
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA successful search for new, efficient, and silver-free manufacturing processes for key platinum(II) intermediates applied in antibody–drug conjugate (ADC) production†‡
Eugen
Merkul
 *a,
Niels J.
Sijbrandi
a,
Ibrahim
Aydin
a,
Joey A.
Muns
a,
Ruud J. R. W.
Peters
b,
Paul
Laarhoven
b,
Hendrik-Jan
Houthoff
a and
Guus A. M. S.
van Dongen
c
*a,
Niels J.
Sijbrandi
a,
Ibrahim
Aydin
a,
Joey A.
Muns
a,
Ruud J. R. W.
Peters
b,
Paul
Laarhoven
b,
Hendrik-Jan
Houthoff
a and
Guus A. M. S.
van Dongen
c
aLinXis BV, De Boelelaan 1085c, Amsterdam 1081 HV, The Netherlands. E-mail: merkul@linxispharmaceuticals.com
bChemConnection BV/Ardena Oss, Kloosterstraat 9, Oss, 5349 AB, The Netherlands
cDepartment of Radiology and Nuclear Medicine, Amsterdam UMC, Location VU Medical Center, Amsterdam, The Netherlands
First published on 5th February 2020
Abstract
A silver-free amination procedure, here called “complexation”, was developed to obtain a class of Pt(II) complexes bearing a payload (such as a diagnostic or a therapeutic moiety). These complexes are crucial intermediates for the efficient development and production of antibody–drug conjugates (ADCs) based on a novel Pt(II)-based linker technology. We termed this metal–organic linker, [ethylenediamineplatinum(II)]2+, “Lx”®. The present, newly developed procedure is a greener alternative for the classically applied activation reaction of Pt-halido complexes with silver salts, followed by amination. The crucial finding is that the leaving ligand of the classical process, chloride, can now be replaced by its higher homologue iodide. This not only decisively improved the manufacturing process of the intermediate, but also was found to be key to a more efficient conjugation procedure, i.e. a subsequent step in which this intermediate is coupled to an antibody. The new process allowed upscaling to be readily realized and the desired intermediate was successfully manufactured on a multigram scale. The obtained Ag-free procedure can be generalized and has a great potential to be applied for other Pt(II) complexes of high importance, such as anti-cancer therapeutics.
1. Introduction
Antibody–drug conjugates (ADCs) represent a targeted approach to treat cancer since they allow the delivery of toxic anti-cancer therapeutics selectively to the malignant cells, thus avoiding damage to healthy cells. Currently, six ADCs are approved by the FDA (Adcetris®,1a Kadcyla®,1b Mylotarg®, Besponsa®,1c the recent Polivy™, and the just approved Padcev™) and >80 ADCs are under clinical evaluation.1d ADCs comprise three components: a disease-selective monoclonal antibody (mAb), a small-molecule therapeutic payload, and a linker which connects mAb and payload to form a conjugate. Linkers are the most modifiable part of an ADC and can form a critical factor in the efficiency and costs of the production process, as well as in the therapeutic efficacy and tolerability of an ADC.2a–c Recently, we described a “plug-and-play” approach to an improved ADC linker design by introducing a cationic metal–organic Pt(II)-based linker, [ethylenediamineplatinum(II)]2+, which we termed “Lx”®.3a–e The Lx concept consists of two key steps (Scheme 1).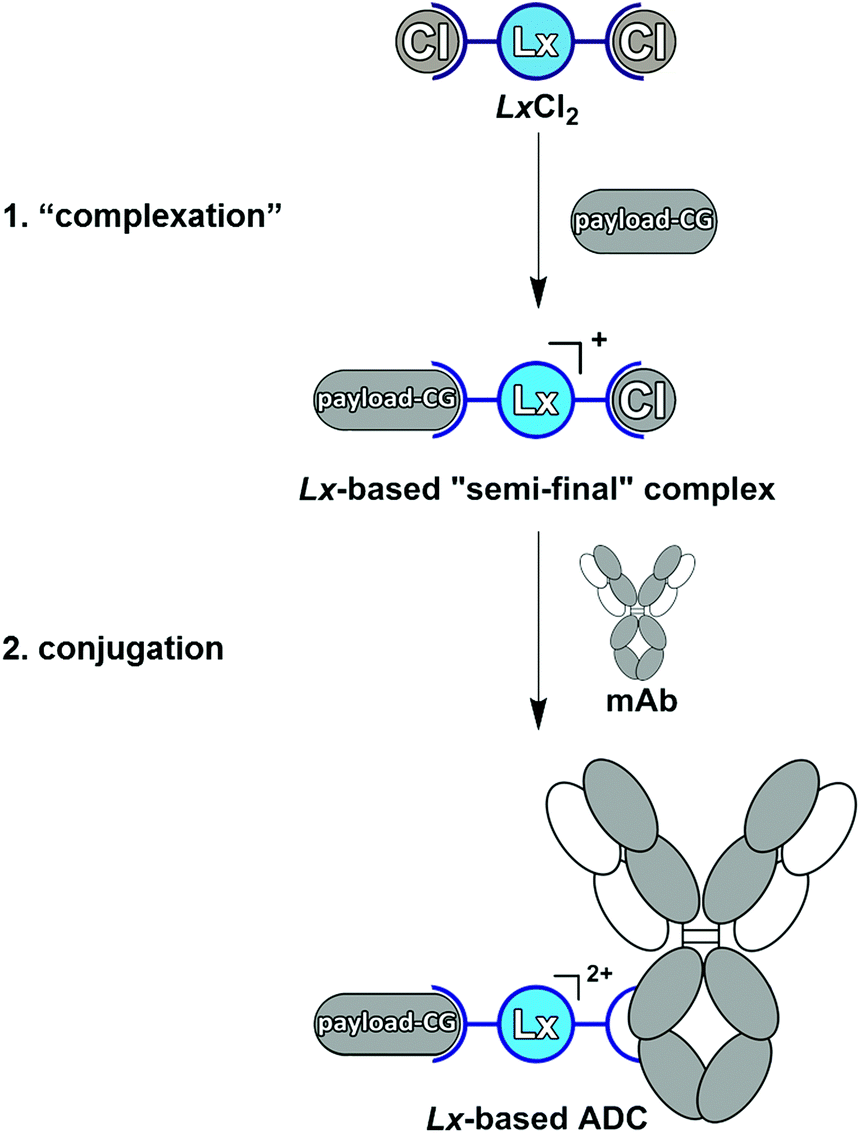 | ||
| Scheme 1 The Pt(II)-based “Lx” approach to ADCs with its two crucial steps: “complexation” and conjugation. mAb = monoclonal antibody, CG = coordination group. | ||
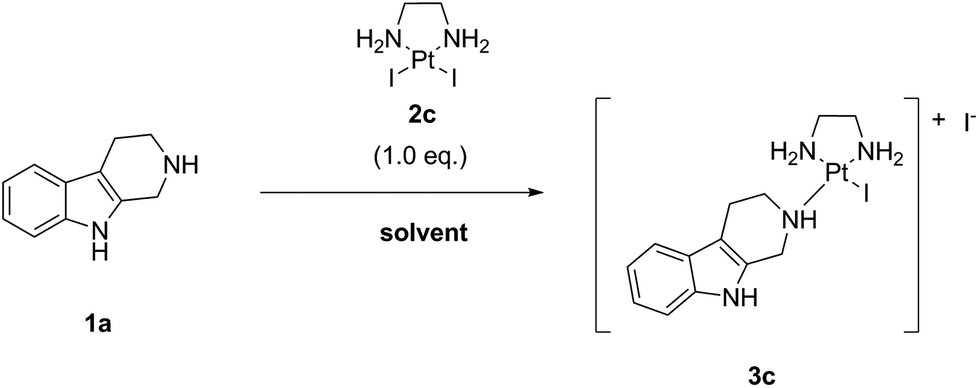
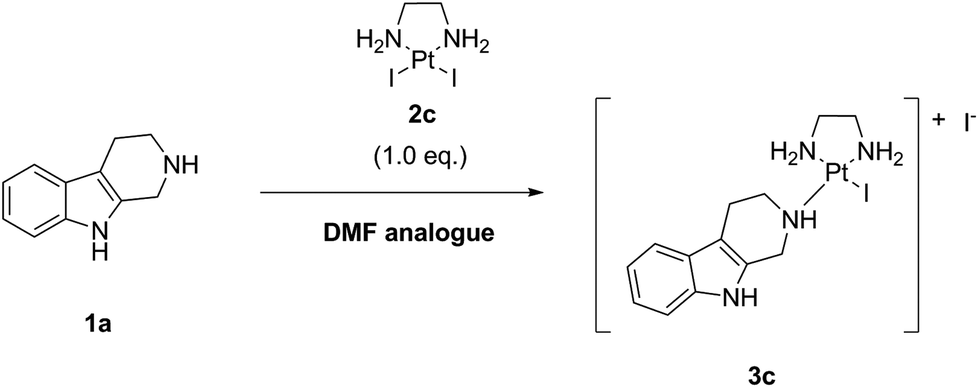
In the first step (“complexation”), the commercially available precursor complex LxCl2 bearing two chloride leaving ligands can be coordinated to payloads bearing a suitable coordination group (payload-CG; CG typically is an N-heterocycle such as piperidine (pip)), to provide storable intermediate products that we termed “semi-final” complexes. In the second step (conjugation), the obtained “semi-final” complexes are conjugated to histidine residues of unmodified mAbs. This novel ADC linker technology could become a game changer in the field of next-generation ADCs.
We have also described earlier the distinguished in vitro and preclinical in vivo targeting and therapeutic properties of Lx-based ADCs.3e From the results that we obtained it can be concluded that the Lx-based ADCs are stable in serum and suitable for optimal tumor targeting.3b Results also revealed that Lx follows the biodistribution of an antibody and, unlike platinum-based chemotherapeutics such as cisplatin, remains an inert component of the ADC after in vivo administration during the time frame of the investigation.3c This information is important with respect to the tolerability and safety of Lx-based ADCs. Currently we are developing a clinical candidate ADC trastuzumab-Lx-pip-AF3b bearing the highly potent tubulin inhibitor auristatin F (AF).4a,b Preclinical studies in tumor bearing mice showed that this anti-HER2 Lx-based ADC outperformed the benchmark ADC Kadcyla®, which is approved by the FDA for the treatment of patients with HER2-positive metastatic breast cancer.3b
The crucial intermediates, which we call “semi-final” products to indicate that they are direct precursors of our “final” Lx-ADC products, have a general formula [Cl-Lx-CG-payload]+ (Scheme 2), where “payload-CG” typically stands for a diagnostic or a therapeutic payload modified with a coordination group suitable for coordination to Pt(II).
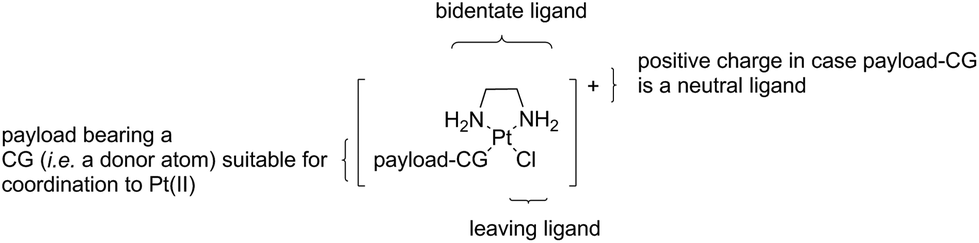 | ||
| Scheme 2 General structure of a “semi-final” product or complex. CG = coordination group. | ||
These storable “semi-final” complexes were initially produced on a laboratory scale using a classical approach via activation of a sparingly soluble LxCl2 compound (2a) by AgNO3 in DMF (e.g. a typical procedure can be found in ESI, 4.2.,‡ for the synthesis of Cl-Lx-pip-AF (3d), and is represented in Scheme 3).3b This activation step, in which ∼2 eq. of LxCl2 (2a) are activated with 1 eq. of AgNO3 – this to avoid a double activation of the precursor 2a leading to a double addition product in which both chlorido ligands are substituted by the payload –, was followed by the addition of a payload-CG compound to achieve coordination of the modified payload moiety to Pt(II).
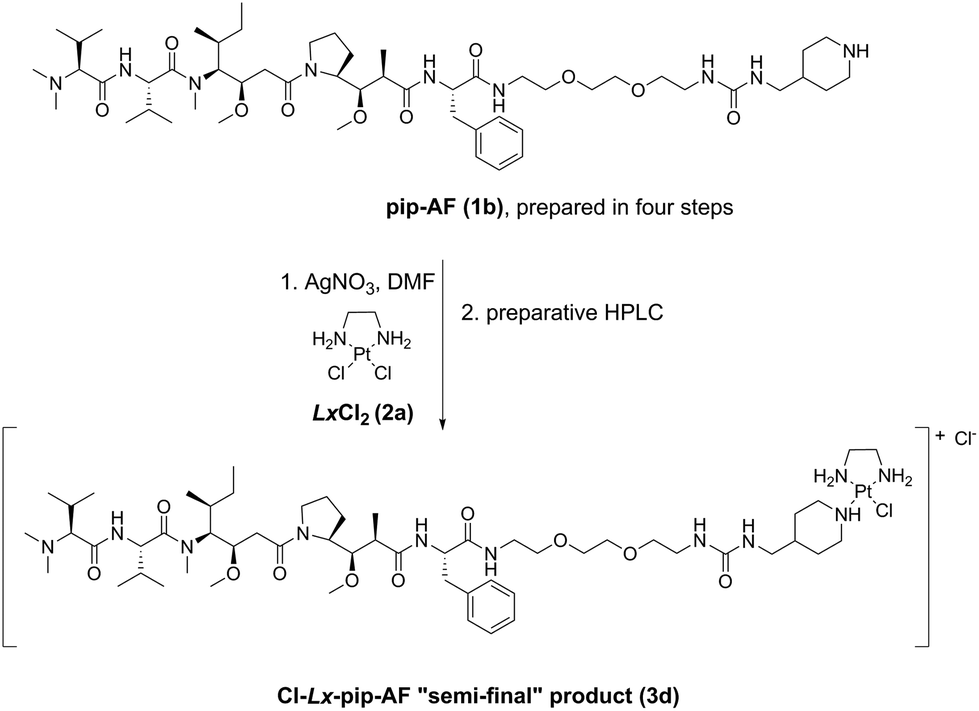 | ||
| Scheme 3 The originally used classical Ag-mediated synthetic route towards the crucial synthetic intermediate Cl-Lx-pip-AF (3d).3b pip = piperidinyl. | ||
This method has several drawbacks. First, Ag is a route-dependent human toxicant5 added intentionally during the manufacturing process, and therefore a risk assessment of such a process is necessary. Moreover, Ag+ has environmental issues6 and needs to be removed from the waste water, as it is highly toxic to a wide variety of organisms including bacteria.7 Second, large amounts of DMF are used: these “complexation” reactions are typically <0.01 M in substrate, and thus a large amount of the high boiling point solvent DMF needs to be removed under reduced pressure after completion of the reaction in order to concentrate the reaction mixtures before the purifications. This DMF removing step can be very tedious on scale. Third, despite the use of an excess of LxCl2 (2a) for activation with AgNO3, some double activation does always occur resulting in complexes bearing two payloads, and thus such double addition products need to be separated from the desired mono-addition products. And finally, the desired products need to be purified by preparative HPLC, known to be very difficult in large scale production.
Since we aimed to manufacture our lead candidate trastuzumab-Lx-pip-AF, which is obtained from the “semi-final” complex Cl-Lx-pip-AF (3d), for toxicological and clinical trials, it appeared necessary to improve the classical synthetic route (Scheme 3) to this crucial intermediate. We therefore aimed to develop a scalable and greener route that would (1) avoid usage of a Ag salt, (2) replace DMF or at least strongly reduce its amount, and (3) avoid preparative HPLC for product purification. Further, omission of a Ag salt would facilitate the product purification because Ag belongs to elements of class 2B category according to the ICH Q3D guideline on elemental impurities (parenteral intake limit of 10 μg day−1) and would therefore require stringent removal. The analytical product release would also be simpler since no Ag determination would be required in case no Ag salt additive is used. Also for the sake of making the process as green as possible, an additive such as a Ag salt, if it is not absolutely necessary, has to be avoided. Therefore, a straightforward Ag-free and environmentally benign production route appeared to be highly desirable.
2. Results and discussion
2.1 Reaction of the model compound noreleagnine with the dihalido Pt(II) complexes LxHal2
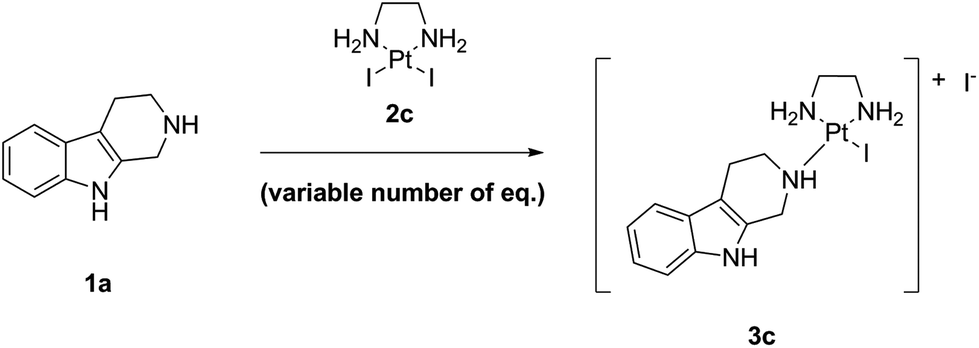
Having a clear goal to eliminate a Ag salt auxiliary and to achieve direct complex formation with the platinum(II) complex LxCl2 (2a), we first tried to react a model compound bearing the same coordination group as our lead candidate, i.e. a piperidine (pip) moiety. For this purpose, we chose the commercially available indole natural product noreleagnine (1a) and reacted it with an excess (1.5 eq.) of the complex LxCl2 (2a) under thermal conditions without AgNO3 (Scheme 4) in dry DMF (0.1 M in substrate 1a). It should be underlined that no further additives or reagents were added to the reaction mixture.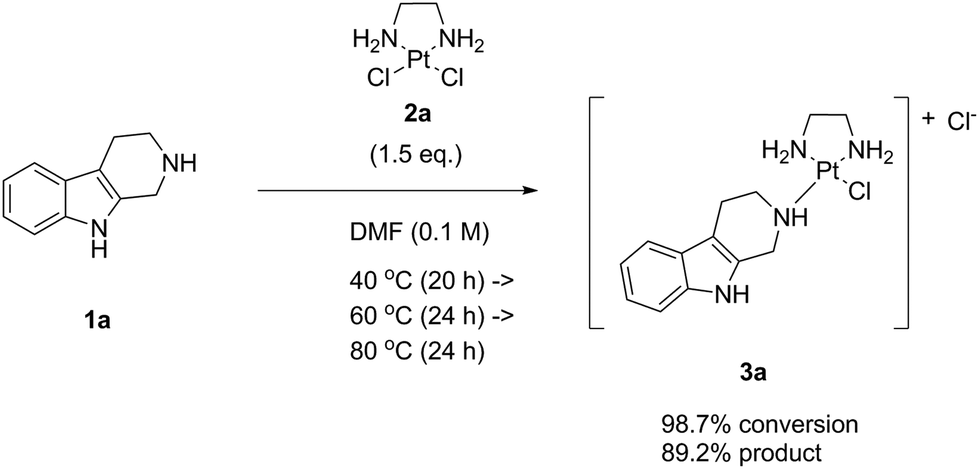 | ||
| Scheme 4 The Ag-free “complexation” reaction of noreleagnine (1a) with LxCl2 (2a). | ||
We started the reaction at 40 °C and increased the temperature step-wise up to 100 °C with increments of 20 °C monitoring the conversion of the reaction and the product formation in the reaction mixture by HPLC (ESI, 3.1.1.‡). The product formation is represented by the product peak area (a/a% value of the product 3a; a = area) on an HPLC chromatogram and the conversion is defined as product peak area divided by the sum of starting material and product peak areas [a/a% (product 3a)/a/a% (starting material 1a + product 3a)].
We observed that the reaction did occur but was not particularly clean. Because of the very low solubility of LxCl2 (2a), the reaction mixture remained heterogeneous during the whole course of the reaction until the end of the temperature program. Nevertheless, a high maximal conversion was achieved after 24 h at 80 °C (98.7%), while the product formation was 89.2%. Although this result already indicated that the process can be run without activation by Ag salts, the temperature at which the conversion became efficient (80 °C) was rather high considering our desire to develop a general procedure that would also be applicable for more sensitive substrates. Interestingly, at 100 °C we observed an increase of the noreleagnine (1a) peak compared to 80 °C, indicating that the product 3a apparently started to undergo a “de-complexation” process by which the chloride ligands that have been released into the mixture during the reaction started to convert the product back to noreleagnine 1a.
After this initial positive result, we reasoned that a replacement of the chlorido leaving ligand in our key complex Cl-Lx-CG-payload by its higher halido homologs, i.e. bromido or iodido leaving ligands, might be the key to achieve our goal. That would mean to perform the reaction of noreleagnine 1a with the corresponding complexes LxBr2 (2b) and LxI2 (2c), which we expected to be more soluble in DMF or other organic solvents then the classical hardly soluble complex LxCl2 (2a) (Scheme 5).
 | ||
| Scheme 5 The Ag-free “complexation” reaction of noreleagnine (1a) with LxHal2 complexes (2a–c). | ||
Indeed, since the halide ligand (Hal) will be lost anyway during the course of the subsequent conjugation reaction of the “semi-final” complex of compound class 3 to an antibody, chloride can potentially be replaced by another leaving ligand, provided that the nature of such a new leaving ligand will not negatively affect the conjugation efficiency. Alternatively, since the literature suggested that those bromido and iodido containing “semi-final” complexes could become less reactive towards conjugation,8 we assumed that we could also achieve a ligand exchange from the bromido or iodido leaving ligands back to the chlorido leaving ligand after the completed “complexation” reaction, simply by addition of an excess of a chloride salt.
To this end, as next we reacted noreleagnine 1a with an excess of the complex LxBr2 (2b) (Scheme 6). Similar to the experiment with LxCl2 (2a), we started the reaction at 40 °C and increased the temperature step-wise up to 80 °C with increments of 20 °C (ESI, 3.1.2.‡). The reaction time for each temperature step was the same as in the previous experiment with LxCl2 (2a). Remarkably, due to a higher solubility of LxBr2 (2b) compared to LxCl2 (2a), the reaction mixture became homogeneous already at 40 °C after stirring for several hours. Accordingly, we achieved a higher conversion (99.8%) and a better product formation (95.7%) at a lower temperature of 60 °C compared to the reaction with LxCl2 (2a). The “de-complexation” reaction in this experiment started to occur at 80 °C.
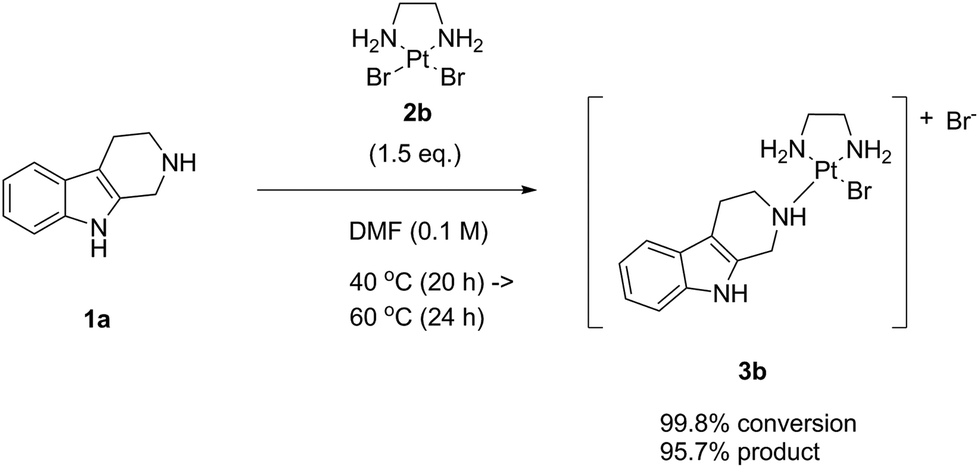 | ||
| Scheme 6 The Ag-free “complexation” reaction of noreleagnine (1a) with LxBr2 (2b). | ||
Finally, we performed the same experiment as performed with LxCl2 (2a) and LxBr2 (2b) with the synthetically readily available (ESI, 2.2. and 5.2.‡), also on a large scale (ESI, 6.5.‡), diiodido complex LxI2 (2c) (Scheme 7). Since this complex has a very high solubility in DMF, the reaction mixture was homogeneous already at room temperature just after mixing the components and adding the solvent.
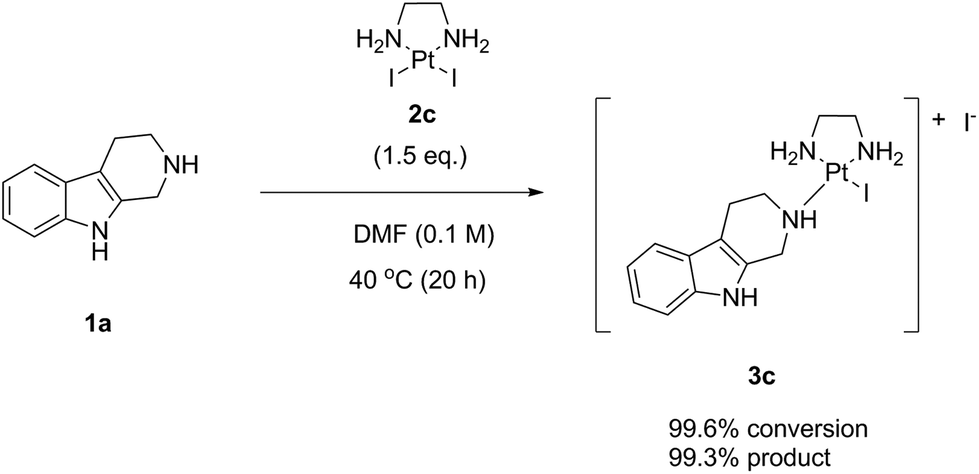 | ||
| Scheme 7 The Ag-free “complexation” reaction of noreleagnine (1a) with LxI2 (2c). | ||
The reaction with the complex 2c impressed with its almost quantitative conversion (99.6%) and high purity (99.3% product 3c, along with 0.4% starting material 1a) that were observed already at 40 °C (ESI, 3.1.3.‡). Its high efficiency and essentially lack of impurities (0.2%) suggested that the product could be isolated without preparative HPLC, which is a further improvement towards a greener alternative of the classical Ag-mediated “complexation” method. Thus, the complex 2c continued to follow the trend in the halide series, giving the highest reaction efficiency among the homologous complexes 2a–c at even lower temperature (40 °C). The “de-complexation” reaction started to occur at a lower temperature as well and was detectable already at 60 °C.
Because the reaction between noreleagnine (1a) and LxI2 (2c) was so efficient and clean, it was then repeated at 25 °C to investigate its efficiency already at ambient temperature (Fig. 1).
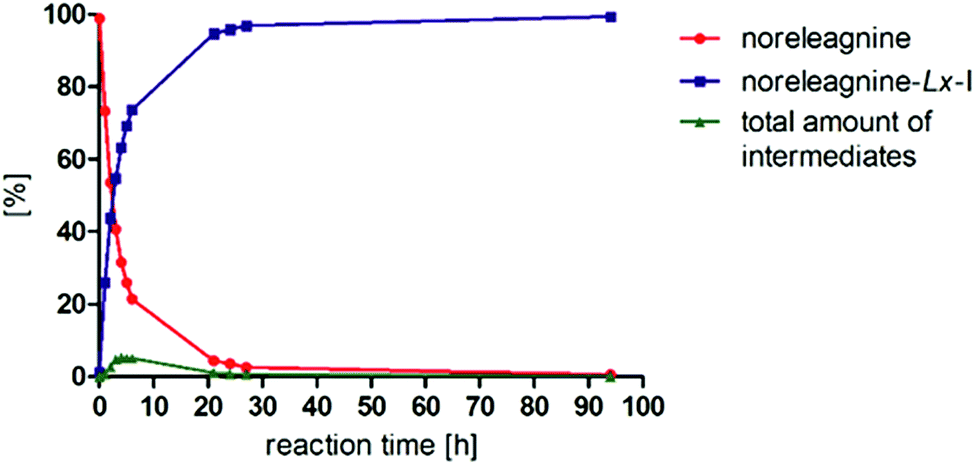 | ||
| Fig. 1 Reaction kinetics of noreleagnine (1a) with the diiodido complex LxI2 (2c) at 25 °C. | ||
We were pleased to find that the reaction proceeded smoothly and was almost complete after 24–27 h. At 24 h reaction time, 95.8% product 3c, 3.5% starting material 1a, and 0.7% intermediates or impurities were observed on HPLC. After that, the conversion improved further giving 99.4% product, 0.5% starting material, and only 0.1% intermediates or impurities after 94 h reaction time. Interestingly, the total a/a% of peaks that did not belong to the starting material 1a or product 3c increased to ∼5% in the first 4–6 h but decreased later to only 0.1% after 94 h. That indicated that initially some intermediates rather than impurities were formed, which were later transformed to the desired product 3c to give a very clean HPLC chromatogram (Fig. 2).
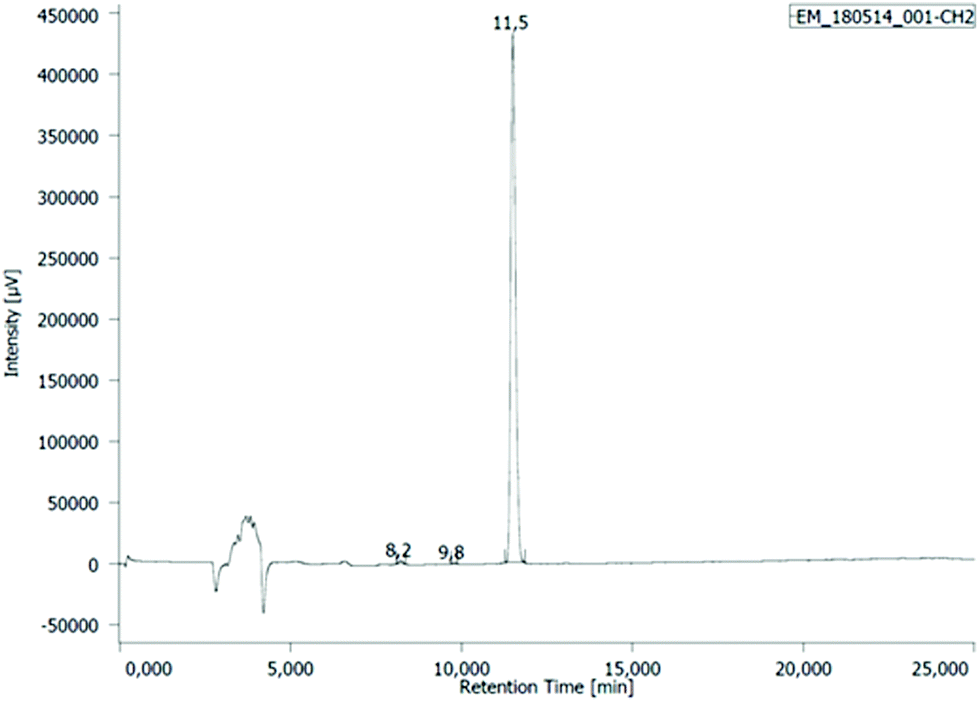 | ||
| Fig. 2 The HPLC chromatogram of the reaction mixture of noreleagnine (1a) with LxI2 (2c) at 25 °C after 94 h reaction time, measured at 273 nm. tR (1a) = 8.2 min (0.5 a/a%), tR (unidentified intermediate) = 9.8 min (0.1 a/a%), tR (3c) = 11.5 min (99.4 a/a%). | ||
The isolation of the desired product 3c was possible without using preparative HPLC simply by diluting the reaction mixture with water, followed by filtration of the precipitated unreacted/excessive LxI2 (2c) and applying the filtrate on the reverse phase silica gel RP-C18 (Merck LiChroprep (15–25 μm)). A yield of 85% was obtained in total, with the purest fraction (53% yield) having the product purity of 98.5% after addition of NaI (to improve the stability of the lyophilizate, vide infra) and lyophilization. This result indicated that manufacturing without the use of preparative HPLC for product purification is possible.
To look into the “de-complexation” reaction more closely, we performed a reaction of noreleagnine (1a) with LxI2 (2c) at 25 °C in the presence of different amounts of KI (0.2–5.0 eq.). After 69 h reaction time, it was observed that the amount of KI did not influence the conversion (ESI, 3.2.‡). Then, the temperature was increased step-wise to 40 °C, 60 °C, and 80 °C. It was observed that the conversion slightly decreased – also in the absence of KI due to the previously observed thermal “de-complexation” – but was again hardly influenced by the amount of KI (at 80 °C after 6 h reaction time, conversions were 87% and 82% for no KI added and 5.0 eq. KI added, respectively).
It should be noted that no undesired double addition product could be identified by HPLC for the “complexation” reactions using all three complexes LxHal22a–c, in contrast to the Ag-promoted procedure that is always plagued by the formation of the double-substituted product, which then needs to be separated from the desired mono-substituted product by preparative HPLC. To further challenge the formation of only the desired product 3c, we performed the reaction between 1.0 eq. of LxI2 (2c) and 3.0 eq. of noreleagnine (1a) at 40 °C. HRMS analysis indicated that no traces of the double addition product were formed even after 50 h reaction time.
2.2 Optimization of the “complexation” reaction between noreleagnine and LxI2
After these very encouraging results which indicated that the diiodido complex LxI2 (2c) might become the new Pt(II)-precursor of choice for the synthesis of Lx “semi-final” complexes of compound class 3, we decided to optimize this direct “complexation” reaction using noreleagnine (1a) and LxI2 (2c). Thus, we screened the following parameters that we considered to be of importance for the process upscaling: solvents, addition of an organic base, stoichiometry of the LxI2 complex (2c), and reaction molarity (amount of DMF used).For the initial solvent screening we made the following selection (we used dry solvents to avoid possible hydrolysis of the product 3c leading to formation of the hydrolyzed complex [H2O-Lx-noreleagnine]2+): DMF, DMA, MeOH, EtOH, MeCN, DMSO, acetone, ethyl acetate, 1-methyl-2-pyrrolidinone (NMP), 1,3-dimethyl-2-imidazolidinone (DMI), and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU). This initial screening was performed at 25 °C, the concentration was 0.1 M in noreleagnine (1a), and a strictly stoichiometric amount (i.e. 1.0 eq.) of LxI2 (2c) was used (Table 1, ESI, 3.3.‡).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Conversion (%) | ||
| 24 h | 48 h | 158 h | ||
| a The reaction time was 136 h instead of 158 h. | ||||
| 1 | DMF | 82.1 | 91.1 | 95.4 |
| 2 | DMF + 1 vol% H2O | 80.7 | 89.9 | 94.5 |
| 3 | DMF + 10 vol% H2O | 76.9 | 87.9 | 94.3 |
| 4 | DMA | 77.3 | 88.7 | 95.2a |
| 5 | MeOH | 1.3 | 2.6 | — |
| 6 | EtOH | 0.4 | 0.9 | — |
| 7 | MeCN | 23.8 | 23.6 | — |
| 8 | DMSO | 73.5 | 75.5 | 81.3 |
| 9 | Acetone | 12.0 | 16.1 | — |
| 10 | Ethyl acetate | 0.6 | 0.5 | — |
| 11 | NMP | 73.5 | 84.0 | — |
| 12 | DMI | 73.8 | 82.7 | — |
| 13 | DMPU | 65.8 | 79.6 | — |
Only in DMF, DMA, DMSO, NMP, DMI, and DMPU the mixtures became homogeneous; in the other solvents LxI2 (2c) did not dissolve and the reaction mixtures remained heterogeneous until the end of the experiment. However, reactions in NMP, DMI, and DMPU (entries 11–13) were found not clean. Correspondingly, the best conversions and cleanest reaction mixtures were observed in DMF, DMA, and DMSO (entries 1–4, and 8); in almost all of the solvents that remained heterogeneous the reactions showed either no conversion at all (e.g. in MeOH (entry 5), EtOH (entry 6), and ethyl acetate (entry 10) after 48 h reaction time) or a low conversion (16% in acetone (entry 9) and 24% in MeCN (entry 7) after 48 h reaction time). It should be noted that DMF that contained water performed similarly well compared with dry DMF (entries 1–3). To summarize, fastest, cleanest, and highest conversions were achieved in DMF (91%; entry 1), closely followed by the reaction in DMA (89%; entry 4) and DMSO (76%; entry 8) after 48 h reaction time. DMSO performed surprisingly well compared with DMF and DMA; however, at longer reaction times the reaction mixture became unclean. It is also known that DMSO, although being a very popular solvent for compounds to be tested in biological systems, is strongly discouraged to be used as a solvent for dissolution of cisplatin analogues.9a,b In all three solvents the formation of the desired product was confirmed by HRMS analyses. Therefore, at the end of this experiment, we decided to isolate product 3c from the reaction mixtures of the three best solvents DMF, DMA, and DMSO, and the isolated yields/purities of product 3c were 71%/97%, 71%/97%, and 47%/77% after preparative chromatographic purification and lyophilization, respectively.
Next, we decided to screen the “complexation” reaction in DMF analogues (other than DMA), i.e. solvents structurally similar to DMF. Thus, the following solvents were screened: N-formyl pyrrolidine, N-formyl piperidine, N-formyl morpholine, diethyl formamide, diisopropyl formamide, methyl formamide, formamide, and tetramethyl urea. Similarly to the previous experiment, this screening was performed at 25 °C, the concentration was 0.1 M in noreleagnine (1a), and a strictly stoichiometric amount (i.e. 1.0 eq.) of LxI2 (2c) was used (Table 2, ESI, 3.4.‡).
|
|
||||
|---|---|---|---|---|
| Entry | DMF analogue | Conversion (%) | ||
| 24 h | 45 h | 136 h | ||
| 1 | DMF (control) | 83.6 | 87.7 | 95.3 |
| 2 | N-Formyl pyrrolidine | 78.5 | 85.3 | 94.6 |
| 3 | N-Formyl piperidine | 82.5 | 87.7 | 93.6 |
| 4 | N-Formyl morpholine | 87.0 | 90.6 | 95.2 |
| 5 | Diethyl formamide | 55.3 | 66.7 | — |
| 6 | Diisopropyl formamide | 29.8 | 37.4 | — |
| 7 | Methyl formamide | 76.5 | 79.3 | 91.3 |
| 8 | Formamide | 14.2 | 18.9 | — |
| 9 | Tetramethyl urea | 70.1 | 80.3 | 89.8 |
In none of the solvents tested was the reaction clearly better than in DMF (that was used as a standard solvent in this experiment and showed a conversion of 95% after 136 h reaction time; entry 1), although five solvents showed conversions comparable to DMF (95%, 94%, 95%, 91%, and 90% in N-formyl pyrrolidine, N-formyl piperidine, N-formyl morpholine, methyl formamide, and tetramethyl urea, respectively; entries 2–4, 7, and 9). In diethyl formamide the reaction was much less efficient when compared to DMF (conversion of 55% was observed after 24 h reaction time (entry 5), compared to 84% in DMF (entry 1)). In diisopropyl formamide and in formamide the reactions showed low conversions (37% (entry 6) and 19% (entry 8) after 24 h reaction time, respectively) and the reaction mixtures remained heterogeneous until the end of the experiment. Thus, after this intensive solvent screening, DMF remained the solvent of choice for the “complexation” reaction.
Since payloads bearing amines or functionalized with amines as coordination groups are sometimes used in the form of their salts (e.g. as a TFA salt) rather than as free bases, we decided to investigate the influence of organic bases on the “complexation” reaction. Thus, we tested conversions in the presence of an excess (5.0 eq.) of following bases: triethylamine (TEA), diisopropylethylamine (DIPEA), N-methyl piperidine, N-methyl morpholine (NMM), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1,4-diazabicyclo[2.2.2]octane (DABCO), 2,6-lutidine, and 2,4,6-collidine. The screening was performed at 25 °C, the concentration was 0.1 M in noreleagnine (1a), and a strictly stoichiometric amount (i.e. 1.0 eq.) of LxI2 (2c) was used (Table 3, ESI, 3.5.‡).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Conversion (%) | ||
| 24 h | 48 h | 96 h | ||
| 1 | No base (control) | 83.9 | 91.2 | 94.6 |
| 2 | TEA | 62.6 | 77.0 | 85.6 |
| 3 | DIPEA | 44.4 | 64.6 | 78.0 |
| 4 | N-Me piperidine | 70.9 | 81.6 | 87.0 |
| 5 | NMM | 82.3 | 89.8 | 93.8 |
| 6 | DBU | 0.0 | 0.0 | 0.0 |
| 7 | DABCO | 66.0 | 78.9 | 78.1 |
| 8 | 2,6-Lutidine | 82.0 | 89.3 | 92.7 |
| 9 | 2,4,6-Collidine | 82.0 | 89.5 | 93.7 |
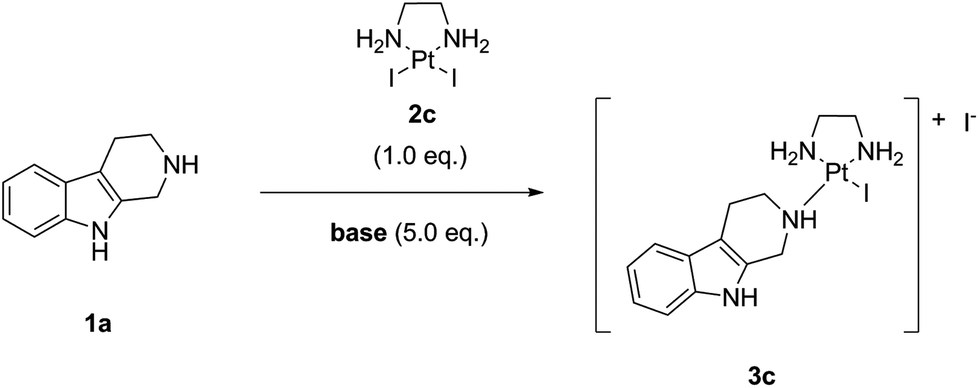
We found that the base had a pronounced effect on the “complexation” reaction with none of them improving the conversion. E.g. DBU completely prevented the formation of the desired product (entry 6). Compared to the “complexation” reaction without a base, only three bases showed a similar conversion and no additional impurities: N-methyl morpholine, 2,6-lutidine, and 2,4,6-collidine (entries 5, 8, and 9). TEA retarded the conversion (by ∼10% after 96 h reaction time, entry 2); however, it is also a stronger base compared with the ones above. So, for some salts this base will need to be used. DIPEA showed an even more pronounced retardation of the conversion (by ∼20% after 96 h reaction time, entry 3). Thus, its use in the “complexation” reaction is not recommendable.
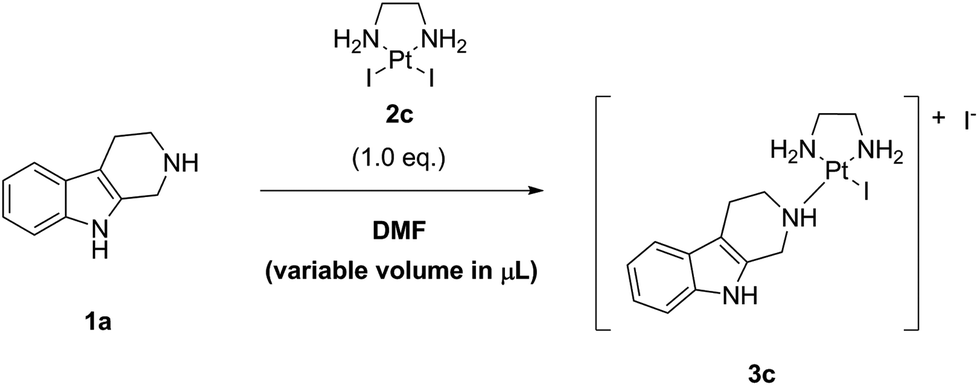
As next, we examined the stoichiometry of the reaction regarding LxI2 (2c), using different stoichiometries of LxI2 (2c), ranging from 1.0 to 5.0 eq. The screening was performed at 25 °C and the concentration was 0.1 M in noreleagnine (1a) (Table 4, ESI, 3.6.‡).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Eq. of LxI2 (2c) | Conversion (%) | |||
| 2 h | 24 h | 48 h | 72 h | ||
| 1 | 1.0 | 27.5 | 84.1 | 92.4 | 95.1 |
| 2 | 1.1 | 29.9 | 86.3 | 93.9 | 96.5 |
| 3 | 1.2 | 32.6 | 89.6 | 96.1 | 98.1 |
| 4 | 1.5 | 40.5 | 95.1 | 98.9 | 99.7 |
| 5 | 2.0 | 51.3 | 98.4 | 99.7 | 99.8 |
| 6 | 3.0 | 66.9 | 99.7 | 99.9 | 99.9 |
| 7 | 4.0 | 76.7 | 99.8 | 99.9 | 99.9 |
| 8 | 5.0 | 82.5 | 99.8 | 100 | 100 |
Expectedly, increasing amounts of LxI2 (2c) increased and accelerated the conversions. We found that for long reaction times, e.g. >72 h, already at 25 °C a conversion of >95% was possible using all the stoichiometries examined, even 1.0 eq. of LxI2 (2c). Using 1.5–2.0 eq. of LxI2 (2c), which is a preparatively reasonable amount also for working on large scale, conversions of >95% and of ∼100% were achieved after 24 h and 72 h reaction time, respectively (entries 4 and 5). It is noteworthy mentioning that the excess of LxI2 (2c) can be easily removed and recovered by simply diluting the reaction mixture with water and filtration of the thus formed precipitate (which also can be reused if it is desirable, vide infra).
As an indicator for the robustness of manufacturing, we also tested if there is a difference if the complex LxI2 (2c) is added as one batch (2.0 eq.) or e.g. in two batches (2 × 1.0 eq.; the second batch being added 24 h after the first batch). Two reactions performed in parallel revealed no difference in the final outcome, despite of course that the reaction with 2.0 eq. of LxI2 (2c) was faster for shorter reaction times (after 24 h the conversions were 98% (2.0 eq. LxI2 (2c)) and 81% (1.0 eq. LxI2 (2c))). Both reactions showed similar conversions (99.7% and 99.3%, respectively) and purity of the reaction mixtures (∼0.3–0.7% starting material, ∼99.3–99.7% product, and ∼0.0–0.1% intermediate) after 48 h reaction time (ESI, 3.7.‡). This result shows that if the conversion is not satisfactory, simply more LxI2 (2c) can be added; this observation can be of relevance for a large-scale manufacturing.
As next, we studied the influence of the concentration of noreleagnine (1a) and LxI2 (2c), i.e. the influence of the amount of solvent DMF (the standard concentration used in all the previous experiments was 0.1 M in noreleagnine 1a), on the reaction conversion. To this end, we used 11 different concentrations between 0.05 M and 1 M. The screening was performed at 25 °C and a strictly stoichiometric amount (i.e. 1.0 eq.) of LxI2 (2c) was used (Table 5, ESI, 3.8.‡).
|
|
||||
|---|---|---|---|---|
| Entry | V (DMF)/concentration | Conversion (%) | ||
| 24 h | 48 h | 144 h | ||
| 1 | 50 μL/1 M | 95.1 | 97.1 | 97.3 |
| 2 | 100 μL/0.5 M | 95.8 | 97.1 | 97.8 |
| 3 | 200 μL/0.25 M | 92.8 | 95.3 | 97.1 |
| 4 | 300 μL/0.167 M | 89.5 | 93.3 | 96.2 |
| 5 | 400 μL/0.125 M | 87.6 | 93.1 | 97.0 |
| 6 | 500 μL/0.1 M | 84.6 | 91.2 | 96.2 |
| 7 | 600 μL/0.083 M | 81.3 | 89.0 | 94.9 |
| 8 | 700 μL/0.071 M | 79.0 | 87.8 | 94.5 |
| 9 | 800 μL/0.063 M | 76.8 | 86.4 | 94.3 |
| 10 | 900 μL/0.056 M | 74.0 | 84.7 | 93.3 |
| 11 | 1000 μL/0.05 M | 71.8 | 83.4 | 92.8 |
We found that, as expected, the reaction was considerably faster at higher concentrations (95%, 85%, and 72% at 1 M, 0.1 M, and 0.05 M, respectively, after 24 h; entries 1, 6, and 11) but the conversions became comparable after a prolonged reaction time (97%, 96%, and 93% at 1 M, 0.1 M, and 0.05 M, respectively, after 144 h). It was comparably clean at all concentrations tested. However, at higher concentrations also a higher amount of an unidentified reactive intermediate was formed at short reaction times (5.5%, 0.7%, and 0.3% at 1 M, 0.1 M, and 0.05 M, respectively, after 24 h), which disappeared again during the course of the reaction. Therefore, the standard concentration of 0.1 M or higher concentrations up to 1 M (entries 1–6) appear reasonable to be used on the laboratory as well as on multigram scale, whereas working at concentrations lower than 0.1 M seems to be of no use (entries 7–11).
Finally, we performed an experiment in which the excess (∼1.0 eq.) of the Pt-precursor complex LxI2 (2c) was recovered and reused in three subsequent runs. The runs were performed at 25 °C (Table 6, ESI, 3.9.‡).
|
|
||||
|---|---|---|---|---|
| Run | Scale (2c)/[mg] | Conversion to 3c/(%) | Product purity of 3c/(%) | Recovery of 2c/(%) |
| 1 | 509 | 99.3 | 99.0 | 80 |
| 2 | 202 | 98.8 | 98.8 | 83 |
| 3 | 83 | 99.3 | 99.3 | 73 |
| 4 | 30 | 99.3 | 99.3 | 53 |
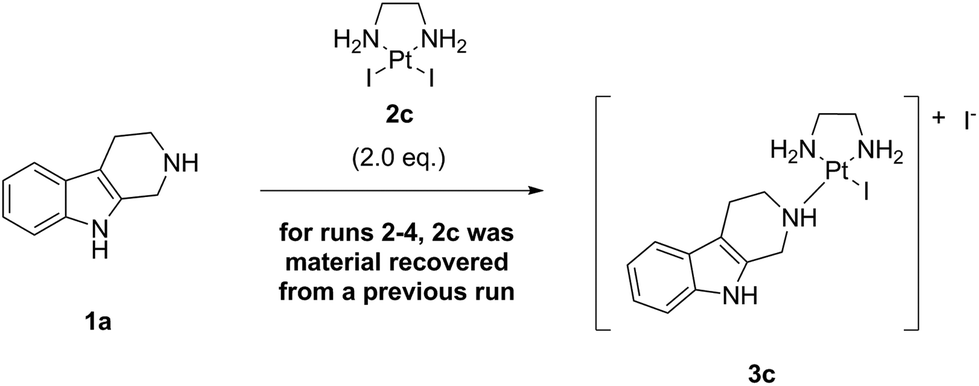
We could demonstrate that just by diluting the “complexation” reaction mixture with water (DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = 1:1) after 24 h reaction time and filtration of the thus formed precipitate, followed by washing and drying, ∼80% of LxI2 (2c) could be easily recovered (except for the last run 4, in which ∼53% 2c were recovered probably due to small amount of remaining material and thus a relatively high loss of material during the filtration step). The recovery can be further improved by applying a larger amount of an aqueous solution, as was demonstrated later during the manufacturing campaign: the complex LxI2 (2c) could be recovered quantitatively (ESI, 6.6.‡). The model experiment described above showed that the recovered LxI2 (2c) possessed a similar quality compared to the fresh material (used in run 1), as demonstrated by similar conversions (∼99%) and product purities (∼99%) obtained in these runs after 24 h at 25 °C. No Pt black was observed, even after the last run 4. The recovered material LxI2 (2c) only contained a small amount of the product 3c, as detected by analytical HPLC. Thus, our procedure allows to recover the excessive Pt-precursor 2c quantitatively from the reaction mixture, giving an opportunity to recycle or reuse the complex easily.
:water = 1:1) after 24 h reaction time and filtration of the thus formed precipitate, followed by washing and drying, ∼80% of LxI2 (2c) could be easily recovered (except for the last run 4, in which ∼53% 2c were recovered probably due to small amount of remaining material and thus a relatively high loss of material during the filtration step). The recovery can be further improved by applying a larger amount of an aqueous solution, as was demonstrated later during the manufacturing campaign: the complex LxI2 (2c) could be recovered quantitatively (ESI, 6.6.‡). The model experiment described above showed that the recovered LxI2 (2c) possessed a similar quality compared to the fresh material (used in run 1), as demonstrated by similar conversions (∼99%) and product purities (∼99%) obtained in these runs after 24 h at 25 °C. No Pt black was observed, even after the last run 4. The recovered material LxI2 (2c) only contained a small amount of the product 3c, as detected by analytical HPLC. Thus, our procedure allows to recover the excessive Pt-precursor 2c quantitatively from the reaction mixture, giving an opportunity to recycle or reuse the complex easily.
2.3 Application of the improved “complexation” reaction for the large scale synthesis of I-Lx-pip-AF
The described Ag-free amination procedure was successfully applied in our laboratory on a milligram scale (ESI, 5.‡) and 18.0 mg of the “semi-final” complex I-Lx-pip-AF (3e) × 2.0 TFA in 98.9% purity were obtained. Then, it was applied without issues on a multigram scale (Scheme 8, ESI, 6.‡) to produce 16.0 g of I-Lx-pip-AF (3e), formulated as a lyophilizate (ESI, 6.7.‡) with a composition I-Lx-pip-AF (3e) × 2.0 TFA × 2.5 NaI (in total, 22.6 g of this lyophilizate were obtained). It should be noted that NaI was added as an excipient in order to increase the product purity (the HPLC purity increased from 98.2%, which was the average purity of five combined batches, to 99.5%) and to ensure product stability during the solvent evaporation, presumably by preventing hydrolysis and/or converting the already hydrolysed product H2O-Lx-pip-AF back into the desired product 3e.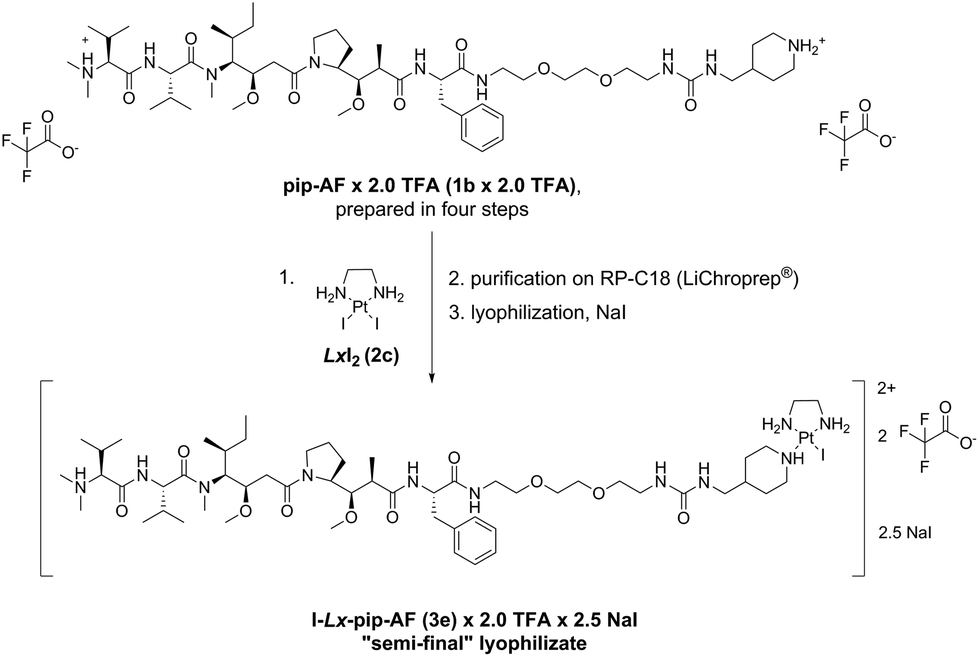 | ||
| Scheme 8 The new large scale manufacturing route towards the lyophilizate I-Lx-pip-AF (3e) × 2.0 TFA × 2.5 NaI. pip = piperidinyl, TFA = trifluoroacetic acid. | ||
2.4 Results of the conjugation of the next-generation “semi-final” complex I-Lx-pip-AF with trastuzumab
With the lyophilizate 3e in hands, we successfully synthesized the corresponding ADC 4 (Scheme 9) with unchanged properties compared with an ADC 4 synthesized from the chlorido “semi-final” complex 3d.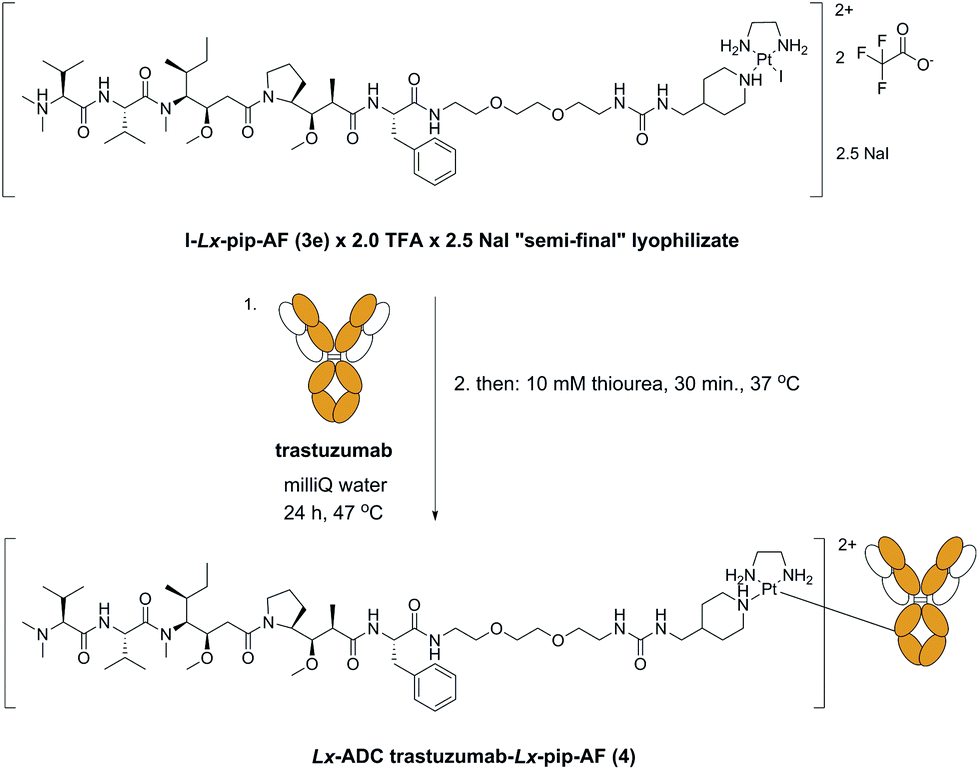 | ||
| Scheme 9 Synthesis of the Lx-ADC trastuzumab-Lx-pip-AF 4 from the lyophilizate I-Lx-pip-AF (3e) × 2.0 TFA × 2.5 NaI. pip = piperidinyl, TFA = trifluoroacetic acid. | ||
This result means that the kind of the leaving group does not affect the properties of the obtained Lx-ADCs of compound class 4. In fact, we found that conjugation of I-Lx-pip-AF (3e) to mAbs is much more efficient than the conjugation of Cl-Lx-pip-AF (3d); this unexpected improvement was found to be general and forms a counterintuitive separate finding10 that will be described in details elsewhere (manuscript in preparation). Thus, the whole Lx approach to ADCs improved greatly, with respect to the economics of manufacturing as well as to environmental and safety aspects, from this seemingly subtle change of the leaving ligand from chloride to iodide (Scheme 10).
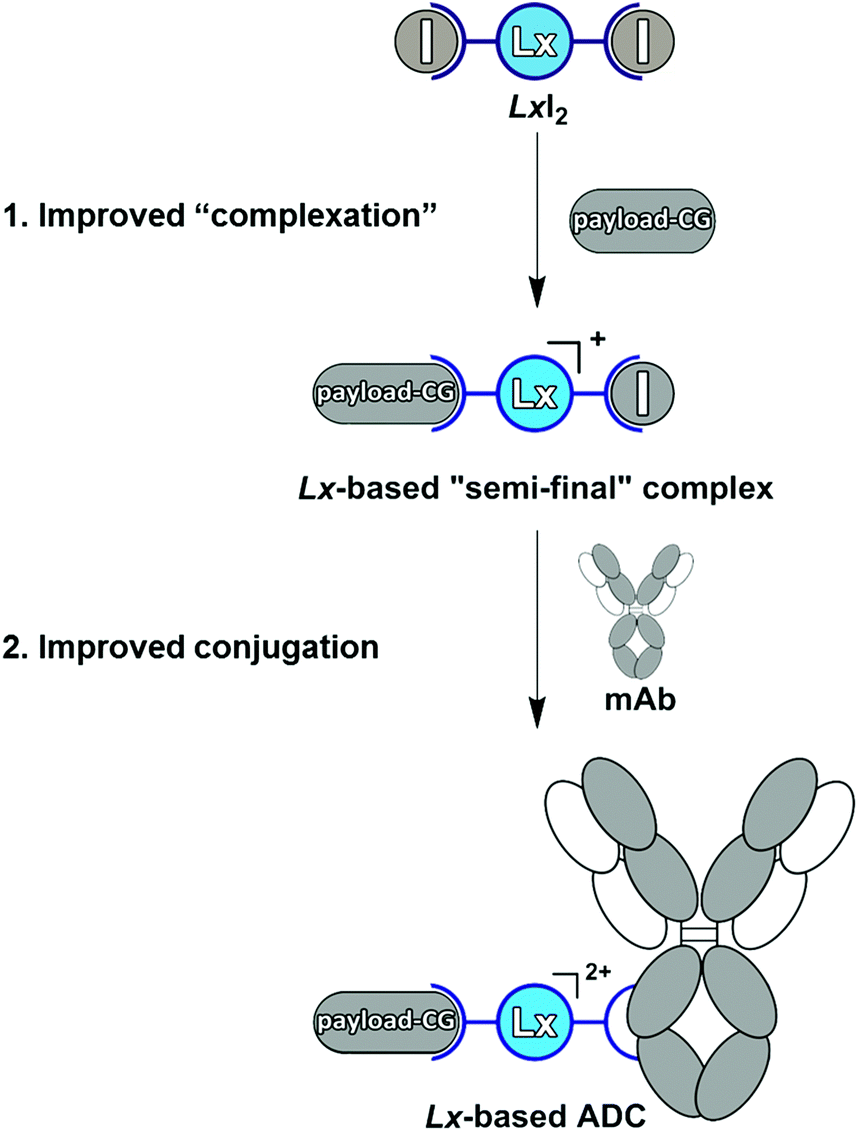 | ||
| Scheme 10 The improved Lx approach to ADCs operating with LxI2 (2c) instead of LxCl2 (2a). mAb = monoclonal antibody, CG = coordination group. | ||
The change of the Pt-precursor complex from LxCl2 (2a) to LxI2 (2c) not only gave an improvement of the overall Lx technology and allowed to omit Ag additives, as outlined above, but also allowed to reduce the costs of the Pt-precursor, thus making the technology even greener. Although LxCl2 (2a) is commercially available, its price is rather high: 367 € per 1 g (Aldrich, product code 244929). On the other hand, LxI2 (2c), although not commercially available, can be made very easily, in high yield (97.5%) and purity (the complex batches are consistently pure by elemental analysis), from the commercially available K2PtCl4, which price is much lower than that of LxCl2 (2a): 392 £ per 10 g (Fluorochem, product code 053555). LxI2 (2c) could be easily prepared on a 12 g scale during the first manufacturing campaign. Moreover, preparation of the diiodido complexes introduced modularity in respect to the Lx variants: the bidentate ligand can now be varied easily, thus giving access to a family of diiodido complexes, and is not limited to ethylenediamine as it is the case with the commercial LxCl2 (2a). These new Lx family members are currently under investigation and the results will be published elsewhere.
We are now planning to apply the described “complexation” method in the GMP manufacturing of the “semi-final” product 3e, followed by a GMP manufacturing of the corresponding Lx-ADC 4 for our ongoing clinical trials.
3. Conclusions
A greener (environmentally less demanding) and more practical route towards a crucial Pt(II) intermediate for ADC production bearing the cytotoxic payload auristatin F and an iodido leaving ligand, has been developed. This synthesis was successfully implemented on a multigram scale and the thus obtained important synthetic intermediate was successfully used in a subsequent bioconjugation reaction to obtain the Lx-based ADC trastuzumab-Lx-pip-AF. Additionally, we demonstrated that the excess of the Pt-diiodido complex that was used to achieve good conversions could be completely recovered, thus avoiding waste. The process demonstrated the following noteworthy improvements over the classical Ag-mediated route: (1) Ag-free direct and operationally simple (one operational step of Ag activation is omitted) amination of the complex Pt(en)I2 (LxI2); (2) improved chemical conversion (>95%), isolated yield, and product purity; (3) drastical reduction of solvent volume; (4) recovery and recyclability of the precursor LxI2 complex; and (5) formation of only the desired mono-substituted product with no double substitution, as confirmed by HPLC and HRMS. The product obtained during the described manufacturing campaign is being used successfully for the production of an anti-cancer antibody–drug conjugate (ADC) which will undergo toxicological and clinical studies in the near future. It turned out that changing the leaving ligand from chloride to iodide not only was a successful strategy in the “complexation” reaction but was also found to be a crucial invention11 for an improved conjugation efficiency.10 It should be mentioned that the Lx conjugation using the new generation of the iodido “semi-final” complexes described herein works generally and reliably with various payloads and mAbs and is not restricted to the herein presented noreleagnine and AF based “semi-final” complexes and trastuzumab as a mAb. This conjugation work will be described later elsewhere in a great detail. Finally, the obtained Ag-free procedure can be potentially applied in the synthesis of other Pt(II) or other transition metal complexes of high importance as anti-cancer therapeutics.12a–iConflicts of interest
E. Merkul, N. J. Sijbrandi, J. A. Muns, and I. Aydin are employed by LinXis B. V., H.-J. Houthoff is Scientific Director of LinXis B. V. and has ownership (including patents) in LinXis B. V., G. A. M. S. van Dongen is a non-profit scientific advisory board member of LinXis B. V. R. J. R. W. Peters and P. Laarhoven are employed by Ardena/ChemConnection B. V., which is a CDMO contracted by LinXis B. V. for the large-scale manufacturing.Acknowledgements
The authors thank Prof. Dr Jan Reedijk (Leiden University) for critical reading of the manuscript.Notes and references
- (a) P. D. Senter and E. L. Sievers, Nat. Biotechnol., 2012, 30, 631–637 CrossRef CAS PubMed; (b) J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed; (c) J. M. Lamb, Drugs, 2017, 77, 1603–1610 CrossRef PubMed; (d) P. M. Drake and D. Rabuka, BioDrugs, 2017, 31, 521–531 CrossRef PubMed.
- (a) H. Saber and J. K. Leighton, Regul. Toxicol. Pharmacol., 2015, 71, 444–452 CrossRef CAS PubMed; (b) M. Acchione, H. Kwon, C. M. Jochheim and W. M. Atkins, mAbs, 2012, 4, 362–372 CrossRef PubMed; (c) S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- (a) D. C. J. Waalboer, J. A. Muns, N. J. Sijbrandi, R. B. M. Schasfoort, R. Haselberg, G. W. Somsen, H.-J. Houthoff and G. A. M. S. van Dongen, ChemMedChem, 2015, 10, 797–803 CrossRef CAS PubMed; (b) N. J. Sijbrandi, E. Merkul, J. A. Muns, D. C. J. Waalboer, K. Adamzek, M. Bolijn, V. Montserrat, G. W. Somsen, R. Haselberg, P. J. G. M. Steverink, H.-J. Houthoff and G. A. M. S. van Dongen, Cancer Res., 2017, 77, 257–267 CrossRef CAS PubMed; (c) J. A. Muns, V. Montserrat, H.-J. Houthoff, K. Codée-van der Schilden, O. Zwaagstra, N. J. Sijbrandi, E. Merkul and G. A. M. S. van Dongen, J. Nucl. Med., 2018, 59, 1146–1151 CrossRef CAS PubMed; (d) E. Merkul, N. J. Sijbrandi, J. A. Muns, I. Aydin, K. Adamzek, H.-J. Houthoff, B. Nijmeijer and G. A. M. S. van Dongen, Expert Opin. Drug Delivery, 2019, 16, 783–793 CrossRef CAS PubMed; (e) V. del Solar and M. Contel, J. Inorg. Biochem., 2019, 199, 110780 CrossRef CAS PubMed.
- (a) J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed; (b) S. O. Doronina, T. D. Bovee, D. W. Meyer, J. B. Miyamoto, M. E. Anderson, C. A. Morris-Tilden and P. D. Senter, Bioconjugate Chem., 2008, 19, 1960–1963 CrossRef CAS PubMed.
- P. L. Drake and K. J. Hazelwood, Ann. Occup. Hyg., 2005, 49, 575–585 CAS.
- P. D. Howe and S. Dobson, Silver and silver compounds: environmental aspects, WHO Concise International Chemical Assessment Document 44, Geneva, 2002 Search PubMed.
- O. Choi, K. K. Deng, N.-J. Kim, L. Ross Jr., R. Y. Surampalli and Z. Hu, Water Res., 2008, 42, 3066–3074 CrossRef CAS PubMed.
- I. Berger, A. A. Nazarov, C. G. Hartinger, M. Groessl, S.-M. Valiahdi, M. A. Jakupec and B. K. Keppler, ChemMedChem, 2007, 2, 505–514 CrossRef CAS PubMed.
- (a) J. Josephsen, Inorg. Chim. Acta, 2018, 478, 54–58 CrossRef CAS; (b) M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed.
- E. Merkul, N. J. Sijbrandi, J. A. Muns, G. A. M. S. van Dongen, P. J. G. M. Steverink and H.-J. Houthoff, WO2019/125153A1, 2019.
- E. Merkul, N. J. Sijbrandi, J. A. Muns, G. A. M. S. van Dongen, P. J. G. M. Steverink and H.-J. Houthoff, WO2019/125154A2, 2019.
- (a) D.-L. Ma, C. Wu, S.-S. Cheng, F.-W. Lee, Q.-B. Han and C.-H. Leung, Int. J. Mol. Sci., 2019, 20, 341 CrossRef PubMed; (b) T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed; (c) M. Fanelli, M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2016, 310, 41–79 CrossRef CAS; (d) X. Han, J. Sun, Y. Wang and Z. He, Med. Res. Rev., 2015, 35, 1268–1299 CrossRef CAS PubMed; (e) J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS; (f) R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280–291 RSC; (g) J. Reedijk, Platinum Metals Rev., 2008, 52, 2–11 CrossRef CAS; (h) J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS PubMed; (i) J. Reedijk, Chem. Commun., 1996, 801–806 RSC.
Footnotes |
| † Dedicated to Lidia Aurelia Merkul on the occasion of her 5th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/C9GC03130H |
| This journal is © The Royal Society of Chemistry 2020 |