
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous photochemical benzylic bromination using in situ generated Br2: process intensification towards optimal PMI and throughput†
Alexander
Steiner
ab,
Jason D.
Williams
 ab,
Oscar
de Frutos
c,
Juan A.
Rincón
c,
Carlos
Mateos
c and
C. Oliver
Kappe
*ab
ab,
Oscar
de Frutos
c,
Juan A.
Rincón
c,
Carlos
Mateos
c and
C. Oliver
Kappe
*ab
aCenter for Continuous Flow Synthesis and Processing (CC FLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at; Web: http://goflow.at
bInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria
cCentro de Investigación Lilly S. A. Avda. de la Industria 30, 28108 Alcobendas-Madrid, Spain
First published on 10th December 2019
Abstract
The detailed development of photochemical benzylic brominations using a NaBrO3/HBr bromine generator in continuous flow mode is reported. Optimization of the bromine generator enables highly efficient mass utilization by HBr recycling, coupled with fast interphase transfer within a microstructured photochemical reactor (405 nm LEDs). Intensification of the reaction system, including complete removal of organic solvent, allowed a reduction in PMI from 13.25 to just 4.33. The photochemical transformation achieved exceptionally high throughput, providing complete conversion in residence times as low as 15 s. The organic solvent-free preparation of two pharmaceutically relevant building blocks was demonstrated with outstanding mass efficiency, by monobromination (1.17 kg scale in 230 min, PMI = 3.08) or dibromination (15 g scale in 20 min, PMI = 3.64).
Introduction
Benzyl bromides serve as important and frequently used building blocks towards target molecules in the pharmaceutical, agrochemical and materials industries (Fig. 1a). Their synthesis is generally performed by radical bromination of a toluene derivative, proceeding via thermally or photochemically generated bromine radicals.1 Carrying out these chemistries in a cost-effective, sustainable and safe manner is a topic of significant interest for both small and large scale applications. | ||
| Fig. 1 (a) A selection of Active Pharmaceutical Ingredients (APIs) containing building blocks accessible using photochemical benzylic bromination reactions; (b) the concept described here, for the photochemical benzylic bromination of toluene derivatives, using a Br2 generator in flow. | ||
The application of photochemistry can achieve the desired reactivity without the need for additional reagents (such as radical initiators) or high temperatures, as has been commonly employed.2 The scalability and productivity of photochemical methods such as these can be significantly enhanced by flow processing, which has the major advantage of providing uniform irradiation of a shorter path length, when compared to large scale batch processes.3 Indeed, many examples of photochemical benzylic bromination reactions have been reported in flow.4
The most frequently used bromine source in these reactions is N-bromosuccinimide (NBS), owing to its safe and convenient handling as a crystalline solid.4b–e These procedures are then, however, limited to comparatively low concentrations, restricted by the solubility of NBS, particularly when an excess of this reagent is required. The resulting reaction conditions can inevitably lead to significantly heightened solvent wastage, which is often a leading contributor to high process mass intensity (PMI)5 for an isolated reaction step.6 A multitude of other reagents have been employed for these reactions, such as CBrCl3,7 CBr48 and tribromoisocyanuric acid,9 but these generally suffer from similar issues of poor solubility and/or low reactivity.
The use of molecular bromine relieves these restrictions, allowing a faster rate of reaction and more concentrated operating conditions.4f Molecular bromine is, however, an exceedingly undesirable reagent to transport and handle.10 In keeping with the principles of green chemistry, a multitude of protocols report the formation of Br2in situ – generally by mixing an oxidant with a source of bromide ions, under acidic conditions. This generator concept has been demonstrated to provide excellent mass efficiency, in many cases even comparable to direct use of Br2.11
The most common oxidant for this role is H2O2,12 which is stored and used as an aqueous solution. However, there are safety concerns associated with the decomposition of such peroxides in storage. Alternatively, NaBrO3 is a crystalline solid, with a very high decomposition temperature of 310 °C,13 allowing safer transport and storage. Reports using this oxidant have hypothesized BrOH as an intermediate, which further reacts with bromide ion to form Br2.14
The oxidation of bromide is a highly exothermic process, particularly when concentrated conditions are used (achieving a lower PMI). Consequently, the use of flow processing is desirable for its vastly improved heat and mass transfer, which is especially important when considering larger volume processes.15 Surprisingly, considering its excellent suitability, only scarce reports exist in which a Br2 generator is utilized in flow.16
According to our interests in flow photochemistry, reaction intensification and chemical generator concepts, we sought to develop a highly optimized bromine generator system, for use in continuous flow photochemical benzylic bromination reactions (Fig. 1b). By conducting a thorough investigation with a focus on minimizing PMI, whilst maximizing throughput, we aimed to develop a safe protocol suitable for implementation at production scale.
Results and discussion
Initial optimization
Reaction optimization was initiated using 4-fluorotoluene (1) as a model substrate, which is a structural component of multiple Active Pharmaceutical Ingredients (APIs) such as the HIV treatment, raltegravir (Fig. 1a).17 It has previously been demonstrated to undergo photochemical benzylic bromination in flow, with a short residence time.4f Furthermore, the reaction performance could be quantified rapidly and effectively using benchtop 19F NMR (Magritek Spinsolve Ultra 43 MHz).18 After a screen of reaction solvents in batch, with 400 nm irradiation, it was found that aprotic solvents in general were favored.18 Chlorobenzene was selected as the most suitable solvent at this point, due to its good solubilizing properties and favorable health and environmental characteristics when compared to other chlorinated solvents.19Flow experiments were performed using a commercial glass plate-based flow photochemical reactor (Corning Lab Photo Reactor) with an irradiated volume of 2.8 mL, an internal mixing structure and a jacket for heat exchange fluid.20 The initial Br2 generator conditions were adapted from a previous flow methodology.16a Here, NaBr was added to solubilize the generated Br2 in the aqueous phase, as a 1 M solution, prior to mixing with the organic substrate stream. Inside the reactor plate, Br2 is extracted into the organic phase, whilst irradiation by 405 nm LEDs (56.8 W output power) initiates the radical bromination reaction (Fig. 2).
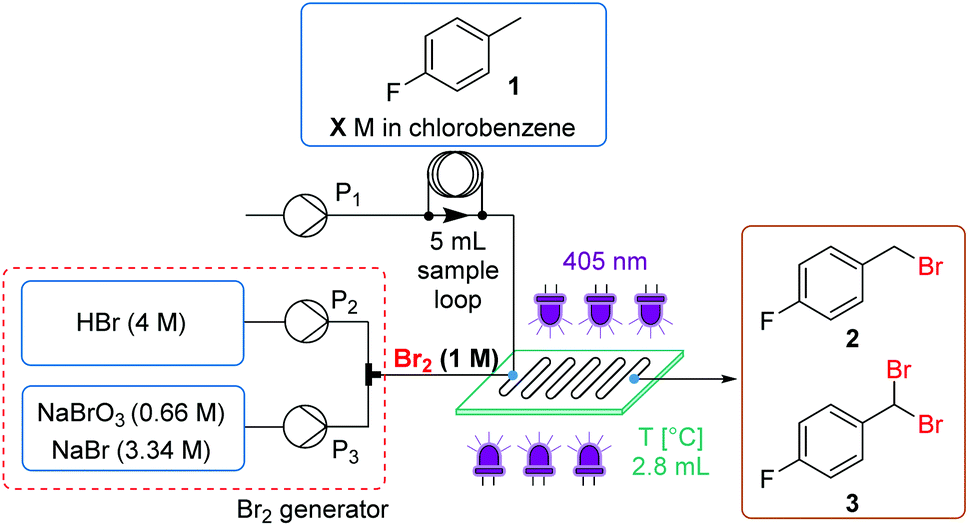 | ||
| Fig. 2 Initial experimental setup using 1st generation Br2 generator. | ||
Using this setup, the influence of residence time, concentration, temperature and Br2 equivalents on the product ratio was established (Table 1). High flow rates (short residence time) were expected to be required, to allow good mass transfer between the aqueous and organic phases.21 The result at 1 M concentration, 20 °C, 15 s (entry 1) showed a very similar product distribution to the optimal seen in batch experiments.18 Conversion of 81% starting material 1 was observed, yielding 73% of the desired monobrominated product 2 and 8% of the dibrominated side product 3.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Conc. [M] | t Res [sec] | Temp. [°C] | Br2 [equiv] |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [%] a [%] |
2a [%] |
3a [%] |
| a Product ratios were determined by benchtop 19F NMR. b Reaction was run without irradiation. | |||||||
| 1 | 1 | 15 | 20 | 1 | 19 | 73 | 8 |
| 2 | 2 | 15 | 20 | 1 | 57 | 42 | 1 |
| 3 | 4 | 15 | 20 | 1 | 93 | 7 | 0 |
| 4 | 4 | 30 | 20 | 1 | 69 | 31 | 0 |
| 5 | 4 | 60 | 20 | 1 | 20 | 74 | 6 |
| 6 | 4 | 60 | 5 | 1 | 47 | 52 | 1 |
| 7 | 4 | 30 | 50 | 1 | 23 | 70 | 7 |
| 8 | 4 | 60 | 50 | 2 | 0 | 25 | 75 |
| 9b | 4 | 60 | 50 | 1 | 100 | 0 | 0 |
It was found that the reaction is significantly slower under more concentrated conditions (entries 1–3). This is proposed to be because a higher substrate concentration results in a higher proportion of aqueous phase (larger aqueous slugs), which hinders mass transfer between the phases. In order to continue working at higher concentration, the residence time was extended to 30 s (entry 4), then to 60 s (entry 5), by which point the same optimal product ratio was observed again (entry 1 vs. entry 5).
A lower temperature of 5 °C resulted in a slower reaction, which still yielded a small amount of dibrominated product 3, implying that no significant improvement in selectivity is possible (entry 6). Increasing the temperature to 50 °C permitted the residence time to be reduced back to 30 s (entry 7). Upon increasing the quantity of Br2 to 2 equivalents (by altering the flow rate ratio of generator to substrate), the dibrominated product 3 could be favored, implying that under forcing conditions this could be obtained as the sole product (entry 8). The tribrominated product was not observed at any point, nor was any extent of (electrophilic) ring bromination. Finally, a control experiment was performed without light, to confirm that the reaction could not be promoted by the elevated temperature alone (entry 9).14a
The mono- versus dibromination selectivity of this transformation appears to be intrinsic to the substrate, as determined by the low energy barrier to reaction for both the first and second functionalization steps.4c Nevertheless, the most favorable conditions up until this point (Table 1, entry 7) provided an excellent productivity of 35 g h−1 (space–time yield = 12.5 kg L−1 h−1). On the other hand, a substantial quantity of waste is generated, with a PMI of 13.55 – the major contribution (10.73) arising from the Br2 generator.
Br2 generator optimization
With the aim of minimizing PMI, the bromine generator was identified as a major source of wastage. Accordingly, efforts were initiated to streamline the generator (Fig. 3a). The initial system had been developed to provide a homogeneous aqueous solution of bromine for extraction at a later stage (Fig. 3b). By using a reactor with an internal mixing structure,21 this was expected to be unnecessary. Consequently, NaBr could be entirely removed from the system, since its main purpose was to improve the aqueous solubility of Br2.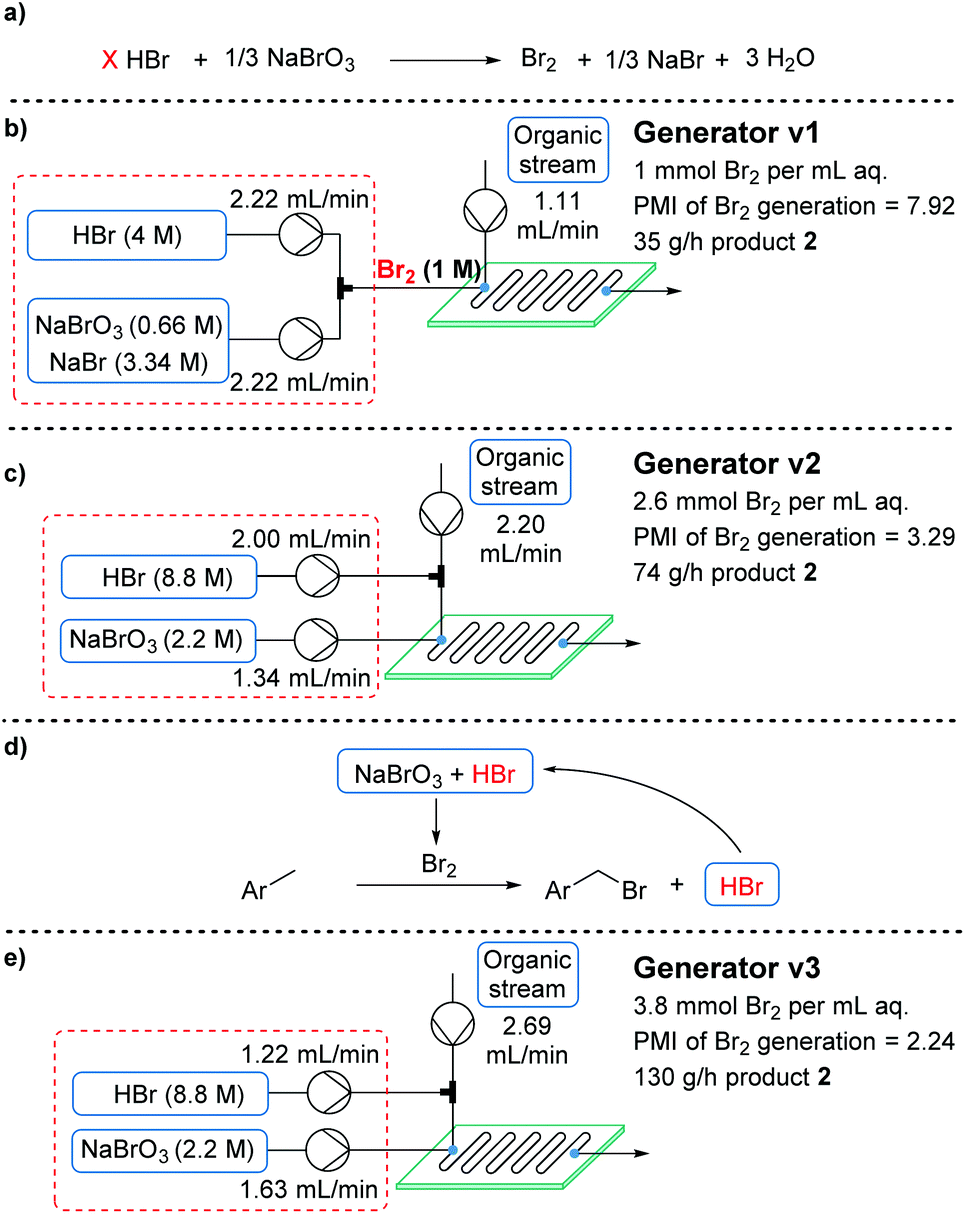 | ||
| Fig. 3 Summary of three “generations” of Br2 generator, including their flow setup, PMI of Br2, aqueous volume efficiency and productivity in the conversion of 1 to monobrominated product 2. The displayed flow rates show the required values for a residence time of 30 s. | ||
Although the efficiency was significantly improved in this manner, the reaction rate appeared to drop, likely due to inefficient transfer of bromine into the organic phase. To further improve interphase mixing, the organic phase was premixed with the HBr stream in a T-piece, prior to entering the reactor plate. As a result, Br2 was generated within the mixing structure, preventing any accumulation (which had been observed previously) and delivering a more uniform quantity of Br2. This uniform delivery, in turn, provided a significantly enhanced rate of reaction (generator v2, Fig. 3c).18 Following a brief intensification study, the generator v2 system provided product 2 in a much improved productivity of 74 g h−1 (space–time yield = 26.7 kg L−1 h−1). Perhaps more importantly, the PMI of the overall bromination process was reduced to 6.93, due to the far higher aqueous feed concentrations.
Upon revisiting the overall reaction stoichiometry, it was noted that each successful bromination releases HBr as a by-product. This can be recycled, reducing the required flow rate of the HBr feed and further decreasing mass waste (Fig. 3d).6 Moreover, it was hypothesized that effective mass transfer within the currently utilized flow reactor would enable the required interphase mixing of produced HBr back into the aqueous phase.21 A short study was carried out, gradually reducing the equivalents of HBr (denoted as X in Fig. 3a), from 2 to 1, which revealed that this alteration is indeed possible (Fig. 4). The reaction performance was maintained when moving from 2 equiv. to 1.5 equiv., yet showed a very minor decrease in yield of desired product 2 at 1 equiv. and a further decrease to 66% using 0.9 equiv. Performance at 1 equiv. could be restored by using more concentrated HBr (48%, 8.8 M). This effect is likely due to the lower aqueous volume, allowing more efficient biphasic mixing.
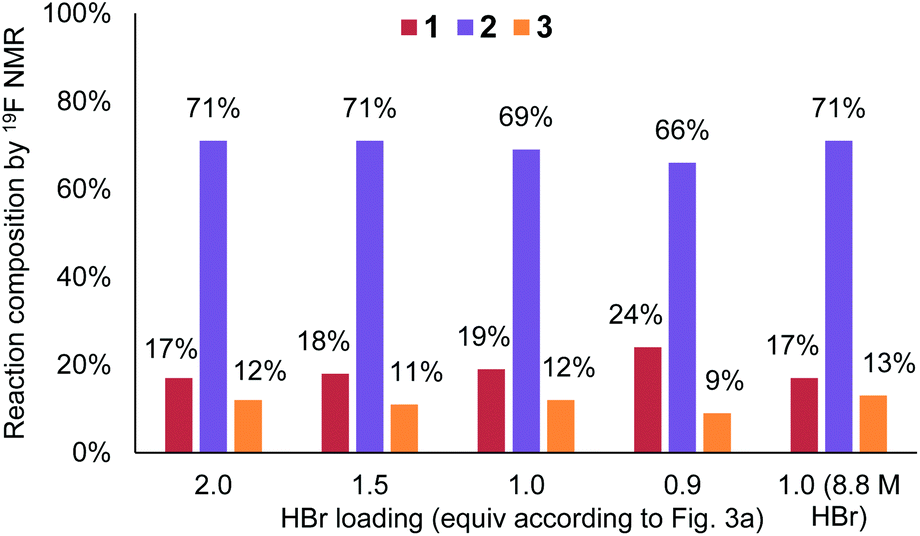 | ||
| Fig. 4 Chart showing the performance of benzylic bromination upon altering equivalents of HBr used in the bromine generator. | ||
The fully optimized generator system (generator v3, Fig. 3e) now produces 3.8 mmol (607 mg) of Br2 per mL of aqueous phase. Furthermore, the productivity of monobrominated product 2 was dramatically increased to 130 g h−1 (space–time yield = 46.9 kg L−1 h−1), due to the reduction in aqueous volume. This system actually incorporates a higher proportion of bromine atoms into the final product than elemental bromine itself (75% vs. 50%, respectively), decreasing the mass of waste, and providing further advantage to using a Br2 generator.
Further intensification using improved Br2 generator
With the optimized generator system in hand, the bromination reaction was revisited to enact further process intensification. More specifically, it was envisaged that this process could be operated with one of two sets of conditions, towards either complete consumption of starting material 1, or complete selectivity for product 2. Complete starting material consumption would invariably yield a significant quantity of dibrominated side product 3, however this can then be readily reduced to the desired product using diethylphosphite.22 Alternatively, operating at incomplete conversion would minimize the quantity of dibrominated product 3 and allow simplified recycling of starting material 1 (Fig. 5).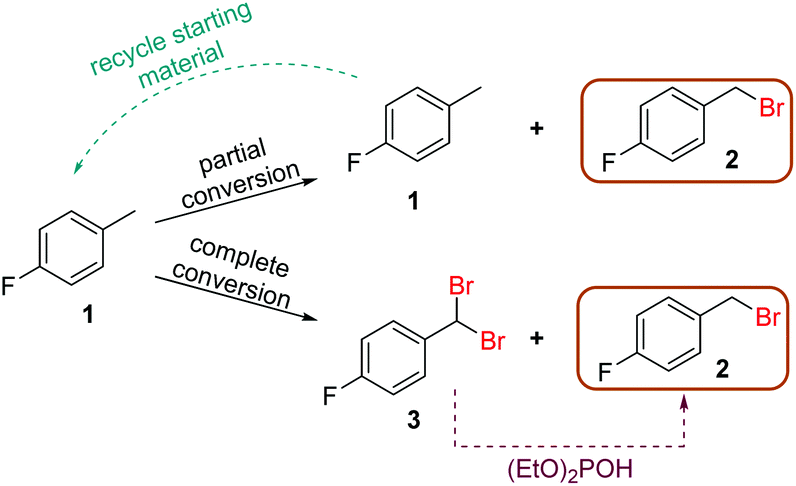 | ||
| Fig. 5 Scheme showing the complementary approaches to obtaining optimal productivity of desired product 2, by starting material recycling, or diethyl phosphite reduction. | ||
To our initial surprise, the bromination of 1 using the improved Br2 generator demonstrated a much improved tolerance for highly concentrated conditions. In fact, the substrate could even be pumped neat (9.1 M) and still achieve an excellent level of conversion at 50 °C, with a residence time of 15 s. This resulted in a significant increase in productivity, to 228 g h−1 (space–time yield = 82.3 kg L−1 h−1), with a low PMI of 4.33. Following functionalization with morpholine,18 the desired product was isolated in 73% yield.
In order to reach close to full conversion whilst maintaining a short residence time, it was found that additional Br2 (1.4 equiv.) and a higher reaction temperature (55 °C) was required.18 The resulting conditions afforded 146 g h−1 of the desired product, with 3% starting material remaining. Following diethylphosphite reduction, these conditions should provide the desired product 2 in higher purity – an approach likely favored by lower volume applications, such as pharmaceutical production.
Conversely, lower temperature (30 °C) and Br2 loading (0.6 equiv.)18 provided a reasonable yield of 2 (50%) with only 3% of side product 3 (Fig. 6). This equates to a throughput of 234 g h−1. The productivity does increase when working at partial conversion, due to the lower relative aqueous flow rates (substrate flow rate = 4.53 mL min−1 at 0.6 equiv. Br2vs. 2.11 mL min−1 at 1.4 equiv. Br2). Overall, however, there is not a significant productivity advantage to working at partial conversion (unlike in many other cases), since the reaction kinetics are fast enough to allow high flow rates under the standard reaction conditions.
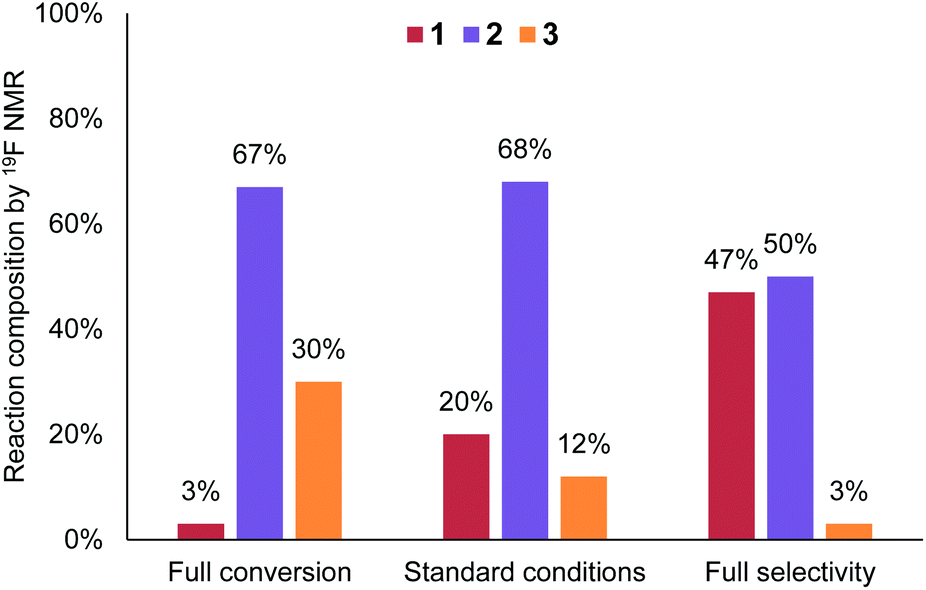 | ||
| Fig. 6 Graph comparing conditions optimized towards full conversion or full selectivity, versus the general conditions. | ||
It should be emphasized that under these highly intensified conditions, the formation of Br2 and the photochemical reaction are both highly exothermic processes. Accordingly, the use of a jacketed flow reactor with sufficient heat transfer is vital. Attempts to reproduce this chemistry in a tubing-based photoreactor resulted in a substantial exotherm within the first minutes of processing. Furthermore, carrying out this chemistry in batch would require a significantly extended reagent addition period to prevent thermal runaway.
Monobromination of 2,6-dichlorotoluene
We sought to demonstrate this methodology in the synthesis of 2,6-dichlorobenzyl bromide (5) – a commonly-occurring building block in medicinal chemistry, notably in the APIs vilanterol and isoconazole (Fig. 1a).23 In particular, vilanterol is an important molecule, used in three inhaled combination therapies marketed by GlaxoSmithKline.24In reactions of this specific substrate, dibromination was not observed, likely due to the steric hindrance imparted by two chlorine atoms flanking the reactive benzylic position. Despite this, the monobromination was still observed to occur at a fast rate, under organic solvent-free conditions, achieving full conversion within 15 s residence time (Fig. 7a).18 In the absence of fluorine atoms, the final reaction composition could also be reliably determined by benchtop 1H NMR spectroscopy.
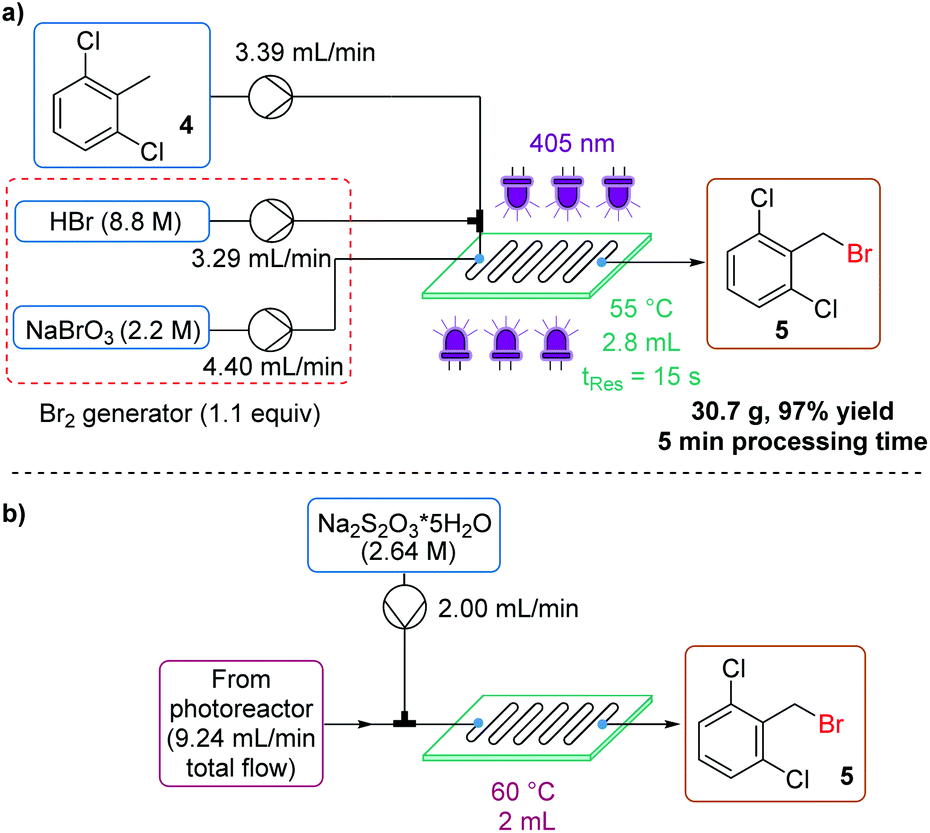 | ||
| Fig. 7 (a) Schematic representation of the experimental setup for the synthesis of 2,6-dichlorobenzyl bromide 5 using the 3rd generation Br2 generator; (b) schematic representation of the in-line sodium thiosulfate quench setup, ensuring that no Br2 remains. | ||
The desired product 5 is a low melting solid, with a melting point of 55 °C – below the reactor temperature, which ensured no solid formation. However, this meant that upon reaching the collection vessel containing Na2S2O3 quench, the product immediately solidified, encapsulating the small amount of excess Br2 before it could be effectively quenched. This could be remedied by heating the collection vessel to 60 °C, which allows mixing between the liquid product and aqueous phase. The product could now be isolated as white crystals in 97% yield, on a 30.7 g scale, in just 5 minutes processing time (Fig. 7a). An inline quench using a second mixing chip was also possible, provided residence time and mixing intensity were sufficient (Fig. 7b).18
A scale-out run was also performed, but with a slightly lengthened residence time (18 s) to minimize the effect of pump pulsation. In this case, 1.17 kg (97% yield) of the desired product 5 was isolated in less than 4 h (230 min) processing time, corresponding to a productivity of 300 g h−1 (space–time yield = 108.3 kg L−1 h−1).18 The PMI of this transformation (including aqueous mass) was calculated to be 3.08 – an exceptional value, particularly for a photochemical transformation.
Dibromination of 2,4-dichlorotoluene
Although less frequently reported, photochemical benzylic dibromination can provide a facile route to masked aldehyde compounds. For example, dibromination of 2,4-dichlorotoluene (6) results in 2,4-dichlorobenzal bromide (8).16c The equivalent aldehyde is also an intermediate in the synthesis of isoconazole (Fig. 1a). Again, benchtop NMR was sufficient to determine the ratio of starting material, mono- and dibrominated products (6, 7 and 8, respectively).As expected, a longer residence time, in combination with a slightly increased temperature, was required to achieve completion of the second bromination reaction (Fig. 8a). The quantity of Br2 was maintained at 2.2 equiv., in order to avoid wastage. Thus, using neat conditions, the masked aldehyde 8 was obtained in 99% isolated yield, with a residence time of 100 s (Fig. 8b). This meant that the productivity was lower than in monobromination reactions, yet was still a comparatively high value of 44 g h−1 (space–time yield = 15.9 kg L−1 h−1).
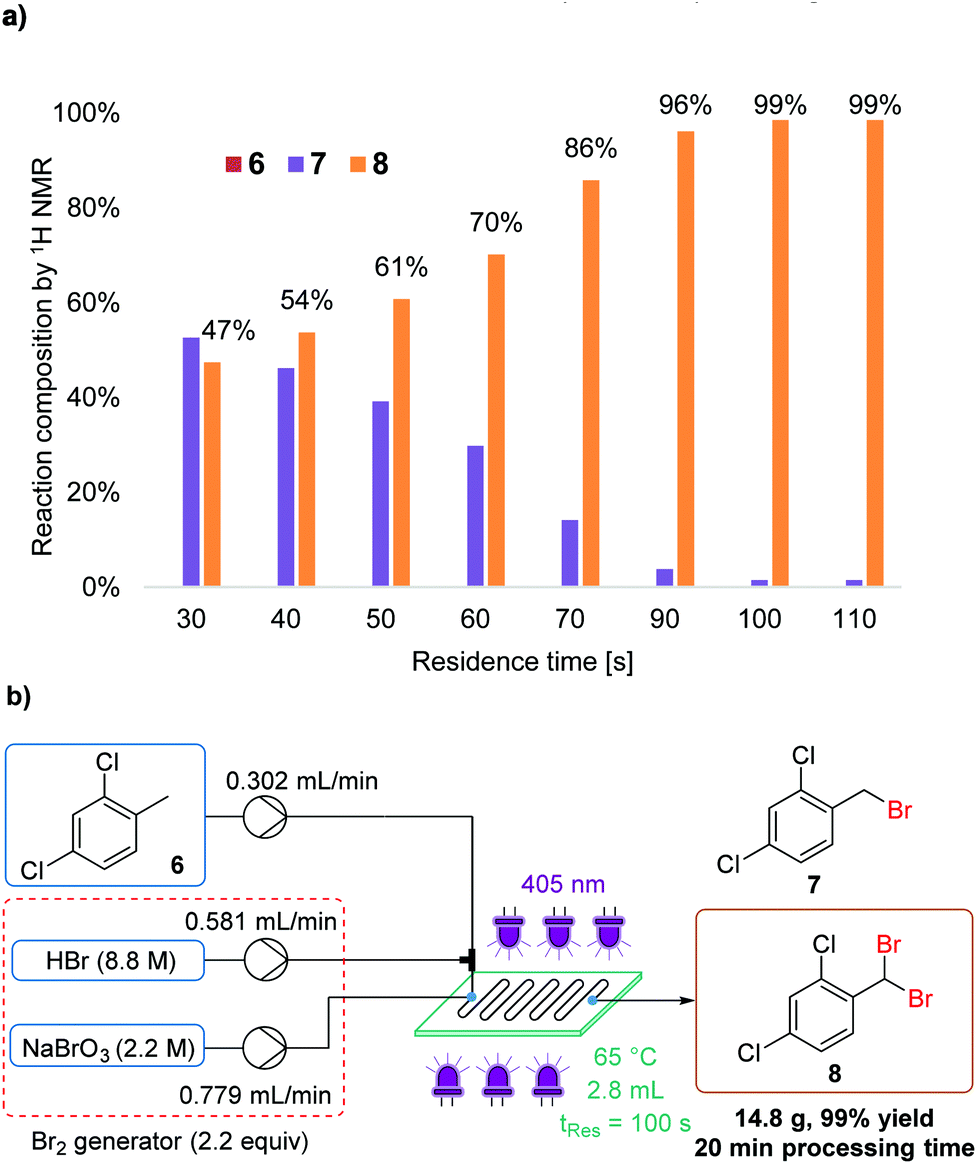 | ||
| Fig. 8 (a) Graph showing the effect of residence time upon the mono- (7) to dibrominated (8) product ratio; (b) experimental setup using 3rd generation Br2 generator for the synthesis of 2,4-dichlorobenzal bromide 8. | ||
Conclusions
The development of a highly intensified NaBrO3/HBr-based Br2 generator has been reported and exemplified. A detailed study of the benzylic bromination reaction was initially carried out, determining the effects of temperature, residence time, concentration and Br2 loading. This model substrate was then used for further optimization of the Br2 generator, which was found to be mixing sensitive, and could be intensified for exceptional mass efficiency (Table 2). Key prerequisites for the success of this transformation are: effective mass transfer for interphase mixing, but also heat transfer to control the exothermic nature of both Br2 generation and the photochemical reaction itself.| Entry | Product | Yield (%) | Productivity (g h−1) | PMI |
|---|---|---|---|---|
| a Reaction carried out using Br2 generator v3. | ||||
| 1 | 2 (Br2 generator v1) | 69 | 35 | 13.55 |
| 2 | 2 (Br2 generator v2) | 69 | 74 | 6.93 |
| 3a | 2 (Br2 generator v3) | 68 | 228 | 4.33 |
| 4a | 2 (full conversion) | 67 | 146 | 5.51 |
| 5a | 2 (full selectivity) | 50 | 234 | 4.12 |
| 6a | 5 | 97 | 300 | 3.08 |
| 7a | 8 | 99 | 44 | 3.64 |
Using the optimized Br2 generator, it was found that the reaction concentration could be increased further, even permitting pumping the starting material neat, which drastically reduces mass wastage. This could be optimized towards full conversion or full selectivity by tuning the temperature and equivalents of Br2 used. Ultimately, the desired product could be isolated in 73% yield, following derivatization with morpholine. The optimized Br2 generator system could also be applied to other bromination processes in flow.
Finally, the protocol was demonstrated in the synthesis of two further API building blocks: monobromination of 2,6-dichlorotoluene (4) was exemplified on a 1.17 kg scale in 97% yield. Using a linear scale-up strategy for this chemistry, the exceptional productivity and PMI can be maintained by preserving mass transfer, heat transfer and irradiation characteristics. This would allow access to a throughput of 4.8 kg h−1 in a 5-plate Corning G1 Photo Reactor (45 mL), or 32 kg h−1 in a 5-plate G3 Photo Reactor (300 mL).18 Dibromination of 2,4-dichlorotoluene 6 was less productive, due to its longer residence time. However, a 14.8 g batch could be isolated in 20 min processing time, which would also achieve high throughput when performed in a larger scale reactor.
Experimental
Safety notes
The Br2 generation and quenching of Br2 by Na2S2O3 are exothermic processes. HBr and NaBrO3 should be stored separately. Extreme caution is advised, particularly when quenching waste or unused solutions – this should be carried out with care and under dilute conditions where possible.This reaction under the intensified conditions described should be carried out only in a reactor system with sufficient temperature control. When attempting this reaction in a tubing-based photoreactor, the temperature was observed to increase extremely rapidly, due to the exothermic nature of the photochemical reaction.
The benzyl bromide products of these reactions have lachrymatory properties (not listed on their MSDS data), so care should be taken to ensure their containment within a ventilated space.
Procedure for the monobromination of 2,6-dichlorotoluene (4)
A waste collection flask was charged with a small volume of saturated sodium thiosulfate solution and was stirred with a magnetic stirrer. A 3-necked flask was charged with ∼50 mL sodium thiosulfate solution (2.64 M) and equipped with a stirrer bar. The reactor was turned on (405 nm LEDs and thermostats) and the reactor thermostat was set to 55 °C. After the temperature of the reactor stabilized, the pumps were turned on. Flow rates: substrate 3.39 mL min−1, HBr 3.29 mL min−1 and NaBrO3 4.40 mL min−1. Solutions: 2,6-Dichlorotoluene (neat, 7.79 M), HBr (8.8 M, 1.1 equiv.) and NaBrO3 (2.2 M, 0.37 equiv.). The system was allowed to equilibrate for ∼5 min, then the reactor output was collected into the stirred 3-necked flask for 5 min (heated to 60 °C during product collection). The collection flask was then allowed to cool gradually for 16 h, before the product was filtered, washed with water (∼200 mL) and then dried under reduced pressure to afford the desired product as a white crystalline solid (30.7 g, 97% yield).Procedure for the dibromination of 2,4-dichlorotoluene 6
A waste collection flask was charged with a saturated solution of sodium thiosulfate and was stirred with a magnetic stirrer. The reactor was turned on (405 nm LEDs and thermostats) and the reactor thermostat was set to 65 °C. After the temperature of the reactor had stabilized, the pumps were turned on. Flow rates: substrate 0.302 mL min−1, HBr 0.581 mL min−1 and NaBrO3 0.779 mL min−1. Solutions: 2,4-Dichlorotoluene (neat, 7.74 M, 46.7 mmol in 20 min), HBr (48%, 103 mmol, 2.2 equiv.) and NaBrO3 (2.2 M, 34.3 mmol, 0.73 equiv.). The system was allowed to equilibrate for ∼15 min, then for 20 min the mixture was collected in a stirred round bottom flask containing sodium thiosulfate solution (2.2 M). The quenched reaction mixture was extracted with DCM (3 × 20 mL) and the organic phase was then washed with sodium bicarbonate solution (1 × 20 mL) and dried over Na2SO4. After removal of the solvent under reduced pressure, product 8 was obtained as a pale yellow oil (14.8 g, 99% yield).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CC Flow Project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Science, Research and Economy (BMWFW), and by the State of Styria (Styrian Funding Agency SFG). The authors gratefully acknowledge Corning SAS for the generous loan of the Corning Advanced-Flow Lab Photo Reactor used in this study.Notes and references
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed.
- C. Djerassi, Chem. Rev., 1948, 43, 271–317 CrossRef CAS PubMed.
- (a) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (b) T. Noël, J. Flow Chem., 2017, 7, 87–93 CrossRef; (c) J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed; (d) F. Politano and G. Oksdath-Mansilla, Org. Process Res. Dev., 2018, 22, 1045–1062 CrossRef CAS.
- (a) D. Cantillo and C. O. Kappe, React. Chem. Eng., 2017, 2, 7–19 RSC; (b) D. Cantillo, O. De Frutos, J. A. Rincon, C. Mateos and C. O. Kappe, J. Org. Chem., 2014, 79, 223–229 CrossRef CAS PubMed; (c) H. E. Bonfield, J. D. Williams, W.-X. Ooi, S. G. Leach, W. J. Kerr and L. J. Edwards, ChemPhotoChem, 2018, 2, 938–944 CrossRef CAS; (d) D. Šterk, M. Jukič and Z. Časar, Org. Process Res. Dev., 2013, 17, 145–151 CrossRef; (e) Y. Chen, O. de Frutos, C. Mateos, J. A. Rincon, D. Cantillo and C. O. Kappe, ChemPhotoChem, 2018, 2, 906–912 CrossRef CAS; (f) Y. Manabe, Y. Kitawaki, M. Nagasaki, K. Fukase, H. Matsubara, Y. Hino, T. Fukuyama and I. Ryu, Chem. – Eur. J., 2014, 20, 12750–12753 CrossRef CAS PubMed.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- (a) H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC; (b) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- Y. Otake, J. D. Williams, J. A. Rincón, O. de Frutos, C. Mateos and C. O. Kappe, Org. Biomol. Chem., 2019, 17, 1384–1388 RSC and references cited therein.
- T. Hou, P. Lu and P. Li, Tetrahedron Lett., 2016, 57, 2273–2276 CrossRef CAS.
- L. S. De Almeida, P. M. Esteves and M. C. S. De Mattos, Tetrahedron Lett., 2015, 56, 6843–6845 CrossRef CAS.
-
(a) Br2 has an exceptionally high vapor pressure:toxicity ratio, so its handling should be avoided wherever possible: J. K. Niemeier and D. P. Kjell, Org. Process Res. Dev., 2013, 17, 1580–1590 CrossRef CAS;
(b) “H330 – Fatal if inhaled, H400 – Very toxic to aquatic life” Br2 MSDS, https://www.sigmaaldrich.com/catalog/product/aldrich/470864, accessed 8th October 2019.
- M. Eissen and D. Lenoir, Chem. – Eur. J., 2008, 14, 9830–9841 CrossRef CAS PubMed.
- (a) A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef PubMed; (b) R. Mestres and J. Palenzuela, Green Chem., 2002, 4, 314–316 RSC; (c) A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Tetrahedron, 2009, 65, 4429–4439 CrossRef; (d) A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Green Chem., 2007, 9, 1212–1218 RSC.
- S. M. K. Nair and P. D. Jacob, Thermochim. Acta, 1991, 181, 269–276 CrossRef CAS.
- (a) D. Kikuchi, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1998, 63, 6023–6026 CrossRef CAS PubMed; (b) S. Adimurthy, G. Ramachandraiah, A. V. Bedekar, S. Ghosh, B. C. Ranu and P. K. Ghosh, Green Chem., 2006, 8, 916–922 RSC; (c) S. Adimurthy, S. Ghosh, P. U. Patoliya, G. Ramachandraiah, M. Agrawal, M. R. Gandhi, S. C. Upadhyay, P. K. Ghosh and B. C. Ranu, Green Chem., 2008, 10, 232–237 RSC; (d) M. Dinda, M. K. Agrawal, M. R. Gandhi, S. C. Upadhyay, S. Adimurthy, S. Chakraborty and P. K. Ghosh, RSC Adv., 2012, 2, 6645–6649 RSC; (e) M. Dinda, S. Samanta, S. Eringathodi and P. K. Ghosh, RSC Adv., 2014, 4, 12252–12256 RSC.
- (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (b) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (c) B. Gutmann and C. O. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS; (d) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- (a) G. Glotz, R. Lebl, D. Dallinger and C. O. Kappe, Angew. Chem., Int. Ed., 2017, 56, 13786–13789 CrossRef CAS PubMed; (b) R. Van Kerrebroeck, P. Naert, T. S. A. Heugebaert, M. D'hooghe and C. V. Stevens, Molecules, 2019, 24, 2116 CrossRef CAS PubMed; (c) W. Bin Yu, D. P. Yu, M. M. Zheng, S. T. Shan, Y. J. Li and J. R. Gao, J. Chem. Res., 2012, 36, 258–260 CrossRef.
- E. Serrao, S. Odde, K. Ramkumar and N. Neamati, Retrovirology, 2009, 6, 25 CrossRef PubMed.
- See ESI for further details.†.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- For examples of photochemistry performed previously using this reactor type, see: (a) J. D. Williams, M. Nakano, R. Gérardy, J. A. Rincon, O. de Frutos, C. Mateos, J.-C. M. Monbaliu and C. O. Kappe, Org. Process Res. Dev., 2019, 23, 78–87 CrossRef CAS; (b) N. Emmanuel, C. Mendoza, M. Winter, C. R. Horn, A. Vizza, L. Dreesen, B. Heinrichs and J.-C. M. Monbaliu, Org. Process Res. Dev., 2017, 21, 1435–1438 CrossRef CAS.
- For detailed studies of biphasic mixing within this type of mixing structure, see: (a) M. J. Nieves-Remacha, A. A. Kulkarni and K. F. Jensen, Ind. Eng. Chem. Res., 2012, 51, 16251–16262 CrossRef CAS; (b) K. J. Wu, V. Nappo and S. Kuhn, Ind. Eng. Chem. Res., 2015, 54, 7554–7564 CrossRef CAS.
- S. Xu, Q. Hao, H. Li, Z. Liu and W. Zhou, Org. Process Res. Dev., 2017, 21, 585–589 CrossRef CAS.
- S. Veraldi, Mycoses, 2013, 56, 3–15 CrossRef CAS PubMed.
- (a) J. Shur, R. Price, D. Lewis, P. M. Young, G. Woollam, D. Singh and S. Edge, Int. J. Pharm., 2016, 514, 374–383 CrossRef CAS PubMed; (b) S. Hickey, Br. J. Health Care Manag., 2018, 24, 558–564 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03662h |
| This journal is © The Royal Society of Chemistry 2020 |