
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen production from formic acid catalyzed by a phosphine free manganese complex: investigation and mechanistic insights†
Alexander
Léval
a,
Anastasiya
Agapova
a,
Christoph
Steinlechner
a,
Elisabetta
Alberico
 ab,
Henrik
Junge
a and
Matthias
Beller
*a
ab,
Henrik
Junge
a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein Straße 29a, Rostock, 18059, Germany. E-mail: matthias.beller@catalysis.de
bIstituto di Chimica Biomolecolare, Consiglio Nazionale delle Ricerche, tr. La Crucca 3, 07100 Sassari, Italy
First published on 20th January 2020
Abstract
Formic acid dehydrogenation (FAD) is considered as a promising process in the context of hydrogen storage. Its low toxicity, availability and convenient handling make FA attractive as a potential hydrogen carrier. To date, most promising catalysts have been based on noble metals, such as ruthenium and iridium. Efficient non-noble metal systems like iron were designed but manganese remains relatively unexplored for this transformation. In this work, we present a panel of phosphine free manganese catalysts which showed activity and stability in formic acid dehydrogenation. The most promising results were obtained with Mn(pyridine-imidazoline)(CO)3Br yielding >14 l of the H2/CO2 mixture and proved to be stable for more than 3 days. Additionally, this study provides insights into the mechanism of formic acid dehydrogenation. Kinetic experiments, Kinetic Isotopic Effect (KIE), in situ observations, NMR labeling experiments and pH monitoring allow us to propose a catalytic cycle for this transformation.
Introduction
A vast majority of today's energy sources are consumed much faster than it takes to produce them naturally on earth. In this context, transition to environmentally benign renewable sources is a growing field advocated by both academics and industries. Thus, several alternatives such as wind, solar and hydroelectric technologies are of growing importance. Unfortunately, they have an Achilles’ heel as they constitute intermittent energy sources.1 On the other hand, hydrogen, which can be easily produced by water electrolysis, is seriously considered as a better storable chemical energy vector. In this respect, safe storage and easy handling of H2 are of general interest for industrial supplying chains. Hitherto, Liquid Organic Hydrogen Carriers, also known as LOHCs, attracted much interest because of their convenient storage and transport.2 More specifically, cyclohexanes and heterocycles,3 ammonia,4 hydrazine,5 alcohols6 and formic acid (FA)7 showed encouraging results.8 The latter compound presents advantageous physicochemical properties and low toxicity enabling safe and easy handling, transportation and storage. Notably, FA can be reversibly generated by hydrogenation of carbon dioxide and subsequently dehydrogenated, thus making it an attractive carrier candidate. For the dehydrogenation process under mild conditions, homogeneous noble metal-based complexes, e.g., ruthenium,9 iridium10 and rhodium,11 constitute the state-of-the-art catalysts. For such molecularly defined systems, it has been shown that the structure of the ligand strongly influences the (de)hydrogenation reactions. Indeed, pH-tunable metal complexes bearing hydroxyl substituted bipyridine ligands led to significantly higher performance and stability.12 Additionally, complexes bearing tridentate phosphine–amine–phosphine pincer type ligands (PNP) reached very high TONs and TOFs.13 Obviously, regarding costs and availability, the use of precious metals and ligands possesses significant drawbacks for eventual applications. Consequently, in the last decade catalysts based on 3d metals attracted significant attention. As an example, our group reported the first iron based catalysts for FA dehydrogenation (DH) in 2010.14 In 2011, Milstein and co-workers described the first PNP pincer ligand with an iron metal center15 which initiated intensive further studies in this area. Apart from this, other systems including cobalt,16 aluminum,17 and boron18 have been subject to investigations. However, except for Boncella's recently reported complex (iPrPNiPrP)Mn(OOCH)(CO)2 for FA DH19 and the very recent studies on [(tBuPNNOP)Mn(CO)3] [Br],20 manganese remained quite unexplored for this transformation despite its abundance in the Earth crust. Furthermore, in the very recent year manganese has shown good results in other dehydrogenation transformations such as amine–alcohol coupling21 or methanol DH.22 Notably, all these complexes vide supra still make use of special, often highly sophisticated ligands. In fact, the costs of tailor-made PNP systems are determined by the ligand and not the metal. In nature, multi-dentate nitrogen ligands, such as porphyrins, are applied in combination with Fe (Heme A, B, C and O) or Co (vitamin B12) for various enzymatic redox processes. Inspired by this and the versatility of Mn based catalysts in DH reactions, as well as the very recent results in carbon dioxide hydrogenation using bipy-Mn systems;23 we became interested towards the development of biomimetic inspired Mn systems for the DH of FA. Thus, in this article, we present a panel of phosphine free, easily accessible and cheap manganese catalysts applicable for efficient DH of FA.Results and discussion
Investigations of the catalytic performance
In the past decade, noble metal complexes with heterocyclic ligands, especially those bearing an N–H moiety, showed high efficiency in the DH of formic acid24–27 or the hydrogenation of carbon dioxide.28,29 In this context, a panel of manganese complexes was synthesized and tested for the DH of FA (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Cat. | H2 + CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (mL) b (mL) |
TON (3 h) | TOF (h−1) |
|
a Reaction conditions: HCOOH (37 mmol), KOH (40 mmol), Mn catalyst (0.005 mmol), H2O (9 mL), triglyme (4 mL), Tset (92.5 °C).
b Time (180 min), light exclusion. Gas evolution monitored with manual burettes, corrected by the blank volume (2.7 mL) and content of the gas phase analyzed by gas chromatography (GC). Ratio H2:CO2 in all cases 1:1; CO content in between 78 ppm (quantification limit) and 2632 ppm. Experiments were performed at least twice (except entries 1–3) with reproducibility differences between 0.8 and 10%.
|
||||
| 1 | 1 | 4.1 | 17 | 6 |
| 2 | 2a | 12 | 50 | 17 |
| 3 | 2b | 4.7 | 19 | 6 |
| 4 | 3a | 37 | 151 | 50 |
| 5 | 3b | 43 | 175 | 58 |
| 6 | 3c | 54 | 220 | 73 |
| 7 | 4a | 138 | 564 | 188 |
| 8 | 4b | 134 | 549 | 183 |
| 9 | 4c | 141 | 577 | 192 |
| 10 | 5a | 136 | 556 | 185 |
| 11 | 5b | 116 | 473 | 158 |

Little gas evolution was noted when Mn(CO)5Br was used as a catalyst (entry 1). Similarly, bipyridine based complexes showed negligible activities (entries 2 and 3). In contrast, ligands containing an N–H moiety as a part of an aromatic heterocycle – such as pyrazole or imidazole – clearly showed improved gas evolution (entries 4, 5 and 6). Modification of the imidazole based ligand (entry 4) to the imidazoline based ligand (entry 7) resulted in further enhanced gas production. In the case of catalyst 3a, the electronic nonbonding pair on the nitrogen is involved in the aromaticity of the imidazole and the proton in the N–H moiety is more acidic (entry 4) unlike catalyst 4a (entry 7), where this electron doublet is localized on the nitrogen and thus not involved in the aromaticity. Extending the heterocycle from a 5 to a 6 membered ring did not benefit the reaction (entry 8). Interestingly, the N-methylated version of this pyridine-imidazoline based complex yielded almost identical results (entry 9). Therefore, catalysts 4a (entry 7) and 4c (entry 9) led to equally efficient activity, suggesting that H in N–H is not mandatory. Ultimately, systems bearing non-aromatic ligands such as 5a and 5b (entries 10 and 11) gave higher initial gas evolution but were prone to deactivation yielding lower activities (Fig. S5†). Finally, at the end of this iterative catalyst screening, the in situ generated complex 4a was tested, which yielded identical results compared to the use of the defined complex (Fig. S6†). We should note that despite the fact that catalyst 4c performed nearly the same as catalyst 4a, the latter was chosen for further optimization as the cost of ethylenediamine used to synthesize it is significantly cheaper than that of N-methylethylenediamine, used to prepare the former (entries 7 and 9). It is noteworthy to mention that Mn–CO bond cleavage induced by light deactivates the catalyst, as previously observed by our group.30 Thus, special care is needed to minimize light exposure.
Next, the influence of important reaction parameters was investigated (Table 2). As expected, the observed gas evolution increased along with the catalytic loading (entries 1–5). KOH base can be successfully changed for HCOOK (entry 6). Performing the DH of FA in various organic solvents showed that triglyme remained superior (entry 6) to dioxane (entry 9), NMP (entry 10), DMSO (entry 7) or DMF (entry 11). Interestingly, plotting the gas evolution over time indicated that the triglyme based reaction led to much higher initial gas evolution (Fig. S7†). Thus, the amount of triglyme was varied, but no significant impact was noted, except that in the absence of triglyme the catalyst was poorly soluble (Fig. S8†). Finally, different working temperatures were investigated (entries 11–14). Although the activity increased, it should be noted that heating the system would favor the dehydration of FA, leading to water and carbon monoxide, which is detrimental for fuel cell applications (Fig. S9†). Regarding the CO content, we observed that an increase in the catalytic loading and higher reaction temperatures lead to more significant amounts in the gas phase (Fig. S10,†Tables 1 and 2). Furthermore, variation of the catalyst and the solvent also leads to changes in the CO amounts.
| Entry | Loading (μmol) | Base | Co-solvent | T (°C) | H2 + CO2c,d (mL) |
|---|---|---|---|---|---|
|
a Reaction conditions: HCOOH (37 mmol), KOH (40 mmol), [Mn(imidazoline-pyridine)(CO)3Br], H2O (9 mL), triglyme (4 mL).
b Reaction conditions: HCOOH (5 mmol), HCOOK (32 mmol), [Mn(pyridine-imidazoline)(CO)3Br] (50 μmol), H2O (9 mL), Solvent (4 mL).
c Time (180 min), light exclusion. Gas evolution monitored with manual burettes, corrected by the blank volume (2.7 mL). Content of the gas phase analyzed by GC, ratio H2:CO2 in all cases 1:1, CO content in between 78 ppm (quantification limit) and 3908 ppm. Experiments were performed at least twice (except entries 5, 7–10) with reproducibility differences between 0.7 and 9.3% except entry 2 (21%).
d TONs, TOFs and conversions calculated based on the ratio of H2:CO2 between 1.3:1 and 2.4:1 (entries 2–6, 9–10 and 14) (ESI section 4†: “Calculation of the hydrogen volume, the TON and the TOF by the ratio of H2:CO2”).
|
|||||
| 1a | 5 | KOH | Triglyme | 92.5 | 138 |
| 2a | 12.5 | KOH | Triglyme | 92.5 | 175 |
| 3a | 25 | KOH | Triglyme | 92.5 | 223 |
| 4a | 50 | KOH | Triglyme | 92.5 | 337 |
| 5a | 370 | KOH | Triglyme | 92.5 | 721 |
| 6b | 50 | HCOOK | Triglyme | 92.5 | 373 |
| 7b | 50 | HCOOK | DMSO | 92.5 | 214 |
| 8b | 50 | HCOOK | Dioxane | 92.5 | 181 |
| 9b | 50 | KCOOK | NMP | 92.5 | 12 |
| 10b | 50 | KCOOK | DMF | 92.5 | 306 |
| 11b | 50 | KCOOK | Triglyme | 60 | 12 |
| 12b | 50 | KCOOK | Triglyme | 80 | 211 |
| 13b | 50 | KCOOK | Triglyme | 85 | 263 |
| 14b | 50 | KCOOK | Triglyme | 120 | 622 |
Impact of the pH
An interesting point was observed when plotting the gas evolution over time. As shown in Fig. S11† after 30 minutes, which corresponds to 8% conversion in FA, the reaction rate dramatically decreased. This unusual behavior is explained by a sudden pH change. To investigate this effect in more detail, we modified the initial conditions by varying the KOH concentration to modulate the starting pH (Fig. S12†). The results clearly showed that the system is most active at neutral pH. This points out a tricky parameter that needs to be tackled: the catalyst is efficiently active at neutral pH, while the conversion of FA will obviously lead to a pH increase in the system. In order to maintain the initial high kinetic rate the pH has to be imperatively kept lower than 7. To confirm this hypothesis, a pH monitoring experiment was carried out (Fig. 1). | ||
| Fig. 1 pH monitoring for the DH of FA. Reaction conditions: HCOOH (37 mmol), KOH (40 mmol), Mn(imidazoline-pyridine)(CO)3Br (50 μmol), H2O (9 mL), triglyme (4 mL), Tset (92.5 °C), time (180 min), light exclusion, gas evolution recorded with manual burettes, corrected by the blank volume (2.7 mL) and content of the gas phase analyzed by GC. | ||
As shown in Fig. 1, the initial phase of the reaction took place at pH 6 with a high gas evolution rate. After 30 minutes, the pH rose. Hence, maintaining the starting pH is necessary to maintain the original activity. Consequently, experiments with a phosphate buffer were carried out, but unfortunately a stable system could not be designed. The best results were obtained using a phosphate buffer with a strength of 1.5 mol L−1 reaching a TON of 338 at 46% conversion in 180 minutes (Fig. S13†). Higher concentrated phosphate buffers were not investigated since the concentration could not be increased. Even though the use of an organic base such as dimethyloctylamine (DMOA) or triethylamine (TEA) proved to be more suitable for hydrogen release, in these cases the attractive aqueous feature of the reaction was lost (Fig. S14†). Thus, alternatively we considered continuous flow experiments. Controlled dosage of FA in our system should permanently maintain the pH at a desired value and therefore lead to a steady and fast gas evolution over an extended period of time.
Towards a continuous flow process
In a preliminary investigation, FA was added to the system after 180 minutes, when the deactivation occurred (Fig. S14†). To our delight, we noted that gas evolution was resumed (1:1 H2/CO2 analyzed by GC). This result suggested that our catalyst did not irreversibly deactivate, but was rather out of its optimum pH zone as proposed above. Next, we concentrated our effort on designing a FA continuous dosage system. The initial experiments showed that injecting FA every 30 minutes yielded a stable and steady gas evolution for the given reaction time. Satisfying results were obtained both in a buffer system (Fig. S15†) and in water (Fig. S16†) reaching TONs of 584 (47% conversion, 1425 mL of H2:CO2) and 508 (41% conversion, 1240 mL of H2:CO2), respectively. Those periodic dosage experiments were transposed into a continuous flow, carried out on a longer time scale.
Finding an optimal rate to steadily maintain the rapid initial gas evolution (7 mL min−1) was challenging. Indeed, an imprecise dosage would irremediably lead to an accumulation of FA and the inhibition of the reaction. In this context, our best result – a TON of 5763 (67% conversion, 14073 mL of H2:CO2 mixture), was obtained after 45 hours. Unfortunately, even under these conditions deactivation slowly occurred and the gas evolution stopped (Fig. S17†). As pointed out before, accumulation of FA acidified the solution and inhibited the reaction as shown by the final pH (3.5). As a result, we performed a long term stability experiment with a lower dosage rate to tackle FA accumulation. In this context, a TON of 2637 was reached (78% conversion, 6442 mL of H2:CO2) within 87 hours. We should note that the gas evolution throughout the reaction is found to be constant on average (Fig. 2).
 | ||
| Fig. 2 Steady continuous dosage of FA. Reaction conditions: HCOOH (5 mmol), HCOOK (32 mmol), Mn(pyridine-imidazoline)(CO)3Br (0.05 mmol), water (9 mL), triglyme (4 mL), Tset (92.5 °C), time (87 h), HCOOH (133 mmol) added via an Infors Precidor Type 8003© syringe pump using a 50 mL Luer-lock© Hamilton© syringe connected to the reactor via a GL14 valve and a PTFE cannula. Rate of the addition: 0.002 mL min−1. Light exclusion, gas evolution monitored with automatic burettes and content of the gas phase analyzed by GC. The rate is calculated as follows: ΔV/Δt with t = 30 min. CO content: 732 ppm. | ||
Mechanistic studies
The dehydrogenation of formic acid has been widely explored for numerous metal complexes including iron,15,31,32 rhenium33 or ruthenium25,34,35 based PNP systems. More recently, mechanistic investigations of iridium21,26,27 and ruthenium25,36,37 catalysts bearing bisheterocyclic ligands revealed interesting features. Notably, the role of the ligand pointing out a proton relay transfer through an N–H moiety was believed to be crucial for this transformation.38,39 Based on these elegant studies, a proposal for the mechanism is shown in Fig. 3. First, dissociative substitution of I by HCOO− leads to the formate complex [LnMn–OOCH]II. Himeda and coworkers reported the formate complex as a starting point, for their highly active iridium catalyst.24 Furthermore, many other authors proposed this species as the resting state.38 Additionally, substitution of the leaving group (LG) by water has been proposed as an intermediate step.39 β-Hydride elimination releasing CO2 forms the hydride complex [LnMn–H]III.38 Protonation of the hydride results in the hydrogen complex [LnMn–H2]IV. This step is accepted as a protonation done by H2O, HCOOH or H3O+ species in solution.40 The active catalyst II is regenerated and H2 is released during the last step of the catalytic cycle. Milstein and co-workers41 followed by other groups32,40 reported the protonation of the hydride complex leading to [LnM–H2] which undergoes liberation of H2 and regeneration of [LnM–OOCH].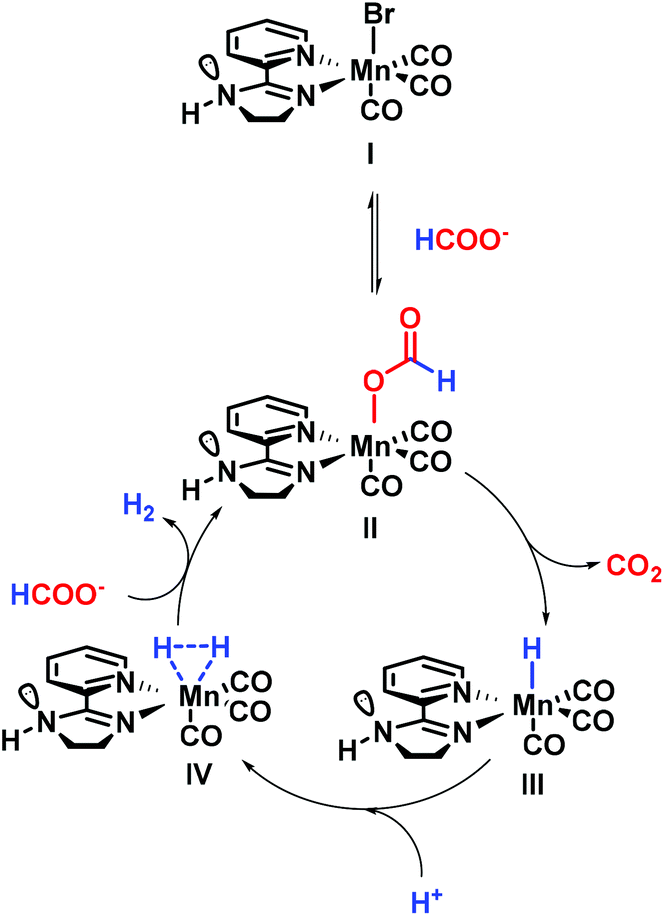 | ||
| Fig. 3 Proposed mechanism. | ||
Regarding the role of manganese in this transformation, it is important to consider the work of Gonsalvi's group, who reported the synthesis of a [Mn(PNPNH-iPr)(κ1-(OCOH))(CO)2] complex in their studies on the reverse reaction (CO2 hydrogenation42). In addition, the group of Boncella synthesized a similar complex [(iPrPNiPrP)Mn(OOCH)(CO)2] dehydrogenating FA.19 Additionally, manganese hydride complexes have previously been synthesized and characterized.43
Last but not least, previous investigations on FA dehydrogenation using noble metal complexes pointed out several subtle aspects that should be mentioned here. The computational studies performed by Zhang et al.44 showed the capacity of formic acid to form stable dimers. As a result, the transition state leading to the hydride protonation would be assisted by the N–H moiety. In a similar manner, J. Xiao, N. G. Berry and co-workers have proposed a mechanism involving formic acid pre-positioned, thanks to the N–H moiety, able to protonate the hydride complex.45 The role of this amine function was furthermore demonstrated by Ikariya and Kayaki where the N–H moiety assists in [M − H] protonation via a proton relay mechanism involving water, leading to the [M − H2] complex.46
Kinetics experiments and crystal structure of intermediate II
To gain additional insights into our specific transformation, investigations have been carried out to consolidate the mechanistic proposal presented in Fig. 3. First, general kinetic experiments were performed. Both reaction orders in manganese catalysts and formic acid/formate were found to be 1 (Fig. S21 and S22†). This means that one manganese complex and one entity of formate are involved in the catalytic cycle.We therefore cross out the eventuality of a dimer type mechanism. Then, a standard catalytic experiment was carried out and the reaction solution was extracted with an organic solvent. Liquid diffusion at −32 °C yielded X-ray crystals of the formate complex suitable for X-ray analysis (Fig. 4) (crystal data: Fig. S18, NMR data: Fig. S20†).
 | ||
| Fig. 4 Crystal structure of the formate complex [Mn-OOCH]. ORTEP representation of Mn(pyridine-imidazoline)(CO)3OOCH. Displacement ellipsoids correspond to 30% probability. | ||
To further confirm the hypothesis of the formate complex as the active species in this transformation, NMR studies of the reaction mixture were performed showing the major presence of the formate complex in solution (Fig. 5).
 | ||
| Fig. 5 In situ observation of [Mn-OOCH] by NMR. Reaction conditions: HCOOH (5 mmol), HCOOK (32 mmol), Mn(pyridine-imidazoline)(CO)3Br (0.05 mmol), water (9 mL), triglyme (4 mL). Tset (92.5 °C), time (3 h) Light exclusion, gas evolution monitored with manual burettes and the content of the gas phase analyzed by GC. NMR sample preparation: 0.3 mL of the triglyme phase was transferred to a GC vial and completed with 0.4 mL of DMSO-d6. 1H and 13C NMR spectra are recorded but nothing was observed in the carbon NMR due to dilution factors. NMR calibrated on the residual DMSO signal at 2.50 ppm for 1H and 39.52 ppm for 13C. | ||
The obtained spectrum (Fig. 5) revealed the characteristic signals of the pyridine at 9.04, 8.23, 8.12 and 7.75 ppm, respectively. Additionally, the broad signal at 8.91 ppm was assigned to the imidazoline proton. The singlet at 8.07 ppm is likely to be the formate signal. The imidazoline ring signals are not shown since they are overlapped by the triglyme signals. All those signals match the NMR data of the formate complex crystalized (Fig. S20†).
Labeling NMR scale experiment using H13COONa
Next, labeling experiments were carried out, using H13COONa as a formate source, on the NMR scale coupled to GC analysis. 1H NMR (Fig. S23†) showed the starting complex with several signals of a much lower intensity that are assigned to the formate complex [Mn–OO13CH], but accurate interpretation was not possible. Additionally, a hydride signal was observed at −17.3 ppm, which might be [Mn–H]. In the corresponding 13C NMR (Fig. S25†) the main signals of the starting material were observed along with side signals of a complex of much lower intensity that should belong to the formate complex. Additionally, 3 intense singlets, due to the labeling, were observed at 161.34 (13CO2 trapped as H13CO3−),47 172.2 and 171.6 ppm. The last two signals are attributed to the formate complex and free formate. 13C no dec. NMR (Fig. S26†) showed that the singlet at 161.3 ppm did not couple to any proton, which is in agreement with 13CO2 trapped as H13CO3−. Two formate signals (H13COO) were observed at 172.2 (J = 197.6 Hz) and 171.6 (J = 190.2 Hz). The signal at 171.6 ppm is identified as the H13COONa formate reagent (Fig. S27†), while [Mn–OO13CH] appears at 172.2 ppm. 2D correlation NMR indicated the correlation between Cformate (172.2 ppm) and Hformate (7.99 ppm) (Fig. S28 and S29†). 1H NOESY NMR (Fig. S30†) was performed to investigate the eventual correlation between Hformate and Himidazoline but nothing could be observed. Therefore, the signal at 7.99 ppm (172.2 ppm) corresponds to the formate complex. Ultimately, GC analysis showed both H2 and CO2 in the gas phase. Carbon monoxide is also detected resulting from the dehydration of formic acid. We can conclude that this experiment allowed us to directly and indirectly observe and confirm each step of the catalytic cycle proposed above (Fig. 2).Kinetic isotope effect
Ultimately, kinetic isotope effect (KIE) experiments have been carried out to investigate which is the rate limiting step for our catalyst. It has been suggested before that the rate limiting step in the presence of an Ir-catalyst is the β-hydride elimination or the hydride protonation.24 Our results are shown in Table 3.| Entry | Substrate | Solvent | TONb | KIEc |
|---|---|---|---|---|
| a Reaction conditions: HCOOH or DCOOD (1 mmol), HCOOK or DCOOK (6.4 mmol), Mn(pyridine-imidazoline)(CO)3Br (10 μmol), H2O/D2O (1.8 mL), triglyme (0.8 mL). Light exclusion, Tset (92.5 °C), time (180 min), gas evolution measured with manual burettes and gas mixture analyzed by GC. b TON calculated at 10 minutes. c KIE calculated at 10 minutes. | ||||
| 1 | HCOOH + HCOOK | H2O | 40 | — |
| 2 | HCOOH + HCOOK | D2O | 39 | 1.02 |
| 3 | DCOOD + DCOOK | H2O | 21 | 1.89 |
| 4 | DCOOD + DCOOK | D2O | 19 | 2.08 |
Using D2O instead of H2O (entry 2) does not really impact the outcome of the reaction. Therefore, we can conclude that the role of water is rather minor in this process. Hence, the idea of the hydride complex protonation by water being the rate limiting step could be excluded. However, using DCOOD and DCOOK instead of HCOOH and HCOOK showed a significant KIE effect (entry 3). This suggests that the β-hydride elimination of the formyl proton leading to the hydride complex might be the rate limiting step for this transformation.
Conclusions
A panel of manganese complexes bearing biomimetic κ2-NN type ligands was synthesized. Compared to previous manganese systems, catalyst 4a showed improved activity with a maximum TON of 5746 (corresponding to 14073 mL of H2:CO2 mixture; 67% conversion) under optimized conditions. The present complex is highly stable as shown by continuous hydrogen production for >3 days. Mechanistic investigations showed that, under aqueous conditions, consumption of FA led to the pH increase which can directly be correlated with a significant drop in the activity. Additional experiments such as varying the starting pH, using a buffer or periodically/continuously adding HCOOH, led us to the conclusion that those manganese type catalysts are efficient under neutral conditions. Crystallization of the formate complex directly from a crude reaction, NMR (13C labeling coupled to GC, in situ NMR) and kinetic experiments (reaction order, KIE) suggested β-hydride elimination of intermediate II as the rate limiting step for this transformation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the state of Mecklenburg-Vorpommern and the German Ministry of Education and Research (BMBF). Anastasiya Agapova is thankful for financial support provided by the German Federal Ministry for Economic Affairs and Energy (BMWi) within the project “Metha-Cycle” (03ET6071C). Christoph Steinleichner acknowledges financial support from EU fund H2020-MSCA-ITN-2015 in Horizon 2020 as part of the NoNoMeCat (Grant Agreement Number: 675020). We thank all members of the research group “catalysis for energy” (LIKAT) for scientific discussion and valuable suggestions. We thank PD Dr W. Baumann, Dr A. Fischer, Dr A. Spannenberg, S. Buchholz, S. Schareina, A. Lehmann, and K. Schubert for their technical and analytical support (all from LIKAT).Notes and references
-
V. Balzani and N. Armaroli, Energy for a Sustainable World: From the Oil Age to a Sun-Powered Future, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed
.
-
(a) W. Leitner, E. Dinjus and F. Gaßner, Activation of Carbon Dioxide: IV. Rhodium-Catalysed Hydrogenation of Carbon Dioxide to Formic Acid, J. Organomet. Chem., 1994, 475, 257–266 CrossRef CAS
- Q.-L. Zhu and Q. Xu, Liquid Organic and Inorganic Chemical Hydrides for High-Capacity Hydrogen Storage, Energy Environ. Sci., 2015, 8, 478–512 RSC
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, Ammonia for Hydrogen Storage: Challenges and Opportunities, J. Mater. Chem., 2008, 18, 2304–2310 RSC
- B. Peng and J. Chen, Ammonia Borane as an Efficient and Lightweight Hydrogen Storage Medium, Energy Environ. Sci., 2008, 1, 479–483 CAS
- M. Andérez-Fernandez, L. K. Vogt, S. Fisher, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, A Stable Manganese Pincer Catalyst for the Selective Dehydrogenation of Methanol, Angew. Chem., Int. Ed., 2017, 56, 559–562 CrossRef PubMed
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acids and Alcohols, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Formic Acid as a Hydrogen Storage Material − Development of Homogeneous Catalysts for Selective Hydrogen Release, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC
- Y. Gao, J. Kuncheria, R. J. Puddephatt and G. P. A. Yap, An Efficient Binuclear Catalyst for Decomposition of Formic Acid, Chem. Commun., 1998, 2365–2366 RSC
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Long-Range Metal−Ligand Bifunctional Catalysis: Cyclometallated Iridium Catalysts for the Mild and Rapid Dehydrogenation of Formic Acid, Chem. Sci., 2013, 4, 1234–1244 RSC
- Z. Wang, S.-M. Lu, J. Wu, C. Li and J. Xiao, Iodide-Promoted Dehydrogenation of Formic Acid on a Rhodium Complex, Eur. J. Inorg. Chem., 2016, 4, 490–496 CrossRef
-
(a) Y. Himeda, S. Miyazawa and T. Hirose, Interconversion between Formic Acid and H2/CO2 Using Rhodium and Ruthenium Catalysts for CO2 Fixation and H2 Storage, ChemSusChem, 2011, 4, 487–493 CrossRef CAS PubMed
-
(a) T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, Secondary Coordination Sphere Interactions Facilitate the Insertion Step in an Iridium(III) CO2 Reduction Catalyst, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed
- A. Boddien, B. Loges, F. Gärtner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, Iron-Catalyzed Hydrogen Production from Formic Acid, J. Am. Chem. Soc., 2010, 132, 8924–8934 CrossRef CAS PubMed
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Low-Pressure Hydrogenation of Carbon Dioxide Catalyzed by an Iron Pincer Complex Exhibiting Noble Metal Activity, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed
- X. Yang, Hydrogenation of Carbon Dioxide Catalyzed by PNP Pincer Iridium, Iron, and Cobalt Complexes: A Computational Design of Base Metal Catalysts, ACS Catal., 2011, 1, 849–854 CrossRef CAS
- T. W. Myers and L. A. Berben, Aluminium−Ligand Cooperation Promotes Selective Dehydrogenation of Formic Acid to H2 and CO2, Chem. Sci., 2014, 5, 2771–2777 RSC
- C. Chauvier, A. Tlili, C. Das Neves Gomes, P. Thuéry and T. Cantat, Metal-Free Dehydrogenation of Formic Acid to H2 and CO2 Using Boron-Based Catalysts, Chem. Sci., 2015, 6, 2938–2942 RSC
- A. M. Tondreau and J. M. Boncella, 1,2-Addition of Formic or Oxalic Acid to –N{CH2CH2(PiPr2)}2-Supported Mn(I) Dicarbonyl Complexes and the Manganese-Mediated Decomposition of Formic Acid, Organometallics, 2016, 35, 2049–2052 CrossRef CAS
- N. Anderson, J. Boncella and A. N. Tondreau, Manganese-Mediated Formic Acid Dehydrogenation, Chem. – Eur. J., 2019, 25, 1–5 CrossRef
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, B. Y. David, N. A. E. Jalapa and D. Mistein, Manganese-Catalyzed Environmentally Benign Dehydrogenative Coupling of Alcohols and Amines to Form Aldimines and H2: A Catalytic and Mechanistic Study, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed
- M. Andérez-Fernandez, L. Vogt, S. Fischer, W. Zhou, H. Jao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, A Stable Manganese Pincer Catalyst for the Selective Dehydrogenation of Methanol, Angew. Chem., Int. Ed., 2017, 56, 559–562 CrossRef PubMed
- A. Dubey, L. Nencini, R. Fayzullin, C. Nervi and J. R. Khusnutdinova, Bio-Inspired (Mn) Complexes for the hydrogenation of CO2 to Formate and Formamide, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS
- W. H. Wang, M. Z. Ertem, S. Xu, O. Naoya, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, Highly Robust Hydrogen Generation by Bioinspired Ir Complexes for Dehydrogenation of Formic Acid in Water: Experimental and Theoretical Mechanistic Investigations at Different pH, ACS Catal., 2015, 5, 5496–5504 CrossRef CAS
- C. Guan, D.-D. Zhang, Y. Pan, M. Igushi, M. J. Ajitha, J. Hu, H. Li, C. Yao, M.-H. Huang, S. Min, J. Zheng, Y. Himeda, H. Kawanami and K.-W. Huang, Dehydrogenation of Formic Acid Catalyzed by a Ruthenium Complex with an N,N′-Diimine Ligand, Inorg. Chem., 2017, 56, 438–445 CrossRef CAS PubMed
- A. Iturmendi, L. Rubio-Perez, J.-J. Perez-Torrente, M. Iglesias and A. L. Oro, Impact of Protic Ligands in the Ir-Catalyzed Dehydrogenation of Formic Acid in Water, Organometallics, 2018, 37, 3611–3618 CrossRef CAS
- P. Zhang, Y.-J. Guo, J. Chen, Y.-R. Zhao, J. Chang, H. Junge, M. Beller and Y. Li, Streamlined hydrogen production from biomass, Nat. Catal., 2018, 1, 332–338 CrossRef CAS
- S.-M. Lu, Z. Wang, J. Li, J. Xiao and C. Li, Base-free hydrogenation of CO2 to formic acid in water with an iridium complex bearing a N,N′-diimine ligand, Green Chem., 2016, 18, 4553–4558 RSC
- S.-M. Lu, Z. Wang, J. Wang, J. Li and L. Can, Hydrogen generation from formic acid decomposition on a highly efficient iridium catalyst bearing a diaminoglyoxime ligand, Green Chem., 2018, 20, 1835–1840 RSC
- C. Steinleichner, A. F. Roesel, E. Oberem, A. Päpcke, N. Rockstroh, F. Gloagen, S. Lochbrunner, R. Ludwig, A. Spannenberg, H. Junge, R. Francke and M. Beller, Selective Earth-Abundant System for CO2 Reduction: Comparing Photo- and Electrocatalytic Processes, ACS Catal., 2019, 9, 2091–2100 CrossRef
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Efficient Dehydrogenation of Formic Acid Using an Iron Catalyst, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed
- E. A. Bielinsky, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, P. O. Bernskoetter, N. Hazari and S. Schneider, Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef PubMed
- M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Reversible CO2 binding triggered by metal–ligand cooperation in a rhenium(I) PNP Pincer-type complex and the reaction with dihydrogen, Chem. Sci., 2014, 5, 2043–2051 RSC
- S.-F. Hsu, S. Rommel, P. Eversfield, K. Muller, E. Klemm, W. R. Thiel and B. Plietker, A Rechargeable Hydrogen Battery Based on Ru Catalysis, Angew. Chem., Int. Ed., 2014, 53, 7074–7078 CrossRef CAS PubMed
- S. Y. De Boer, T. J. Korstanje, S. R. La Rooij, R. Kox, J. N. H. Reek and J. I. Van der Vlugt, Ruthenium PNN(O) Complexes: Cooperative Reactivity and Application as Catalysts for Acceptorless Dehydrogenative Coupling Reactions, Organometallics, 2017, 36, 1541–1549 CrossRef CAS PubMed
- A. Boddien, B. Loges, H. Junge, F. Gärtner, J. R. Noyes and M. Beller, Continuous Hydrogen Generation from Formic Acid: Highly Active and Stable Ruthenium Catalysts, Adv. Synth. Catal., 2009, 351, 2517–2520 CrossRef CAS
- P. Prichatz, M. Trincado, L. Tan, F. Casas, A. Kammer, H. Junge, M. Beller and H. Grützmacher, Highly Efficient Base-Free Dehydrogenation of Formic Acid at Low Temperature, ChemSusChem, 2018, 11, 1–5 CrossRef PubMed
- M. Iglesias and L. A. Oro, Mechanistic Considerations on Homogeneously Catalyzed Formic Acid Dehydrogenation, Eur. J. Inorg. Chem., 2018, 20, 2125–2138 CrossRef
- C. Guan, D.-D. Zhang, Y. Pan, M. Iguchi, M. J. Ajitha, J. Hu, H. Li, C. Yao, M.-H. Huang, S. Min, J. Zheng, Y. Himeda, H. Kawanami and K.-W. Huang, Dehydrogenation of Formic Acid Catalyzed by a Ruthenium Complex with an N,N′-Diimine Ligand, Inorg. Chem., 2017, 56, 438–445 CrossRef CAS PubMed
- Z. Wang, S. M. Lu, J. Li, J. Wang and C. Li, Unprecedentedly High Formic Acid Dehydrogenation Activity on an Iridium Complex with an N,N′-Diimine Ligand in Water, Chem. – Eur. J., 2015, 21, 12592–12595 CrossRef CAS PubMed
- T. Zell, B. Butschke, Y. Ben-David and D. Milstein, Efficient Hydrogen Liberation from Formic Acid Catalyzed by a Well-Defined Iron Pincer Complex under Mild Conditions, Chem. – Eur. J., 2013, 19, 8068–8072 CrossRef CAS PubMed
- F. Bertini, M. Glatz, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Carbon dioxide hydrogenation catalysed by well-defined Mn(I) PNP pincer hydride complexes, Chem. Sci., 2017, 8, 5024–5029 RSC
- M. Mastali, M. Glatz, M. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros and K. Kirchner, Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride Mn(I) and Fe(II) PNP Pincer Complexes, Chem. – Eur. J., 2016, 22, 12316–12320 CrossRef PubMed
- J. Li, J. Li, D. Zhang and C. Liu, DFT Study on the Mechanism of Formic Acid Decomposition by a Well-Defined Bifunctional Cyclometalated Iridium(III) Catalyst: Self-Assisted Concerted Dehydrogenation via Long-Range Intermolecular Hydrogen Migration, ACS Catal., 2016, 6, 4746–4754 CrossRef CAS
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Long-range metal–ligand bifunctional catalysis: cyclometallated iridium catalysts for the mild and rapid dehydrogenation of formic acid, Chem. Sci., 2013, 4, 1234–1244 RSC
- A. Matsunami, Y. Kayaki and T. Ikariya, Enhanced Hydrogen Generation from Formic Acid by Half-Sandwich Iridium(III) Complexes with Metal/NH Bifunctionality: A Pronounced Switch from Transfer Hydrogenation, Chem. – Eur. J., 2015, 21, 13513–13517 CrossRef CAS PubMed
- J. Seravalli and S. W. Ragsdale,
13C NMR Characterization of an Exchange Reaction between CO and CO2 Catalyzed by Carbon Monoxide Dehydrogenase, Biochemistry, 2008, 47, 6770–6781 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: General methods and equipment, DH of FA procedures, calculation of TON and TOF, gas evolution plots, mechanistic elucidations experiments along with analytical data, synthesis of the ligand and catalysts. Crystallographic data for complex 4a and crystallographic data for the intermediate Mn(pyridine-imidazoline)(CO)3OCOH. CCDC 1922221 and 1922222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9gc02453k |
| This journal is © The Royal Society of Chemistry 2020 |