
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel on nitrogen-doped carbon pellets for continuous-flow hydrogenation of biomass-derived compounds in water†
Francesco
Brandi
a,
Marius
Bäumel
a,
Valerio
Molinari
a,
Irina
Shekova
a,
Iver
Lauermann
b,
Tobias
Heil
a,
Markus
Antonietti
 a and
Majd
Al-Naji
*a
a and
Majd
Al-Naji
*a
aMax Planck Institute of Colloids and Interfaces, Department of Colloids Chemistry, 14476 Potsdam, Germany. E-mail: majd.al-naji@mpikg.mpg.de
bPVcomB, Helmholtz-Zentrum Berlin für Materialien and Energie GmbH, Schwarzschildstr. 3, 12489, Berlin, Germany
First published on 27th January 2020
Abstract
Hydrogenation reactions in water at elevated temperatures are challenging for heterogeneous catalysts. Thus, we present a simple, cheap, scalable, and sustainable approach for synthesizing an efficient and stable Ni catalyst supported on highly porous nitrogen-doped carbon (NDC) in pellet form. The performance of this catalyst was evaluated in the aqueous-phase hydrogenation of lignocellulosic biomass-derived compounds, i.e., glucose (Glu), xylose (Xyl), and vanillin (V), using a continuous-flow system. The as-prepared 35 wt% Ni on NDC catalyst exhibited a high catalytic performance in all three aqueous-phase hydrogenation reactions, i.e., the conversion of Glu, Xyl, and V was 96.3 mol%, 85 mol%, and 100 mol% and the yield of sorbitol (Sor), xylitol (Xyt), and 2-methoxy-4-methylphenol (MMP) was 82 mol%, 62 mol%, and 100 mol%, respectively. This high activity was attributed to the high specific surface area of NDC and mainly to the heterojunction effects stabilizing and adjusting the homogenously and highly dispersed Ni nanoparticles (ANi = 20 m2 g−1) on the surface of NDC. Changing the electron density in the nickel nanoparticles allows the high performance of the catalyst for a long time on stream (40 h) with minimized Ni leaching and without the loss in catalytic performance.
Introduction
A biorefinery is an engineering concept for producing a wide range of valuable bio-based building blocks from renewable resources such as lignocellulosic biomass.1–5 As about 80 wt% of lignocellulosic biomass comprises sugars (Fig. 1) and the remaining 20 wt% being lignin-based aromatics (Fig. 1), it possesses high potential to be catalytically upgraded to a wide range of platform chemicals.6–10 For this purpose, different catalytic reactions have been implemented such as hydrolysis, hydrogenation, dehydration, oxidation, isomerization, aldol condensation, and reforming.11–14 Among these reactions, catalytic hydrogenation is an important reaction towards diverse compounds with a wide range of applications.2,15–17 Most of the reported studies dealing with the hydrogenation of lignocellulosic biomass-derived compounds utilized noble metal-based catalysts in batch systems.16,18 However, to be economically efficient, a biorefinery must be based on a robust process, which can operate for a long period at a large scale with minimal downstream processes.19 The development of heterogeneous catalysts for the continuous flow system can contribute to these needs. Moreover, the mass and heat control guaranteed by the flow process can allow the tunability of the reaction products.20,21 Additionally, the development of the flow process at the lab scale facilitates seamless scalability to industrial purposes.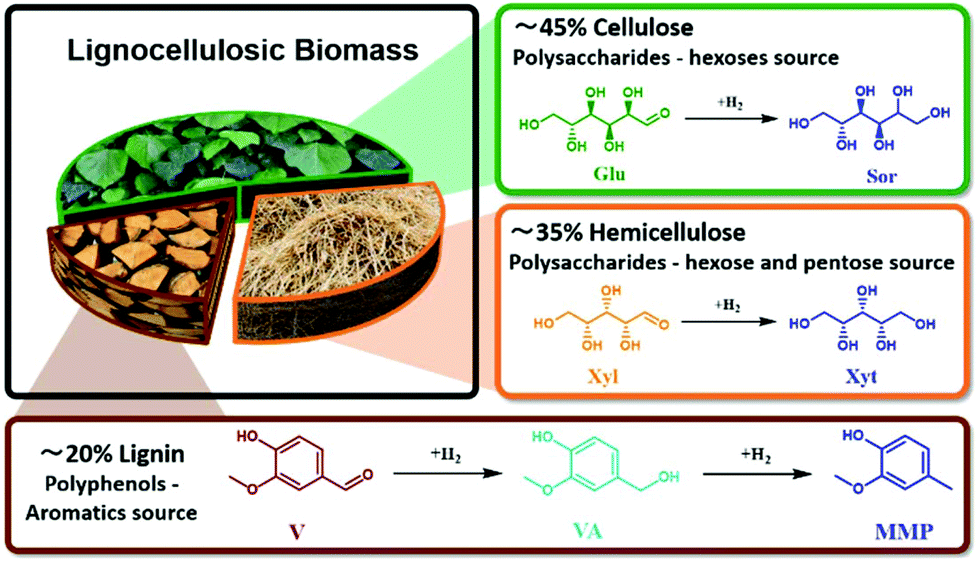 | ||
| Fig. 1 Lignocellulosic biomass composition and hydrogenation of its derivatives, i.e., glucose (Glu) hydrogenation to Sorbitol (Sor) as cellulose-derived process, hemicellulose derived compounds, Xylose (Xyl) conversion to Xylitol (Xyt), and the hydrogenation of Vanillin (V), from lignin, to 2-methoxy-4methylphenol (MMP) through Vanillyl alcohol (VA). | ||
Generally, the exposure of metal-based catalysts to water at high temperatures leads to fast deactivation due to metal leaching and loss of textural properties.22 The leaching effect in the case of noble metals (Au, Pd, Pt, and Ru) is lower than that with non-noble metals, e.g., Ni, due to their high oxidation potential. However, if compared to noble metals, Ni is preferable due to its lower cost and higher sustainability. Additionally, Ni is a classical hydrogenation catalyst.23 For Ni, it has been reported that slightly aqueous acidic conditions (pH < 6) as well as the presence of alcohol species in the solution lead to the rapid formation of Ni2+ species, which accelerates the leaching process.24,25 Traditionally, heterogeneously-catalyzed reactions are conducted using supports (Al2O3, SiO2, and zeolite) that also barely survive in hot water for longer operational times. These support materials undergo phase changes and collapse of the porous structure in the aqueous phase above 150 °C.26–28 On the other hand, carbonaceous materials are well known to be stable under harsh hydrothermal conditions (350–550 °C).29 In addition, it is possible to tune the electrochemical properties of carbon materials via doping with heteroatoms, i.e., N and S.30,31 Therefore, utilizing nitrogen-doped carbon (NDC) as a support for Ni nanoparticles allows charge transfer from Ni to the support, i.e., NDC, which leads to Ni with more positive oxidation potentials, i.e., higher “nobility”.30,31 In this regard, it has already been described that Ni supported on NDC presents higher activity when compared with a pristine carbon support.32
Based on the valorisation of the cellulosic biomass fraction, the hydrogenation of glucose (Glu) to sorbitol (Sor) is an essential reaction and the worldwide demand of Sor is already higher than 500 kton per year, mainly as an additive in cosmetics, food, or pharmaceuticals.33–35 Moreover, Sor dehydration leads to the production of isosorbide, which can be used as a diol building block for a novel class of bio-polymers.36–38 Industrially, Sor is synthesized from the hydrogenation of glucose, utilizing skeletal Ni catalysts.33,39 Additionally, several noble metal-based catalytic systems have been reported for Glu hydrogenation in water, such as Ru (Ru/Al2O3, Ru/C, Ru/zeolite, and Ru/SiO2)35,40–45 as well as Pt (Pt/Al2O3, Pt/SiO2, and Pt/C).45–48 Also, non-noble metals were used, such as Cu (Cu/SiO2)49 and Ni (Ni/SiO2, Ni/TiO2, Ni/Al2O3).35,40,49 Furthermore, rapid deactivation of Ni catalysts was observed due to the significant leaching of Ni under the reaction conditions in the initial hours of reaction.35,40,49 Also, 5.0 wt% Ru supported on MCM-48, carbon, and TiO2 each provided Glu conversion of 90–95% with 100% selectivity for Sor in a batch system at 220 °C, 2.5 MPa of H2, and 25 min of reaction time.2,44 Pan et al.42 demonstrated that Ru on multi-wall carbon nanotubes (MWNT) has a higher efficiency than the RANEY® Ni catalyst in the batch setup at 220 °C, 4.0 MPa, and 120 min of reaction time.
Similarly, xylose (Xyl) is the major sugar in the hemicellulose fraction. Therefore, the hydrogenation of Xyl to xylitol (Xyt)—a natural 5-carbon polyalcohol—is an important reaction due to its properties such as low caloric content, anti-cariogenic action, solubility in water, and stability at high temperatures.38 These properties allow Xyt to be utilized in a wide range of consumer products. Analogous to the process for Sor production, Xyt can be synthesized via hydrogenation of Xyl using supported Ni (RANEY® Ni, Ni/Al2O3, Ni/SiO2) or Ru (Ru/Al2O3, Ru/C, Ru/TiO2, Ru/ion exchange resins, and Ru/HY zeolite catalysts).38,50–55 Most of these studies reported a high conversion yield of Xyl and Xyt in a batch system, temperature range from 120 °C–190 °C, H2 pressure of 2.0 MPa–8.0 MPa at longer reaction times of 2–6 h.50–55
From the lignin fraction, vanillin (V), for instance, is used in food, beverages, pharmaceuticals, and fragrances but also fuel additives.56,57 The vanillin hydrogenation product, 2-methoxy-4-methylphenol (MMP), is used as a fuel additive or food flavour and also as a sustainable source of bisphenolic compounds.58 Typically, the hydrogenation of V to vanillyl alcohol (VA) or MMP is conducted using supported precious metal (Pd, Pt, Ru, and Rh) catalysts in batch systems and high pressure of H2 (0.5 MPa–10 MPa).59 Nevertheless, few studies have investigated the utilization of supported Ni or Cu as an active species. In this regard, Bindwal et al.57 showed efficient conversion of V to VA and MMP using Ru/C (complete V conversion with VA yield up to 90 mol% and MMP yield up to 10 mol% at 65 °C and 2.1 MPa of H2).57 Furthermore, investigations about the performance of noble metals catalysts (Ru, Rh, Pd, Au, and Pt) supported on C in the aqueous phase hydrogenation of V in a batch system were reported.60 All these catalysts showed complete V conversion, whereas Pd/C gave the highest yield of MMP (95%) H2.60 Moreover, bimetallic Cu–Pd catalysts anchored on organic porous polymer were found to be able to convert V to MMP (yield 93 mol%) in i-propanol as the solvent.61 Nevertheless, few studies have been reported utilizing non-noble metal based catalysts for V hydrogenation. For instance, Nie et al.62 obtained V conversion of 90% and MMP yield of 66% using 20 wt% Ni on nitrogen-doped carbon in a batch system at 150 °C, 0.5 MPa of H2, and 2 h of reaction time.62
From the abovementioned survey, it can be seen that the continuous flow process for aqueous-phase hydrogenation of lignocellulosic biomass-derived compounds namely Glu, Xyl, and V has barely been investigated. Furthermore, most of the used catalysts are not sustainable for large scale operation, i.e., noble metal-based catalysts.
To establish a sustainable biorefinery process, a catalyst in the pellet form is highly required, should be based on a non-noble metal, and must be able to work in the aqueous phase for a long time on stream at a temperature above 100 °C. Therefore, this study presents a simple, cheap, sustainable, and scalable approach for the preparation of a stable Ni catalyst supported on porous nitrogen-doped carbon (NDC). The performance of the as-prepared catalyst (Ni/NDC) was assessed in the aqueous-phase hydrogenation reactions, namely, Glu to Sor, Xyl to Xit, and V to MMP in the continuous flow process (Fig. 1). Furthermore, the effect of reaction temperature and residence time on the catalyst performance and product distribution was addressed. Finally, the performance of the catalysts as a function of time on stream (TOS) was evaluated.
Experimental
Preparation of Ni supported on nitrogen-doped carbon (NDC)
The porous nitrogen-doped carbon (NDC) pellets were synthesized based on a physical mixture of semolina as a sustainable carbon source, urea, glucose, water, and ZnO nanoparticles (20 nm) as a porogen. Firstly, the glucose, urea, and water mixture as a nitrogen source and binder with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:11 was prepared at 90 °C for 2 h with continuous stirring. Then, a 2 kg mixture composed of semolina (48 wt% of the mixture) and the previously prepared glucose, urea, and water mixture (8 wt% of the mixture) and ZnO nanoparticles (20 nm) as a porogen (24 wt% of the mixture) were mixed together using a commercial kitchen kneader (Bosch, professional home series, model MUMVH48BCN). To provide a consistency that allows for extrusion, 160 mL water (20 wt% of the mixture) was slowly added to the mixture as a plasticizer. Finally, the low-moisture mixture was extruded and cut in the pellet form with dimensions of 1 mm (diameter) and 1.5 mm (length) using a commercial noodle extruding machine (La Monferrina P3) equipped with a 1 mm PTFE die and automatic cutting knife, cf. Fig. S1 in the ESI† and the video of the extrusion process. The extruded pellets were dried at room temperature for 12 h. The dried pellets were carbonized at 950 °C under N2 atmosphere using a three step program: (i) purging the oven atmosphere with N2 at room temperature for 30 min; (ii) increasing the temperature to 120 °C with the heating rate of 3 °C min−1 and maintaining it for 2 h; this step is to promote crosslinking;63 (iii) elevating the temperature to 950 °C with the heating rate of 3 °C min−1 and maintaining it for 2 h, then cooling it down to room temperature. Finally, the carbonized pellets (Fig. S2 in the ESI†) were washed using 0.1 M HCl solution (15 mL g−1 of NDC) under continuous stirring for 10 h to remove the residual Zn from the pellet.
:3:11 was prepared at 90 °C for 2 h with continuous stirring. Then, a 2 kg mixture composed of semolina (48 wt% of the mixture) and the previously prepared glucose, urea, and water mixture (8 wt% of the mixture) and ZnO nanoparticles (20 nm) as a porogen (24 wt% of the mixture) were mixed together using a commercial kitchen kneader (Bosch, professional home series, model MUMVH48BCN). To provide a consistency that allows for extrusion, 160 mL water (20 wt% of the mixture) was slowly added to the mixture as a plasticizer. Finally, the low-moisture mixture was extruded and cut in the pellet form with dimensions of 1 mm (diameter) and 1.5 mm (length) using a commercial noodle extruding machine (La Monferrina P3) equipped with a 1 mm PTFE die and automatic cutting knife, cf. Fig. S1 in the ESI† and the video of the extrusion process. The extruded pellets were dried at room temperature for 12 h. The dried pellets were carbonized at 950 °C under N2 atmosphere using a three step program: (i) purging the oven atmosphere with N2 at room temperature for 30 min; (ii) increasing the temperature to 120 °C with the heating rate of 3 °C min−1 and maintaining it for 2 h; this step is to promote crosslinking;63 (iii) elevating the temperature to 950 °C with the heating rate of 3 °C min−1 and maintaining it for 2 h, then cooling it down to room temperature. Finally, the carbonized pellets (Fig. S2 in the ESI†) were washed using 0.1 M HCl solution (15 mL g−1 of NDC) under continuous stirring for 10 h to remove the residual Zn from the pellet.
Ni with 35 wt% as the aimed loading was incorporated on NDC via incipient wetness impregnation. Later, the 35Ni/NDC was calcined in the presence of N2 atmosphere. Finally, and prior to the catalytic test, the catalyst was reduced under the flow of the forming gas (more details of the incipient wetness impregnation can be found in the ESI†).
Characterization of NDC and 35Ni/NDC
The prepared parent NDC, and fresh and used 35Ni/NDC were characterized using N2 sorption, elemental analysis, scanning electron microscopy (SEM), transmission electron microscopy (TEM), thermogravimetric analysis (TGA), X-ray diffraction (XRD), X-ray photoemission spectroscopy (XPS), and CO-temperature programmed desorption (CO-TPD). The details of characterization procedures are reported in the ESI.†Catalytic evaluation
Catalytic experiment
In a typical experiment, 1 wt% of an aqueous solution of glucose (CGlu = 56 mM), xylose (CXyl = 64 mM), or vanillin (CV = 72 mM) were fed via the HPLC pump at different liquid flow rates of 0.3, 0.5, 0.7, 1.0, or 3.0 mL min−1, mixed with an excess amount of H2 (QH2 = 15 mL min−1 at a pressure of 2.5 MPa), and introduced into the preheating unit prior to the fixed bed reactor. 1 g of the catalyst pellets were fixed between 0.1 g of quartz wool. The reactor temperature and pressure were kept at ambient values for 15 min. Later, the system pressure was adjusted to 2.5 MPa and the temperature was increased to the targeted values, i.e., 80 °C, 100 °C, 125 °C, 135 °C, and 150 °C. The samples (ca. 2 mL) were collected once the steady state was reached after ca. 30 min.Caution: Each time of stream (TOS) experiment was conducted in 4 consecutive days (ca. 10 h each day) without removing the catalyst from the tubular reactor and applying any post treatment on the catalyst.
Product analysis was performed using HPLC Agilent 1200 series equipped with a quaternary pump, diode array detector (DAD), and refractive index detector (RID). The analysis procedure and quantification of the reaction products are described in detail in the ESI.†
Results and discussion
Catalyst preparation and characterization
The Ni catalyst (35 wt%) supported on porous nitrogen-doped carbon (NDC) pellets was prepared via incipient wetness impregnation. The parent support (NDC) and 35Ni/NDC were characterized and compared via elemental analysis, N2 sorption, scanning electron microscopy (SEM), transmission electron microscopy (TEM), powder X-ray diffraction (XRD), thermogravimetric analysis (TGA), temperature programmed desorption (TPD), and X-ray photoemission spectroscopy (XPS).Ni is a sustainable metal for hydrogenation reactions, which, supported on NDC, possesses a high catalytic performance.32,65,66 NDC, as a support, has already been reported to change the chemical behaviour of metal nanoparticles compared to pristine carbon supports.31,32,66–68 The presence of N makes the work function of the support more positive and promotes a partial charge transfer from the metal to the support, establishing a Mott–Schottky heterojunction.67 This effect is proposed to result in strong Ni nanoparticles-support interaction, as shown in Fig. 2. Moreover, the charge transfer between Ni nanoparticles (δ+) leads to strongly “bonded” nanoparticles on the support, which is due to Coulomb interaction but also due to better wetting of the metal to the support. This, as such, gives to the Ni a higher reductive character and resistance against oxidation against leaching.30,32,67,69
 | ||
| Fig. 2 Proposed charge-transfer stabilization effect between Ni nanoparticles and NDC (left) in comparison to a pristine carbon support (Ni/C). | ||
Herein, we present the first application of pelletized “Mott–Schottky” catalyst for the hydrogenation of biomass-derived compounds in water. Beyond this specific application, this type of catalyst is considered to be suitable for biorefinery processes at a large scale and in a broader context, as it is cheap and scalable and, in the present case, even based on largely re-grown sustainable carbon sources. In the novel “kitchen-lab” synthesis of 35Ni/NDC, we directly produce pellets that are suitable for continuous flow systems. The synthetic protocol follows traditional pasta manufacturing (kg scale) and starts with a cheap and abundant carbon source, viz., semolina, and ZnO nanoparticles (broadly available as a UV pigment or baby powder) that are used as a porogen. Semolina and a mixture of urea/glucose have been selected as “publicly available” carbon and nitrogen precursors. Semolina is relatively rich in protein (12 wt%) and carbohydrates (71 wt%), cf.Table 1.
| Catalyst | C/wt% | N/wt% | C/N ratio | Nia/wt% | Zna/wt% | A BET/m2 g−1 |
A
mesob/m2 g−1 |
V
mesob/cm3 g−1 |
V p/cm3 g−1 |
A
Nic/m2 g−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| n.a.: Not applicable.a Measured via ICP-OES.b Calculated via QSDFT.c Measured via CO-TPD.d Normalized to C. | ||||||||||
| Dried NDC Precursor | 38 | 5.2 | 13 | n.a. | 34 | n.a. | n.a. | n.a | n.a | n.a. |
| NDC | 80 | 3.5 | 23 | n.a. | <0.2 | 827 | 99 | 0.30 | 0.57 | n.a. |
| 35Ni/NDC | 53 | 2.9 | 18 | 35 | <0.2 | 700 | 92 | 0.24 | 0.42 | 21 |
| 35Ni/NDC_UGlu | 56 | 2.8 | 20 | 31 | <0.2 | 730 | 90 | 0.25 | 0.51 | 14 |
| 35Ni/NDC_UXyl | 52 | 2.7 | 19 | 29 | <0.2 | 799 | 113 | 0.26 | 0.55 | 14 |
| 35Ni/NDC_UV | 59 | 2.4 | 24 | 30d | <0.2 | 125 | 70 | 0.08 | 0.14 | 14 |
In addition, the glucose–urea mixture has been reported as a low price, processable liquid precursor for carbon.70 Moreover, ZnO has been previously utilized as a versatile templating agent since it can be simply removed via thermal treatment at high temperatures (>900 °C) through ZnO carbothermal reduction to Zn, which evaporates at that temperature (Fig. 3).71,72 After the carbonization step, porous NDC pellets were obtained with a mass yield of 30 wt%. The elemental composition of porous NDC is listed in Table 1. After the Ni impregnation, a content of 35 wt% of Ni was obtained (characterized via ICP-OES). Upon carbonization and catalyst activation, the C/N ratio was found to be constant (∼20), cf., Table 1. The N2 sorption isotherm of the parent carbonized NDC and 35Ni/NDC is presented in Fig. 4, representing type IV, which is characteristic of mesoporous materials. As expected upon Ni impregnation, the specific surface area (ABET) and specific pore volume (Vp) decreased from 827 m2 g−1 to 700 m2 g−1 and 0.57 cm3 g−1 to 0.42 cm3 g−1, respectively. The reduction in the textural properties is attributed to the change in the mass density of the sample, as well as the coverage of the support surface and pores with Ni.73 However, the specific surface area of the mesoporous material was found to be (∼100 m2 g−1), cf.Table 1.
 | ||
| Fig. 3 Schematic representation of the catalyst synthesis protocol starting from the extrusion, carbonization, calcination, and reduction steps. | ||
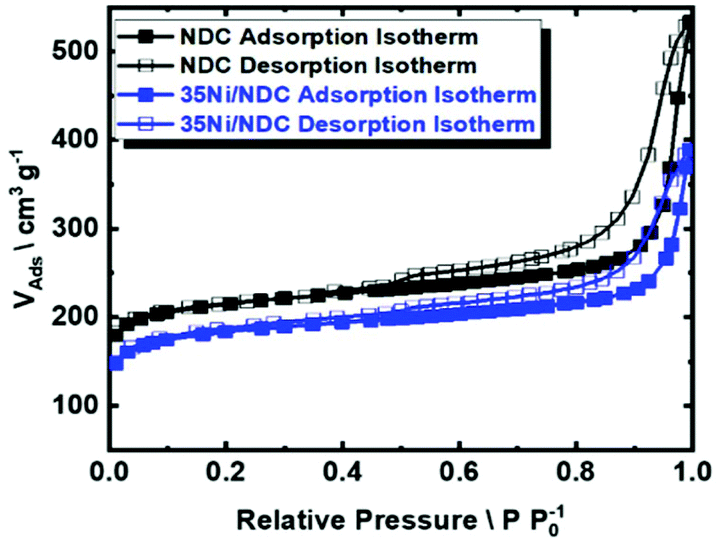 | ||
| Fig. 4 N2 sorption isotherm at 77 K of the parent NDC and 35Ni/NDC. | ||
The XRD patterns of parent NDC and freshly reduced 35Ni/NDC showed a main broad reflection at 2θ of 25°, which corresponds to the typical reflection of turbostratic carbon (002), as shown in Fig. 5.74 The absence of any Zn phase indicates the efficient removal of the template. The freshly reduced 35Ni/NDC presents only typical reflections of Ni0 at 2θ of 44° and 51°, which indicate the complete reduction of NiO after calcination and reduction processes.73
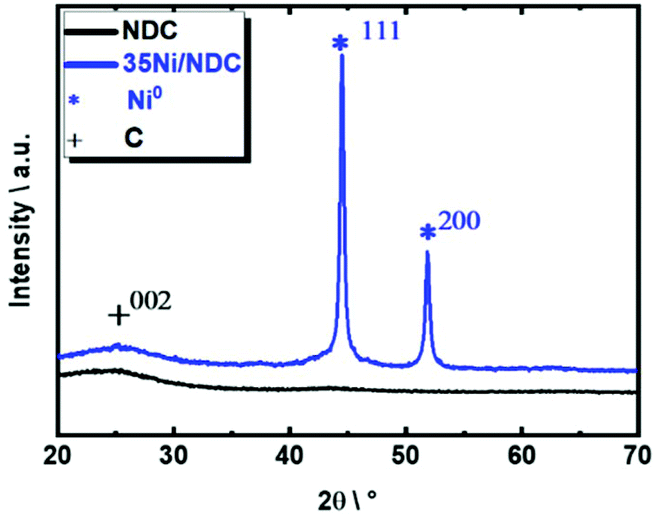 | ||
| Fig. 5 XRD patterns recorded for the parent NDC and freshly reduced 35Ni/NDC. | ||
The images obtained via high resolution scanning transmission election microscopy (HR-STEM) and scanning electron microscopy (SEM) are shown in Fig. 6 and Fig. S5–S7 ESI† show the support morphology and the Ni nanoparticles on the parent NDC and 35Ni/NDC. The comparison between NDC and 35Ni/NDC revealed that the Ni nanoparticles are homogenously dispersed on the surface of NDC with an average nanoparticle size of 25 nm. This is in good agreement with the crystallite diameter calculated using the Scherrer equation from the XRD pattern. In addition, the EDX mapping for C, N, and Ni, as shown in Fig. 7 and S6,† shows that N is homogeneously distributed on the NDC support. The XRD results and microscopy imaging results are in good agreement with the results obtained from CO-TPD showing a high Ni surface area (21 m2 g−1) with crystallite size of 21 nm. Furthermore, X-ray photoelectron spectroscopy (XPS) results of the binding state of N (1s) bond for both parent NDC and 35Ni/NDC showed the presence of N-pyridinic, N-pyrrolic, N-graphitic, and N-oxide species (Fig. S8 in the ESI†). However, due to overlap of different N contributions, a clear quantitative deconvolution of these peaks was not possible. The XPS of Ni (2p3/2) for freshly reduced 35Ni/NDC exhibited the presence of Ni0 and partly oxidized Ni (Fig. S9 in the ESI†). The presence of NiO on the outer layer of Ni0 is attributed to the exposure of the catalyst to air after the reduction step. This assumption is in good agreement with HR-STEM coupled with EDX in which low O amount (11 at%) was found on the core of Ni nanoparticles (cf. EDX quantification in Table S1 in the ESI†). However, an accurate surface quantitative analysis of the Ni species by XPS was not possible due to signal interference from different Ni species contributions.
 | ||
| Fig. 6 (A) HR-STEM and (B) SEM picture of 35Ni/NDC. | ||
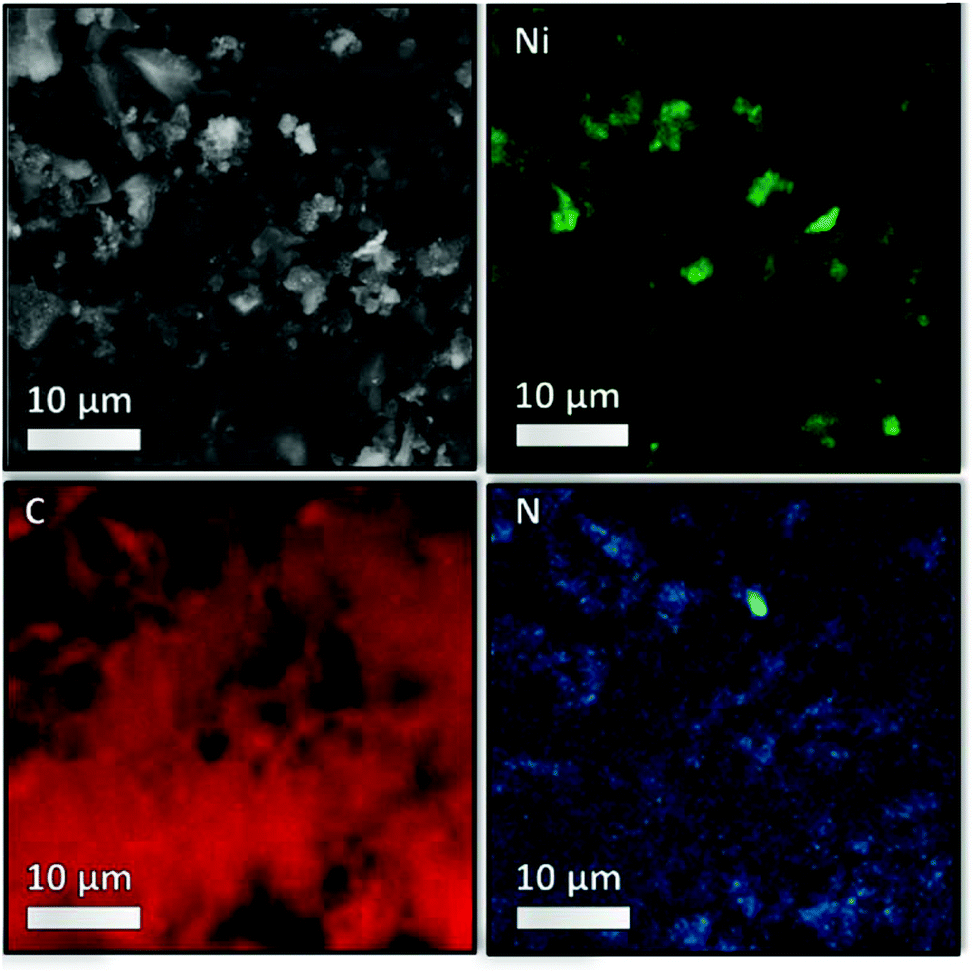 | ||
| Fig. 7 EDX mapping of 35Ni/NDC crushed pellets placed on a conductive carbon tape. In black and white, the selected area for mapping, in green, red, and blue are Ni, C, and N, respectively. The black area in the C map is the shadowing effect due to the sample thickness. | ||
Performance of 35Ni/NDC in the aqueous-phase hydrogenation of biomass-derived compounds
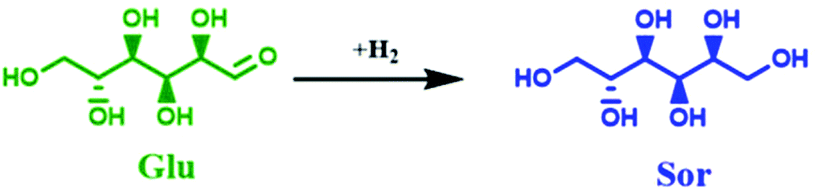 | ||
| Scheme 1 The aqueous-phase hydrogenation of Glucose (Glu) into Sorbitol (Sor). | ||
Firstly, a blank experiment with no catalyst at 150 °C showed a low XGlu (9.6 mol%), cf. ESI, Fig. S10.† In addition, fructose (Fru) was the only detected product with YFru of 5.3 mol%, which is formed via Glu isomerization in water under the utilized reaction conditions (Scheme 1). The unidentified products (∼4.3 mol%) can be correlated to the formation of insoluble humins in the presence of water under these reaction conditions.75,76 Furthermore, only the presence of the support (NDC) improved XGlu from 9.6 mol% to 29.3 mol% without a significant change in YFru (6.2 mol%), cf. ESI, Fig. S10,† which is related to the increased formation of insoluble humins (∼23 mol%). Accordingly, the presence of porous NDC accelerates the dehydration and humification process of Glu even in the absence of active Ni0 hydrogenation sites. The effect of the reaction temperature on the performance of 35Ni/NDC as well as on the formation of insoluble humins was investigated at three different reaction temperatures, i.e., 100 °C, 125 °C, and 150 °C. As expected, the presence of 35Ni/NDC improved Glu conversion at 150 °C with respect to the experiment conducted with no catalyst and with only the use of NDC (Fig. 8 and Fig. S10 in the ESI†), yielding a XGlu of 97 mol%, which corresponds to a glucose space time conversion (STCGlu) of 0.23 mol h−1 molNi−1. Also, in the presence of Ni0 on NDC, Sor was the only identified product with a Sor space yield (STYSor) of 0.21 mol h−1 molNi−1 (Ysor = 89 mol%). Moreover, the formation of insoluble humins was drastically reduced (from ∼23 to ∼8 mol%) in comparison with the experiments conducted in the absence of Ni (only NDC).
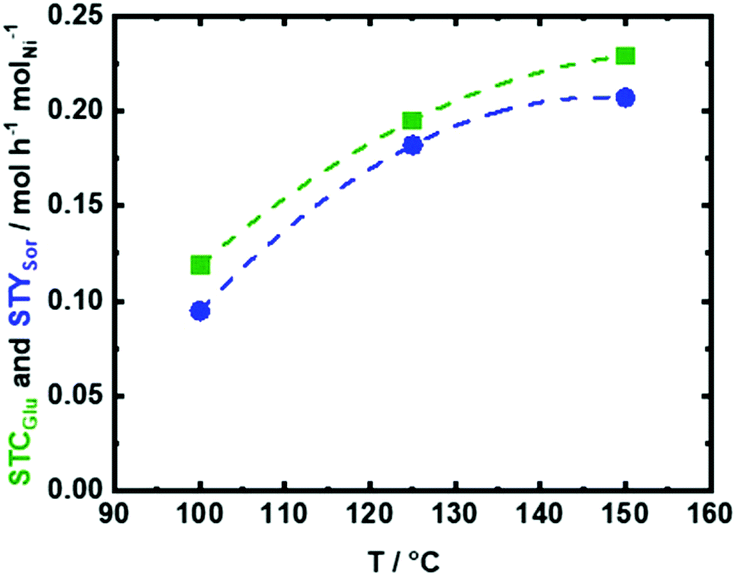 | ||
| Fig. 8 The space time conversion of Glu (STCGlu) and yield of Sor (STYSor) as a function of reaction temperature (T), using 35Ni/NDC in the aqueous-phase hydrogenation of Glu to Sor; reaction conditions: cGlu = 56 mM (1.0 wt%), T = 100 °C, 125 °C and 150 °C, Qeduct = 0.3 mL min−1 (τ = 4.2 h molNi molGlu−1), QH2 = 15 mL min−1 and p = 2.5 MPa. | ||
As expected that STCGlu and STYSor increase with temperature, elevating the reaction temperature from 100 °C to 125 °C and then to 150 °C is combined with an increase in STCGlu from 0.11 mol h−1 molNi−1 (XGlu = 51 mol%) to 0.19 mol h−1 molNi−1 (XGlu = 89 mol%) and to 0.23 mol h−1 molNi−1 (XGlu = 97 mol%), respectively (Fig. 8). Similarly, STYSor increased from 0.10 mol h−1 molNi−1 (YSor = 46 mol%) to 0.18 mol h−1 molNi−1 (YSor = 84 mol%) and to 0.21 mol h−1 molNi−1 (YSor = 89 mol%), respectively (Fig. 8). These results showed that at the lower reaction temperature, i.e., 100 °C, the conversion is very low and hydrogenation does not proceed with high efficiency, while at 125 °C, there is an improvement in STCGlu from 0.11 mol h−1 molNi−1 to 0.19 mol h−1 molNi−1. A further increase in the temperature to 150 °C leads to almost complete conversion (XGlu = 97 mol%) and to the highest STYSor of 0.21 mol h−1 molNi−1. On the other hand, at low temperature, such as 100 and 125 °C, the formation of insoluble humins is very low (<5 mol%), while at 150 °C, the humification appears to be more pronounced (∼8 mol%). However, the amount of insoluble humins that were formed using a space time (τ) of 4.2 h molNi molGlu−1, i.e., a residence time of 15 min, is still very low in comparison to the reported data using a batch system (over 50 mol% at similar conditions).76
Due to its high STYSor and STCGlu, the reaction temperature of 150 °C was selected for further investigation corresponding to the aqueous-phase hydrogenation of Glu to Sor. The high STYSor and STCGlu values using 35Ni/NDC are attributed to the high specific surface area (700 m2 g−1) and specific pore volume (0.42 cm3 g−1) of the 35Ni/NDC catalyst. Moreover, it is correlated to the high amount of Ni (35 wt%, as proved by ICP-OES), which is homogenously and highly dispersed on the surface of NDC with a high specific Ni surface area (ANi) of 20 m2 g−1 derived from CO-TPD (see the SEM, TEM, and EDX images in Fig. 6, 7, and Table 1). Moreover, experiments were performed using 35Ni/NDC at 150 °C and at different space time (τ), i.e., 2.0, 2.8, and 4.2 h molNi molGlu−1, which correspond to the residence time of 6.4, 9.0, and 15 min, respectively (Fig. S11 and S12 in the ESI†). These experiments intend to demonstrate the key advantage in the continuous flow process, namely to control the contact times, which allows to avoid follow up reactions, e.g., formation of humins, and to maximise the STCGlu and STYSor as well as the selectivity towards the desired product, i.e., Sor. Shortening the space time from 4.2 to 2.8 and to 2.0 h molNi molGlu−1, the STCGlu is drastically increased from 0.22 to 0.34 and to 0.46 molGlu h−1 molNi−1 (Fig. S11†), analogously resulting increase in STYSor from 0.21 to 0.31 and to 0.39 molsor h−1 molNi−1. Despite the higher STCGlu and STYSor, the glucose conversion XGlu slightly decreased from 97 mol% to 95 mol% and then to 84 mol% (Fig. S12†).
This decrease in XGlu is attributed to the very short contact time between the reactant and the catalyst. A maximized XGlu (97 mol%) and YSor (89 mol%) was found at the space time (τ) of 4.2 h molNi molGlu−1. A shorter space time (τ), i.e., 2.0 h molNi molGlu−1 and 2.8 h molNi molGlu−1, showed only a linear decrease in the XGlu and YSor (Fig. S12†). Thus, it can be deduced that a longer residence time can be applied to ensure efficient catalytic performance. Interestingly, we observed only a little drop in the selectivity towards sorbitol from 91% to 89% and to 85%, when the residence time was shortened, from 4.2 to 2.8 and 2.0 h molNi molGlu−1, respectively. This corresponded to the presence of traces of unreacted Fru of ∼1 mol% and ∼4 mol% at 2.8 and 2.0 h molNi molGlu−1, respectively.
Therefore, the optimum reaction conditions namely (150 °C, 4.2 h molNi molGlu−1 of space time, 1.0 g of catalyst, H2 flow of 15 mL min−1, and pressure of 2.5 MPa) were applied to study the catalyst performance as a function of time on stream (TOS) for the conversion of Glu to Sor (Fig. 9). The catalyst performance from the perspective of XGlu was retained without any change within 35 h of TOS (STCGlu and STYSor > 0.20 mol h−1 molNi−1). In addition, only a slight drop in the selectivity of Sor was observed after 35 h of TOS (Fig. 9). Nevertheless, the STYSor persisted above 0.20 molsor h−1 molNi−1 (YSor > 79 mol%) after 40 h of TOS (Fig. 9). The observed decrease in STYSor can be correlated to the deposition of insoluble humins (∼10 wt%, as derived from TGA, viz., in Fig. S13 at ESI† and elemental analysis shown in Table 1) on the Ni active sites. Changing the work function of carbon by introducing nitrogen to the framework is intended to create a more stable catalyst under hydrothermal conditions. This promotes a charge transfer from Ni to carbon, which as a side effect strongly attracts Ni to the surface of NDC. Under the utilized reaction conditions and after 40 h on TOS, the Ni amount decreased from 35 wt% to only 31 wt%, which is relatively low. This leaching of Ni could be because the loading of 35 wt% is relatively high in which slight amount of Ni0 are weakly bonded to the NDC, i.e., not via Mott–Schottky transfer. However, it is a well-known phenomenon that Ni on other support materials is completely and rapidly leached off under similar hydrothermal conditions.40 We attribute this low amount of Ni leaching to the strong attraction of Ni to the support caused by the introduction of nitrogen into the NDC framework.
 | ||
| Fig. 9 The space time conversion of Glu (STCGlu) and yield of Sor (STYSor) as a function of time on stream (TOS) using 35Ni/NDC in the aqueous-phase hydrogenation of Glu to Sor; reaction conditions: cGlu = 56 mM (1.0 wt%), T = 150 °C, Qeduct = 0.3 mL min−1 (τ = 4.2 h molNi molGlu−1), QH2 = 15 mL min−1 and p = 2.5 MPa. | ||
The spent 35Ni/NDC_UGlu system was studied using powder XRD to investigate the crystal structure (Fig. S14 at ESI†). Although the main reflections that are typical of Ni0 at 2θ of 44° and 51° were preserved, an additional pattern showed reflections at 2θ of 19°, 33°, 38°, and 52°, which indicates the formation of hydroxide species as Ni(OH)2 and NiOOH.77 This oxidation of nickel is due to the exposure of the catalyst to hot water within the washing process and cooling of the system after reaction completion. The same can also be due to drying of the catalytic system in air at 80 °C before post characterization. The XPS analysis for Ni species of the spent catalyst, which is labelled as 35Ni/NDC_UGlu, confirmed the presence of mostly oxidized species on the surface of the catalyst. This is attributed to the hydroxide formation. This is a common observation for Ni supported catalysts that are present in moisture conditions (Fig. S9 in the ESI†).77 Additionally, it showed no change in the position of N (1s) in the spent 35Ni/NDC with respect to that in Fig. S7.†
The morphology of the spent catalyst represents similar morphology of the fresh catalyst (Fig. S15 in the ESI†). In spite of formation of humins, no significant changes in the textural properties were observed after 40 h of TOS with respect to the fresh catalyst (Table 1 and Fig. S16 in the ESI†). The specific surface area ABET slightly increased from 700 m2 g−1 to 730 m2 g−1 but only due to contribution from micropores (Fig. S16 in the ESI†). This can be correlated to the higher availability of micropores due to minimal leaching of Ni and/or to the change in the material density. After 40 h of TOS, CO-TPD showed a slight reduction in the Ni specific surface area from 21 m2 g−1 to 14 m2 g−1 that can be correlated to the slight Ni leaching from 35 wt% to 31 wt%, deposition of carbonaceous species, and agglomeration of weakly bonded Ni (Table 1).
After 40 h of TOS, the XRD pattern and XPS of the spent catalyst showed the presence of Ni oxidized species on the surface of NDC. Therefore, an attempt to regenerate Ni0 in the spent catalyst was performed (see the detailed reactivation procedure in the ESI†). Indeed, Ni0 was successfully restored via the regeneration procedure, which was confirmed by XRD and HR-STEM. These characterization results showed that Ni0 was the dominant phase on the surface of the catalyst (Fig. S17 and S18 in the ESI†). Throughout the manuscript, the regenerated catalyst is given the code 35Ni/NDC_Rx.
Finally, as a proof of concept, the catalytic activity of 35Ni/NDC and synthesized 35 wt% Ni in commercially available carbon black (CB) in Glu hydrogenation to Sor under the optimum reaction conditions was compared. The results of using 35Ni/CB were worse with respect to 35Ni/NDC (Fig. S19 in the ESI†). This can be an advantage of using Ni on heteroatom doped carbon. This is consistent with the findings of Nie et al.62 for the aqueous-phase hydrogenation of biomass-derived compounds.
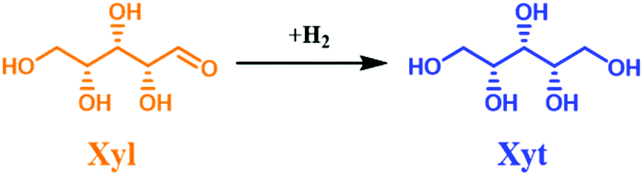 | ||
| Scheme 2 Hydrogenation of xylose (Xyl) to xylitol (Xyt). | ||
The influence of reaction temperature and space time (τ) on the catalyst performance was studied. Also, the catalyst performance at longer TOS was evaluated. Similar to the glucose experiment, a blank experiment was conducted at 150 °C without the catalyst (Fig. S20 in the ESI†), which yielded 18 mol% loss of Xyl and no Xit was detected. This indicated the occurrence of a humification process. A higher conversion of XXyl (40 mol%) was found using parent NDC, while no Xyt was formed. In this case, Xyl simply converted into insoluble humins, the process being catalyzed by the available high specific surface area in the parent NDC. The formed humins’ fractions are larger than in the case of glucose (Fig. S10 and S20 in the ESI†). This is due to the fact that under hydrothermal conditions and at high temperature (150 °C), Xyl is less stable than Glu. However, the humins’ formation in both cases in the continuous flow system remains lower than similar experiments conducted in a batch system.76 After adding 35 wt% Ni to NDC, the catalytic hydrogenation performance improved drastically, yielding a STCXyl of 0.28 molXyl h−1 molNi−1 and STYXyt of 0.27 molXyt h−1 molNi−1, which correspond to XXyl = 99 mol% and YXit = 97 mol%, respectively, cf. ESI, Fig. S20.† In other words, we found almost quantitative conversion of Xyl to Xyt. The effect of reaction temperature on the catalytic performance of 35Ni/NDC was studied at 100 °C, 125 °C, and 150 °C (Fig. S21†). A linear increase in the STCXyl and STYXyt was found by increasing the reaction temperature to 100 °C, 125 °C, and 150 °C (Fig. S21 in the ESI†). The higher reactivity of Xyl with respect to Glu is attributed to its higher instability when exposed to high temperature.
Also, the space time study confirmed the high activity of 35Ni/NDC for Xyl reduction to Xyt. In fact, even at a short space time (1.6 h molNi molXyl−1), XXyl was 92 mol% and YXyt was 88 mol% (cf. Fig. S22 in the ESI†). Shortening the space time from 3.6 to 2.2 h molNi molXyl−1 did not affect the selectivity towards Xyt (>99%). Applying a shorter contact time (1.6 h molXyl molNi−1), a negligible drop in the Xyt yield was observed from >99 mol% to 95 mol%. This is attributed to traces of some undetected isomerization product such as xylulose. Therefore, likewise for glucose, the optimized conditions were 150 °C and 3.6 h molXyl molNi−1, corresponding to a residence time of 15 min.
35Ni/NDC was exposed for 42 h of TOS at the abovementioned optimum reaction conditions (Fig. 10), which showed high catalytic performance with excellent stability. This exceptional stability is attributed to the heterojunction effect due to the presence of N doping in carbon. Identical to the reported characterization of the spent catalyst in Glu hydrogenation, the textural properties of 35Ni/NDC_UXyl were preserved over long TOS. Thus, only a little Ni leaching from 35 wt% to 29 wt% was found (Table 1). In addition, the specific surface area showed a slight increase due to the deposition of a slight amount of humin (>4 wt% derived from TGA) on the surface of the catalyst (viz. Fig. S17† and Table 1). Also, in this case, the XRD pattern of the spent 35Ni/NDC_UXyl and the regenerated 35Ni/NDC_RXyl showed the formation of Ni-hydroxide species after the reaction and their further reduction to Ni0 (Fig. S14 and S18 in the ESI†). Moreover, the CO-TPD analysis yielded the reduction of the specific Ni surface area ANi from 20 m2 g−1 to 14 m2 g−1 (Table 1) with preserved crystallite size (19 nm). This very similar behaviour of the two 35Ni/NDC_UXyl and 35Ni/NDC_UGlu confirmed the robustness of 35Ni/NDC at such long TOS and after regeneration treatment.
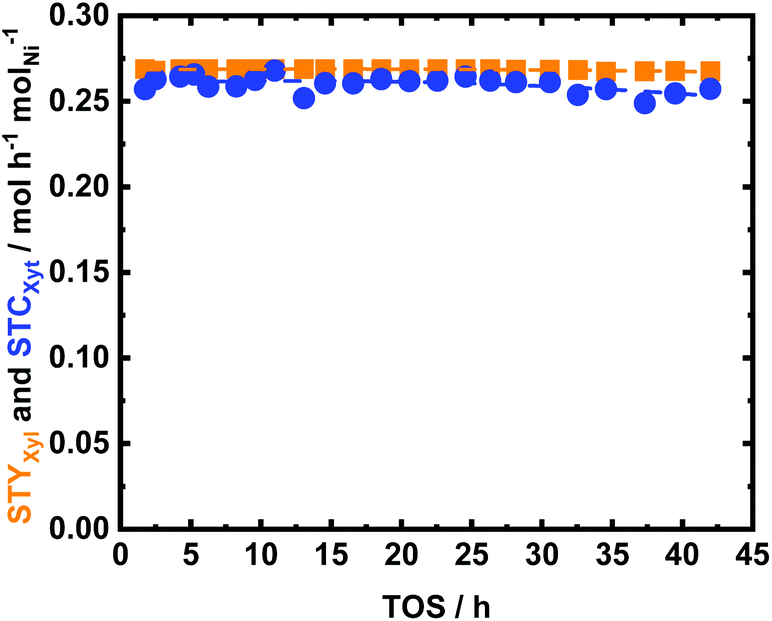 | ||
| Fig. 10 The space time conversion of Xyl (STCXyl) and space time yield of Xyt (STYXyt) as a function of time on stream (TOS) using 35Ni/NDC in the aqueous-phase hydrogenation of Xyl to Xyt; reaction conditions: cXyl = 64 mM (cXyl = 1 wt%), T = 150 °C, Qeduct = 0.3, mL min−1 (τ = 3.6 h molNi molXyl−1), QH2 = 15 mL min−1 and p = 2.5 MPa. | ||
 | ||
| Scheme 3 Hydrogenation of vanillin (V) to 2-methoxy-4-methylphenol (MPP) through vanillyl alcohol (VA). | ||
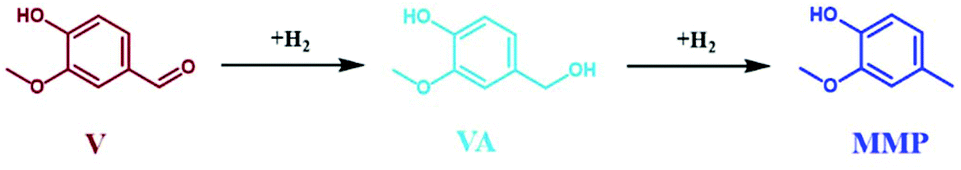
The influence of reaction temperature on the space time conversion of V (STCV) and on the product distribution was investigated by applying five different reaction temperatures (80 °C, 100 °C, 125 °C, 135 °C, and 150 °C). The complete STCV to VA and MMP was obtained at over 100 °C (Fig. 11). It can be clearly seen that at the reaction temperature of 125 °C and above, the reaction equilibrium is shifted towards the formation of MMP, which is the consecutive product of VA hydrogenation (Scheme 3). Interestingly, the experiment utilized a low reaction temperature (80 °C), resulting in a high space time yield of VA (STYVA) of 0.22 molVA h−1 molNi−1 (YVA = 77 mol%) and low STYMMP of 0.04 molMMP h−1 molNi−1 (YMMP = 16.9 mol%), cf., Fig. 11. A similar interplay between VA and MMP is in agreement with similar studies reported in a batch system using Ni, Pd, and Ru catalyst by altering the reaction time.59,62,80 Therefore, it can be deduced that the prepared catalyst exhibited high efficiency on V hydrogenation to VA and/or MMP. Since varying the temperature has been reported to show a change in the main product from MMP to VA, the same has been researched by varying the space time at 150 °C (Fig. S24†). In this context, the space time was found to be less effective in tuning the reaction products with respect to the reaction temperature. However, it is possible to see that by altering the space time by 1 order of magnitude, i.e., from 3.4 h molNi molV−1, which is equal to 15 min of residence time, to 0.35 h molNi molV−1 that corresponds to 1 min of residence time; the STYVA increased to 0.95 molVA h−1 molNi−1 (YVA from 0 to 33 mol%), cf. Fig. S24 in the ESI.† These results showed that the tunability of reaction products is also possible by reducing the residence time from minutes to potentially seconds in a low reaction temperature range. In addition, this indicates that using a flow system also allows the optimization and potential isolation of the intermediate product.
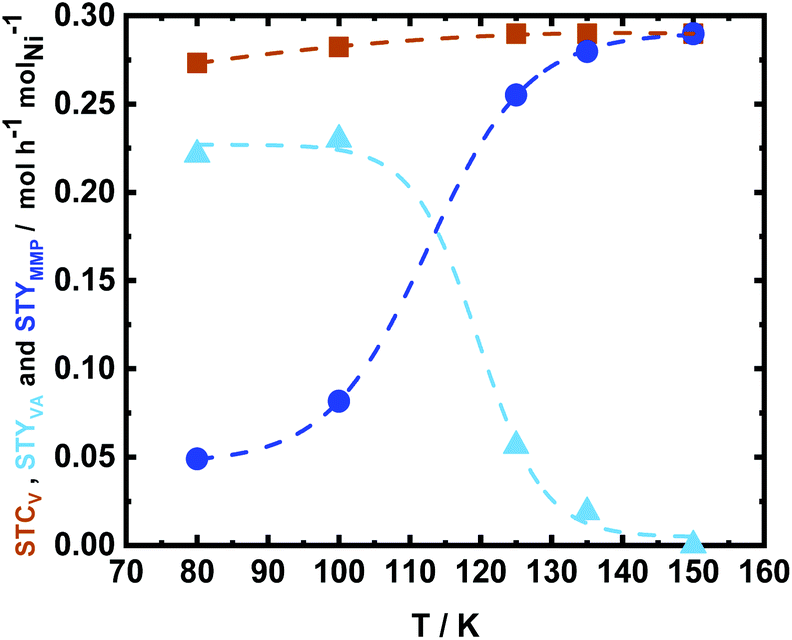 | ||
| Fig. 11 The space time conversion of V (STCV), the space time yield of VA (STYVA) and space time yield of MMP (STYMMP) as a function of temperature (T) using 35Ni/NDC in the aqueous-phase hydrogenation of V to VA and MMP; reaction conditions: cV = 72 mM (1.0 wt%), T = 80 °C, 100 °C, 125 °C, 135 °C, and 150 °C, Qeduct = 0.3 mL min−1 (τ = 3.4 h molNi molV−1), QH2 = 15 mL min−1, and p = 2.5 MPa. | ||
The stability of the catalyst was investigated under the optimized reaction conditions, in which V was quantitatively converted to MMP. Also, in this case, the catalyst was sustained after 40 h of time of stream (Fig. 12). In fact, the utilized catalyst represented a high catalytic performance, i.e., STCV of 0.29 molV h−1 molNi−1 (XV = 100 mol%) and STYMMP of 0.28 molV h−1 molNi−1 (YMMP > 98 mol%) after 40 h of TOS. Contrary to what was observed in the case of Glu and Xyl hydrogenations, the textural properties of the spent catalyst revealed a significant decrease in the specific surface area (ABET) from 700 m2 g−1 to 125 m2 g−1. This reduction is mainly attributed to a drop in the micropores, as shown in Fig. S17 in the ESI.† This is a common phenomenon for porous carbon in the presence of aromatic species.32 Therefore, the decrease in the specific surface area can be explained as pore blocking due to strong interaction between the aromatics and the NDC surface. This is confirmed from the elemental analysis of the spent catalyst in which increase in C from 53 wt% to 59 wt% was noted when compared to the fresh 35Ni/NDC (cf.Table 1). However, this deposition did not affect the catalyst performance since the STCV and STYMMP were extremely stable over 40 h of TOS. This constant activity is correlated to the presence of highly dispersed Ni and the remaining efficient mesopores that allow this reaction to proceed without noticeable deactivation.
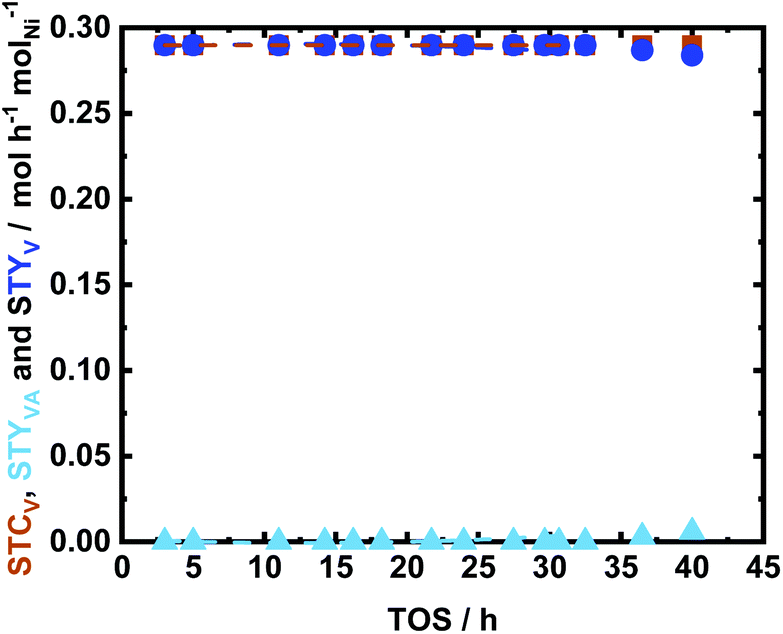 | ||
| Fig. 12 The space time conversion of V (STCV), the space time yield of VA (STYVA) and space time yield of MMP (STYMMP) as a function of time on stream (TOS) using 35Ni/NDC in the aqueous-phase hydrogenation of V to MMP; reaction conditions: cV = 72 mM (1.0 wt%), T = 150 °C, Qeduct = 0.3 mL min−1 (τ = 3.4 h molNi molV−1), QH2 = 15 mL min−1, and p = 2.5 MPa. | ||
Moreover, Ni leaching was minimal and found to be 30% after 40 h TOS, which is similar to the values reported for 35Ni/NDC_UGlu and 35Ni/NDC_UXyl (Table 1). Consistent with that of the spent catalyst in Glu and Xyl hydrogenation, a slight reduction in the Ni specific surface area from 21 m2 g−1 to 14 m2 g−1 was observed, which is correlated to slight Ni leaching from 35 wt% to 31 wt%, deposition of carbonaceous species, and agglomeration of weakly bonded Ni (Table 1). Again, Ni0 was successfully regenerated from the used catalyst and this was confirmed by XRD reflections, which showed the re-reduction of Ni from Ni2+ to Ni0 (Fig. S14 and S18 in the ESI†). In addition, both XRD and CO-TPD of the regenerated sample showed similar Ni crystallite size of ∼20 nm.
Finally, the robustness of this catalyst was confirmed one more time, extending its usage from the valorization of sugar fraction of the biomass to the aromatic fraction. In all the cases, the catalyst was sustained after 40 h of TOS with high catalytic performance and was successfully regenerated.
Conclusions
A simple, cheap, sustainable, scalable, efficient, and stable Ni catalyst supported on highly porous nitrogen-doped carbon was produced in the pellet form. This simple approach requires only sustainable household starting products, a noodle machine, and an oven. The as-prepared catalyst exhibits high catalytic performance in the aqueous-phase hydrogenation of glucose, xylose, and vanillin in a continuous-flow system. The utilization of a continuous-flow system allows high control on the reactant conversion and product selectivity. In the case of aqueous-phase sugar hydrogenation, the reaction temperature is the most important factor that moderates the undesired humification process. In vanillin hydrogenation, the reaction temperature and residence time are the key factors that control the product selectivity between either vanillyl alcohol or 2-methoxy-4-methylphenol. In this reaction and under optimized conditions, the catalyst system could perform for 40 h on flow with practically no loss in its catalytic performance, which corresponds to 120 batch cycles with direct catalyst reuse. In other words, paraphrasing the space time yield concept, 1 kg of our catalyst can produce 7.7 kg of the targeted product (sorbitol, xylitol or 2-methoxy-4-methylphenol) in a day, without losing its activity. The phenomenon of rapid Ni leaching in water was massively slowed down by changing the electron density of the carbon by introducing nitrogen to the framework, which provides enhanced catalytic performance for long time on stream. Nevertheless, further development on the long-term operation of the catalyst, i.e., optimizing the support synthesis to increase the N content on the support and introduction of Ni on the support, are greatly required.Conflicts of interest
The authors have no conflicts of interests to declare.Acknowledgements
The authors are grateful for the financial support from Max Planck Society and Unifying System in Catalysis “UniSysCat” Cluster of Excellence Berlin-Potsdam. Thanks are also extended to Jessica Brandt, Antje Völkel, Rona Pitschke, and Heike Runge from Max Planck Institute of Colloids and Interfaces for ICP-OES, elemental analysis, TGA, and SEM and TEM measurements, respectively. The effort of the Electrical and Mechanical Workshops at the Max Planck Institute of Colloids and Interfaces is greatly acknowledged. Open Access funding provided by the Max Planck Society.References
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- N. Brun, P. Hesemann and D. Esposito, Chem. Sci., 2017, 8, 4724–4738 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- T. P. Vispute and G. W. Huber, Green Chem., 2009, 11, 1433–1445 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- X. L. Tong, Y. Ma and Y. D. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- R. Gérardy, R. Morodo, J. Estager, P. Luis, D. P. Debecker and J.-C. M. Monbaliu, Top. Curr. Chem., 2018, 377, 1 CrossRef.
- A. Hommes, H. J. Heeres and J. Yue, ChemCatChem, 2019, 11, 4671–4708 CrossRef CAS.
- R. Gérardy, N. Emmanuel, T. Toupy, V.-E. Kassin, N. N. Tshibalonza, M. Schmitz and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef.
- I. Sádaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- J. L. Fajín, M. N. D. Cordeiro, F. Illas and J. R. Gomes, J. Catal., 2010, 276, 92–100 CrossRef.
- T. Van Haasterecht, C. Ludding, K. De Jong and J. Bitter, J. Catal., 2014, 319, 27–35 CrossRef CAS.
- U. K. Singh, S. W. Krska and Y. Sun, Org. Process Res. Dev., 2006, 10, 1153–1156 CrossRef CAS.
- H. N. Pham, A. E. Anderson, R. L. Johnson, K. Schmidt-Rohr and A. K. Datye, Angew. Chem., Int. Ed., 2012, 51, 13163–13167 CrossRef CAS.
- R. M. Ravenelle, J. R. Copeland, W.-G. Kim, J. C. Crittenden and C. Sievers, ACS Catal., 2011, 1, 552–561 CrossRef CAS.
- R. M. Ravenelle, F. Schüβler, A. D'Amico, N. Danilina, J. A. Van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582–19595 CrossRef CAS.
- A. H. Van Pelt, O. A. Simakova, S. M. Schimming, J. L. Ewbank, G. S. Foo, E. A. Pidko, E. J. Hensen and C. Sievers, Carbon, 2014, 77, 143–154 CrossRef CAS.
- M. Antonietti, N. Lopez-Salas and A. Primo, Adv. Mater., 2019, 31, 1805719 CrossRef.
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 30, 1706836 CrossRef.
- S. M. G. Lama, J. Pampel, T. P. Fellinger, V. P. Beskoski, L. Slavkovic-Beskoski, M. Antonietti and V. Molinari, ACS Sustainable Chem. Eng., 2017, 5, 2415–2420 CrossRef CAS.
- H. Schiweck, A. Bär, R. Vogel, E. Schwarz, M. Kunz, C. Dusautois, A. Clement, C. Lefranc, B. Lüssem and M. Moser, Ullmann's encyclopedia of industrial chemistry, Wiley-VCH Verlag GmbH & Co, KGaA, 2000 Search PubMed.
- C. Marques, R. Tarek, M. Sara and S. Brar, in Platform Chemical Biorefinery, Elsevier, 2016, pp. 217–227 Search PubMed.
- M. Ahmed and B. Hameed, J. Taiwan Inst. Chem. Eng., 2019, 96, 341–352 CrossRef CAS.
- C. Dussenne, T. Delaunay, V. Wiatz, H. Wyart, I. Suisse and M. Sauthier, Green Chem., 2017, 19, 5332–5344 RSC.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- P. Barbaro, F. Liguori and C. Moreno-Marrodan, Green Chem., 2016, 18, 2935–2940 RSC.
- B. Garcia, J. Moreno, J. Iglesias, J. A. Melero and G. Morales, Top. Catal., 2019, 62, 570–578 CrossRef CAS.
- B. Kusserow, S. Schimpf and P. Claus, Adv. Synth. Catal., 2003, 345, 289–299 CrossRef CAS.
- E. Crezee, B. W. Hoffer, R. J. Berger, M. Makkee, F. Kapteijn and J. A. Moulijn, Appl. Catal., A, 2003, 251, 1–17 CrossRef CAS.
- J. Pan, J. Li, C. Wang and Z. Yang, React. Kinet. Catal. Lett., 2007, 90, 233–242 CrossRef CAS.
- D. K. Mishra, A. A. Dabbawala, J. J. Park, S. H. Jhung and J.-S. Hwang, Catal. Today, 2014, 232, 99–107 CrossRef CAS.
- A. Romero, E. Alonso, Á. Sastre and A. Nieto-Márquez, Microporous Mesoporous Mater., 2016, 224, 1–8 CrossRef CAS.
- P. A. Lazaridis, S. Karakoulia, A. Delimitis, S. M. Coman, V. I. Parvulescu and K. S. Triantafyllidis, Catal. Today, 2015, 257, 281–290 CrossRef CAS.
- R. M. Ravenelle, J. R. Copeland, A. H. Van Pelt, J. C. Crittenden and C. Sievers, Top. Catal., 2012, 55, 162–174 CrossRef CAS.
- D. Messou, L. Vivier, C. Canaff and C. Especel, Catalysts, 2019, 9, 146 CrossRef.
- X. Zhang, L. J. Durndell, M. A. Isaacs, C. M. Parlett, A. F. Lee and K. Wilson, ACS Catal., 2016, 6, 7409–7417 CrossRef CAS.
- L. Silvester, F. Ramos, J. Thuriot-Roukos, S. Heyte, M. Araque, S. Paul and R. Wojcieszak, Catal. Today, 2019, 338, 72–80 CrossRef CAS.
- M. Yadav, D. K. Mishra and J. S. Hwang, Appl. Catal., A, 2012, 425, 110–116 CrossRef.
- J. P. Mikkola, H. Vainio, T. Salmi, R. Sjoholm, T. Ollonqvist and J. Vayrynen, Appl. Catal., A, 2000, 196, 143–155 CrossRef CAS.
- J. C. Lee, Y. Xu and G. W. Huber, Appl. Catal., B, 2013, 140, 98–107 Search PubMed.
- D. K. Mishra, A. A. Dabbawala and J. S. Hwang, J. Mol. Catal. A: Chem., 2013, 376, 63–70 CrossRef CAS.
- H. M. Baudel, C. A. M. de Abreu and C. Z. Zaror, J. Chem. Technol. Biotechnol., 2005, 80, 230–233 CrossRef CAS.
- C. Hernandez-Mejia, E. S. Gnanakumar, A. Olivos-Suarez, J. Gascon, H. F. Greer, W. Z. Zhou, G. Rothenberg and N. R. Shiju, Catal. Sci. Technol., 2016, 6, 577–582 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- A. B. Bindwal and P. D. Vaidya, Energy Fuels, 2014, 28, 3357–3362 CrossRef CAS.
- H. A. Meylemans, T. J. Groshens and B. G. Harvey, ChemSusChem, 2012, 5, 206–210 CrossRef CAS.
- F. M. Zhang, S. Zheng, Q. Xiao, Y. J. Zhong, W. D. Zhu, A. Lin and M. S. El-Shall, Green Chem., 2016, 18, 2900–2908 RSC.
- J. L. Santos, M. Alda-Onggar, V. Fedorov, M. Peurla, K. Eranen, P. Maki-Arvela, M. A. Centeno and D. Y. Murzin, Appl. Catal., A, 2018, 561, 137–149 CrossRef CAS.
- S. C. Shit, R. Singuru, S. Pollastri, B. Joseph, B. S. Rao, N. Lingaiah and J. Mondal, Catal. Sci. Technol., 2018, 8, 2195–2210 RSC.
- R. F. Nie, H. H. Yang, H. F. Zhang, X. L. Yu, X. H. Lu, D. Zhou and Q. H. Xia, Green Chem., 2017, 19, 3126–3134 RSC.
- J. Gerrard, Trends Food Sci. Technol., 2002, 13, 391–399 CrossRef CAS.
- M. Al-Naji, B. Puertolas, B. Kumru, D. Cruz, M. Baumel, B. Schmidt, N. V. Tarakina and J. Perez-Ramirez, ChemSusChem, 2019, 12, 2628–2636 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS.
- Z. B. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. S. Yan, Nat. Commun., 2016, 7, 10141 CrossRef CAS.
- X. H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- Z. H. Xue, J. T. Han, W. J. Feng, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, Angew. Chem., Int. Ed., 2018, 57, 2697–2701 CrossRef CAS.
- H. Su, K. X. Zhang, B. Zhang, H. H. Wang, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, J. Am. Chem. Soc., 2017, 139, 811–818 CrossRef CAS.
- R. Rothe, M. Antonietti and N. Fechler, J. Mater. Chem. A, 2017, 5, 16352–16358 RSC.
- R. Yan, K. Leus, J. P. Hofmann, M. Antonietti and M. Oschatz, Nano Energy, 2019, 104240 Search PubMed.
- P. Strubel, S. Thieme, T. Biemelt, A. Helmer, M. Oschatz, J. Bruckner, H. Althues and S. Kaskel, Adv. Funct. Mater., 2015, 25, 287–297 CrossRef CAS.
- C. M. Mani, M. Braun, V. Molinari, M. Antonietti and N. Fechler, ChemCatChem, 2017, 9, 3388–3394 CrossRef CAS.
- K. P. Gierszal, M. Jaroniec, T. W. Kim, J. Kim and R. Ryoo, New J. Chem., 2008, 32, 981–993 RSC.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- N. Shi, Q. Y. Liu, R. M. Ju, X. He, Y. L. Zhang, S. Y. Tang and L. L. Ma, ACS Omega, 2019, 4, 7330–7343 CrossRef CAS PubMed.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 Search PubMed.
- Y. D. Arcaño, O. D. V. García, D. Mandelli, W. A. Carvalho and L. A. M. Pontes, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.07.060.
- T. Vangeel, W. Schutyser, T. Renders and B. F. Sels, Top. Curr. Chem., 2018, 376, 30 CrossRef.
- M. M. Li, J. Deng, Y. K. Lan and Y. Wang, ChemistrySelect, 2017, 2, 8486–8492 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03826d |
| This journal is © The Royal Society of Chemistry 2020 |