
Pyrenediones as versatile photocatalysts for oxygenation reactions with in situ generation of hydrogen peroxide under visible light†
Yuannian
Zhang
a,
Xin
Yang
a,
Haidi
Tang
b,
Dong
Liang
 a,
Jie
Wu
*b and
Dejian
Huang
*a
a,
Jie
Wu
*b and
Dejian
Huang
*a
aFood Science and Technology Program, Department of Chemistry, National University of Singapore, Singapore 117543. E-mail: chmhdj@nus.edu.sg
bDepartment of Chemistry, National University of Singapore, Singapore 117543. E-mail: chmjie@nus.edu.sg
First published on 19th November 2019
Abstract
Pyrenediones (PYDs) are efficient photocatalysts for three oxygenation reactions: epoxidation of electron deficient olefins, oxidative hydroxylation of organoborons, and oxidation of sulfides via in situ generation of H2O2 under visible light irradiation, using oxygen as a terminal oxidant and IPA as a solvent and a hydrogen donor.
Oxygenation reactions are of crucial importance in the syntheses of fine chemicals, natural products, and pharmaceutically active molecules.1–3 The preparation of O-containing molecules in a highly selective, environmentally friendly, and sustainable manner is challenging in both laboratory and industrial settings. Light-induced oxygenation (or photooxygenation) using molecular oxygen as a stoichiometric oxidant is appealing compared to the conventional use of strong oxidants, such as hydrogen peroxide (H2O2), ozone, hypervalent iodine, and tert-butyl hydroperoxide (TBHP), which is usually associated with the risk of explosion and adverse environmental impact. In general, photooxygenation with O2 could be accomplished through two pathways, namely singlet oxygen (1O2) generated via energy transfer from 3O24 or a superoxide radical anion (O2˙−) and hydrogen peroxide (H2O2) produced via a single electron transfer (SET) process.5 Appropriate choice of the photocatalytic system is the key to achieve efficient incorporation of an oxygen atom from molecular oxygen. Thus, a great number of photocatalysts have been developed for this purpose, the majority of which are transition-metal catalysts6–8 and inorganic semiconductors.9,10
In the field of photocatalysis, metal-free organophotocatalysts have recently gained momentum because they are more environmentally benign than metal catalysts.11–13 In particular, Zhang14 and Chen15 groups have reported rose Bengal-mediated oxidative hydroxylation of organoborons with in situ generated H2O2. Photocatalytic hydroxylation of alkenes and sulfide oxidation were achieved by using 9-mesityl-10-methylacridinium ions (Acr+-Mes) or rose Bengal.16,17 However, most of these approaches require sacrificial electron donors or additives to complete the photocatalytic cycle. Therefore the development of organophotocatalysts for versatile oxygenation reactions with a milder and more economical protocol is still highly desired. Notably, Itoh and co-workers have disclosed elegant studies on H2O2 generation using anthracene or anthraquinone as a photocatalyst by aerobic photo-oxidation of IPA, which was further applied for the efficient synthesis of gem-dihydroperoxides,18,19 epoxidation of unsaturated ketones,20 and oxidation of arylboronic acids.21
We were intrigued by conjugated molecules containing the quinone hydroquinone-like redox system that absorbs visible light,22 which may act as photocatalysts for hydrogen atom transfer.11 One family of such molecules is pyrenedione (PYD). Two isomers 1,6-PYD and 1,8-PYD were conveniently prepared by directly oxidizing pyrene using potassium dichromate (Fig. 1a).23 With the extended conjugated structure, both isomers exhibited strong absorption bands in the visible region (λmax = 428 and 461 nm, ε = 8.3 × 103 M−1 cm−1 and 6.1 × 103 M−1 cm−1, respectively, Fig. 1b). Cyclic voltammetry (CV) scans revealed two half wave potentials at −0.30 V and −0.52 V for 1,6-PYD and −0.35 V and −0.59 V for 1,8-PYD, indicating their ability to undergo two electron reductions (Fig. 1c). Peaks Ia/Ic and IIa/IIc correspond, respectively, to the reversible redox couples of pyrenedione/radical anion (1,6-PYD/1,6-PYD˙−) and radical anion/dianion (1,6-PYD˙−/1,6-PYD2−). The same phenomenon applies to 1,8-PYD. The excited state reductive potentials (E1/2 (P*/P˙−)) were calculated to be 2.28 V for 1,6-PYD and 2.12 V for 1,8-PYD based on the Rehm–Weller equation (see Tables S1 and S2 for details†). The high oxidizing ability of their excited states enables them potentially efficient hydrogen abstractors. The excited state lifetime of one isomer, 1,8-PYD, has been reported to be 29 μs (benzene), which is sufficient for the following hydrogen atom transfer (HAT) process.24
 | ||
| Fig. 1 (a) Preparation reaction of 1,6-PYD and 1,8-PYD. (b) Normalized UV-Vis absorption (solid) and fluorescence emission spectra (dash) of 1,8-PYD and 1,6-PYD in MeCN/THF (1/1). (c) Cyclic voltammetry of 1,8-PYD and 1,6-PYD with a scan rate of 100 mV s−1 in MeCN. E1/2 (P*/P˙−) represents the excited state reductive potential. | ||
Upon irradiation of a solution of 1,6-PYD in IPA using an 18 W blue LED strip for 10 min, the yellowish solution turned colourless (Fig. S1†). The dynamic photochemical reaction of 1,6-PYD and IPA was monitored by UV-Vis spectra. Irradiation of deaerated acetonitrile (MeCN) solution containing 1,6-PYD and IPA with an 18 W blue LED strip at room temperature resulted in decreased absorbance at 457 nm and 428 nm and in the appearance of new absorption bands at shorter wavelengths (Fig. 2a). 1H-NMR and HRMS-ESI spectra revealed the product to be 1,6-pyrenediol (1,6-PYDH2).
 | ||
| Fig. 2 (a) Changes of the UV-Vis absorbance spectra of 1,6-PYD (64 μM) with IPA (100 mM) in deaerated MeCN under 18 W blue LED light. Concentration of 1,6-PYD over time (Inset). (b) Changes in the UV-Vis absorbance spectra of 1,6-PYDH2 (60 μM) in MeCN under an aerobic atmosphere in the dark. Concentration of 1,6-PYD over time (Inset). (c) Proposed photocatalytic cycle of 1,6-PYD-promoted H2O2 generation in IPA under an aerobic atmosphere. (d) H2O2 generation under different conditions. Reaction conditions: 1,6-PYD (0.01 mmol), IPA (4 mL), irradiated by an 18 W blue LED strip. The H2O2 amount was determined by iodometric assay. | ||
Mechanistically, the process likely started from the photo-excited 1,6-PYD accepting sequentially two hydrogen atoms from IPA, which was converted to acetone (Fig. 2c and Fig. S2†). Molecular oxygen then acted as the hydrogen acceptor and was converted to hydrogen peroxide, confirmed by iodometric experiments and 1H-NMR spectra (Fig. S3†), to complete the catalytic cycle. The oxidation of 1,6-PYDH2 to 1,6-PYD was feasible due to the low oxidative potential of 1,6-PYDH2 (E1/2 = +0.50 V vs. SCE, Fig. S4†). An irreversible electrochemical behaviour of 1,6-PYDH2 is shown in Fig. S4,† which corresponded to the two electron oxidation to [1,6-PYDH2]2+.25 The different shapes of 1,6-PYDH2 and 1,6-PYD were due to the influence of the protons in 1,6-PYDH2, because the CV of 1,6-PYD in the presence of excess amounts of acids, shows only one oxidation peak similar to that of 1,6-PYDH2 (Fig. S4†). An analogous situation has been observed with anthraquinone.26 The changes in the UV-Vis spectra of 1,6-PYDH2 was observed in aerobic MeCN in the dark (Fig. 2b). The peak at 457 nm increased over time, suggesting the formation of 1,6-PYD. Alternative to a second HAT, the possibility of two monoreduced pyrenedione radicals undergoing disproportionation to give PYD and PYDH2 cannot be ruled out. Several control experiments were performed (Table S3†). Significantly decreased yield of H2O2 in the presence of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) supported a radical process. Addition of a singlet oxygen quencher, NaN3, only gave a slightly decreased yield of H2O2, thus excluding the energy transfer pathway.27
It was observed that the H2O2 amount increased over time under an oxygen atmosphere (Fig. 2d). The amount of H2O2 generated increased 52% after 48 hours of irradiation by the addition of aqueous Na2CO3 solution (TON = 54). This could be explained by the deprotonation of 1,6-PYDH2 in alkaline solution and the anion species formed is more prone to be oxidized by oxygen. The H2O2 production rate in the presence of a base was calculated to be 6.75 mM h−1 within the first 10 hours. Nearly no H2O2 was detected when H2SO4 was added. The H2O2 generation in an air atmosphere was less efficient than that in an oxygen atmosphere. A negligible amount of H2O2 was detected when the reaction was conducted under argon. The industrial production of H2O2 is currently based on the anthraquinone oxidation (AO) process in which hydrogen gas and oxygen in air are reacted indirectly in the presence of anthraquinone to yield H2O2, which is further extracted with water to obtain 30–37% H2O2 solution.28,29 Although economical and relatively stable, aqueous solution of H2O2 is not always ideal for organic reactions and the high concentration of H2O2 may cause side-reactions. Our method provides a mild pathway for the slow release of H2O2 with IPA as the solvent.
Making use of the in situ generated H2O2 by the 1,6-PYD/IPA photocatalytic system, we achieved several environmentally benign oxidative transformations. Our initial study found that the epoxidation of electron deficient alkene 1a in IPA with 5 mol% 1,6-PYD under blue LED irradiation in an oxygen atmosphere delivered epoxide 2a in 68% yield after 24 h (Table 1, entry 1). Prolonging the reaction to 48 h afforded excellent yield (93%, entry 2). The reaction was less effective in air (entry 3). Increasing the temperature to 50 °C resulted in a decreased yield of the product, which was likely attributed to the instability of epoxide 2a (entry 4). Decreasing the loading of the photocatalyst to 2.5 mol% or 1 mol% afforded much lower product yields (55% and 45%, respectively, Table S6†). The screening of several alcohols as solvent indicated the decreased hydrogen donating ability from secondary alcohols to primary alcohols (entries 6–8, Fig. S5†). A negligible amount of 2a was observed in tert-butanol due to the absence of hydridic proton (entry 9). Control experiments confirmed that oxygen, light and photocatalyst were all essential for an efficient transformation (entries 5, 10, and 11). Several other common photocatalysts were also evaluated (Table S4†). No epoxide product was formed with eosin Y and [Ru(bpy)3]2+, and only moderate yields of the products were obtained with anthraquinone as the photocatalyst. Both 1,6-PYD and 1,8-PYD afforded excellent yields (93% and 90%, respectively). Due to the better isolation yield of 1,6-PYD (Scheme 1a), it was utilized as the photocatalyst in our subsequent studies.
 | ||
| Scheme 1 . Substrate scope of epoxidation with electron-deficient alkenesa. Reaction conditions: dicyanide (0.2 mmol), 1,6-PYD (0.01 mol, 5 mol%), IPA (4 mL), irradiated by a blue LED strip under an oxygen atmosphere at room temperature for 24 h. Unsaturated ketone (0.2 mmol), 1,6-PYD (0.01 mmol, 5 mol%), IPA (4 mL), aqueous Na2CO3 solution (1 mmol mL−1, 0.4 mL), irradiated by a blue LED strip under an oxygen balloon at room temperature for 18 h. aIsolated yields. bReaction time was 40 h instead of 24 h. cNa2CO3 powder was added. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Atmosphere | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 1,6-PYD (0.01 mmol, 5 mol%), solvent (4 mL), irradiated under an 18 W blue LED strip at room temperature. b Yields were determined by the 1H-NMR spectra of the crude product using 1,3,5-trimethoxybenzene as an internal standard. c Reaction performed at 50 °C. d Without light. e Without photocatalyst. | ||||
| 1 | IPA | O2 | 24 | 68 |
| 2 | IPA | O2 | 48 | 93 |
| 3 | IPA | Air | 48 | 62 |
| 4c | IPA | O2 | 48 | 60 |
| 5 | IPA | Argon | 48 | 13 |
| 6 | MeOH | O2 | 24 | 0 |
| 7 | EtOH | O2 | 24 | 21 |
| 8 | n-Butanol | O2 | 24 | 21 |
| 9 | tert-Butanol | O2 | 48 | Trace |
| 10d | IPA | O2 | 48 | 0 |
| 11e | IPA | O2 | 48 | 0 |

To rule out the possibility of singlet oxygen as the reactive oxygen species (ROS) in this reaction process, 1O2 quenching experiment was conducted (see the ESI†). In the presence of NaN3, a commonly used singlet oxygen quencher,14 the epoxidation was still efficient (80% yield). This excluded the energy transfer process from excited PYD to 3O2. In contrast, a small amount of epoxide 2a was obtained in the presence of 1,4-benzoquinone, a superoxide quencher,30 suggesting that the formation of a superoxide radical was crucial for the reaction to take place.
With the optimized reaction conditions in hand, the scope for epoxidation of various electron-deficient alkenes was evaluated (Scheme 1). As shown in Scheme 1a, 1,6-PYD effectively promoted the epoxidation of methylene-malononitriles bearing a phenyl or alkyl group with good to excellent yields (51% to 98%, 2b–2k). Functionalities including chloride, bromide, trifluoromethyl, and nitro compounds and acids were well-tolerated under the reaction conditions. However, no epoxidation product was observed with weaker Michael acceptors such as 2-cyclohexen-1-one. We envisioned that a base might be required to promote the formation of hydroperoxide anions, which would facilitate the nucleophilic addition to alkenes.31 Indeed, in the presence of 2 equivalents of sodium carbonate, the reaction proceeded effectively to afford 3a in excellent yield (91%) within 18 h. Among the bases screened, Na2CO3 was the optimal one (Table S5†) and was employed in the substrate scope study. The photocatalytic protocol was extended to other cyclic ketones, esters and chalcones (3a–3g), in each case affording the corresponding epoxides in good yields.
This PYD/IPA photocatalytic system was effective for oxidative hydroxylation of organoborons as well. Phenylboronic acid (4a) was converted to the desired phenol 5a in excellent yield (90%) after 40 h in the presence of 10 mol% PYD and IPA under blue LED irradiation (Table 2, entry 1). Again, light, oxygen and photocatalysts were all essential for this transformation (see the ESI†). Other phenylboronic acids bearing electron-donating or electron-withdrawing substituents (entries 2–6) were all good candidates for this oxidative hydroxylation, affording the corresponding phenols in excellent yields ranging from 90% to 98% (5b–5f). Moreover, 1,6-PYD could also effectively promote the hydroxylation of alkylboronic acids and boronic pinacol esters in excellent yields (88–98%, entries 7–10).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) | Entry | Substrate | Product | Yieldb (%) |
| a Reaction conditions: boronic acids or boronic acid pinacol esters (0.2 mmol), 1,6-PYD (0.02 mmol, 10 mol%), IPA (4 mL), irradiated by a blue LED strip under an oxygen atmosphere at room temperature for 40 h. b Isolated yields. | |||||||
| 1 | 4a | 5a | 90 | 6 | 4f | 5f | 95 |
| 2 | 4b | 5b | 90 | 7 | 4g | 5g | 98 |
| 3 | 4c | 5c | 92 | 8 | 4h | 5h | 88 |
| 4 | 4d | 5d | 98 | 9 | 4i | 5a | 86 |
| 5 | 4e | 5e | 98 | 10 | 4j | 5b | 90 |

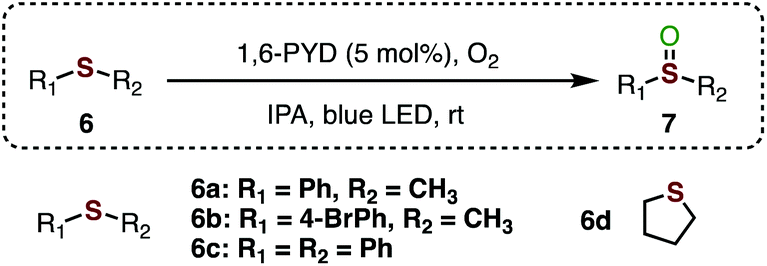
Lastly, the ability of 1,6-PYD to catalyse the photo-oxidation of sulfides was investigated. The oxygenation of sulfides through the visible-light-promoted approach has attracted considerable interest in recent years.32 However, a mixture of sulfoxides and sulfones was normally produced, which caused downstream isolation problems. Notably, oxidation of thioanisole (6a) using the 1,6 PYD/IPA photocatalytic system selectively afforded sulfoxide 7a exclusively in 90% yield after 24 hours (Table 3, entry 1). Oxidation of substrates 6b and 6c afforded sulfoxides 7b and 7c in good yields, respectively (entries 2–3). An alkyl sulfide such as tetrahydrothiophene (6d) was also well-tolerated, and excellent yield of the product was obtained (entry 4).
A comparative study of the PYD/IPA photocatalytic system with direct treatment of commercially available 30% H2O2 solution as an oxidant was further conducted to demonstrate the merits of our protocol (Scheme 2). Apart from clean epoxidation of alkene 1a with the photocatalytic method, oxidation of 1a using 30% of aqueous solution of H2O2 gave a complex mixture of products, with hydrolyzed product 2a′ and aldehyde 2a′′being detected, illustrating the side-effect of water content (Scheme 2a). Oxidative hydroxylation of phenylboronic acid 4a and oxidation of sulfide 6a with 30% H2O2 exhibited lower efficiency compared to the PYD/IPA photocatalytic system (Schemes 2b and c). The superoxide radical anion was formed based on quenching experiments (see the ESI†), which may be another reason for the observed enhanced reactivity associated with the PYD/IPA photocatalytic system (see Scheme S1† for the proposed mechanisms for the three photocatalytic reactions).
 | ||
| Scheme 2 . Comparison studies between the 1,6-PYD/IPA photocatalytic system and commercial 30% H2O2 aqueous solution. | ||
Conclusions
In conclusion, we discovered 1,6-PYD as an organophotocatalyst under visible-light irradiation for oxygenation reactions. H2O2 was smoothly generated by 1,6-PYD in IPA in the presence of O2 under visible light, and this catalytic system can be applied to achieve effective epoxidation of electron-deficient alkenes, oxidative hydroxylation of organoborons, and selective oxidation of sulfides to sulfoxides. The results agreed well with those obtained using commercial aqueous H2O2. This mild and metal-free protocol, which uses oxygen as a terminal oxidant and IPA as a hydrogen donor and green solvent, is promising for its wide applications in oxidative transformations, especially where the reaction components suffer from poor robustness in the presence of excess H2O2 or water.33Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank for the financial support provided by the National University of Singapore and the Ministry of Education (MOE) of Singapore (grant no: MOE2014-T2-1-134, MOE2017-T2-2-081) and Natural Science Foundation of Jiangsu, China (grant no: BK20141219).Notes and references
- Y. F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS.
- M. Milan, M. Salamone, M. Costas and M. Bietti, Acc. Chem. Res., 2018, 51, 1984–1995 CrossRef CAS.
- G. J. Ten Brink, I. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- X. J. Lang, J. C. Zhao and X. D. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC.
- Z. Z. Shi, C. Zhang, C. H. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- X. J. Lang, W. H. Ma, C. C. Chen, H. W. Ji and J. C. Zhao, Acc. Chem. Res., 2014, 47, 355–363 CrossRef CAS PubMed.
- A. Savateev, I. Ghosh, B. Konig and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. Da Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- W.-Z. Weng, H. Liang and B. Zhang, Org. Lett., 2018, 20, 4979–4983 CrossRef CAS.
- H. Wang, W. G. Li, K. Zeng, Y. J. Wu, Y. Zhang, T. L. Xu and Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 561–565 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, Chem. Commun., 2011, 47, 8515–8517 RSC.
- X. Gu, X. Li, Y. Chai, Q. Yang, P. Li and Y. Yao, Green Chem., 2013, 15, 357 RSC.
- L. Cui, N. Tada, H. Okubo, T. Miura and A. Itoh, Green Chem., 2011, 13, 2347 RSC.
- N. Tada, L. Cui, H. Okubo, T. Miura and A. Itoh, Adv. Synth. Catal., 2010, 352, 2383–2386 CrossRef CAS.
- L. Cui, S. Furuhashi, Y. Tachikawa, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2013, 54, 162–165 CrossRef CAS.
- K. Matsui, T. Ishigami, T. Yamaguchi, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Synlett, 2014, 25, 2613–2616 CrossRef CAS.
- Y. Zhang, W. Yao, D. Liang, M. Sun, S. Wang and D. Huang, Sens. Actuators, B., 2018, 259, 768–774 CrossRef CAS.
- M. Yasutake, T. Fujihara, A. Nagasawa, K. Moriya and T. Hirose, Eur. J. Org. Chem., 2008, 2008, 4120–4125 CrossRef.
- R. S. Becker and L. V. Natarajan, J. Phys. Chem., 1993, 97, 344–349 CrossRef CAS.
- X. Ji, C. E. Banks, D. S. Silvester, A. J. Wain and R. G. Compton, J. Phys. Chem. C, 2007, 111, 1496–1504 CrossRef CAS.
- D. P. Valencia, P. D. Astudillo, A. Galano and F. J. González, Org. Biomol. Chem., 2013, 11, 318–325 RSC.
- M. Bancirova, Luminescence, 2011, 26, 685–688 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- T. Nishimi, T. Kamachi, K. Kato, T. Kato and K. Yoshizawa, Eur. J. Org. Chem., 2011, 2011, 4113–4120 CrossRef CAS.
- G. Kibriya, A. K. Bagdi and A. Hajra, Org. Biomol. Chem., 2018, 16, 3473–3478 RSC.
- R. J. Mayer, T. Tokuyasu, P. Mayer, J. Gomar, S. Sabelle, B. Mennucci, H. Mayr and A. R. Ofial, Angew. Chem., Int. Ed., 2017, 56, 13279–13282 CrossRef CAS.
- C. L. Su, R. Tandiana, B. B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- W. Zhang, B. O. Burek, E. Fernández-Fueyo, M. Alcalde, J. Z. Bloh and F. Hollmann, Angew. Chem., Int. Ed., 2017, 56, 15451–15455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03152a |
| This journal is © The Royal Society of Chemistry 2020 |