
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCycloamination strategies for renewable N-heterocycles
Hu
Li
 *a,
Haixin
Guo
b,
Zhen
Fang
*a,
Taku Michael
Aida
c and
Richard Lee
Smith
Jr
*b
*a,
Haixin
Guo
b,
Zhen
Fang
*a,
Taku Michael
Aida
c and
Richard Lee
Smith
Jr
*b
aBiomass Group, College of Engineering, Nanjing Agricultural University, 40 Dianjiangtai Road, 210031, Nanjing, Jiangsu, China. E-mail: hli17@njau.edu.cn; zhenfang@njau.edu.cn
bGraduate School of Environmental Studies, Tohoku University, 6-6-11, Aoba, Aramaki, Aoba-ku, Sendai, 980-8579, Japan. E-mail: smith@scf.che.tohoku.ac.jp
cDepartment of Chemical Engineering, Faculty of Engineering, Fukuoka University, 8-19-1, Nanakuma Jonan-ku, Fukuoka 814-0180, Japan
First published on 9th January 2020
Abstract
Biomass resources have infinite possibilities for introducing nitrogen, sulfur, or phosphorus heteroatoms into their structures by virtue of controllable carbon–heteroatom bond formation. In this review, cycloamination approaches for thermal (catalyst-free) and catalytic transformation of biomass feedstocks into N-heterocyclic molecules including mechanistic pathways are analyzed. Bottom-up (small molecule substrates) and top-down (large molecule substrates) are considered. Sustainable routes for synthesis of five-membered (pyrroles, pyrrolidones, pyrazoles, imidazoles), six-membered (pyridines, pyrazines), fused (indoles, benzimidazoles), and other relevant azaheterocycles are critically assessed. Production of biomass-derived six-, seven-, and eight-membered as well as fused N-heterocyclic compounds with present approaches have relatively low selectivities. Attention to methods for forming analogous sulfur or phosphorus heteroatom compounds from biomass resources using either bottom-up or top-down strategies appear to have been greatly overlooked. Synthetic auxiliaries (heating modes, nitrogen sources) that enhance reaction efficiency and tunability of N-heterocyclic ring size/type are considered and plausible reaction mechanisms for pivotal pathways are developed.
1. Introduction
Renewable biomass is a future replacement for fossil resources being used to produce energy and chemicals.1–6 Due to the presence of abundant oxygen species, biomass derivatives have been considered as consummate intermediates or platform molecules for the synthesis of oxygen-containing chemicals such as esters, acids, aldehydes, ketones, phenolics, and alcohols,7–12 that contain unsaturated hydrocarbon moieties (e.g., alkenyl, alkynyl, aryl groups),13–18 as shown in Fig. 1. The specific functional groups of biomass-derived compounds provide infinite possibilities in the introduction of heteroatoms (e.g., N, S, P) and even construction of heterocycles by virtue of controllable carbon–heteroatom bond formation,19–22 which are not readily available from conventional fossil resources. Over the past several decades, there has been increasing interest in development of sustainable processes with simple and green approaches to accessing value-added heteroatom-containing chemicals.23–26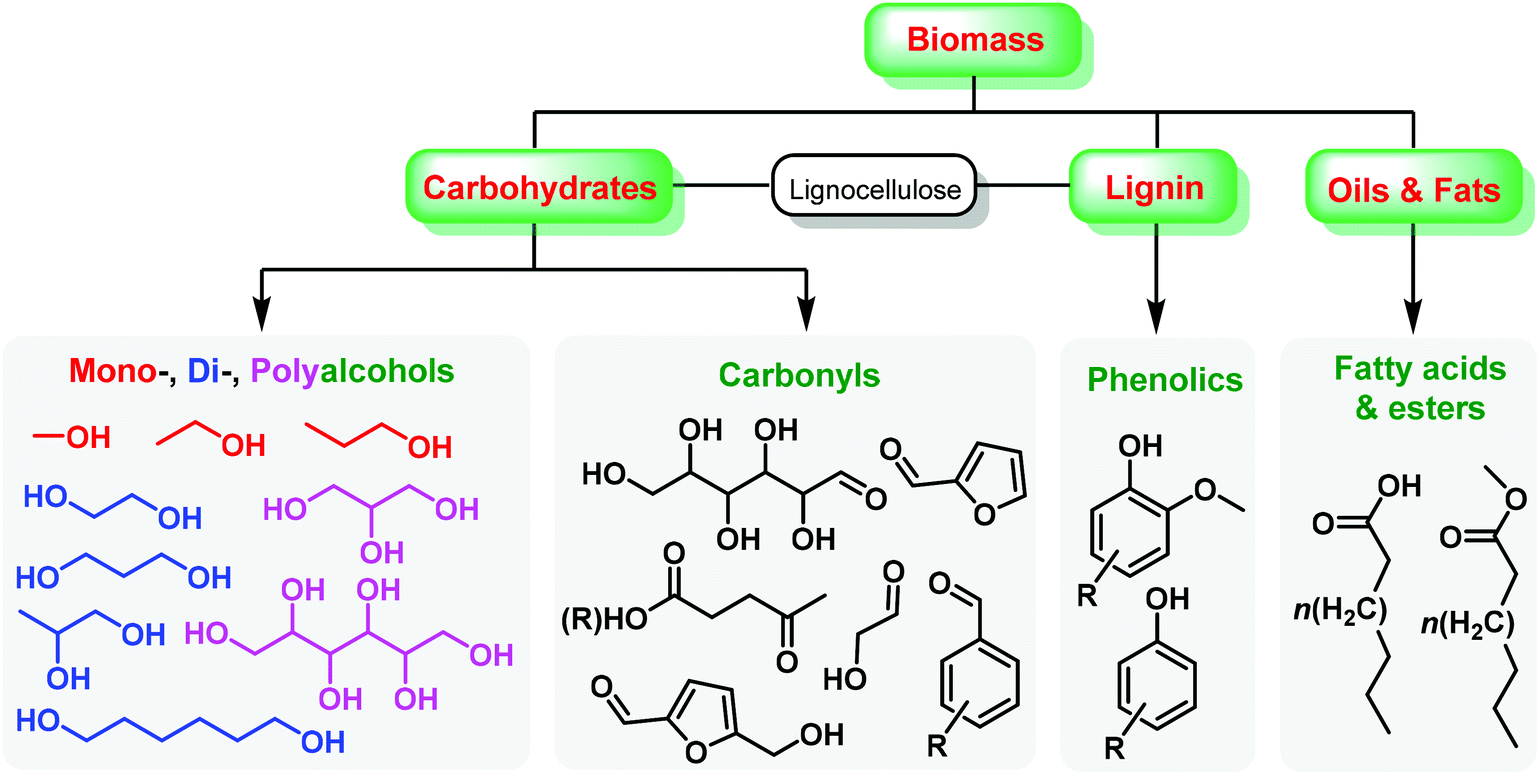 | ||
| Fig. 1 General pathways for upgrading biomass to oxygen-containing chemicals. | ||
Nitrogenous compounds hold a privileged position in the preparation of drugs, agrochemicals, polymers, and other functional materials.27,28 In particular, nitrogen species are presented in more than 80% of the top 200 pharmaceuticals,29,30 and two thirds of these N-containing medicines contain N-heterocyclic skeletons.31 Given the above considerations, urgent attention is needed to develop green and efficient approaches to task-specific azaheterocycles from renewable biomass feedstocks via sustainable chemistry. The employed nitrogen sources can be derived from ammonia, amines/amides pre-prepared by C–N coupling reactions (e.g., reductive amination, aminolysis, and amidation),32–36 or natural N-reservoirs (e.g., chitin, proteins) for producing amines and derivatives (e.g., glucosamine, N-acetyl-D-glucosamine, amino acids).37–41 Beginning from the accessible bio-based functional molecules, N-heterocycles can be constructed by C–N and/or C–C bond formation typically via three dominant synthetic routes including intramolecular cyclization, cycloaddition (e.g., aza-Diels–Alder), and multicomponent condensation reactions (Fig. 2).42,43 Relevant reaction processes and pathways in the presence or absence of designed catalysts have been investigated under optimized thermal conditions,42,43 for which these cycloamination strategies are attractive for the sustainable synthesis of bio-based N-heterocycles, with much development needed to further expand substrate scope and generality.
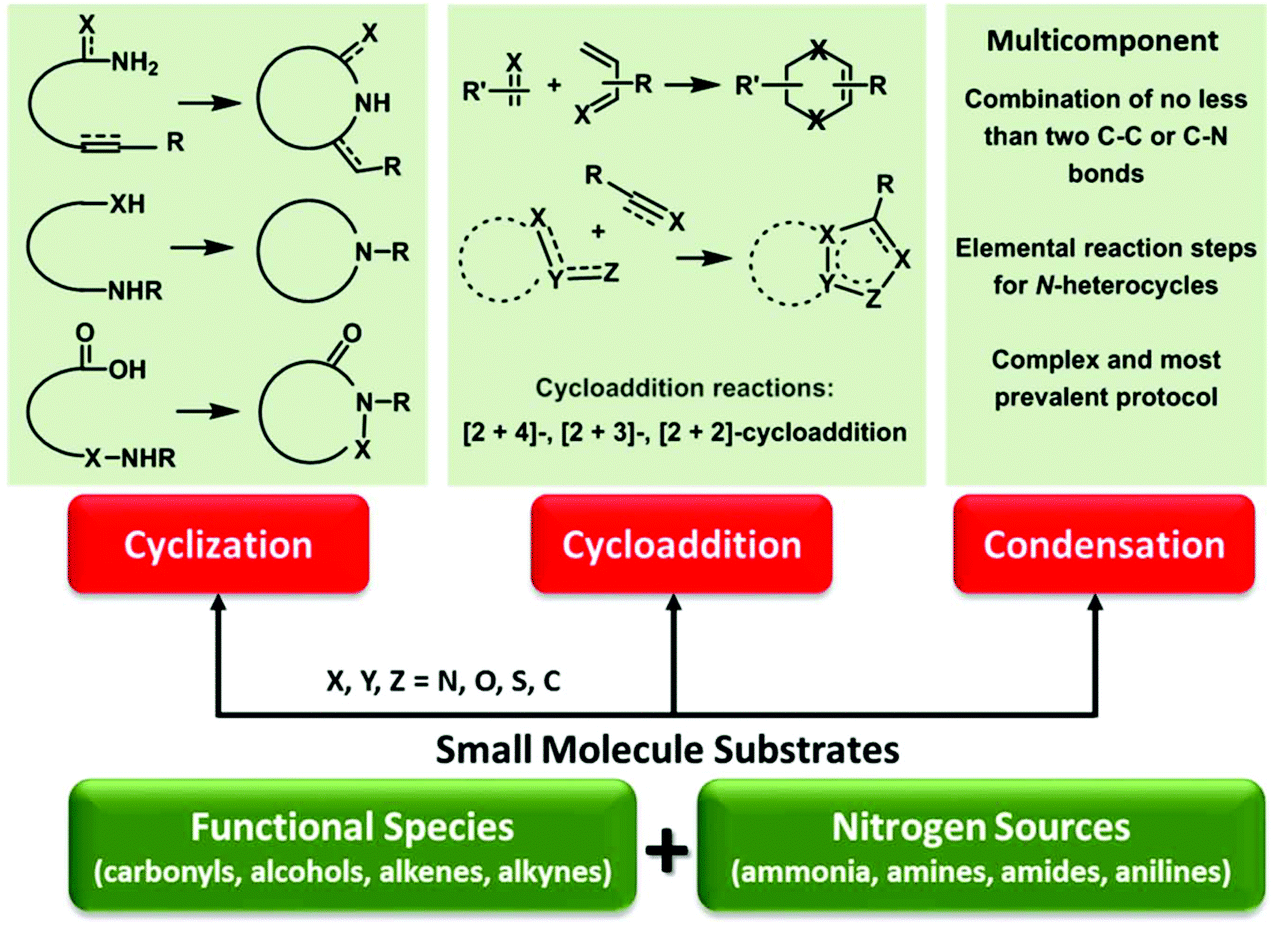 | ||
| Fig. 2 Bottom-up strategy to N-heterocycles via small molecule substrates. | ||
Previously published review articles have focused on design of functional catalytic materials and conversion means for efficient production of specific/desired platform molecules and biofuels from lignocellulosic biomass.44–55 However, increasing attention is on the synthesis of heteroatom-containing chemicals via green and sustainable routes, with emphasis on the exploitation of approaches to acyclic amines and correlative N-containing commodities or fine chemicals.56–58 In view of the much higher versatility of azaheterocycles, this review aims to critically assess the cycloamination strategies developed for the preparation of five-membered (section 2), six-membered (section 3), fused (section 4), and other (section 5) biomass-derived N-heterocyclic products. Auxiliaries such as heating modes and solvent or substrate types optimized to overcome reaction barriers are discussed, with reaction mechanisms and pathways being outlined for representative processes and systems.
2. Five-membered N-heterocycles
2.1. Pyrroles
Pyrrole-containing N-heterocycles are important structural motifs and are widely applied in pharmaceuticals, pesticides, catalysts, functional materials, and supramolecular chemistry.59–61 The Hantzsch, Knorr, Paal–Knorr, Van Leusen, Barton–Zard, and Piloty–Robinson reactions are typical approaches toward the synthesis of pyrroles.62–68 In view of the versatility of substituted pyrroles, many renewed synthetic methods have been developed to construct these types of unique N-heterocycles, such as metal-catalyzed cyclization, cycloaddition, rearrangement, aza-Wittig, multicomponent/oxidative coupling, hydroamination/cyclization, and isocyanide-based reactions.69–79 Instead of fossil-based resources, synthesis of pyrroles by full or partial use of renewable resources via newly-developed reaction routes is a highly desirable goal in sustainable chemistry. However, in these methods, leaching of metal species may cause serious environmental issues that should be taken into consideration for metal-mediated catalytic systems, especially with respect to the catalyst stability and reusability.Beginning from 1,4-dicarbonyl compounds 1, especially since 2,5-hexadione is derivable from hexose sugars,80N-substituted pyrroles 4 can be synthesized by Paal–Knorr condensation with primary amines 2 over acid catalysts or with nitro compounds 3 over metal catalysts (Scheme 1). The combination of a heterogeneous cobalt–nitrogen catalyst (Co–Nx/C-800-AT) with formic acid serving as both hydrogen donor and an acid catalyst is efficient for one-pot synthesis of 2,5-dimethyl-1-phenylpyrrole (95.2% yield, 110 °C, 12 h) from nitrobenzene and 2,5-hexadione in ethanol.81 Nitrobenzene is subjected to transfer hydrogenation with formic acid over the Co–Nx/C-800-AT catalyst, affording aniline (Scheme 1). The carboxide species of 2,5-hexadione is protonated by formic acid to yield the intermediate (IM), which is then attacked by aniline, followed by cascade cyclization and dehydration to afford the product.81 The reaction system is applicable to heterocyclization of substituted nitrobenzenes with 2,5-hexadione to produce corresponding N-substituted pyrroles with yields of up to 100%. Notably, the developed cobalt–nitrogen catalyst is tolerable to the acidic liquid H-donor (HCOOH), which may be ascribed to the highly dispersed metal particles that are coordinated and stabilized by nitrogen species of the solid carbonaceous supports. This unique heterogeneous feature of the non-noble metal catalyst is able to not only remarkably reduce the loss of metal species in the reaction processes and the overall production cost, but also provides an example of using sustainable HCOOH instead of flammable hydrogen gas as H-donor.
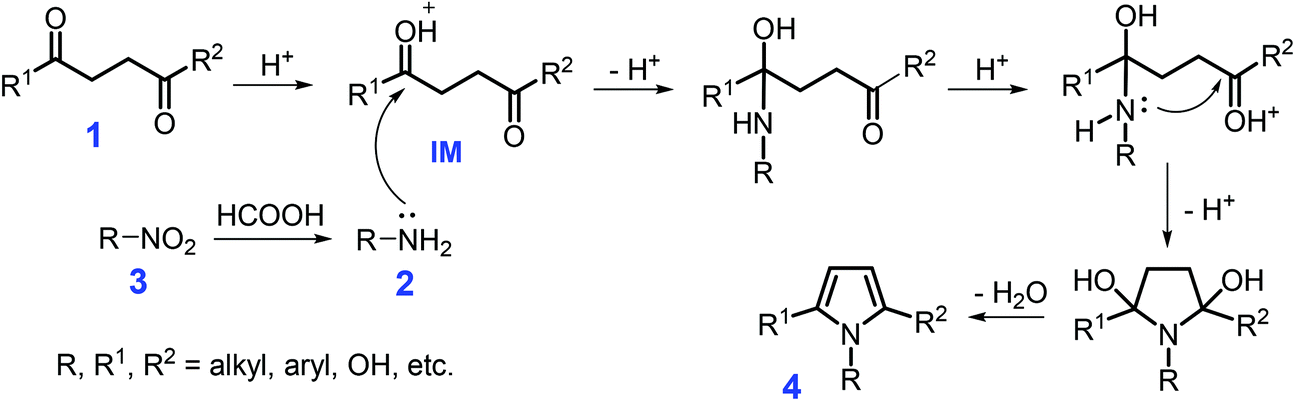 | ||
| Scheme 1 Synthesis of N-substituted pyrroles 4 from 1,4-dicarbonyls 1 with amines 2 or nitro compounds 3. Adapted with permission from ref. 81 and 82, Copyright © 2017 Royal Society of Chemistry; Copyright © 2010, American Chemical Society. | ||
Lignocellulosic biomass-derived alcohols are another green feedstock for synthesis of pyrroles, although complex byproducts (e.g., cyclic imides, pyrrolidines, and lactones) were often frequently obtained by coupling of carbonyl intermediates 1in situ formed from catalytic dehydrogenation of alcohols.82–85 With hydrogen and water as co-products, catalytic acceptorless dehydrogenative coupling of 1,4-butanediol or 1,4-substituted 1,4-butanediols 5 and amines 2 over base-metal complexes (e.g., cobalt or manganese pincer complex) generate 1 or 1,2,5-substituted pyrroles 4, respectively, at 150 °C for 24 h with 90% yield (Scheme 2).86,87 In the catalytic process, an aldehyde or ketone intermediate 1 is initially formed by dehydrogenation of alcohol to liberate H2, followed by coupling with the primary amine 2 to produce the N-substituted pyrrole 4 and water via Paal–Knorr condensation (Scheme 2).86,87 A synergic effect between the metal and ligand species is observed in the dehydrogenative coupling reaction, which mainly contributes to the enhanced selectivity towards the product pyrrole.87 Although these homogeneous catalytic systems exhibit pronounced performance in the Paal–Knorr condensation reaction, difficulty in the catalyst recovery will lead to additional cost and negative impact on the environment.
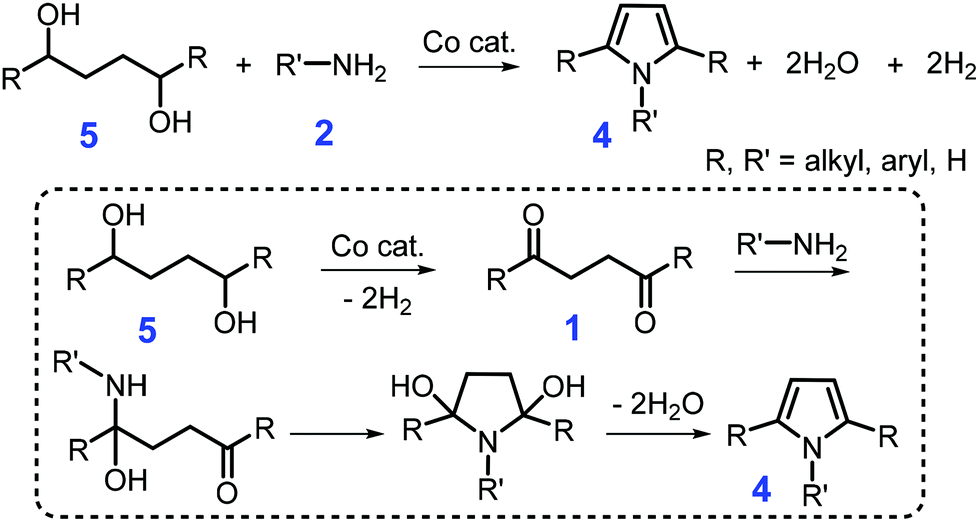 | ||
| Scheme 2 Catalytic coupling of primary or secondary diols 5 with primary amines 2. Adapted with permission from ref. 86 and 87, Copyright © 2016 Wiley-VCH; Copyright © 2019, American Chemical Society. | ||
Unsaturated diols, such as butene-1,4-diol or butyne-1,4-diol, can be used to construct substituted pyrroles by reaction with primary amines (Scheme 3) over precious metals (e.g., Ru, Pd) in the presence of phosphine ligands, although they afford relatively low selectivities.88,89 The application of earth-abundant base metals (e.g., Ni, Co, Fe, and Mn) to catalysis with specific nitrogen ligands is able to overcome the long-standing problem of high catalyst cost and low productivity.90,91 It is postulated by the authors that the nonprecious metal (e.g., Fe and Ni) catalyze dehydrogenation or isomerization of cis-butene-1,4-diol or butyne-1,4-diol (6) to afford 7 that is then condensed with primary amine 2, leading to the formation of allylic amine 9via hydrogenation of imine 8.92,93 In a one-pot operation, subsequent dehydrogenation of 9 gives intermediate 10, which is further subjected to cascade intermolecular cyclization and dehydration to construct N-substituted pyrroles 4 (Scheme 3). In parallel, N-substituted pyrroles may also be generated by sequential metal-catalyzed isomerization and intermolecular cyclization of the imine 8via intermediate 10.92,93 Due to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C
C or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond in the diols, the reaction system is tolerant of free halides and alcohols, leading to pyrrole yields of up to 90%.90,91
C bond in the diols, the reaction system is tolerant of free halides and alcohols, leading to pyrrole yields of up to 90%.90,91
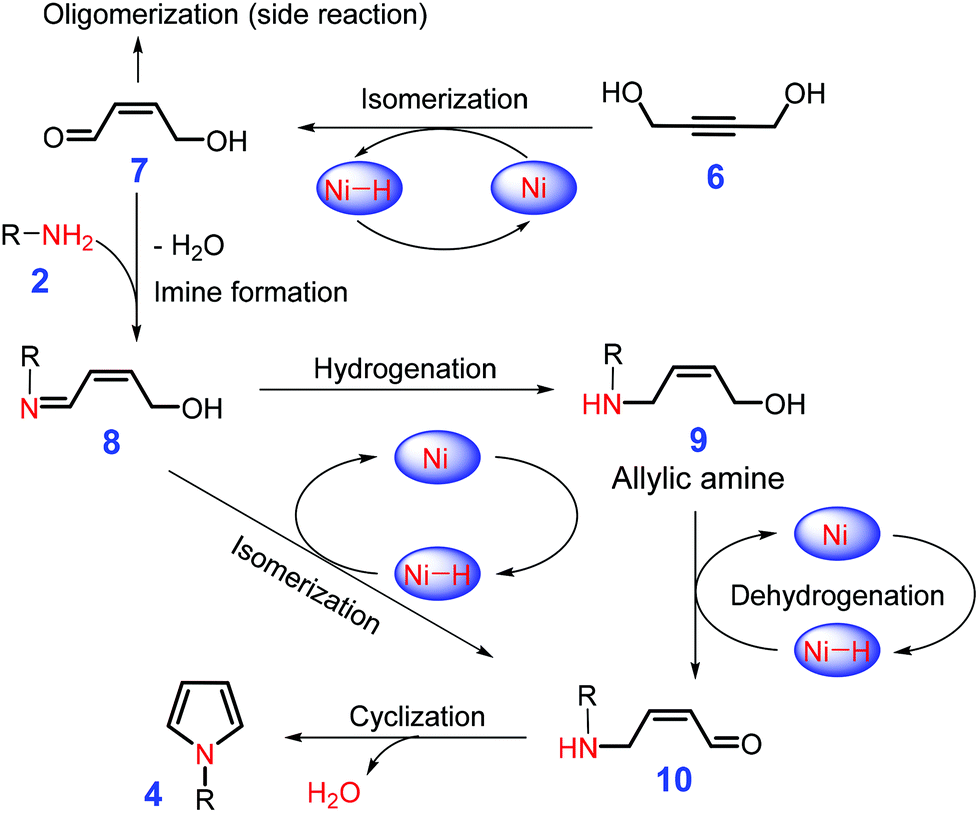 | ||
| Scheme 3 Intermolecular cyclization of butene-1,4-diol or butyne-1,4-diol 6 with primary amines 2 to pyrroles 4 catalyzed by base metal (e.g., Ni). Adapted with permission from ref. 92 and 93, Copyright © 2017, American Chemical Society; Copyright © 2016 Wiley-VCH. | ||
Metal-catalyzed hydrogen autotransfer or the borrowing of hydrogen is capable of converting alcohols to carboxides together with the liberation of H2, followed by condensation to give imine species that could be further reduced to amines by the in situ formed hydrogen (Scheme 3).94,95 Michlik and Kempe reported on the synthesis of 2,5-disubstituted pyrroles 4 from sustainable secondary alcohols 11 and amino alcohols 12 initiated by alcohol dehydrogenation in the presence of sodium tert-butoxide (NaO-t-Bu) and an organoiridium catalyst via sequential formation of C–N and C–C bonds (Scheme 4).96 This synthetic protocol was implemented under relatively mild reaction conditions (90 °C, 24 h) with ketone 13 and imine 14 as key intermediates, and could tolerate a wide range of functional groups like olefins, Cl, Br, NH2, OH, and organometallic moieties with moderate to high yields of pyrroles (42–97%).96 Other types of N/P-ligands stabilized Ir and Ru complexes are demonstrated to be efficient for the synthesis of pyrrole derivatives,97–101 although it would be desirable to develop catalysts based on earth-abundant base metals in view of recycle and reuse requirements. Kallmeier et al. reported that pyrrole could be synthesized in isolated yields of up to 93% over Mn PN5P-pincer catalysts, whereas no significant activity was observed for related Fe and Co complexes.102 The Mn-mediated reaction smoothly progressed at moderate reaction temperatures (78 °C) with 2-methyltetrahydrofuran as solvent, which is a lower temperature than that used for Ir- and Ru-catalyzed processes (≥90 °C),96–102 and is an inspiring example of nonprecious metal-based catalysis for replacing noble metal complexes in the synthesis of heterocycles. The release of H2 from the used reagent alcohols over the metal catalysts can eliminate the use of flammable and high-pressure hydrogen gas, while the development of active heterogeneous counterparts will be helpful for practical applications.
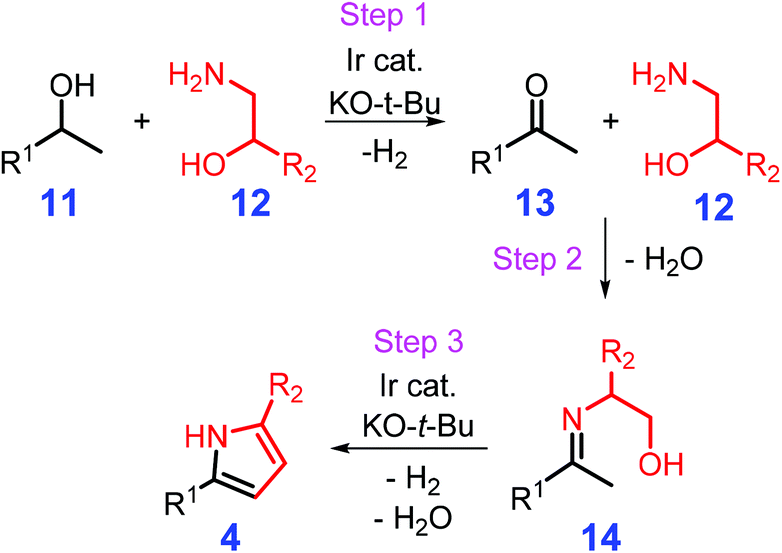 | ||
| Scheme 4 Catalytic synthesis of pyrroles from secondary alcohols and amino alcohols via cascade dehydrogenation (step 1), imine formation (step 2), intramolecular C–C coupling and isomerization (step 3). Adapted with permission from ref. 96, Copyright © 2013, Springer Nature. | ||
Another method to synthesize pyrroles (Scheme 5) is via catalytic amination of bio-derived furanic compounds 15 with primary amines 2 in the presence of an acid catalyst (e.g., Al2O3 and TiO2) that affords pyrrole derivatives at 20–60% yields at high-temperature (250 °C to 400 °C).103,104 Under relatively mild reaction conditions (150 °C, 5 bar N2, 0.7 h), Tao et al. demonstrated that a solid acid H-form zeolite H-Y(2.6) gave N-(m-tolyl)-2,5-dimethylpyrrole 4a in nearly quantitative yields (99%) from the condensation of 2,5-dimethylfuran 15a and m-methylaniline 2a.105 Besides the porous structure, moderate Lewis/Brønsted acid strength of H-Y(2.6) as determined by NH3-TPD and pyridine-adsorbed FT-IR, contribute to the optimized pyrrole yield. In contrast, H-MOR(12.5) and H-ZSM-5(18) with strong acidity significantly inhibit the desired condensation reaction (9–34% yields), possibly due to deactivation of the active sites by a strong binding interaction with N-containing intermediates.105 Rather than from the direct reaction between aniline and 2,5-dimethylfuran, 2,5-hexanedione 16 was identified to be the key intermediate that reacts with different anilines via Paal–Knorr reaction to afford pyrrole derivatives. With water as the only co-product, this atom-economic H-Y(2.6)-catalytic system (Scheme 5) shows good generality for synthesis of polysubstituted pyrroles 4 (62–93% yields) from anilines 2 and bio-derived furans 15 (e.g., 2,5-dimethylfuran, 2-methylfuran, and furan) at 150 °C or 180 °C for 0.5 h to 6 h.105 Unlike conventional Paal–Knorr reactions that typically use 1,4-diketones as substrates, this catalytic approach based on biofurans and commercial zeolites is a promising method to construct pyrrole-based scaffolds.
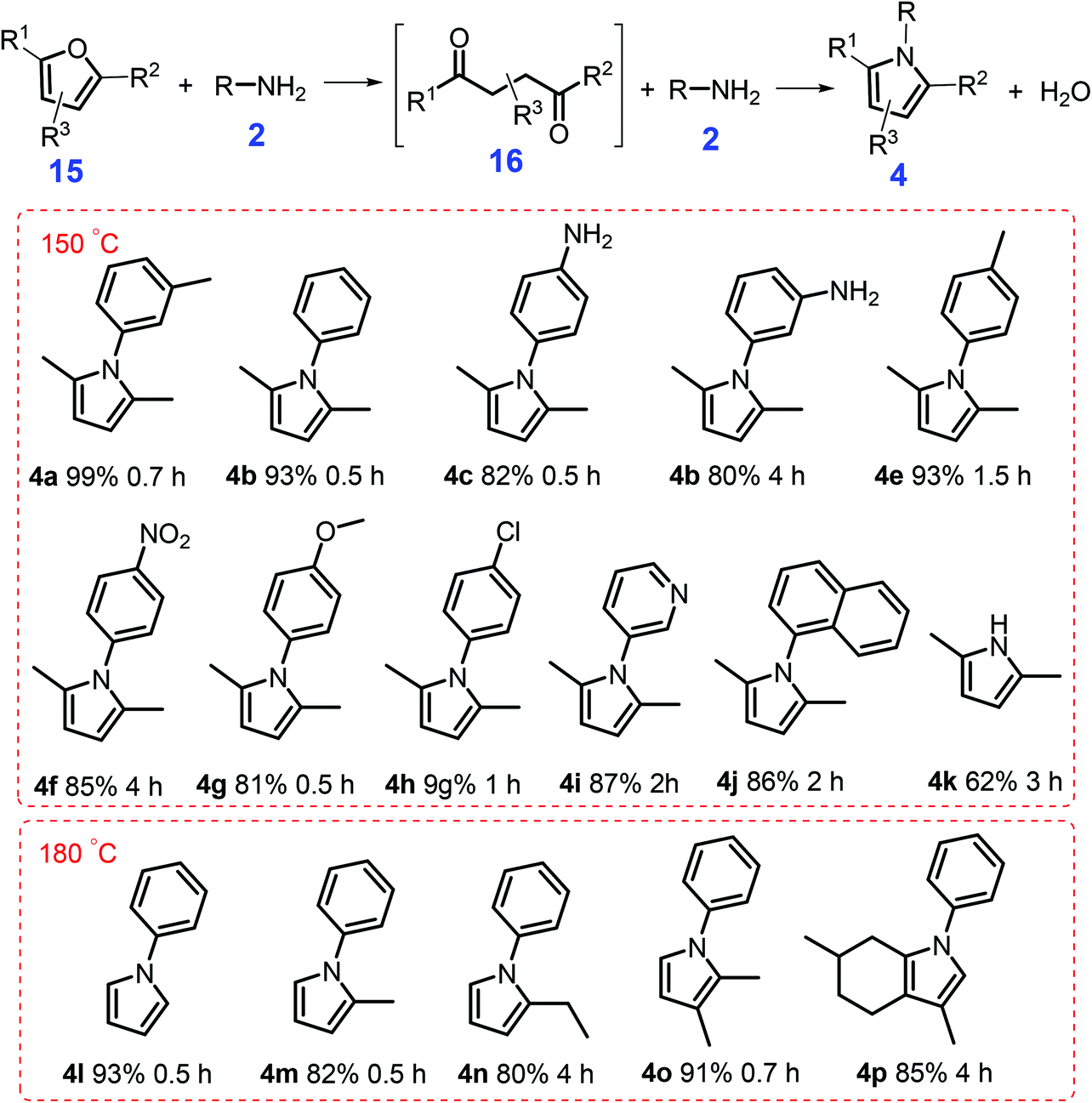 | ||
| Scheme 5 Catalytic condensation of furans 15 with anilines 2 to give pyrroles 4. Reaction conditions: 1 mmol furan, 1 mmol aniline, 2 mL toluene, 150 mg of H-Y(2.6), pN2 = 5 bar. Adapted with permission from ref. 105, Copyright © 2017, American Chemical Society. | ||
Starting from biomass-derived 5-hydroxymethyl-furfural (HMF, 17), N-substituted 2-hydroxymethyl-5-methylpyrroles 4 (74–99% yields) can be synthesized using ethanol as solvent at room temperature after reaction for 10 min to 48 h via a two-step catalyst-free process, involving the hydrogenation of HMF (17) to 1-hydroxyhexane-2,5-dione (HHD, 18) over Ir-complex 19, followed by Paal–Knorr reaction with amines and anilines (2) bearing both electron-withdrawing and electron-donating groups (Scheme 6).106 The presence of hydroxyl functionality has the possibility for further modification to afford bioactive compounds and to introduce desired ligating groups, with no detailed purification steps being required to obtain the desired products, except for simple evaporation of the solvent (ethanol) and vacuum drying.
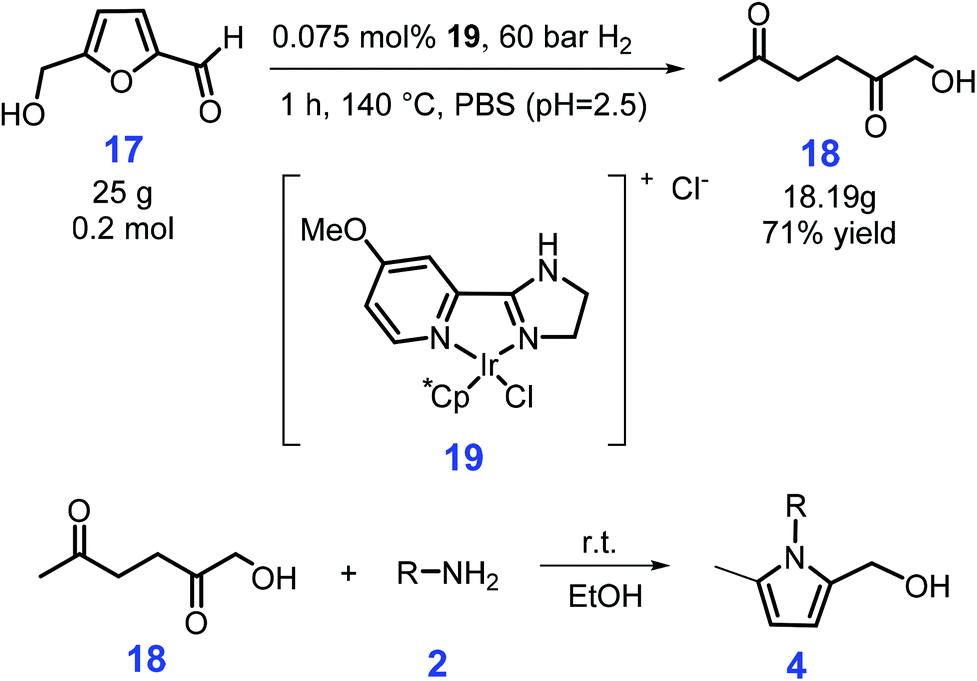 | ||
| Scheme 6 Synthesis of N-substituted 2-hydroxymethyl-5-methylpyrroles from 5-hydroxymethyl-furfural (HMF) via 1-hydroxyhexane-2,5-dione (HHD). Adapted with permission from ref. 106, Copyright © 2018, Wiley-VCH. | ||
Conventionally, reaction of hexose sugars (e.g., glucose and fructose) directly with amines 2 are able to proceed either Amadori rearrangement of glycosylamine 20 or Maillard reaction, affording the equilibrium mixture of furanose 21a, pyranose 21b, and the open chain isomer 21c in solution107 or a complex mixture of poorly characterized molecules at 200 °C or higher,108 respectively (Scheme 7). In the presence of an organic acid (oxalic acid), Adhikary et al. found that glucose directly reacting with N-benzylamine gives 1-benzyl-5-(hydroxymethyl)pyrrole-2-carbaldehyde with a maximum yield of 40% in DMSO at 90 °C for 0.5 h.109 In contrast, acetic acid (pKa 4.7), trifluoroacetic acid (pKa 0.23), and sulfuric acid (pKa −3.0) afford the pyrrole in yields of 8%, 28%, and 16%, respectively. The optimal acidity of oxalic acid (pKa 12.5) most likely contributed to its superior reactivity in the cascade process involving nonaqueous Maillard reaction, ring-opening, dehydration, cyclization, and hydrolysis to produce the pyrrole 4 and regenerate the amine 2 (Scheme 7).109 The hydroxyl configuration of sugars does not remarkably affect the reaction efficiency, and corresponding N-substituted 5-(hydroxymethyl)pyrrole-2-carbaldehydes in comparable yields (21–53%) is obtained from galactose, mannose, ribose, or xylose by reacting with primary amines. Due to the existence of active species (–OH, –CHO), the pyrrole-2-carbaldehyde skeleton shows great potential for production of pyrrole alkaloid natural products (Scheme 7). The use of more easily available bio-based feedstocks can remarkably reduce production cost, and improve development of correlated reaction routes with recoverable and lost-cost catalysts in the presence of low-pressure liquid H-donor sources (e.g., HCOOH, alcohols) and is thus highly desirable for the synthesis of pyrroles.
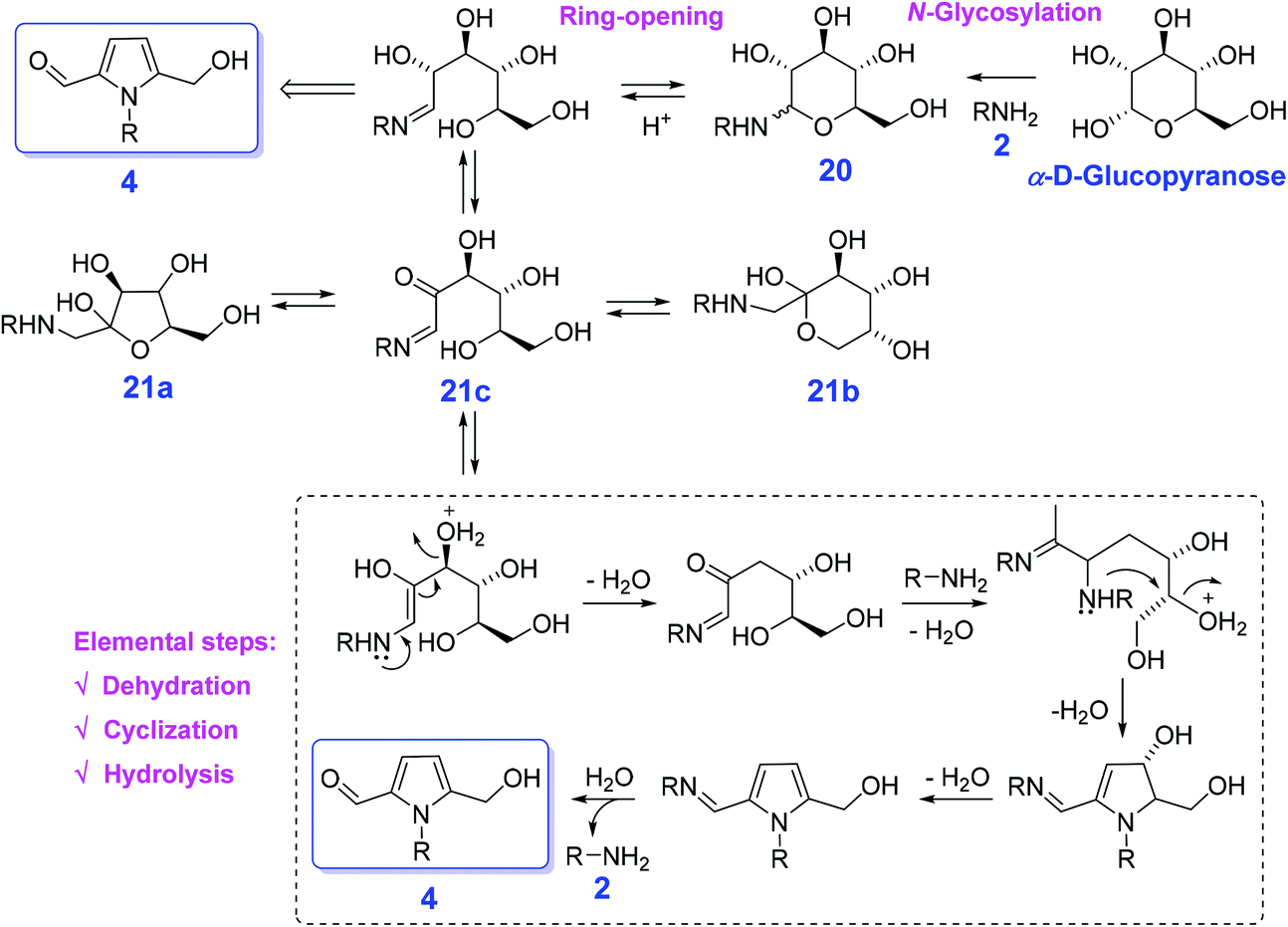 | ||
| Scheme 7 Direct conversion of hexose sugars by reacting with amines 2 to N-substituted 5-(hydroxymethyl)pyrrole-2-carbaldehydes 4. Adapted with permission from ref. 109, Copyright © 2015, American Chemical Society. | ||
2.2. Pyrrolidones
Pyrrolidones are important core scaffolds widely applied in pharmaceutical products, printing inks and fiber dyes, which are also directly employed as surfactants and solvents.110,111 In the presence of homogeneous or heterogeneous metal catalysts, pyrrolidones 23 could be produced from biomass-derived levulinic acid (LA, 22a) or keto acids (22) through either reductive amination or amidation processes with H2, formic acid or hydrosilane as hydrogen source (Scheme 8).112,113 In most cases, imines 24 are reported to be first formed from amination of LA (22a) with primary amines 2, followed by hydrogenation to give γ-amino-pentanoic acid 25 that is finally subjected to intramolecular amidation to yield pyrrolidones 23 (path 1, Scheme 8).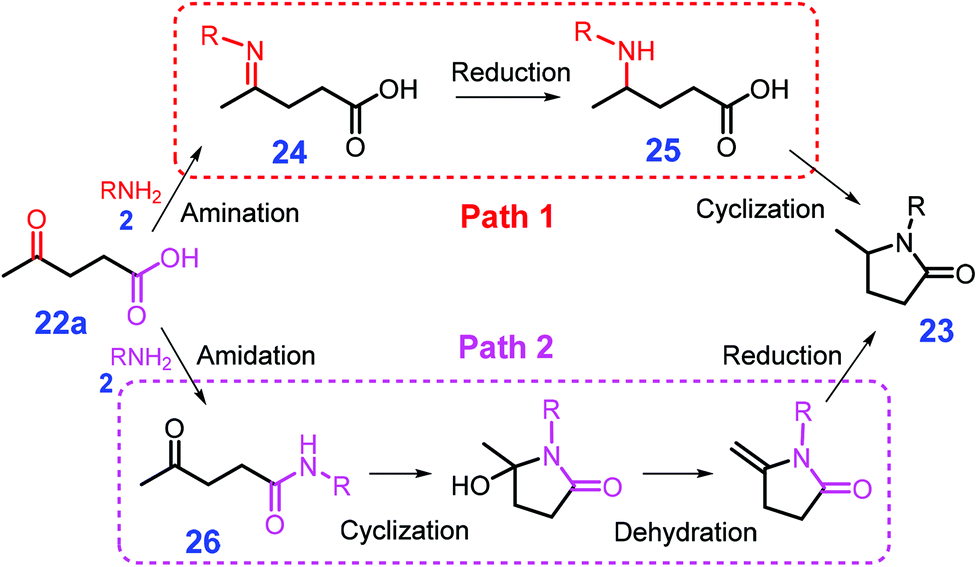 | ||
| Scheme 8 Catalytic reductive amination of levulinic acid (LA, 22a) with primary amines 2 to pyrrolidones via amination–reduction–cyclization (path 1) or amidation–cyclization–dehydration–reduction (path 2). Adapted with permission from ref. 112 and 113, Copyright © 2017 & 2018, Royal Society of Chemistry. | ||
Molecular hydrogen gas is frequently used as H-donor in schemes for cascade reductive amination and amidation of LA (22a) or its ester to synthesize pyrrolidones. Among the developed catalytic systems, noble metal catalysts (e.g., Pt/P-TiO2, Pt-MoOx/TiO2, Pt/TiO2D, Ru/TiO2, Ir/SiO2-SO3H, and iridium complexes) are highly efficient for the cascade reaction process (up to 99% yield of pyrrolidones) under mild conditions (room temperature to 120 °C, P(H2) ≲1 MPa).114–120 Apart from LA (22a) and primary amines, carbohydrates (e.g., glucose) and nitro compounds can be employed as the precursor of LA (22a) and nitrogen sources for producing pyrrolidones via additional acid-catalyzed hydrolysis and in situ hydrogenation steps, respectively.121,122 The replacement of precious elements in Scheme 8 with abundant and low-cost metals is highly desirable, for which several well-designed base metal catalysts, such as Cu15/Al2O3 doped with Pr (Cu15Pr3/Al2O3),123 carbon-supported FeNi nanoparticles,124 and carbon nanotubes supported porous-carbon-coated Ni (CNFx@Ni@CNTs)125 have been explored for synthesis of N-substituted pyrrolidones (up to 99% yield) from LA (22a) and amines although they require relatively harsh reaction conditions (130 °C to 175 °C, 1 to 5 MPa H2). Well dispersed metal particles (e.g., Cu and Ni) on porous solid supports (e.g., γ-Al2O3, carbon nanotubes) and the use of continuous fixed-bed reactors enhance catalytic performance of prepared non-noble metals.123–125 Solid supports not only increase metal stability and improve reusability, but also provide acid sites with appropriate acid strength and density that enhances substrate adsorption onto active sites of the catalyst that facilitates tandem condensation and hydrogenation processes.
In comparison with non-noble metals (e.g., Ni), precious metals (e.g., Pt) typically show stronger chemisorption of the imine generated from LA (22a) and an amine (e.g., benzyl amine) via condensation, but they have lower affinity for H2.126,127 With respect to non-noble metal catalysts, weak chemisorption towards the intermediate leads to difficulty in activation of the imine, while strong H2 adsorption results in excessive blocking of the metal surface. These two factors might eventually result in the non-noble metal catalyst to have relatively low hydrogenation activity based on the Sabatier principle.128 In good agreement with the above statement, Gao et al. elaborated an unconventional pathway for the reductive amination of LA (22a) over a base metal catalyst, in which Ni/C first promotes the formation of amides 26 followed by undergoing cascade cyclization, dehydration, and reduction to yield the target product pyrrolidone 23 (path 2, Scheme 8),125 in accordance with the Sabatier principle.
Instead of using flammable H2 gas, air-stable and safe hydrosilane (e.g., PhSiH3 and (EtO)3SiH) can be used as a mild reductant for reductive cycloamination of LA (22a) with primary amines to produce pyrrolidones 23 in moderate to good yields (up to 99%) over B(C6F5)3,129 Fe-complex,130 or transition metal salts such as In(OAc)3 and AlCl3·6H2O131,132 at 30 to 120 °C for 1 to 24 h, as in Scheme 9. Particularly, by changing the transition metal salt from In(OAc)3 or AlCl3·6H2O to either InI3 or RuCl3·3H2O that bear stronger Lewis acidity, affords pyrrolidines 27 in good yields (55–93%) under similar reaction conditions to those used in the synthesis of pyrrolidones 23.131,132 Namely, the removal of oxygen from the lactam (pyrrolidone, 23) with hydrosilane (PhSiH3) to give the cyclic amine (pyrrolidine, 27) and corresponding siloxane needs to be activated by a Lewis acidic metal salt that is stronger than In(OAc)3 or AlCl3·6H2O. In other words, the selectivity toward pyrrolidones can be controlled by design of the catalyst with appropriate Lewis acidity or basicity. Similar to metal salts, a pharmaceutically acceptable and nontoxic ionic liquid 1-butyl-3-methylimidazolium lactate ([BMIm][Lac]), in combination with (EtO)3SiH, is efficient for production of pyrrolidones 23 (36–96% yields) at 80 °C within 1 h to 3 h.133 Serving as a multifunctional catalyst, [BMIm][Lac] with both acidic (–OH) and basic (–COO−) sites concurrently promotes activation of hydrosilane and cycloamination of LA (22a) or keto acids (22) successively to 24 and 28, to finally afford pyrrolidones 23 (Scheme 9B). However, the use of hydrosilanes as H-donor may co-produce waste due to the formation of silicon resins, and the spent homogeneous catalysts suffer from issues regarding reusability and potential environmental contamination. Exploration of benign and eco-friendly alternatives for both aspects is thus highly desirable for sustainable synthesis of pyrrolidones and other relevant azaheterocycles.
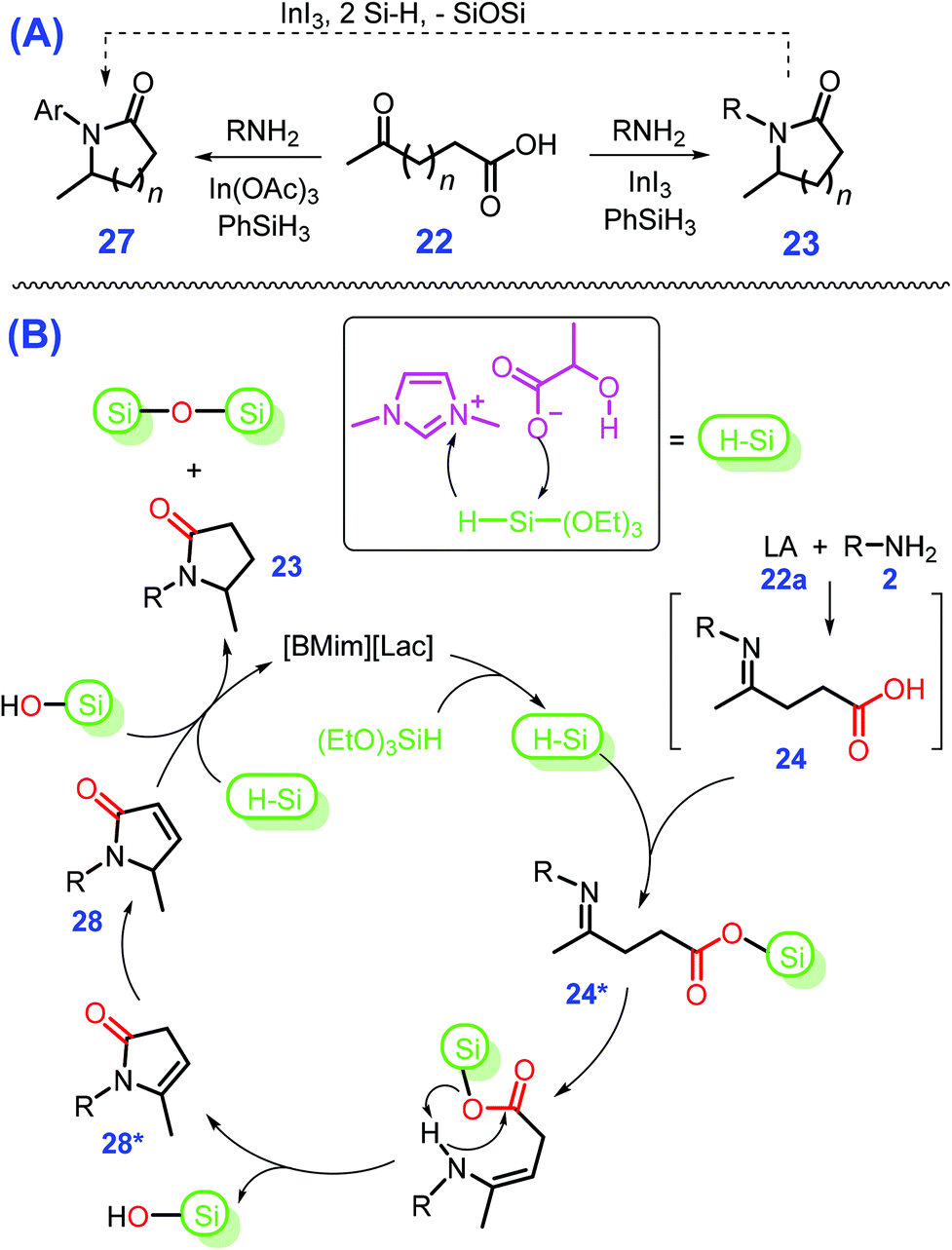 | ||
| Scheme 9 Schematic illustration for (A) reductive cycloamination of LA (22a) or keto acids (22) to pyrrolidones/lactams 23 or pyrrolidines/cyclic amines 27 (30 °C to 120 °C for 1 h to 24 h) and (B) detailed reaction mechanism. Adapted with permission from ref. 131–133, Copyright © 2016, Wiley-VCH; Copyright © 2017, Royal Society of Chemistry; Copyright © 2017, American Chemical Society. | ||
Formic acid (HCOOH) was a co-product of LA (22a) in the synthesis of LA from biomass sugars, and its use as a hydrogen source minimizes the generation of chemical waste in biorefineries.134,135 Metal catalysts such as Au/ZrO2-VS, Ir-complex, Ru-complex, Ru3(CO)12, and Fe3(CO)12 allow selective decomposition of HCOOH to CO2 and H2, while they were normally unable to tolerate CO that forms in the dehydration of HCOOH.136–140 In the reductive cycloamination of LA (22a), either straightforward hydrogenation with H2 generated in situ from HCOOH or via transfer hydrogenation process occurs as the rate-determining step in the synthesis of pyrrolidones.136–140 Instead of using high-pressure hydrogen or acidic HCOOH and basic ammonia or amines for the reductive amination reaction, ammonium formate (HCOONH4) is a convenient nitrogen and hydrogen source. Amarasekara and Lawrence found that RANEY® Ni efficiently catalyzes cycloamination of LA (22a) with HCOONH4 in water, affording 94% yield of 5-methyl-2-pyrrolidone (MPD) at 180 °C for 3 h,141 and their protocol has authentic safety advantages over the conventional methods using high-pressure H2–NH3 gas mixture. Sun et al. showed that NHC (N-heterocyclic carbene)-based Ru(II)-coordination polymer is active for the synthesis of MPD (up to 99% yield) from LA (22a) and HCOONH4 at 80 °C for 12 h, with TON value of 6.7 × 104 being reported,142 with both catalysts being recyclable without apparent activity loss, and being applicable to producing N-substituted 5-methyl-2-pyrrolidones from amines and LA (22a).141,142
Under non-catalytic conditions, Wei et al. reported that LA (22a) reacts with benzylamine to give 1-benzyl-5-methylpyrrolidin-2-one (BMP) to afford a moderate yield of 72% using DMSO as solvent at 100 °C for 4 h.143 Upon addition of equivalent triethylamine relative to LA (22a), BMP yield increases to 89% under otherwise identical conditions. Thus, an appropriate balance of system acidity and basic additive (e.g., triethylamine) is used to obtain favorable reaction progress, and the catalyst-free system is suitable for reductive amination of LA (22a) with electron-deficient and -rich amines to afford a variety of N-substituted 5-methyl-2-pyrrolidones (up to 93% yield).143 The use of a continuous-flow microreactor heated at 140 °C remarkably accelerated the reaction rate, and comparable yields of pyrrolidones could be achieved with a residence time of 10 min.144 To further improve the greenness of the reaction, Ledoux et al. shows that the thermal treatment of LA (22a), HCOOH and amine mixtures allow access to a series of 5-methylpyrrolidone derivatives with >80% yields in most cases.145 Due to the absence of catalysts, solvents or additives, this sustainable and efficient reaction system has an exceptionally low E-factor of 0.2 and has good potential for application on the industrial scale.
Li et al. developed a generic strategy that does not require catalyst or external hydrogen by involving in situ controlled-release of HCOOH from N-formyl species (e.g., HCONH2) with H2O for the cycloamination of LA (22a) and other keto-acids (22), that provides access to 5-methylpyrrolidones and relevant N-(un)substituted lactams,146 that is elucidated by a combination of model experiments and density functional theory calculations. An unconventional pathway via cyclic imines (5-methyl-3,4-dihydro-2-pyrrolone (29) and its tautomeric structures) as key intermediates is elucidated by density functional theory (DFT) calculations show yields of 5-methyl-2-pyrrolidone (30) from LA (22a) and HCONH2, which is different from the conventional approaches encompassing cascade reductive amination and cyclization (Scheme 10).146 The simple and eco-friendly protocol of Li et al.146 may open an avenue for direct synthesis of N-(un)substituted pyrrolidones/lactams by cyclic diamination of keto acids without external hydrogen gas. Besides the above-discussed reductive amination of LA (22a) and N-containing compounds, a number of other renewed synthetic routes such as ketoamides proceeding via cascade cyclization and ionic hydrogenation mediated by Al(OTf)3 and Et3SiH,147 reductive transformation of itaconic acid and NH3 over Ru/C and H2,148 reductive N-alkylation and decarboxylation of glutamic acid catalyzed by Pd/Al2O3 with and H2,149 and Zr-catalyzed N-acylation of lactams150 have been explored for efficient construction of pyrrolidone-type motifs. The catalyst-free reaction system seems more sustainable and economic for producing pyrrolidones, while its reaction rate is much lower than metal- or acid-catalyzed processes. In this regard, the design of appropriate continuous flow reactors may facilitate the rapid thermal conversion routes to the synthesis of pyrrolidones.
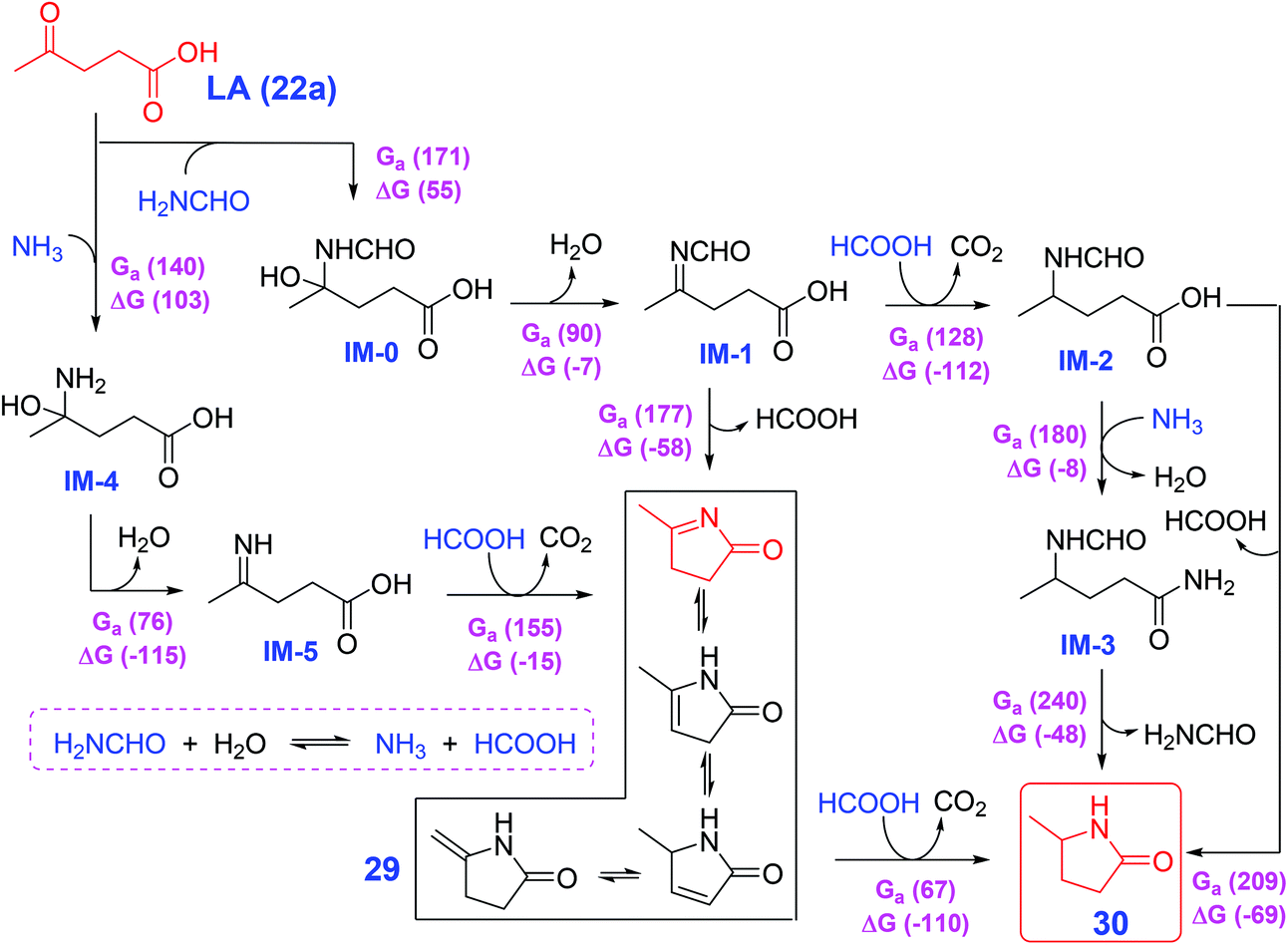 | ||
| Scheme 10 Reaction pathways for cycloamination of LA (22a) and H2NCHO in water with computed free energies and enthalpies in parentheses (kJ mol−1). IM: intermediate; MDPY: 5-methyl-3,4-dihydro-2-pyrrolone; MPD: 5-methyl-2-pyrrolidone. Adapted with permission from ref. 146, Copyright © 2019, Wiley-VCH. | ||
2.3. Pyrazoles
Pyrazoles are a class of structural motifs prevalent in biologically active agents and medicines, notably, Lexiscan, Xalkori, Celebrex, and Viagra,151 and these motifs are also present in dyes and they are utilized as ligands for metal catalysts.152,153 The synthesis of pyrazole derivative 32 from acetic anhydride reacting with glucose phenylosazone 31 which is readily derivable from glucose and phenylhydrazine in the presence of acetic acid was first reported by El Khadem et al. (Scheme 11).154–156 Under microwave irradiation, cyclic addition of glucose phenylosazone goes to completion in 5 min, affording the pyrazole derivative 32 in good yields (86%).157 The microwave-assisted reaction system154–156 is applicable to synthesis of pyrazoles (up to 96% yield) from respective osazones derived from galactose, arabinose, and xylose, and although comparable yields of pyrazoles are obtained using conventional heating modes, more than 1 h is required.158,159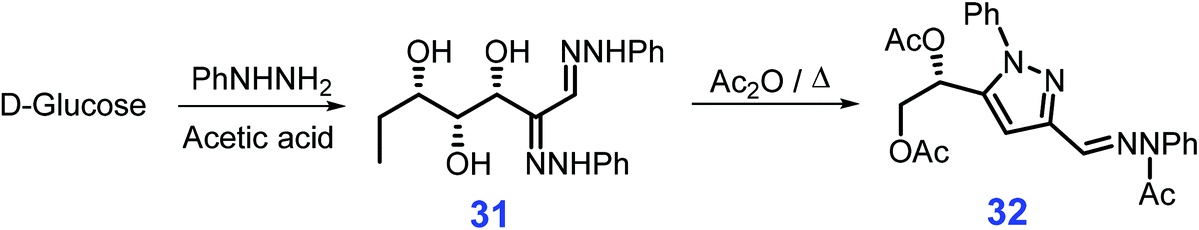 | ||
| Scheme 11 Representative synthetic routes to pyrazole derivative 32 from glucose and phenylhydrazine via glucose phenylosazone 31. Adapted with permission from ref. 157, Copyright © 2007, Taylor & Francis. | ||
As shown in Scheme 12, starting from phenylhydrazine 33, β-ketoesters 34, and aromatic aldehydes 35 (e.g., vanillin and syringaldehyde) pre-prepared from lignin by thermal treatment with 2 mol L−1 NaOH and nitrobenzene (170 °C, autogenous pressure 1 MPa, 2 h) in a stainless steel autoclave, a diversity of 4,4′-arylmehtylenebis(1H-pyrazole-5-ol)s 36 with 79–88% yields are obtained in water under microwave irradiation (300 W, 100 °C, 10 min).160 In comparison with standard drug (Trolox), all the prepared bispyrazoles exhibit good radical scavenging activities of 2,2′-azino-bis(3-ethylenzothiazoline-sulphonic acid) diammonium salt (ABST+) and N,N-diphenyl-N′-picrylhydrazyl (DPPH). Given that the environmentally benign synthetic procedures developed by Yang et al.,160 these bispyrazoles derivatives have promising potential in the therapy of free radical-related diseases (e.g., tumors).
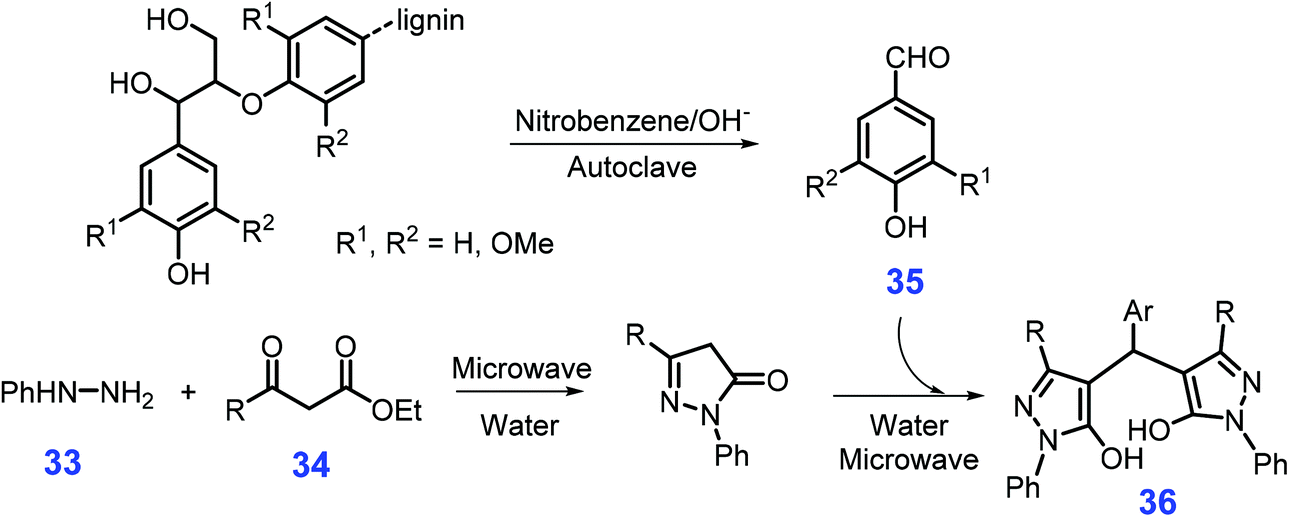 | ||
| Scheme 12 Synthetic routes to 4,4′-arylmehtylenebis(1H-pyrazole-5-ol)s 36 from phenylhydrazine 33, β-ketoesters 34, and lignin-derived aromatic aldehydes 35. Adapted with permission from ref. 160, Copyright © 2012, Wiley-VCH. | ||
Other bio-based molecules (e.g., 1,3-dicarbonyl compounds, vinylogous formamides, and 1,3-diols) besides sugars or lignin derivatives can be used to produce pyrazoles.161,162 The 1,3-dicarbonyls are commonly employed as feedstocks to construct pyrazoles by condensation with a aryl or alkyl hydrazine, while their low stability restricts their applicability for obtaining substituted pyrazoles.163,164 On the other hand, 1,3-diacetals or vinylogous formamides 37 by reaction with hydrazines 38 allow access to substituted pyrazole motifs 39 (path A, Scheme 13), while multistep synthesis procedures were prerequisite due to the existence of sensitive functional groups.165 Much attention has been paid to the development of more efficient routes for substituted pyrazoles. For instance, Schmitt et al. reported that 1,3-diols 40 used instead of the typical masked dialdehydes enable the production of pyrazoles via hydrogen transfer process promoted by a Ru-complex, RuH2(PPh3)3CO/xantphos (path B, Scheme 13).166 The scope of both the diol and hydrazine components can be extended with good compatibility, and satisfactory pyrazole yields (84%) from scaled-up isopropyl diol (1 g, 8.5 mmol) and phenylhydrazine (8.5 mmol) in toluene at 110 °C after 24 h,166 however, unreliable regioselectivity of the obtained 3-alkyl pyrazoles needs to be resolved in follow-up studies. Also, the initial acquisition of starting materials from biomass generally involves complex processes, which would be another primary task needing to take into consideration.
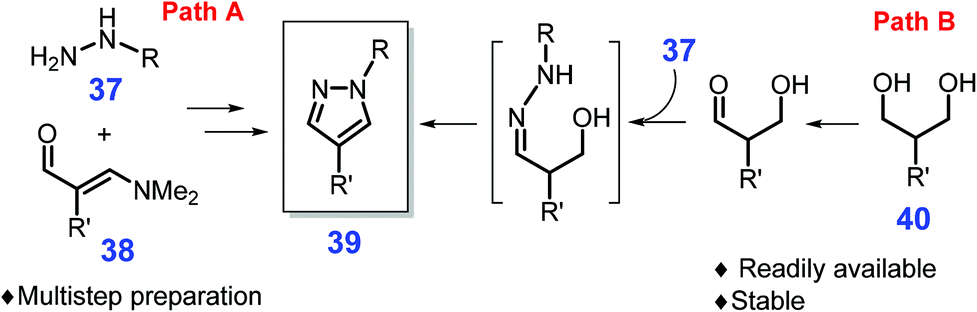 | ||
| Scheme 13 Synthetic routes to pyrazoles from masked dialdehydes 38 or diols 40. Adapted with permission from ref. 166, Copyright © 2015, American Chemical Society. | ||
2.4. Imidazoles
Like other five-membered N-heterocycles, substituted imidazoles occur in natural products bearing broad-spectrum biological activities, and are target-oriented in the preparation of functional molecules such as ionic liquids or N-heterocyclic carbenes (NHCs).167–169 Industrially, simple imidazoles are synthesized from the condensation of 1,2-dicarbonyls with ammonia and aldehydes via the Radziszewski reaction.170 In the presence of basic catalysts (e.g., CuCO3/Cu(OH)2), 4-hydroxymethyl imidazole is obtained by thermal treatment of formaldehyde and concentrated ammonia with fructose or glucose that underwent retro-aldol fission to in situ release of dihydroxyacetone and glyceraldehyde.171 If hexose sugar (e.g., fructose) is heated in a pressure vessel with formamidinium acetate and liquid ammonia, the retro-aldol fission is remarkably inhibited and instead affords 4-tetrahydroxy-butyl imidazole 41 as the dominant product (Scheme 14).172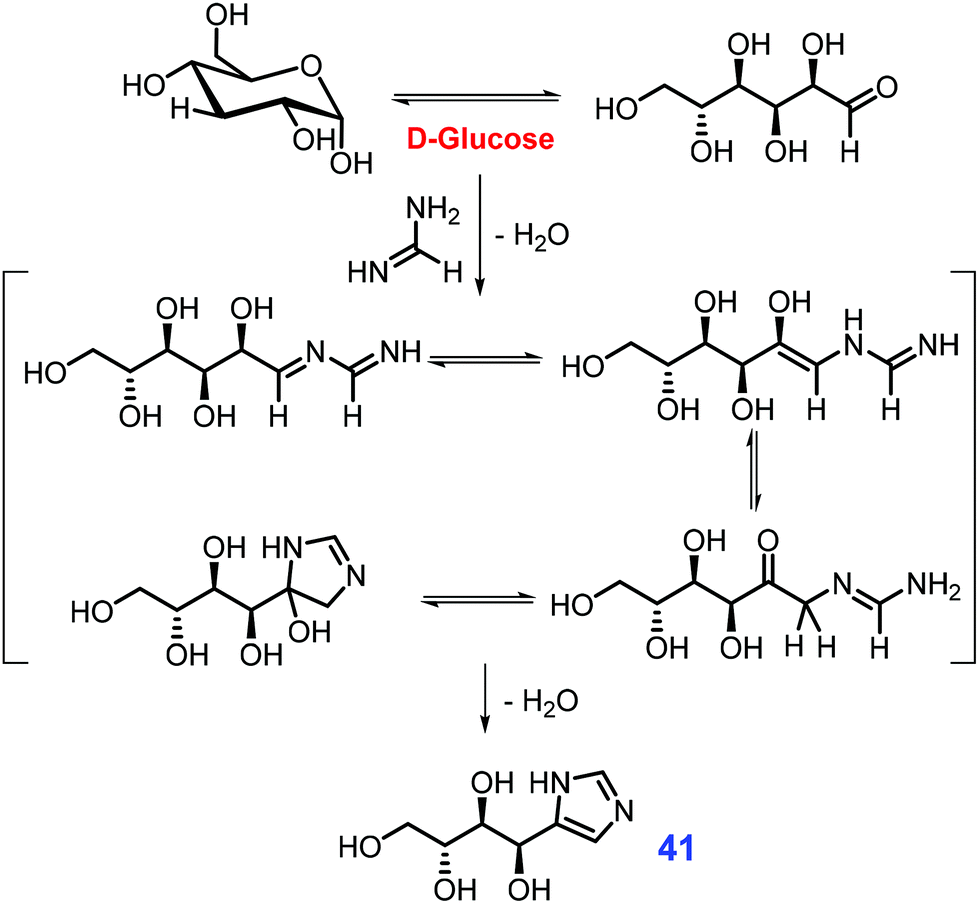 | ||
| Scheme 14 Synthetic routes to 4-tetrahydroxy-butyl imidazole 41 from glucose and formamidine. Adapted with permission from ref. 172, Copyright © 2016, Thieme Chemistry. | ||
In a one-pot process, a variety of mono- and disaccharides react with amidines 42 in molten ammonium carbonate to give tetrahydroxybutyl substituted imidazoles 43 with varying glycosylation patterns, depending on the type of saccharide substrates (Table 1).173 After purification by silica gel chromatography (eluent, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ethanol:ammonia), moderate isolated yields (25–50%) of the imidazole products 43 are obtained from sugars like fructose, glucose, isomaltulose, melibiose, leucrose, maltose, cellobiose, and lactose (Table 1) after heating at 65 °C to 80 °C until reaction completion as monitored by thin-layer chromatography.173 The reaction system uses a benign solvent (molten ammonium carbonate), it does not require protection groups in its chemistry, and is thus sustainable and has great potential to replace methods based on petroleum resources.
:1 (v/v) ethanol:ammonia), moderate isolated yields (25–50%) of the imidazole products 43 are obtained from sugars like fructose, glucose, isomaltulose, melibiose, leucrose, maltose, cellobiose, and lactose (Table 1) after heating at 65 °C to 80 °C until reaction completion as monitored by thin-layer chromatography.173 The reaction system uses a benign solvent (molten ammonium carbonate), it does not require protection groups in its chemistry, and is thus sustainable and has great potential to replace methods based on petroleum resources.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Substrate | R | Product | R4 | R3 | R2 | R1 | Yield (%) |
| a Formamidine acetate. b Ethylacetimidate. c Benzamidine hydrochloride. | |||||||
| Fructose | Ha | 2 | H | H | H | H | 50 |
| Glucose | H | 2 | H | H | H | H | 47 |
| Isomaltulose | H | 3 | α-D-Glc | H | H | H | 49 |
| Isomaltulose | Meb | 4 | α-D-Glc | H | H | H | 30 |
| Isomaltulose | Phc | 5 | α-D-Glc | H | H | H | 5 |
| Melibiose | H | 6 | α-D-Glc | H | H | H | 38 |
| Melibiose | Me | 7 | α-D-Glc | H | H | H | 27 |
| Leucrose | H | 8 | H | α-D-Glc | H | H | 38 |
| Leucrose | Me | 9 | H | α-D-Glc | H | H | 26 |
| Maltose | H | 10 | H | H | α-D-Glc | H | 28 |
| Cellobiose | H | 11 | H | H | β-D-Glc | H | 25 |
| Lactose | H | 12 | H | H | β-D-Glc | H | 40 |

Several other synthetic approaches were also developed to produce substituted imidazoles from sugars (Scheme 15).174–176 For example, arabinose-derived aldehyde 44 undergoes condensation with glyoxals 45 in methanol/NH3 mixtures to yield linear imidazole sugars that can be further converted to the imidazole 46 after the removal of the trityl group with HCl in dioxane (Scheme 15A).174 The reaction of O-acetylated glucoseamine 47 with o,o′-disubstituted arylisothiocyanates 48 gives 49, which when followed by treatment with acetic anhydride in pyridine and subsequent elimination of acetic acid affords the imidazoline thion 50 (Scheme 15B).175 In another manner, direct introduction of imidazole ring is achieved through adding 2-lithio1-[(dimethylamino)methyl]-1H-imidazole 52 to 2,3,4,6-O-benzyl-glucono-1,4-lactone 51, yielding sugar-derived imidazole 53 (Scheme 15C).176
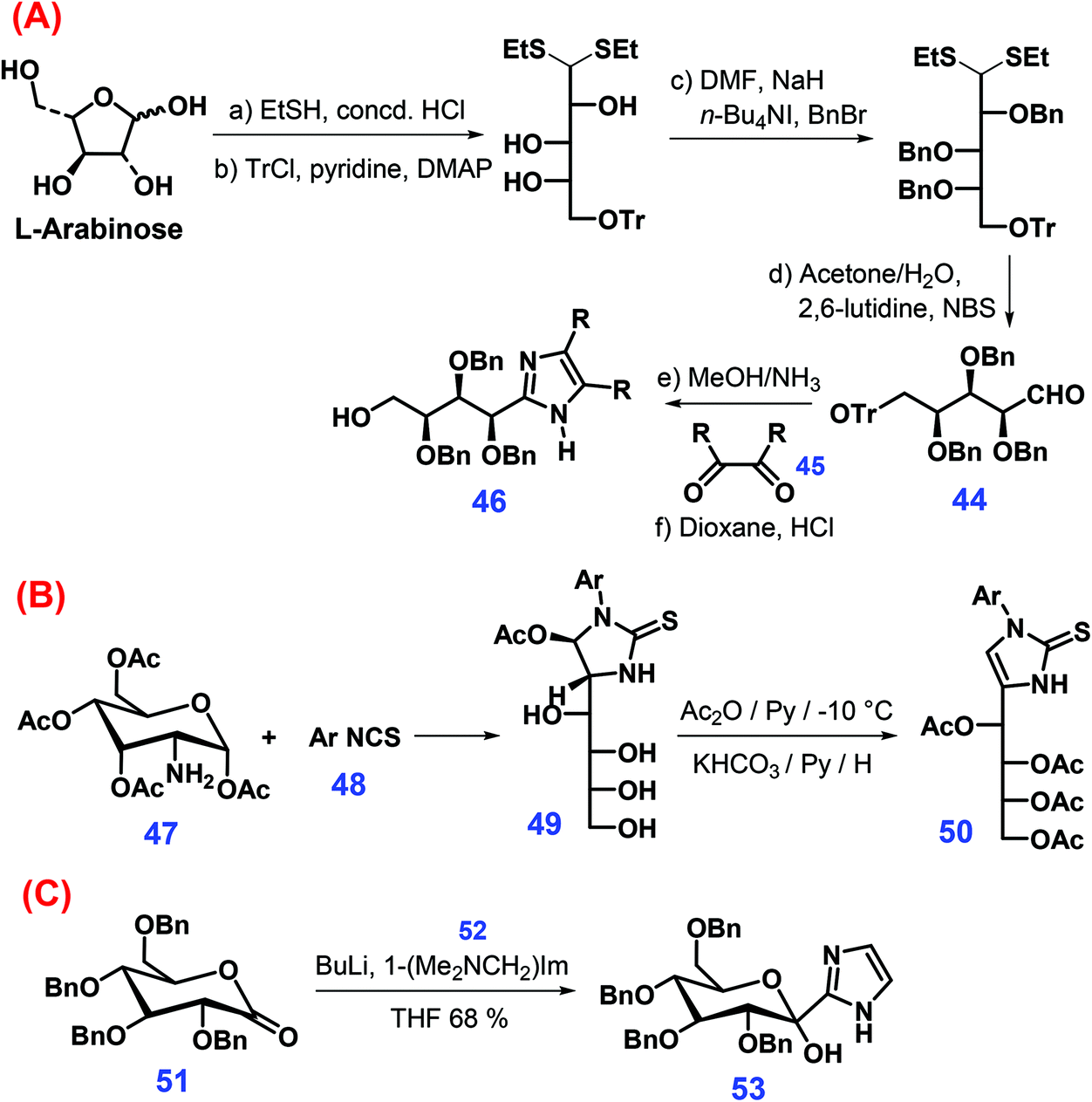 | ||
| Scheme 15 Synthetic routes to imidazole derivatives from sugars: (A) L-arabinose, (B) O-acetylated glucoseamine 47, and (C) 2,3,4,6-O-benzyl-glucono-1,4-lactone 51. Adapted with permission from ref. 174–176, Copyright © 1995 & 2006, Wiley-VCH; Copyright © 2015, Elsevier. | ||
The 2-haloenones 54 derivable from sugars were deemed as crucial intermediates for construction of heterocycles.177,178 In the absence of any ligand or metal catalyst at ambient temperature, Mal and Das showed that a diversity of chiral hydroxy imidazoles 55 and furans 56 were able to be synthesized from 2-haloenones 54 with amidines or 1,3-dicarbonyl compounds, respectively, over K2CO3 in DMSO at room temperature (Scheme 16) through sequential Michael addition, cyclization, and sugar-ring opening reactions.179 Notably, the benign reaction system is tolerant to the substrates with variable substituents, affording chiral substituted imidazoles 55 with moderate to good yields (43–88%) and excellent regioselectivity (single in most cases) at room temperature within 45–120 min. It is worth mentioning that the yields of imidazoles obtained from sugars are relatively low (typically <50%), possibly due to the complicated reaction processes, which has to be improved by developing more efficient catalyst systems.
 | ||
| Scheme 16 Synthetic routes to either chiral hydroxy imidazoles 55 or furans 56 from 2-haloenones 54. Adapted with permission from ref. 179, Copyright © 2016, American Chemical Society. | ||
2.5. Other five-membered N-heterocycles
Furfural, as one of five-membered oxygen-containing heterocycles, could be readily produced from xylose.180 Through condensation of cysteine 57 with 2-cyanofuran 58 that is pre-synthesized from furfural, aqueous ammonia and iodine over basic K2CO3 in a mixture of methanol/water at 60 °C, 4-carboxy-2-furylthiazoline 59 could be obtained, followed by alkylation with MeI over K2CO3 in N,N-dimethylformamide (DMF) to produce furylthiazoline 60 in a three-step overall yield of 63% (Scheme 17).181 Finally, 2-furylthiazole 61 (97% yield) is formed by activated carbon-promoted the thermal aromatization of the thiazoline ring of 60 in an oxygen atmosphere (1 bar, O2 balloon) at 100 °C in toluene.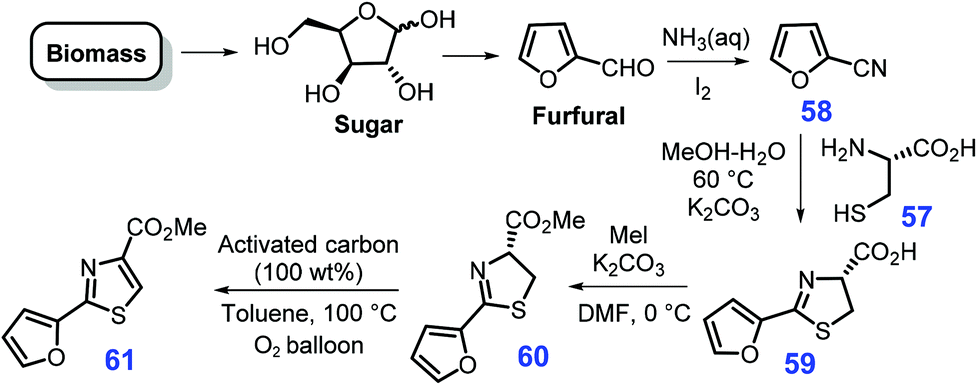 | ||
| Scheme 17 Synthetic routes to 2-furylthiazole 61 from bio-based furfural. | ||
Similar to the construction of the furan-thiazole conjugated scaffold, furyloxazole 63 can be obtained (52% yield) via a one-pot cascade dehydrative condensation and oxidation of bio-based furfural and serine methyl ester 62 (Scheme 18).182 At room temperature, the condensation of furfural with serine methyl ester 62 mediated by K2CO3 and MgSO4 in N,N-dimethylacetamide (DMA) affords oxazolidine 64 in ring-chain tautomers (64a and 64b), followed by oxidation over BrCCl3/DBU (1,8-diazabicyclo(5.4.0)undec-7-ene) to give 2,5-dihydrooxazole 65. The resulting intermediate is then subjected to isomerization and a second oxidation, ultimately yielding the furyloxazole 63.182
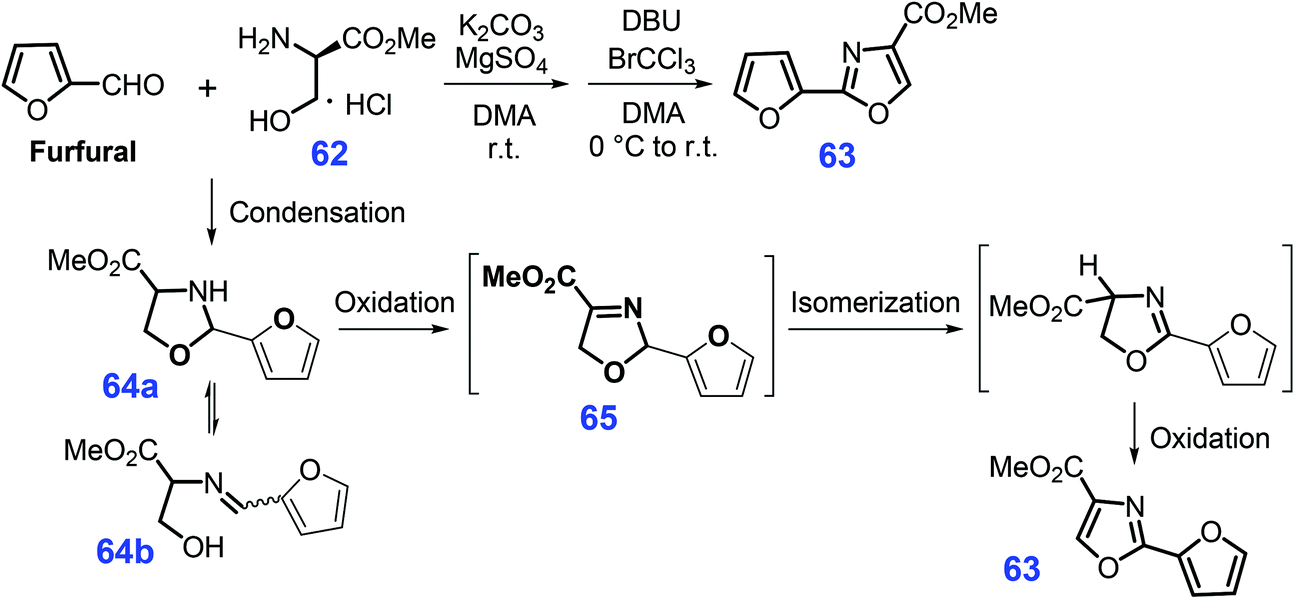 | ||
| Scheme 18 Synthetic routes to furyloxazole 63 from serine methyl ester 62 and furfural. Adapted with permission from ref. 182, Copyright © 2010, American Chemical Society. | ||
The readily available feedstock 2,5-furandicarboxylic acid (FDCA, 66) can be derived from cellulosic biomass-derived HMF (17) via oxidation.183 Selvakumar et al. demonstrated that FDCA (66) can be used for producing chiral bisoxazolines 69 (Scheme 19).184 Initially, FDCA (66) is transformed into its acyl chloride 67, followed by condensation with chiral substituted amino alcohols to yield relevant amides 68 (32–69% yields), which are eventually cyclized to generate the bisoxazolines 69. Furthermore, other five-membered N-heterocycles such as 1,2,3- and 1,2,4-triazoles, oxadiazoles, thiadiazoles, and tetrazoles can be synthesized from sugars in modest yields.185 However, most reaction systems involved lack stereo- or enantioselectivity, and need to be updated to use green synthetic methods (e.g., photo- and electroinduction) as such pathways are highly desirable for concise synthesis of these N-heterocycles.186–188
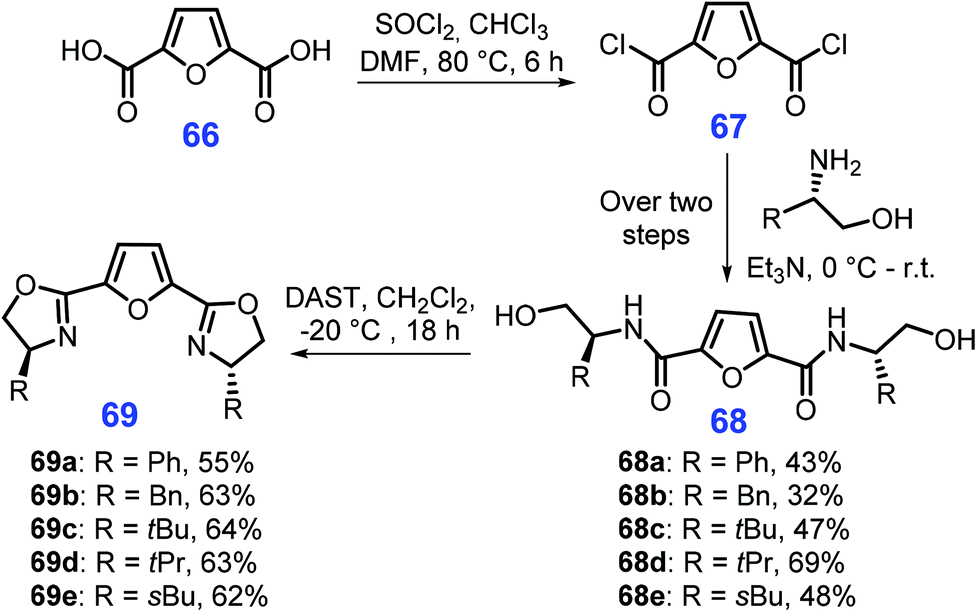 | ||
| Scheme 19 Synthetic routes to chiral bisoxazolines 69 from 2,5-furandicarboxylic acid (FDCA, 66). | ||
3. Six-membered N-heterocycles
3.1. Pyridines
Pyridines, which have high biological activity, are prevalent in many agrochemicals and pharaceuticals,189,190 as well as directly employed as catalysts and solvents in chemical syntheses.191,192 Industrially, pyridines (pyridine and picolines) are mainly produced by Chichibabin condensation of aldehydes (e.g., formaldehyde and acetaldehyde) with ammonia in fixed-bed reactors.193–196 A more effective method for producing pyridines than Chichibabin to react 70 with NH3 to afford pyridine 71 in high yields (ca. 70%) with up to ca. 60% 3-picoline 72 yield upon addition of propanal or acetaldehyde.197 Typically, several elemental steps including C–N condensation, Michael addition, hydrogenation, and dehydrogenation involve in the synthesis of pyridine and 3-picoline from acrolein (Scheme 20), whereas acetaldehyde formed in situ from acrolein via water-promoted retro-aldol reaction is important in the formation of pyridine while acrolein hydrogenation to propionaldehyde is necessary for 3-picoline.198,199 Considering that acrolein is a key intermediate in the synthesis of pyridines, glycerol as the co-product of biodiesel industry and waste polylactic acid are also explored as starting material for the synthesis of pyridines despite of relatively low yields.200–204 Zeolite-based catalysts (e.g., HZSM-5) have unique features such as shape selectivity, high surface area, and good thermal stability, and they are highly active for the production of pyridines by promoting C–N condensation and Michael addition.205–207 In most cases, the pore size and acidity of ZSM-5 have to be regulated by treatment with alkali or acids or introduction of metal oxide into their structure, so as to increase picoline yield with increased basicity or acidity in the modified catalysts.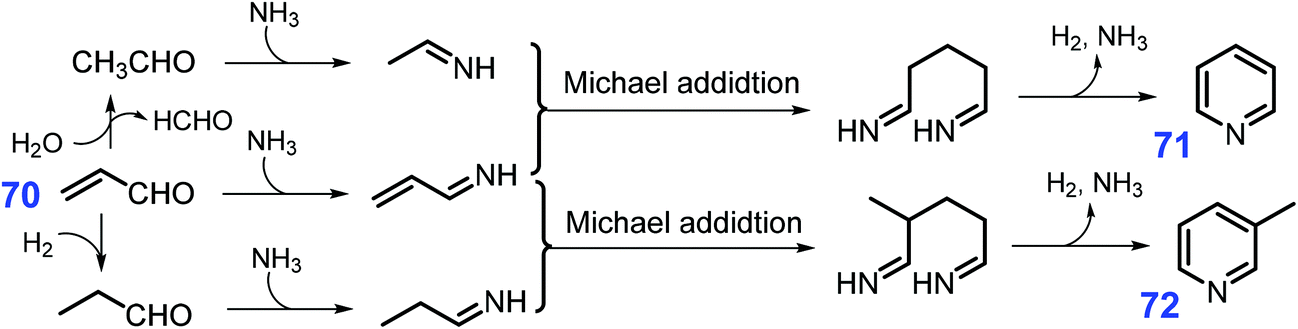 | ||
| Scheme 20 Synthetic routes to pyridines (71 and 72) from acrolein 70 and NH3. Adapted with permission from ref. 198 and 199, Copyright © 2019, Springer Nature; Copyright © 2011, Wiley-VCH. | ||
Via a three-step sequential reaction process, 3-pyridinol 74 can be synthesized from xylose or even pentosan involving acid-catalyzed hydrolysis/dehydration to furfural, reductive amination to furfurylamine 73, and succedent 30% H2O2-mediated oxidation in 3 mol L−1 HCl (Scheme 21A).208 In contrast, more laborious procedures are required for the upgrading fructose (or hexosan) to 6-hydroxymethyl-3-pyridinol 77 (Scheme 21B), where N- and O-blocking (75 → 76) of HMF 17 are prerequisite prior to proceeding with electrocatalytic oxidation in methanol, thus leading to tandem deacetylation, acetal-hydrolysis, and cyclization to form the pyridine framework (85% yield of 77).208 In related ways, 2-(hydroxymethyl)-5-(aminomethyl)-furan 75 can be converted to 6-methyl-3-pyridinol 78 with a high yield of 88% via cascade hydrolysis and cyclization catalyzed by HCl under reflux (Scheme 21C).209
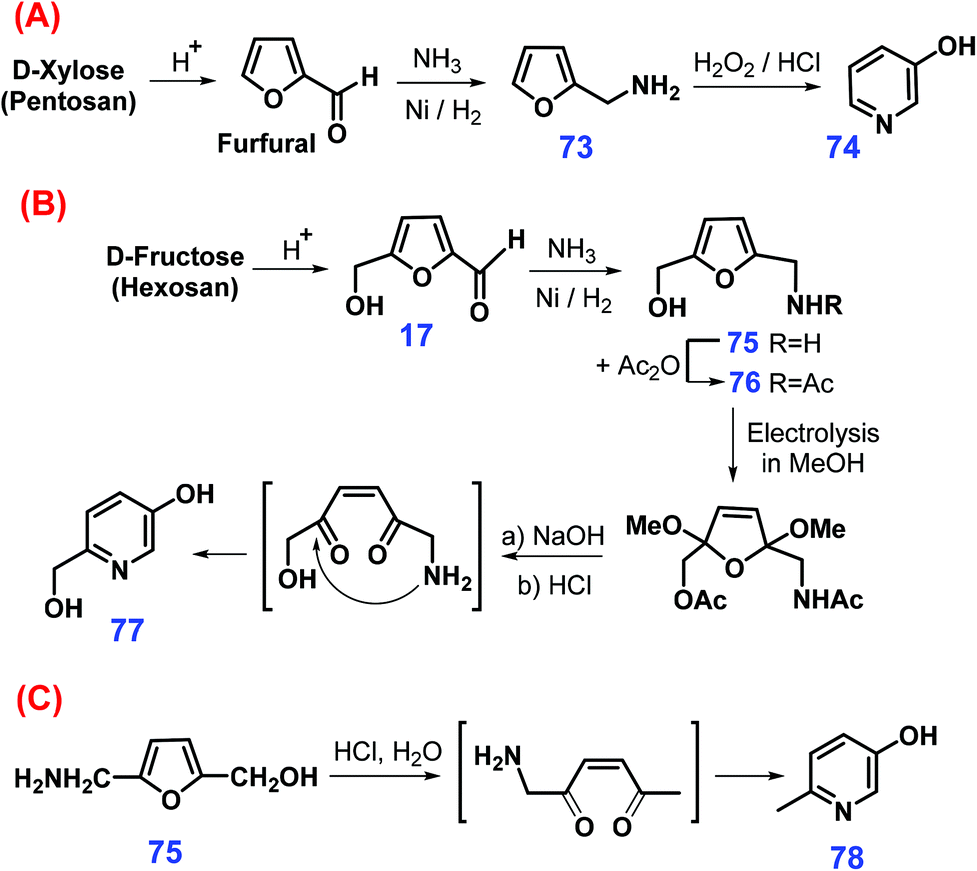 | ||
| Scheme 21 Synthetic routes to pyridinols (74, 77 & 78) from furfuryl amines: (A) 73, (B) 75 & 76, and (C) 75 derived from pentose and hexose sugars. Adapted with permission from ref. 208, Copyright © 1998, Elsevier. | ||
3.2. Pyrazines
Pyrazines have two nitrogen atoms in a 6-memebered aromatic ring and exhibit good antitumor, antibacterial, and antibiotic activities, which are extensively applied in commercial medicines and in the polymer industry.210–213 Three major routes commonly employed for the synthesis of pyrazines 81 are (a) intermolecular dehydrogenative coupling of ethylenediamine 79 with 1,2-propanediol 80 or dehydrogenative self-coupling of 2-amino alcohols 12 (Scheme 22A),214,215 (b) intramolecular cyclization–dehydrogenation of hydroxyl diamines such as hydroxypropyl ethylenediamine 82 (Scheme 22B),216 and (c) Maillard condensation of sugars with amino acids.217 However, these developed approaches suffer from environmental issues due to the use of toxic organic ligands and low selectivity toward pyrazines that can be accompanied with unwanted side reactions.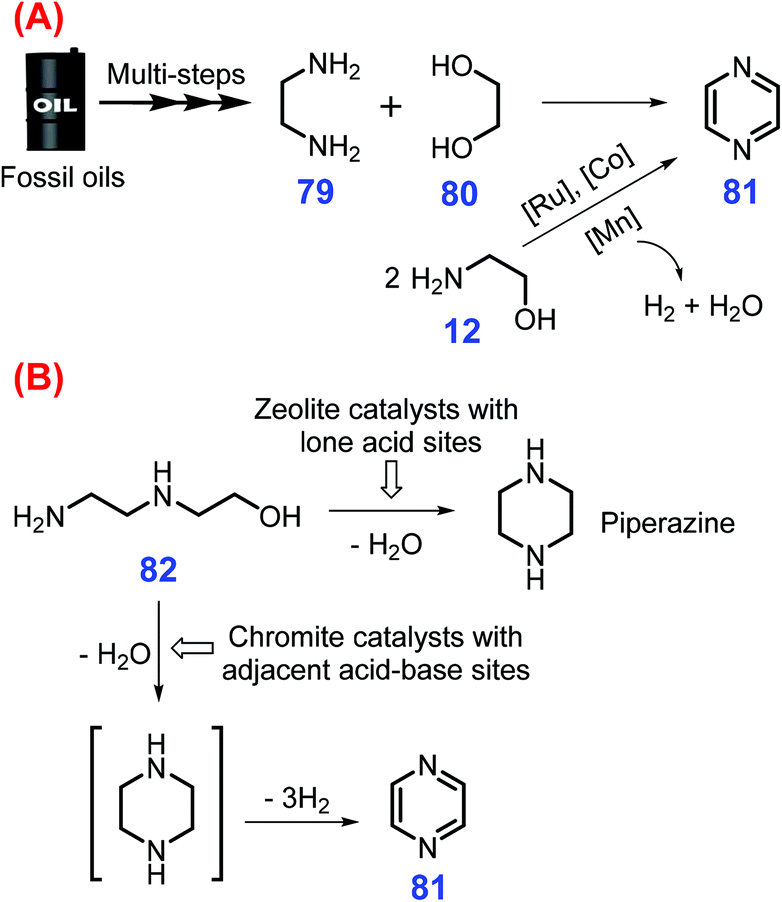 | ||
| Scheme 22 Synthetic routes to pyrazines 81via (A) inter- and intramolecular dehydrogenative self-coupling of amino alcohols and (B) intramolecular cyclization dehydrogenation of hydroxyl diamines. Adapted with permission from ref. 214, Copyright © 2003, Elsevier. | ||
Starting from glyceraldehyde 83 or 1,3-dihydroxyacetone 84 which can be derived from biomass, Song et al. found that 2-hydroxymethyl-5-methylpyrazine 87 is obtained in good yields (ca. 72%) by reacting 87 in the presence of diammonium phosphate at 90 °C for 1 h in basic mixture solvent of water and dioxane (pH = 8.0–9.1).218 The 2-imino-1,3-propanediol 85 was identified to be a key reaction intermediate, which might be formed by ketimine condensation of 1,3-dihydroxyacetone 84 with NH3 generated in situ from diammonium phosphate, followed by cyclization to 86 and final dehydration to yield the pyrazine product 87 (Scheme 23).218 Among these three elemental steps, the rate-determining step is the cyclization reaction, which is possibly ascribed to the lack of active catalytic sites.
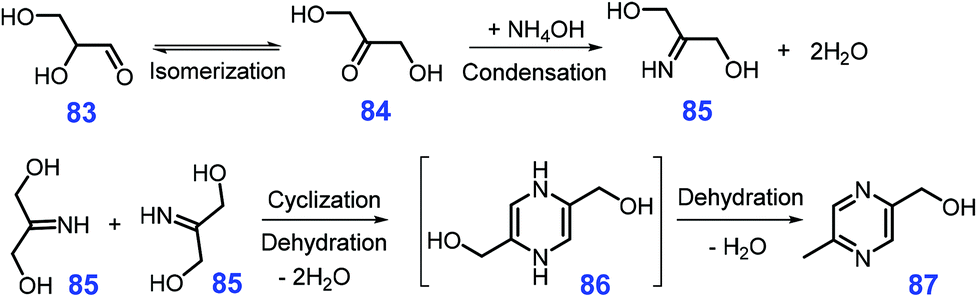 | ||
| Scheme 23 Plausible pathways for synthesis of 2-hydroxymethyl-5-methylpyrazine 87 from diammonium phosphate and 1,3-dihydroxyacetone 84 at hydrothermal conditions. Adapted with permission from ref. 218, Copyright © 2017, Royal Society of Chemistry. | ||
In the presence of a tungsten-based catalyst (ammonium metatungstate), Chen et al. reported that glucose in 25% aqueous ammonia is transformed directly into 2-methyl pyrazine 89 in moderate yields (ca. 25.6%) within 15 min at 180 °C via tandem fragmentation and cyclization in a single pot (Scheme 24).219 Prior to fragmentation, β-D-glucopyranosylamine 88 formed from condensation of glucose and NH3 was identified as one of the important intermediates. The reaction system was remarkably facilitated by tungsten clusters (e.g., [HW2O7]− and [W4O13]2−), with 7.2–23.3% yields of 2-methyl pyrazine 89 being achieved from monosaccharides (e.g., fructose, xylose, chitin monomer, glucosamine) and several disaccharides (e.g., maltose and cellulose) under otherwise identical reaction conditions.219 From the viewpoint of sustainability, it would highly desirable to further develop more robust and active catalysts for using tandem fragmentation and cyclization methodologies.
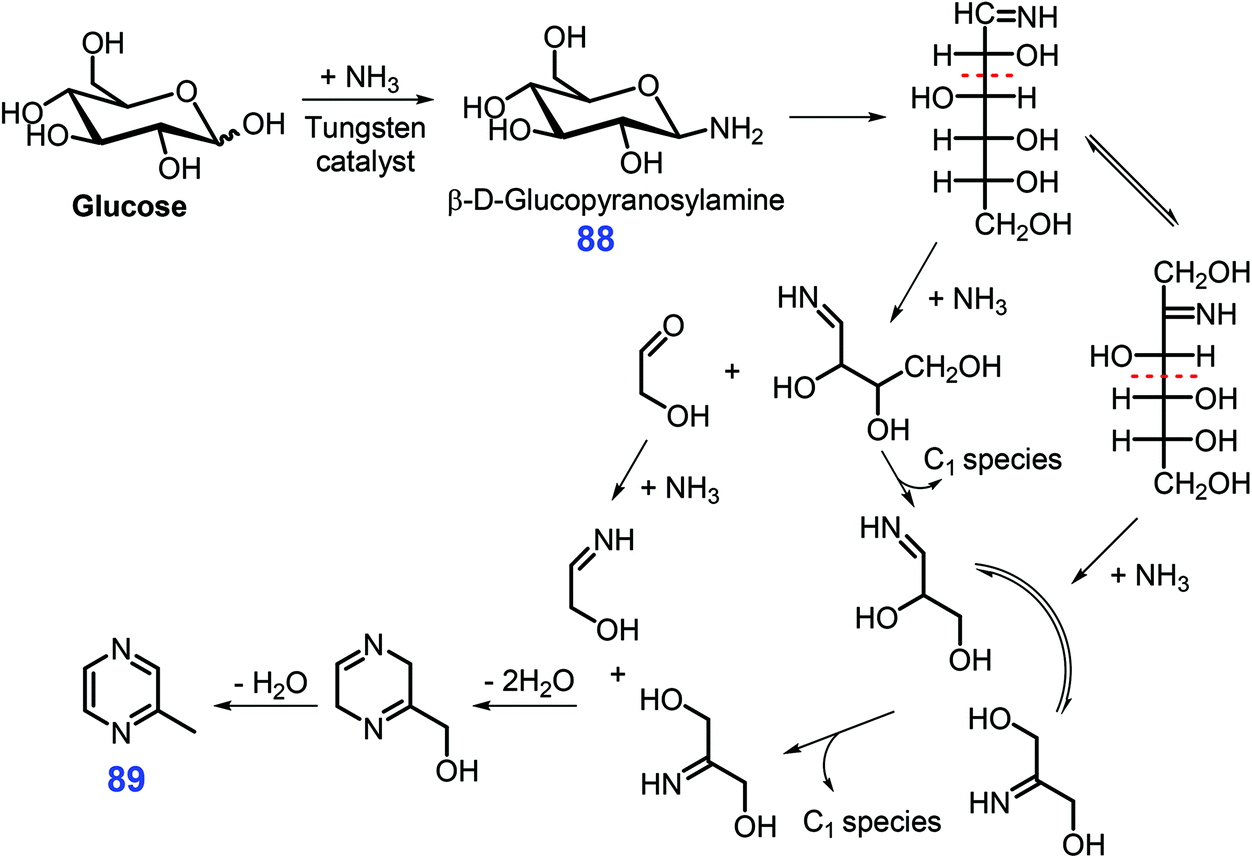 | ||
| Scheme 24 Possible pathways for the synthesis of 2-methyl pyrazine 89 from glucose and NH3 over ammonium metatungstate. Adapted with permission from ref. 219, Copyright © 2017, American Chemical Society. | ||
3.3. Other six-membered N-heterocycles
Apart from pyridines and pyrazines, other six-membered N-heterocycles such as pyridazines, pyrimidines, and triazines can be synthesized from bio-based feedstocks in spite of being reported for only a few works. For instance, 2,5-bis(alkoxymethyl)furan 90 undergoes oxidative ring-opening by metachloroperbenzoic acid (MCPBA) to give the hexene dione 91, followed by action with hydrazine to furnish the pyridazine 92 (Scheme 25).185,220 For producing enantiomerically pure pyridazines, β-keto ester functionalization of epoxy pyranosides is an effective way.221 Other relevant N-heterocyclic synthons can be constructed via these synthetic strategies.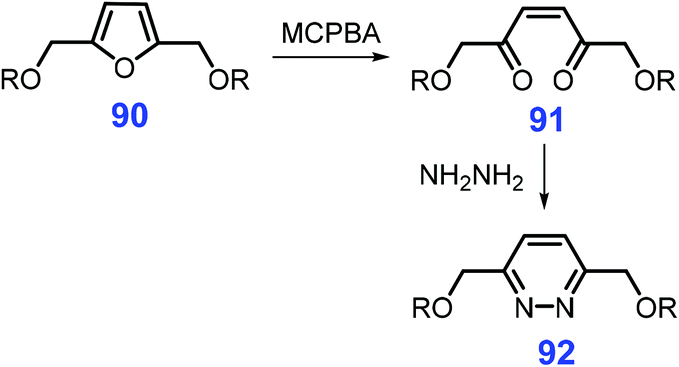 | ||
| Scheme 25 Synthetic routes to pyridazine 92 from 2,5-bis(alkoxymethyl)furan 90. | ||
Under basic conditions (MeONa in methanol), nucleophilic attack of the amidinium reagent (i.e., acetamidinium, benzamidinium and guanidinium salts) takes place at the C1 position of 2-formyl-galactal 93, followed by cyclization to give the substituted 5-(1,2,4-tri-O-benzyl-D-lyxo-1,2,3,4-tetrahydroxy-butyl)pyrimidines 94 (Scheme 26A).222 Likewise, substituted triazines 97 can be prepared at 37 °C by condensation of aminoguanidine 96 with two 3-deoxy-1,2-dicarbonyl derivatives 95, including 4-O-acetyl-1-deoxy-5,6-O-isopropylidene-2,3-D-threo-hexodiulose 95a, and 4-O-acetyl-1-deoxy-5,6-O-isopropylidene-2,3-D-erythro-hexodiul 95b (Scheme 26B).223 In contrast to their significance, development of more pragmatic strategies for the sustainable production of six-membered N-heterocycles from biomass resources is urgently required.
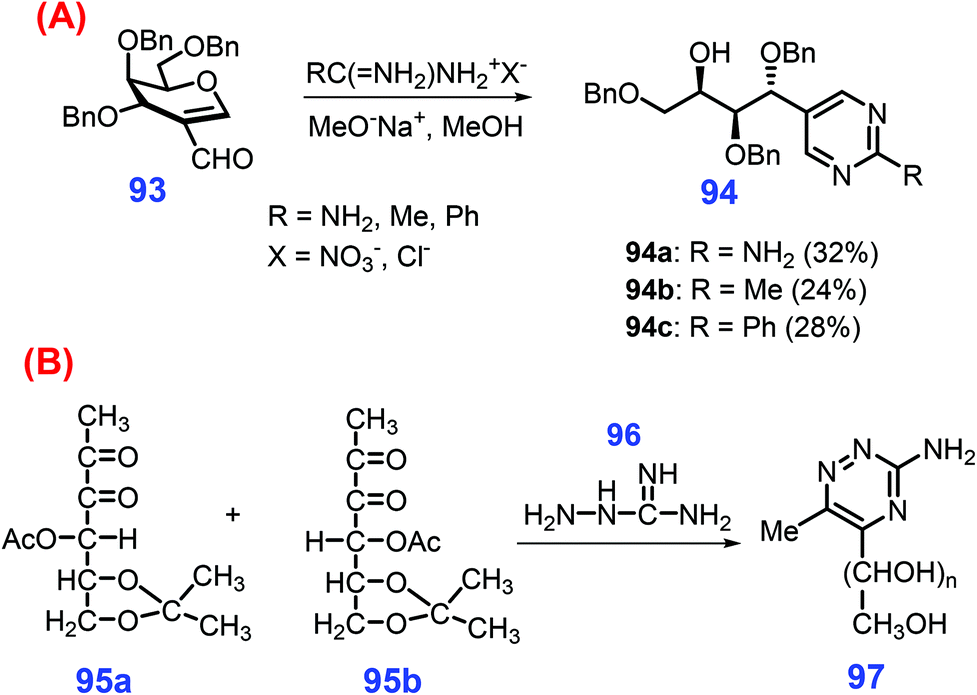 | ||
| Scheme 26 Synthesis of (A) pyrimidines 94 from 93, and (B) triazines 97 from dicarbonyl derivatives 95. Adapted with permission from ref. 222 and 223, Copyright © 1995 & 2002, Taylor & Francis. | ||
4. Fused N-heterocycles
4.1. Indoles
As one of the most important classes of azaheterocycles, indoles are used in agricultural chemicals (e.g., pesticide), pharmaceuticals, dyes, and other related chemicals.224–227 Indoles can be extracted directly from fungal biomass,228 even though a large number of conventional methods such as Fischer, Fukuyama, Gassman, and Leimgruber–Batcho reactions have been explored for their synthesis.229,230 With respect to upgrading “furan platform”, various N-heterocycles (e.g., pyrroles and indoles) can be synthesized via respective processes like Yuriev, Butin, and Achmatowicz reactions (Scheme 27A–C).231 Specifically, biomass-derived 2-(2-aminobenzyl)furans 98 undergoes oxidative rearrangement successively with m-chloroperbenzoic acid (m-CPBA) at 0 °C and then application of trifluoroacetic acid (TFA) at room temperature gives 2-(2-acylvinyl)indoles 99 (up to >90% yield) in exclusive E- or Z-isomers, closely dependent on the presence or absence of electron-donating alkoxy substituents in the phenyl ring, respectively (Scheme 27D).231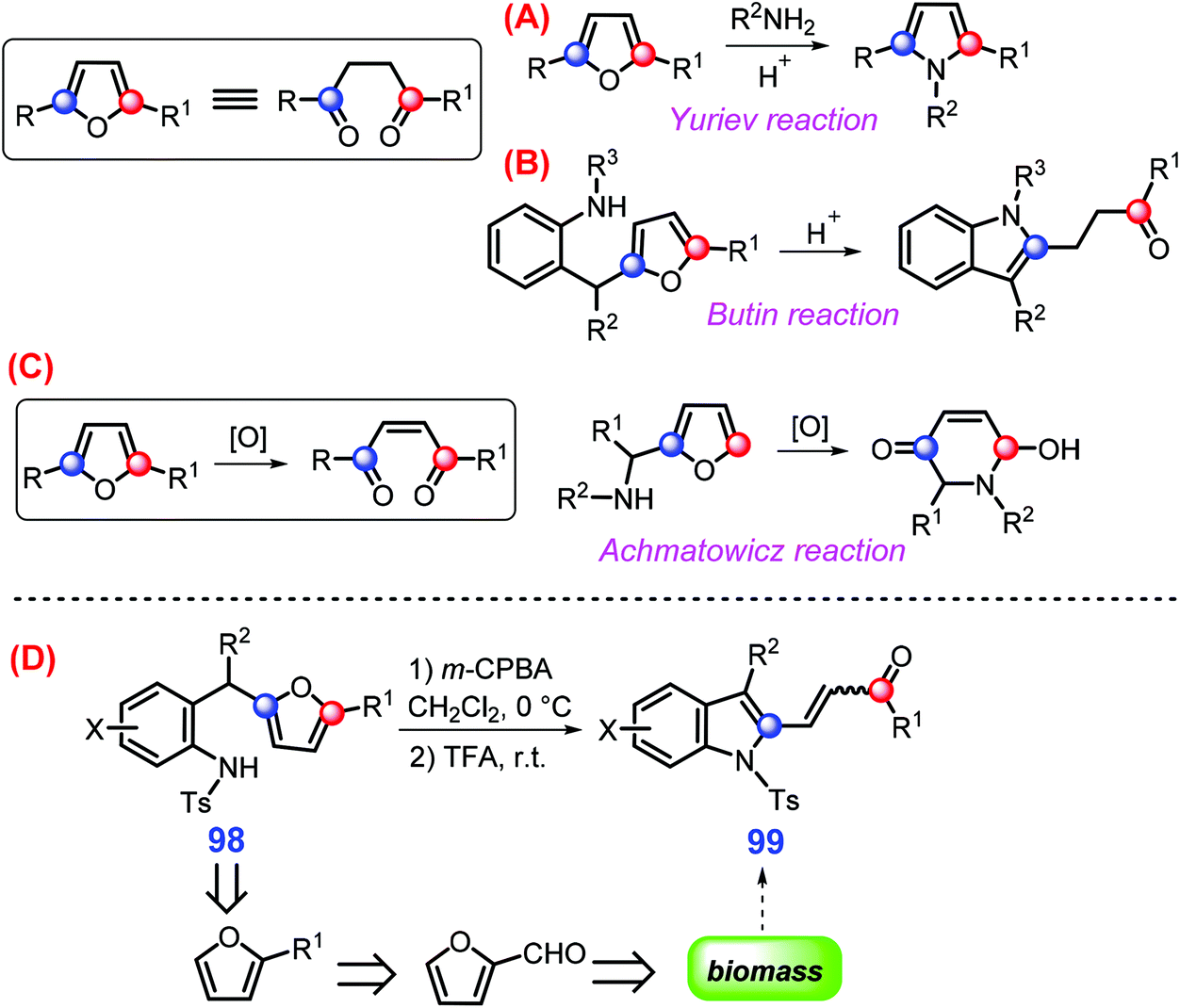 | ||
| Scheme 27 Representative pathways for upgrading furans to azaheterocycles via (A) Yuriev reaction, (B) Butin reaction, (C) Achmatowicz reaction, and (D) oxidative rearrangement. Adapted with permission from ref. 231, Copyright © 2016, American Chemical Society. | ||
Zeolite HZSM-5 (Si/Al = 25) was illustrated to efficiently catalyze direct gas-phase conversion of furfural via cascade thermal conversion and ammonization with NH3 to give indoles (yield 20.79%) at 650 °C with a weight hourly space velocity (WHSV) of 1.0 h−1 and NH3/furfural molar ratio of 2. It was proposed that the reaction between furfural and ammonia gives furfuryl imine 100, followed by cracking reaction to furan 101 which is considered as the key intermediate leading to pyrrole 102 and ultimately to indoles 103, with pyridines, aniline, and benzenes being concurrently generated as byproducts (Scheme 28).232 At moderately high temperatures (500 °C), moderate yields of indoles 103 (ca. 32%) are obtained from furan 101 under otherwise identical reaction conditions,233 indicating that the elemental step involving decarbonylation of furfural to furan 101 is crucial to the overall reaction. It is interesting that NH3 diluted by N2 is able to inhibit the formation of coke derived from 2-furonitrile 104, thus significantly increasing the yield of indoles 103 (33%) through enhancement of the HZSM-5 stability.234
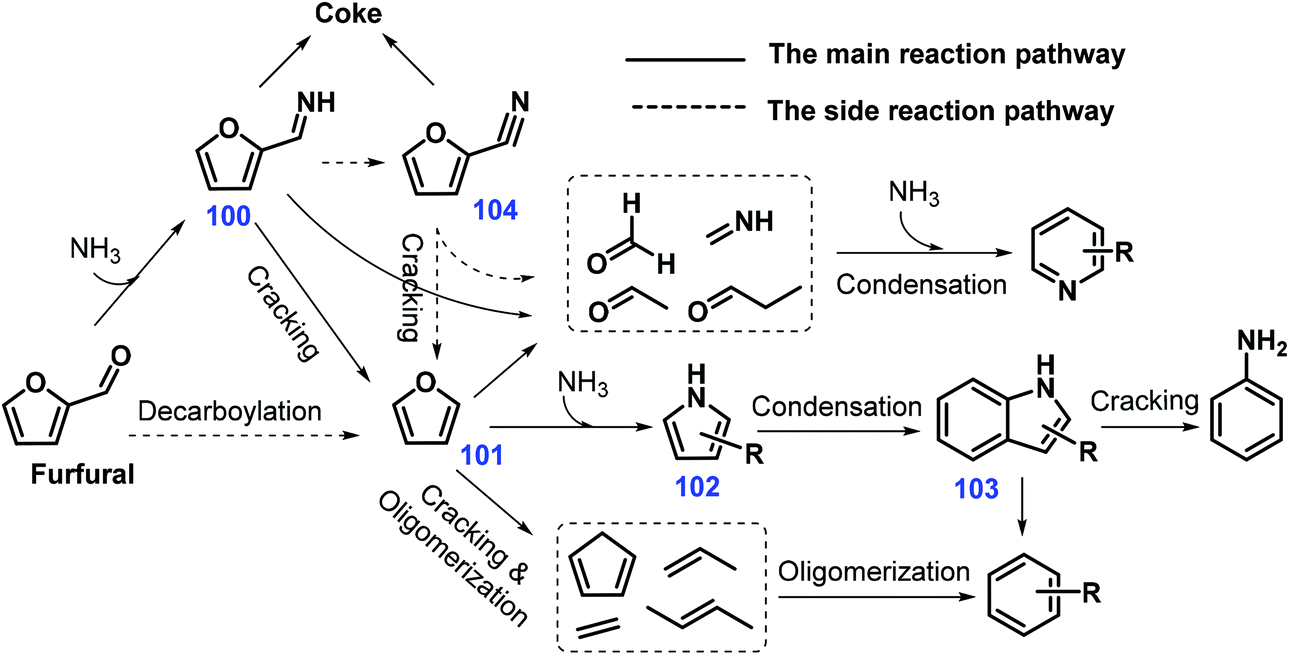 | ||
| Scheme 28 Possible pathways toward indoles 103 directly from furfural over HZSM-15. Adapted with permission from ref. 232, Copyright © 2015, Elsevier. | ||
Bio-based diols and anilines are applicable to the production of indoles under thermal conditions (e.g., 350 °C) or with metal catalysts at relatively low temperatures (e.g., 175 °C).235,236 The combined use of Pt/Al2O3 and ZnO catalyzes the dehydrogenation of ethylene glycol 105 to glycolaldehyde 106, followed by condensation with anilines to the imines 107 that undergo tautomerism and acylation/elimination reaction to afford relevant pyrrole-ring unsubstituted indoles 108 (Scheme 29).236 In view of H2 and H2O being the sole co-products, this type of atom-efficient reaction systems, in combination with stable and lost-cost catalysts, represents an efficient and promising synthetic approach toward indoles.
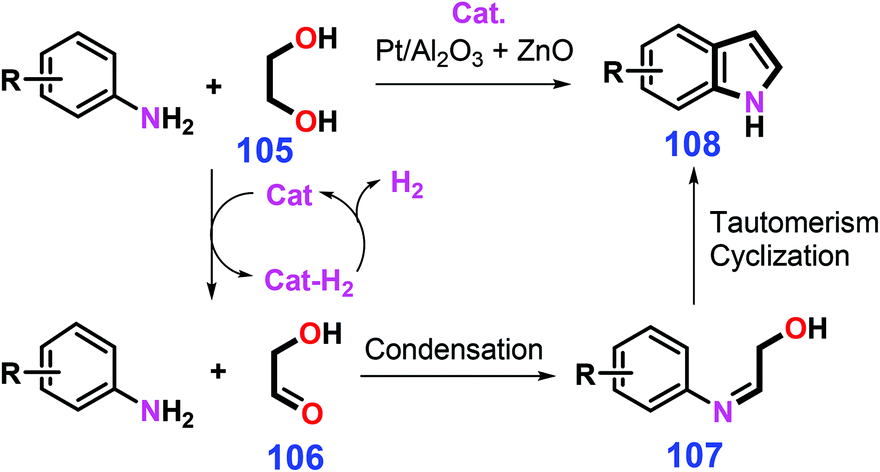 | ||
| Scheme 29 Synthetic routes to indoles from anilines and ethylene glycol 105 involving acceptorless dehydrogenative condensation. Adapted with permission from ref. 236, Copyright © 2018, American Chemical Society. | ||
4.2. Benzimidazoles
Benzimidazoles exhibit broad-spectrum bioactivities such as antifungal, antiulcer, antihelmintic, anticancer, and antiviral (e.g., HIV and herpes) activities, and they are used in a number of industrial chemicals such as UVB filters, thermostable membranes (fuel cells), optical brighteners (coatings), and pigments that include the benzimidazole motif.237–239 Classical approaches to synthesis of benzimidazoles 111 are the strong acid-catalyzed coupling of 1,2-diaminobenzene 109 with carboxylic acids, anhydride, or acyl chloride 110 (Scheme 30A).240 As another approach, the use of aldehydes as substrates with hydrogen peroxide catalyzed by iodine or ultrasmall ZnO nanoparticles leads to enhanced benzimidazole yields (up to >90%) under benign reaction conditions (room temperature to 40 °C).241,242 Sulfonated graphitic carbon nitride is able to efficiently catalyze direct conversion of xylose in the presence of 1,2-phenylenediamine in water to benzimidazole derivatives with good yields (ca. 84%) at 100 °C for 30 min, where xylose dehydration to furfural followed by cycloamination with 1,2-phenylenediamine occurs consecutively.243 Similarly, primary alcohols, which are readily available by either fermentation or chemocatalytic valorization of lignocellulose,244–246 are promising starting materials for in situ classical or photochemical oxidation or dehydrogenation to liberate more reactive aldehydes for producing benzimidazoles.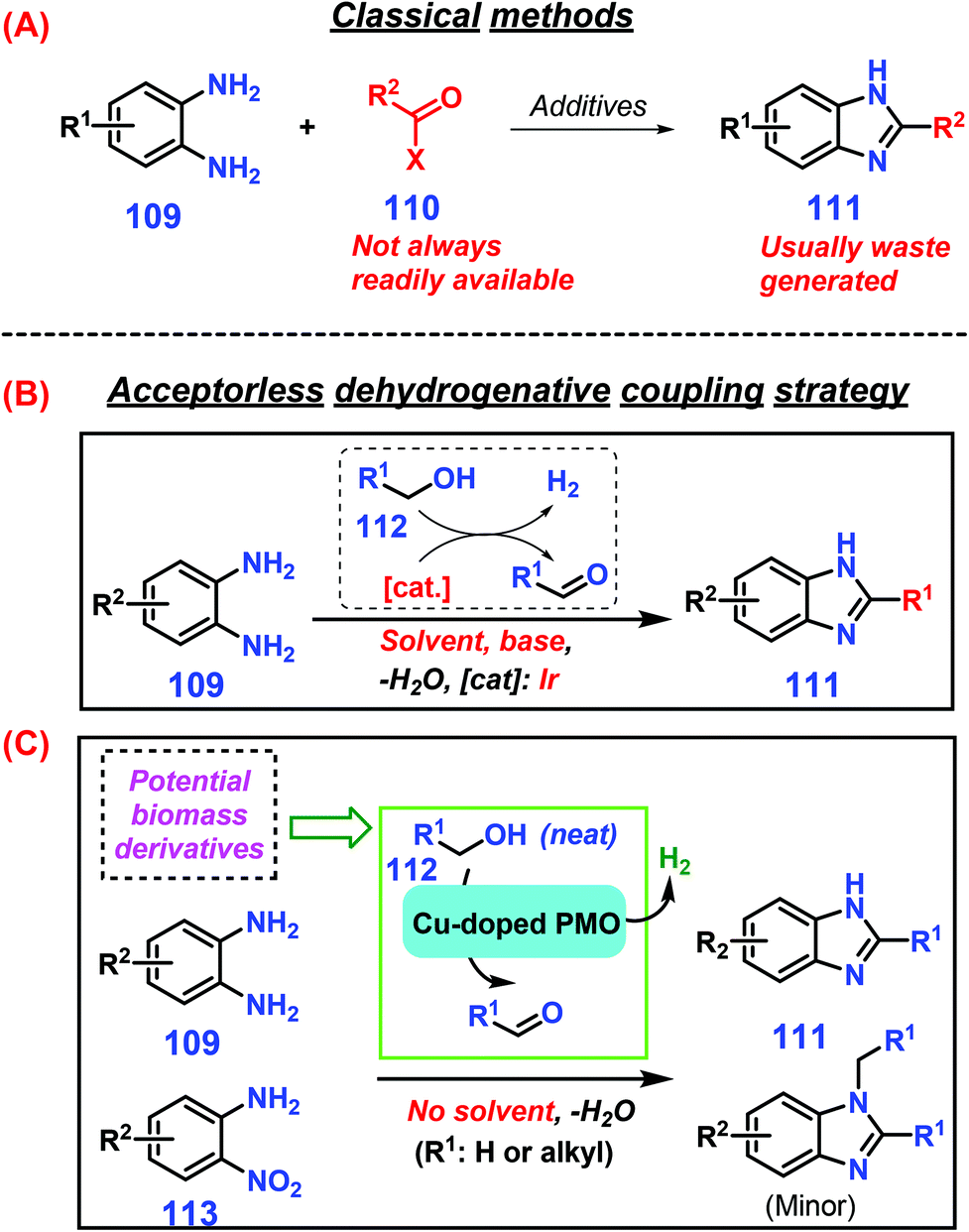 | ||
| Scheme 30 Representative synthetic routes to benzimidazoles 111via (A) classical methods or (B) acceptorless dehydrogenative coupling strategies with solvent or (C) acceptorless dehydrogenative coupling strategies without solvent. Adapted with permission from ref. 247, Copyright © 2013, American Association for the Advancement of Science. | ||
Among the developed strategies for condensation of diamines 109 with alcohols 112 to construct azaheterocycles like benzimidazoles 111, acceptorless dehydrogenative coupling (ADC) has emerged as an attractive protocol with only molecular H2 and innocuous H2O formed as the co-products (Scheme 30B & C).247,248 However, the requirement of a homogeneous noble-metal-based complex with the co-addition of a strong base are prerequisite to ensure smooth reaction progress.249,250 To make the ADC chemistry sustainable, several earth-abundant metal catalysts such as copper-doped porous metal oxides, cobalt–pincer complexes, and magnetic nanofibers confined in carbon nanotubes have been explored, and are capable of promoting efficient production of 2-substituted benzimidazoles 111 (up to 99% yield) from dehydrogenative coupling of primary alcohols 112 and aromatic diamines 109 under base-free conditions.251–253 Sun et al. disclosed that readily available 2-nitroanilines 113 can be used as nitrogen sources despite the requirement of an additional hydrogenation step prior to cyclization, such that comparable yields of benzimidazoles 111 are obtained by reaction with primary alcohols 112 at 250 °C for 3 h to 5 h reaction time.251
Besides monohydric alcohols, polyhydric alcohols have the possibility to form azaheterocycle frameworks together with the benzimidazole ring. Climent et al. showed that glyceraldehyde 83 (or glycerol) undergoes oxidation–cyclization twice with o-phenylenediamine derivatives 109 to give various substituted benzimidazoylquinoxalines 115 (24–80% yields) catalyzed by ceria supported gold nanoparticles (Au/CeO2) using air as the oxidant at 140 °C for 24 h (Scheme 31).254 In the reaction pathway, glyceraldehyde 83 (possibly in situ generated from glycerol by oxidation) first proceeds coupling and cyclization with o-phenylenediamine derivatives 109 to produce the intermediate benzimidazol 114, followed by cascade oxidation–cyclization with another o-phenylenediamine 109 to afford the substituted benzimidazoylquinoxalines 115. The electron-donating substituents in the phenyl ring favor the competitive oxidative cleavage of the diol 114, and lead to a decrease in the product selectivity by formation of other azaheterocycles 116–119 (Scheme 31).254
 | ||
| Scheme 31 Possible pathways for the formation of benzimidazoylquinoxalines 115 and byproducts 116–119. Adapted with permission from ref. 254, Copyright © 2013, Wiley-VCH. | ||
4.3. Other fused N-heterocycles
Quinolines can be synthesized by conventional Knorr, Skraup, and Camps chemistry, although the protocols typically involve multiple steps.255–257 Friedländer reaction is one of the most convenient and versatile ways to access quinolines, but the major drawback is that the 2-aminobenzaldehyde feedstock is too reactive to avoid self-condensation.258 Instead, 2-aminobenzyl alcohols 120 are stable, and readily undergo oxidative cyclization with either ketones 121 or secondary alcohols 122 catalyzed by transition metals (e.g., Au, Pd, Ir, Rh, Ru) along with stoichiometric amount of bases to give quinolines 123 with improved performance (Scheme 32A).259–263 Development of low-cost non-noble metal (e.g., Fe, Mn, Co, Cu) catalytic systems under basic additive-free conditions that promote hydrogen borrowing processes are needed for production of quinolines.264–269 Interestingly, when alcohols 122 are replaced with nitriles 124 for the annulation of 2-aminobenzyl alcohols 120, 2-amino-quinoline derivatives 125 (ca. 95% yields) are obtained at 140 °C for 24 h (Scheme 32B).258 Furthermore, the post-addition of primary alcohol 112 into the reaction mixture of amino alcohol 120 and nitrile 126 in a single pot furnishes 2-alkylaminoquinolines 127 by in situ N-alkylation (Scheme 32C).270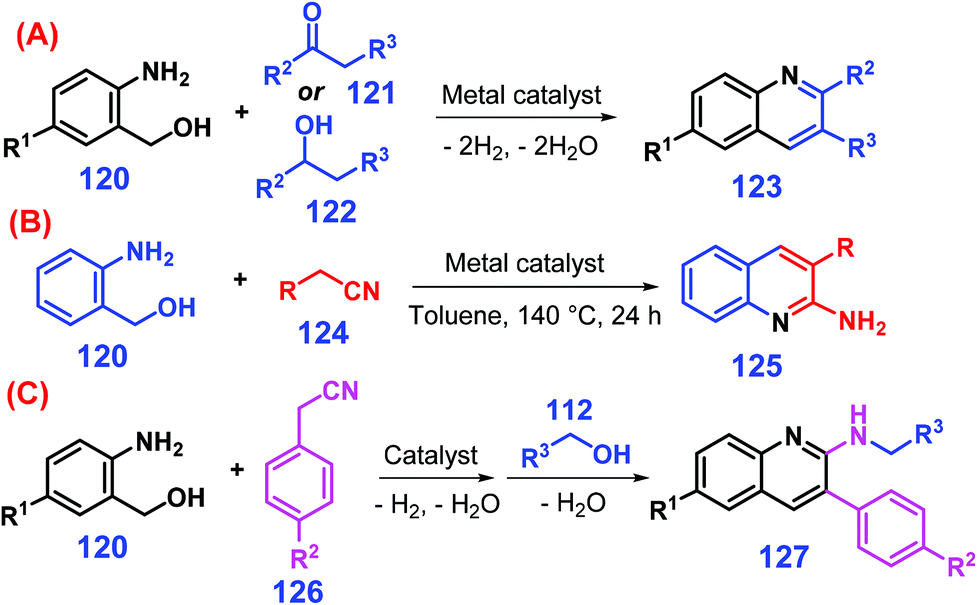 | ||
| Scheme 32 Synthetic routes to quinolines from amino alcohol 120 reacting with (A) ketone 121 or secondary alcohol 122, (B) nitrile 124, or (C) nitrile 124 and alcohol 112. Adapted with permission from ref. 258, 259 and 270, Copyright © 2018 & 2019, Wiley-VCH; Copyright © 2007, Elsevier. | ||
Synthesis of quinoxalines was traditionally performed by double-coupling of 1,2-carbonyls with 1,2-phenylenediamines, while the employed 1,2-carbonyls were highly reactive and cause undesired self-condensation.271,272 Several other synthetic approaches such as oxidative coupling of α-hydroxycarbonyls and 1,2-diamines,273,274 1,4-addition of diazenylbutenes with 1,2-diamines,275 oxidation trapping of epoxides and ene-1,2-diamines,276 and cyclization–oxidation of phenacyl bromides and 1,2-phenylenediamines277 can provide substituted quinoxalines, but with moderate or low yields in most cases. Using biomass-derived glycols or vicinal diols 128 with 1,2-phenylenediamine derivatives 109 as starting materials, Climent et al. reported that quinoxalines 130 (35–91% yields) are efficiently synthesized in a one-pot two-step oxidative coupling reaction process over ceria supported gold nanoparticles (Au/CeO2) at 140 °C after 24 h in the absence of any homogeneous base with air as oxidant (Scheme 33).278 Notably, the oxidative cleavage of the substrate diol 128 or the hydroxycarbonyl intermediate 129 inevitably took place to form aldehydes 131 and 132, which further undergo condensation with the phenylenediamine 109 affording the benzimidazole derivatives 133, 134, 135, and 136. Similarly, starting from 1,2-dinitrobenzene and 1,2-propanediol, yields of up to 83% of 2-methylquinoxaline are realized, where the nitro-to-amino reduction is performed at 80 °C under 1 MPa H2, followed by oxidative coupling at 140 °C for 30 h in the presence of atmospheric air.278 The quinoxaline skeleton can also be established by tandem cyclization/hydrosilylation of 1,2-phenylenediamines and keto esters,279 and coupling of 2-(1H-pyrrol-1-yl)anilines with DMSO.280 Both of these latter methods provide sustainable approaches to quinoxalines since they use bio-based feedstocks although the protocols could be improved by possibly using earth-abundant metal catalysts or safe or renewable solvents.
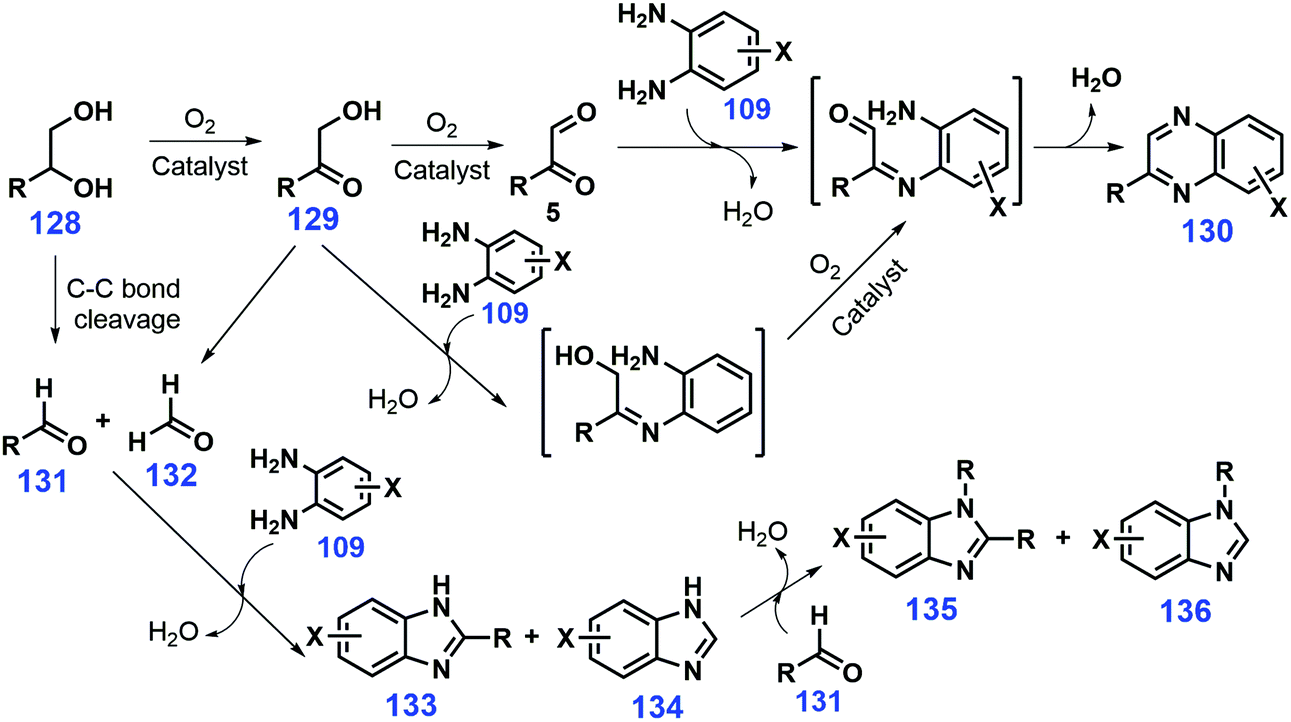 | ||
| Scheme 33 Reaction pathways for the formation of quinoxalines 130 and byproducts 133–136 from vicinal diols 128 and 1,2-phenylenediamines 109. Adapted with permission from ref. 278, Copyright © 2012, Elsevier. | ||
As illustrated in Scheme 34, carbohydrate substrates (e.g., glucose, galactose, arabinose, xylose, maltose, mannose, lactose) combined with an amine (e.g., anilines, glucosamine) and barbituric acid 137 or thio-barbituric acid 138 as reagents can be used to form pyrimidine-fused heterocycles 139 (with up to >90% yield) via a one-pot multicomponent condensation reaction in the presence of an acid catalyst (e.g., para-toluenesulfonic acid, nanocrystalline cellulose sulfuric acid) under mild conditions (e.g., 50 °C).281–283 The active aldehyde group of the sugar takes part in the condensation reaction, which gives the resulting pyrimidine-fused heterocycles hydrophilicity and potentially good bioactivity.281–283 All together with glucosamine, aldehyde, and barbituric acid, addition of malononitrile is able to allow a four-component condensation reaction, giving polyhydroxy-substituted pyrido[2,3-d]pyrimidines in good yields (89–94%) catalyzed by para-toluenesulfonic acid in ethanol heated at 50 °C.284
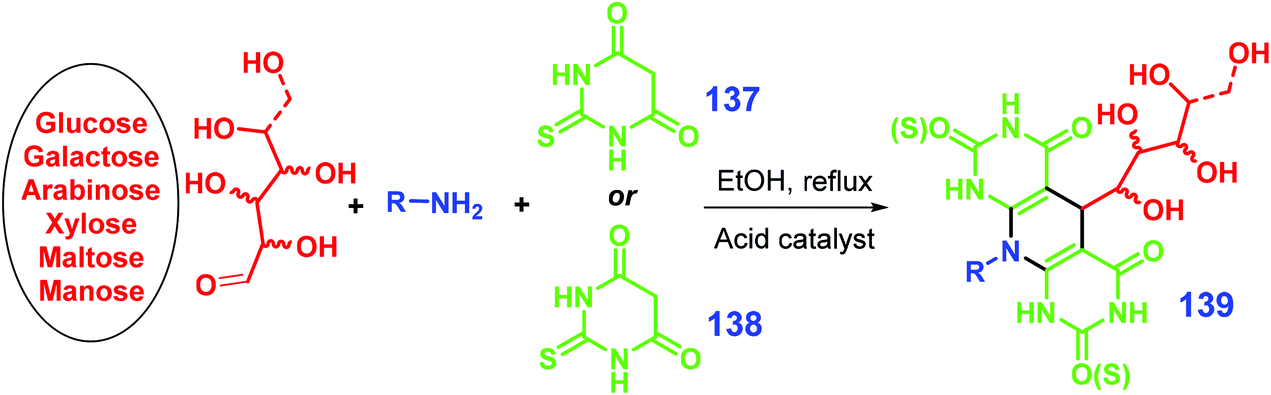 | ||
| Scheme 34 Synthesis of pyrimidine-fused heterocycles 139via multicomponent condensation of sugar, amine, and barbituric acid 137 or thio-barbituric acid 138. Adapted with permission from ref. 281, Copyright © 2018, Springer Nature. | ||
Multicomponent condensation reactions can be used for the synthesis of pyridine-fused heterocycles. For example, Shpuntov et al. showed that a three-component Mannich-type reaction of 2-alkylfuran, methyl 2-formylbenzoate, and carbamate in the presence of iodine at 0 °C for 2 h affords N-Boc-1-[2-(carbomethoxy)aryl]furfurylamines in moderate yields (51–89%), and further application of a two-step reaction process of oxidative furan-ring cleavage and N-Boc deprotection catalyzed by meta-chloroperoxybenzoic acid and HCl, respectively, gives 6H-isochromeno-[4,3-b]pyridin-6-ones in moderate yields (61–68%).285 The coupling of chitin-derived 3-acetamido-5-acetylfuran 140 with ketones 141 catalyzed by HCl provides dihydrodifuropyridines (up to 64% yield) after reaction at 70 °C for 16 h (Scheme 35).286 The ketone 141 initially reacts with 140 to form difurylmethane 142, followed by sequential acid-mediated acetamide hydrolysis, tautomerism, and intramolecular cyclisation, eventually giving dihydrodifuropyridine 143 after removal of ammonia (Scheme 35), as proposed by those authors.286
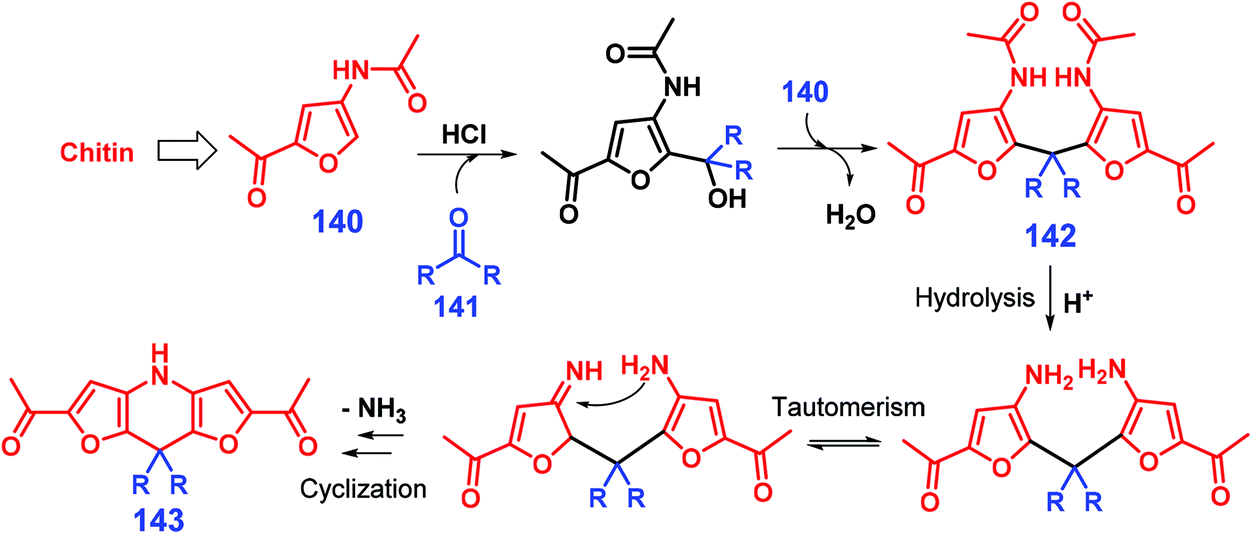 | ||
| Scheme 35 Reaction pathways for the synthesis of pyridine-fused heterocycles 143 from chitin-derived 3-acetamido-5-acetylfuran 140 and aliphatic ketones 141. Adapted with permission from ref. 286, Copyright © 2018, Springer Nature. | ||
Considering that aryl aldehydes are available from both lignin and carbohydrate components, efforts have been made to prepare dihydropyrano[2,3-c]pyrazoles from biomass-derived aldehydes via multicomponent condensation reactions. For example, Yang et al. reported a one-pot, two-step catalyst-free protocol for synthesis of 2H,4H- and 1H,4H-dihydro-pyrano[2,3-c]pyrazoles from lignin-derived aromatic aldehydes 35 (Scheme 36A).287 In the first stage, microwave-assisted condensation of hydrazines 37 and β-ketoesters 144 in water at 80 °C in 2 min affords intermediates (pyrazolones, 145). To the resulting mixture, malononitrile 146 and aromatic aldehydes 35 promptly added and heated under the identical irradiation conditions for another 3 min gives dihydropyrano[2,3-c]pyrazoles 147 (48–95% yields) by direct precipitation from the reaction mixture after cooling to room temperature.287 In the replacement of β-ketoesters with acetylene ester, Ambethkar et al. illustrated that dihydropyrano[2,3-c]pyrazoles (65–93% yields) are directly synthesized through four-component condensation of diethylacetylene dicarboxylate, hydrazine hydrate, aryl aldehydes, and malononitrile in the presence of L-proline under solvent-free and mechano (grinding) conditions.288 In the single pot one-step reaction process (Scheme 36B), diethylacetylene dicarboxylate 148 is initially condensed with hydrazine hydrate 149 to give intermediate 150, followed by Michael addition to intermediate 151 that is in situ formed from Knoevenagel condensation between aryl aldehyde 131 and malononitrile 146, while the dihydropyrano[2,3-c]pyrazole product 152 is afforded after undergoing subsequent intramolecular cyclization and tautomerization.288 On the whole, this synthetic protocol seems to be facile, efficient, and eco-friendly for producing dihydropyrano[2,3-c]pyrazole derivatives.
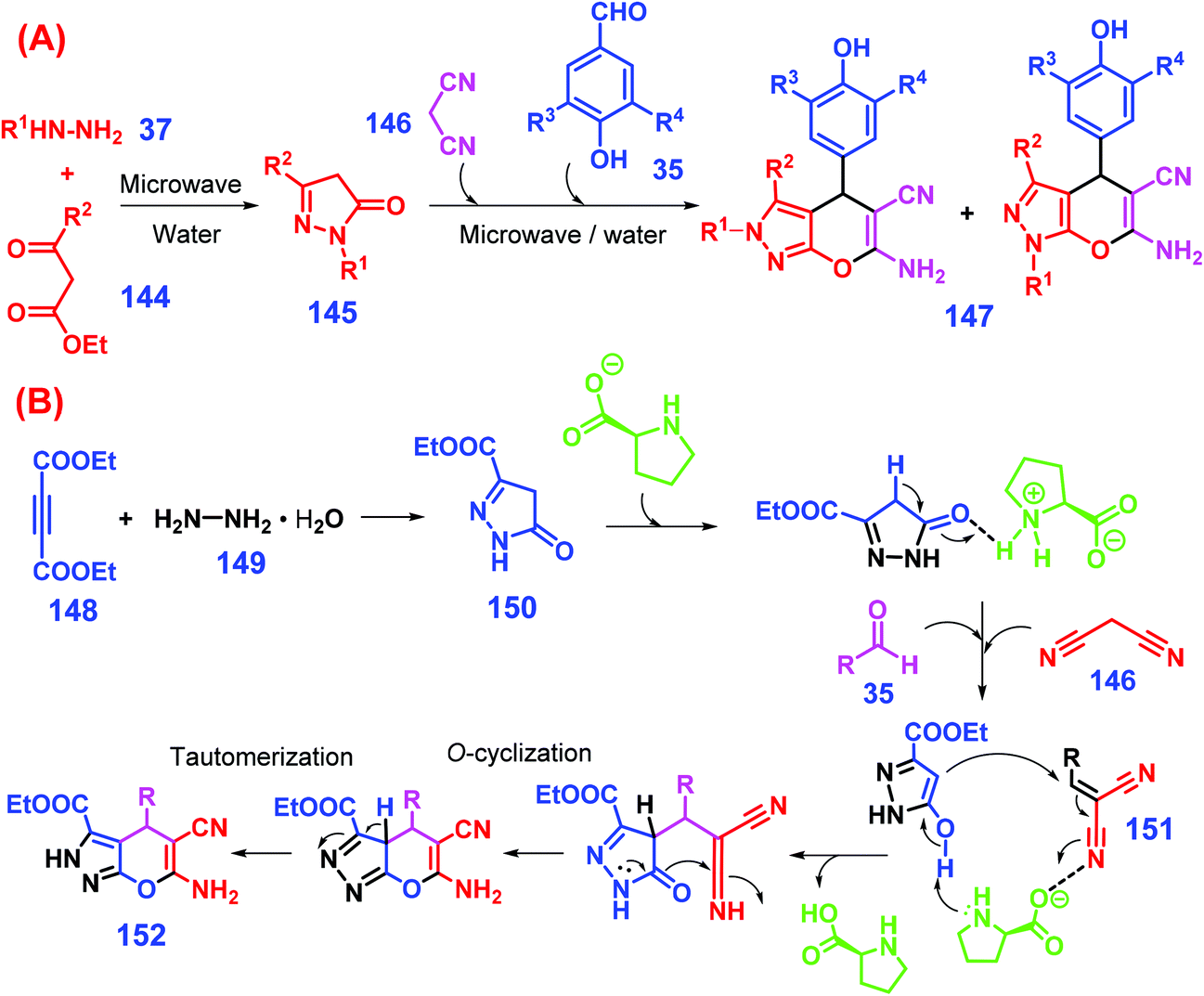 | ||
| Scheme 36 Synthesis of dihydropyrano[2,3-c]pyrazoles 152 from biomass-derived aldehydes 35via a (A) two- or (B) one-step approach. Adapted with permission from ref. 287 and 288, Copyright © 2014 & 2015, Elsevier. | ||
Besides the above-mentioned synthetic methods employed for synthesis of fused azaheterocycles, several other strategies have been developed with the aim of constructing more specific N-heterocyclic skeletons amendable to biomass feedstocks. For instance, a furan ring opening-pyrrole ring closure strategy has been adopted for the synthesis of pyrrole-fused heterocycles such as pyrrolo[1,2-a][1,4]diazepines and 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazin-3-ones.289,290 In view of the feasibility of furan-ring opening, biofuranics and their derivatives (e.g., levulinic acid) are extensively used for the construction of complex azaheterocycles like indolo[3,2-c]quinolines and bicyclic heterocycles.291,292 Direct transformation of biomass sugars to azaheterocycles has been investigated with profound results,293,294 while most studies of upgrading lignin are more focused on its model molecules such as 2-phenoxy acetophenone and aromatic aldehydes via modified approaches based on conventional reactions.295,296
5. Other N-heterocycles
5.1. CO2 participating N-heterocycles
Carbon dioxide is a promising C1 source for organic synthesis with unique characteristics like abundance, low toxicity, and sustainability, while it is thermodynamically stable and kinetically inert.297,298 Three major CO2-incorporated cyclization strategies have been developed to construct azaheterocycles (Scheme 37): (1) carboxylative cyclization via cascade nucleophilic attack on CO2 and intramolecular cyclization, (2) carbonylative cyclization via twice nucleophilic attack on CO2, and (3) reductive cyclization via nucleophilic attack on reduced CO2 and subsequent cyclization.299 To a certain degree, the type of obtained N-heterocyclic compounds is highly dependent on the employed N-containing nucleophiles. For example, oxazolidinones can be synthesized by nucleophilic attack of CO2 with aziridines or propargylic amines via carboxylative cyclization in the presence of a metal or base catalyst.300–302 Similarly, benzoxazinones are formed by base-catalyzed three-component coupling of imines, benzyne, and CO2.303 In another case, the carbonylation using CO2 takes place without any reductant via two possible pathways: (a) successive nucleophilic attack of CO2 with the nitrogen-containing compound bearing two nucleophilic sites, and (b) initial cyclization to form an unstable cyclic intermediate followed by immediate rearrangement to a stable counterpart.299 For example, base-catalyzed double coupling (i.e., carbonylative cyclization) of 2-aminobenzonitriles with CO2 gives quinazoline-2,4(1H,3H)-diones.304 In the carbonylation process, either cyclization or nucleophilic attack was proposed to be initialized, which needs to be elaborated in-depth with in situ characterization techniques.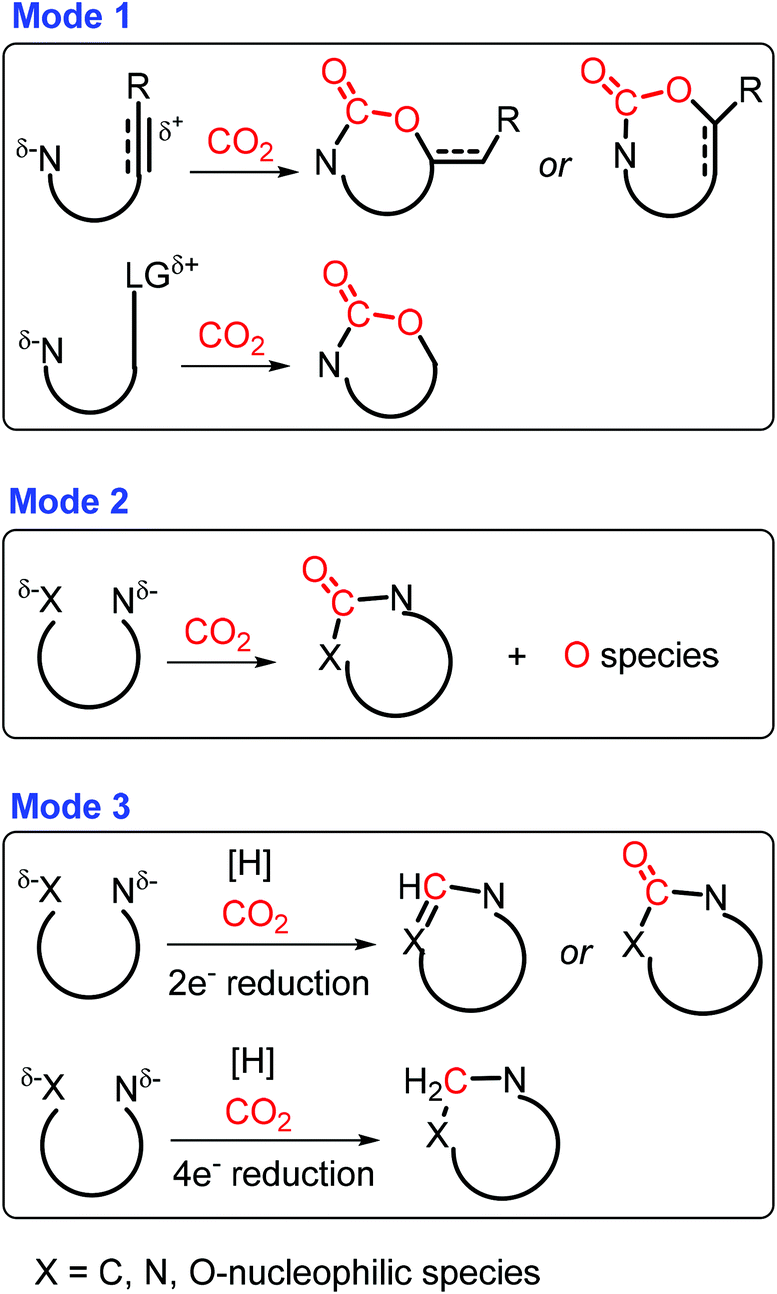 | ||
| Scheme 37 CO2-incorporated annulation triggered by C, N, or O nucleophiles. Adapted with permission from ref. 299, Copyright © 2019, American Chemical Society. | ||
In the presence of a hydrogen source (e.g., H2, boranes, and silanes), CO2 can be subjected to in situ reduction followed by cyclization with N-containing nucleophiles to afford a wide range of azaheterocycles.299 The 2-electron reduction is able to promote the hydrogenation of CO2 to formates, followed by condensation with substrates containing two nucleophilic sites to furnish quinazolinones, benzimidazoles, formamidines and their derivatives.305–307 On the other hand, CO2 may also serve as the methylene species via 4-electron reduction, giving saturated N-heterocycles after successive nucleophilic attacks.299 Although studies on CO2 reduction have been investigated extensively, controllable CO2 hydrogenation combined with successive cyclization still remains a challenge.
5.2. N-heterocycles with controllable ring size/type
As discussed above, five-membered, six-membered, and fused N-heterocycles can be selectively obtained from bio-based feedstocks with specific functional groups and carbon-chain length using developed strategies. Attention is being placed on developing integrated synthetic approaches to control the ring size of N-heterocycles (Fig. 3). Using imine esters 153 as three-atom units (Fig. 3), (a) five-membered heterocyclic compounds (e.g., pyrrolidines) can be acquired by [3 + 2]-cycloaddition with two-atom dipolarophiles (e.g., electron-deficient alkenes);308 (b) fused and bridged six-membered piperidines can be acquired via [6 + 3]-cycloaddition reactions of 6-π dipolarophiles (e.g., fulvenes, 2-acyl cycloheptatrienes, tropone) with azomethine,309–311 while six-membered heterocyclic frameworks can be acquired by cross 1,3-dipolar [3 + 3]-cycloaddition of azomethine with pyrazolidinium ylides;312 and (c) seven-membered heterocyclic azepines can be acquired from methyl coumalate via tandam [4 + 3]-cycloaddition/decarboxylation/isomerization.308 By changing the chain length of the employed substrates, other five, six, seven, and eight membered N-heterocycles are obtained using appropriate catalysts or catalytic systems.313–315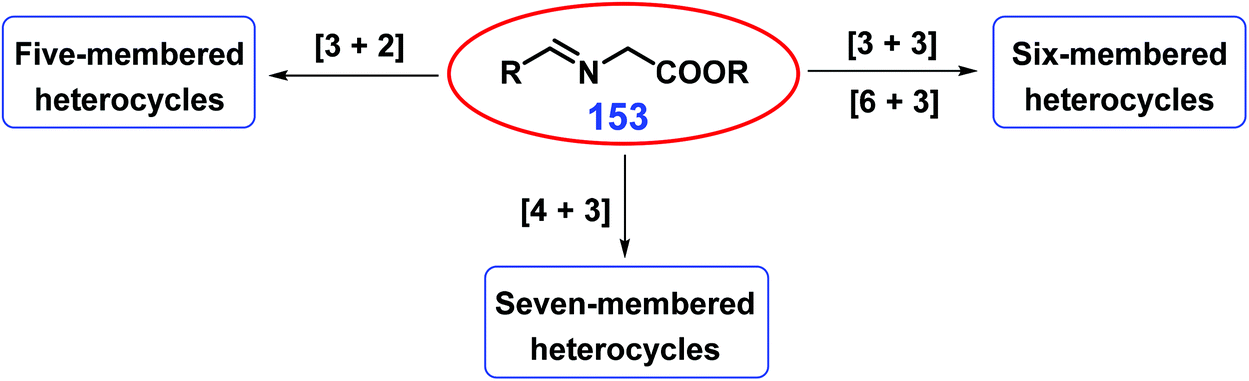 | ||
| Fig. 3 Schematic illustration for the synthesis of N-heterocycles with controllable ring size from imine esters 153. | ||
Cycloamination strategies are commonly used for the construction of azaheterocycles that can be well controlled by adjusting the type of nitrogen source. Starting from 1,4-dicarbonyl compounds 1 that are readily available from oxidative opening of the furan ring, pyrroles 154, pyridazines 155, and diazepines 156 can be selectively obtained by diamination with NH3, N2H4, and o-phenylenediamine, respectively (Scheme 38).316 In a similar way, reduced sugars proceeding via 1,2-dicarbonyl intermediates by reaction with different nitrogen sources furnish the quinoxaline, 1,2,4-triazine, pyrazine and pyrazolo[3,4-b]quinoxaline skeletons.317 In these strategies, protection group chemistry is not needed for the in situ multi-step transformations. With respect to the synthesis of azaheterocycles via multicomponent reactions, selection of subcomponents allows control of the product distributions. For instance, coupling cyclization of amino alcohols and alcohols with desired functionalities with suitable spatial distances is able to selectively afford pyrrole, pyridine, quinoline, pyrazine, carbazole, or acridine derivatives.318–322
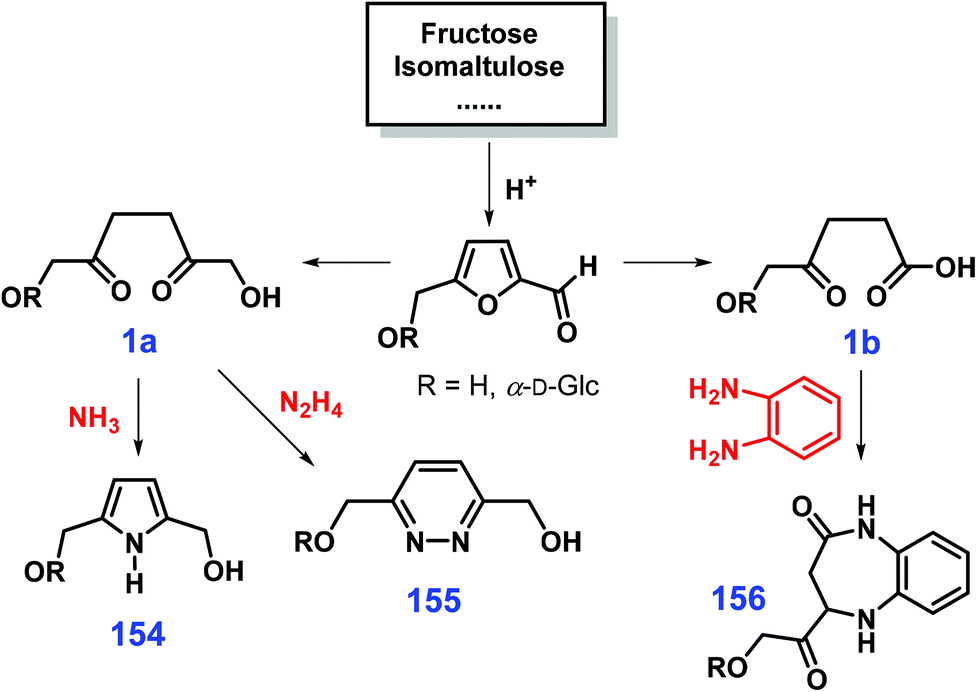 | ||
| Scheme 38 Reaction routes to pyrroles 154, pyridazines 155 and diazepines 156 from sugars by adjusting the type of nitrogen source. Adapted with permission from ref. 316, Copyright © 2001, Royal Society of Chemistry. | ||
Chiral N-heterocyclic compounds have a privileged role in pharmaceuticals.323 A specific chiral source of asymmetric induction (i.e., chiral catalyst) is typically required for the enantioselective synthesis of N-heterocycles,324,325 through which stereoselective upgrading of thermodynamically stable CO2 to optically pure azaheterocycles can be realized.326 Biomass derivatives (e.g., carbohydrates, amino acid, and levoglucosenone) are inherently in enantiopure form, thus they have high feasibility for use in simple production of N-heterocycles with appropriate configuration. For example, in the absence of any chiral catalyst, cellulose-derived (−)-levoglucosenone can undergo well-controlled reactions like 1,3-dipolar cycloaddition, aza-Michael addition or isomerization, being enantioselectively converted into polysubstituted pyrrolidines or 1,2,3-triazoles.327–329 A versatile protocol via multicomponent Ugi-type reactions of unprotected saccharides and L- or D-configured amino acids is accessible to 1,2-syn or anti configured products, respectively.330 In this regard, the exploitation of synthetic protocols to biomass-derived chiral molecules, especially those with potent bioactivities, is promising with respect to green manufacturing processes.
5.3. Direct thermal amination and hydrothermal approaches
The majority of studies focus on the transformation of small platform molecules (e.g., carbonyl compounds, furans, unsaturated/polybasic carboxylic acids) or simple biomass derivatives (e.g., sugars, lignin model molecules like aromatic aldehydes/alcohols) into azaheterocycles.331–335 However, direct valorization of raw biomass materials to N-heterocycles and relevant value-added chemicals could bring many opportunities to practical production (Fig. 4).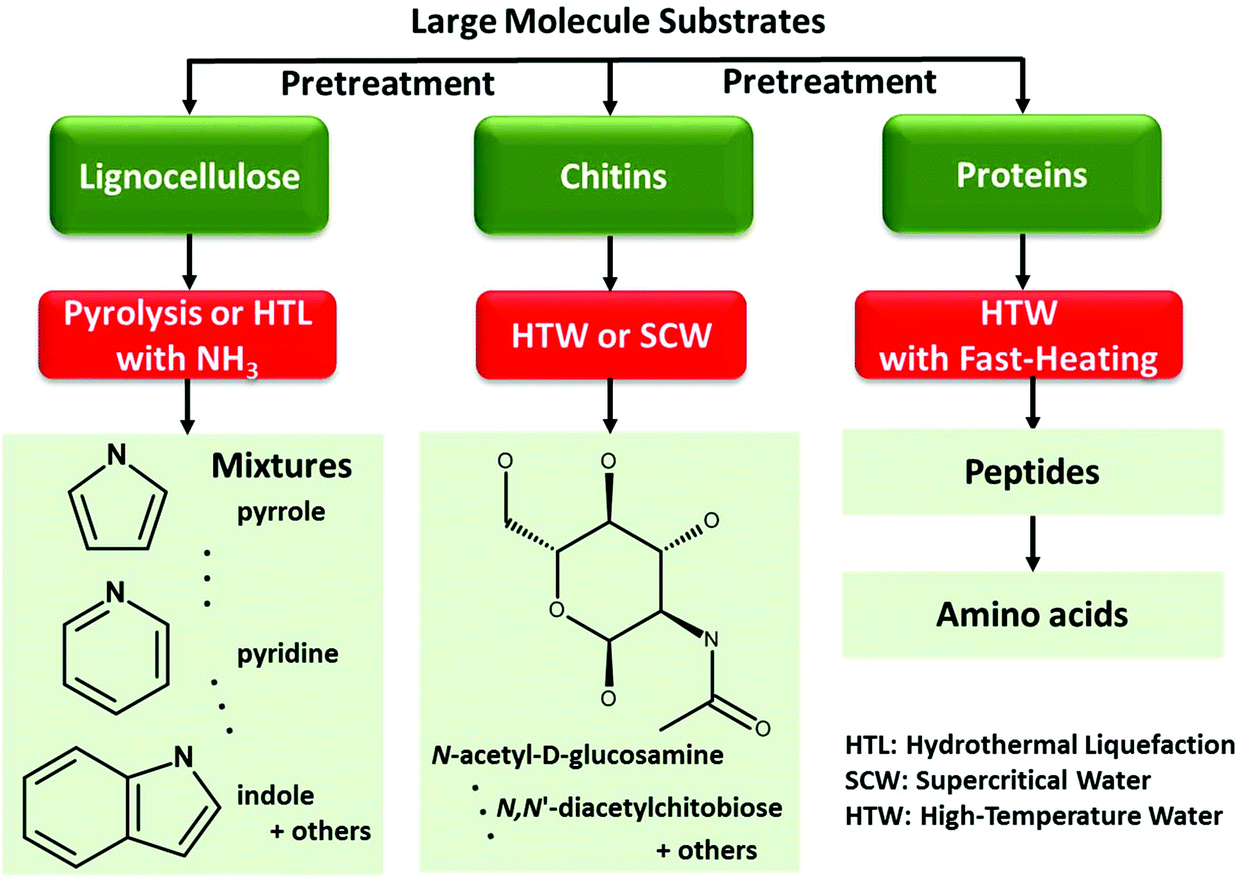 | ||
| Fig. 4 Top-down strategy to N-heterocycles via large molecule substrates. | ||
Pyrolysis or hydrothermal liquefaction of lignocellulosic biomass with NH3 has been developed for the production of nitrogenous heterocyclic compounds, while obtained products are mixtures of pyrrole, pyridine, indole and other nitrogen-containing compounds with relatively low overall yields (<30%).336–343
Direct thermal conversion of nitrogenous biopolymers (e.g., chitin and proteins) can be implemented by sub- and supercritical water treatment, where the overall hydrolysis process including surface hydrolysis and destruction of hydrogen and glycosidic bond is controlled by solvent conditions.344 Mechanochemical treatment (e.g., ball mill) of chitin enhances its solubility in supercritical water (400 °C) allowing rapid (1 min) transformation, while raw chitin with high crystallinity shows poor dissolution in high-temperature water (HTW).345 After pretreatment, chitin could be transformed into N-acetyl-D-glucosamine (the monomer) and/or N,N′-diacetylchitobiose (the dimer) with yields of no more than 8%.346,347 At relatively high temperatures of (200 to 260) °C, treatment of proteins with HTW and fast heating rates (135 to 180) K s−1 inhibits initial protein aggregation that occurs during the reaction process at slow heating rates (ca. 0.25 K s−1), affording high-molecular-weight peptides (1500 to 8300) Da as dominant products with the transformation advantageously occurring as random-scission.348
On the other hand, with hydrothermal liquefaction (HTL) at temperatures between 250 °C and 350 °C, several heterocycles could originate from the components of proteins, such as piperidines and quinolines from lysine in bio-oils, while pyrazines form from the mixture of disaccharides and lysine.349 Besides organic matter derived from HTL with liquid-N accounting for the majority of nitrogenous products,350–353 the HTL conditions can affect the concentrations of metals (e.g., Fe, Na, Mg, Ca) and inorganic species (e.g., P) in algal biocrude.354 It is worth noting that N-containing heterocycles partition undesirably into bio-crude oils after hydrothermal treatment.355,356 leading to a future challenge for the reaction system or product separation.
The use of catalysts (e.g., HZSM-5) in the reaction system remarkably increases the selectivity toward the N-containing compounds composed of up to 79% pyrroles, 63% pyridines, and 57% indoles, respectively.357 By selecting crude shrimp shells as the starting feedstock, pyrrole is detected as the dominant nitrogen-containing product after hydrothermal treatment with NH3 in the presence of NaOH,358 while the co-production of N-heterocyclic bio-char and other aromatic amines are unavoidable in most cases.359 Appropriate design of reaction processes and functional catalysts opens avenues to other elegant N-containing chemicals like β-lactam, nitrile, and isoxazole.360–362 Last, but not least, adopting new strategies for C–N bond formation are required for sustainable and efficient valorization of raw biomass materials.
6. Conclusions and outlook
An overview of amination strategies is given for synthesis of N-heterocycles from biomass feedstocks. The C–N bond formation is typically established by reductive amination, multicomponent coupling reactions, or acceptorless dehydrogenative coupling reactions, which will furnish the N-containing cyclic skeletons by double C–N bonding processes or C–N coupling combined with other elemental C–C bond formation reactions like condensation. Much attention is on the construction of five-membered N-heterocycles such as pyrroles, pyrrolidones, pyrazoles, imidazoles, thiazoles, oxazoles, triazoles, and tetrazoles from either bio-based platform molecules or directly from raw biomass materials. However, studies on the production of biomass-derived six-, seven-, and eight-membered as well as fused N-heterocyclic compounds are still in their infancy, with target products being obtained with relatively low selectivities. More effort should be devoted to the design of reaction systems specifically, by matching synthesis routes. Several points may be made for access to azaheterocycles:(1) Organic synthesis and synthetic methodologies may provide efficient references for rationally devising effective and renewed reaction routes toward specific N-heterocycles with satisfactory stereo- or enantioselectivity, while the reaction processes can be further simplified by following the principles of green engineering and green chemistry. In contrast, studies on sustainable production of other heteroatom (S, P)-containing compounds are relatively scarce, indicating the necessity to explore synthetic approaches in the construction of S–C or P–C bonds.
(2) The presence of a suitable catalyst with designated chiral induction center can remarkably lower reaction barriers with enhanced overall rate and optical purity. Basic nitrogen sources or used together with acidic substrates can themselves act as catalyst, and most of biomass derivatives naturally bearing absolute/spatial configurations have the greatest potential to furnish the desired N-containing enantiomers without any catalyst.
(3) Hydrothermal treatment of raw biomass materials with ammonia has been demonstrated to be capable of giving N-containing compounds but with low selectivity, let alone the desired relevant azaheterocycles. In this regard, biomass pretreatment with sustainable solvents and/or ecofriendly catalysts can be a promising auxiliary to enhance reaction efficiency with respect to both selectivity and yield of azaheterocycles.
(4) Definite reaction mechanisms have been explicitly illustrated for some of the above-mentioned reactions, while a majority of reaction schemes have been revealed without neither elucidation of the reaction pathway nor the catalyst structure–activity relationship. There is no doubt that the disclosure of detailed reaction routes using modern characterization techniques will be helpful for both catalyst preparation and design of reaction processes for producing N-heterocycles.
(5) Raw biomass materials are solid-state and are not soluble in thermal reaction processes, which often results in the formation of plentiful solid biochars rather than the desired N-heterocyclic compounds. In this respect, the design of compatible reactors is needed to facilitate production in overall cycloamination processes.
In conclusion, cycloamination coupled with C–C bond formation processes is efficient for the synthesis of N-heterocycles especially five-membered azaheterocycles from biomass feedstocks. Many endeavors have been made with great achievements being made that improve the chemical avenues to bio-based N-heterocycles. Studies on the diversity of the obtained N-containing compounds need to be further strengthened. Development of efficient and renewed synthesis approaches to task-specific N-heterocyclic compounds with emphasis on sustainable chemistry will be required for comprehensive biomass valorization.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is financially supported by National Natural and Science Foundation of China (21878161), Nanjing Agricultural University (68Q-0603), International Postdoctoral Exchange Fellowship Program of China (20170026), Postdoctoral Science Foundation of China (2016M600422), Jiangsu Postdoctoral Research Funding Plan (No. 1601029A, for H. L. to study at Tohoku University), the Japan Society for the Promotion of Science (JSPS) Grantin-Aid for Early-Career Scientists (No. 19K15347, Japan), and the financial support from Tohoku University Center for Gender Equality Promotion (H. G., Tohoku University, Japan).References
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- R. Gérardy, R. Morodo, J. Estager, P. Luis, D. P. Debecker and J. C. M. Monbaliu, Top. Curr. Chem., 2019, 377, 1 CrossRef PubMed.
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS PubMed.
- H. Li, Z. Fang, R. L. Smith and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef.
- J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- H. Li, S. Yang, A. Riisager, A. Pandey, R. S. Sangwan, S. Saravanamurugan and R. Luque, Green Chem., 2016, 18, 5701–5735 RSC.
- N. Brun, P. Hesemann and D. Esposito, Chem. Sci., 2017, 8, 4724–4738 RSC.
- S. H. Krishna, K. Huang, K. J. Barnett, J. He, C. T. Maravelias, J. A. Dumesic, G. H. Huber, M. De bruyn and B. M. Weckhuysen, AIChE J., 2018, 64, 1910–1922 CrossRef CAS.
- H. Li, Z. Fang, J. He and S. Yang, ChemSusChem, 2017, 10, 681–686 CrossRef CAS PubMed.
- F. Liu, Q. Liu, J. Xu, L. Li, Y. T. Cui, R. Lang, L. Li, Y. Su, S. Miao, H. Sun, B. Qiao, A. Wang, F. Jérôme and T. Zhang, Green Chem., 2018, 20, 1770–1776 RSC.
- H. Li, X. He, Q. Zhang, F. Chang, W. Xue, Y. Zhang and S. Yang, Energy Technol., 2013, 1, 151–156 CrossRef CAS.
- K. S. Arias, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2016, 4, 6152–6159 CrossRef CAS.
- H. Zhang, Y. T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 RSC.
- Y. T. Cheng and G. W. Huber, ACS Catal., 2011, 1, 611–628 CrossRef CAS.
- H. Li, W. Zhao, A. Riisager, S. Saravanamurugan, Z. Wang, Z. Fang and S. Yang, Green Chem., 2017, 19, 2101–2106 RSC.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed.
- H. Li, T. Yang and Z. Fang, Appl. Catal., B, 2018, 227, 79–89 CrossRef CAS.
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, Appl. Catal., A, 2014, 469, 490–511 CrossRef CAS.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- C. G. S. Lima, N. M. Moreira, M. W. Paixão and A. G. Corrêa, Curr. Opin. Green Sustainable Chem., 2019, 15, 7–12 CrossRef.
- H. Li, A. Riisager, S. Saravanamurugan, A. Pandey, R. S. Sangwan, S. Yang and R. Luque, ACS Catal., 2017, 8, 148–187 CrossRef.
- B. Ganem, Acc. Chem. Res., 1996, 29, 340–347 CrossRef CAS.
- I. V. Trushkov, M. G. Uchuskin and A. V. Butin, Eur. J. Org. Chem., 2015, 2999–3016 CrossRef CAS.
- M. A. R. Meier, Macromol. Rapid Commun., 2019, 40, 1800524 CrossRef PubMed.
- V. S. C. de Andrade and M. C. S. de Mattos, Curr. Green Chem., 2018, 5, 68–85 CrossRef CAS.
- M. O. Sydnes, Curr. Green Chem., 2018, 5, 22–39 CrossRef CAS.
- A. M. Medway and J. Sperry, Green Chem., 2014, 16, 2084–2101 RSC.
- M. Platon, R. Amardeil, L. Djakovitch and J. C. Hierso, Chem. Soc. Rev., 2012, 41, 3929–3968 RSC.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- H. Li, H. Guo, Y. Su, Y. Hiraga, Z. Fang, E. J. M. Hensen, M. Watanabe and R. L. Smith, Nat. Commun., 2019, 10, 699 CrossRef PubMed.
- N. J. Race, I. R. Hazelden, A. Faulkner and J. F. Bower, Chem. Sci., 2017, 8, 5248–5260 RSC.
- I. L. Simakova, A. V. Simakov and D. Y. Murzin, Catalysts, 2018, 8, 365 CrossRef.
- M. Pelckmans, T. Mihaylov, W. Faveere, J. Poissonnier, F. Van Waes, K. Moonen, G. B. Marin, J. W. Thybaut, K. Pierloot and B. F. Sels, ACS Catal., 2018, 8, 4201–4212 CrossRef CAS.
- T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123 CrossRef PubMed.
- X. Li, J. Ma, X. Jia, F. Xia, Y. Huang, Y. Xu and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 8048–8054 CrossRef CAS.
- M. Pelckmans, W. Vermandel, F. Van Waes, K. Moonen and B. F. Sels, Angew. Chem., Int. Ed., 2017, 56, 14540–14544 CrossRef CAS PubMed.
- M. J. Hülsey, Green Energy Environ., 2018, 3, 318–327 CrossRef.
- J. Yu, K. Maliutina and A. Tahmasebi, Bioresour. Technol., 2018, 270, 689–701 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- K. Techikawara, H. Kobayashi and A. Fukuoka, ACS Sustainable Chem. Eng., 2018, 6, 12411–12418 CrossRef CAS.
- V. Bragoni, R. K. Rit, R. Kirchmann, A. S. Trita and L. J. Gooßen, Green Chem., 2018, 20, 3210–3213 RSC.
- S. Chassaing, V. Bénéteau and P. Pale, Curr. Opin. Green Sustainable Chem., 2018, 10, 35–39 CrossRef.
- M. M. Khan, R. Yousuf, S. Khan and Shafiullah, RSC Adv., 2015, 5, 57883–57905 RSC.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118(2), 505–613 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- P. Sudarsanam, R. Zhong, S. V. den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664 RSC.
- A. Sharma, V. Pareek and D. Zhang, Renewable Sustainable Energy Rev., 2015, 50, 1081–1096 CrossRef CAS.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- S. H. Y. S. Abdullah, N. H. M. Hanapi, A. Azid, R. Umar, H. Juahir, H. Khatoon and A. Endut, Renewable Sustainable Energy Rev., 2017, 70, 1040–1051 CrossRef CAS.
- K. A. Rogers and Y. Zheng, ChemSusChem, 2016, 9, 1750–1772 CrossRef CAS PubMed.
- G. Kumar, S. Shobana, W. H. Chen, Q. V. Bach, S. H. Kim, A. E. Atabani and J. S. Chang, Green Chem., 2017, 19, 44–67 RSC.
- M. Pelckmans, T. Renders, S. Van de Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed.
- J. A. Joule and K. Mills, in Heterocyclic Chemistry, Wiley, 5th edn, 2010 Search PubMed.
- D. Forberg, J. Obenauf, M. Friedrich, S. M. Hühne, W. Mader, G. Motz and R. Kempe, Catal. Sci. Technol., 2014, 4, 4188–4192 RSC.
- V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC.
- V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Commun., 2013, 49, 591–593 RSC.
- V. Chandrashaker, M. Taniguchi, M. Ptaszek and J. S. Lindsey, Tetrahedron, 2012, 68, 6957–6967 CrossRef CAS.
- A. Kornienko and J. J. La Clair, Nat. Prod. Rep., 2017, 34, 1051–1060 RSC.
- X. L. He, H. R. Zhao, X. Song, B. Jiang, W. Du and Y. C. Chen, ACS Catal., 2019, 9, 4374–4381 CrossRef CAS.
- B. C. Milgram, K. Eskildsen, S. M. Richter, W. R. Scheidt and K. A. Scheidt, J. Org. Chem., 2007, 72, 3941–3944 CrossRef CAS PubMed.
- V. F. Ferreira, M. C. B. de Souza, A. C. Cunha, L. O. Pereira and M. L. Ferreira, Org. Prep. Proced. Int., 2001, 33, 411–454 CrossRef CAS.
- M. A. Yurovskaya and R. S. Alekseyev, Chem. Heterocycl. Compd., 2014, 49, 1400–1425 CrossRef CAS.
- Z. Su, W. Gu, S. Qian, S. Xue and C. Wang, Eur. J. Org. Chem., 2018, 1019–1025 CrossRef CAS.
- V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC.
- J. Vaitla, A. Bayer and K. H. Hopmann, Angew. Chem., Int. Ed., 2017, 56, 4277–4281 CrossRef CAS PubMed.
- P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 14373–14377 CrossRef CAS PubMed.
- V. Estevez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC.
- M. Gao, C. He, H. Chen, R. Bai, B. Cheng and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 6958–6961 CrossRef CAS PubMed.
- B. B. Thompson and J. Montgomery, Org. Lett., 2011, 13, 3289–3291 CrossRef CAS PubMed.
- T. J. Donohoe, J. F. Bower and L. K. M. Chan, Org. Biomol. Chem., 2012, 10, 1322–1328 RSC.
- B. Ramanathan, A. J. Keith, D. Armstrong and A. L. Odom, Org. Lett., 2004, 6, 2957–2960 CrossRef CAS PubMed.
- M. S. T. Morin, D. J. St-Cyr and B. A. Arndtsen, Org. Lett., 2010, 12, 4916–4919 CrossRef CAS PubMed.
- X. Qi, H. Xiang, Y. Yang and C. Yang, RSC Adv., 2015, 5, 98549–98552 RSC.
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, Appl. Catal., A, 2015, 504, 664–671 CrossRef CAS.
- Z. Gong, Y. Lei, P. Zhou and Z. Zhang, New J. Chem., 2017, 41, 10613–10618 RSC.
- G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- G. Chelucci, Coord. Chem. Rev., 2017, 331, 1–36 CrossRef CAS.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- N. D. Schley, G. E. Dobereiner and R. H. Crabtree, Organometallics, 2011, 30, 4174–4179 CrossRef CAS.
- P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 14373–14377 CrossRef CAS PubMed.
- J. C. Borghs, Y. Lebedev, M. Rueping and O. El-Sepelgy, Org. Lett., 2019, 21, 70–74 CrossRef CAS PubMed.
- S. J. Pridmore, P. A. Slatford, J. E. Taylor, M. K. Whittlesey and J. M. J. Williams, Tetrahedron, 2009, 65, 8981–8986 CrossRef CAS.
- S. I. Murahashi, T. Shimamura and I. Moritani, J. Chem. Soc., Chem. Commun., 1974, 931–932 RSC.
- K. Singh, L. M. Kabadwal, S. Bera, A. Alanthadka and D. Banerjee, J. Org. Chem., 2018, 83, 15406–15414 CrossRef CAS PubMed.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- B. Emayavaramban, M. Sen and B. Sundararaju, Org. Lett., 2017, 19, 6–9 CrossRef CAS PubMed.
- T. Yan and K. Barta, ChemSusChem, 2016, 9, 2321–2325 CrossRef CAS PubMed.
- G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140–144 CrossRef CAS PubMed.
- D. Forberg, J. Obenauf, M. Friedrich, S. M. Hühne, W. Mader, G. Motz and R. Kempe, Catal. Sci. Technol., 2014, 4, 4188–4192 RSC.
- D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012–4015 CrossRef CAS PubMed.
- K. Iida, T. Miura, J. Ando and S. Saito, Org. Lett., 2013, 157, 1436–1439 CrossRef PubMed.
- M. Zhang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 597–601 CrossRef CAS PubMed.
- M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384–11388 CrossRef CAS PubMed.
- F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 7261–7265 CrossRef CAS PubMed.
- D. C. Hargis and R. L. Shubkin, Tetrahedron Lett., 1990, 31, 2991–2994 CrossRef CAS.
- K. Hatada, M. Shimada, K. Fujita, Y. Ono and T. Keii, Chem. Lett., 1974, 3, 439–442 CrossRef.
- L. Tao, Z. J. Wang, T. H. Yan, Y. M. Liu, H. Y. He and Y. Cao, ACS Catal., 2017, 7, 959–964 CrossRef CAS.
- B. Wozniak, Y. Li, S. Hinze, S. Tin and J. G. de Vries, Eur. J. Org. Chem., 2018, 2009–2012 CrossRef CAS.
- V. V. Mossine, C. L. Barnes, D. L. Chance and T. P. Mawhinney, Angew. Chem., Int. Ed., 2009, 48, 5517–5520 CrossRef CAS PubMed.
- M. Hellwig and T. Henle, Angew. Chem., Int. Ed., 2014, 53, 10316–10329 CrossRef CAS PubMed.
- N. D. Adhikary, S. Kwon, W. J. Chung and S. Koo, J. Org. Chem., 2015, 80, 7693–7701 CrossRef CAS PubMed.
- C. Moreno-Marrodan, F. Liguori and P. Barbaro, Mol. Catal., 2019, 466, 60–69 CrossRef CAS.
- H. Wu, W. Dai, S. Saravanamurugan, H. Li and S. Yang, ACS Sustainable Chem. Eng., 2019, 7, 10207–10213 CrossRef CAS.
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527–5547 RSC.
- Z. Xue, Q. Liu, J. Wang and T. Mu, Green Chem., 2018, 20, 4391–4408 RSC.
- C. Xie, J. Song, H. Wu, Y. Hu, H. Liu, Z. Zhang, P. Zhang, B. Chen and B. Han, J. Am. Chem. Soc., 2019, 141, 4002–4009 CrossRef CAS PubMed.
- A. S. Touchy, S. M. A. Hakim Siddiki, K. Kon and K. Shimizu, ACS Catal., 2014, 4, 3045–3050 CrossRef CAS.
- S. M. A. H. Siddiki, A. S. Touchy, A. Bhosale, T. Toyao, Y. Mahara, J. Ohyama, A. Satsuma and K. Shimizu, ChemCatChem, 2018, 10, 789–795 CrossRef CAS.
- J. D. Vidal, M. J. Climent, P. Concepcion, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2015, 5, 5812–5821 CrossRef CAS.
- D. Rodríguez-Padrón, A. R. Puente-Santiago, A. M. Balu, A. A. Romero, M. J. Muñoz-Batista and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 16637–16644 CrossRef.
- Z. Xu, P. Yan, H. Jiang, K. Liu and Z. C. Zhang, Chin. J. Chem., 2017, 35, 581–585 CrossRef CAS.
- J. J. Martíneza, L. Silva, H. A. Rojas, G. P. Romanelli, L. A. Santos, T. C. Ramalho, M. H. Brijaldo and F. B. Passos, Catal. Today, 2017, 296, 118–126 CrossRef.
- S. Wang, H. Huang, C. Bruneau and C. Fischmeister, ChemSusChem, 2017, 10, 4150–4154 CrossRef CAS PubMed.
- J. D. Vidal, M. J. Climent, A. Corma, P. Concepcion and S. Iborra, ChemSusChem, 2017, 10, 119–128 CrossRef CAS PubMed.
- P. Cao, T. Ma, H. Y. Zhang, G. Yin, J. Zhao and Y. Zhang, Catal. Commun., 2018, 116, 85–90 CrossRef CAS.
- G. Chieffi, M. Braun and D. Esposito, ChemSusChem, 2015, 8, 3590–3594 CrossRef CAS PubMed.
- G. Gao, P. Sun, Y. Li, F. Wang, Z. Zhao, Y. Qin and F. Li, ACS Catal., 2017, 7, 4927–4935 CrossRef CAS.
- M. H. Tang, S. J. Mao, M. M. Li, Z. Z. Wei, F. Xu, H. F. Li and Y. Wang, ACS Catal., 2015, 5, 3100–3107 CrossRef CAS.
- G. Papoian, J. K. Nørskov and R. Hoffmann, J. Am. Chem. Soc., 2000, 122, 4129–4144 CrossRef CAS.
- G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiely-VCH, New York, 2nd edn, 2008, vol. 1, p. 3 Search PubMed.
- M. C. Fu, R. Shang, W. M. Cheng and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 9042–9046 CrossRef CAS PubMed.
- D. Wei, C. Netkaew and C. Darcel, Adv. Synth. Catal., 2019, 361, 1781–1786 CrossRef CAS.
- Y. Ogiwara, T. Uchiyama and N. Sakai, Angew. Chem., 2016, 128, 1896–1899 CrossRef.
- C. Wu, X. Luo, H. Zhang, X. Liu, G. Ji, Z. Liu and Z. Liu, Green Chem., 2017, 19, 3525–3529 RSC.
- C. Wu, H. Zhang, B. Yu, Y. Chen, Z. Ke, S. Guo and Z. Liu, ACS Catal., 2017, 7, 7772–7776 CrossRef CAS.
- L. Deng, Y. Zhao, J. Li, Y. Fu, B. Liao and Q. X. Guo, ChemSusChem, 2010, 3, 1172–1175 CrossRef CAS PubMed.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC.
- X. L. Du, L. He, S. Zhao, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., 2011, 123, 7961–7965 CrossRef.
- Y. Wei, C. Wang, X. Jiang, D. Xue, J. Li and J. Xiao, Chem. Commun., 2013, 49, 5408–5410 RSC.
- Y. B. Huang, J. J. Dai, X. J. Deng, Y. C. Qu, Q. X. Guo and Y. Fu, ChemSusChem, 2011, 4, 1578–1581 CrossRef CAS PubMed.
- Y. B. Huang, J. J. Dai, X. J. Deng, Y. C. Qu, Q. X. Guo and Y. Fu, ChemSusChem, 2011, 4, 1578–1581 CrossRef CAS PubMed.
- G. Metzker, R. M. P. Dias and A. C. B. Burtoloso, ChemistrySelect, 2018, 3, 368–372 CrossRef CAS.
- A. S. Amarasekara and Y. M. Lawrence, Tetrahedron Lett., 2018, 59, 1832–1835 CrossRef CAS.
- Z. Sun, J. Chen and T. Tu, Green Chem., 2017, 19, 789–794 RSC.
- Y. Wei, C. Wang, X. Jiang, D. Xue, Z. T. Liu and J. Xiao, Green Chem., 2014, 16, 1093–1096 RSC.
- T. Ma, H. Y. Zhang, G. Yin, J. Zhao and Y. Zhang, J. Flow Chem., 2018, 8, 35–43 CrossRef CAS.
- A. Ledoux, L. Sandjong Kuigwa, E. Framery and B. Andrioletti, Green Chem., 2015, 17, 3251–3254 RSC.
- H. Li, H. Wu, H. Zhang, Y. Su, S. Yang and E. J. M. Hensen, ChemSusChem, 2019, 12, 3778–3784 CrossRef CAS PubMed.
- J. Qi, C. Sun, Y. Tian, X. Wang, G. Li, Q. Xiao and D. Yin, Org. Lett., 2014, 16, 190–192 CrossRef CAS PubMed.
- Y. Louven, K. Schute and R. Palkovits, ChemCatChem, 2019, 11, 439–442 CrossRef CAS.
- F. De Schouwer, S. Adriaansen, L. Claes and D. E. De Vos, Green Chem., 2017, 19, 4919–4929 RSC.
- J. Hulsbosch, L. Claes and D. E. De Vos, Tetrahedron Lett., 2018, 59, 1646–1650 CrossRef CAS.
- H. Batchu, S. Bhattacharyya, R. Kant and S. Batra, J. Org. Chem., 2015, 80, 7360–7374 CrossRef CAS PubMed.
- H. Lee, M. Y. Berezin, R. Tang, N. Zhegalova and S. Achilefu, Photochem. Photobiol., 2013, 89, 326–331 CrossRef CAS PubMed.
- E. C. Constable and P. J. Steel, Coord. Chem. Rev., 1989, 93, 205–223 CrossRef CAS.
- H. El Khadem and M. M. Mahammed-Aly, J. Chem. Soc., 1963, 4929–4932 RSC.
- H. El Khadem, J. Org. Chem., 1964, 29, 3072–3074 CrossRef CAS.
- H. El Khadem, Z. M. El-Shafei and M. M. Mohamed-Aly, J. Org. Chem., 1964, 29, 1565–1567 CrossRef.
- E. S. H. El Ashry, K. F. Atta, S. Aboul-Ela and R. Beldi, J. Carbohydr. Chem., 2007, 26, 429–437 CrossRef CAS.
- V. Diehl, E. Cuny and F. W. Lichtenthaler, Heterocycles, 1998, 48, 1193–1201 CrossRef CAS.
- N. Oikawa, C. Müller, M. Kunzb and F. W. Lichtenthaler, Carbohydr. Res., 1998, 309, 269–279 CrossRef CAS.
- X. Yang, P. Zhang, Y. Zhou, J. Wang and H. Liu, Chin. J. Chem., 2012, 30, 670–674 CrossRef CAS.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. Mabkhot, F. Al-aizari and M. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1883, 16, 2597–2599 CrossRef.
- L. Wu, L. Feng, H. Zhang, Q. Liu, X. He, F. Yang and H. Xia, J. Org. Chem., 2008, 73, 2883–2885 CrossRef CAS PubMed.
- D. L. Reger, J. R. Gardinier, T. Christian Grattan, M. R. Smith and M. D. Smith, New J. Chem., 2003, 27, 1670–1677 RSC.
- D. C. Schmitt, A. P. Taylor, A. C. Flick and R. E. Kyne, Org. Lett., 2015, 17, 1405–1408 CrossRef CAS PubMed.
- L. Zhang, X. M. Peng, G. L. V. Damu, R. X. Geng and C. H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed.
- J. C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A. C. Gaumont, Chem. Rev., 2008, 108, 5035–5060 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010, vol. 3, p. 518 Search PubMed.
- W. J. Darby, H. B. Lewis and J. R. Totter, J. Am. Chem. Soc., 1942, 64, 463–464 CrossRef CAS.
- J. Streith, A. Boiron, A. Frankowski, D. Le Nouen, H. Rudyk and T. Tschamber, Synthesis, 1995, 944–946 CrossRef CAS.
- A. Brust and E. Cuny, Green Chem., 2013, 15, 2993–2998 RSC.
- E. Dubost, D. Le Nouën, J. Streith, C. Tarnus and T. Tschamber, Eur. J. Org. Chem., 2006, 610–626 CrossRef CAS.
- M. Avalos, R. Babiano, P. Cintas, M. B. Hursthouse, J. L. Jimenez, M. E. Light, J. C. Palacios and G. Silvero, Tetrahedron, 2005, 61, 7931–7944 CrossRef CAS.
- T. Graier and A. Vasella, Helv. Chim. Acta, 1995, 78, 1738–1746 CrossRef.
- G. R. Peh and P. E. Floreancig, Org. Lett., 2015, 17, 3750–3753 CrossRef CAS PubMed.
- K. Mal, A. Sharma and I. Das, Chem. – Eur. J., 2014, 20, 11932–11945 CrossRef CAS PubMed.
- K. Mal and I. Das, J. Org. Chem., 2016, 81, 932–945 CrossRef CAS PubMed.
- H. Li, Y. Li, Z. Fang and R. L. Smith Jr., Catal. Today, 2019, 319, 84–92 CrossRef CAS.
- S. Tanaka, K. Ashida, G. Tatsuta and A. Mori, Synlett, 2015, 26, 1496–1500 CrossRef CAS.
- T. H. Graham, Org. Lett., 2010, 12, 3614–3617 CrossRef CAS PubMed.
- H. Li, T. Yang and Z. Fang, Appl. Catal., B, 2018, 227, 79–89 CrossRef CAS.
- S. Selvakumar, A. Fairweather, A. Ugrinov and M. P. Sibi, Heterocycles, 2018, 97, 151–157 CrossRef CAS.
- E. S. H. El Ashry, Y. El Kilany and N. M. Nahas, Top. Heterocycl. Chem., 2007, 7, 1–30 CAS.
- H. Yang, J. Guo, Z. Gao, J. Gou and B. Yu, Org. Lett., 2018, 20, 4893–4897 CrossRef CAS PubMed.
- L. Guillemard, F. Colobert and J. Wencel-Delord, Adv. Synth. Catal., 2018, 360, 4181–4190 CrossRef.
- Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS.
- K. S. K. Reddy, C. Srinivasakannan and K. V. Raghavan, Catal. Surv. Asia, 2012, 16, 28–35 CrossRef.
- M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096–10097 CrossRef CAS PubMed.
- S. Shimizu, N. Abe, A. Iguchi and H. Sato, Catal. Surv. Asia, 1998, 2, 71–76 CrossRef CAS.
- S. Shimizu, N. Abe, A. Iguchi, M. Dohba, H. Sato and K. I. Hirose, Microporous Mesoporous Mater., 1998, 21, 447–451 CrossRef CAS.
- K. R. S. K. Reddy, I. Sreedhar and K. V. Raghavan, Appl. Catal., A, 2008, 339, 15–20 CrossRef CAS.
- Y. Liu, H. Yang, F. Jin, Y. Zhang and Y. Li, Chem. Eng. J., 2008, 136, 282–287 CrossRef CAS.
- F. Jin, Y. Tian and Y. Li, Ind. Eng. Chem. Res., 2009, 48, 1873–1879 CrossRef CAS.
- F. Jin, Y. Cui and Y. Li, Appl. Catal., A, 2008, 350, 71–78 CrossRef CAS.
- X. Zhang, Z. Wu, W. Liu and Z. Chao, Catal. Commun., 2016, 80, 10–14 CrossRef CAS.
- W. Zhang, S. Duan and Y. Zhang, React. Kinet., Mech. Catal., 2019, 127, 391–411 CrossRef CAS.
- F. Jin, G. Wu and Y. Li, Chem. Eng. Technol., 2011, 34, 1660–1666 CrossRef CAS.
- Y. Zhang, X. Yan, B. Niu and J. Zhao, Green Chem., 2016, 18, 3139–3151 RSC.
- L. Xu, Q. Yao, Y. Zhang and Y. Fu, RSC Adv., 2016, 6, 86034–86042 RSC.
- L. Xu, Z. Han, Q. Yao, J. Deng, Y. Zhang, Y. Fu and Q. Guo, Green Chem., 2015, 17, 2426–2435 RSC.
- C. W. Luo, C. Huang, A. Li, W. J. Yi, X. Y. Feng, Z. J. Xu and Z. S. Chao, Ind. Eng. Chem. Res., 2016, 55, 893–911 CrossRef CAS.
- L. Xu, Q. Yao, Z. Han, Y. Zhang and Y. Fu, ACS Sustainable Chem. Eng., 2016, 4, 1115–1122 CrossRef CAS.
- C. W. Luo and A. Li, React. Kinet., Mech. Catal., 2018, 125, 365–380 CrossRef CAS.
- X. Zhang, C. W. Luo, C. Huang, B. H. Chen, D. G. Huang, J. G. Pan and Z. S. Chao, Chem. Eng. J., 2014, 253, 544–553 CrossRef CAS.
- C. W. Luo, A. Li, J. F. An, X. Y. Feng, X. Zhang, D. D. Feng and Z. S. Chao, Chem. Eng. J., 2015, 273, 7–18 CrossRef CAS.
- C. Müller, V. Diehl and F. W. Lichtenthaler, Tetrahedron, 1998, 54, 10703–10712 CrossRef.
- N. I. E. L. S. Elming and N. Clauson-Kaas, Acta Chem. Scand., 1956, 10, 1603–1605 CrossRef CAS.
- J. S. Dickschat, H. Reichenbach, I. Wagner-Dobler and S. Schulz, Eur. J. Org. Chem., 2005, 4141–4153 CrossRef CAS.
- D. F. Taber, P. W. DeMatteo and K. V. Taluskie, J. Org. Chem., 2007, 72, 1492–1494 CrossRef CAS PubMed.
- Z. Yin, H. Zeng, J. Wu, S. Zheng and G. Zhang, ACS Catal., 2016, 6, 6546–6550 CrossRef CAS.
- C. Y. Zhang and J. M. Tour, J. Am. Chem. Soc., 1999, 121, 8783–8790 CrossRef CAS.
- I. Park, J. Lee, Y. Rhee, Y. Han and H. Kim, Appl. Catal., A, 2003, 253, 249–255 CrossRef CAS.
- P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 7734–7741 CrossRef CAS PubMed.
- M. Subrahmanyam, S. J. Kulkarni and B. Srinivas, React. Kinet. Catal. Lett., 1993, 49, 455–459 CrossRef CAS.
- T. Shibamoto and R. A. Bernhard, J. Agric. Food Chem., 1977, 25, 609–614 CrossRef CAS.
- L. Song, M. Zheng, J. Pang, J. Sebastian, W. Wang, M. Qu, J. Zhao, X. Wang and T. Zhang, Green Chem., 2017, 19, 3515–3519 RSC.
- X. Chen, H. Yang, M. J. Hülsey and N. Yan, ACS Sustainable Chem. Eng., 2017, 5, 11096–11104 CrossRef CAS.
- P. Merino, S. Franco, F. L. Merchan and T. Tejero, in Recent Research Development in Synthetic Organic Chemistry, 2000, vol. 3, p. 65 Search PubMed.
- R. A. Al-Qawasmeh, T. H. Al-Tel, R. J. Abdel-Jalil and W. Voelter, Chem. Lett., 1999, 28, 541–542 CrossRef.
- A. Montero, H. Feist, M. Michalik, J. Quincoces and K. Peseke, J. Carbohydr. Chem., 2002, 21, 305–312 CrossRef CAS.
- J. Hirsch, E. Petrakova and M. S. Feather, J. Carbohydr. Chem., 1995, 14, 1179–1186 CrossRef CAS.
- G. E. Üstün, S. K. A. Solmaz, T. Morsünbül and H. S. Azak, J. Hazard. Mater., 2010, 180, 508–513 CrossRef PubMed.
- P. V. Thanikachalam, R. K. Maurya, V. Garg and V. Monga, Eur. J. Med. Chem., 2019, 180, 562–612 CrossRef CAS.
- X. H. Zhang, Y. Cui, R. Katoh, N. Koumura and K. Hara, J. Phys. Chem. C, 2010, 114, 18283–18290 CrossRef CAS.
- M. Zhang, G. Qin, J. Liu, Z. Zhen, A. A. Fedorchuk, G. Lakshminarayana, A. A. Albassamf, A. M. El-Naggar, K. Ozgah, I. V. Kitykh and I. V. Kityk, Chem. Phys. Lett., 2017, 681, 105–109 CrossRef CAS.
- J. Gartz, J. Basic Microbiol., 1994, 34, 17–22 CrossRef CAS.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- J. Siu, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2004, 2, 160–167 RSC.
- A. S. Makarov, A. A. Merkushev, M. G. Uchuskin and I. V. Trushkov, Org. Lett., 2016, 18, 2192–2195 CrossRef CAS PubMed.
- Q. Yao, L. Xu, Z. Han and Y. Zhang, Chem. Eng. J., 2015, 280, 74–81 CrossRef CAS.
- L. Xu, Y. Jiang, Q. Yao, Z. Han, Y. Zhang, Y. Fu, Q. Guo and G. W. Huber, Green Chem., 2015, 17, 1281–1290 RSC.
- Q. Yao, L. Xu, Y. Zhang and Y. Fu, J. Anal. Appl. Pyrolysis, 2016, 121, 258–266 CrossRef CAS.
- M. Campanati, S. Franceschini, O. Piccolo and A. Vaccari, J. Catal., 2005, 232, 1–9 CrossRef CAS.
- P. J. Llabres-Campaner, R. Ballesteros-Garrido, R. Ballesteros and B. Abarca, J. Org. Chem., 2018, 83, 521–526 CrossRef CAS PubMed.
- S. Ali, N. Ali, B. Ahmad Dar, V. Pradhan and M. Farooqui, Mini-Rev. Med. Chem., 2013, 13, 1792–1800 CrossRef PubMed.
- A. V. Karchava, F. S. Melkonyan and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 48, 391–407 CrossRef CAS.
- T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed.
- S. S. Panda, R. Malik and S. C. Jain, Curr. Org. Chem., 2012, 16, 1905–1919 CrossRef CAS.
- C. Zhu and Y. Wei, ChemSusChem, 2011, 4, 1082–1086 CrossRef CAS.
- B. Chen, C. Zhang, L. Niu, X. Shi, H. Zhang, X. Lan and G. Bai, Chem. – Eur. J., 2018, 24, 348–3487 Search PubMed.
- S. Verma, R. B. Nasir Baig, M. N. Nadagouda, C. Len and R. S. Varma, Green Chem., 2017, 19, 164–168 RSC.
- M. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720–722 CrossRef CAS PubMed.
- A. Yamaguchi, O. Sato, N. Mimura and M. Shirai, Catal. Today, 2016, 265, 199–202 CrossRef CAS.
- Q. Liu, G. Xu, X. Wang, X. Liu and X. Mu, ChemSusChem, 2016, 9, 3465–3472 CrossRef CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef.
- A. V. Karchava, F. S. Melkonyan and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 48, 391–407 CrossRef CAS.
- A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi and J. M. Williams, Org. Lett., 2009, 11, 2039–2042 CrossRef CAS PubMed.
- T. Hille, T. Irrgang and R. Kempe, Chem. – Eur. J., 2014, 20, 5569–5572 CrossRef CAS PubMed.
- Z. Sun, G. Bottari and K. Barta, Green Chem., 2015, 17, 5172–5181 RSC.
- P. Daw, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 7456–7460 CrossRef CAS.
- M. Shaikh, R. Yadav, P. K. Tyagi, L. Mishra and K. V. Ranganath, ChemNanoMat, 2018, 4, 542–545 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and S. Martínez-Silvestre, ChemCatChem, 2013, 5, 3866–3874 CrossRef CAS.
- L. Knorr, Justus Liebigs Ann. Chem., 1886, 236, 69–115 CrossRef.
- Z. H. Skraup, Monatsh. Chem. Verw. Teile Anderer Wiss., 1880, 1, 316–318 CrossRef.
- R. Camps, Arch. Pharm., 1899, 237, 659–691 CrossRef CAS.
- D. Wei, V. Dorcet, C. Darcel and J. B. Sortais, ChemSusChem, 2019, 12, 3078–3082 CrossRef CAS PubMed.
- S. Atechian, N. Nock, R. D. Norcross, H. Ratni, A. W. Thomas, J. Verron and R. Masciadri, Tetrahedron, 2007, 63, 2811–2823 CrossRef CAS.
- B. W. J. Chen, L. L. Chng, J. Yang, Y. Wei, J. Yang and J. Y. Ying, ChemCatChem, 2013, 5, 277–283 CrossRef CAS.
- R. Wang, H. Fan, W. Zhao and F. Li, Org. Lett., 2016, 18, 3558–3561 CrossRef CAS PubMed.
- C. S. Cho, H. J. Seok and S. O. Shim, J. Heterocycl. Chem., 2005, 42, 1219–1222 CrossRef CAS.
- B. Pan, B. Liu, E. Yue, Q. Liu, X. Yang, Z. Wang and W. H. Sun, ACS Catal., 2016, 6, 1247–1253 CrossRef CAS.
- S. Elangovan, J. B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS PubMed.
- S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- M. Glatz, B. Stöger, D. Himmelbauer, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 4009–4016 CrossRef CAS PubMed.
- R. Fertig, T. Irrgang, F. Freitag, J. Zander and R. Kempe, ACS Catal., 2018, 8, 8525–8530 CrossRef CAS.
- S. Shee, K. Ganguli, K. Jana and S. Kundu, Chem. Commun., 2018, 54, 6883–6886 RSC.
- C. S. Cho, W. X. Ren and N. S. Yoon, J. Mol. Catal. A: Chem., 2009, 299, 117–120 CrossRef CAS.
- M. Maji, K. Chakrabarti, B. Paul, B. C. Roy and S. Kundu, Adv. Synth. Catal., 2018, 360, 722–729 CrossRef CAS.
- S. V. More, M. N. V. Sastry and C. F. Yao, Green Chem., 2006, 8, 91–95 RSC.
- Z. Zhao, D. D. Wisnoski, S. E. Wolkenberg, W. H. Leister, Y. Wang and C. W. Lindsley, Tetrahedron Lett., 2004, 45, 4873–4876 CrossRef CAS.
- S. Sithambaram, Y. Ding, W. Li, X. Shen, F. Gaenzler and S. L. Suib, Green Chem., 2008, 10, 1029–1032 RSC.
- S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Org. Biomol. Chem., 2004, 2, 788–796 RSC.
- D. Aparicio, O. A. Attanasi, P. Filippone, R. Ignacio, S. Lillini, F. Mantellini, F. Palacios and J. M. De los Santos, J. Org. Chem., 2006, 71, 5897–5905 CrossRef CAS PubMed.
- S. Antoniotti and E. Duñach, Tetrahedron Lett., 2002, 43, 3971–3973 CrossRef CAS.
- S. K. Singh, P. Gupta, S. Duggineni and B. Kundu, Synlett, 2003, 2147–2150 CAS.
- M. J. Climent, A. Corma, J. C. Hernández, A. B. Hungría, S. Iborra and S. Martínez-Silvestre, J. Catal., 2012, 292, 118–129 CrossRef CAS.
- Y. Pan, C. Chen, X. Xu, H. Zhao, J. Han, H. Li, L. Xu, Q. Fan and J. Xiao, Green Chem., 2018, 20, 403–411 RSC.
- C. Xie, Z. Zhang, D. Li, J. Gong, X. Han, X. Liu and C. Ma, J. Org. Chem., 2017, 82, 3491–3499 CrossRef CAS PubMed.
- K. Nikoofar, H. Heidari and Y. Shahedi, Cellulose, 2018, 25, 5697–5709 CrossRef CAS.
- S. Gupta and N. K. Khare, J. Mol. Struct., 2017, 1127, 309–313 CrossRef CAS.
- M. Nourisefat, F. Panahi and A. Khalafi-Nezhad, Org. Biomol. Chem., 2014, 12, 9419–9426 RSC.
- S. K. Dangolani, F. Panahi, Z. Tavaf, M. Nourisefat, R. Yousefi and A. Khalafi-Nezhad, ACS Omega, 2018, 3, 10341–10350 CrossRef PubMed.
- P. M. Shpuntov, A. A. Kolodina, M. G. Uchuskin and V. T. Abaev, Eur. J. Org. Chem., 2018, 461–469 CrossRef CAS.
- T. T. Pham, X. Chen, N. Yan and J. Sperry, Monatsh. Chem., 2018, 149, 857–861 CrossRef CAS.
- X. H. Yang, P. H. Zhang, Z. M. Wang, F. Jing, Y. H. Zhou and L. H. Hu, Ind. Crops Prod., 2014, 52, 413–419 CrossRef CAS.
- S. Ambethkar, V. Padmini and N. Bhuvanesh, J. Adv. Res., 2015, 6, 975–985 CrossRef CAS PubMed.
- A. V. Butin, T. A. Nevolina, V. A. Shcherbinin, I. V. Trushkov, D. A. Cheshkov and G. D. Krapivin, Org. Biomol. Chem., 2010, 8, 3316–3327 RSC.
- I. V. Trushkov, T. A. Nevolina, V. A. Shcherbinin, L. N. Sorotskaya and A. V. Butin, Tetrahedron Lett., 2013, 54, 3974–3976 CrossRef CAS.
- M. G. Uchuskin, A. S. Pilipenko, O. V. Serdyuk, I. V. Trushkovc and A. V. Butin, Org. Biomol. Chem., 2012, 10, 7262–7265 RSC.
- C. Lambruschini, A. Basso, L. Moni, A. Pinna, R. Riva and L. Banfi, Eur. J. Org. Chem., 2018, 5445–5455 CrossRef CAS.
- F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728–737 CrossRef CAS PubMed.
- S. I. Sadraei, B. S. Onge and J. F. Trant, Phys. Sci. Rev., 2018, 4, 20180074 Search PubMed.
- J. Zhang, X. Lu, T. Li, S. Wang and G. Zhong, J. Org. Chem., 2017, 82, 5222–5229 CrossRef CAS PubMed.
- S. A. Ali, S. K. Mondal, T. Das, S. K. Manna, A. Bera, D. Dafadar, S. Naskar, M. R. Molla and S. Samanta, Org. Biomol. Chem., 2019, 17, 4652–4662 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- T. Sakakura, J. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382–404 RSC.
- N. A. Tappe, R. M. Reich, V. D'Elia and F. E. Kühn, Dalton Trans., 2018, 47, 13281–13313 RSC.
- Q. Wu, J. Chen, X. Guo and Y. Xu, Eur. J. Org. Chem., 2018, 3105–3113 CrossRef CAS.
- S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS.
- H. Yoshida, H. Fukushima, J. Ohshita and A. Kunai, J. Am. Chem. Soc., 2006, 128, 11040–11041 CrossRef CAS PubMed.
- W. Lu, J. Ma, J. Hu, J. Song, Z. Zhang, G. Yang and B. Han, Green Chem., 2014, 16, 221–225 RSC.
- Z. Zhang, Q. Sun, C. Xia and W. Sun, Org. Lett., 2016, 18, 6316–6319 CrossRef CAS PubMed.
- S. Shyshkanov, T. N. Nguyen, F. M. Ebrahim, K. C. Stylianou and P. J. Dyson, Angew. Chem., Int. Ed., 2019, 58, 5371–5375 CrossRef CAS PubMed.
- O. Jacquet, C. D. N. Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117–120 CrossRef CAS.
- K. Liu, H. L. Teng and C. J. Wang, Org. Lett., 2014, 16, 4508–4511 CrossRef CAS PubMed.
- Z. L. He, H. L. Teng and C. J. Wang, Angew. Chem., Int. Ed., 2013, 52, 2934–2938 CrossRef CAS PubMed.
- H. L. Teng, L. Yao and C. J. Wang, J. Am. Chem. Soc., 2014, 136, 4075–4080 CrossRef CAS PubMed.
- Q. H. Li, L. Wei and C. J. Wang, J. Am. Chem. Soc., 2014, 136, 8685–8692 CrossRef CAS PubMed.
- M. C. Tong, X. Chen, H. Y. Tao and C. J. Wang, Angew. Chem., Int. Ed., 2013, 52, 12377–12380 CrossRef CAS PubMed.
- Y. Shi, P. C. J. Kamer, D. J. Cole-Hamilton, M. Harvie, E. F. Baxter, K. J. C. Lim and P. Pogorzelec, Chem. Sci., 2017, 8, 6911–6917 RSC.
- C. Lambruschini, A. Basso, L. Moni, A. Pinna, R. Riva and L. Banfi, Eur. J. Org. Chem., 2018, 5445–5455 CrossRef CAS.
- E. Zhang, X. Zhang, W. Wei, D. Wang, Y. Cai, T. Xu, M. Yan and Y. Zou, RSC Adv., 2015, 5, 5288–5294 RSC.
- F. W. Lichtenthaler, A. Brust and E. Cuny, Green Chem., 2001, 3, 201–209 RSC.
- A. Brust and E. Cuny, RSC Adv., 2014, 4, 5759–5767 RSC.
- S. P. Midya, V. G. Landge, M. K. Sahoo, J. Rana and E. Balaraman, Chem. Commun., 2018, 54, 90–93 RSC.
- D. Deng, B. Hu, M. Yang and D. Chen, Organometallics, 2018, 37, 2386–2394 CrossRef CAS.
- K. Singh, M. Vellakkaran and D. Banerjee, Green Chem., 2018, 20, 2250–2256 RSC.
- H. Chai, L. Wang, T. Liu and Z. Yu, Organometallics, 2017, 36, 4936–4942 CrossRef CAS.
- D. Forberg, T. Schwob and R. Kempe, Nat. Commun., 2018, 9, 1751 CrossRef PubMed.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC.
- M. Sawa, S. Miyazaki, R. Yonesaki, H. Morimoto and T. Ohshima, Org. Lett., 2018, 20, 5393–5397 CrossRef CAS PubMed.
- N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- G. G. Gerosa, N. Grimblat, R. A. Spanevello, A. G. Suárez and A. M. Sarotti, Org. Biomol. Chem., 2017, 15, 426–434 RSC.
- Y. Tsai, C. M. B. Etichetti, C. D. Benedetto, J. E. Girardini, F. T. Martins, R. A. Spanevello, A. G. Suárez and A. M. Sarotti, J. Org. Chem., 2018, 83, 3516–3528 CrossRef CAS PubMed.
- S. W. Kim, E. T. Ledingham, S. Kudo, B. W. Greatrex and J. Sperry, Eur. J. Org. Chem., 2018, 2028–2038 CrossRef CAS.
- B. Voigt, M. Linke and R. Mahrwald, Org. Lett., 2015, 17, 2606–2609 CrossRef CAS PubMed.
- R. P. Bhusal and J. Sperry, Green Chem., 2016, 18, 2453–2459 RSC.
- O. Sato, Y. Ikushima and T. Yokoyama, J. Org. Chem., 1998, 63, 9100–9102 CrossRef CAS.
- A. A. Jarrahpour, M. Shekarriz and A. Taslimi, Molecules, 2004, 9, 29–38 CrossRef CAS PubMed.
- X. Yang, Q. Shang, C. Bo, L. Hu and Y. Zhoua, Org. Chem., 2018, 5, 184–193 Search PubMed.
- D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima and R. Kempe, Nat. Commun., 2016, 7, 13201 CrossRef CAS PubMed.
- K. Li, C. Zhu, L. Zhang and X. Zhu, Bioresour. Technol., 2016, 209, 142–147 CrossRef CAS PubMed.
- W. Chen, K. Li, M. Xia, Y. Chen, H. Yang, Z. Chen, X. Chen and H. Chen, Bioresour. Technol., 2018, 263, 350–357 CrossRef CAS PubMed.
- W. Chen, Y. Chen, H. Yang, K. Li, X. Chen and H. Chen, Bioresour. Technol., 2018, 249, 247–253 CrossRef CAS PubMed.
- Z. X. Xu, J. H. Cheng, Z. X. He, Q. Wang, Y. W. Shao and X. Hu, Bioresour. Technol., 2019, 278, 311–317 CrossRef CAS PubMed.
- B. M. E. Chagas, C. Dorado, M. J. Serapiglia, C. A. Mullen, A. A. Boateng, M. A. F. Melo and C. H. Ataíde, Fuel, 2016, 179, 124–134 CrossRef CAS.
- W. Chen, H. Yang, Y. Chen, X. Chen, Y. Fang and H. Chen, J. Anal. Appl. Pyrolysis, 2016, 120, 186–193 CrossRef CAS.
- M. Schnitzer, C. M. Monreal and E. E. Powell, J. Environ. Sci. Health, Part B, 2014, 49, 51–67 CrossRef CAS PubMed.
- Z. X. Xu, L. Xu, J. H. Cheng, Z. X. He, Q. Wang and X. Hu, Fuel Process. Technol., 2018, 182, 37–44 CrossRef CAS.
- W. Yang, H. Wang, J. Zhou and S. Wu, J. Supercrit. Fluids, 2018, 135, 254–262 CrossRef CAS.
- T. M. Aida, K. Oshima, C. Abe, R. Maruta, M. Iguchi, M. Watanabe and R. L. Smith Jr., Carbohydr. Polym., 2014, 106, 172–178 CrossRef CAS PubMed.
- G. Margoutidis, V. H. Parsons, C. S. Bottaro, N. Yan and F. M. Kerton, ACS Sustainable Chem. Eng., 2018, 6, 1662–1669 CrossRef CAS.
- M. Osada, C. Miurab, Y. S. Nakagawa, M. Kaihara, M. Nikaido and K. Totani, Carbohydr. Polym., 2015, 134, 718–725 CrossRef CAS PubMed.
- T. M. Aida, M. Oshima and R. L. Smith Jr., ACS Sustainable Chem. Eng., 2017, 5, 7709–7715 CrossRef CAS.
- Y. Fan, U. Hornung, N. Dahmen and A. Kruse, Biomass Convers. Biorefin., 2018, 8, 909–923 CrossRef CAS.
- X. Zhuang, Y. Huang, Y. Song, H. Zhan, X. Yin and C. Wu, Bioresour. Technol., 2017, 245, 463–470 CrossRef CAS PubMed.
- T. M. Aida, T. Nonaka, S. Fukuda, H. Kujiraoka, Y. Kumagai, R. Maruta, M. Ota, I. Suzuki, M. M. Watanabe, H. Inomata and R. L. Smith Jr., Algal Res., 2016, 18, 61–68 CrossRef.
- T. M. Aida, R. Maruta, Y. Tanabe, M. Oshima, T. Nonaka, H. Kujiraoka, Y. Kumagai, M. Ota, I. Suzuki, M. M. Watanabe, H. Inomata and R. L. Smith Jr., Bioresour. Technol., 2017, 228, 186–192 CrossRef CAS PubMed.
- A. Gollakota and P. E. Savage, ACS Sustainable Chem. Eng., 2018, 6, 9018–9027 CrossRef CAS.
- J. Jiang and P. E. Savage, Energy Fuels, 2017, 32, 4118–4126 CrossRef.
- J. D. Sheehan and P. E. Savage, ACS Sustainable Chem. Eng., 2017, 5, 10967–10975 CrossRef CAS.
- J. D. Sheehan, A. Abraham and P. E. Savage, React. Chem. Eng., 2019, 4, 1237–1252 RSC.
- L. Xu, Q. Yao, J. Deng, Z. Han, Y. Zhang, Y. Fu, G. W. Huber and Q. Guo, ACS Sustainable Chem. Eng., 2015, 3, 2890–2899 CrossRef CAS.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- Y. Zheng, Z. Wang, C. Liu, L. Tao, Y. Huang and Z. Zheng, J. Energy Inst., 2020, 93, 210–223 CrossRef.
- S. B. Kim, C. Park and S. W. Kim, Bioresour. Technol., 2014, 172, 194–200 CrossRef CAS PubMed.
- Y. Zhang, Z. Yuan, B. Hu, J. Deng, Q. Yao, X. Zhang, X. Liu, Y. Fu and Q. Lu, Green Chem., 2019, 21, 812–820 RSC.
- H. Li, M. Wang, H. Liu, N. Luo, J. Lu, C. Zhang and F. Wang, ACS Sustainable Chem. Eng., 2018, 6, 3748–3753 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |