOH into the corresponding methyl esters. 1H NMR spectra (available in the ESI, Fig. S2†) confirm the disappearance of phenolic protons, but are unable to provide further information, since the protons of the new methoxyl groups overlap with those initially present in the lignin. SEC of HW-OL1 and HW-OL2 after methylation gives results close to those of their acetylated counterparts (ESI, Fig. S3 and Table S1†), indicating the absence of significant side reactions. The solubility of WS-SL in THF was enhanced by methylation, but it was however not completely soluble and thus could not be analyzed by SEC.
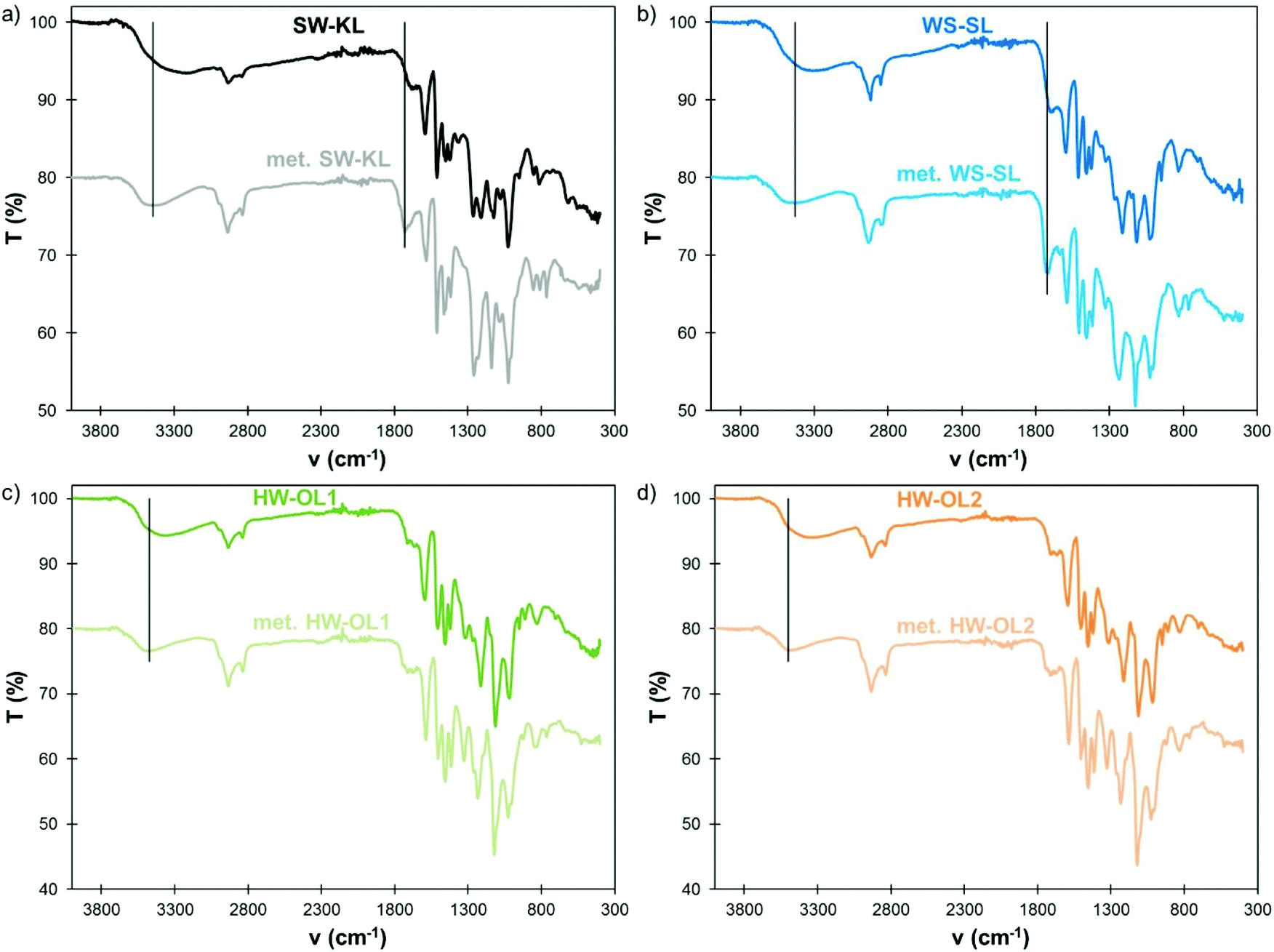 | ||
| Fig. 4 FTIR spectra of initial and methylated lignins (10 eq. TMP, 1 eq. K2CO3, 120 °C, 1 h): SW-KL (a), WS-SL (b), HW-OL1 (c) and HW-OL2 (d). | ||
The reproducibility of the process was evaluated by performing 3 distinct methylations of SW-KL under similar reaction conditions. As seen in Table 3, the reproducibility is excellent regarding the conversion calculated by 31P NMR. 1H NMR and FTIR spectra and SEC traces also confirm the optimal reproducibility (data are available in the ESI, Fig. S4 to S6†).
Insights into the reaction mechanism
As TMP possesses 3 methyl groups, a single molecule would potentially be able to methylate up to 3 phenol groups. However, in their seminal work on alkyl phosphates as methylating agents, Noller and Dutton used 1 eq. of alkyl phosphate per phenol and recovered only moderate yields of phenol ethers.23 Saidi and Rajabi used a slight excess of TMP (1.1 eq.) to methylate various phenols with BF3 as a catalyst under microwave activation.26 In a recent study on the methylation of aliphatic polyols with TMP, Duclos et al. also reported the need for 1 eq. of alkyl phosphate.27 They proposed a reaction mechanism under acid catalysis that leads to the formation of a dialkyl phosphate, and further showed that the dialkyl phosphate is unable to alkylate aliphatic alcohols.To gain insights into the reaction mechanism, aliquots of the crude reaction mixture of the methylation of SW-KL with TMP were taken at regular intervals and 1H and 31P NMR spectra were recorded (Fig. 5). The 1H NMR spectra showed the characteristic doublet of TMP at 3.67 ppm. During the course of the reaction, a new doublet appeared upfield at 3.26 ppm. The same phenomenon was observed in the 31P NMR spectra, in which a new peak appeared upfield at 1.28 ppm in addition to the TMP peak at 2.33 ppm. No additional signals were visible on the 31P NMR spectra, indicating that a single phosphorus-containing byproduct is formed. The new signal can be attributed to dimethyl phosphate (DMP),37 thus showing that only one methyl group of TMP is transferred to the phenols.
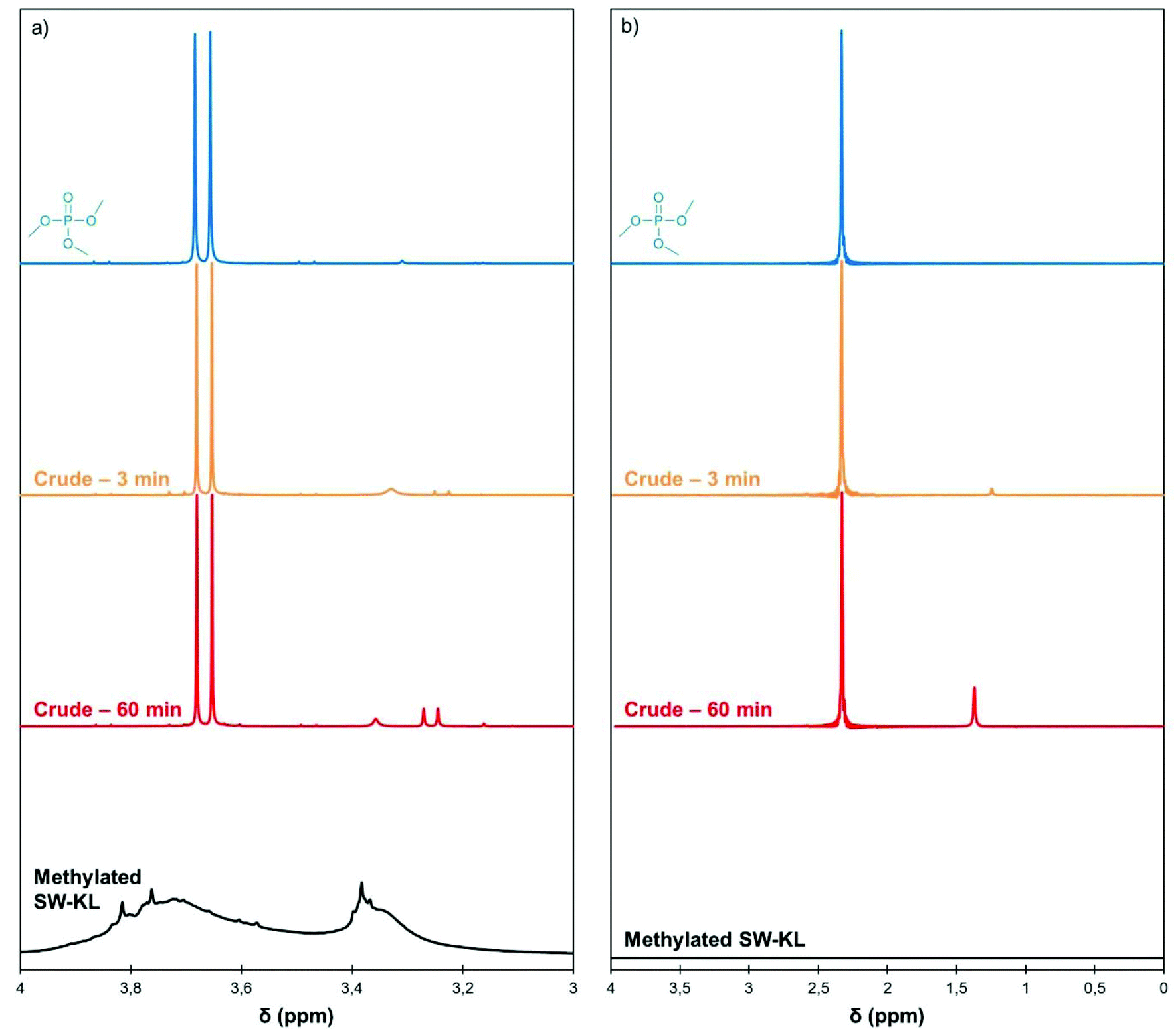 | ||
| Fig. 5 Details of 1H (a) and 31P NMR spectra (b) of TMP, the crude reaction mixture of SW-KL methylation (10 eq. TMP, 1 eq. K2CO3, 120 °C) after 3 and 60 min of reaction, and the isolated methylated SW-KL. Full spectra are available in the ESI (Fig. S7†). | ||
A tentative reaction mechanism is shown in Scheme 3, by analogy with the mechanism of the base-catalyzed methylation of phenols with dimethyl sulfate. It involves first the deprotonation of Ph-OH groups by K2CO3, followed by the nucleophilic attack of the phenolate anions on the methyl group of TMP. Since K2CO3 has a relatively low pKa value (10.3), it is unable to deprotonate Al-OH groups, which can thus not be activated and remain largely unaffected under these reaction conditions.
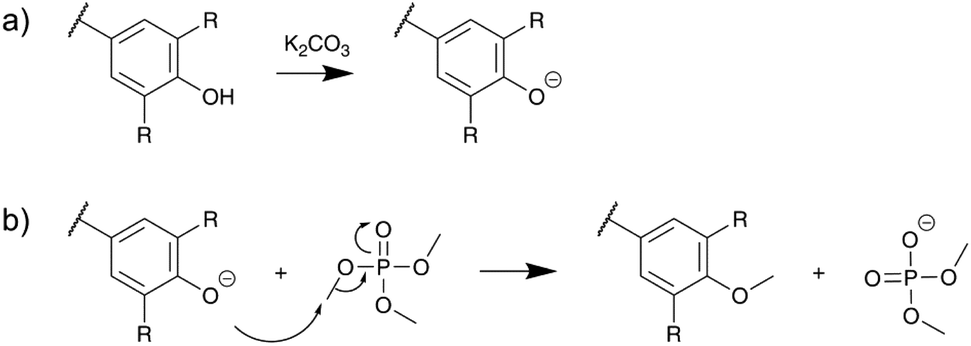 | ||
| Scheme 3 Proposed reaction mechanism for the base-catalyzed methylation of the Ph-OH group of lignin with TMP: deprotonation of Ph-OH groups (a) and nucleophilic attack of the phenolate anion on the methyl group of TMP (b). | ||
Al-OH groups can potentially be methylated by TMP, but this involves the use of strong Brønsted (sulfuric or trifluoromethanesulfonic acid) or Lewis acids (transition metals or lanthanide triflates) as a catalyst.27 Their methylation under neutral conditions has also been reported, but only at high temperature (T > 160 °C) and for extended reaction times.38 In our case, the use of relatively low temperature (120 °C) and short reaction time (1 h) prevents the reaction of most of the Al-OH groups, leading to a good selectivity towards the methylation of Ph-OH groups.
The 1H and 31P spectra of the isolated methylated SW-KL do not show signals for TMP or DMP (Fig. 5), indicating that the workup procedure allows the recovery of a pure product. No other signals are detected in the 31P NMR spectrum of methylated SW-KL, confirming that side reactions, such as the formation of phosphate esters, do not occur on lignin.
1H and 31P NMR spectra recorded at regular intervals allow the quantification of DMP formed during the reaction (spectra are available in the ESI, Fig. S8†). Fig. 6 shows the evolution of the DMP content in the reaction mixture, as measured by 1H NMR. The experiment was replicated three times and the results are presented as average values with the corresponding standard deviations. At the end of the reaction, the content of DMP is around 15 mol%, which is slightly higher than expected considering that all Ph-OH and COOH groups and part of the Al-OH groups have reacted. A blank reaction was performed in the absence of lignin, and it did not show any degradation of TMP within 1 h at 120 °C in the presence of K2CO3 (Fig. S10†).
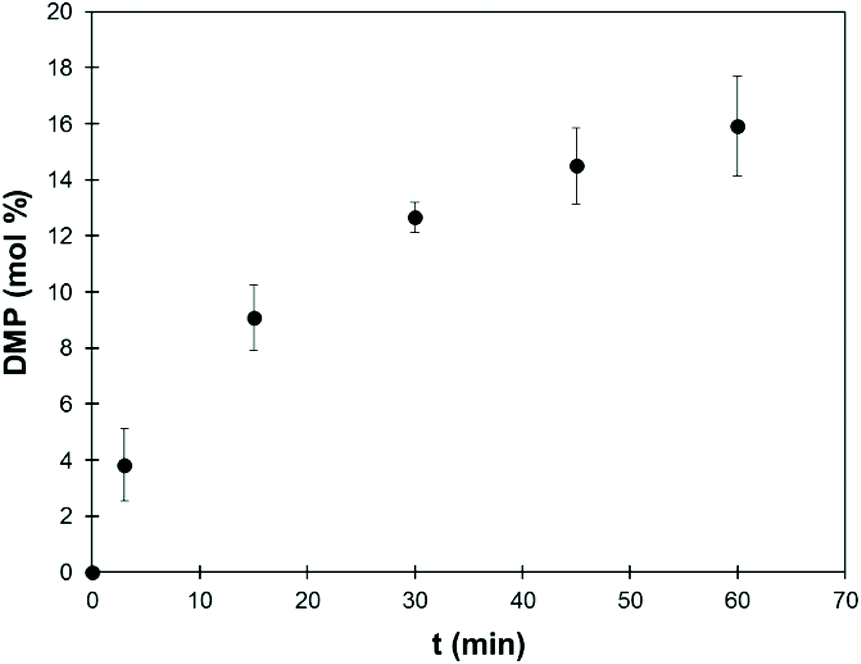 | ||
| Fig. 6 Mole percent of DMP in the reaction mixture depending on the reaction time (SW-KL, 10 eq. TMP, 1 eq. K2CO3, 120 °C), as measured by 1H NMR. Error bars represent the standard deviations of 3 replicates of the experiment. Similar data obtained by 31P NMR are given in the ESI (Fig. S9†). | ||
Toward a precise control of lignin functionality through partial methylation
The increasing use of lignins as building blocks in polymer science or for the synthesis of new materials requires a better control of their chemical structure and functionality. For instance, the high number of reactive Ph-OH groups per lignin macromolecule can be a drawback for the synthesis of polymer networks with controlled properties. It thus appears necessary to be able to tune the content of Ph-OH groups of lignins. For this purpose, methylation reactions were performed under the predetermined experimental conditions (120 °C for 1 h) but with varying contents of K2CO3 as a catalyst (Fig. 7).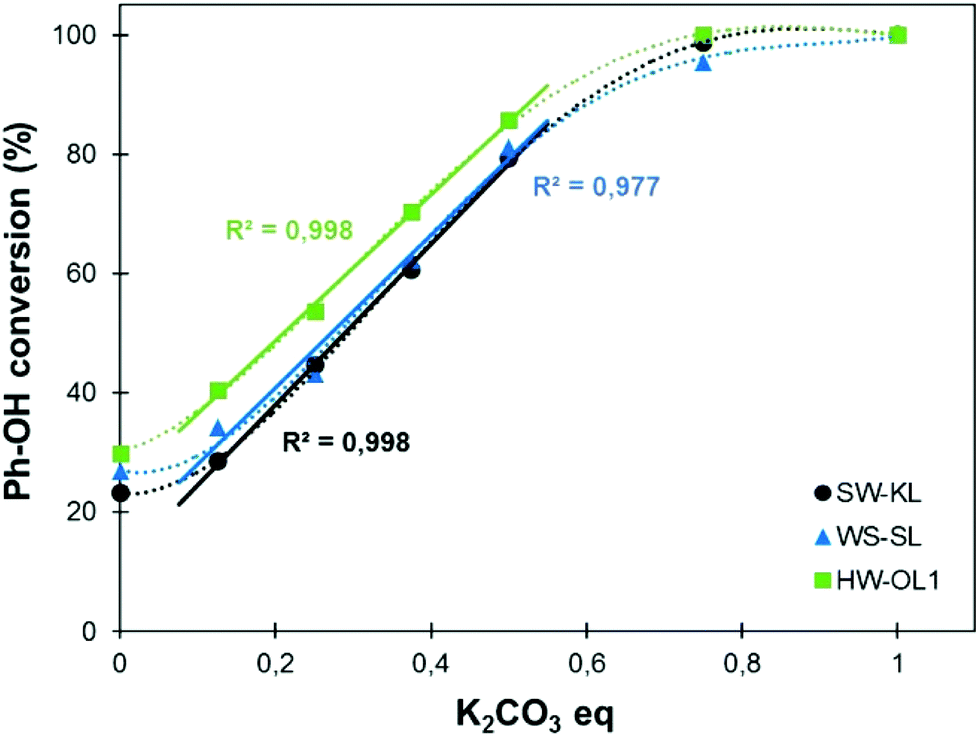 | ||
| Fig. 7 Conversion of phenolic OH groups depending on the catalyst content (10 eq. TMP, 120 °C, 1 h). Linear regression plotted between 0.125 and 0.5 eq. K2CO3 and the corresponding R2 values are reported. | ||
Fig. 7 shows that in the absence of a catalyst, 20–30% of the Ph-OH groups are converted into methoxyl groups. The conversion of the Ph-OH groups depends then on the catalyst content, and all kinds of lignins follow similar trends. Remarkably, the conversion varies linearly with the catalyst content between 0.125 and 0.5 eq., to yield a Ph-OH conversion in the range of 20–80%. This evolution gives the opportunity to finely tune the content of Ph-OH groups of a lignin sample with a controlled and quick methylation reaction.
A detailed study of 31P NMR spectra shows that the reactivities of the different kinds of Ph-OH groups (H-type, G-type and 5-substituted, Scheme 2) are very similar (data are available in the ESI, Fig. S11†). It confirms that this protocol is applicable to all kinds of lignins for the preparation of lignin samples with tailored content of Ph-OH groups. The possibility to control the content of Ph-OH groups is also a key to control the thermal properties of lignins. Tg depends on the content of Ph-OH groups, as shown in Fig. 8. Methylation reduces the hydrogen bonding ability and increases the free volume, leading to a gradual reduction of Tg.
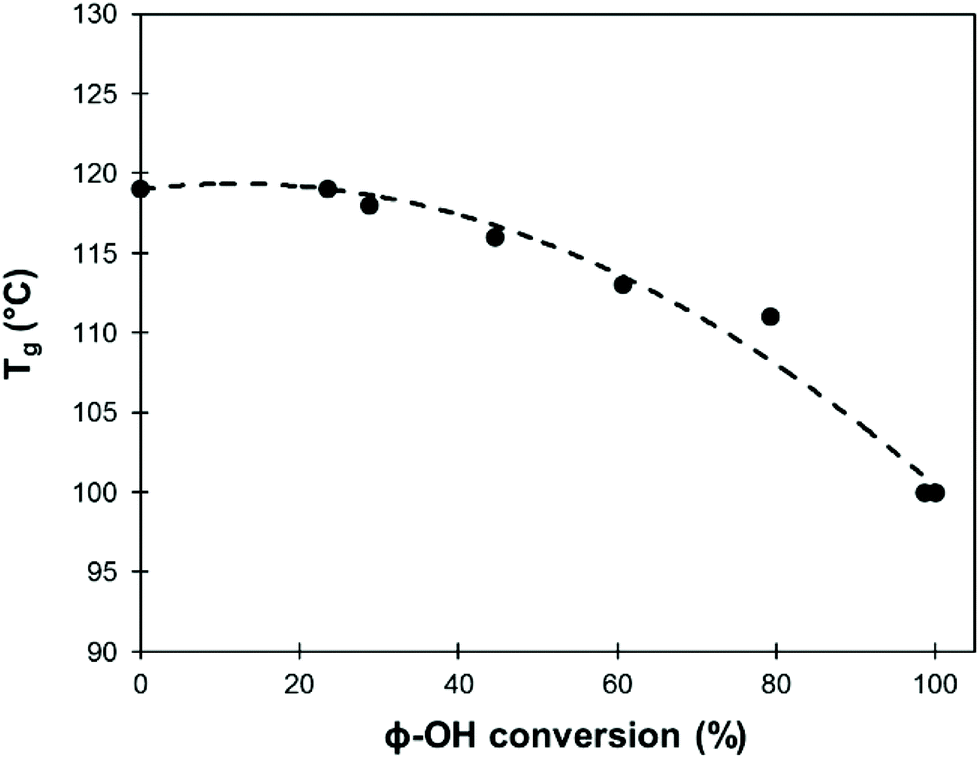 | ||
| Fig. 8 Evolution of Tg of SW-KL depending on the degree of methylation. | ||
Comparison of our method with other methylation procedures
Methylation of lignin is a very common reaction. Its importance to modify lignin in a materials science perspective has emerged more recently, leading researchers to focus much more on the process (Table 4). It has been particularly studied in the past few years by Argyropoulos’ group.22,39 They first compared methylations with dimethyl sulfate (DMS) and methyl iodide (MeI), two common reagents for the methylation of phenols.39 The methylation with DMS afforded good results: complete and selective methylation of phenol groups was achieved within 2.5 h at room temperature in water. However, continuous addition of 0.7 M NaOH was mandatory to maintain the pH above the pKa value of the phenols during the reaction. The main drawback of this process is the very high toxicity and the carcinogenicity of DMS (group 2A carcinogen according to IARC classification).21| Methylating agent (MA) | Amount of MA | Solventa | Catalyst | Conditions | Ph-OH | Al-OH | Ref. |
|---|---|---|---|---|---|---|---|
| a Volume of solvent per gram of lignin. | |||||||
| Dimethyl sulfate (DMS) | 2.5 eq. | 0.7 M NaOH (15 mL) | NaOH | RT, 30 min, then 80 °C, 2 h | 100% | 4% | 39 |
| Methyl iodide (MeI) | 3 eq. | DMF (15 mL) | K2CO3 | RT, 18 h | 61% | 38% | 39 |
| Dimethyl carbonate (DMC) | 10 eq. | DMSO (19 mL) | NaOH (2 eq.) | 150 °C, 15 h | 100% | 75% | 22 |
| Trimethyl phosphate (TMP) | 10 eq. | None | K2CO3 (1 eq.) | 120 °C, 1 h | 100% | 12% | This work |
The replacement of DMS by MeI is not completely satisfactory. Only 61% of the phenol groups of a softwood Kraft lignin were methylated despite using an excess of MeI (3 eq.), and the selectivity was quite poor, since 38% of the Al-OH groups were also affected.39 Furthermore, the reaction was performed in DMF, and MeI also has acute toxicity and is suspected to be carcinogenic (group 3 in the IARC classification).40
The replacement of these toxic chemicals by dimethyl carbonate (DMC) appears then as a valuable option. The ability of DMC to methylate phenols has been largely studied in organic chemistry,41,42 but it has been applied to lignin only recently. Sen et al.22 showed that softwood Kraft lignin could be fully methylated using 10 eq. DMC in DMSO. However, the reaction requires an excess of an inorganic base as a catalyst (2 eq. NaOH) and has to be performed in a pressurized reactor at high temperature (150 °C) for a long reaction time (15 h). In addition, the methylation with DMC lacks selectivity, since 75% of the Al-OH groups also react, either via carboxymethylation with DMC or via a side reaction with DMSO.22 The latter leads to an increase in the sulfur content of the lignin,20 which can be a strong issue for several applications.
The use of TMP as a methylating agent solves several issues encountered with the other protocols for lignin methylation (Table 4). TMP has much lower acute toxicity than DMS and MeI.43 It has also been reported to show considerably lower mutagenic activity than DMS.25 In accordance with green chemistry principles, the reaction is performed without organic solvents, and only water is used for the workup and purification. Complete methylations can be achieved with a wide variety of lignin substrates in only 1 h at 120 °C under atmospheric pressure, making it the quickest and simplest methylation procedure described for lignins, and considerably improving the energy efficiency as compared to DMC methylations. Furthermore, its selectivity is better than with DMC since only 12% of the Al-OH groups of a softwood KL are converted into methoxyls.
Conclusion
A new greener and safer protocol for the methylation of lignins has been developed. It uses trimethyl phosphate (TMP) as a mild methylating agent. The reactions were performed using an excess of TMP (10 equivalents), which acts as both solvent and reagent, in the presence of a stoichiometric amount of K2CO3 as a catalyst. After 1 h of reaction at 120 °C, a quantitative conversion of Ph-OH groups into methoxyls was observed for all kinds of lignins under study (Kraft, soda and organosolv lignins from softwood, hardwood and annual plants). Carboxylic acids were also quantitatively converted into methyl esters, whereas Al-OH groups remained largely unaffected. The proposed methylation thus converts lignins into purely aliphatic polyols.The final conversion of the Ph-OH groups can be easily tuned by lowering the catalyst content. This way, it is possible to reduce precisely the content of the Ph-OH groups of lignin samples, leading to an unprecedented control of their functionality. It is a key for their application in high value-added applications in polymer science.
The developed methylation procedure solves several issues encountered with the other common protocols. TMP is less toxic than dimethyl sulfate and methyl iodide, the reaction is fast, organic solvents are not required for the reaction or workup, the reaction uses only a cheap and benign catalyst, and the reaction shows a good selectivity towards Ph-OH groups.
Further studies will now be focused on the scale-up of the reaction, on the recovery of the excess of TMP used during the reaction, and on the use of modified lignins to develop new macromolecular architectures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Bio Based Industries Joint Undertaking (JU) under grant agreement no 792004. The JU receives support from the European Union's Horizon 2020 research and innovation program and the Bio Based Industries Consortium. We are grateful to Marlen Verges and Dr Moritz Leschinsky (Fraunhofer CBP, Leuna, Germany) for providing the beech and birch organosolv lignins obtained from scale-up runs of the FABIOLA™ process. Arjan Smit and Dr André Van Zomeren (ECN-TNO, Petten, Netherlands) are kindly acknowledged for helpful discussions on the lignins obtained by the FABIOLA™ process. Céline Piras (ICPEES) is acknowledged for performing the SEC experiments.References
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- L. Kouisni, A. Gagné, K. Maki, P. Holt-Hindle and M. Paleologou, ACS Sustainable Chem. Eng., 2016, 4, 5152–5159 CrossRef CAS.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- F. E. Brauns, J. Am. Chem. Soc., 1939, 61, 2120–2127 CrossRef CAS.
- Y.-Z. Lai, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Berlin Heidelberg, 1992, pp. 423–434 Search PubMed.
- C. W. Dence, in Methods in Lignin Chemistry, ed. D. S. Y. Lin and P. E. D. C. W. Dence, Springer Berlin Heidelberg, 1992, pp. 33–61 Search PubMed.
- K. Freudenberg and A. C. Neish, Constitution and Biosynthesis of Lignin, Springer-Verlag, Berlin Heidelberg, 1968 Search PubMed.
- G. Gellerstedt, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Berlin Heidelberg, 1992, pp. 322–333 Search PubMed.
- G. Gellerstedt, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Berlin Heidelberg, 1992, pp. 487–497 Search PubMed.
- C.-L. Chen, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Berlin Heidelberg, 1992, pp. 527–548 Search PubMed.
- Y. Li and S. Sarkanen, Macromolecules, 2002, 35, 9707–9715 CrossRef CAS.
- Y. Li and S. Sarkanen, Macromolecules, 2005, 38, 2296–2306 CrossRef CAS.
- J. F. Kadla and S. Kubo, Macromolecules, 2003, 36, 7803–7811 CrossRef CAS.
- D. Barana, M. Orlandi, L. Zoia, L. Castellani, T. Hanel, C. Bolck and R. Gosselink, ACS Sustainable Chem. Eng., 2018, 6, 11843–11852 CrossRef CAS.
- C. Cui, H. Sadeghifar, S. Sen and D. S. Argyropoulos, BioResources, 2013, 8, 864–886 Search PubMed.
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, Appl. Catal., A, 2014, 469, 490–511 CrossRef CAS.
- J.-Y. Kim, S. Heo and J. W. Choi, Fuel, 2018, 232, 81–89 CrossRef CAS.
- J. A. Barrett, Y. Gao, C. M. Bernt, M. Chui, A. T. Tran, M. B. Foston and P. C. Ford, ACS Sustainable Chem. Eng., 2016, 4, 6877–6886 CrossRef CAS.
- J.-Y. Kim, P. Hafezi-Sefat, S. Cady, R. G. Smith and R. C. Brown, Energy Fuels, 2019, 33, 1248–1255 CrossRef CAS.
- International Agency for Research on Cancer, IARC monographs on the evaluation of carcinogenic risks to humans, 1999, vol. 71, pp. 575–588 Search PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 1077–1087 RSC.
- C. R. Noller and G. R. Dutton, J. Am. Chem. Soc., 1933, 55, 424–425 CrossRef CAS.
- J. M. Fevig, in Encyclopedia of Reagents for Organic Synthesis, American Cancer Society, 2001 Search PubMed.
- R. B. Nelson, United States, US4453017A, 1984 Search PubMed.
- M. R. Saidi and F. Rajabi, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 2343–2348 CrossRef CAS.
- M.-C. Duclos, A. Herbinski, A.-S. Mora, E. Métay and M. Lemaire, ChemSusChem, 2018, 11, 547–551 CrossRef CAS PubMed.
- K. Damen, A. Smit, W. Huijgen and J. Van Hal, Fabiola: fractionation of biomass using low-temperature acetone, RRB-13 (13th International Conference on Renewable Resources and Biorefineries), Wroclaw, Poland, 2017.
- A. Smit, W. Huijgen, J. Van Hal, L. Lanting and K. Damen, Effective Fractionation of Lignocellulose Using a Mild Acetone-based Organosolv Process, 25th European Biomass Conference & Exhibition (EUBCE), Stockholm, Sweden, 2017.
- A. Smit and W. Huijgen, Green Chem., 2017, 19, 5505–5514 RSC.
- A. Duval and L. Avérous, ChemSusChem, 2017, 10, 1813–1822 CrossRef CAS PubMed.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2017, 5, 7334–7343 CrossRef CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2016, 4, 3103–3112 CrossRef CAS.
- M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 CrossRef CAS.
- S. Kubo and J. F. Kadla, Biomacromolecules, 2005, 6, 2815–2821 CrossRef CAS PubMed.
- A. M. Modro and T. A. Modro, J. Phys. Org. Chem., 1989, 2, 377–382 CrossRef CAS.
- A. D. F. Toy, J. Am. Chem. Soc., 1944, 66, 499–499 CrossRef CAS.
- H. Sadeghifar, C. Cui and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2012, 51, 16713–16720 CrossRef CAS.
- International Agency for Research on Cancer, IARC monographs on the evaluation of carcinogenic risks to humans, 1999, vol. 71, pp. 1503–1510 Search PubMed.
- P. Tundo and A. Perosa, Chem. Rec., 2002, 2, 13–23 CrossRef CAS PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- Z.-T. He, H. Li, A. M. Haydl, G. T. Whiteker and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 17197–17202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03890f |
| This journal is © The Royal Society of Chemistry 2020 |