
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile method for the simultaneous recovery of rare-earth elements and transition metals from Nd–Fe–B magnets†
Xuan
Xu
 *ab,
Saso
Sturm
ab,
Zoran
Samardzija
a,
Janez
Scancar
bc,
Katarina
Markovic
c and
Kristina
Zuzek Rozman
ab
*ab,
Saso
Sturm
ab,
Zoran
Samardzija
a,
Janez
Scancar
bc,
Katarina
Markovic
c and
Kristina
Zuzek Rozman
ab
aDepartment for Nanostructured Materials, Jožef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia. E-mail: xuan.xu@ijs.si
bJožef Stefan International Postgraduate School, Jamova 39, SI-1000 Ljubljana, Slovenia
cDepartment of Environmental Sciences, Jožef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
First published on 29th January 2020
Abstract
A green and facile electrochemical method for Nd–Fe–B PM recycling was developed. The proposed procedure enables a selective REE recovery and a simultaneous Fe metal deposition in only two processing steps, with the total re-use of the electrolyte. The proposed procedure thus avoids the formation of solid Fe-based waste and wastewater discharge.
Rare-earth elements (REEs) which are the essential components of permanent magnets (PMs) are considered by the European Commission to be the most critical materials in terms of their economic importance and supply risk.1 The use of PMs, especially neodymium–iron–boron (Nd–Fe–B) types, will expand rapidly, as e-vehicles, e-scooters and e-bikes have become increasingly widespread and add to the huge quantities of magnets already being used in electricity-generating wind turbines.2 A Nd–Fe–B PM typically contains 28–35 wt% REEs (Pr, Nd, Tb, and Dy), which means that end-of-life (EoL) magnets are an important secondary resource for REEs. Recovering REEs from EoL Nd–Fe–B PMs is, therefore, going to be the key strategy for overcoming the serious supply risks associated with REEs and making REEs available for other applications, e.g., solid–oxide fuel cells and nuclear reactors.3
Various approaches for REE recovery, such as glass-slag extraction,4 gas-phase extraction,5 and hydrometallurgical methods,6 are currently at various technology-readiness levels. Among them, hydrometallurgical methods are seen as the most promising. They require relatively simple equipment to extract REEs with high purity and are generally applicable to both non-oxidised and oxidised Nd–Fe–B magnets. With hydrometallurgy, however, the Nd–Fe–B magnets are completely dissolved in HCl or H2SO4 solutions. For instance, a H2SO4 solution is used to leach the Nd–Fe–B magnets to form REE sulphate, followed by precipitating REE-sodium–sulphate double salts, (RE, Na)2(SO4)2·xH2O.7 When put quantitatively, for every 1 kg of Nd–Fe–B, 10 L of 2 mol L−1 H2SO4 is required (solid-to-liquid (S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L) ratio of 10% w/v) to dissolve the magnets and keep the pH low enough to prevent the precipitation of Fe(OH)3. The pH is then raised to 1.5 with NaOH, at which point (Nd, Na)2(SO4)2·xH2O is deposited. The iron remains in solution as long as the pH stays below 2.0. The sulphate double salt can then be converted to either NdF3 by leaching in a HF solution, or Nd oxalate, by adding an aqueous oxalic acid solution. The REE oxalate can then be calcined to form REE oxides (REOs). Both the REF3 and REOs can then be reduced to the corresponding REE metal (e.g., molten-salt electrolysis8). After removal of the REE-sodium–sulphate double salts, oxygen gas or a H2O2 solution is added to fully oxidise Fe2+ to Fe3+ that precipitates as yellow jarosite, NaFe3(SO4)2(OH)6, which is easier to filter than Fe(OH)3. Alternatively, a roasting pre-treatment of the PMs at 900 °C can completely oxidise Fe to Fe2O3 before the acid leaching step, which would favour the Fe removal step.
:L) ratio of 10% w/v) to dissolve the magnets and keep the pH low enough to prevent the precipitation of Fe(OH)3. The pH is then raised to 1.5 with NaOH, at which point (Nd, Na)2(SO4)2·xH2O is deposited. The iron remains in solution as long as the pH stays below 2.0. The sulphate double salt can then be converted to either NdF3 by leaching in a HF solution, or Nd oxalate, by adding an aqueous oxalic acid solution. The REE oxalate can then be calcined to form REE oxides (REOs). Both the REF3 and REOs can then be reduced to the corresponding REE metal (e.g., molten-salt electrolysis8). After removal of the REE-sodium–sulphate double salts, oxygen gas or a H2O2 solution is added to fully oxidise Fe2+ to Fe3+ that precipitates as yellow jarosite, NaFe3(SO4)2(OH)6, which is easier to filter than Fe(OH)3. Alternatively, a roasting pre-treatment of the PMs at 900 °C can completely oxidise Fe to Fe2O3 before the acid leaching step, which would favour the Fe removal step.
Although hydrometallurgical processing of Nd–Fe–B magnets is effective, many time- and chemical-consuming steps are needed to obtain REEs. When put quantitatively, treating 1000 kg of Nd–Fe–B magnets requires 2172 kg of H2SO4, of which 272 kg is for Nd and 1900 kg is for Fe. The consumption of alkali for Fe removal as jarosite and the effluent neutralization is correspondingly 1600 kg of NaOH or 1100 kg of CaO.9 Jarosite is considered as problematic waste within the European Union;10 therefore, further hydrometallurgical treatment of the solid waste is required that adds to the overall chemical consumption and wastewater generation.11
There have been efforts towards finding more efficient routes for Nd–Fe–B recycling; however, the aspect of either solid waste or wastewater was not yet elaborated. For example, the REEs from EoL PMs can be selectively leached based on the principle of the pH-dependent dissolution of the calcined magnets using the potential-pH diagram.12 Koyama and Tanaka13 reported that the selective leaching of REEs could be realised by using 0.02 mol L−1 HCl in an autoclave at 180 °C for 2 h or at a lower temperature by increasing the initial acid concentration and having a prolonged etching time. An oxidative roasting of the magnet at 900 °C was needed to fit within the potential-pH diagram and the overall process operates at elevated temperatures. In the end, the REEs were precipitated out via conventional hydrometallurgy.11b
Electrochemistry can be alternatively used as an integral part of REE-based PM recycling as it is often used to recover metals at room temperature. A patent14 describes an electrochemical dissolution of Nd–Fe–B magnet slags and recovery of the REEs. HF was added as the precipitating agent that however endangers the selective REEs precipitation as Nd3+, Fe3+, and Fe2+ precipitate as insoluble NdF3, FeF315 and FeF2.16 P. Venkatesan et al.17 were able to selectively recover the REEs, using membrane electrolysis to treat the partially HCl-leached Nd–Fe–B magnet within four steps. More than 95% of the REEs were leached into the solution and simultaneously Fe2+ was anodically oxidised and precipitated as Fe(OH)3 that was removed by filtration. The remaining REEs in the electrolyte were precipitated with oxalic acid. This process facilitates the oxidation of Fe2+ to Fe3+ without extra addition of oxidants. The same group18 further developed a two-anode system to leach Nd–Fe–B scraps and convert Nd and Fe into the respective hydroxides without pretreatment of partial acid-leaching. The REEs were recovered using oxalic acid, followed by calcination to obtain REOs with high purity (99.2%). The electrochemical process precipitated >90% of Fe in the form of solid akaganeite (FeOOH).
Despite the progress made in the electrochemical recycling of the REEs, the methods though efficient in REE recovery still end up with Fe-based solid waste and the electrolyte that has to be treated.
Here, we develop a greener and more facile electrochemical method for Nd–Fe–B PM recycling. The proposed procedure enables a selective REE recovery and a simultaneous Fe metal deposition in only two steps with the total re-use of the electrolyte, thus avoiding the formation of solid Fe-based waste and wastewater discharge.
Our initial investigations focused on the electrochemical leaching of Nd–Fe–B scraps and the Fe deposition. Linear sweep voltammetry (LSV) was performed for a Pt-wire working electrode (black curve) and the initial magnet (blue curve) in the bath solution containing 0.6 mol L−1 FeSO4, 0.4 mol L−1 (NH4)2SO4, 0.175 mol L−1 Na3Cit and 0.4 mol L−1 H3BO3 (pH 4.0) at a scan rate of 100 mV s−1 (Fig. 1a). In the potential segment AB of the black curve, the current density is stable at around 0 mA cm−2. From point B on, where the potential is 0.4 V, the current density starts to increase along the line BC due to the oxidation of Fe2+ on the Pt electrode (reaction (1)).
| Fe2+ ⇌ Fe3+ + e− | (1) |
| Nd2Fe14B ⇌ 2Nd3+ + 14Fe2+ + B3+ + 37e− | (2) |
| H2O ⇌ 4H2+ + O2↑ + 4e− | (3) |
 | ||
| Fig. 1 (a) LSV of a Pt-wire working electrode (black curve) and a sintered Nd–Fe–B magnet (blue curve) with 0.6 mol L−1 FeSO4, 0.4 mol L−1 (NH4)2SO4, 0.175 mol L−1 Na3Cit and 0.4 mol L−1 H3BO3 in the electrolyte, at a scan rate of 100 mV s−1. (b) BSE–SEM images of the magnet before and after LSV. | ||
When the magnet was used as the working electrode, the current density starts to increase at point D with a potential of −0.70 V, shown by the blue curve in Fig. 1a. The initial magnet generally consists of metallic Nd2Fe14B (matrix phase, accounting for ∼90% in terms of volume19), metallic NdFe4B4, metallic Nd and Nd2O3 phases, and the increase in current density from −0.7 V is caused by the oxidation/leaching of the metallic phases, e.g., Nd2Fe14B (reaction (2)), in the magnet. No pronounced oxidation peaks are observed in the DE portion, although a steady increase in the current density can be observed, indicating that all the metallic phases in the magnet were oxidised/leached without significant selectivity. A similar result was reported in our recent study,20 where the fast leaching kinetics at high current density leads to non-selective leaching of all the metallic phases in the Nd–Fe–B magnet. The current density increases continuously along EF with the presence of kinks that are caused by oxygen bubbles formed on the magnet surface. These bubbles were due to the side reaction of water decomposition (reaction (3)). To avoid the water decomposition that decreases the leaching efficiency of the magnet, the applied potential should be more negative than 0 V (corresponding current density of 323 mA cm−2). Fig. 1b shows back-scattered electron (BSE)-SEM images of the polished magnet before and after LSV. The initial magnet before LSV consists of Nd2Fe14B grains surrounded by the Nd-rich grain boundaries that are supposed to be leached preferentially, due to the more negative electrochemical potential of Nd than that of Fe.21 After LSV, the remaining Nd2Fe14B grains (grey phase) were progressively leached, indicated by the pores and pits on their surfaces. However, these Nd2Fe14B grains connect with each other through the remaining grain boundaries (yellow dashed lines), further demonstrating the non-selective leaching of the magnet.
Fig. 2a shows the kinetics of leaching the magnets with applied currents of 50, 100, 150 and 200 mA (current densities of 25, 50, 75 and 100 mA cm−2, respectively) on the anode. An increased leaching rate of the REEs (the slope values in Fig. 2a) from 300.06 to 1073.92 mg L−1 h−1 with the increasing current density from 25 to 100 mA cm−2 is observed. The leaching of REEs into the solution increased with the current density and their concentration increased linearly with the time of electrolysis. The anodic leaching efficiency of the magnets at current densities of 25, 50, 75 and 100 mA cm−2 was 99.9%, 99.8%, 98.7% and 92.4% with corresponding energy consumptions of 0.82, 0.96, 1.21 and 1.51 kW h per kilogram of magnets, respectively (Table 1). For aiming at a leaching efficiency of ∼100%, the current density applied to the anode should not exceed 50 mA cm−2 to avoid the water decomposition that can consume the supplied charge. Taking this into consideration we have chosen a current density of 25 mA cm−2 for later experiments, as this is the current density that leads to the leaching efficiency ∼100%, and low energy consumption, however at a low leaching rate. For practical purposes, when aiming at a 100% leaching efficiency, a constant current could be applied using the current density even below 25 mA cm−2; however a case to case study needs to be done taking into account the particular energy consumption and the time scale of the whole process.
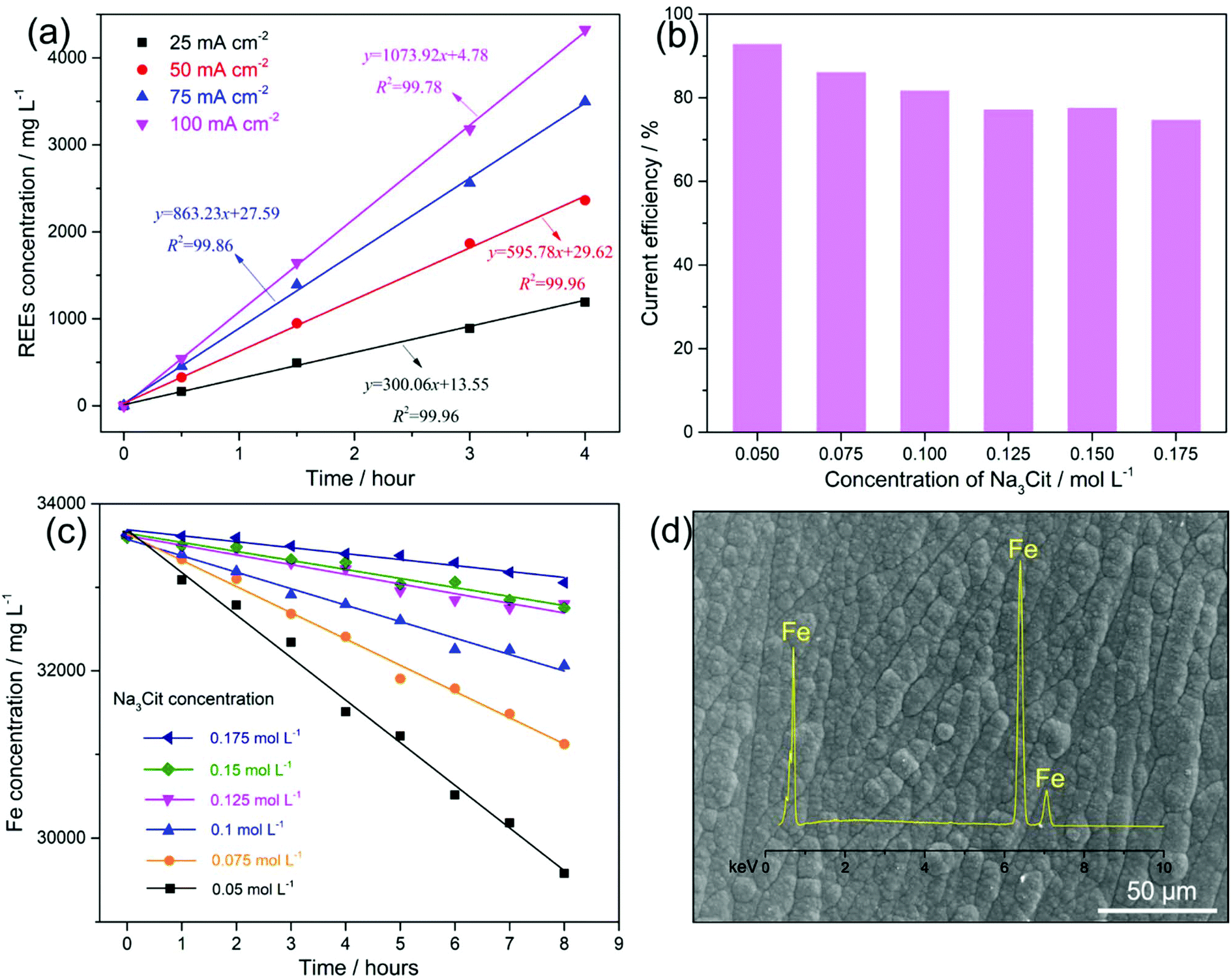 | ||
| Fig. 2 (a) Influence of the current density of 25–100 mA cm−2 on the leaching of the REEs from a Nd–Fe–B magnet anode with 0.6 mol L−1 FeSO4, 0.4 mol L−1 (NH4)2SO4, 0.4 mol L−1 H3BO3 and 0.175 mol L−1 Na3Cit in the initial electrolyte. (b) Effect of Na3cit concentration on the current efficiency of the Fe deposition in the electrolyte (0.6 mol L−1 FeSO4, 0.4 mol L−1 (NH4)2SO4 and 0.4 mol L−1 H3BO3 in the initial electrolyte); (c) effect of Na3cit concentration on the Fe concentration in the electrolyte (0.6 mol L−1 FeSO4, 0.4 mol L−1 (NH4)2SO4 and 0.4 mol L−1 H3BO3 in the initial electrolyte) at an anodic current density of 25 mA cm−2 and a cathodic current density of 12.5 mA cm−2. (d) The SE–SEM image of the Fe deposit with an EDS spectrum (0.6 mol L−1 FeSO4, 0.4 mol L−1 (NH4)2SO4, 0.175 mol L−1 Na3Cit and 0.4 mol L−1 H3BO3 in the initial electrolyte) at a cathodic current density of 12.5 mA cm−2. | ||
| Current density (mA cm−2) | Mass of magnet dissolved (g) | Anodic leaching efficiency (%) | Energy consumption (kW h kg−1) |
|---|---|---|---|
| 25 | 0.220 | 99.9 | 0.82 |
| 50 | 0.439 | 99.8 | 0.96 |
| 75 | 0.651 | 98.7 | 1.21 |
| 100 | 0.813 | 92.4 | 1.51 |
Simultaneously, the Fe metal was deposited (reaction (4)) on the cathode at a current density of 12.5 mA cm−2. As hydrogen evolution (reaction (5)) occurs simultaneously with Fe deposition, this leads to an increase in pH > 7.0 at the cathode interface.22 Consequently, both Fe2+ and Fe3+ (pH > 3.0)22,23 and REE3+, e.g., Nd3+ (pH > 6.0),24 are expected to hydrolyse and form metal hydroxides on the cathode, which means that Fe deposition would be limited and the overall REE recovery would be reduced. Upon that (NH4)2SO4 was added as a complexing agent to maintain the Fe2+ and Fe3+ as soluble species22 at increased pH, and another complexing agent, Na3Cit, was added to prevent the formation of REE hydroxides on the cathode.25 However, citrate ions (Cit3−) can also chelate with Fe2+ (Fe(Cit)−), which decreases the concentration of unchelated Fe2+. As a result, the over-potential for Fe deposition increases, lowering its current efficiency.26Fig. 2b shows that with the increasing concentration of Na3Cit from 0.05 to 0.175 mol L−1, the current efficiency of Fe deposition decreased from 92.9% to 74.9%.
| Fe2+ + 2e− ⇌ Fe | (4) |
| 2H+ + 2e− ⇌ H2↑ | (5) |
It should be noted that in a long run, the increasing thickness of the Fe deposit will largely change the overall surface area of the cathode, leading to the change of the current efficiency. This can be avoided by removing the deposited Fe on the cathode surface using heat treatment27 that increases the internal stress between the cathode material and deposited Fe, resulting in the detachment of the Fe deposit from the cathode material.
For a leaching efficiency of Nd–Fe–B magnets close to 100% under constant current, approximately 70.9–75.7% of the current contributes to the leaching of Fe, which can be regarded as the Fe leaching efficiency from the anode (see SEI “Calculations”). The concentration of total dissolved Fe (including Fe2+ and Fe3+) in the electrolyte is a result of the Fe leaching from the anode and the Fe consumption on the cathode. Because the current efficiency is either higher than or within the Fe leaching efficiency range, the concentration of total dissolved Fe in the electrolyte decreased with leaching time (Fig. 2c). A more rapid decline in Fe concentration is observed with a lower concentration of Na3Cit in the electrolyte. It should be noted that the reducing Fe concentration in the electrolyte, in turn, results in a decrease of current efficiency.28 Therefore, the current efficiency of the Fe deposition in the electrolyte with the Na3Cit concentration from 0.05 to 0.175 mol L−1, in the long term, is expected to decrease to an Fe leaching efficiency range of 70.9–75.7%, where the Fe leaching from the anode is balanced by the Fe consumption on the cathode. Consequently, a stable concentration of total dissolved Fe (balanced concentration) in the electrolyte will be achieved. Therefore, a lower concentration of Na3Cit will result in a lower balanced concentration of the total dissolved Fe, favouring the next steps of selective REE precipitation and purification. In contrast, if the current efficiency of Fe deposition were to be lower than 70.9%, a continuous increase in the total dissolved Fe concentration in the electrolyte would occur. This would have adverse effects on the next steps of selective REE precipitation and purification. Moreover, the lower current efficiency resulted in more added H2SO4 to keep the pH of electrolyte between 3.5 and 4.5, due to the side reaction of hydrogen evolution (reaction (5)).
Alternatively, instead of lowering down the current efficiency of Fe deposition by adding more Na3Cit, supplying a certain amount of Fe salt like FeSO4 into the electrolyte to compensate for the net consumption of Fe is another option to achieve a balanced Fe concentration in the electrolyte, if the current efficiency of Fe deposition is maintained higher than 75.7%.
Nevertheless, using 0.4 mol L−1 (NH4)2SO4 and 0.175 mol L−1 Na3Cit as complexing agents, it was possible to keep the current efficiency of the Fe deposition on the cathode close to the Fe leaching efficiency from the anode (70.9–75.7%), and thus a balanced Fe (Fe2+ and Fe3+) concentration in the electrolyte could be achieved that enables repeated electrolyte recycling. With this electrolyte bath, the Fe metal was obtained on the cathode (Fig. 2d) avoiding the usage of alkali, e.g., NaOH, to remove Fe as akaganeite or jarosite that needs further treatment. As the initial magnet contains a small amount of other transition metals, e.g., Co (2.13 wt% in this study), transition metal alloys, e.g., Fe–Co, are expected to be deposited on the cathode with repeated recycling of the electrolyte. After electrolysis for 8 hours at an applied current of 200 mA, 1.76 g of magnet was leached with an efficiency of 99.9% and an energy consumption of 1.36 kW h per kilogram of the magnet. Around 1.5 mL of 4 mol L−1 H2SO4 was used to adjust the pH of the electrolyte between 3.5 and 4.5, from which the H2SO4 consumption is calculated to be 0.34 kg per kilogram of the magnets, which saves 84.4% of the acid usage, compared to the conventional hydrometallurgical process.9 Combining all the reactions occurring on both the anode (reaction (2)) and the cathode (reactions (4) and (5)) in a balanced Fe (Fe2+ and Fe3+) concentration electrolyte, the net reaction of the whole electrolysis can be expressed using reaction (6), which realises the “selective leaching” of REEs, leaving the major component of metallic Fe “untouched”.
| Nd2Fe14B + 9H+ → 2Nd3+ + 14Fe + B3+ + 4.5H2↑ | (6) |
After the electrolysis step, a purple solution was obtained as the leachate with the pH adjusted to 3.5, in which the REEs were present as sulphate salts. It is known that the sulphate double salt of light REEs, including La, Ce, Pr, Nd and Sm, has very low solubility in an aqueous solution.29 REEs, e.g., Nd, can be precipitated as (Nd, Na)(SO4)2·xH2O by introducing Na2SO4, with the following reaction:
| Nd2(SO4)3 + Na2SO4 + 2xH2O ⇌ 2(Nd, Na)(SO4)2·xH2O | (7) |
Generally, more than 90% of REEs in the solution can be precipitated, when the addition of Na2SO4 at a molar ratio of Na2SO4 to REEs is higher than 2:1.30 In this study, the addition of Na2SO4 at a molar ratio of Na2SO4 to REEs was fixed to 1:1 to prevent the excess of Na2SO4 during repeated recycling of the electrolyte. If the molar ratio of Na2SO4 to REEs was higher than 2:1 for each recycling cycle, although high REE recovery would be achieved, the concentration of Na2SO4 would reach saturation (13.9 g per 100 mL at 20 °C (ref. 31)) at around the eighteenth cycle, leading to the recrystallization of Na2SO4 as a precipitate together with REE-sodium–sulphate double salts. Thus, an extra washing step would be required.
As both Fe2+ and Fe3+ in the leachate stayed soluble due to the addition of (NH4)2SO4 and Na3Cit, with the addition of Na2SO4 (0.57 g) at a molar ratio of Na2SO4 to REEs of 1:1, followed by heating the solution at 70 °C for 2 hours, REEs were selectively precipitated as shown in Fig. 3a. The X-ray diffraction (XRD) analyses show that the diffraction peaks are well indexed to the (Nd, Na)(SO4)2·H2O (PDF# 01-076-2597) phase (Fig. 3b). The inductively coupled plasma mass spectrometry (ICP-MS) analyses of the precipitates revealed that the recovered REEs were Nd (93.7%), Dy (3.1%) and Pr (2.6%), with high purity (99.4%) and trace levels of Fe. Both the XRD and ICP-MS analyses prove that the REEs in the leachate were successfully precipitated as REE-sodium–sulphate double salts with high selectivity.
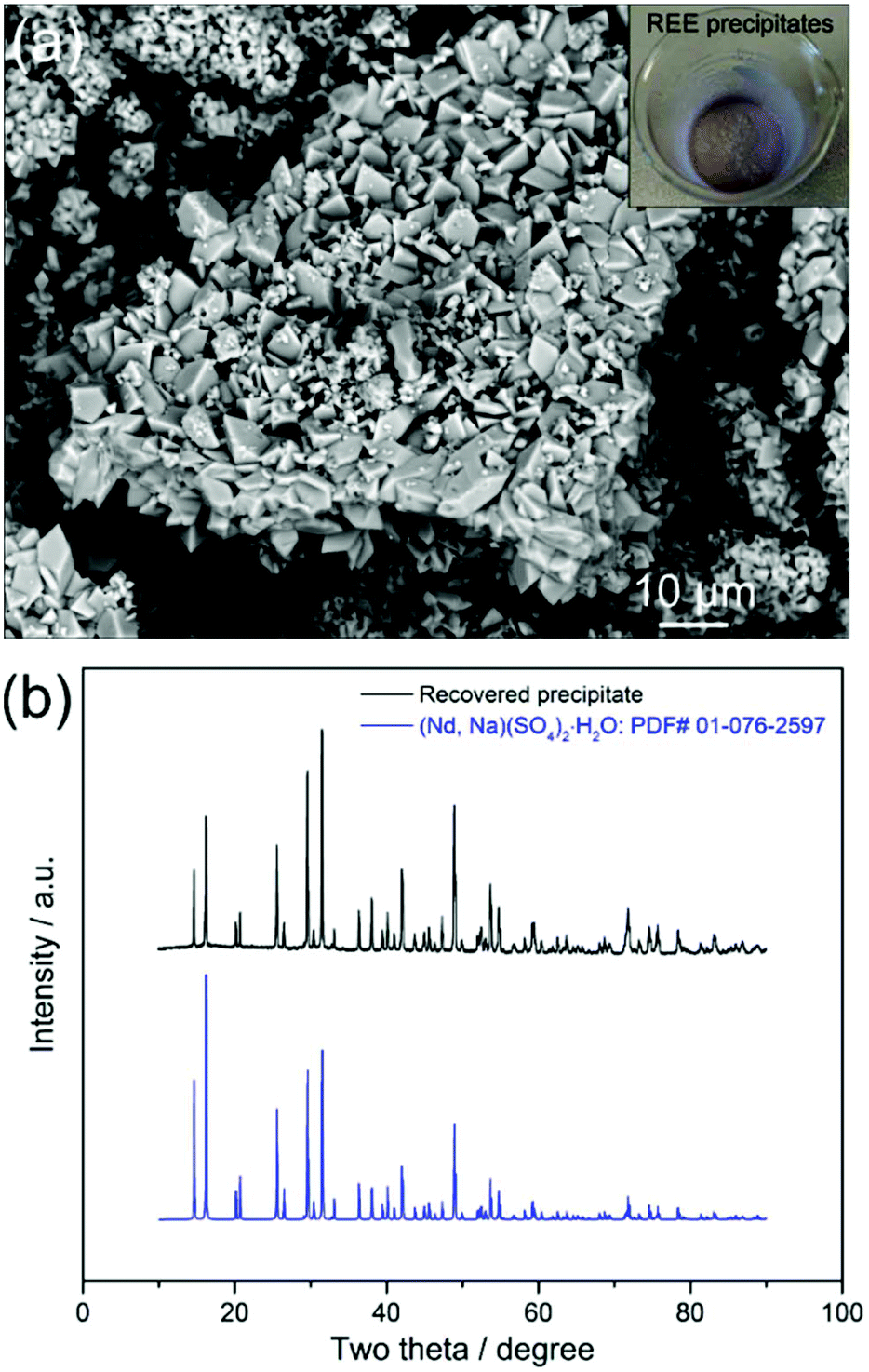 | ||
| Fig. 3 (a) BSE–SEM images with an inset of a real image and (b) XRD pattern of the REE precipitates. | ||
After filtration of the REE precipitates, the filtrate was sent back for the next electrolysis cycle. The overall REE recovery for 9 cycles is shown in Fig. 4a. The REE recovery from the fresh electrolyte was only 56.8% due to the insufficient addition of Na2SO4, keeping the molar-ratio of Na2SO4 to REEs of 1:1, compared to 2:1 in a mole of Na2SO4 to REEs used in the conventional hydrometallurgical process.30 However, with the repeated recycling, the concentrations of both unreacted Na2SO4 and REEs in the solution accumulated and drove reaction (7) to the right. As a result, around 92.5% of the REEs were recovered by the fourth cycle. From the fourth cycle on, 92.5% of the leached REEs were precipitated, leaving 7.5% of Na2SO4 and REEs in the electrolyte for the next cycle. The accumulated concentrations of Na2SO4 and REEs in the electrolyte after REE-sodium–sulphate double salt filtration for the next cycle increased by only 7.5%, leading to a stable recovery of the REEs for all subsequent cycles. It has been reported that REEs such as Nd3+ can form a complex with Cit−, according to the following equilibrium, and the existence of NdCit, (NdHCit)+, (NdHCit2)2− and (NdCit2)3+ was confirmed by the solution in the pH range of 2–5.32
| nNd3+ + jH+ + kCit3+ ⇌ NdnHjCitk(3n+j−3k) | (8) |
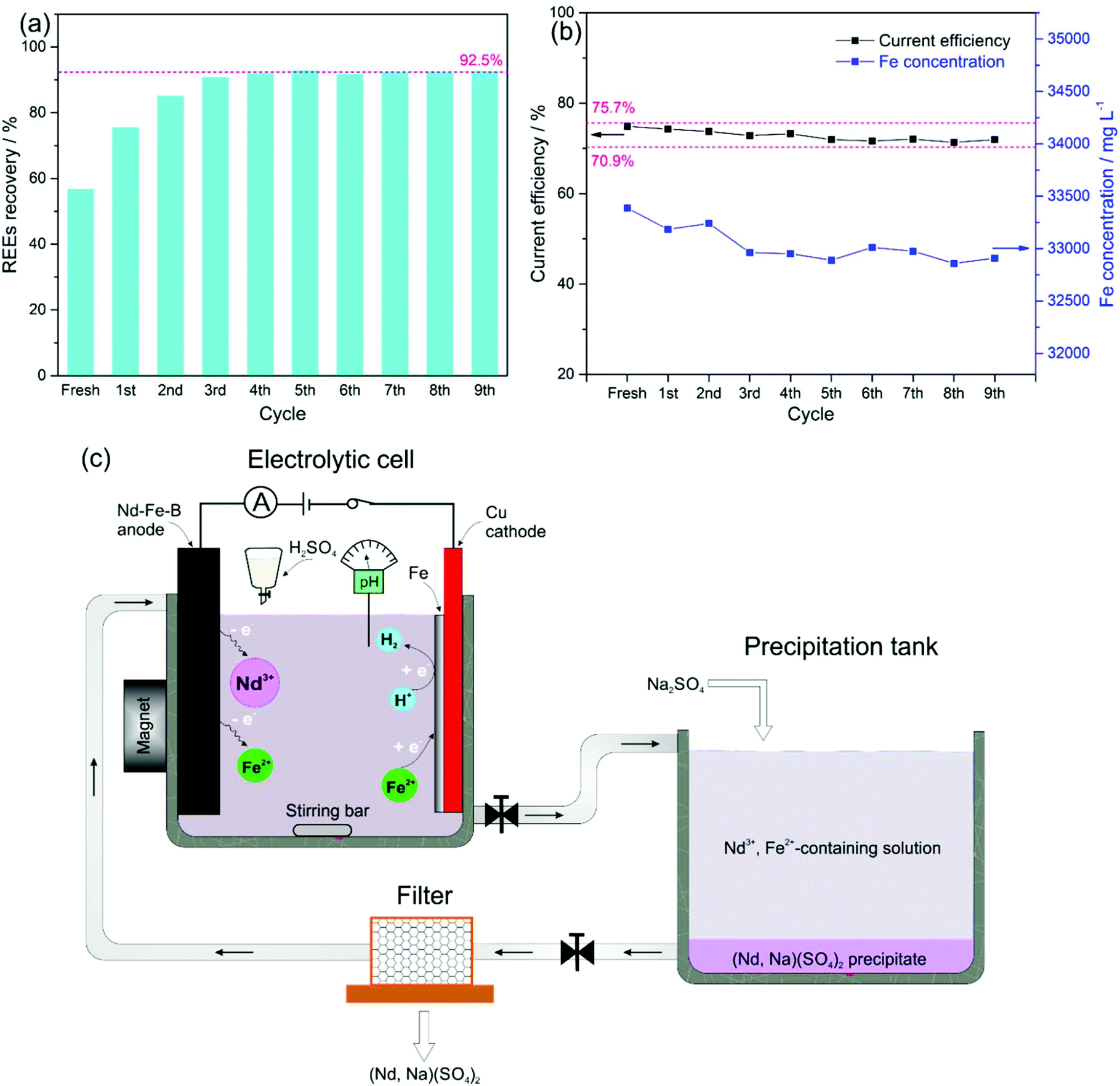 | ||
| Fig. 4 (a) Percentage extraction of REEs at an anodic current density of 25 mA cm−2 and a cathodic current density of 12.5 mA cm−2 and (b) current efficiency of Fe deposition and total dissolved Fe concentration over 9 recycling cycles. (c) Schematic of the proposed closed-loop recycling route for recovering REEs from the sintered Nd–Fe–B magnets. | ||
However, the addition of Na2SO4 caused the precipitation of the REEs as REE-sodium–sulphate double salts from these REE-Cit complexes. This is probably due to the higher stability of REE-sodium–sulphate double salts than that of REE-Cit complexes in the solution with a high sodium sulphate concentration.33 Therefore, the constant citrate concentration in this study does not influence the REE recovery in a long run.
Correspondingly, with the initial electrolyte bath of 0.6 mol L−1 FeSO4 + 0.4 mol L−1 (NH4)2SO4 + 0.175 mol L−1 Na3Cit + 0.4 mol L−1 H3BO3, the current efficiency of the Fe deposition was kept within 70.9–75.7% (indicated with pink-dashed lines in Fig. 4b), which guaranteed a relatively stable concentration of total dissolved Fe around 0.6 mol L−1 for 9 repeating cycles. We expect that a REE recovery of 92.5% with a relatively stable composition of the electrolyte can be achieved over the long term. Therefore, repeated re-use of the electrolyte can avoid wastewater release by using the current two-step procedure, i.e., electrolysis-selective precipitation, for the REE recovery from Nd–Fe–B magnets (Fig. 4c).
It should be noted that boron (B) was also leached out from Nd–Fe–B magnets in the form of H3BO3 electrochemically, according to the half-reaction:34
| B + 3H2O ⇌ H3BO3 + 3H+ + 3e− | (9) |
In the initial electrolyte, H3BO3 was added as a buffer to favour the Fe electrodeposition on the cathode, because it can prevent the precipitation of metal hydroxides on the cathode surface that hinder the Fe deposition.35 The concentration of boron increased from 0.402 mol L−1 in the initial electrolyte to 0.428 mol L−1 after the electrochemical leaching for 8 h at 200 mA (25 mA cm−2), which is ascribed to the leaching of boron from the Nd–Fe–B magnet in the form of H3BO3. The increased concentration of H3BO3 in the electrolyte can strength the buffer capacity, facilitating the Fe deposition.34 It is expected that in the long term of reusing the electrolyte, the concentration of H3BO3 will reach a saturation (∼0.92 mol L−1 at 25 °C (ref. 36)) where H3BO3 will crystallise. In the REE precipitation step, H3BO3 will precipitate together with the REE-sodium–sulphate double salts. The precipitated H3BO3 can be easily washed away with water and then recovered as zinc borate hydrate37 by adding zinc and raising the pH. The zinc borate hydrate could be commercially used as a flame retardant.37
Additionally, the recovered REEs from the Nd–Fe–B magnets in the form of REE-sodium–sulphate double salts can be readily converted to either REOs or REF3 with negligible losses of REEs.29 However, unlike the fluoride precipitation for REE separation in the conventional hydrometallurgical process where NdF3 is readily gelatinous and is, therefore, difficult to separate from the solution by filtration,29 the NdF3 obtained from the sulphate double salt is easy to filter, which is a significant advantage of this route. Because of this, REE-sodium–sulphate double salts are generally used for the precipitation and storage of REEs in industry today.29
Conclusions
We have demonstrated a green and facile route for Nd–Fe–B PM recycling, showing that both the REEs and Fe metal can be recovered in only two steps: electrolysis and selective precipitation. Compared to the state-of-the-art methods, the proposed method largely reduces the chemical consumption and avoids Fe-based solid waste generation and wastewater discharge, making it economically and environmentally attractive for industrial applications.Conflicts of interest
X. Xu, S. Sturm, K. Zuzek Rozman and Jozef Stefan Institute have filed a patent on the presenting results. All other authors declare no competing financial interests.Acknowledgements
This work was supported by the European Union's EU Framework Programme for Research and Innovation Horizon 2020 under Grant Agreement No. 674973 (DEMETER). The authors thank the COST Action e-MINDs community for sharing the knowledge.Notes and references
- K. M. Goodenough, F. Wall and D. Merriman, Nat. Resour. Res., 2018, 27, 201–216 CrossRef CAS.
- M. V. Reimer, H. Y. Schenk-Mathes, M. F. Hoffmann and T. Elwert, Metals, 2018, 8, 867 CrossRef.
- J. H. L. Voncken, in The rare earth elements: an introduction, Springer, 2016 Search PubMed.
- (a) T. Saito, H. Sato, S. Ozawa, J. Yu and T. Motegi, J. Alloys Compd., 2003, 353, 189–193 CrossRef CAS; (b) Y. Bian, S. Guo, L. Jiang, K. Tang and W. Ding, J. Sustainable Metall., 2015, 1, 151–160 CrossRef.
- (a) J. Jiang, T. Ozaki, K.-I. Machida and G.-Y. Adachi, J. Alloys Compd., 1997, 260, 222–235 CrossRef CAS; (b) K. Murase, K.-I. Machida and G.-Y. Adachi, J. Alloys Compd., 1995, 217, 218–225 CrossRef CAS.
- (a) C. Tunsu, in Waste Electrical and Electronic Equipment Recycling, ed. F. Vegliò and I. Birloaga, Woodhead Publishing, 2018, pp. 175–211 Search PubMed; (b) N. Swain and S. Mishra, J. Cleaner Prod., 2019, 220, 884–898 CrossRef CAS.
- (a) J. W. Lyman and G. R. Palmer, US Pat, 5129945, 1992 Search PubMed; (b) T. Ellis, F. Schmidt and L. Jones, Methods and opportunities in the recycling of rare earth based materials, Ames Lab., IA (United States), 1994 Search PubMed; (c) D. D. München, A. M. Bernardes and H. M. Veit, J. Sustainable Metall., 2018, 4, 288–294 CrossRef.
- H. Vogel and B. Friedrich, in Proceedings of the 8th European Metallurgical Conference, Düsseldorf, 2015 Search PubMed.
- M. Tanaka, T. Oki, K. Koyama, H. Narita and T. Oishi, in Handbook on the physics and chemistry of rare earths, Elsevier, 2013, pp. 159–211 Search PubMed.
- P. Kangas, P. Koukkari, B. Wilson, M. Lundström, J. Rastas, P. Saikkonen, J. Leppinen, V. Hintikka and P. C. Oy, in Conference in Minerals Engineering 2017, 2017 Search PubMed.
- (a) T. Palden, M. Regadío, B. Onghena and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 4239–4246 CrossRef CAS; (b) K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- (a) N. Takeno, in Geological survey of Japan open file report, 2005, 419, 102 Search PubMed; (b) M. Pourbaix, in Atlas of electrochemical equilibria in aqueous solutions, Pergamon Press, New York, 1966 Search PubMed.
- K. Koyama, A. Kitajima and M. Tanaka, Kidorui, 2009, 54, 36–37 CAS.
- B. Greenberg, US Pat, 5362459, 1994 Search PubMed.
- D. L. Perry, in Handbook of inorganic compounds, CRC Press, 2016 Search PubMed.
- P. Patnaik, in Handbook of inorganic chemicals, McGraw-Hill, 2002 Search PubMed.
- P. Venkatesan, T. Vander Hoogerstraete, T. Hennebel, K. Binnemans, J. Sietsma and Y. X. Yang, Green Chem., 2018, 20, 1065–1073 RSC.
- P. Venkatesan, T. Vander Hoogerstraete, K. Binnemans, Z. Sun, J. Sietsma and Y. Yang, ACS Sustainable Chem. Eng., 2018, 6, 9375–9382 CrossRef CAS.
- J. Holc, S. Beseničar and D. Kolar, J. Mater. Sci., 1990, 25, 215–219 CrossRef CAS.
- X. Xu, S. Sturm, Z. Samardzija, J. Vidmar, J. Scancar and K. Zuzek Rozman, ChemSusChem, 2019, 12, 4754–4758 CrossRef CAS PubMed.
- L. Schultz, A. El-Aziz, G. Barkleit and K. Mummert, Mater. Sci. Eng., A, 1999, 267, 307–313 CrossRef.
- K. Osseo-asare, M. E. Deelo and K. Weil, ECS Trans., 2006, 2, 293–302 Search PubMed.
- M. Schlesinger and M. Paunovic, in Modern electroplating, John Wiley & Sons, 2011 Search PubMed.
- R. Mesmer and C. Baes, in The hydrolysis of cations, Wiley, EUA, 1976 Search PubMed.
- M. A. Brown, A. J. Kropf, A. Paulenova and A. V. Gelis, Dalton Trans., 2014, 43, 6446–6454 RSC.
- C. Chu and C. Wan, J. Mater. Sci., 1992, 27, 6700–6706 CrossRef CAS.
- S. Yoshimura, S. Yoshihara, T. Shirakashi, E. Sato and K. Ishii, IEEE Transl. J. Magn. Jpn., 1994, 9, 112–117 Search PubMed.
- E. Zhelibo, K. Aryupina and É. Natanson, Powder Metall. Met. Ceram., 1973, 12, 97–101 CrossRef.
- H. Zhongsheng, in Sustainable Inorganic Chemistry, ed. D. A. Aatwood, John Wiley & Sons, Lexington, USA, 2016 Search PubMed.
- H.-S. Yoon, C.-J. Kim, J.-Y. Lee, S.-D. Kim, J.-S. Kim and J.-C. Lee, J. Korean Inst. Resour. Recycl., 2003, 12, 57–63 CAS.
- Wikipedia, in Sodium sulfate, https://en.wikipedia.org/wiki/Sodium_sulfate.
- M. A. Brown, A. J. Kropf, A. Paulenova and A. V. Gelis, Dalton Trans., 2014, 43, 6446–6454 RSC.
- G. Senanayake, S. Jayasekera, A. Bandara, E. Koenigsberger, L. Koenigsberger and J. Kyle, Miner. Eng., 2016, 98, 169–176 CrossRef CAS.
- D. C. Harris, in Quantitative chemical analysis, W. H. Freeman, 8th edn, 2010 Search PubMed.
- N. Zech and D. Landolt, Electrochim. Acta, 2000, 45, 3461–3471 CrossRef CAS.
- Wikipedia, in Boric acidhttps://en.wikipedia.org/wiki/Boric_acid.
- J. Lyman and G. Palmer, High Temp. Mat. Pr-ISR., 1993, 11, 175–188 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03325d |
| This journal is © The Royal Society of Chemistry 2020 |