
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(Non-)Kolbe electrolysis in biomass valorization – a discussion of potential applications
F. Joschka
Holzhäuser
,
Joel B.
Mensah
 and
Regina
Palkovits
*
and
Regina
Palkovits
*
Chair of Heterogeneous Catalysis and Chemical Technology, Institut für Technische und Makromolekulare Chemie, RWTH Aachen University, Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
First published on 20th December 2019
Abstract
Driven by the goal of a circular economy, the importance of renewable energies and sustainable sources of raw materials is steadily increasing. The electrochemical conversion of biomass-based compounds into liquid energy sources and basic chemicals enables the direct combination of renewable energies with sustainable carbon sources. A variety of carboxylic acids are efficiently accessible from biomass and represent important platform chemicals in a future bioeconomy. With the help of Kolbe and Non-Kolbe electrolysis, these raw materials offer great potential for electrified value chains as part of bio-refinery concepts. This contribution highlights current developments in these areas as well as the open challenges in order to gain deeper scientific insights and to develop technically viable processes. Moreover, aspects of green chemistry with regard to (Non-)Kolbe electrolysis are discussed. Last but not least, electrochemical conversions are an attractive approach to implementing modular and dynamic production plants.
 F. Joschka Holzhäuser | F. Joschka Holzhäuser obtained his PhD in chemistry from RWTH Aachen University in 2019. He was a visiting researcher in the lab of Prof. Rob Liskamp (Chair of Chemical Biology and Medicinal Chemistry) working on cysteine protease inhibitors in 2015. Since 2016 he has been a PhD student in the group of Prof. Regina Palkovits (Chair of Heterogeneous Catalysis and Technical Chemistry), studying the electrochemical valorization of biogenic compounds into fuels and commodities. Recently he was promoted to project leader with a focus on green protocols in electrochemical conversions and the development of novel electrode materials. |
 Joel B. Mensah | Joel B. Mensah received his MSc in chemistry from RWTH Aachen University in 2016, specialising in catalysis and synthesis. During his studies, he was also a visiting research student in the group of Dr. Christian Hulteberg (Department of Chemical Engineering) at Lund University, Sweden in 2015. Currently, he is a PhD student under the supervision of Prof. Regina Palkovits (Chair of Heterogeneous Catalysis and Technical Chemistry), working on the heterogeneously catalyzed upgrading of hydroxy fatty acids into fuels and fine chemicals. His main research interest is focused on heterogeneous catalysis and electrochemistry in biomass upgrading besides the valorisation of plastic waste streams. |
 Regina Palkovits | R. Palkovits is a full professor for Heterogeneous Catalysis & Chemical Technology at RWTH Aachen University. She graduated in Chemical Engineering from Technical University Dortmund in 2003 and finished her PhD under the supervision of Prof. Ferdi Schüth at the Max-Planck-Institut für Kohlenforschung in 2006. Afterwards, she joined the group of Prof. Bert Weckhuysen at Utrecht University as a postdoctoral fellow. In 2008, she returned as a group leader to the Max-Planck-Institut für Kohlenforschung and since 2010 she has been Professor at RWTH Aachen University. |
Introduction
In the light of the steady retreat from finite fossil resources due to their contribution to the greenhouse effect and the rising average global temperature, the search for sustainable alternative feedstocks for the supply of energy, fuels, and commodities is in full swing. Electric power harvested from wind, water, or the sun allows for a more sustainable energy supply.1,2 The use of abundant and renewable biomass feedstocks for the production of fuels and fine chemicals provides a promising route avoiding the perturbation of the atmospheric carbon cycle.3–5 The conversion of biomass into building blocks for chemical industries can be performed through a broad range of technologies e.g. chemocatalytic, enzymatic, and thermochemical reactions, starting from various polymeric and monomeric biomass feedstocks such as lignocellulose, saccharides, furans, alcohols, aldehydes, and acids.5–10 In recent years the electrochemical conversion of bio-derivable compounds has found growing implementation, thus enriching the portfolio of available technologies for biomass down-streaming.11–13 The growing interest in electrochemistry in the context of biomass utilization and renewable electricity can be traced down to several advantages: due to fluctuations in the increased share of renewable feeds, efficient ways for the use and storage of over-produced green electricity during peak hours are required in order to ensure a steady supply. Here, electrochemistry shows great potential to close this technological gap through the valorisation of excess renewable electricity in bio-refineries. The efficiency and simplicity with which electrochemistry can be conducted makes it highly attractive (Fig. 1). Conventional catalytic hydrogenation or oxygenation processes typically require elevated temperatures well above 100 °C and high pressures of hydrogen or oxygen.14–17 | ||
| Fig. 1 Ecological and economic context of the (Non-)Kolbe bio-refinery. | ||
The corresponding electrochemical route, however, is usually performed at ambient pressure and temperature in aqueous or mildly hazardous solutions. Moreover, typically no additional pressures are required and expensive noble metal catalysts often can be avoided.13,18–22 Reduction or oxidation takes place on an electrode surface where electrons are provided or taken up by a solid electrode. Another advantage is the possibility of a direct integration of electrochemistry into biotechnology.23 Harnisch and co-workers have reported on biotechnological fatty acid production with further electrochemical upgrading into alkanes after an integrated separation step.24 Moreover, electrochemical conversions of biogenic acids in fermentation broths to value added products were demonstrated by our group.25,26
One of the most relevant electrochemical reactions is the Kolbe electrolysis. It comprises the anodic decarboxylation of organic acids leading to the formation of prolonged hydrocarbons and CO2 as the co-product.27 For more than 150 years, research groups have investigated the most important reaction parameters and screened a broad substrate scope.21,28–31 Many of these studies were reviewed in detail by Schäfer in 1990 which is the last thorough description of studies regarding all types of anodic decarboxylation of organic acids.29 Most of these scientific reports focused on gaining a more fundamental understanding i.e. mechanistic studies on (Non-)Kolbe electrolysis and extending the substrate scope to more complex heteroaromatic acids.29,32 Today, in view of climate change, the motivation for further investigations has shifted; global research activities target the transition to renewable energy systems as well as alternative carbon feedstocks. In this regard, the renaissance of (Non-)Kolbe electrolysis allows building a bridge between the use of renewable electrical energy and the conversion of bio-derived acids for the production of fuels and chemicals.
This perspective comprises three main parts. The first section Background of (Non-)Kolbe electrolysis describes the general mechanism and reaction conditions. Furthermore, a review of the latest state of the art for (Non-)Kolbe electrolysis in green chemistry is provided in the second part Green (Non-)Kolbe electrolysis. Here, special attention is devoted to the conversion of biogenic compounds and recent studies since 2000. Finally, the last section Perspective evaluates the economic feasibility and discusses current challenges besides future research directions and opportunities for improved reaction set-ups.
Background of (Non-)Kolbe electrolysis
Although Michael Faraday discovered the Kolbe electrolysis in 1834 by studying the electrolysis of aqueous acetic acid solutions – it was Hermann Kolbe who recognized 15 years later that the electrolysis of acetic acid and valeric acid over Pt electrodes yields ethane and octane besides CO2 as the co-product.27 Different theories for the mechanism of Kolbe reactions have been proposed and discussed in the last 100 years. Today, there is much consensus about the concept shown in Scheme 1.29 It is supposed that the carboxylic acid is deprotonated by the base present. In the following the carboxylate adsorbs on the anode surface.33–35 The critical potential for a sufficient coverage of alkoxy or alkyl radicals on the surface lies between 2.1 and 2.4 V vs. NHE (normal hydrogen electrode). Below this value, water oxidation will mainly occur. Thus, it is assumed that the coverage of the metal (oxide) surface with alkoxy and alkyl radicals inhibits the oxygen evolution reaction or solvent oxidation, while the Kolbe electrolysis is promoted.36–42 After the adsorption, an irreversible single electron transfer from the carboxylate to the anode takes place, whereas the simultaneous decarboxylation leads to the formation of an alkyl radical and CO2.36,37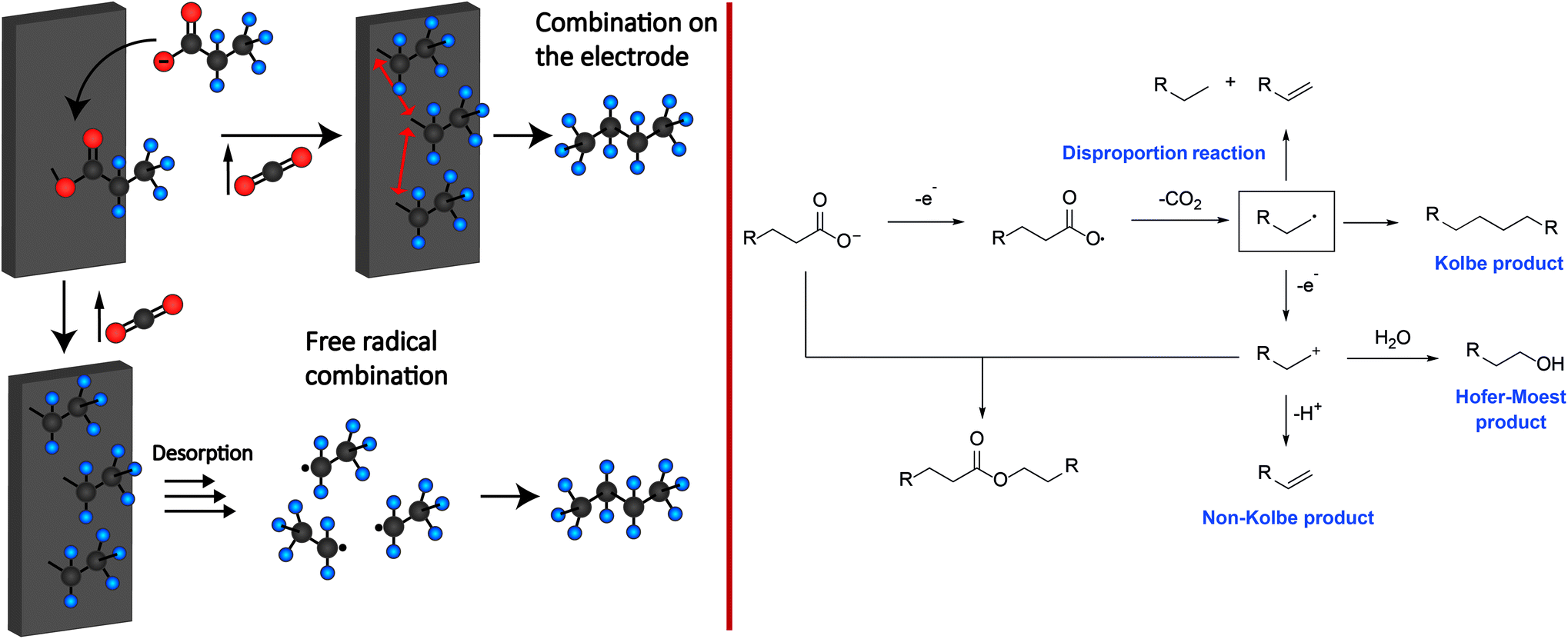 | ||
| Scheme 1 Left: Two different proposed electrode mechanisms for Kolbe electrolysis. Right: Reaction mechanism for the formation of Kolbe, Non-Kolbe and Hofer-Moest products. | ||
Subsequently, the formed alkyl radicals can follow four different reaction pathways depending on the applied reaction conditions:
The first pathway comprises the disproportionation of two alkyl radicals leading to the formation of an alkane and alkene. Moreover, the dimerization of the alkyl radicals will yield the respective Kolbe-product. Furthermore, oxidation at the anode through electron transfer may occur. The obtained carbenium ion can either react with an alkene via β-H-elimination or with an alcohol or ether (Hofer-Moest-product), in the case of an aqueous or alcoholic solution. Additionally, the carbenium ion may react with a deprotonated acid leading to the formation of an ester as a side product. The experimental parameters that have been identified to impact the reaction outcome include the pH, temperature, solvent, electrode material, current density, electrolyte, and the shape as well as functional groups of the substrate.29
In order to achieve increased yields of the dimerized Kolbe-products, high current densities and carboxylic acid concentrations are favourable using an undivided electrolysis cell. High current densities are necessary to overcome the aforementioned critical potential. However, the dimerization of saturated carboxylic acids, e.g. valeric acid, does not occur on the electrode surface which commonly consists of a Pt bulk layer. Several studies state that only very weakly or non-adsorbed radicals dimerize.36,37 Interestingly, for unsaturated and aromatic acids a rather non-statistical combination of dimers is obtained, indicating that these functional groups favour dimerization for their respective adsorbed counterparts.43,44 However, detailed information on the role of the interaction with the electrode surface yet remains unexplained (Scheme 1).
Foreign anions should be avoided for the Kolbe coupling as they can interfere with the alkoxy and alkyl radical layer.45 The same applies to foreign cations as they may form oxide layers on the anode surface. Moreover, temperatures above 65 °C seem to promote disproportion reactions while elevated pressures lead to more coupling of the intermediate radical. The pH of the media should be between neutral and weakly acidic; however, it is favoured to add small amounts of alkali metal hydroxide or alkoxide in order to form a sufficient concentration of deprotonated carboxylate reactants.
The quantity of the carboxylate ions will be constant during electrolysis as the base is formed at the cathode while the carboxylate is consumed at the anode. This is usually confirmed by a shift to more basic pH values with proceeding electrolysis.29 The most suitable solvent for Kolbe dimerization reactions is methanol due to the widely inhibited oxidation because of the carboxylate layer. Aqueous solutions and dimethylformamide or acetonitrile may also be used as the solvent, however, at the expense of lower conversions and yields, respectively. The addition of water to organic solvents leads to a decreased product yield e.g. already for amounts below 5 vol%. Of note, few examples show that Pt, RuO2, PdO2 and RhO2 on Ti can be used as electrodes in aqueous–organic solutions although dimer yields are still much higher in pure methanol.29,46 For cross-coupled Kolbe dimers, the use of acids with antagonistic reactivities (nucleophilic vs. electrophilic) appears to be essential.47 Additionally, ultrasonic mixing proved to be advantageous which is assumed to enhance the adsorption of organic acids on the electrode surface also increasing mass transport.48 Nevertheless, studies elucidating the rate determining step in Kolbe electrolysis are not available.
The standard Kolbe electrolysis set-up consists of an undivided beaker cell, adequately cooled, using high currents over bulk Pt as the working electrode preferably (Fig. 2). The optimal solution for Kolbe electrolysis is methanol-based with small amounts of an alkali metal hydroxide and high reactant concentrations.
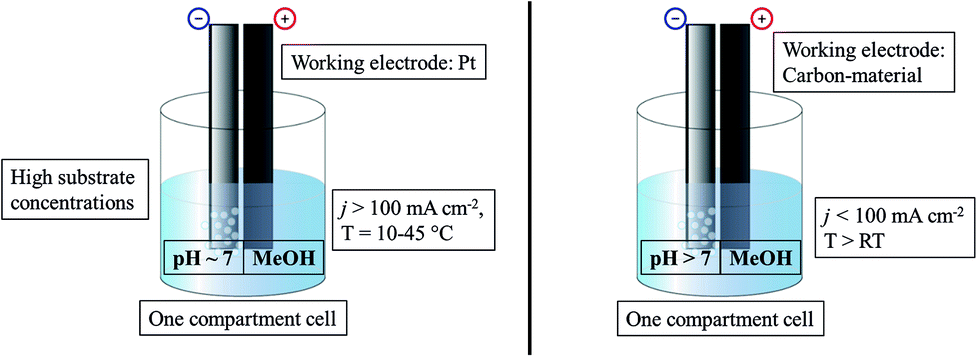 | ||
| Fig. 2 Illustration of commonly used conditions for Kolbe electrolysis (left) and Non-Kolbe electrolysis (right). | ||
If the carboxylic acid possesses a group that is prone to elimination such as SiMe3, SPh, COOH or hydrogen, the formation of respective olefins (Non-Kolbe pathway) is favoured over dimerization (Kolbe pathway). As depicted in Scheme 1, the formed radical at the anode surface is further oxidized to a carbenium ion which undergoes subsequent β-elimination yielding the olefin.
Depending on the solvent, the carbenium ion can react to esters and alcohols in aqueous solutions or to olefins and ethers in alcoholic solutions. The reactions leading to ethers or esters are often referred to as Hofer-Moest reactions while the production of olefins is called Non-Kolbe electrolysis.29,49–52
Contrary to the Kolbe dimerization reaction, lower current densities lead to the formation of carbenium ions and promote the Hofer-Moest or Non-Kolbe electrolysis.29,53 The yields are further shifted to Non-Kolbe and Hofer-Moest products when using a carbon electrode. However, various studies reported that the surface shape and functionalization of the electrode have a crucial influence on the activity for Non-Kolbe reactions.54–56 Water/pyridine mixtures57,58 and also pure methanol with small amounts of sodium perchlorate are the solvent mixtures of choice for Non-Kolbe electrolysis.53 Similar to the Kolbe dimerization, higher pH values favour carbocation formation.29 Nevertheless, a high deprotonation degree of the organic acid facilitates the Non-Kolbe pathway, most likely due to an increased ionization strength of the electrolyte.59 Finally, the substrate is a further substantial factor: electron releasing and sterically shielding groups at the α-position, for example, promote the Non-Kolbe pathway while the opposite leads to Kolbe dimerization.53,57,59,60
The Non-Kolbe electrolysis is usually performed in a one compartment electrochemical cell (Fig. 2) using rough or porous carbon paper as the working electrode. The commonly used solvent is methanol (similar to Kolbe dimerization) and acidic pH values also promote the formation of carbocations. Contrary to the dimerization reaction foreign anions, neutralizing the acid to a certain degree, increase the selectivity towards esters, ethers, and olefins. However, the choice of the substrate is the most important factor: a sterically demanding acid which features an electron donor group at the α-position represents an optimal starting material.
Green (Non-)Kolbe electrolysis
Since depleting fossil resources and climate change were less pressing issues before 2000 most scientific reports covering (Non-)Kolbe reactions dealt with mechanistic concepts, the possible scope of (Non-)Kolbe electrolysis and process optimisation. However, this has significantly changed; today, a growing number of studies focus on the necessary understanding and applicability in bio-refineries.18,25,26,61–63This chapter aims to recapitulate the literature concerning (Non-)Kolbe electrolysis since 2000, devoting special attention to green and economical approaches, in particular the conversion of biogenic acids into fuels, platform chemicals and value-added compounds. The chapter will be organized with respect to general feasibility e.g. economic electrode development from expensive to low-cost. Furthermore, the ecological impact will be taken into account e.g. the prevention of hazardous additives. Table 1 summarizes selected contributions and the respective reaction conditions.
| Electrode | Most important products (highest yield) | Faradaic eff. | General set-up | Conditions | Ref. |
|---|---|---|---|---|---|
| Pt or B doped diamond | C10 (40 ± 5%), C12 (10 ± 5%), C18 alkanes | 40 ± 5% (C6), 10 ± 5% (C7) | Ultrasonic bath one compartment cell | Aq./org. solution, carboxylic acids, 1 M NaOH, 293 K, 0.18 A cm−2 (Pt) or 0.35 A cm−2 B doped diamond, ultrasonic frequency of 20 kHz | Marken64 |
| B doped diamond | Ethane, methanol, methyl acetate | 43.3% acetic acid | One compartment cell | Aq. solution, 0.5 M acetic acid, 1 M HClO4, 298 K, 2.85 V vs. RHE, 0.59 A cm−2 | Comninellis66 |
| Pt on Nafion® 117 | Alkanes, aromatic compounds | 75% | Polymer electrolyte membrane reactor | Using gaseous substrates, using an electrode simultaneously as an electrolyte and separator, 315 K, liquid water between the electrodes | Law67 |
| Pt electrode | Several alkanes 45–95% (95% for C16) | n.a. | One compartment cell | Using thermomorphic systems with pyridine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:MeCN and cyclohexane as the electrolyte, different organic acids as the substrate (0.125 M), KOH saturated, 0.25–0.60 A cm−2 :MeOH:MeCN and cyclohexane as the electrolyte, different organic acids as the substrate (0.125 M), KOH saturated, 0.25–0.60 A cm−2 |
Chiba74 |
| Pt electrode | Several alkanes | n.a. | Bio-membrane-Kolbe reactor | Using an aqueous solution, different carboxylic acids mainly caproic acid, pH 12 adjusted with NaOH, 0.6 farad equivalents | Harnisch24 |
| Graphite | C9-Oxygenates (alcohols, esters, ketones, aldehydes and acetals) | 70% | One compartment cell | MeOH or MeOH/fermentation broth, 0.1 M model substrate 3-hydroxy decanoic acid, 0.1 M KOH, 0.1 A cm−2 | Palkovits26 |
| Graphite, RuxTiyO2/Ti or Pt | Diethyl adipate (57%) and ethylacrylate (50%) | 57%, 50% | One compartment cell | Using different H2O–MeOH mixture with 0.1 M KOH or NEt3 as the electrolyte, 1 M hydrogen ethyl succinate, 0.1 A cm2 | Palkovits61 |
| RuxTiyO2/Ti or Pt | Esters (42–61%) and alkanes (42–52%) | 43–47%, 39–44% | One compartment cell | Using a different H2O–MeOH mixture with 0.1 M KOH or NEt3 as the electrolyte, 1 M hydrogen ethyl succinate, hydrogen methyl methylsuccinate, 2,5-methylhexanoic acid, co-acid: isovaleric acid, 0.1 A cm−2 | Palkovits62 |
| Ir, Pt, glassy carbon, pyrographite, graphite, Pt-10% Ir alloy | Various alkanes, olefins and alcohols, and 3-methyl dodecane (58%) | 15% | One compartment cell | EtOH, 0.017–0.027 M 10-undeclenic acid, 0.078–0.08 M acetic acid, 0.0078 M sodium acetate, 323 K, 0.015–0.025 A cm−2 | Stepanov70 |
| Pt | Different linear or branched oils (>50% regarding examples) | n.a. | One compartment cell | Different fatty acids (0.8–1.4 M), 1–25 mol% base (regarding examples sodium methoxide), 273–342 K, 0.1–1.0 A cm−2 | Dierker90 |
| Pt or graphite | Non-Kolbe products for sodium butyrate (21–21.5%), hexadecane for sodium octanoate (19.8–37.6%) | n.a. | One compartment cell | Using a H2O–MeOH mixture (40 vol% water), 0.66 M substrate, no additional base was used, 0.2–0.6 A cm−2 | Noel102 |
| Pt | Octadecane (conversion 90–100% decanoic acid) | n.a. | One compartment cell | MeOH, 7.5–10.5% decanoic acid, 1.4–1.8% KOH (pH 6), 0.15–0.05 A cm−2, 10–60 min | Joshi95 |
| Pt | C6–C54 (5–79.5%) | n.a. | One compartment cell | MeOH, different fatty acids (9.4%) and oleic acid (15.7–16.5%), co-acid: acetic acid (0.1–6.7%), 0.2–0.3 A cm−2 | Joshi72 |
| Pt | Different alkanes (hexane + dodecane) | n.a. | Two compartment micro reactor | MeOH, sodium oleate, sodium linoleate, 0.05–0.2 A cm−2 | Bhavaraju119 |
| Graphite | Ethylene, propylene | n.a. | One compartment cell | Fermentation broth (0.29 M propionic acid) or 0.12 M butyric acid, 10 M KOH (for butyric acid only), 298 K, 0.01 A cm−2 | Pereira98 |
| Graphite | Various alkanes and alkenes, including branched alkanes (C1–C20) | n.a. | Two compartment micro reactor | Fermentation broth (C1–C18 linear acids), 283–298 K, 0.06–0.08 A cm−2, pH adjustments | Kuhry101 |
| Pt | Various alkanes and olefins (among others 90% decane, caproate pentyl) | n.a. | One compartment cell | Mono and cross coupling of caproic, valeric and butyric acids, 1 M NaOH, | Dupre100 |
| Pt | Decane (48% in MeOH), dodecane | One compartment cell | Aqueous solution or MeOH, 1 M hexanoic acid, heptanoic acid, 0.5 M KOH | Haas99 | |
| Pt | 1,4-Butanediol (80%), acetoin (80%), 2,3-butanediol (27%) | 80%, 80%, 34% | One compartment cell | MeOH, sodium 3-hydroxy propionate (10 wt%), sodium lactate (10%), NaOH (10 wt%), 323 K, 0.009–0.0185 A cm−2 | Mosby120 |
| Pt | Various alkanes from fatty acids (14–100%) | 24–99% | One compartment cell | Different solvents mostly based on MeOH, oleic acid, fatty acids, aromatic acids, carboxylic tri or diacids bases: sodium methoxide or triamines, 0.15–0.25 A cm−2 | Wang93 |
| Pt | 2,7-Octanedione (68.3%) and 5-acetyl-2,9-decanedione (22.1%) | 32–83% | One compartment cell | MeOH/H2O (0–100 mol%), 0.5 M levulinic acid, 0.01 M Na leading to a pH of 5, 303 K, 0.25–0.4 A cm−2 | Yuan96 |
| Pt, graphite sheet or foil | 2,7-Octanedione (28.2%), n-octane (n.a.) | 4–86% | One compartment cell and membrane-separated flow cell | MeOH, H2O, 0.1–1 M levulinic acid, valeric acid, 0.1–0.75 M NaOH, Na2SO4, 0.1–1 M NaClO4 | Schröder18,19 |
| Graphite | Different isomers of heptadecadiene, heptadecene, different ethers (conversion >95%) | 20–50% | Ultrasonic bath one compartment cell | MeOH, EtOH, H2O, 0.025–0.25 M oleic acid, 0.025–0.25 M stearic acid, 0.025–0.25 M triglycerides, equimolar amounts KOH, 17 mA cm−2 in MeOH or EtOH, 3 V in H2O, ultrasonic frequency of 24 kHz | Schröder105 |
| Pt | Adiponitrile (78%) | 29% | One compartment cell | MeOH (acetone addition after the first step 1:1), 0.4 M 3-cyanopropanic acid methyl ester, 0.6 M KOH, 333 K, 0.18 A cm−2 |
Fu86 |
| Pt foil | C30, N-triacontane (69%) | n.a. | One compartment cell | Biphasic system (water/MeOH/petroleum ether (3:3:4, v/v/v), 0.085 mM palmitic acid, 0.068–0.128 mM KOH, 313–338 K |
Wu77 |
| Graphite sheet | Olefins (45% carbon yield), ethers (36% carbon yield) and epoxy resins (99% carbon yield) | n.a. | One compartment cell | MeOH, tall oil fatty acids (0.27 M), sodium methanolate (0.27 M), 323 K, 0.02 A cm−2 | Waldvogel63 |
| Pt | 2,7-Octanedione (65%), 2,5-dimethyladipate (60%) | 65%, 60% | One compartment cell | MeOH, 0.4 M levulinic acid or 2-methylsuccinic acid 1-methyl ester, 0.075 M KOH, 295 or 273 K, 0.178 A cm−2 | Mascal104 |
| Pt | n-Octane (40%) | 75% | NMR-tube cell | MeOH, 1 M valeric acid, 0.2 M KOH, 298 K, 0.046 A cm−2, in situ NMR study, tube was placed in an 1 Tesla spectrometer | Blümich73 |
In 2001 the influence of ultrasound on the Kolbe electrolysis of aliphatic acids over Pt and B-doped diamond working electrodes was investigated by the group of Ryley in cooperation with other institutes. For high faradaic equivalents a yield of 75% of n-decane was achieved using a Pt disc electrode. The reactions were conducted in aqueous solutions with only low solubilities of the used organic acids. However, the application of ultrasound led to the emulsification of the organic acids and the formation of an alkane layer on the electrode surface (Fig. 3). While this configuration allowed high yields, insights with regard to surface coverage under reaction conditions and rate determining steps appear to be essential for further process optimization.
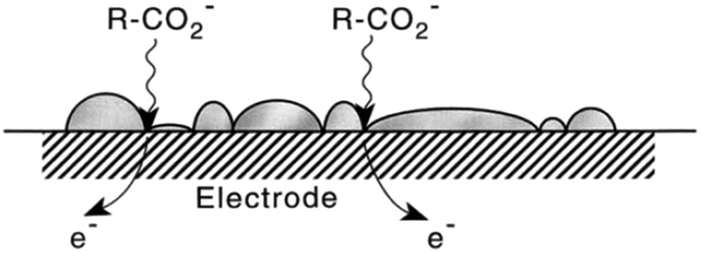 | ||
| Fig. 3 Schematic illustration of the potential formation of an insoluble product layer on the electrode surface and the transfer of the substrate to the electrode surface supported by sono-emulsion.64 Reprinted from ref. 64, Copyright (2001), with permission from Elsevier. | ||
One of the few alternatives reported for Pt electrodes are B-doped diamond (BDD) electrodes. They possess a higher resistance to corrosion and surface erosion than Pt under the demanding conditions of Kolbe electrolysis.64,65 However, Kapałka et al. employed a BDD electrode for the Kolbe reaction of acetic acid to ethane in acidic media and only observed small amounts of the desired product.66 Instead water is discharged at the electrode surface and forms OH radicals which abstract the hydrogen of the methyl groups mainly leading to a consecutive formation of CO2, methanol and methyl acetate. Only at high current densities the electrode surface is sufficiently covered by acetic acid to enable Kolbe dimerization.66 In fact, the coverage of the electrode surface by the carboxylate seems to be an essential element for the reaction.
Another alternative for the use of neat Pt electrodes was patented by Law et al. in 2001.67 Nafion® 117 is coated with Pt and phenylic and alkylic substrates with a maximum of six carbon atoms, which are fed to the cell reactor in gaseous form. These conditions lead to less erosion of the Pt particles on the electrode. The system acts as a kind of membrane-electrode-assembly and is connected via a liquid water phase.67 Despite the absence of solvents or salts, current efficiencies of 75% could be obtained.
Nevertheless, not only the material is important for the successful conversion of organic acids. The surface of the electrode should be rather rough than smooth as this prevents the blocking of the electrode by large CO2 bubbles that are formed by Kolbe electrolysis.68 Moreover, owing to the predominantly hydrophobic properties of the formed Kolbe products, they are prone to adhere on flat electrode surfaces e.g. on conventional Pt electrodes leading to a gradual blocking of the active material and full deactivation eventually.69 The incorporation of multiscale structures on the Pt surface leads to rough surfaces with oil-repelling properties. Hence, the durability of the Kolbe electrolysis can be significantly enhanced. Peng et al. have recently shown that rough and surface modified superoleophobic Pt electrodes possess improved working times of up to 12000 s compared to the unmodified flat Pt electrode with 5000–7500 s for a multitude of organic acids.69 A rougher electrode surface can also facilitate superior current densities. In a recent study by our group RuxTiyO2/Ti was synthesised and screened for Kolbe electrolysis. The electrode materials with rather rough surfaces and different Ru loadings were compared to the commercial Pt electrode.61,62 Dimensionally stable RuxTiyO2 electrodes for Kolbe electrolysis were first described in 1980.46 However, in the latest study, they have been employed for the conversion of biogenic acids and the Ru amount could be further reduced by titania dilution without significant losses in activity.61,62 Andreev et al. investigated Pt, Ir, pyrographite, graphite, glassy carbon and a Pt–Ir alloy (1:9) in the Kolbe mono- and cross-coupling reaction of 10-undecylenic and acetic acid.70 The corresponding voltammograms of the different electrodes for the electrooxidation of a mixture of 10-undecylenic and acetic acid are shown in Fig. 5. All voltammograms possess high current densities after reaching the critical potential (2.5 V). However, for graphite electrode passivation for potentials above 2.75 V can be observed. The highest current density vs. voltage slope was observed for Ir electrodes with a maximum of 45 mA cm−2 at 3.0 V while the highest activity for Kolbe dimerization was achieved over the Pt–Ir alloy.70
With regard to the electrolyte, it is crucial to avoid costly additives and to enable a facile product separation after the reaction, which will also make the process economically more attractive. A recent study by the group of Harnisch suggests that electrolytic basic additives like KOH do not promote Kolbe electrolysis but rather the use of high substrate concentrations.71 An application described by Joshi et al. recommends the use of acetic acid to improve conductivity.72 An alternative way to improve conductivity for Kolbe electrolysis was recently revealed for the model reaction of valeric acid into n-octane. In case the reaction is conducted in situ in a low-field-NMR spectrometer the intermixing can be improved and the charge resistance is strongly reduced by the interaction of the magnetic field with the electric field of the electrolysis. This interaction leads to much higher current densities for in situ experiments in comparison with ex situ experiments. Clearly, a thorough understanding of the effects of mass (especially charge) transport which is most probably strongly related to conductivity as well as the electrode–electrolyte interface is required.73
Another interesting aspect relates to applying thermomorphic solvent systems, which can change from biphasic to monophasic as a function of temperature (Fig. 4) and thus allow for a reduction of work-up steps.74 Considering that different studies assigned major energy demands in chemical industries in the US to thermal separation (10–15%), a facilitated separation and process intensification are of utmost importance.75,76
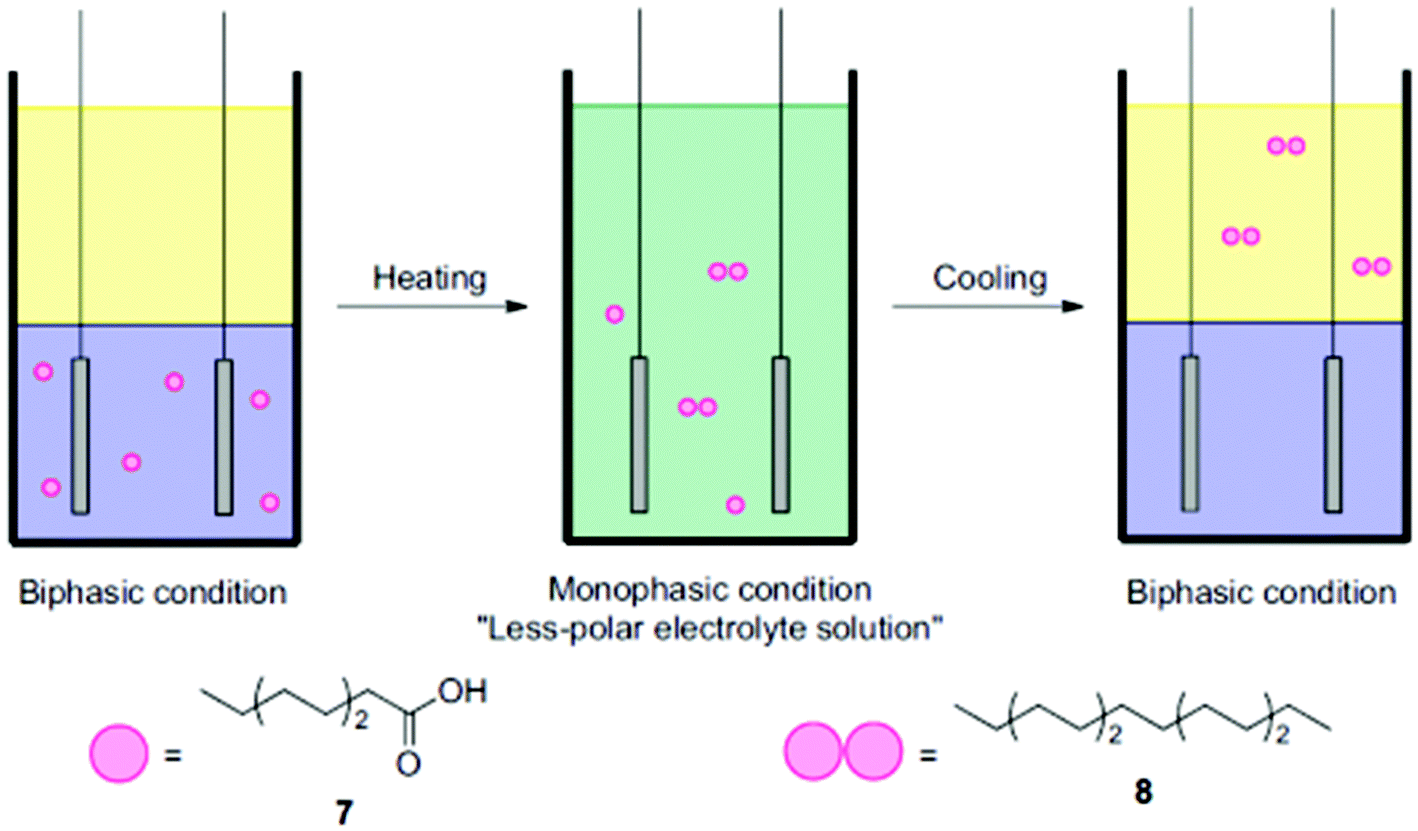 | ||
| Fig. 4 Illustration of the coupling of octanoic acid using a thermomorphic system.74 Reprinted from ref. 74, Copyright (2012), with permission from Elsevier. | ||
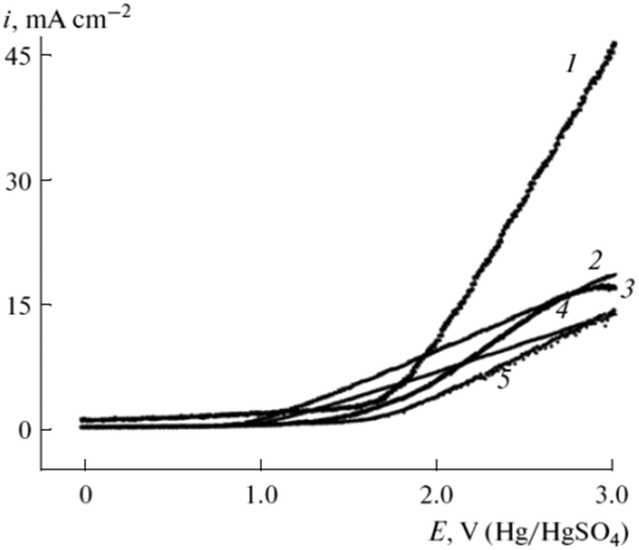 | ||
| Fig. 5 Voltammograms of the electrooxidation of a mixture of 10-undecylenic and acetic acid and sodium acetate in EtOH on different electrodes: (1) Ir, (2) pyrographite, (3) glassy carbon SU-2000, (4) graphite, (5) Pt.70 Reprinted from ref. 70, Copyright (2013), with permission from Springer Nature. | ||
The group of Chiba employed a thermomorphic solvent mixture, in the dimerization of different organic acids.74 The polar phase consisted of a pyridine/MeOH/MeCN (1:1:2) mixture, while cyclohexane was used as the electrolyte (1:1). At 48 °C, the biphasic mixture started to switch to monophasic and with the support of a non-polar co-electrolyte a yield of up to 95% was achieved. Using only the polar phase facilitated a yield of 93%. Of note, in this case significantly lower substrate amounts of only a tenth were used. Thermomorphic systems provide two great advantages: the simple separation of non-polar products such as alkanes and the preservative conditions which can prolong the electrode lifetime.74 The biphasic thermomorphic system could also be implemented for the Kolbe electrolysis, leading to a maximum yield of >69%.77 Urban et al. also demonstrated a separation of respective alkane products in their proof-of-concept study. A combined process consisting of a bio-reactor, a membrane-module and an electrochemical cell was applied in order to produce drop-in fuels.24 The fermentation-derived acids were separated from the broth via a liquid–liquid extraction (pertraction system). The partition equilibrium of the protonated acids in the liquid phase determines the extraction of the carboxylic acids. A transfer facilitates extraction efficiently for non-polar medium-chain acids such as caproic acid. In addition, only the protonated acid is effectively extracted requiring a low pH of the fermentation broth (two hollow fibre modules). Direct Kolbe electrolysis of the different acids in the fermentation broth is hampered by intermediates formed during fermentation which might be oxidized or reduced electrochemically lowering the faradaic efficiency. The non-polar Kolbe dimers can be easily skimmed off from the reaction mixture. Although other studies worked at the interface of biotechnology and electrochemistry25,78,79 the presented case study covers a feasible process design combining microbial and electrochemical steps along the value chain to fuels.24
With regard to process development, the group of Wirth demonstrated continuous electrochemical set-ups in the Kolbe dimerization of di- and trifluoro acids as well as diphenylacetic acid. Interestingly, the operated micro reactor enabled superior yields compared to the corresponding batch experiments.80,81 Despite the potential of electrochemistry to operate at moderate temperature and pressure, there are still challenges for (Non-)Kolbe electrolysis related to both reducing waste and preventing the use of toxic substances. Tajima et al. for instance developed an alternative electrolytic system where commonly used soluble bases e.g. NaOH or KOH were substituted by piperidine immobilised on silica.82 Using this electrolytic system not only Kolbe but also Non-Kolbe products could be obtained in excellent yields.82,83 In particular, the Kolbe process enables alternative reaction routes which reduce or prevent waste and toxic compounds for certain industrially relevant products. In this regard, the group of Yadav found a possibility to reduce the number of reaction steps in the production of macrocyclic alkanones.84,85 Recently, we successfully reported that adipic acid is also accessible via an electrochemical coupling of the bio-derivable mono-ethyl succinic acid, thus avoiding the use of toxic nitric acid which is used in the classical route for the oxidation of cyclohexene (Scheme 2).61 Adipic acid and hexamethylenediamine are valuable starting materials for the production of Nylon 66. The precursor for hexamethylenediamine is petro-based adiponitrile and its industrial production requires external nitrogen sources that are associated with serious environmental concerns (e.g. NH3, NaCN or HCN). The Kolbe dimerization of naturally occurring glutamic acid overcomes these issues in a remarkable way (Scheme 2).86
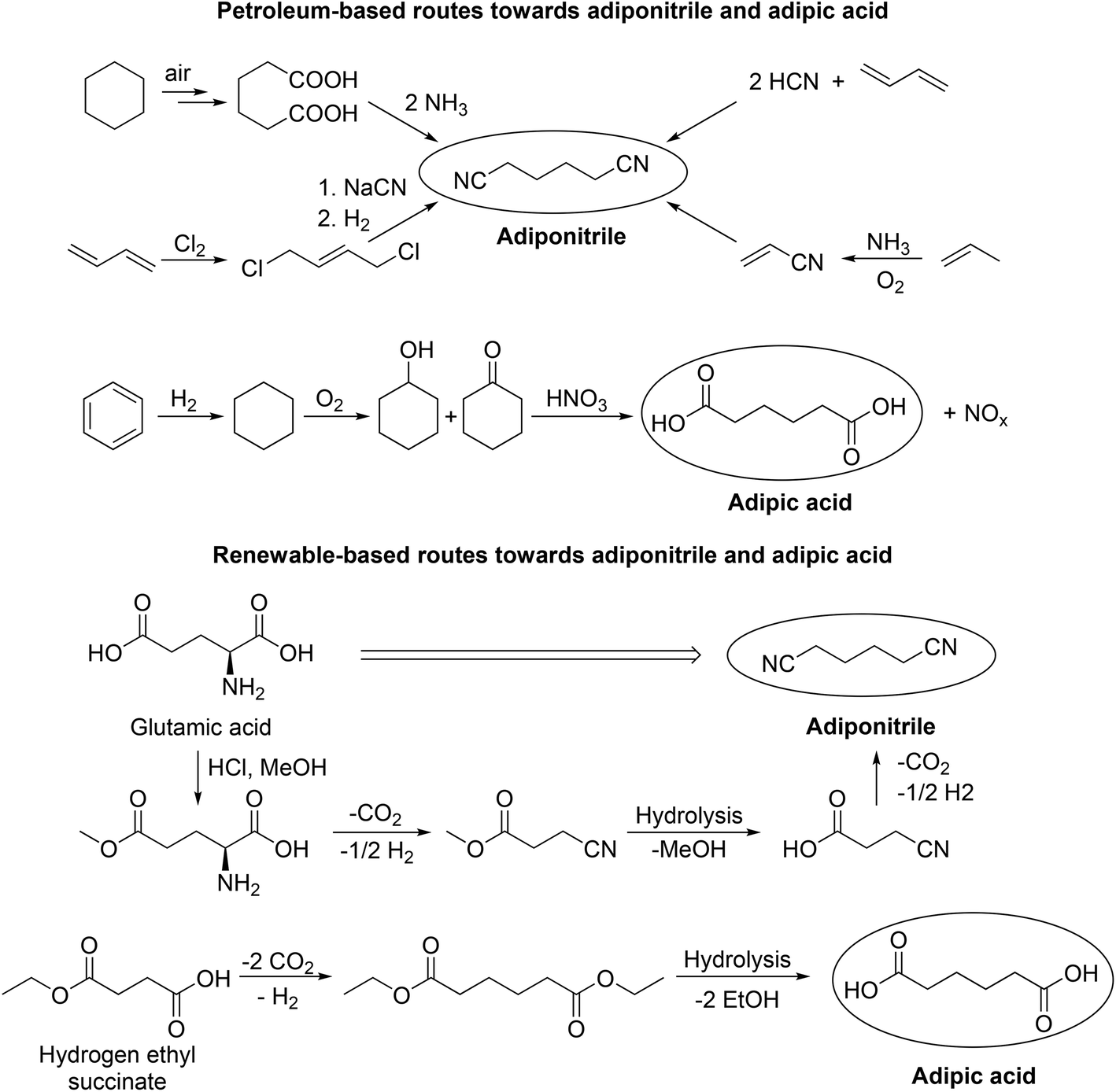 | ||
| Scheme 2 Petroleum- and bio-based routes for the production of adiponitrile and adipic acid. The renewable routes include Kolbe electrolysis steps. Redrawn from ref. 86 (Copyright (2012), with permission from John Wiley & Sons Ltd) and ref. 61 (Copyright (2018), with permission from the American Chemical Society). | ||
The green potential of Kolbe electrolysis even goes beyond the mere elimination of wastes and hazardous compounds. It may also find application in the efficient degradation of persistent toxic pollutants. Lin et al. used the decarboxylation mechanism of Kolbe for the degradation of perfluorooctanoic acid – a substance that is toxic to reproduction and carcinogenic. However, in this case the target is not the dimerization reaction but the decomposition of the entire acid into CO2. Here, the usual electrodes like dimensionally stable anodes (Ti/RuO2, Ti/IrO2, Ti/IrO2–Ti/RuO2) and Pt do not work properly for degradation.87 Lin et al. employed Ti/SnO2Sb, Ti/SnO2Sb/PbO2 and Ti/SnO2Sb/MnO2 anodes while a very recent study shows excellent decomposition rates for perfluorocarboxylic acids (C4–C8) over Ce-doped porous nanocrystalline PbO2 electrodes.88 Furthermore, Ahad and de Klerk investigated Kolbe dimerization for the processing of acidic waste water accumulated during the Fischer–Tropsch synthesis.89 However, the current efficiency for the acetic acid conversion to methane or ethane remained below 3%. Thus, the technology readiness level of Kolbe electrolysis for acid waste handling e.g. in small-scale remote Fischer–Tropsch facilities is still considerably low. Nevertheless, Kolbe electrolyses ideally combined with new oleophobic electrodes are expected to facilitate applications in the treatment of surfactant contaminated domestic wastewater streams.69
Besides the reports on new electrocatalytic systems and the potential of (Non-)Kolbe reactions for more environmentally conscious chemistry, several patents and publications deal with bio-derivable materials and their conversion into fuels, platform compounds and value-added chemicals via (Non-)Kolbe.
Dierker patented the production of cosmetics and pharmaceutics through the dimerization of branched or linear fatty acids (C6–26 or respectively C6–22) in 2006.90 The synthesised oils might find applications in skin and hair care. The electrolysis was conducted in a standard Kolbe cell with a current density between 100–1000 mA cm−2 and an alkaline solution with high fatty acid concentrations.90 Furthermore, Bradin claimed the production of fuels from sugars and triglycerides.91,92 The latter originated from vegetable oil or animal fat. After hydrolysis to fatty acids, subsequent Kolbe dimerization to diesel products took place. Since the efficiency of the Kolbe electrolysis is highly dependent on the conductivity of the system, the choice of the electrolyte and solvent has to be considered with caution. In this regard, Bradin et al. proposed the use of ionic liquids for the transformation of acids via Kolbe electrolysis.91,92 Kolbe electrolysis is sensitive to water due to potential oxygen evolution as the competing reaction. Thus, substituting water with ionic liquids might shift the dimerization to a higher efficiency. The alkane products from triglycerides can be used as fuel or lube base stock oil. However, an alternative reaction route enables metathesis in order to combine relatively short with longer olefins after dehydrogenation of the alkanes. In this case, the resulting product stream after metathesis has an averaged molecular weight.93 Palmitic acid, which is a bio-derived compound produced through the saponification of its respective triglyceride found in palm oil, can be converted into wax in the form of n-triacontane using Kolbe dimerization as recently reported by Zhang et al.77
In addition to the production of fuels and waxes from triglycerides, biodiesel might be purified from remaining free fatty acids via Kolbe electrolysis as patented by Bradin.92,94 However, here no detailed description of the conducted Kolbe electrolysis was provided. Nevertheless, a very similar approach was published by Joshi et al. demonstrating the conversion of triglycerides into fatty acids followed by Kolbe electrolysis to produce fuels.72,95
The inventors observed 90% conversion of decanoic acid in methanol yielding octadecane as the only product. Moreover, the production of heavy fuels like kerosene and aviation fuels from fatty acids has been patented. In another work, Bradin presented the production of n-hexane and gasoline components from fermentable sugars. After delignification and depolymerization of lignocellulosic materials, the as derived glucose, xylose, sucrose and fructose can be fermented using yeast bacteria predominantly producing butyric acid and CO2. Butyric acid is Kolbe or photo-Kolbe electrolysed into n-hexane, while the accumulated lignin may be converted into synthesis gas. Since n-hexane possesses insufficient gasoline properties due to its low octane rating, reforming of the dimer product is also included by Bradin, yet no experimental specifications concerning the Kolbe dimerization are mentioned.97
In addition to the reports of Bradin et al. several other groups claimed the fermentation of biogenic materials such as carbohydrates, proteins or fat into short-chained organic acids (butyric acid and propionic acid)98,99 or fatty acids100,101 and their use in the (Non-)Kolbe conversion to fuel candidates and fine chemicals (olefins and alkanes). The group of Harnisch also reported the electrochemical production of drop-in diesel fuels along with integration to a microbiological fermentation process and pertraction of the herein synthesised carboxylic acids.24 Another drop-in fuel can be produced from bio-derivable 3-hydroxy decanoic acid.26 The long-chain acid can be obtained from bio-feedstocks after the fermentation of glucose and xylose into 3-(3-hydroxy-alkanoyloxy)alkanoates (HAAs) and subsequent hydrolysis. The Non-Kolbe electrolysis of 3-hydroxy decanoic acid or slightly modified fermentation broth leads to high amounts of C9-oxygenates (95% yield) with excellent fuel properties (e.g. a cetane number of 63).
Mosby and others reported the synthesis of diols and dienes from oxygen containing acids (with two or more functional oxygen groups) derived through the fermentation of biomass. The produced diols might be used as solvents and monomers, whereas the dienes might find applications as substrates for elastic polymer materials. In addition to this brief summary of publications and patents involving (Non-)Kolbe conversion of fatty acids to petrochemicals and fuel candidates, at this point reference to the review of Andreev et al. is made, who have thoroughly reviewed the state of the art in this regard.21
The group of Noel screened different alkane- and perfluoroalkane carboxylates for (Non-)Kolbe electrolysis.102 The authors also tested sodium butanoate over graphite and Pt electrodes in aqueous methanol (40 vol%). However, they could not identify the side product in both cases and only obtained Non-Kolbe products mainly 3- and 1-propyl butyrate (10% and 24% selectivity for Pt and 10% and 20% selectivity for graphite). The electrochemical oxidation of perfluorobutyrate only gave heptafluorobutanol in low yields for aqueous methanol irrespective of the electrode material. The yields of the dimer could be increased when the electrolytic system was changed to a MeOH–MeONa mixture. When using octanoic acid and higher applied current densities (200–600 mA), the conversion could be increased from 30 to 60%. However, when Pt was used as the electrode the selectivity of Kolbe products decreased from 66 to 57%. Interestingly, this trend was not observed for perfluorooctanoic acid, where only the dimer was formed.102
A novel compound was synthesised via the Kolbe mechanism starting from levulinic acid. Besides the coupling product 2,7-octanedione, also an increased formation of 3-buten-2-one was observed which led to the formation of 5-acetyl-2,9-decanedione. This route was only possible when using a water–methanol mixture as the solvent for Kolbe dimerization. The 3-buten-2-one successively couples twice with the 3-oxobutyl radical (Scheme 3). The obtained triketone possesses a unique Y-structure which as a ligand can dissolve Li salts and could be a solvent for Li-ion batteries. According to computational studies, the solvation energy (ΔH°) reaches a value of approximately 350 kJ mol−1 for 5-acetyl-2,9-decanedione while the values for conventional solvents such as propylene carbonate or ethylene carbonate are clearly lower.96 The group of BeMiller successfully decarboxylated biogenic D-glucuronic acid and D-glucuronosides via the Hofer-Moest mechanism into dialdehydes showing that sugar acids are suitable substrates for Kolbe electrolysis.103
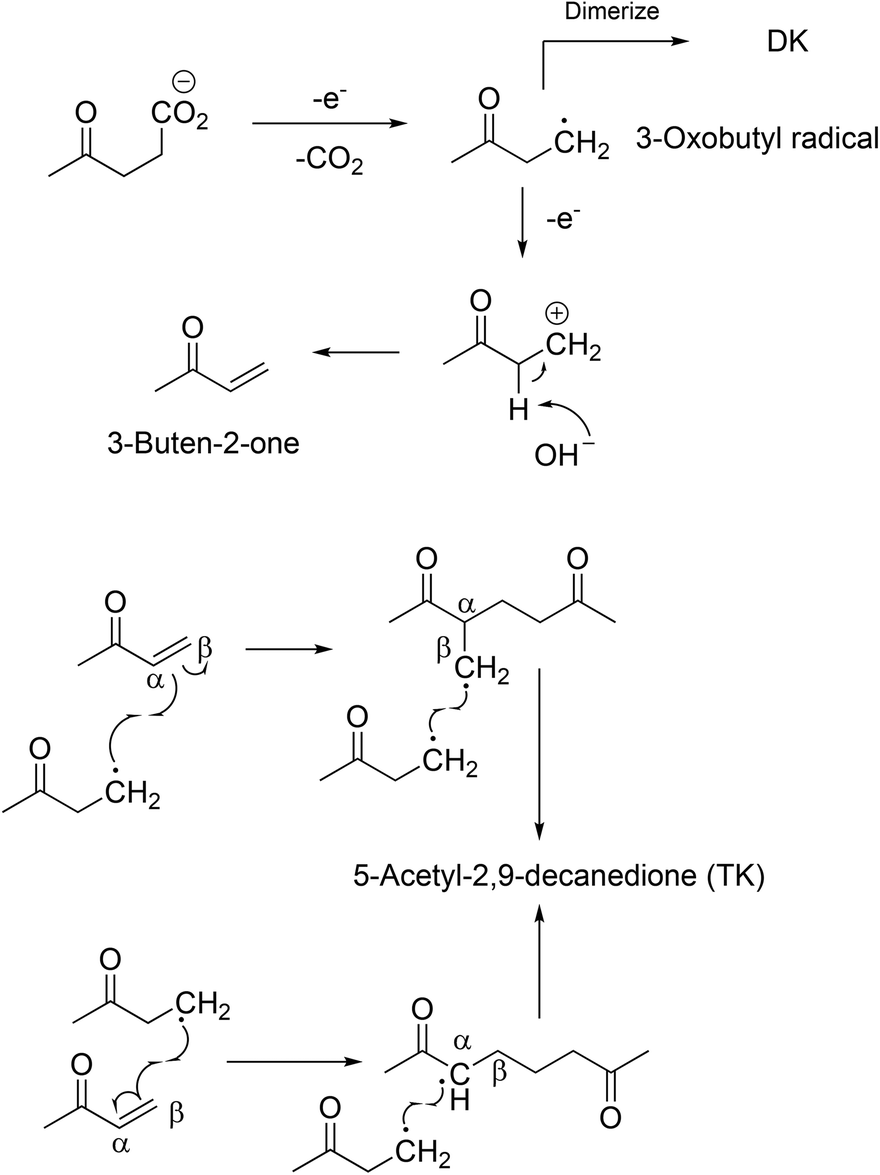 | ||
| Scheme 3 Proposed synthesis pathways toward 5-acety-2,9-decane-dione.96 Reprinted from ref. 96, Copyright (2012), with permission from the Royal Society of Chemistry. | ||
According to a recent study, the coupling product of levulinic acid 2,7-octanedione (65% yield) can be hydrogenated into 2,7-octanediol (94% yield) – a potential monomer for the synthesis of polyesters.104 The copolyesters poly(2,7-octanediol)terephthalate and poly(2,7-octanediol)-2,5-furanoate were obtained in fair yields (54–63%) with a medium chain length (3978–8531 Da), showing sufficient stability and a low polydispersity index (Mw/MN). The synthesised 2,7-octanedione can also be further reacted in an aldol condensation to form 1-methylcyclopentyl ketone or 3-methylcycloheptanone. The latter can be transformed into different cycloalkanes (85% yield over two steps) after subsequent hydrogenation. The calculated octane rating for a blend of the obtained cycloalkanes is 86.8 (for comparison n-octane has an octane rating of −11). The study also mentions the Kolbe electrolysis of methyl hydrogen methylsuccinate into 2,5-dimethyladipate (60% yield), which because of its similarity to adipic acid, might represent a promising monomer for polymer industries.104 The production of industrially relevant monomers via the Kolbe electrolysis of bio-derived acids was also shown by Dai et al. The synthesis of adopinitrile (78% yield) from glutamic acid is an important intermediate step for the production of hexamethylenediamine which is the starting material of Nylon besides adipic acid.86
Moreover, very recently our group reported the direct production of diethyl adipate starting from the renewable substrate succinic acid in its monoethylester form.61 A new Ti/(RuxTi1−x)O2 electrode material with different Ru loadings was developed to replace the expensive commonly used bulk Pt material (Fig. 6).
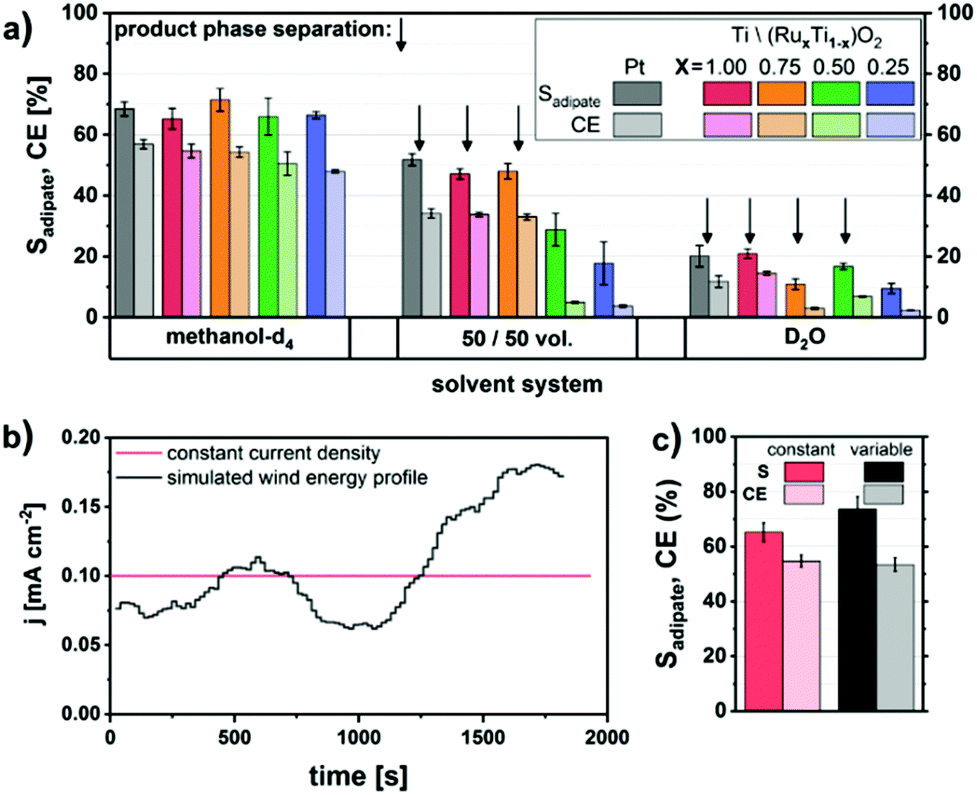 | ||
| Fig. 6 (A) Solid Pt and (RuxTi1−x)O2-coated Ti electrodes in various water–methanol solvent systems, (B) graph showing a constant and simulated wind-energy current density profile and (C) comparison of the efficiency and selectivity for the respective electrolysis using RuO2-coated Ti electrodes.61 Reprinted from ref. 61, Copyright (2018), with permission from the American Chemical Society. | ||
The conversion of methyl hydrogen succinate was also conducted by applying a transient wind-energy current density profile. The obtained selectivities and faradaic efficiencies for constant and fluctuating current densities were comparable both lying between 65 and 75%. Furthermore, the separation of the obtained ester could be achieved easily when water or a methanol–water mixture was used as the solvent.61 Combining the work of Dai et al.86 with our recent contribution,61 the electrochemical production of Nylon 66 via Kolbe is already fully feasible from biogenic compounds. Additionally, the production of yet another industrially relevant monomer was discovered by using the same substrate methyl hydrogen succinate using carbon as the electrode material under slightly different reaction conditions. Thus, the unsaturated monomer ethyl acrylate was obtained (50% yield and faradaic efficiency). In both cases the use of NEt3 instead of KOH is recommended as the deprotonating electrolyte for (Non-)Kolbe. Due to its high basicity KOH leads to the undesired hydrolysis of the ester group in the product at high temperatures and thus to a mixture of acid and ester increasing the expenditure for product separation.61
In 2012, the group of Schröder reported the synthesis of renewable fuel candidates from levulinic acid. The authors showed the efficient electrochemical hydrogenation of levulinic acid into valeric acid and the possible use of this compound for the production of n-octane in a second step. The separation of n-octane was promoted by the use of aqueous solutions.18 Three years later, a more extensive study explained different reaction pathways and tuneable conditions for the dimerization and Non-Kolbe electrolysis of valeric and levulinic acid. Furthermore, a reason for the reduced mass balances during Kolbe reactions was given: a complete anodic degradation of the carboxylic acid was contemplated. Moreover, energetic considerations of the authors lead to the conclusion that the sustainable synthesis of fuels or petrochemicals such as n-octane or methyl vinyl ketone is also possible from renewable energy forms e.g. solar or wind power. The electrochemically synthesized compounds can be considered for energy storage. An advantage compared to numerous conventional processes relates to the use of ambient pressure, aqueous electrolyte solutions, and room temperature following the principles of green chemistry.19 The work of Schröder,18,19 Mascal,104 and Yuan96 shows that levulinic acid offers great potential for electrochemical processes. The (Non-)Kolbe electrolysis is especially suitable to convert levulinic acid into fuels or fine chemicals. Scheme 4 shows a summary of the most important products that can be obtained by Kolbe electrolysis of levulinic acid itself or its derivative valeric acid which is obtained by simple electrochemical reduction.
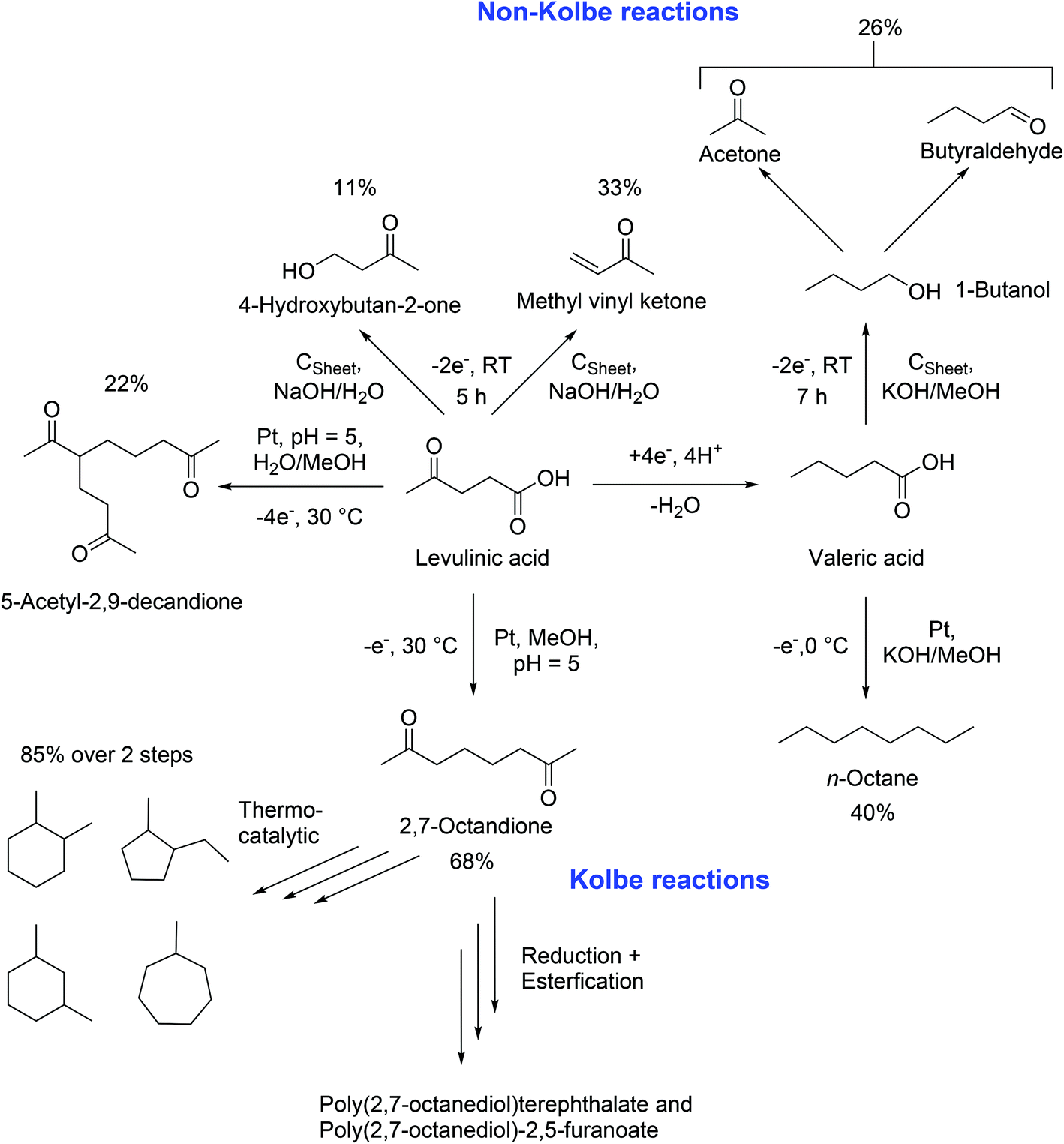 | ||
| Scheme 4 Synthesis possibilities starting from levulinic acid LA and valeric acid VA, based on the research results of Schröder18,19 Yuan96 and Mascal.104 The reaction conditions were selected according to the maximal achieved yield. | ||
The group of Schröder also published the electrochemical transformation of triglyceride rapeseed oil and stearic- and oleic acids as model fatty acids into olefins and ethers. The fatty acid conversion only worked in methanol, while triglyceride only formed methyl esters of the corresponding fatty acids in methanolic solutions. de Kruijff et al. reported a suitable replacement for the harmful bisphenol A resins (bisphenol A diglycidyl ether) through the production of epoxy resins from lignocellulosic biomass.63 The novel resins could be obtained from tall oil fatty acids via Non-Kolbe electrolysis (81% ethers and olefins) and subsequent epoxidation with oxone (99%). The Non-Kolbe operation mode enabled a scale up of up to three orders of magnitude (5 mL to 1.5 L). The obtained epoxy resins exhibited good mechanical properties and storage moduli, particularly if the curing was conducted with MTHPA.63
When using an aqueous electrolyte in combination with ultrasound, triglycerides were converted into olefins, glycerol, and oleic acid. However, high conversions (>95%) of oleic- and stearic acid could be only achieved when applying four equivalents of Farad charge (20% faradaic efficiency) (Fig. 7). A higher faradaic efficiency is achieved when applying only one Farad equivalent leading to conversions of 40–70% and efficiencies between 35 and 45%. Therefore, the authors suggest that the decreasing substrate concentration over time might lower the efficiency and could be overcome through the application of a continuous set-up. For triglycerides the charge transfer efficiency only reached 20%. The olefins obtained via Non-Kolbe electrolysis may be suitable for biofuel applications as e.g. heptadecene with a boiling point of 300 °C lies in the boiling range of conventional diesel. Furthermore, the mixture of heptadecene and heptadecane methyl ether has a higher heating value (42.3 kJ g−1) than biodiesel (37 kJ g−1) and is close to that of fossil diesel (42.8 kJ g−1).105
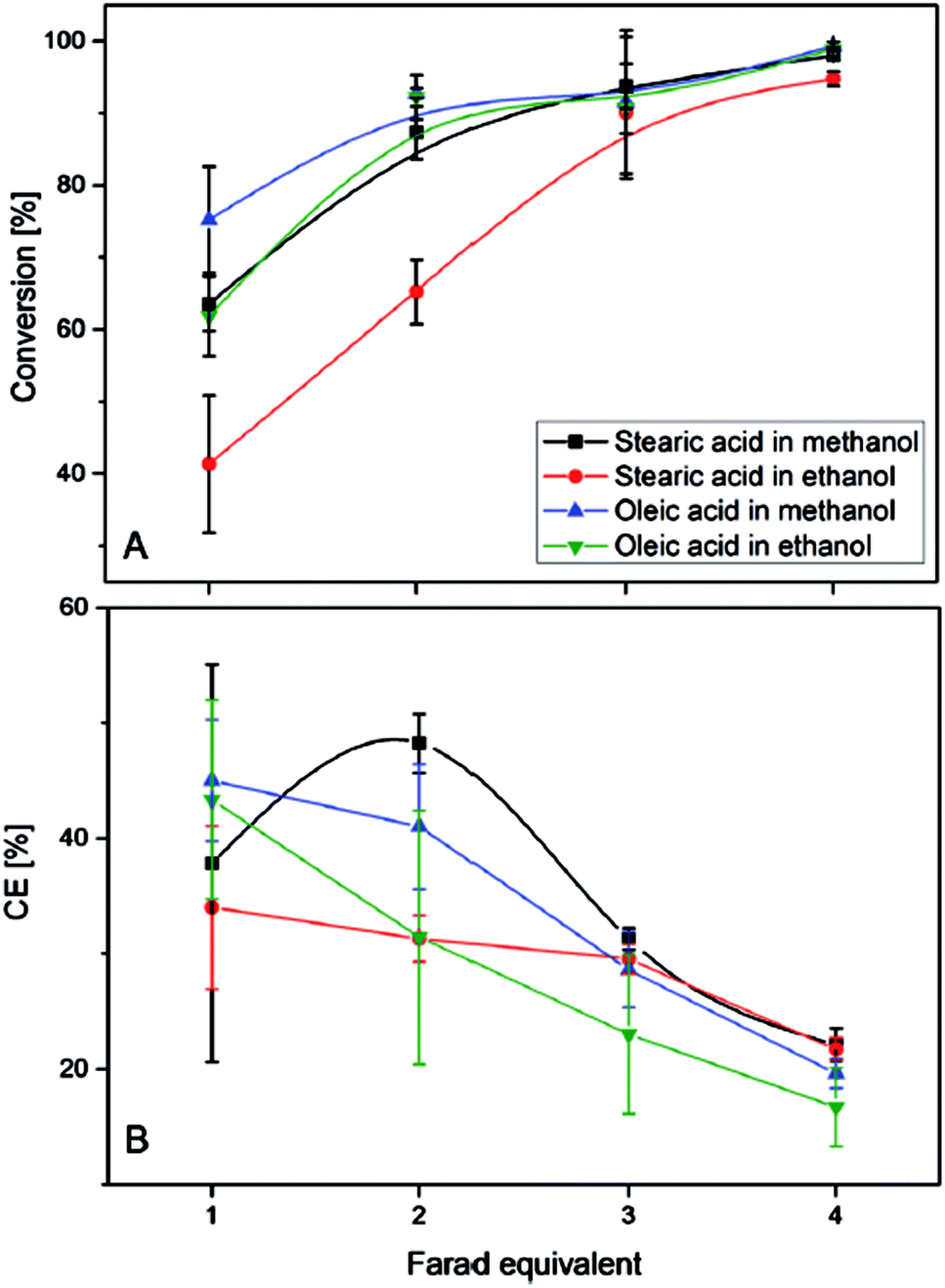 | ||
| Fig. 7 (A) Degree of the electrochemical decarboxylation of stearic acid and oleic acid in MeOH and EtOH as a function of the applied charge. (B) The faradaic efficiency vs. the flown charge.105 Reprinted from ref. 105, Copyright (2015), with permission from John Wiley & Sons Ltd. | ||
In a recently published study of our group potential diesel or gasoline fuels have been synthesised by cross and mono-coupling of different acids. In order to allow for a more economic process the previously described RuxTiy(O2)/Ti electrode was employed as a cheaper alternative. The bio-based starting materials ethyl hydrogen succinate, methyl hydrogen ethylsuccinate and isovaleric acid were used to produce different esters and alkanes aiming for tailored fuel structures. The monoester diacids were cross-coupled with isovaleric acid and hydrolysed afterwards. In a second step the obtained methylhexanoic acid was cross-coupled again with isovaleric acid or with itself. By optimizing different process parameters such as electrolyte, solvent, electrode and ratio of the acids, a tuneable product distribution and a facilitated product separation was successfully achieved. The formed esters showed promising octane ratings (71–99) for application in gasoline. Furthermore, the calculated cetane numbers of the obtained alkanes (45–60) are comparable or even superior to benchmark biodiesel fuels (45–55). In conclusion, the different bio-derivable mono- and diacids can be reassembled to a desired composition like single bricks.62
A selection of the most relevant types of bio-based carboxylic acids and their conversion into the respective main products via the herein reviewed (Non-)Kolbe electrolysis is summarized in Scheme 5.
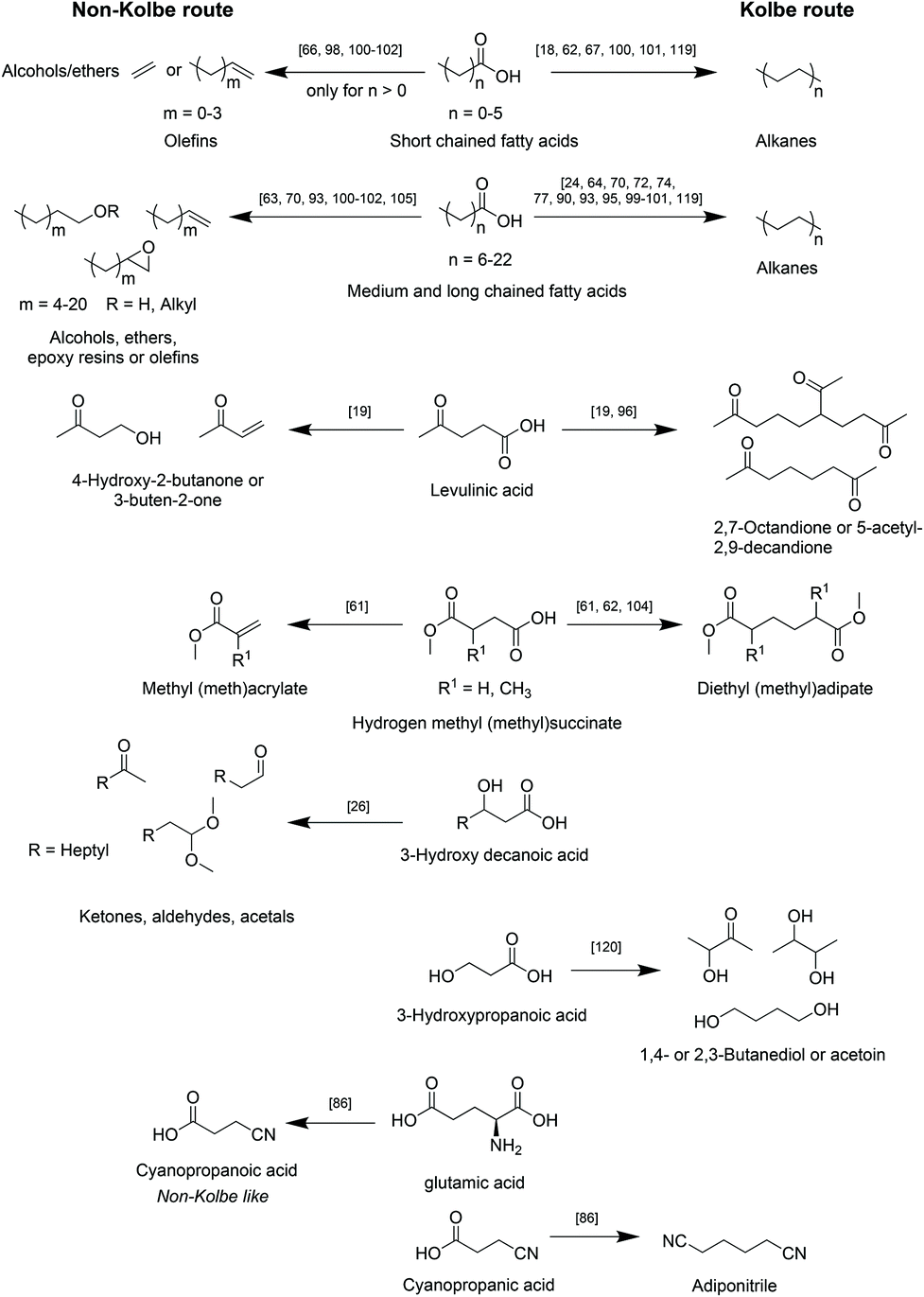 | ||
| Scheme 5 Overview of reported classes of biogenic acids to corresponding main products after upgrading via (Non-)Kolbe electrolysis. | ||
Perspective
The bio-availability of several carboxylic acids as platform chemicals and the potential of electrochemical transformations to directly integrate renewable energy into chemical value chains drive (Non-)Kolbe electrolysis to become an attractive tool in future electrified bio-refineries. Recent contributions highlight the concept of electro-bio-refineries.11,23,106 Several studies on biofuel synthesis using (Non-)Kolbe electrolysis emphasize this approach as a pathway towards chemical energy storage of renewable energies. Considering the modular and flexible nature of electrochemical reactions, dynamic operation is within reach. In addition to the versatile product portfolio of (Non-)Kolbe electrolysis, both anodic decarboxylations could be combined with important cathodic half-cell reactions such as hydrogen generation or CO2 reduction to the so-called 200% reactions. They also offer an ideal interface for biotechnological carboxylic acid production potentially reducing the need for separation steps. Interestingly, (Non-)Kolbe electrolysis also holds potential as a technology in the purification of product streams through the decompostion of undesired compounds. In the past, the use of biotechnologically contaminated solutions has not necessarily been a disadvantage in (Non-)Kolbe reactions. Through a clever reactor design with an associated pertraction system24 or the direct conversion of the fermentation broth in the electrolysis cell,26 it was possible to achieve moderate yields and selectivities despite high contamination of the substrate solution. Particularly Pt, the standard working electrode for Kolbe dimerization, is known to be very resistant to electrochemical deactivation. However, the presence of impurities in the reaction solution can provoke undesired side reactions or increase the solution resistance leading to a more energy intensive electrolysis. In this regard, special attention should be devoted to the counter electrode of (Non-)Kolbe reactions in one-compartment cells. Depending on the type, the counter electrode may lead to product decomposition or side reactions (reduction of functional groups). In Non-Kolbe electrolysis, carbon is usually employed as the working electrode. However, carbon materials are much more prone to deactivation at high anodic potentials (e.g. via decomposition into carbonates) and therefore must be replaced more frequently. A more stable substitute could have a decisive influence on the service life in the processes. Another potential drawback but also an opportunity relates to CO2 which is the principal co-product in (Non-)Kolbe electrolysis. CO2 could be further valorized in case the process provides a clean and pressurized stream.A thorough understanding of the mechanism with special regard to the reaction steps on the electrodes will allow for a more rational design of tailored surfaces. Thus far, no consensus on the exact (Non-)Kolbe mechanism could be established in the literature.29,32 Moreover, several studies have examined the influence of the electrolyte on the reaction outcome. However, especially the role of the solvent remains the subject of current research. In previous studies, numerous organic and aqueous solvents have been tested, establishing methanol as the most suitable solvent for (Non-)Kolbe chemistry.26,29,61,104,105 The utilization of less conventional solvents e.g. ionic liquids or deep eutectic solvents remains almost untouched. Future research devoted to the application of these new solvents could reveal unprecedented activities and selectivities. Two major issues that will determine the future course of (Non-)Kolbe transformations are the economic feasibility and the role of CO2 which is formed as the co-product. Hydrogen evolution presents the parallel reaction on the cathode side. However, the second half-cell could also serve for the reduction of bio-based platform chemicals considering the increased economic value of the resulting products (i.e. levulinic acid to γ-valerolactone or furfural and 5-hydroxymethylfurfural to furans). This would enable the electrochemical upgrading of bio-based compounds to value added platform chemicals at both the cathode and anode. However, the production of different products in one or two half cells also requires efficient product separation from the reaction solution. This circumstance favours the use of aqueous systems over alcoholic solvents, as it allows direct in situ separation of the commonly non-polar (Non-)Kolbe products (through the formation of two phases). The successful separation has already been demonstrated in the past by subsequent extraction with suitable non-polar organic solvents.18,24,61,62 Despite several alternative half-cell reactions with different valuable products, the high relevance of hydrogen for numerous industrial processes makes a parallel hydrogen production attractive. For instance, H2 in combination with the CO2 originating from (Non-)Kolbe reactions can be applied in molten carbonate fuel cells. Levy et al. published in the report of the Department of Energy (DoE, USA) in 1980 that almost 50% of the necessary energy consumption of Kolbe electrolysis could be recovered by using H2 and CO2 in fuel cells.107 The molten carbonate fuel cell approach also directly addresses the further usage of the co-produced CO2. Other possibilities involve the capture of CO2 and further utilization as a C1 building block.108–111 In this regard, the electrocatalytic reduction of CO2112 or the thermocatalytic reverse water–gas shift113 display two promising paths.
Not yet well explored are challenges and potential opportunities of an up scaling and continuous operation. Case studies on both the techno-economic analysis and overall life cycle assessment are urgently needed to evaluate the viability and sustainability of these approaches, respectively. The key enabler is certainly the fundamental understanding of the role of electrode, electrolyte, substrate structure and process parameters on the (surface) reaction mechanism and overall reaction outcome. Accordingly, tailored electrode materials – especially noble metal free systems in the case of Kolbe electrolysis – are of utmost importance. In addition, specifically designed electrolytes could add to reaction performance, enable facilitated product recovery and reduce the environmental foot print of (Non-)Kolbe electrolysis. Novel value chains based on carboxylic acids readily available in the form of plant based fatty acids63,100,101,105 and via the fermentation of various feedstocks such as biomass,24–26,114–118 COx and even plastic wastes remain yet to be explored.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Cluster of Excellence Tailor-Made Fuels for Biomass (EXC236) and Excellenzcluster 2186 “The Fuel Science Center” ID: 390919832. J. B. M. and R. P. acknowledge financial support from the European Regional Development Fund (ERDF) and the state of North-Rhine Westphalia, Germany, under the operational program ‘Regional Competitiveness and Employment’ Project ‘Sustainable Chemical Synthesis’. We thank M. G. Al Shaal for the idea of the graphical abstract. Further thanks go to S. Kerner who helped with graphical designs and to J. Artz, J. Meyers, Y. Louven, T. Janke, N. Kurig, K. Birkelbach, M. Fischer, J. Deischter and Mona Felis-Catus for fruitful discussions.References
- IEA, World Energy Outlook 2018, OECD, Paris, 2018 Search PubMed.
- V. Quaschning, Understanding Renewable Energy Systems, Routledge, London, 2016 Search PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC.
- W. de Jong and J. R. van Ommen, Biomass as a Sustainable Energy Source for the Future: Fundamentals of Conversion Processes, Wiley, 2014 Search PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I - Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Golden, USA, 2004 Search PubMed.
- E. de Jong and R. J. A. Gosselink, in Bioenergy Research: Advances and Applications, ed. V. K. Gupta, M. G. T. P. Kubicek and J. S. Xu, Elsevier, Amsterdam, 2014, pp. 277–313 Search PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- P. N. Amaniampong, J. Clément, D. Gigmes, C. Ortiz Mellet, J. M. García Fernández, Y. Blériot, G. Chatel, K. De Oliveira Vigier and F. Jérôme, ChemSusChem, 2018, 11, 2673–2676 CrossRef CAS PubMed.
- J. Deischter, K. Schute, D. S. Neves, B. E. Ebert, L. M. Blank and R. Palkovits, Green Chem., 2019, 21, 1710–1717 RSC.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328 CrossRef CAS PubMed.
- Y. Kwon, K. J. P. Schouten, J. C. van der Waal, E. de Jong and M. T. M. Koper, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS PubMed.
- J. Artz and R. Palkovits, ChemSusChem, 2015, 8, 3832–3838 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574 RSC.
- P. Nilges, T. R. dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235 RSC.
- T. R. dos Santos, P. Nilges, W. Sauter, F. Harnisch and U. Schröder, RSC Adv., 2015, 5, 26634–26643 RSC.
- Y. Kwon, E. de Jong, S. Raoufmoghaddam and M. T. M. Koper, ChemSusChem, 2013, 6, 1659–1667 CrossRef CAS PubMed.
- V. N. Andreev, V. A. Grinberg, A. G. Dedov, A. S. Loktev, I. I. Moiseev and A. Y. Tsivadze, Prot. Met. Phys. Chem. Surf., 2013, 49, 32–39 CrossRef CAS.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925 RSC.
- F. Harnisch and C. Urban, Angew. Chem., Int. Ed., 2018, 57, 10016–10023 CrossRef CAS PubMed.
- C. Urban, J. Xu, H. Sträuber, T. R. dos Santos Dantas, J. Mühlenberg, C. Härtig, L. T. Angenent and F. Harnisch, Energy Environ. Sci., 2017, 10, 2231–2244 RSC.
- F. J. Holzhäuser, J. Artz, S. Palkovits, D. Kreyenschulte, J. Büchs and R. Palkovits, Green Chem., 2017, 19, 2390–2397 RSC.
- J. Meyers, J. B. Mensah, F. J. Holzhäuser, A. Omari, C. C. Blesken, T. Tiso, S. Palkovits, L. M. Blank, S. Pischinger and R. Palkovits, Energy Environ. Sci., 2019, 12, 2406–2411 RSC.
- H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- H.-J. Schäfer, in Electrochemistry IV, Springer, Berlin/Heidelberg, 1990, pp. 91–151 Search PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 CrossRef.
- A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623–664 CrossRef CAS.
- A. G. Dubinin, L. A. Mirkind, V. E. Kazarinov and M. Y. Fioshin, Elektrokhimiya, 1979, 15, 1337–1340 CAS.
- A. A. Yakovleva, S. N. Kaidalova, Y. B. Skuratnik and V. I. Veselovskii, Elektrokhimiya, 1972, 8, 1799–1802 CAS.
- J. W. Shaw, T. Nonaka and T. C. Chou, J. Chin. Inst. Chem. Eng., 1985, 16, 245–250 CAS.
- L. Eberson, Acta Chem. Scand., 1963, 17, 2004–2018 CrossRef CAS.
- L. Eberson, S. Granse and B. Olofsson, Acta Chem. Scand., 1968, 22, 2462–2470 CrossRef CAS.
- P. R. Nadebaum and T. Z. Fahidy, Electrochim. Acta, 1972, 17, 1659–1681 CrossRef CAS.
- B. E. Conway and M. Dzieciuch, Can. J. Chem., 1963, 41, 21–37 CrossRef CAS.
- B. E. Conway and M. Dzieciuch, Can. J. Chem., 1963, 41, 38–54 CrossRef CAS.
- B. E. Conway and M. Dzieciuch, Can. J. Chem., 1963, 41, 55–67 CrossRef CAS.
- S. N. Shukla and O. J. Walker, Trans. Faraday Soc., 1931, 27, 722–730 RSC.
- G. E. Hawkes, J. H. P. Utley and G. B. Yates, J. Chem. Soc., Perkin Trans. 2, 1976, 1709–1716 RSC.
- J. H. P. Utley and G. B. Yates, J. Chem. Soc., Perkin Trans. 2, 1978, 395–400 RSC.
- S. S. Khidirov and K. S. Khibiev, Russ. J. Electrochem., 2005, 41, 1176–1179 CrossRef CAS.
- C. Iwakura, F. Goto and H. Tamura, Denki Kagaku, 1980, 48, 21–25 CAS.
- M. O. F. Goulart and H.-Y. Schafer, J. Braz. Chem. Soc., 1999, 10, 153–162 CrossRef CAS.
- H. Fujiwara, M. Atobe, H. Nanetsuna and T. Onaka, J. Chin. Chem. Soc., 1998, 45, 175–181 CrossRef CAS.
- M. Friedrich, Angew. Chem., 1943, 56, 167 Search PubMed.
- Y. M. Tyurin and G. N. Afon'shin, Elektrokhimiya, 1969, 5, 1198–1202 CAS.
- A. K. Vijh, J. Electrochem. Soc., 1972, 119, 679–683 CrossRef CAS.
- C. P. Andrieux, F. Gonzalez and J. M. Savéant, J. Electroanal. Chem., 2001, 498, 171–180 CrossRef CAS.
- J. P. Coleman, R. Lines, J. H. P. Utley and B. C. L. Weedon, J. Chem. Soc., Perkin Trans. 2, 1974, 1064–1069 RSC.
- D. L. Muck and E. R. Wilson, J. Electrochem. Soc., 1970, 117, 1358–1362 CrossRef CAS.
- M. P. J. Brennan and R. Brettle, J. Chem. Soc., Perkin Trans. 1, 1973, 257–261 RSC.
- M. Rueffer, D. Bejan and N. J. Bunce, Electrochim. Acta, 2011, 56, 2246–2253 CrossRef CAS.
- P. G. Gassman and F. V. Zalar, J. Am. Chem. Soc., 1966, 88, 2252–2257 CrossRef CAS.
- S. D. Ross and M. Finkelstein, J. Org. Chem., 1969, 34, 2923–2927 CrossRef CAS.
- E. Klocke, A. Matzeit, M. Gockeln, H. J. Schaefer and H. J. Schäfer, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS.
- T. Shono, I. Nishiguchi, S. Yamane and R. Oda, Tetrahedron Lett., 1969, 1965–1968 CrossRef CAS.
- G. Creusen, F. J. Holzhäuser, J. Artz, S. Palkovits and R. Palkovits, ACS Sustainable Chem. Eng., 2018, 6, 17108–17113 CrossRef CAS.
- F. J. Holzhäuser, G. Creusen, G. Moos, M. Dahmen, A. König, J. Artz, S. Palkovits and R. Palkovits, Green Chem., 2019, 21, 2334–2344 RSC.
- G. H. M. de Kruijff, T. Goschler, L. Derwich, N. Beiser, O. M. Türk and S. R. Waldvogel, ACS Sustainable Chem. Eng., 2019, 7, 10855–10864 CrossRef CAS.
- J. D. Wadhawan, F. J. Del Campo, R. G. Compton, J. S. Foord, F. Marken, S. D. Bull, S. G. Davies, D. J. Walton and S. Ryley, J. Electroanal. Chem., 2001, 507, 135–143 CrossRef CAS.
- J. D. Wadhawan, F. Marken, R. G. Compton, S. D. Bull and S. G. Davies, Chem. Commun., 2001, 87–88 RSC.
- A. Kapałka, B. Lanova, H. Baltruschat, G. Fóti and C. Comninellis, J. Electrochem. Soc., 2008, 155, E96–E100 CrossRef.
- C. G. Law Jr., P. S. Fedkiw and M. T. Hicks, US6238543B1, 2001.
- D. Koutsaftis, D. Marinis and A. Karantonis, Electrochim. Acta, 2012, 59, 376–381 CrossRef CAS.
- Y. Peng, Y. Ning, X. Ma, Y. Zhu, S. Yang, B. Su, K. Liu and L. Jiang, Adv. Funct. Mater., 2018, 28, 1800712 CrossRef.
- V. N. Andreev, V. I. Bykov, V. A. Grinberg, A. G. Dedov, A. S. Loktev, N. A. Mayorova, I. I. Moiseev and A. A. Stepanov, Russ. J. Electrochem., 2013, 49, 216–220 CrossRef CAS.
- C. Stang and F. Harnisch, ChemSusChem, 2016, 9, 50–60 CrossRef CAS PubMed.
- C. H. Joshi, M. G. Horner, G. T. T. Gibson and D. Malevich, US8961775B2, 2015.
- B. F. Gomes, F. J. Holzhäuser, C. M. S. Lobo, P. Ferreira da Silva, E. P. Danieli, M. Carmo, L. A. Colnago, S. Palkovits, R. Palkovits and B. Blümich, ACS Sustainable Chem. Eng., 2019 DOI:10.1021/acssuschemeng.9b02768.
- Y. Okada, K. Kamimura and K. Chiba, Tetrahedron, 2012, 68, 5857–5862 CrossRef CAS.
- Oak Ridge National Lab, Materials for Separation Technologies. Energy and Emission Reduction Opportunities, USA, 2005 Search PubMed.
- J. L. Humphrey and G. E. Keller, Separation process technology, McGraw-Hill, New York, USA, 1997 Search PubMed.
- Y. Zhang, G. Liu and J. Wu, J. Electroanal. Chem., 2018, 822, 73–80 CrossRef CAS.
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T.-Y. Wu, W. Higashide, P. Malati, Y.-X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596–1596 CrossRef CAS PubMed.
- C. Gimkiewicz, R. Hegner, M. F. Gutensohn, C. Koch and F. Harnisch, ChemSusChem, 2017, 10, 958–967 CrossRef CAS PubMed.
- K. Watts, W. Gattrell and T. Wirth, Beilstein J. Org. Chem., 2011, 7, 1108–1114 CrossRef CAS PubMed.
- K. Arai, K. Watts and T. Wirth, ChemistryOpen, 2014, 3, 23–28 CrossRef CAS PubMed.
- T. Tajima, H. Kurihara and T. Fuchigami, J. Am. Chem. Soc., 2007, 129, 6680–6681 CrossRef CAS PubMed.
- H. Kurihara, T. Fuchigami and T. Tajima, J. Org. Chem., 2008, 73, 6888–6890 CrossRef CAS PubMed.
- A. Singh, N. Singhal, H. Agrawal and A. K. Yadav, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2002, 41, 423–426 Search PubMed.
- A. K. Yadav and A. Singh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2002, 41, 1724–1726 Search PubMed.
- J.-J. Dai, Y.-B. Huang, C. Fang, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 617–620 CrossRef CAS PubMed.
- H. Lin, J. Niu, S. Ding and L. Zhang, Water Res., 2012, 46, 2281–2289 CrossRef CAS PubMed.
- J. Niu, H. Lin, J. Xu, H. Wu and Y. Li, Environ. Sci. Technol., 2012, 46, 10191–10198 CrossRef CAS PubMed.
- N. Ahad and A. de Klerk, Fuel, 2018, 211, 415–419 CrossRef CAS.
- M. Dierker, WO2006094642A1, 2006.
- D. Bradin, US8241881B2, 2012.
- D. Bradin, WO2007027669A1, 2007.
- K. Wang and L. Tan, WO2014137534A2, 2014.
- D. Bradin and G. L. Grune, US8481771B2, 2013.
- C. H. Joshi and M. G. Horner, US20120197050A1, 2012.
- I. Cabasso, M. Li and Y. Yuan, RSC Adv., 2012, 2, 9998–10006 RSC.
- D. Bradin, WO2007095215A3, 2007.
- G. A. G. Pereira, J. Rincones Perez, M. Falsarella Carazzolle, A. L. Ribeiro de Castro Morschbacker, L. Roza and M. H. dos Santos Andrade, US20130203953A1, 2013.
- T. Haas, U. Paulmann and S. Beck, WO2016008979A1, 2016.
- J.-Y. Dupre and O. Y. Rodriguez, WO2014170603A1, 2014.
- A. B. Kuhry and P. J. Weimer, US9663864B2, 2017.
- T. M. Rangarajan, D. Velayutham and M. Noel, Ionics, 2011, 17, 827–833 CrossRef CAS.
- J. A. Stapley and J. N. BeMiller, Carbohydr. Res., 2007, 342, 610–613 CrossRef CAS PubMed.
- L. Wu, M. Mascal, T. J. Farmer, S. P. Arnaud and M.-A. Wong Chang, ChemSusChem, 2017, 10, 166–170 CrossRef CAS PubMed.
- T. R. dos Santos, F. Harnisch, P. Nilges and U. Schröder, ChemSusChem, 2015, 8, 886–893 CrossRef CAS PubMed.
- S. Palkovits and R. Palkovits, Chem. Ing. Tech., 2019, 91, 699–706 CrossRef CAS.
- P. F. Levy, J. E. Sanderson, E. Ashare, D. L. Wise and M. S. Molyneaux, Liquid fuels production from biomass. Final report, Argonne, USA, 1980 Search PubMed.
- J. Leclaire and D. J. Heldebrant, Green Chem., 2018, 20, 5058–5081 RSC.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- M. M.-J. Li and S. C. E. Tsang, Catal. Sci. Technol., 2018, 8, 3450–3464 RSC.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- A. Kuenz, Y. Gallenmüller, T. Willke and K.-D. Vorlop, Appl. Microbiol. Biotechnol., 2012, 96, 1209–1216 CrossRef CAS PubMed.
- T. Willke and K.-D. Vorlop, Appl. Microbiol. Biotechnol., 2001, 56, 289–295 CrossRef CAS PubMed.
- J. Gorden, E. Geiser, N. Wierckx, L. M. Blank, T. Zeiner and C. Brandenbusch, Eng. Life Sci., 2017, 17, 809–816 CrossRef CAS.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I - Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Golden, USA, 2004 Search PubMed.
- H. R. Beller, T. S. Lee and L. Katz, Nat. Prod. Rep., 2015, 32, 1508–1526 RSC.
- S. Bhavaraju, M. Karanjikar, A. V. Joshi, D. J. Hunt and P. Chitta, WO2011011492A3, 2011.
- J. Mosby, S. Bhavaraju and M. Karanjikar, WO2014138631A1, 2014.
| This journal is © The Royal Society of Chemistry 2020 |