
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable catalytic protocols for the solvent free epoxidation and anti-dihydroxylation of the alkene bonds of biorenewable terpene feedstocks using H2O2 as oxidant†‡
William B.
Cunningham
ab,
Joshua D.
Tibbetts
 ab,
Marc
Hutchby
ab,
Katarzyna A.
Maltby
bc,
Matthew G.
Davidson
ab,
Ulrich
Hintermair
ab,
Pawel
Plucinski
bc and
Steven D.
Bull
*a
ab,
Marc
Hutchby
ab,
Katarzyna A.
Maltby
bc,
Matthew G.
Davidson
ab,
Ulrich
Hintermair
ab,
Pawel
Plucinski
bc and
Steven D.
Bull
*a
aDepartment of Chemistry, University of Bath, Bath, BA27AY, UK
bCentre for Sustainable Chemical Technologies, University of Bath, BA27AY, UK
cDepartment of Chemical Engineering, University of Bath, Bath, BA27AY, UK. E-mail: s.d.bull@bath.ac.uk
First published on 26th November 2019
Abstract
A tungsten-based polyoxometalate catalyst employing aqueous H2O2 as a benign oxidant has been used for the solvent free catalytic epoxidation of the trisubstituted alkene bonds of a wide range of biorenewable terpene substrates. This epoxidation protocol has been scaled up to produce limonene oxide, 3-carene oxide and α-pinene oxide on a multigram scale, with the catalyst being recycled three times to produce 3-carene oxide. Epoxidation of the less reactive disubstituted alkene bonds of terpene substrates could be achieved by carrying out catalytic epoxidation reactions at 50 °C. Methods have been developed that enable direct epoxidation of untreated crude sulfate turpentine to afford 3-carene oxide, α-pinene oxide and β-pinene oxide. Treatment of crude epoxide products (no work-up) with a heterogeneous acid catalyst (Amberlyst-15) results in clean epoxide hydrolysis to afford their corresponding terpene-anti-diols in good yields.
Introduction
The development of efficient processes that enable biorenewable feedstocks to be converted into fine chemicals, drugs and polymers that are currently sourced from petroleum feedstocks is a key challenge for the 21st century. Consequently, much attention has been focused on the use of biopolymers (e.g. lignocellulose, chitin, etc.) as potential biorenewable feedstocks for sustainable chemical production.1 However, use of oxygenated biopolymers as chemical feedstocks currently requires the use of chemical/microbial hydrolytic processes to enable their efficient depolymerization.2 Separation methods are then required to purify complex mixtures of highly oxygenated products, which can be difficult to achieve economically on a large scale. Most of the oxygen-rich bioproducts produced are incompatible with industrial catalytic technologies used to upgrade the aryl and alkene functionalities of hydrocarbon-based petrochemical feedstocks. This means that further microbial fermentation is required to convert these oxygenated products into relatively low value organic products (e.g. fermentation of glucose into bioethanol).3 Alternatively, new catalytic transformations (e.g. catalytic deoxydehydration reactions) have been developed to access new chemical products (e.g. conversion of fructose into 5-hydroxymethyl-furfural) for which new chemical supply chains need to be developed.4 However, significant technological barriers still need to be overcome and further investment will be required before oxygenated biopolymers become established as replacement biorenewable feedstocks for the industrial synthesis of chemical products.Monoterpenes (and monoterpenoids) represent a large and diverse set of secondary metabolites produced by plants, insects and microbes that exhibit a wide range of biological activity. They are produced from reactions of C5 isoprene-containing precursors that afford lightly oxygenated unsaturated acyclic/cyclic C10 frameworks containing one or more alkene functionalities.5 The hydrocarbon nature of terpenes means they are easy to purify/derivatize using existing petrochemical technologies and so can be considered as potentially attractive non-polymeric biorenewable feedstocks for the sustainable synthesis of chemical products.6 Global biogenic production of terpenes is estimated to be around 109 tons per year, although most terpenes are currently only available in relatively small amounts and so are not suitable as bulk feedstocks for industrial chemical synthesis. However, large volumes of a defined sub-set of bulk monoterpene feedstocks (e.g. α-pinene, β-pinene, 3-carene and limonene) are currently available as low-cost waste by-products of the agricultural and forestry industries.5
An estimated annual volume of around 330![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of terpene feedstocks are produced worldwide,7 which is sufficient quantity to consider using terpenes as biorenewable feedstocks for the sustainable synthesis of selected chemical products. Turpentine represents the cheapest and largest volume monoterpene feedstock available that is produced by the forestry and paper industries. Annual worldwide turpentine production ranges between 300000–310000 tonnes,8 with turpentine comprised of α-pinene and β-pinene, and different amounts of other terpenes, such as 3-carene, camphene, (−)-limonene and terpinolene, whose ratios are dependent on the geographical origin of the tree extracts.9
000 tonnes of terpene feedstocks are produced worldwide,7 which is sufficient quantity to consider using terpenes as biorenewable feedstocks for the sustainable synthesis of selected chemical products. Turpentine represents the cheapest and largest volume monoterpene feedstock available that is produced by the forestry and paper industries. Annual worldwide turpentine production ranges between 300000–310000 tonnes,8 with turpentine comprised of α-pinene and β-pinene, and different amounts of other terpenes, such as 3-carene, camphene, (−)-limonene and terpinolene, whose ratios are dependent on the geographical origin of the tree extracts.9
Crude sulfate turpentine (CST) is the cheapest source of turpentine available in bulk quantities (two thirds of world supply, $220 per metric tonne),8,9 which is produced as a by-product of the Kraft process used to produce pulp for paper production.10 Current applications of CST involve burning it as a fuel to provide energy for the paper mill, or carrying out desulfurization/processing to enable it to be used as paint-thinner or cleaning fluids (e.g. α-terpineol). Alternatively, fractional distillation can be carried out to afford its individual terpene components (e.g. α-pinene, β-pinene and 3-carene) that can then be sold as biorenewable feedstocks for the synthesis of value-added products (e.g. flavours and fragrances). The remainder of the turpentine that is commercially available is gum turpentine, which is produced from distillation of resin obtained from the renewable tapping of tree plantations (mainly pine), which currently retails at around $1000–1500 per tonne.
Other commercial bulk terpene sources include (+)-limonene which is available in smaller volumes as a by-product of waste fruit peel from the citrus juice industry. Global production levels of (+)-limonene are currently estimated to be around 70000 tonnes per year, which retails at a more volatile price of between $3300–10000 per tonne.11 Promising industrial biotechnology developments have recently revealed the potential of synthetic biology to engineer metabolic pathways into microbes/plants for the industrial production of economically important higher terpenes such as farnesene and squalene.12 Monoterpenes are relatively toxic to microbial cells, which has typically limited their maximum titre levels to around 1 g L−1 or less, which potentially limits their commercial production on a large scale. However, a recent report has described the creation of a recombinant bacteria expressing a metabolic pathway incorporating 27 enzymes that can transform glucose into α-pinene (or limonene) at titres of up to 15 g L−1.13 The impact of these biotechnological processes is potentially significant, because they can potentially provide geographically flexible processes that enable low-cost lignocellulosic waste streams to be converted into terpene based feedstocks on a large scale.
The volumes and prices of commercial turpentine (and derived products) supplies suggest that a scalable biorefinery industry based on biorenewable terpene feedstocks is potentially feasible.8 Terpenes and terpenoids are already widely used for the production of flavours, fragrances, cleaning agents and medicinally active compounds,5 with the widespread use of α-pinene and β-pinene as biorenewable feedstocks for the synthesis of camphor, menthol and rose oil particularly noteworthy. Monoterpenes have often been used as chiral building blocks for impressive natural product and drug syntheses, albeit on a comparatively small scale.14 However, many of the reactions used to transform terpenes into more complex high-value products do not conform to the principles of green chemistry, with many of these transformations relying on waste generating reactions, stoichiometric reagents, expensive catalysts and environmentally unfriendly solvents.14 Therefore, a concerted effort to identify sustainable catalytic transformations that can be used to transform biorenewable monoterpene feedstocks into value-added chemical products on a large scale is urgently required.15
The unsaturated hydrocarbon nature of monoterpenes means that access to catalytic oxidative protocols that enable selective functionalization of their alkene functionalities is necessary. Epoxides are widely used as privileged functional groups in organic synthesis, with many regio- and stereoselective protocols available for their transformation into a wide range of functional groups.16 Indeed, the synthetic utility of many terpene epoxides has already been demonstrated, including their use as chiral building blocks for the synthesis of a wide range of fragrances, natural products, polymers, vitamins, pharmaceuticals, insecticides and natural products.17 However, the development of scalable epoxidation reactions of terpene substrates is challenging, because terpenes (and their epoxides) readily undergo competing ring-opening, rearrangement and hydrolysis reactions to give unwanted product mixtures that are difficult to separate.18 Alkene selectivity is also an important issue, because many key terpene substrates (e.g. limonene) contain trisubstituted and disubstituted alkenes that often need to be selectively epoxidized. Therefore, the development of sustainable catalytic protocols that enable the selective epoxidation of terpene feedstocks on a large scale is challenging, particularly when the catalytic epoxidation process must be capable of tolerating the multiple sulfur compounds (e.g. Me2S, Me2S2) present in untreated CST.
Polyoxometalate (POM) catalysts and H2O2 have been widely used in catalytic epoxidation reactions, because these protocols exhibit low peroxide disproportionation that maximize oxygen atom transfer from H2O2 to the alkene substrate.19 In this respect, tungsten oxide based Venturello phase transfer catalysts (VPTCs) have previously been used to epoxidize trisubstituted, disubstituted and monosubstituted alkenes in a variety of organic solvents.20 These easily prepared catalysts contain lipophilic quaternary counter-ions that facilitate alkene epoxidation at the interface of biphasic aqueous–organic mixtures. Their catalytic epoxidation reactions exhibit similar reactivity profiles to those of stoichiometric peracid epoxidation reactions, with more substituted alkenes normally being epoxidized from their least hindered face. These VPTC systems have been shown to exhibit good functional group compatibility, use H2O2 as a cheap, environmentally friendly oxidant with the use of immobilised catalysts potentially allowing for catalyst recycling.19,21 Consequently, VPTC based systems represent a highly promising candidate for the development of catalytic epoxidation protocols for biorenewable terpene substrates within a biorefinery context.
VPTCs have previously been used for catalytic epoxidation of the alkene bonds of a range of terpene substrates to give their corresponding epoxides in acceptable to good yields.21 These previous terpene epoxidation reactions have employed a wide range of conditions, including the use of homogeneous (preformed and in situ generated) and heterocyclic catalysts, varying catalyst loadings ranging from 1–8 mol%, 30–50% solutions of H2O2 of varying pH (3.0–7.0), different temperatures (rt–90 °C), varying reaction times (1.5–72 h), and different organic co-solvents (e.g. benzene, toluene, CHCl3, t-BuOH, acetone). Furthermore, a number of reports have also described the use of additives [e.g. NH2CH2PO3H2 (1–2 mol%), PhP(O)(OH)2 (4–10 mol%), Na2SO4 (30 mol%), imidazole (5 mol%)] to increase the rate of epoxidation and/or prevent unwanted acid catalysed hydrolysis of terpene epoxides into their corresponding anti-diols.21–28 However, a systematic study to identify optimal conditions for the VPTC mediated epoxidation of a wide range of terpene substrates has not been carried out. Building on previous literature precedent, we now report the development of practically simple and broadly applicable solvent-free epoxidation protocols that employ a cheap preformed homogeneous tungsten based VPTC for the selective epoxidation of the trisubstituted alkene bonds of a wide range of biorenewable terpene/terpenoid substrates. These sustainable epoxidation conditions have been scaled up to safely produce limonene oxide, 3-carene oxide and α-pinene oxide on multigram scales, with recycling studies showing that the catalyst can be recycled three times for the solvent free production of 3-carene oxide. Use of elevated temperatures (up to 50 °C) enables epoxidation of terpenes containing less electron-rich disubstituted alkene functionalities, which enabled both alkene bonds of limonene to be epoxidized in good yield. Optimal VPTC/H2O2 epoxidation conditions have been applied for the first direct epoxidation of the major components of crude CST (non-desulfurized) to afford mixtures of 3-carene oxide, α-pinene oxide and β-pinene oxide that could be separated by distillation. Finally, treatment of crude epoxide mixtures with a heterogeneous Lewis acid catalyst (Amerlyst-15) has enabled a tandem ‘one-pot’ stepwise catalytic epoxidation/epoxide hydrolysis protocol to be developed that enables Prilezhaev dihydroxylation of the alkene bonds of terpenes to give their corresponding anti-diols in good yield.
Results and discussion
Solvent free VPTC/H2O2 mediated epoxidation of the alkene bonds of biorenewable terpenes and CST
Our goal was to develop practically simple protocols that used preformed VPTC/H2O2 system for the scalable catalytic epoxidation of a wide range of terpenes, including untreated CST. Consequently, a preformed VPTC was first prepared by stirring a solution of tungstic acid in 30 wt% aqueous H2O2 at 60 °C for 1.5 h, followed by cooling to rt and addition of 85% orthophosphoric acid. A solution of Aliquat 336 [mixture of MeN+(C8H17)3Cl− and MeN+(C10H21)3Cl−, £0.08 per mL (Merck), phase transfer catalyst (PTC)] in CH2Cl2 was then added and the resultant mixture stirred vigorously for 1 h at rt, before the organic phase was concentrated under vacuum to give a Venturello-A336 catalyst (PW4O24[PTC]3) as a viscous, transparent yellow syrup in 70% yield on a multigram scale.29Limonene was chosen as a model terpene for the initial reaction optimisation phase,28 because it enabled the selectivity of the VPTC for epoxidation of its trisubstituted alkene over its disubstituted alkene to be determined. 1 mol% of PW4O24[PTC]3 was dissolved in neat limonene, followed by dropwise addition of 1.0 equiv. of 30% aqueous H2O2 (pH 4.0) and vigorous stirring of the resultant biphasic reaction mixture at rt. This resulted in complete consumption of limonene after 1 h, affording a ∼1:1 mixture of diastereomeric 1,2-limonene oxides 1a/1b in 71% yield, along with 25% of the corresponding limonene-anti-1,2-diols (1:1 mixture of diastereomers), and a small amount (<5%) of limonene bis-epoxide (mixture of all four stereoisomers). The unwanted 1,2-diols formed in this reaction are formed by competing hydrolysis of their corresponding 1,2-epoxides 1a/1b, which we subsequently discovered was caused by the acidic nature of commercial 30% aqueous H2O2 (pH 3.0–4.0). Consequently, the pH of the hydrogen peroxide solution was preadjusted to pH 7.0 using 0.5 M NaOH solution, and the catalytic epoxidation reaction repeated to give a 57:43 mixture of 1,2-limonene oxides 1a/b, with <5% of their corresponding diols now present (Scheme 1). Isolation of the crude reaction product was achieved by simply decanting off the upper organic layer of the biphasic reaction which 1H NMR spectroscopic analysis revealed was comprised of limonene oxides 1a/b (containing 1 mol% catalyst) in >95% purity. The crude limonene oxides 1a/b were then purified by chromatography (or distillation in vacuo) to afford pure limonene-1,2-oxide 1a/b in 94% isolated yield (average of 5 repeat epoxidation reactions),30 with no diol or bis-epoxide contaminants present.
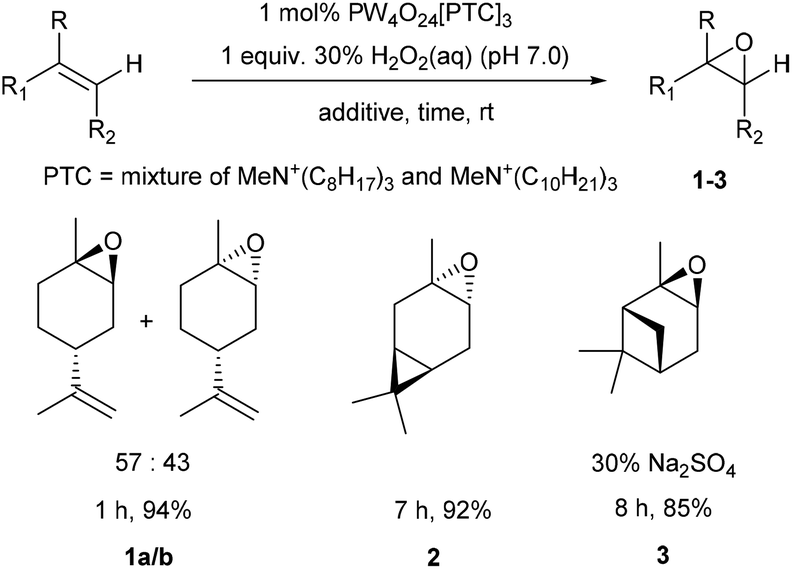 | ||
| Scheme 1 Solvent free catalytic epoxidation of the bulk commodity terpenes – limonene, 3-carene and α-pinene. | ||
We then explored application of this solvent free protocol for the epoxidation of the trisubstituted alkene functionalities of the bulk commodity terpenes 3-carene and α-pinene that are major components of CST. 3-Carene was cleanly epoxidized using 1 mol% VPTC and 30% H2O2 (pH 7.0) under solvent free conditions at rt to afford 3-carene oxide 2 as a single diastereomer in 92% isolated yield after 7 h (Scheme 1).31 Epoxidation reactions of α-pinene are known to be potentially problematic, because its epoxide can undergo competing acid catalysed rearrangement, fragmentation and hydrolysis reactions that result in low yields of α-pinene oxide 3. Indeed, epoxidation of α-pinene using the standard conditions developed for limonene proved unsuccessful, affording complex mixtures of α-pinene and anti-diols, with no α-pinene oxide 3 present. Sato and coworkers, had previously reported that addition of 0.3 equiv. of Na2SO4 to VPTC mediated epoxidation protocol gave good yields of α-pinene oxide 3, suggesting that competing epoxide hydrolysis at the biphasic interface is minimized by the increased ionic strength of the aqueous layer.26 Therefore, we repeated the catalytic epoxidation reaction of α-pinene in the presence of 0.3 equiv. of Na2SO4 which resulted in clean epoxidation to afford α-pinene oxide 3 as a single diastereomer after 8 h in 85% isolated yield (Scheme 1).32 The excellent diastereoselectivities observed for epoxidation of both 3-carene and α-pinene are consistent with both their alkene bonds being epoxidized from their least-hindered faces.
The epoxidation reactions of limonene, α-pinene and 3-carene were then scaled up to demonstrate the potential of using these protocols for the large-scale synthesis of bulk terpene epoxides containing trisubstituted alkenes. Preliminary scale-up reactions revealed that our solvent free epoxidation conditions were associated with a potentially dangerous exotherm that could result in a dangerous delayed onset thermal runaway reaction occurring. However, modification of the experimental conditions enabled safe solvent free epoxidation of limonene to be carried out on a larger scale. This involved slow dropwise addition of a 30% H2O2 solution (pH 7.0) to a rapidly stirred solution of limonene (500 rpm) containing 1 mol% catalyst, whilst ensuring that the temperature of the reaction was maintained between 35 and 50 °C through external cooling. This modified protocol enabled the safe and reproducible solvent free epoxidation of 100 g of limonene to afford 95 g of limonene oxide 1 after 2 h in 85% isolated yield. This scaled-up procedure was then used to epoxidize 15 g of 3-carene to afford 11.4 g of 3-carene oxide 2 in 76% isolated yield and epoxidise 15 g of α-pinene (2 mol% catalyst, 30 mol% Na2SO4) to afford 12.8 g of α-pinene-oxide 3 in 85% isolated yield. A representative range of catalytic epoxidation reactions of limonene using H2O2 as an oxidant are shown in Table 1. These results clearly show that our catalytic solvent and additive free system affords higher yields of limonene oxide in shorter reaction times.28,33
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | H2O2 equiv. | Solv. | Additive | Time (h) | Yield | Ref. |
| PW4O24 (preformed) | 1 | — | — | 1 | 94% | This work |
| PW4O24 (in situ) | 1 | — | Na2SO4 | 1 | 73% | 28 |
| PW4O24 (in situ) | 1 | Limonene (300 mol%) | Na2SO4 | 0.25 | 95% | 28 |
| FeIII cat. | 6 | Acetone | — | 24 | 78% | 33a |
| W-SiO2 | 2 | MeCN | — | 6 | 65% | 33b |
| PW9 on Si | 24 | MeCN | — | 24 | 54% | 33c |
| Candida arctica lipase | 6 | PhMe | Octanoic acid | 1 | 78% | 33d |
| Eco-Mn® | 3 | DMF | NaHCO3 | 4 | 43% | 33e |
| Mn4(PW9)2 | 2 | MeCN | — | 2 | 23% | 33f |
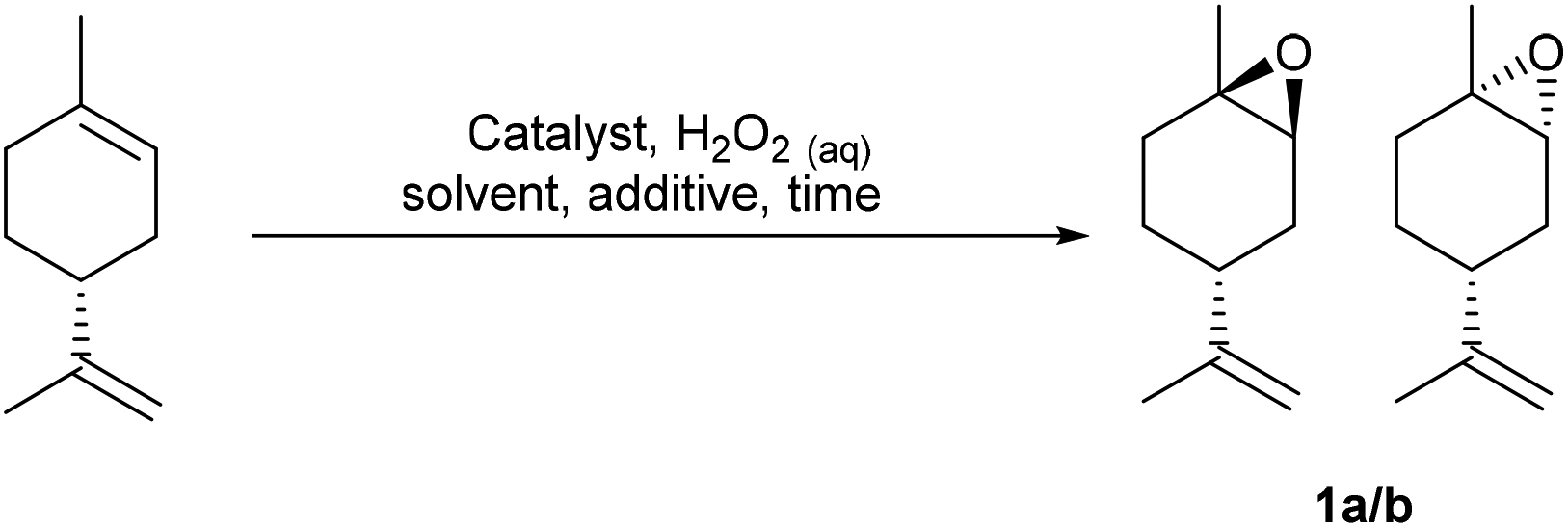
The potential of these protocols to catalyse epoxidation of the trisubstituted alkene bonds of a range of other naturally occurring cyclic monoterpene alcohols was then investigated (Scheme 2). Perillyl alcohol was monoepoxidized to give a clean mixture of its corresponding α-/β-epoxides 4a/b in a 58:42 ratio after 2 h in 71% yield, with no epoxidation of its terminal alkene or oxidation of its primary alcohol occurring. Similarly, myrtenol was cleanly epoxidized (30 mol% Na2SO4) to give a single diastereomeric epoxide 5 after 2 h in 70% yield, with epoxidation occurring exclusively on the face opposite to its more bulky 1,1-dimethyl substituent. The homoallylic alkene bond of carvomenthenol was successfully epoxidized to give a mixture of epoxide diastereomers 6a/b in 77% yield, with the 90:10 selectivity for the cis epoxide due to the directing effect of the tertiary alcohol group. Attempts to epoxidize citronellol under these conditions resulted in a low 35% yield of its corresponding epoxides, however, epoxidation of O-acetyl-citronellol was more successful, affording a 50:50 mixture of epoxides 7a/b in an improved 55% isolated yield. This mixture of epoxides 7a/b could then be methanolyzed (5 equiv. K2CO3, MeOH, 12 h) to afford a 50:50 mixture of their corresponding deprotected citronellol oxides in 80% yield.
 | ||
| Scheme 2 Solvent free catalytic epoxidation of the trisubstituted alkene bonds of a series of cyclic and acyclic terpene alcohols/esters. | ||
Our standard catalytic epoxidation protocol was then applied to a series of monoterpene, sesquiterpene, diterpene and triterpene substrates that contain multiple alkene bonds (Scheme 3). The non-conjugated trisubstituted alkene bond of acyclic myrcene was epoxidized to give the monoepoxide (rac)-8 in 82% yield after 3 h, with its less reactive synthetically useful diene fragment remaining intact. Similarly, selective epoxidation of the fused tricyclic alkene bond of the sesquiterpene valencene gave a 76:24 mixture of exocyclic and endocyclic monoepoxides 9a/b after 24 h, with no epoxidation of its isopropenyl group observed. The non-conjugated trisubstituted alkene functionality of (rac)-α-ionone was selectively epoxidized at 50 °C to afford an 80:20 mixture of its α-/β-mono-epoxides (rac)-10a/b in 79% yield after 4 h. The more electron-rich tetra-substituted γ,δ-alkene of β-ionone was selectively epoxidized at 50 °C to afford its (rac)-epoxide 11 in 84% yield. Use of two equiv. of H2O2 enabled both trisubstituted alkene functionalities of γ-terpinene to be simultaneously epoxidized to afford (rac)-cis–bis-epoxide 12 in 86% yield and >90% de. In this case, unselective epoxidation of one of the trisubstituted alkene bonds of γ-terpinene affords two mono-epoxide intermediates, whose oxygen atoms then direct epoxidation of their remaining trisubstituted alkenes to the same face. The catalytic epoxidation protocol was then used for the epoxidation of farnesene and squalene, because both these terpenes are currently produced on a large scale using industrial biotechnology processes.34,35 Use of 2.0 equiv. of H2O2 resulted in both trisubstituted alkene bonds of farnesene being selectively epoxidized to afford a 50:50 mixture of its diastereomeric bis-epoxides 13a/b in 86% yield after 18 h, with its synthetically valuable diene fragment untouched.
 | ||
| Scheme 3 Solvent free catalytic epoxidation of the trisubstituted alkene bonds of a range of biorenewable terpene substrates. | ||
Even more impressively, treatment of squalene with 1 mol% Ishii–Venturello catalyst and 6 equiv. of H2O2 under solvent free conditions gave a mixture of diastereomeric hexa-epoxides 14 in 89% yield after 12 h. Global epoxidation of all the alkene bonds of squalene was confirmed by HPLC-MS analysis that revealed the presence of a single broad peak (multiple hexaepoxy diastereomers) with a correct HPLC-HRMS molecular ion of 529.3628 m/z.
We next turned our attention to determining whether use of higher temperatures might enable epoxidation of terpenes containing less reactive disubstituted alkenes (Scheme 4). Epoxidation of dihydrocarvone (4:1 mixture of trans:cis isomers) using 1 mol% catalyst and 1 equiv. of H2O2 at 50 °C for 2 h, resulted in formation of a 10:10:1:1 mixture of four diastereomeric epoxides 15a–d in 71% isolated yield. The disubstituted alkene bond of isopulegol was epoxidised at 50 °C to give a 60:40 mixture of diastereomeric epoxides 16a/b in 63% yield after 1 h, whilst acyclic dihydromyrcenol was epoxidised at 50 °C to afford a 50:50 ratio of its corresponding diastereomeric epoxides 17a/b in 75% yield after 4 h.
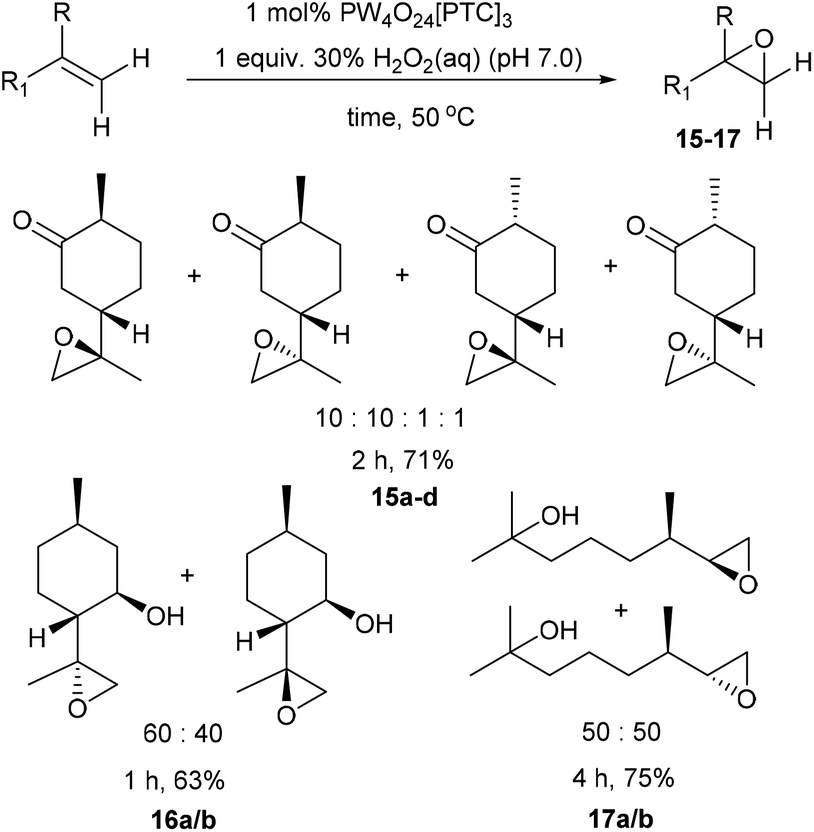 | ||
| Scheme 4 Solvent free catalytic epoxidation of the disubstituted alkene bonds of a series of monoterpene substrates. | ||
Attempts to epoxidise the bulk monoterpene β-pinene were less successful, due to the lower reactivity of its sterically hindered disubstituted alkene bond and the propensity of β-pinene oxide 18 to undergo facile ring-opening reactions.36 For example, treatment of β-pinene with 3 mol% VPTC and 1 equiv. of 30% H2O2 at 20 °C in the presence 30 mol% Na2SO4 for 7 h on a 1 g scale, only gave β-pinene oxide in 47% isolated yield (Scheme 5).37 The mass balance from this epoxidation reaction was found to be a complex mixture of by-products, with previous unsuccessful β-pinene epoxidation reactions reported to produce up to 9 possible by-products (e.g. pinocarveol).38 Consequently, it was decided to modify the epoxidation conditions to try and increase the rate of epoxidation of β-pinene and minimize hydrolysis of β-pinene-oxide 18 at the aqueous interface. Gratifyingly, use of 3 mol% VPTC, 1 equiv. of a more concentrated commercial 50% H2O2 solution, increasing the temperature of the epoxidation reaction to 35 °C and employing 2 equiv. of toluene as an organic cosolvent resulted in β-pinene epoxide 18 being produced to a more satisfactory 62% isolated yield after only 2 h.
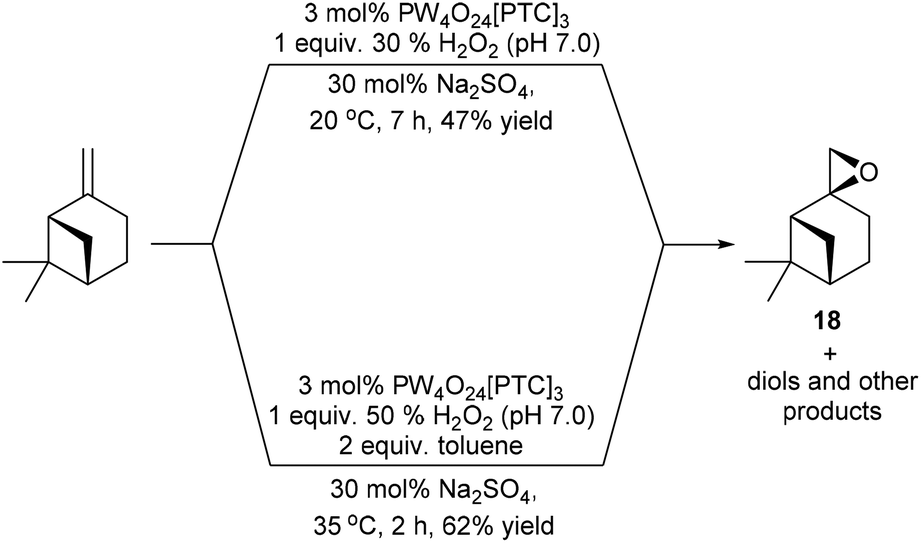 | ||
| Scheme 5 Catalytic epoxidation of β-pinene using modified conditions designed to reduce ring-opening of β-pinene oxide 18. | ||
Limonene has proven to be a popular terpene feedstock for the production of a range of biorenewable polymers, with 1,2-limonene oxide 1a/1b having been copolymerised with carbon dioxide for the production of polycarbonates, or copolymerised with cyclic anhydrides for the production of polyesters.30 The use of limonene bis-epoxide 19 as a bifunctional monomer for biopolymer synthesis is less well explored, however it has recently been reacted with carbon dioxide to afford cyclic bis-carbonates that were polymerised to afford biorenewable polyurethanes and polycarbamates.39,40 Attempts to catalyse the simultaneous epoxidation of both alkene bonds of limonene using our standard mono-epoxidation conditions for longer periods of time (or at higher temperatures) proved unsuccessful, affording complex mixtures of epoxides, diols and tetrol products. However, use of the modified conditions developed to epoxidize β-pinene (50% H2O2, 30 mol% Na2SO4, toluene) resulted in initial formation of a mixture of diastereomeric 1,2-limonene mono-epoxides 1a/1b after 20 minutes, with further epoxidation of their disubstituted alkenes then occurring to afford limonene bis-epoxide 19 (mixture of all four possible stereoisomers) after 7 h in 69% isolated yield (Scheme 6).
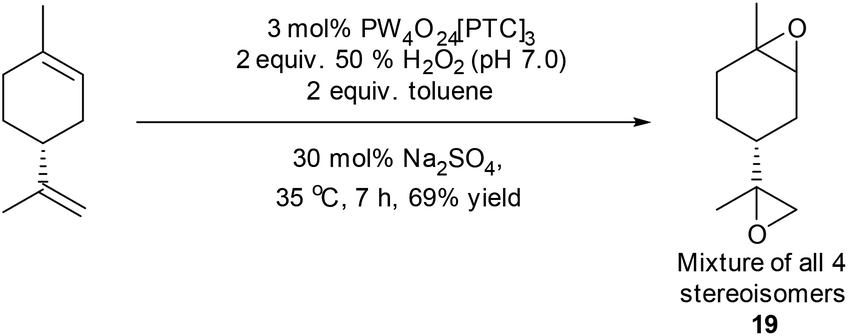 | ||
| Scheme 6 Modified conditions applied to epoxidation of both alkenes in limonene to afford bis-epoxide 19. | ||
We then decided to challenge our catalytic protocols for the direct catalytic epoxidation of the major terpene components of untreated CST, which would require alkene epoxidation in the presence of multiple sulfur contaminants (e.g. Me2S) that are known to deactivate many metal-based catalysts. Non-desulfurized industrial grade CST was obtained from the Södra Forestry Cooperative based in Southern Sweden, whose composition was analysed by 1H NMR spectroscopy and GCMS analysis. The major terpene components of this CST were found to be 49% α-pinene, 39% 3-carene and 12% β-pinene, which is a typical terpene distribution for North European sources of CST that generally exhibit a high 3-carene content. Treatment of this industrial CST with 1 mol% of VPTC and 1.5 equiv. of neutralised 30 wt% H2O2 (pH 7.0) under solvent free conditions at rt resulted in 90% consumption of its three major terpene components after 5 h. Analysis revealed that its 3-carene fraction had been converted into 3-carene oxide 2, whilst α- and β-pinene had been transformed into their corresponding diols (and other ring-opened products). The crude product (no sulfurous odour) was purified by fractional distillation (or chromatography) to afford 3-carene oxide 2 (bp 87 °C at 50 mm Hg) in 30% overall yield (77% theoretical yield) (Scheme 7a). Repeating this CST epoxidation reaction using 1 mol% of catalyst with 1.5 equiv. of 30 wt% H2O2 (pH 7.0) and 30 mol% Na2SO4 for 5 h gave 36% (92% theoretical) and 37% yields (76% theoretical) of 3-carene oxide and α-pinene oxide, respectively, with all the β-pinene component converted into its corresponding diol (and other products) (Scheme 7b).
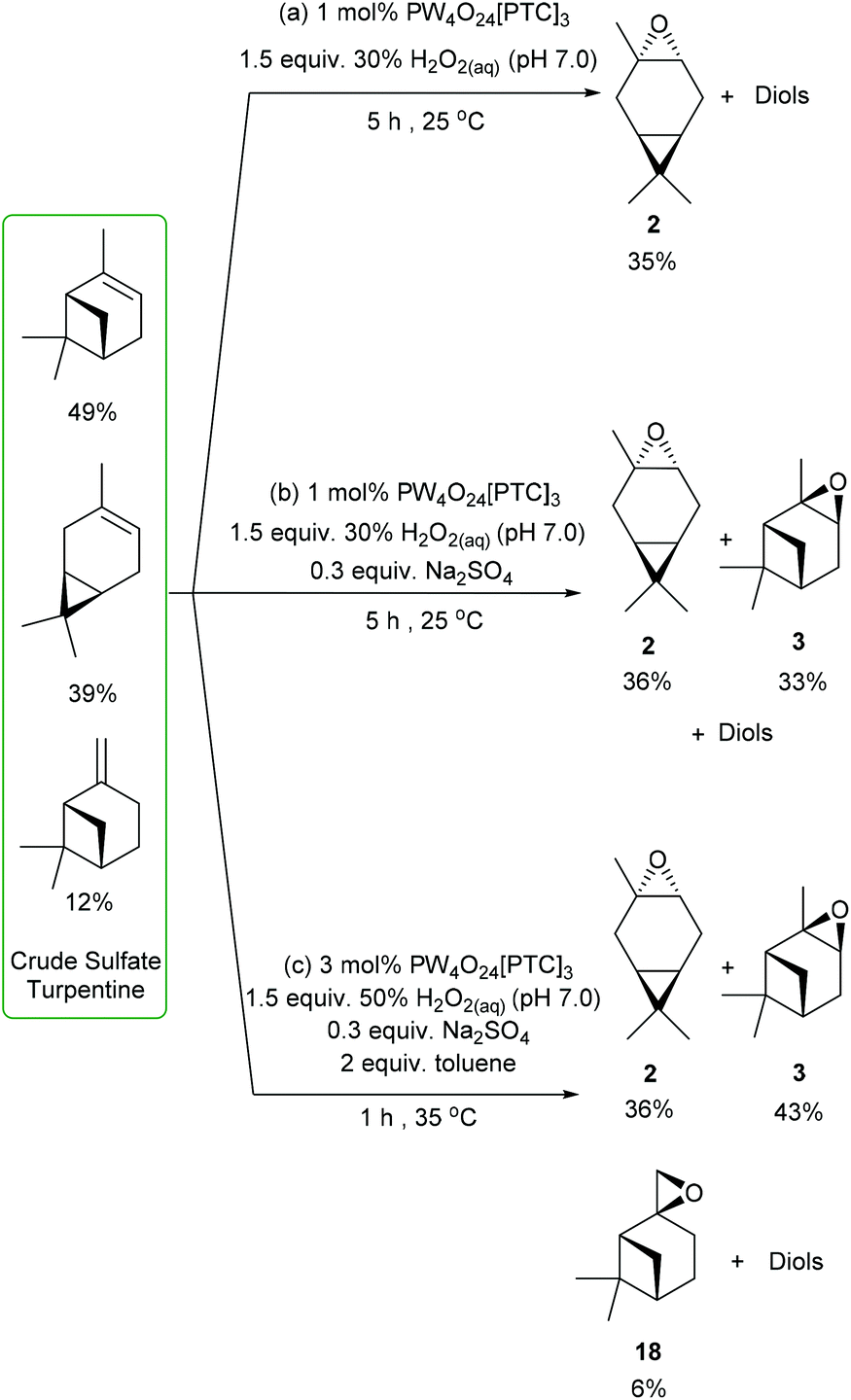 | ||
| Scheme 7 Catalytic epoxidation of CST to selectively afford (a) 3-carene oxide; (b) 3-carene oxide and α-pinene oxide; (c) 3-carene oxide, α-pinene oxide and β-pinene oxide. | ||
Finally, CST was subjected to the more forcing epoxidation conditions (3 mol% catalyst, 50% H2O2, 30 mol% Na2SO4, toluene) developed for β-pinene, which gave 3-carene oxide, α-pinene oxide and β-pinene oxide in 36% (92% theoretical), 43% (88% theoretical) and 6% (50% theoretical) yields, respectively (Scheme 7c). These epoxides could be separated by fractional distillation under reduced pressure to afford 3-carene oxide (bp 87 °C at 50 mm Hg), followed by α-pinene oxide (bp 102 °C at 50 mm Hg) and then β-pinene oxide (bp 112 °C at 50 mm Hg).
Catalyst recycling studies
We next investigated the possibility of recycling the catalyst for use in consecutive epoxidation reactions, with 3-carene chosen as a model substrate because its relatively fast epoxidation rate and the fact that it affords a single epoxide diastereomer that is easily purified by distillation. Epoxidation of 3-carene was carried out using 1 mol% catalyst using our standard solvent/additive free conditions using 1.0 equiv. of 30% H2O2, which resulted in 100% consumption of 3-carene after 7 h. Separation and 1H NMR spectroscopic analysis of the resultant biphasic aqueous/organic layers revealed that the organic layer contained the VPTC species. Distillation of the crude organic layer afforded 3-carene oxide in 73% isolated yield, with the recovered distillation residue containing the catalyst then reused for epoxidation of a fresh batch of 3-carene. This second epoxidation reaction required 24 h to proceed to completion, with distillative purification affording 3-carene oxide in a comparable 76% yield. A third epoxidation run using the recovered catalyst distillate took 100 h to reach 90% conversion, with distillation affording unreacted 3-carene (9%) and 3-carene oxide in 66% yield. Finally, a fourth run using the recycled catalyst distillate only gave 13% conversion of 3-carene after 100 h, indicating that the recycled VPTC had lost most of its activity after the third epoxidation run (Fig. 1). The mass of the recovered distillate residue was found to effectively double after each run, with 1H NMR spectroscopic analysis (see ESI‡ for details) revealing the presence of high boiling/oligomeric/polymeric terpene residues accumulating over time to deactivate the catalyst. Additionally, the POM anion (or its degradation products) was presumed to be leaching into the aqueous phase of each subsequent reaction to some extent, as has been previously observed by other groups.41 This was confirmed by 1H and 31P NMR spectroscopy which revealed retention of PTC species but the absence of any phosphorous-containing POM anion species in the organic layer after the fourth run. However, the first three catalytic recycling runs gave acceptable 66–75% yields of 3-carene oxide under solvent free conditions, suggesting that catalyst recycling is potentially feasible, particularly if distillative purifications of terpene oxides are carried out at lower temperatures/higher pressures that are available on an industrial scale. These recycling results are in accordance with previous studies by other groups using similar homogeneous polyoxotungsten-based epoxidation catalysts, who also described a significant drop in activity after successive epoxidation runs.42,43 Efficient recycling has been achieved using polyoxotungsten-based catalysts immobilised on graphene and polymer supports,44,45 however, these heterogeneous systems generally require longer reaction times and higher temperatures for epoxidation to occur. Finally, we found that the catalyst distillate residue isolated after the fourth run was highly flammable, which affords the opportunity of burning it for energy and recovering its tungsten metal content (Fig. 2).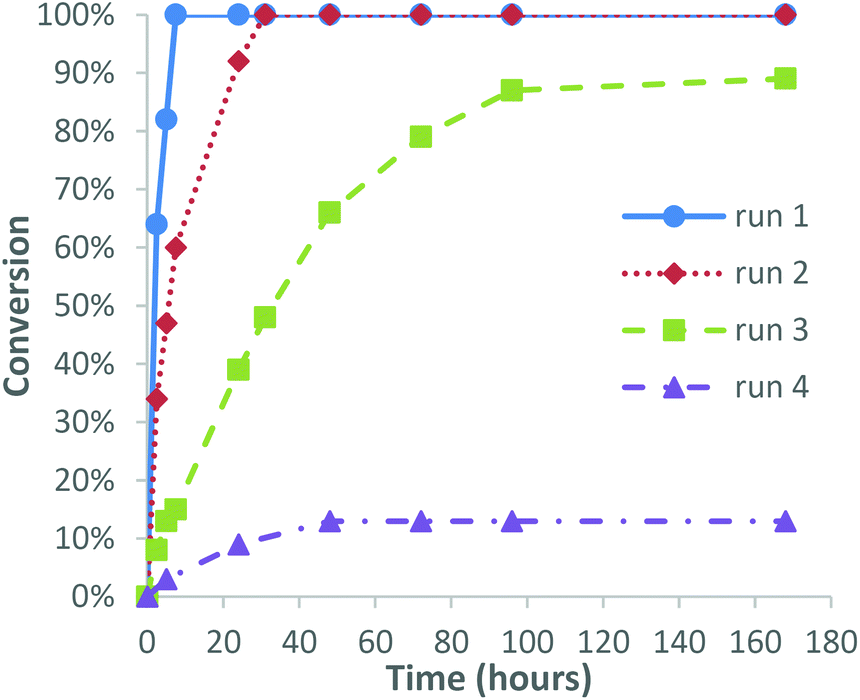 | ||
| Fig. 1 Graph showing the conversion of 3-carene over time with 1 mol% catalyst recycled through four reactions. | ||
 | ||
| Fig. 2 Image of recycled catalyst residue burning vigorously after being ignited using an open flame. | ||
One-pot catalytic epoxidation/hydrolysis protocol for Prilezhaev anti-dihydroxylation of biorenewable terpenes
Since a number of our catalytic epoxidation experiments had resulted in competing in situ hydrolysis of epoxides to afford anti-1,2-diols, it was decided to develop a one-pot stepwise protocol for carrying out Prilezhaev anti dihydroxylation of the alkene bonds of terpenes.38 Venturello et al. have previously reported that low loadings of a preformed Venturello-PTC catalyst could be used to catalyse tandem epoxidation/hydrolysis reactions of aryl- and alkyl–alkene substrates (no terpenes) using benzene as a solvent to afford their corresponding anti-diols.38 Attempts to employ Venturello's conditions38 for the epoxidation/hydrolysis of limonene produced variable results, with replacement of benzene with a range of cosolvents (e.g. toluene) resulting in complex mixtures of limonene, limonene-1,2-oxide and limonene anti-1,2-diols. Consequently, we devised an alternative stepwise one-pot anti-dihydroxylation protocol, involving catalytic epoxidation of limonene (1 mol% catalyst, 30 wt% H2O2 (pH 3–4)) to afford a crude epoxidation product (no work-up) that was then treated with the heterogeneous Lewis acid catalyst Amberlyst-15 (0.1 mol%) at rt. Although initial hydrolysis of the crude mixture of limonene oxide proceeded smoothly, the viscosity of this solvent free system was found to increase dramatically as the percentage of diols 20a/b present exceeded 25%, with inefficient stirring/mixing resulting in the hydrolysis reaction stalling at around 50% epoxide conversion. Consequently, the epoxidation reaction was repeated on a 5 mmol scale, using 1.0 mL of EtOAc (10 mmol, 2.0 equiv.) as a co-solvent, which enabled efficient stirring and complete hydrolysis of the limonene epoxides in the second Amberlyst-15 hydrolysis step. Filtering off the heterogeneous Amberlyst-15 catalyst and removal of the EtOAc solvent in vacuo enabled a mixture of diastereomeric limonene-1,2-anti-diols 20a/b to be obtained in 68% yield (Scheme 8). Attempts to carry out this Prilezhaev reaction by including Amberlyst-15 at the start of the epoxidation reaction produced inferior results, leading to significantly lower rates of epoxidation which resulted in complex mixtures of products. Attempts to recover and recycle the Amberlyst-15 were unsuccessful due to rapid stirring of the biphasic mixture resulting in mechanical degradation of the resin. | ||
| Scheme 8 Solvent free catalytic epoxidation/hydrolysis protocol for the anti-dihydroxylation of selected monoterpenes. | ||
This catalytic one-pot stepwise epoxidation/hydrolysis method was then applied to the dihydroxylation of a range of nine cyclic and acyclic terpene substrates to afford their respective anti-diol derivatives 21–28 in 56 to 77% isolated yields (Scheme 8).46 Reaction of 3-carene gave anti 3,4-carene diol 21 as a single diastereoisomer in 73% isolated yield, with <5% of 3-carene (or its corresponding epoxides) present in the crude reaction product. The anti-dihydroxylation reactions of 3-carene and γ-terpinene were carried out on a larger 10 g scale, allowing their corresponding diol 21 and tetrol (rac)-22 to be obtained in 55% and 62% isolated yields, respectively. Anti-dihydroxylation of the exocyclic disubstituted isopropylidene bond of isopulegol (epoxidation at 50 °C) gave the corresponding triols 23a/b as a 60:40 mixture of diastereomers in 65% yield. Epoxidation/hydrolysis of dihydrocarvone (4:1 mixture of isomers) gave a 10:10:1:1 mixture of diastereomeric diols 24a–d in 62% yield. Anti-dihydroxylation of perillyl alcohol and carvomenthenol gave 58:42 and 90:10 mixtures of triols 25a/b and 26a/b in 56% and 77% yields, respectively. Anti-dihydroxylation of the non-conjugated alkene bond of α-ionone gave α-ionone anti-diols (rac)-27a/b as an 80:20 mixture of cis-/trans-diastereomers in 70% yield, whilst anti-dihydroxylation of the trisubstituted γ,δ-alkene bond of β-ionone gave (rac)-diol 28 in 55% yield.
Unsurprisingly, attempts to apply these epoxidation (0.3 equiv. Na2SO4)/hydrolysis (Amberlyst-15) conditions to α-pinene were only partially successful, affording a mixture of α-pinene-diol 29 (39%) and the ring-opened products – campholenic aldehyde 30 (15%), trans-sobrerol 31 (31%) and cis-sobrerol 32 (13%) (Scheme 9).25 Epoxidation/hydrolysis of β-pinene was not carried out because of the relatively low yield of its epoxidation step and the fact that β-pinene oxide 18 readily undergoes acid catalysed ring-opening reactions to afford mixtures of myrtenal, myrtenol and perillyl alcohol.47 Attempts to apply our epoxidation/hydrolysis protocol to untreated CST resulted in formation of a complex mixture of oxygenated products, with 3-carene-diol 21 (∼20% yield) being the only identifiable product detected by 1H NMR spectroscopic/GCMS analysis. Nevertheless, the anti-dihydroxylation results reported in Scheme 8 clearly demonstrate that Amberlyst-15 can be used as a heterogeneous acid catalyst for catalysing hydrolysis of a of crude terpene epoxide mixtures, without unwanted dehydration/rearrangement reactions occurring.
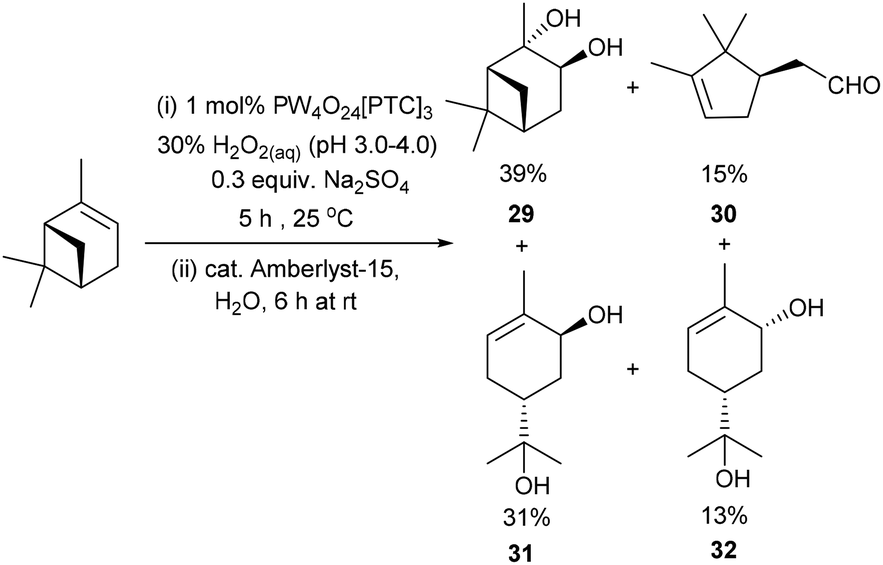 | ||
| Scheme 9 Solvent free catalytic epoxidation/hydrolysis of α-pinene affords a mixture of four products. | ||
Conclusions
An inexpensive and easily prepared tungsten based polyoxometalate VPT catalyst has been used for the solvent free catalytic epoxidation of the trisubstituted alkene bonds of fourteen biorenewable terpene substrates using aqueous 30% H2O2 as a cheap and benign stoichiometric oxidant at rt. These scalable sustainable epoxidation conditions have been applied for the synthesis of α-pinene oxide, 3-carene oxide and limonene oxide (85 g) on a multigram scale. Carrying out this catalytic epoxidation reaction at higher temperatures enabled the less reactive disubstituted alkene bonds of terpenes to be epoxidized in good yields. These catalytic conditions have been used for the direct epoxidation of untreated CST for the first time, affording mixtures of 3-carene oxide, α-pinene oxide and β-pinene oxide that could be separated by distillation. The VPTC could be recycled three times for the catalytic solvent free epoxidation of 3-carene affording 3-carene oxide in 64–75% yield. Treatment of the crude epoxide products (no work-up) with a heterogeneous acid catalyst (Amberlyst-15) results in clean epoxide hydrolysis to afford their corresponding terpene-anti-diols in good yields. Therefore, we believe these green, sustainable and scalable catalytic epoxidation and anti-dihydroxylation protocols provide valuable synthetic tools for transforming cheap biorenewable terpene feedstocks into valuable chemical building blocks for the synthesis of valuable chemical products within a biorefinery context.Experimental
Preparation of modified Ishii–Venturello catalyst
Tungstic acid [H2WO4] (15 g, 60 mmol) was added to 30 wt% aqueous H2O2 (30 mL) in distilled H2O (12 mL) and the bright yellow mixture stirred for 1.5 h at 60 °C until a cloudy pale-yellow solution formed. The solution was cooled to room temperature and a solution of 85% orthophosphoric acid (H3PO4) (1.86 mL) in distilled water (1.86 mL) added, followed by addition of 180 mL of distilled H2O. The reaction mixture was stirred for 30 min at rt, followed by dropwise addition of a solution of Aliquat 336 (13.7 mL, £0.80 per 10 mL (Fisher Scientific)) in CH2Cl2 (240 mL) over a period of 15 min. The resulting mixture was then stirred vigorously at room temperature for 1 h, with the organic phase separated off, washed with distilled H2O, dried (MgSO4), and concentrated under vacuum to give the Venturello-A336 catalyst as a viscous, transparent yellow syrup (16.5 g, 70%).Solvent free catalytic epoxidation of terpene substrates
A terpene (5 mmol, 1.0 equiv.) and PW4O24[PTC]3 (0.11 g, 2259 g mol−1, 0.05 mmol, 1 mol%) were added to a 20 mL glass tube and the resultant suspension stirred rapidly for 10 minutes to fully dissolve the catalyst. 30% aqueous H2O2 solution (0.5 mL, 5 mmol, preadjusted to pH 7.0 using 0.5 M NaOH) was then added dropwise to the neat terpene that was stirred slowly at ∼50 rpm. CAUTION – Exothermic runaway needs to be prevented by controlling the rate of H2O2 addition and cooling the reaction flask using an external water bath. The epoxidation reaction was then stirred (∼100 rpm) at room temperature until tlc indicated that all the terpene had been consumed. The top organic layer of the biphasic reaction was then collected and purified via column chromatography (or fractional distillation under reduced pressure) to produce the desired epoxide as a clear oil.Experimental variants of the general epoxidation protocol are as follows
(a) Incorporation of 0.3 equiv. Na2SO4 into the catalytic epoxidation protocol was achieved by dissolving 0.3 equiv. of Na2SO4 in the hydrogen peroxide solution (pH 7.0) prior to its dropwise addition to the stirred terpene solution.(b) Higher catalyst loadings were used for epoxidation of less reactive terpenes, with the catalyst loadings employed for each terpene substrate reported on their respective reaction schemes.
(c) Elevated temperatures of 35–50 °C were used to epoxidize terpene substrates that contained less reactive disubstituted alkenes.
(d) Incorporation of toluene (2 equiv.) into the catalytic epoxidations was achieved by dissolving the terpene substrate and catalyst in the solvent prior to addition of H2O2.
(e) Higher concentration H2O2 (50 wt%) was used for substrates where hydrolysis of epoxidation products to their corresponding anti-diols occurred under standard conditions.
(f) One equiv. of H2O2 was used to for every alkene bond present in the terpene substrate. For example, the six tri-substituted alkene bonds of squalene were epoxidized using six equiv. of H2O2.
(g) Large scale batch epoxidations were stirred at a faster stirring rate of 500 rpm to take into account the larger volumes of terpene substrate used, with hydrogen peroxide added in small portions using a dropping funnel.
Catalyst recycling study for epoxidation of 3-carene
3-Carene (3.26 g, 24 mmol) and PW4O24[PTC]3 (0.55 g, 2259 gmol−1, 0.24 mmol, 1 mol%) were added to a 20 mL glass tube and the resultant suspension stirred rapidly for 10 minutes to fully dissolve the catalyst. 30% aqueous H2O2 solution (2.5 mL, 24 mmol, preadjusted to pH 7 using 0.5 M NaOH) was then added dropwise to the terpene mixture that was stirred slowly at ∼50 rpm. CAUTION – Exothermic runaway needs to be prevented by controlling the rate of H2O2 addition and cooling the reaction mixture using an external water bath. The reaction was then stirred (∼100 rpm) at room temperature and monitored by 1H NMR spectroscopy, with the epoxidation reaction complete after 7 h. The top organic layer of the biphasic reaction was then collected and purified via fractional distillation under reduced pressure to produce 3-carene oxide as a clear oil (2.65 g, 73%). The same amount of 3-carene (3.26 g, 24 mmol) was then added to the recovered catalyst residue and the epoxidation reaction repeated using the same amount of H2O2 (1 equiv.). This epoxidation/catalyst recycling procedure was carried out for a total of four experiments, giving 3-carene oxide yields of 73% for run 1, 76% for run 2, 66% for run 3 (+9% recovered 3-carene) and 11% for run 4 (+65% recovered 3-carene).Prilezhaev anti-dihydroxylation reaction of terpene substrates
A terpene substrate (5 mmol, 1.0 equiv.), PW4O24[PTC]3 (0.11 g, 2259 g mol−1, 0.05 mmol, 1 mol%) and EtOAc (10 mmol, 1 mL) were stirred in a 20 mL glass tube for 10 minutes to afford a homogeneous solution. 30% aqueous hydrogen peroxide solution (0.5 mL, 5 mmol, pH 3.0–4.0) was then added, with slow stirring carried out at ∼50 rpm for the duration of peroxide addition. CAUTION – exothermic runaway needs to be prevented by controlling the rate of H2O2 addition and cooling the reaction mixture using an external water bath. The reaction was then stirred as a faster rate (∼100 rpm) at room temperature until tlc indicated that the epoxidation reaction was complete. Amberlyst-15 (0.13 mol%, 140 mg, 4.6 mmol g−1) was then added directly to the crude reaction mixture which was then stirred (50 rpm) for 7–8 h at room temperature, with tlc analysis used to determine when epoxide hydrolysis was complete. The reaction mixture was then filtered to remove Amberlyst-15, extracted with EtOAc (3 × 10 mL), the combined organic layers dried (MgSO4) and solvent removed in vacuo to give a crude product that was purified by chromatography to afford the desired terpene-anti-diol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank EPSRC for funding through “Terpene-based Manufacturing for Sustainable Chemical Feedstocks” EP/K014889 and the Centre for Doctoral Training in Sustainable Chemical Technologies (EP/L016354/1). Södra Forestry Cooperative are thanked for supplying an authentic industrial sample of CST.Notes and references
- B. J. Nikolau, M. A. Perera, L. Brachova and B. Shanks, Plant J., 2008, 54, 536–545 CrossRef CAS.
- M. Rastogi and S. Shrivastava, Renewable Sustainable Energy Rev., 2017, 80, 330–340 CrossRef.
- K. A. Gray, L. S. Zhao and M. Emptage, Curr. Opin. Chem. Biol., 2006, 10, 141–146 CrossRef CAS PubMed.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- A. J. D. Silvestre and A. Gandini, Chapter 2 – Terpenes: Major Sources, Properties and Applications in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 17–38 Search PubMed.
- K. A. D. Swift, Top. Catal., 2004, 27, 143–155 CrossRef CAS.
- R. A. Sheldon, I. Arends and U. Hanefield, Green Chemistry and Catalysis, Wiley-VCH, 2007 Search PubMed.
- A. Celli, A. Gandini, C. Gioia, T. M. Lacerda, M. Vannini and M. Colonna, Chapter 12 – Chemicals and Fuels from Bio-Based Building Blocks – Polymers from Pristine and Modified Natural Monomers in Chemical and Fuels from Bio-Based Building Blocks, ed. F. Cavani, S. Albonetti and F. Basile, Wiley-VCH, 2016 Search PubMed.
- (a) R. Höfer, Chapter 3b – The Pine Biorefinery Platform Chemicals Value Chain in Industrial Biorefineries & White Biotechnology, ed. A. Pandey, R. Höfer, M. Taherzaden and K. M. Nampoothiri, Elsevier, 2006 Search PubMed; (b) D. Helmdach, P. Yaseneva, P. K. Heer, A. M. Schweidtmann and A. A. Lapkin, ChemSusChem, 2017, 10, 3632–3643 CrossRef CAS PubMed.
- M. Ragnar, G. Henriksson, M. E. Lindström, M. Wimby, J. Blechschmidt and S. Heinemann, Pulp, in Biorenewable Resources, Ullman's Encyclopedia of Industrial Chemistry, Wiley, 2014 Search PubMed.
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Carà, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 RSC.
- J. Zhou, G. Du and J. Chen, Curr. Opin. Biotechnol., 2014, 25, 17–23 CrossRef CAS PubMed.
- T. P. Korman, P. H. Opgenorth and J. U. Bowie, Nat. Commun., 2017, 8, 15526 CrossRef CAS PubMed.
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed.
- A. Behr and L. Johnen, ChemSusChem, 2009, 2, 1072–1095 CrossRef CAS PubMed.
- R. Reeves and M. Lawrence, Epoxides, Synthesis, Reactions and Uses, Nova Science, 2017 Search PubMed.
- N. Ravasio, F. Zaccheria, M. Guidotti and R. Psaro, Top. Catal., 2004, 27, 157–168 CrossRef CAS.
- For selected applications of some of the terpene epoxides prepared in this study, see: (a) J. A. Marshall, G. S. Bartley and E. M. Wallace, J. Org. Chem., 1996, 61, 5729–5735 CrossRef CAS; (b) D. Tanner, P. G. Andersson, L. Tedenborg and P. Somfai, Tetrahedron, 1994, 50, 9135–9144 CrossRef CAS; (c) G. P. More and S. V. Bhat, Tetrahedron Lett., 2013, 54, 4148–4149 CrossRef CAS; (d) X. Z. Zhao, Y. Q. Tu, L. Peng, X. Q. Li and Y. X. Jia, Tetrahedron Lett., 2004, 45, 3713–3716 CrossRef CAS; (e) J. H. Kim, H. J. Lim and S. H. Cheon, Tetrahedron, 2003, 59, 7501–7507 CrossRef CAS; (f) E. Vyskočilová, J. Dušek, M. Babirádová, J. Krupka, I. Paterová and L. Červený, Res. Chem. Intermed., 2018, 44, 3971–3984 CrossRef; (g) E. Salminen, L. Rujana, P. Mäki-Arvela, P. Virtanen, T. Salmi and J.-P. Mikkola, Catal. Today, 2015, 257, 318–321 CrossRef CAS; (h) K. Mori, Tetrahedron: Asymmetry, 2006, 17, 2133–2142 CrossRef CAS; (i) A. Bosser, E. Paplorey and J. Belin, Biotechnol. Prog., 1995, 11, 689–692 CrossRef CAS; (j) M. Ansari and S. Emami, Eur. J. Med. Chem., 2016, 123, 141–154 CrossRef CAS; (k) J. R. Lowe, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2003–2008 CrossRef CAS PubMed; (l) T.-L. Ho and Z. U. Din, Synth. Commun., 2006, 19, 813–816 CrossRef.
- A. Mouret, L. Leclercq, A. Muhlbauer and V. Nardello-Rataj, Green Chem., 2014, 16, 269–278 RSC.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS.
- A. L. Villa de P, B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Org. Chem., 1999, 64, 7267–7270 CrossRef CAS.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto, D. Panyella and R. Noyori, Bull. Chem. Soc. Jpn., 1997, 70, 905–915 CrossRef CAS.
- S. Sakaguchi, Y. Nishiyama and Y. Ishii, J. Org. Chem., 1996, 61, 5307 CrossRef CAS.
- G. Grigoropoulou and J. H. Clark, Tetrahedron Lett., 2006, 47, 4461–4463 CrossRef CAS.
- Y. Kon, H. Hachiya, Y. Ono, T. Matsumoto and K. Sato, Synthesis, 2011, 1092–1098 CAS.
- K. Sato, Y. Kon, H. Hachiya, Y. Ono, K. Takumi, N. Sasagawa and Y. Ezaki, Synthesis, 2012, 1672–1678 Search PubMed.
- K. Kamata, K. Sugahara, R. Ishimoto, S. Nojima, M. Okazaki, T. Matsumoto and N. Mizuno, ChemCatChem, 2014, 6, 2327–2332 CrossRef CAS.
- M. F. M. G. Resul, A. M. L. Fernández, A. Rehman and A. P. Harvey, React. Chem. Eng., 2018, 3, 747–756 RSC.
- P. Levecque, D. W. Gammon, H. H. Kinfe, P. Jacobs, D. De Vos and B. Sels, Adv. Synth. Catal., 2008, 350, 1557–1568 CrossRef CAS.
- For selected uses of limonene 1,2-oxide see: (a) C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed; (b) K. Geoghegan and P. Evans, Tetrahedron Lett., 2014, 55, 1431–1433 CrossRef CAS; (c) J. D. White, J. F. Ruppert, M. A. Avery, S. Torii and J. Nokami, J. Am. Chem. Soc., 1981, 103, 1813–1821 CrossRef CAS; (d) O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed; (e) R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed; (f) M. Pagliaro, Chim. Oggi, 2018, 36, 57–58 CAS; (g) D. Steiner, S. G. Sethofer, C. T. Goralski and B. Singaram, Tetrahedron: Asymmetry, 2002, 13, 1477–1483 CrossRef CAS; (h) C. Raptis, H. Garcia and M. Stratakis, Angew. Chem., Int. Ed., 2009, 48, 3133–3136 CrossRef CAS PubMed; (i) E. E. Royals and J. C. Leffingwell, J. Org. Chem., 1966, 31, 1937–1944 CrossRef CAS; (j) C. M. Binder, D. D. Dixon, E. Almaraz, M. A. Tius and B. Singaram, Tetrahedron Lett., 2008, 49, 2764–2767 CrossRef CAS PubMed.
- For selected uses of 3-carene-oxide see: (a) V. Srirajan, B. M. Bhawal and A. R. A. Deshmukh, Tetrahedron, 1996, 52, 5585–5590 CrossRef CAS; (b) M. H. Shastri, D. G. Patil and V. D. Patil, Tetrahedron, 1985, 41, 3083–3090 CrossRef CAS; (c) F. Z. Macaev and A. V. Malkov, Tetrahedron, 2006, 62, 9–29 CrossRef CAS; (d) M. Uroos, P. Pitt, L. M. Harwood, W. Lewis, A. J. Blake and C. J. Hayes, Org. Biomol. Chem., 2017, 15, 8523–8528 RSC; (e) L. P. Carrodeguas, C. Martin and A. W. Kleij, Macromolecules, 2017, 50, 5337–5345 CrossRef; (f) G. S. Kauffman, G. D. Harris, R. L. Dorow, B. R. P. Stone, R. L. Parsons, J. A. Pesti, N. A. Magnus, J. M. Fortunak, P. N. Confalone and W. A. Nugent, Org. Lett., 2000, 2, 3119–3121 CrossRef CAS PubMed; (g) N. S. Joshi and S. V. Malhotra, Tetrahedron: Asymmetry, 2003, 14, 1763–1766 CrossRef; (h) M. Kodama, U. S. F. Tambunan and T. Tsunoda, Tetrahedron Lett., 1986, 27, 1197–1200 CrossRef CAS.
- For selected uses of α-pinene-oxide see: (a) Z. Xu and J. Qu, Chem. – Eur. J., 2013, 19, 314–323 CrossRef CAS PubMed; (b) A. Srikrishna, V. Gowri and G. Neetu, Tetrahedron: Asymmetry, 2010, 21, 202–207 CrossRef CAS; (c) J. B. Lewis and G. W. Hedrick, J. Org. Chem., 1965, 30, 4271–4275 CrossRef CAS; (d) J. K. Crandall and L. H. Chang, J. Org. Chem., 1967, 32, 435–439 CrossRef CAS; (e) R. Lakshmi, T. D. Bateman and M. C. McIntosh, J. Org. Chem., 2005, 70, 5313–5315 CrossRef CAS PubMed; (f) J. M. Castro, P. J. Linares-Palomino, S. Salido, J. Altarejos, M. Nogueras and A. Sanchez, Tetrahedron, 2005, 61, 11192–11203 CrossRef CAS; (g) A. Fernandez-Mateos, P. Herrero Teijon and R. Rubio Gonzalez, Tetrahedron, 2011, 67, 9529–9534 CrossRef CAS; (h) S. M. Bruno, A. A. Valente, M. Pillinger, J. Amelse, C. C. Romao, I. S. Goncalves and S. Isabel, ACS Sustainable Chem. Eng., 2019, 7, 13639–13645 CrossRef CAS.
- For comparisons of catalytic limonene epoxidation see: (a) G. Tseberlidis, L. Demonti, V. Pirovano, M. Scavini, S. Capelli, S. Rizzato, R. Vicente and A. Caselli, ChemCatChem, 2019, 11, 4907–4915 CrossRef CAS; (b) C. Bisio, A. Gallo, R. Psaro, C. Tiozzo, M. Guidotti and F. Carniato, Appl. Catal., A, 2019, 581, 133–142 CrossRef CAS; (c) S. S. Balula, I. C. M. S. Santos, L. Cunha-Silva, A. P. Carvalho, J. Pires, C. Freire, J. A. S. Cavaleiro, B. de Castro and A. M. V. Cavaleiro, Catal. Today, 2013, 203, 95–102 CrossRef CAS; (d) M. S. Melchiors, T. Y. Viera, L. P. S. Pereira, B. A. M. Carciofi, P. H. H. de Araújo, D. de Oliveira and C. Sayer, Ind. Eng. Chem. Res., 2019, 58, 13918–13925 CrossRef CAS; (e) V. Escande, E. Petit, L. Garoux, C. Boulanger and C. Grison, ACS Sustainable Chem. Eng., 2015, 3, 2704–2715 CrossRef CAS; (f) I. C. M. S. Santos, J. A. F. Gamelas, T. A. G. Duarte, M. M. Q. Simões, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2017, 593–599 CrossRef CAS; (g) M. F. M. G. Resul, A. M. L. Fernandez, A. Rehman and A. P. Harvey, React. Chem. Eng., 2018, 3, 747 RSC.
- P. P. Peralta-Yahya, F. Zhang, S. B. del Cardayre and J. D. Keasling, Nature, 2012, 488, 320–328 CrossRef CAS.
- M. Spanova and G. Daum, Eur. J. Lipid Sci. Technol., 2011, 113, 1299–1320 CrossRef CAS.
- For previous reports where metal catalysed epoxidation of β-pinene using H2O2 as oxidant have been reported to afford β-pinene oxide in good yield, see: (a) Methyltrioxorhenium catalyst: S. Yamazaki, Org. Biomol. Chem., 2010, 8, 2377–2385 RSC; (b) Iron “helmet” phthalocyanine catalyst: I. Y. Skobelev, E. V. Kudrik, O. V. Zalomaeva, F. Albrieux, P. Afanasiev, O. A. Kholdeeva and A. B. Sorokin, Chem. Commun., 2013, 49, 5577 RSC; (c) [Bmim]5 [PW11ZnO39]·3H2O catalyst: Z. Nadealian, V. Mirkhani, B. Yadollahi, M. Moghadam, S. Tangestaninejad and I. Mohammadpoor-Baltrock, J. Iran. Chem. Soc., 2013, 10, 777–782 CrossRef CAS.
- V. V. Fomenko, O. V. Bakhvalov, V. F. Kollegov and N. F. Salakhutdinov, Russ. J. Gen. Chem., 2017, 87, 1675–1679 CrossRef CAS.
- C. Venturello and M. Gambaro, Synthesis, 1989, 295–297 CrossRef CAS.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, Macromolecules, 2017, 50, 944–955 CrossRef CAS.
- D. C. Duncan, R. C. Chambers, E. Hecht and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 681–691 CrossRef CAS.
- J. Wu, P. Jiang, X. Qin, Y. Ye and Y. Leng, Bull. Korean Chem. Soc., 2014, 35, 1675–1680 CrossRef CAS.
- L. Gharnati, O. Walter, U. Arnold and M. Döring, Eur. J. Inorg. Chem., 2011, 2756–2762 CrossRef CAS.
- M. Masteri-Farahani and M. Modarres, Mater. Chem. Phys., 2017, 199, 522–527 CrossRef CAS.
- S. P. Maradur, C. Jo, D. Choi, K. Kim and R. Ryoo, ChemCatChem, 2011, 3, 1435–1438 CrossRef CAS.
- For selected uses of some of the terpene-anti-diols prepared in this study, see: (a) Y. Shono, K. Watanabe, H. Sekihachi, A. Kakimizu, M. Suzuki and N. Matsuo, 1992, EP476885A219920325; (b) M. Makida and T. Matsunaga, 2000, JP2000072644(A); (c) Z. Muljiani, A. R. A. S. Deshmukh, S. R. Gadre and V. S. Joshi, Synth. Commun., 1987, 17, 25–32 CrossRef CAS; (d) Z. Wang, Y. Cui, Z. Xu and J. Qu, J. Org. Chem., 2008, 73, 2270–2274 CrossRef CAS PubMed; (e) T. S. Kaufman, R. P. Srivastava and R. D. Sindelar, Bioorg. Med. Chem. Lett., 1995, 5, 501–506 CrossRef CAS; (f) W. Skorianetz and G. Ohloff, Helv. Chim. Acta, 1973, 56, 2025–2028 CrossRef CAS.
- O. de La Torre, M. Renz and A. Corma, Appl. Catal., A, 2010, 1–2, 165–171 CrossRef.
Footnotes |
| † We would like to dedicate this paper to the memory of Professor Jonathan M. J. Williams. |
| ‡ Electronic supplementary information (ESI) available: For experimental procedures and other data. See DOI: 10.1039/c9gc03208h |
| This journal is © The Royal Society of Chemistry 2020 |