
Rhodium-terpyridine catalyzed redox-neutral depolymerization of lignin in water†
Yuxuan
Liu
ab,
Changzhi
Li
 *b,
Wang
Miao
a,
Weijun
Tang
a,
Dong
Xue
a,
Jianliang
Xiao
ac,
Tao
Zhang
b and
Chao
Wang
*a
*b,
Wang
Miao
a,
Weijun
Tang
a,
Dong
Xue
a,
Jianliang
Xiao
ac,
Tao
Zhang
b and
Chao
Wang
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an, 710119, China. E-mail: c.wang@snnu.edu.cn
bCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: licz@dicp.ac.cn
cDepartment of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK
First published on 20th November 2019
Abstract
Simple rhodium terpyridine complexes were found to be suitable catalysts for the redox neutral cleavage of lignin in water. Apart from cleaving lignin model compounds into ketones and phenols, the catalytic system could also be applied to depolymerize dioxasolv lignin and lignocellulose, affording aromatic ketones as the major monomer products. The (hemi)cellulose components in the lignocellulose sample remain almost intact during lignin depolymerization, providing an example of a “lignin-first” process under mild conditions. Mechanistic studies suggest that the reaction proceeds via a rhodium catalyzed hydrogen autotransfer process.
Aromatic compounds are important building blocks for the production of value-added chemicals for materials, fine chemicals and pharmaceuticals.1–4 Currently, aromatic chemicals are mainly produced from fossil resources. With diminishing fossil resources and the growing worldwide concern over this environmental problem, it is vital to find renewable approaches for the production of aromatic chemicals. Lignin is the largest aromatic heteropolymer in nature, accounting for 15–30 wt% of biomass.5,6 By virtue of this feature, lignin is considered to be a promising renewable source of aromatics that can alleviate the dependence on fossil resources.7,8 However, due to the complicated amorphous structure and highly heterogeneous nature of lignin, selective depolymerization of lignin remains a challenge.5,6,9–11 One approach is to develop an efficient catalytic system for the selective cleavage of the relatively unreactive C–O bonds in lignin, particularly the most abundant β-O-4 linkages.12–24 Despite the various elegant strategies reported for the chemical disassembly of lignin,19–21 the following major issues have not been well resolved in most cases: (1) in a certain number of processes, external oxidants9,10,17,25–29 or reductants12,13,21,30–37 are required, which are often hazardous and generate waste; (2) severe reaction conditions, such as high temperature and pressure, are usually required;9,14,31 (3) organic solvents, e.g. alcohols, toluene, and dioxane,18,22,23,31,38–41 are normally used, which does not meet the green chemistry criteria. A lignin depolymerization process requiring no external oxidant/reductant in aqueous media under mild conditions is desirable from the green chemistry viewpoint. Lignin contains a large number of hydroxyl groups in its sidechain,6,7,11 which have the potential to be dehydrogenated to provide hydrogen for the hydrogenolysis of C–O bonds. Thus, the redox-neutral cleavage or depolymerization of lignin could be realized, if a catalyst can transfer the hydrogen from the alcohol moieties in lignin to cleave its C–O bonds. This concept has been recently demonstrated by several groups with both homogeneous22,23,39,42–44 and heterogeneous19,20,30,45–48 catalytic systems. Despite these progresses, homogeneous catalytic systems with water as the solvent under mild conditions, which could be used for real lignin depolymerization, are still highly sought after. As native lignin is generally an insoluble solid, homogeneous catalytic systems might be more suitable for its depolymerization compared to heterogeneous catalytic systems in view of its mass transfer efficiency. Recently, we have demonstrated this hypothesis with a water soluble binuclear Rh complex catalytic system.24
In this work, structurally tunable terpyridine mononuclear Rh complexes are found to be efficient catalysts for the selective cleavage of lignin model compounds into ketones and phenols with water as the solvent under mild conditions. In addition, this robust catalytic system can be used for depolymerizing real lignin and raw woody biomass, affording aromatic ketones as the major monomer products. Notably, the lignin component in raw woody biomass could be selectively depolymerized, leaving the (hemi)cellulose components intact, providing an example of a homogeneous “lignin-first”49–51 process.
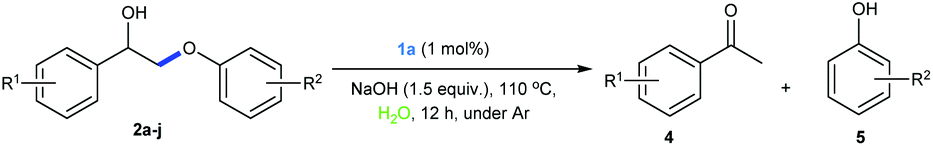
Terpyridine ligands have found broad applications in coordination chemistry and molecular recognition. Terpyridine metal complexes are oxidatively and thermally robust. However, examples of using terpyridine metal complexes as catalysts are few.52–57 Previously, we have shown that a binuclear Rh complex with terpyridine (2,2′:6′,2′′-terpyridine) ligand is an effective catalyst for dehydrogenation of alcohols,58 dehydrogenative coupling reactions,59 borrowing hydrogen reactions60 and aqueous redox-neutral depolymerization of lignin.24 Nevertheless, the structure of this binuclear Rh complex is difficult to modify, as the binuclear structure might not be formed upon changing the substituents on terpyridine. Crabtree and co-workers reported that Ru and Ir terpyridine complexes could catalyze borrowing hydrogen reactions between alcohols.54 We envisioned that structurally tunable simple Rh terpyridine complexes might be also employed as catalysts for hydrogen transfer reactions. We thus synthesized several Rh-terpyridine complexes (Table 1, 1a–1h) with different electronic and hydrophilic properties and tested them as catalysts for the redox-neutral cleavage of a lignin model compound (2a) in water. Gratifyingly, when the reaction was performed with 1 mol% 1a and 1 equivalent of NaOH in water at 110 °C for 12 h, the majority of 2a was cleaved into 4a (4-acetylanisole, 73% yield) and 5a (guaiacol, 77% yield), with a small amount of the dehydrogenation product 3 being obtained (Table 1, entry 1). Catalysts with electron-donating substituents showed better activity than the ones with electron-withdrawing groups (1avs.1c, and 1gvs.1h). During the reaction, we observed that catalyst 1a is insoluble in water. As the model substrate 2a also has low solubility in water, the cleavage reaction might take place in an “on water” manner, differing from our previous binuclear Rh catalytic system.24 We then increased the hydrophobicity of the catalysts by introducing more hydrophobic groups, e.g. 2-naphthyl (1d) and 9-anthracyl (1e). Although good conversion was obtained with 1d (Table 1, entry 4), low yield was observed with 1e (Table 1, entry 5), suggesting the requirement of a balanced hydrophobicity and hydrophilicity. Taken together, the above results suggest that ligands with electron-donating groups and conjugated groups with moderate hydrophobicity are good for this transformation. The amount of NaOH also affected the conversion and yields of the products of the reaction (Table 1, entries 9 and 10). Full conversion of 2a into 4a (84% yield) and 5a (87% yield) was achieved and a small amount of the dehydrogenated intermediate 3 was obtained, when the reaction was conducted with 1 mol% of catalyst 1a in the presence of 1.5 equivalents of NaOH (lower amount of base compared with our previous binuclear Rh system)24 in water at 110 °C for 12 h under an Ar atmosphere (Table 1, entry 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | NaOH(equivalent) | Conversion (%) | Yieldb (%) | ||
| 3 | 4a | 5a | ||||
| a Reaction conditions: 2a (0.2 mmol), catalyst (0.002 mmol), NaOH (0.2 mmol), H2O (1 mL), 110 °C, 12 h, under Ar. After the reaction, hydrochloric acid (1 M) was used to acidify the solution to pH = ca. 1. b Yields were determined by GC-FID with diphenyl as the internal standard. c NaOH (0.1 mmol). d NaOH (0.3 mmol). | ||||||
| 1 | 1a | 1 | 86 | 7 | 73 | 77 |
| 2 | 1b | 1 | 82 | 10 | 67 | 71 |
| 3 | 1c | 1 | 62 | 9 | 48 | 51 |
| 4 | 1d | 1 | 80 | 8 | 65 | 69 |
| 5 | 1e | 1 | 21 | 8 | 11 | 12 |
| 6 | 1f | 1 | 55 | 11 | 38 | 41 |
| 7 | 1g | 1 | 69 | 11 | 46 | 52 |
| 8 | 1h | 1 | 40 | 11 | 23 | 26 |
| 9c | 1a | 0.5 | 65 | 11 | 47 | 51 |
| 10d | 1a | 1.5 | 100 | 8 | 84 | 87 |

|
||||||

A variety of lignin model compounds were then tested to probe the versatility of the catalyst (Table 2). It was found that the methoxy groups at ortho-, meta- and para-positions on both phenyl rings, which are commonly found in natural lignin, are all tolerated under the optimized conditions (Table 2, entries 1–6). Full conversions were observed for these substrates. However, low activities were observed for substrates with a hydroxyl group on the α-phenyl ring; the starting substrates could not be fully consumed (67% conversion for 2g and 79% conversion for 2h) even with higher catalyst loading and prolonged reaction time (Table 2, entries 7 and 8). Delightfully, substrates with the γ-OH functionality (2i and 2j) were fully consumed, albeit affording relatively complex products (Table 2, entries 9 and 10). As the structures of 2i and 2j are closely related to that of real lignin, we then tested the catalytic system for the depolymerization of real lignin samples.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2 | R1 | R2 | Time (h) | Yieldb (%) | |
| a Reaction conditions: 2 (0.4 mmol), 1a (0.004 mmol), NaOH (0.6 mmol), H2O (1 mL), 110 °C, under Ar. After the reaction, hydrochloric acid (1 M) was used to acidify the solution to pH = ca. 1. b Isolated yields. c Yield was determined by GC-FID. d 1a (0.008 mmol). | ||||||
| 1 | 2a | 4-OMe | 2-OMe | 12 | 4a, 84 | 5a, 87 |
| 2 | 2b | — | 2-OMe | 12 | 4b, 76c | 5a, 90 |
| 3 | 2c | — | — | 12 | 4b, 79 | 5b, 76 |
| 4 | 2d | 3,4-OMe | 2-OMe | 12 | 4c, 83 | 5a, 85 |
| 5 | 2e | 3,4-OMe | 2,6-OMe | 12 | 4c, 87 | 5c, 90 |
| 6 | 2f | 3,4-OMe | 3,5-OMe | 12 | 4c, 82 | 5d, 86 |
| 7d | 2g | 4-OH | 2-OMe | 60 | 4d, 48 | 5a, 60 |
| 8d | 2h | 3-OMe, 4-OH | 2-OMe | 60 | 4e, 69 | 5a, 73 |
| 9 | 2i |

|
60 |

|
||
| 10 | 2j |

|
60 |

|
||
Dioxasolv lignin extracted from poplar wood was used to test the feasibility of the depolymerization of real lignin samples with the above developed catalytic system. Delightfully, the lignin can be effectively converted into an oil sample under the optimized catalytic conditions, with a yield of 90 wt%. MALDI-TOF analysis of the oil samples suggested that the molecular weights of the major products are in the range of 50–400 Da (Scheme S1†), indicating monomers, dimers and trimers to be the major products. Further analysis by GC-MS and GC-FID indicated that aromatic ketones were the dominant monomers with a yield of 10.8 wt% based on oil and 9.7 wt% based on the starting lignin solid (Scheme S2 and Table S3†).
The contents of the monomeric units and the percentage of different linkages of lignin were characterized by Two-Dimensional Heteronuclear Single Quantum Coherence NMR (2D HSQC NMR). Fig. 1 (I) shows that β-O-4 aryl ethers (A linkages) are dominant linkages (up to 87.7%), whilst the number of β-5 (B linkages, 6.2%) and β–β (C linkages, 6.1%) is very small in dioxasolv lignin. After the reaction, the 2D NMR spectra show that most of the A linkages have disappeared (Fig. 1, Ivs.III), indicating that the majority of the A linkages were cleaved by the catalytic system. The aromatic regions of the 2D-HSQC NMR spectra show that the S/S′ molar ratio has decreased from 68.3 to 1.2 (Fig. 1, IIvs.IV), suggestive of the dehydrogenation of the Cα–OH group. The dehydrogenation of the Cα–OH group is also supported by the presence of G′2, G′5 and G′6 as shown in Fig. 1 (IV). These results suggest that the depolymerization of real lignin could be achieved with the aqueous redox-neutral catalytic system.
 | ||
| Fig. 1 2D HSQC NMR spectra. I and II: dioxasolv poplar lignin; III and IV: oil obtained from the dioxasolv poplar lignin sample; V and VI: oil obtained from lignocellulose. | ||
To further demonstrate the versatility of this catalytic system, conversion of raw wood powder (poplar, 40 mesh, 1000 mg) without further pretreatment was conducted under the standard conditions using 1a as the catalyst. Impressively, 15 wt% of oil products (based on the starting raw wood powder) was obtained in water at 110 °C for 24 h, and 65 wt% (651 mg) of a solid residue was recovered.61 Noting that the lignin content in poplar wood powder is determined to be 18.6%,62 most of the lignin (80.6%) has been deconstructed into oil products. The solid residue could be easily separated from the oil products via simple filtration. Enzymatic hydrolysis of the solid residue yielded 72% of glucose and 20% of xylose, respectively (based on the weight of solid), which suggests that the solid residue mainly consists of cellulose and hemicellulose. The above results suggest that the Rh catalytic system is capable of “lignin-first” depolymerization49–51 of lignocellulose.
The oil product obtained from the raw wood powder was further analyzed by GC-MS, GC-FID and 2D HSQC NMR. The GC-MS and GC-FID analysis results indicate that the structures of the aromatic monomers are similar to those obtained from dioxasolv lignin oil (Scheme S2 vs. Scheme S4 and Table S3 vs. S5†). The total yield of the monomers is 16.7 wt% based on the oil and 2.5 wt% based on the starting raw poplar wood powder. Further, the 2D HSQC NMR analysis results show that most of the A linkages (β-O-4) have disappeared in the side-chain region (Fig. 1, Ivs.V) and certain amounts of S2,6′, G2′, G5′ and G6′ have appeared in the aromatic region of the oil products (Fig. 1, IIvs.VI), which are in agreement with the results of the dioxasolv lignin sample. The above results suggest that the Rh-terpyridine catalytic system is highly active and selective not only for the cleavage of lignin model compounds and real lignin depolymerization, but also for the woody biomass depolymerization in a “lignin-first” manner, affording aromatic ketones as the major monomer products. As aromatic ketones were obtained as the major monomers in the depolymerization of both real lignin and lignocellulose, the cleavage might follow a mechanism similar to that for the model substrates.
The mechanism for this Rh-terpyridine catalyzed redox neutral depolymerization was then considered. Upon cleavage of the model compound 2a, the dehydrogenated product 3a was obtained. Based on our previous study and the literature, 3a could serve as an intermediate for the cleavage reaction. Indeed, 3a was cleaved into ketone and phenol products in the presence of a hydrogen source (Table 3). These results suggest that the cleavage was initiated by dehydrogenation of the alcohol moiety into a ketone intermediate (with a weaker ether C–O bond compared with 2a, 55.9 vs. 69.2 kcal mol−1),16 followed by the reductive cleavage of the ether bond. Further deuterium labelling studies showed that the deuterium atom adjacent to the hydroxyl group in 2a′ was selectively transferred to the α-methyl group of the ketone product 4a′ (Scheme 1), possibly via a Rh–D intermediate. These observations are similar to our previous studies on binuclear Rh catalyzed lignin depolymerization.24 Thus, the key steps for this mononuclear Rh-terpyridine complex catalyzed lignin cleavage might be similar to those with the binuclear Rh complex.
 | ||
| Scheme 1 Deuterium labelling experiment. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Additive | Conversion (%) | Yieldb (%) | |
| 4a | 5a | |||
| a Reaction conditions: 3a (0.2 mmol), 1a (0.002 mmol), NaOH (0.3 mmol), H2O (1 mL), 12 h, under Ar. After the reaction, hydrochloric acid (1 M) was used to acidify the solution to pH = ca. 1. b Yields were determined by GC-FID with diphenyl as the internal standard. | ||||
| 1 | — | 9 | 4 | 7 |
| 2 | 1-Phenylethan-1-ol(0.2 mmol) | 80 | 73 | 78 |
| 3 | H2 (0.2 mmol) | 26 | 16 | 24 |

Substrates 2i and 2j with the γ-OH functionality gave complex products (Table 2, entries 9 and 10). Based on the above mechanistic studies, a plausible mechanism for the formation of these products as exemplified with 8 is proposed (Scheme 2). 8 is first dehydrogenated to produce a Rh hydride and 9; 9 could undergo a retro-aldol reaction to give 10 and formaldehyde (the formation of formaldehyde is confirmed by 1H NMR studies, see section 8.2 of the ESI for details†) or dehydration to afford 11; 10 could be cleaved by a Rh hydride to give 14 and 15; and 11 could be reduced by a Rh hydride to form 12, which could be further cleaved to give 13 and 14 (Scheme 2). Clearly, the hydrogen source in 8 is not sufficient to cleave itself completely into 13, 14, and 15. Thus, a substantial amount of 12 was produced. Various lines of evidence suggest that 1a remains mononuclear for this reaction (see section 8.3 in the ESI for details†).
 | ||
| Scheme 2 A plausible mechanism for the cleavage of 8. | ||
Conclusions
In conclusion, simple and structurally modular Rh terpyridine complexes are found to be effective catalysts for redox-neutral cleavage of the C–O bonds of β-O-4 lignin model compounds in water under mild conditions. The modularity of the catalyst structure allows us to find a catalyst with optimal electronic and hydrophobic properties for lignin cleavage, which requires less base additives (1.5 equiv. vs. 4 equiv.) and behaves differently (on water vs. in water) compared with our previous binuclear Rh catalytic system. The catalytic system could also be used for depolymerizing real lignin and raw poplar wood powder. The depolymerization of raw poplar wood powder proceeds in a “lignin-first” manner giving high yields of lignin oil products. The solid residue could be easily separated and enzymatically hydrolyzed to produce glucose and xylose.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Xiaozan Dai and Prof. Zongbao Kent Zhao for performing the enzymatic hydrolysis reaction. This research was supported by the 2017 Royal Society International Collaboration Award (IC170044), the National Natural Science Foundation of China (21773145, 21473109, 21690080, 21690083, and 21878288), the Science and Technology Program of Shaanxi Province (2016KJXX-26), Projects for the Academic Leaders and Academic Backbones, Shaanxi Normal University (16QNGG008), the 111 project (B14041), and the DNL cooperation fund CAS (DNL180302).Notes and references
- T. Dai, C. Li, L. Li, Z. K. Zhao, B. Zhang, Y. Cong and A. Wang, Angew. Chem., Int. Ed., 2018, 57, 1808–1812 CrossRef CAS PubMed.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS PubMed.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- Y. Xu, D. Liu and X. Liu, Appl. Catal., A, 2018, 552, 168–183 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- Z. R. Zhang, J. L. Song and B. X. Han, Chem. Rev., 2017, 10, 6834–6880 CrossRef PubMed.
- H. Guo, D. M. Miles-Barrett, A. R. Neal, T. Zhang, C. Li and N. J. Westwood, Chem. Sci., 2018, 9, 702–711 RSC.
- J. A. Melero, J. Iglesias and A. Garcia, Energy Environ. Sci., 2012, 5, 7393–7420 RSC.
- M. V. Galkin, C. Dahlstrand and J. S. M. Samec, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS PubMed.
- M. V. Galkin, S. Sawadjoon, V. Rohde, M. Dawange and J. S. M. Samec, ChemCatChem, 2014, 6, 179–184 CrossRef CAS.
- F. Gao, J. D. Webb, H. Sorek, D. E. Wemmer and J. F. Hartwig, ACS Catal., 2016, 6, 7385–7392 CrossRef CAS.
- S. H. Lim, K. Nahm, C. S. Ra, D. W. Cho, U. C. Yoon, J. A. Latham, D. Dunaway-Mariano and P. S. Mariano, J. Org. Chem., 2013, 78, 9431–9443 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC.
- N. Luo, M. Wang, H. Li, J. Zhang, T. Hou, H. Chen, X. Zhang, J. Lu and F. Wang, ACS Catal., 2017, 7, 4571–4580 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- W. Huo, W. Li, M. Zhang, W. Fan, H. M. Chang and H. Jameel, Catal. Lett., 2014, 144, 1159–1163 CrossRef CAS.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- Y. Liu, C. Li, W. Miao, W. Tang, D. Xue, C. Li, B. Zhang, J. Xiao, A. Wang, T. Zhang and C. Wang, ACS Catal., 2019, 9, 4441–4447 CrossRef CAS.
- S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. P. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- Z. P. Cai, J. X. Long, Y. W. Li, L. Ye, B. L. Yin, L. J. France, J. C. Dong, L. R. Zheng, H. Y. He, S. J. Liu, S. C. E. Tsang and X. H. Li, Chem, 2019, 5, 2365–2377 CAS.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed.
- J. Ji, H. Guo, C. Li, Z. Qi, B. Zhang, T. Dai, M. Jiang, C. Ren, A. Wang and T. Zhang, ChemCatChem, 2018, 10, 415–421 CrossRef CAS.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- I. Klein, C. Marcum, H. Kenttamaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttamaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC.
- H. R. Wu, J. L. Song, C. Xie, C. Y. Wu, C. J. Chen and B. X. Han, ACS Sustainable Chem. Eng., 2018, 6, 2872–2877 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2013, 15, 3049–3056 RSC.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- C. Crestini, M. C. Caponi, D. S. Argyropoulos and R. Saladino, Bioorg. Med. Chem., 2006, 14, 5292–5302 CrossRef CAS PubMed.
- S. C. Chmely, S. Kim, P. N. Ciesielski, G. Jiménez-Osés, R. S. Paton and G. T. Beckham, ACS Catal., 2013, 3, 963–974 CrossRef CAS.
- T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef CAS PubMed.
- D. Weickmann and B. Plietker, ChemCatChem, 2013, 5, 2170–2173 CrossRef CAS.
- X. Zhou, J. Mitra and T. B. Rauchfuss, ChemSusChem, 2014, 7, 1623–1626 CrossRef CAS PubMed.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2016, 9, 2074–2079 CrossRef CAS PubMed.
- C. S. Lancefield, L. W. Teunissen, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2018, 20, 3214–3221 RSC.
- A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed.
- R. G. Harms, I. I. E. Markovits, M. Drees, W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed.
- B. Zhang, C. Li, T. Dai, G. W. Huber, A. Wang and T. Zhang, RSC Adv., 2015, 5, 84967–84973 RSC.
- J. W. Zhang, G. P. Lu and C. Cai, Green Chem., 2017, 19, 4538–4543 RSC.
- Z. Cao, M. Dierks, M. T. Clough, I. B. Daltro de Castro and R. Rinaldi, Joule, 2018, 2, 1118–1133 CrossRef CAS PubMed.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci, 2017, 10, 1551–1557 RSC.
- R. Rinaldi, Joule, 2017, 1, 427–428 CrossRef CAS.
- S. Enthaler, B. Hagemann, G. Erre, K. Junge and M. Beller, Chem. – Asian J., 2006, 1, 598–604 CrossRef CAS PubMed.
- D. Gnanamgari and R. H. Crabtree, Organometallics, 2009, 28, 922–924 CrossRef CAS.
- D. Gnanamgari, C. H. Leung, N. D. Schley, S. T. Hilton and R. H. Crabtree, Org. Biomol. Chem., 2008, 6, 4442–4445 RSC.
- D. D. Joarder, S. Gayen, R. Sarkar, R. Bhattacharya, S. Roy and D. K. Maiti, J. Org. Chem., 2019, 84, 8468–8480 CrossRef CAS PubMed.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS PubMed.
- F. Shi, M. K. Tse and M. Beller, Chem. – Asian J., 2007, 2, 411–415 CrossRef CAS PubMed.
- X. Wang, C. Wang, Y. Liu and J. Xiao, Green Chem., 2016, 18, 4605–4610 RSC.
- J. Cheng, M. Zhu, C. Wang, J. Li, X. Jiang, Y. Wei, W. Tang, D. Xue and J. Xiao, Chem. Sci., 2016, 7, 4428–4434 RSC.
- J. Li, Y. Liu, W. Tang, D. Xue, C. Li, J. Xiao and C. Wang, Chem. – Eur. J., 2017, 23, 14445–14449 CrossRef CAS PubMed.
- The catalytic activity of 1a is superior to our previous Rh–Rh complex for depolymerization of raw poplar wood powder; see Table S4 in the ESI for details.†.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03057c |
| This journal is © The Royal Society of Chemistry 2020 |