
Type I collagen promotes the migration and myogenic differentiation of C2C12 myoblasts via the release of interleukin-6 mediated by FAK/NF-κB p65 activation
Xiaoling
Liu
a,
Yanfang
Gao
a,
Xinyu
Long
a,
Toshihiko
Hayashi
ab,
Kazunori
Mizuno
c,
Shunji
Hattori
c,
Hitomi
Fujisaki
c,
Takayuki
Ogura
c,
Dan Ohtan
Wang
a and
Takashi
Ikejima
 *ad
*ad
aWuya College of Innovation, Shenyang Pharmaceutical University, Shenyang, 110016, China. E-mail: ikejimat@vip.sina.com; Fax: +86-24-81073882; Tel: +86-24-81073882
bDepartment of Chemistry and Life Science, School of Advanced Engineering, Kogakuin University, 2665-1, Nakanomachi, Hachioji, Tokyo 192-0015, Japan
cNippi Research Institute of Biomatrix, Ibaraki, 302-0017, Japan
dKey Laboratory of Computational Chemistry-Based Natural Antitumor Drug Research & Development, Shenyang Pharmaceutical University, Shenyang 110016, Liaoning, P.R. China
First published on 20th November 2019
Abstract
Skeletal muscle regeneration is a complicated process, requiring the proliferation, migration and differentiation of myoblasts whose processes are highly regulated by the extracellular matrix (ECM) surrounding the muscle tissues in vivo. However, the effects of respective ECM components on the regulation of myoblast behaviors are unknown. In this study, we report on the effect of collagen I, a major ECM component in muscle tissue and a popular food supplement, on mouse C2C12 myoblast proliferation, migration and differentiation as well as the underlying mechanisms. Collagen I (col 1) enhances the migration and myogenic differentiation of C2C12 cells, but has no effect on cell proliferation. Col I significantly promotes the production and release of interleukin-6 via nuclear translocation of nuclear factor κB (NF-κB) p65. The release of IL-6 plays a critical role in the col I-enhanced migration and differentiation of C2C12 cells. Furthermore, col I increases phosphorylation of focal adhesion kinase (FAK) that is involved in the nuclear translocation of NF-κB p65. Collectively, col I enhances the migration and differentiation of C2C12 cells through IL-6 release induced by FAK/NF-κB p65 activation.
Introduction
Skeletal muscle tissues have the capacity to repair minor injuries in response to mechanical damage by regeneration. The regeneration of muscle is a complicated and orchestrated process, including the migration and differentiation of myoblasts. Myoblast migration towards injured areas is a key step in the alignment of myoblasts for the subsequent fusion into multinucleated myotubes.1,2 The process of myogenesis is controlled by the spatio-temporal expression of various myogenic regulatory factors (MRFs) such as myogenin and MyoD. Upon initiation of differentiation, the cells elongate, adhere, and fuse into multinucleated myotubes that express the muscle-specific marker, myosin heavy chain (MyHC).3,4 However, the ability to maintain or restore the normal structure and function becomes impaired under pathological conditions including aging and incurable injuries.5,6 Recently, a novel strategy for muscle regeneration has been the transplantation of myogenic progenitor cells to the muscle. But its therapeutic efficacy is restricted by the poor migratory capacity and the differentiation rate of cells.7,8The extracellular matrix (ECM) is a complicated network in terms of higher order structures and also intercellular connections, occupying the space between cells, tissues and organs.9 Skeletal muscle tissue depends heavily upon the ECM for organization, structural support, and mechanical function.10 During skeletal muscle regeneration, ECM components play a key role in the migration, proliferation and differentiation of myoblasts.11–13 In the muscle tissue, the ECM mainly exists in the tendon and endomysial (around the muscle cell), perimysial (around groups of muscle cells), and epimysial (around the whole muscle) connective tissues.14 The major component of the muscle ECM is collagen, accounting for 1–10% muscle mass dry weight.15 It was reported that the major ECM in the perimysium and epimysium of muscle tissue is type I collagen.16 Type I collagen fibers are characterized by thick fibers in vivo, serving as a support structure. Collagen I exhibits considerable biomechanical properties including tensile strength and load bearing in vivo.17 As the major components of the ECM, collagens play a significant role in the regulation of myoblast behaviors. It was reported that an inhibitor of collagen synthesis down-regulates the differentiation of cultured myoblasts.18 A collagen-derived dipeptide, hydroxyprolyl-glycine, promotes C2C12 myoblast differentiation and myotube hypertrophy.19
IL-6 acts as both a pro-inflammatory cytokine and an anti-inflammatory myokine, a cytokine produced from muscle, which is elevated in response to muscle contraction.20,21 Skeletal muscle can produce and contribute to the circulating levels of IL-6 during exercise.22 Many reports demonstrate that IL-6 production is associated with muscle contraction.23 IL-6 may participate in tissue repair after injury and in the generation of muscle tissue in dystrophy and after denervation.24,25 It was reported that the effects of autocrine-secreted IL-6 may promote myogenic differentiation of skeletal muscle cells via the STAT3 pathway.26 However, whether IL-6 is involved in the collagen I-mediated regulation of the migration and myogenic differentiation of C2C12 cells remains largely unknown.
C2C12 myoblasts derived from mouse muscle satellite cells are a unique cell line and have been commonly used in research on cell migration, proliferation, and differentiation.27,28 In this study, we develop a C2C12 cell culture model, where the cell plates are pre-coated with collagen I (col I) to investigate the distinct roles of col I in the migration, proliferation, and differentiation of C2C12 cells and the underlying molecular mechanism.
Materials and methods
Reagents
Primary antibodies against p-FAK and IL-6 were purchased from Cell Signaling Technology (Cell Signaling, Danvers, USA). Primary antibodies against β-actin, lamin B and NF-κB p65 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies against myogenin and MyoD were purchased from Proteintech (Wuhan, Hubei, China). MyHC antibody and primary monoclonal antibody against IL-6 (IL-6 neutralization antibody) were obtained from R&D System (Minneapolis, MN, USA). Rhodamine (TRITC)-conjugated AffiniPure goat anti-rabbit IgG (H + L) was purchased from KeyGEN Biotechnology (Nanjing, Jiangsu, China).Cell culture and differentiation
A mouse myoblast cell line, C2C12, was obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). C2C12 cells were grown in DMEM, 10% fetal bovine serum (FBS) (Beijing Yuanheng Shengma Research Institution of Biotechnology, Beijing, China), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at 37 °C and 5% CO2. When the cell density reached 90%, the growth medium was replaced by differentiation medium or DMEM containing 2% horse serum (Gibco, NY, USA). The cells were cultured for 5 days and the medium was changed every day.Col I-coated culture
Nippi Research Institute of Biomatrix (Toride, Japan) provided col I for the study. The extraction of col I was carried out as described previously.29,30 To prepare col I-coated dishes, col I (0, 10, 50, and 100 μg ml−1) was dissolved in acetic acid (0.5 mM) and coated on the dishes for at least 4 h at 37 °C. Before culturing cells, the solution was discarded and the dishes were washed with PBS three times.Cell proliferation assay
To examine the cell growth, the cell counting kit 8 (CCK-8, EnoGene, Nanjing, China) was used according to the manufacturer's instructions as described previously.29F-actin staining
The cells were cultured for 24 h, then fixed with 4% paraformaldehyde (PFA) for 20 min and permeabilized with 0.5% Triton X-100 for 15 min before blocking with PBS containing 10% NBCS for 30 min. F-actin was stained using Alexa Fluor 488 Phalloidin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500, Ye Sen, Shanghai, China). Fluorescence images were captured with a fluorescence microscope (Olympus, Tokyo, Japan).
:500, Ye Sen, Shanghai, China). Fluorescence images were captured with a fluorescence microscope (Olympus, Tokyo, Japan).
Confocal fluorescence microscopy analysis of NF-κB p65 and MyHC
The staining of NF-κB p65 and MyHC was performed as described previously.30 Briefly, cells were fixed using PFA for 20 min, and then permeabilized by treatment with 0.15% Triton X-100 for 12 min. After blocking for 30 min, the cells were incubated with anti-NF-κB p65 (1:300) or anti-MyHC (1:200) antibodies overnight, and then treated with fluorochrome-conjugated secondary antibodies (1:300) for 2 h. Last, the cells were stained with DAPI for 8 min. The stained cells were observed with a confocal microscope (Nikon, Tokyo, Japan).
Transfection of siRNA
C2C12 cells were transfected with negative control siRNA or siRNA targeting NF-κB p65 (20 nM) and FAK (20 nM) using siRNA-Mate™ transfection reagent (GenePharma, Shanghai, China) according to the manufacturer's instructions. Transfected cells were cultured for another 48 h for further analyses.Western blot analysis
Cells were harvested and then were lysed to obtain the proteins in whole cells using RIPA lysis buffer (Beyotime, Haimen, China) with 1 mM phenylmethanesulfonyl fluoride (PMSF). To extract nuclear proteins, the NE-PER extraction reagent (Thermo Fisher Scientific, Rockford, IL, USA) was used according to the manufacturer's protocol.10–12% SDS-PAGE was used to separate proteins in lysates at the same amount of total protein. Protein bands were transferred to Millipore Immobilon®-P Transfer Membrane (Millipore, Billerica, MA, USA). Then the membranes were blocked using 5% skimmed milk solution. After incubation with primary antibodies, the membranes were treated with the respective horseradish-peroxidase-conjugated secondary antibodies and observed with a SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL, USA).
RNA extraction and real time PCR
The isolation of total RNA was conducted with RNAiso Plus reagent (TaKaRa, Tokyo, Japan) according to the manufacturer's instructions. One μg of total RNA is used for cDNA synthesis using a PrimeScriptTM RT-PCR Kit (TaKaRa). The amplification of cDNA using the PrimeScriptTMRT Master Mix (TaKaRa) was performed to determine the mRNA levels of myogenin, MyoD and MyHC. The levels of targeted genes were calculated as described previously.31 The following primers were used (5′–3′): myogenin: forward: CAATGCACTGGAGTTCGGT; reverse: GCCAGGTTGACATTGGATTG; MyoD: forward: CATTCCAACCCACAGAACCT; reverse: CAAGCCCTGAGAGTCGTCTT; MyHC: forward: CGCAAGAATGTTCTCAGGCT; reverse: GCCAGGTTGACATTGGATTG; IL-6: forward: GAGACTTCCATCCAGTTGCC; reverse: CTGATTATATCCAGTTTGGTAGCATC; GAPDH: forward: TCCCACTCTTCCACCTTC; reverse: CTGTAGCCGTATTCATTGTC. GAPDH was used as the internal control to determine the relative changes in the target samples.Cell migration assays
Cell migration was examined by transwell migration assay and cell scratch migration assay as described previously.29 Relative migration activity in cell scratch assay is obtained as shown below.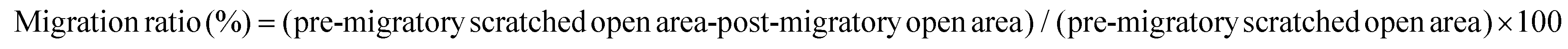
Quantification of IL-6 in the supernatants by ELISA
C2C12 cells were seeded in 6-well dishes at a density of 2.5 × 105 cells per well and cultured until reaching about 90% confluence. The cells and supernatants were harvested from each well. The number of cells in different groups was counted under a microscope. The concentrations of IL-6 were determined by using ELISA kits (DakeweBiotech, Shenzhen, China) according to the manufacturer's protocol.Statistical analysis
At least three independent assays were performed. Student's t test was used to examine the comparisons between groups. P < 0.05 is considered as significant.Results
Effects of col I on the proliferation, migration and differentiation of C2C12 cells
To investigate the effect of col I on the cell growth, the cells were cultured on col I-coated plates (0, 10, 50 and 100 μg mL−1) for 24, 48 and 72 h. The results of CCK-8 assay showed that the number expansion of cells cultured on different doses of col I-coated dishes was similar, indicating that col I had no significant effect on cell proliferation (Fig. 1A). Staining of the cytoskeleton using Alexa Fluor 488 Phalloidin showed that col I changed the cell morphology of C2C12 from a large and flattened morphology to an elongated, bipolar and fibroblast-like one. Compared to the cells cultured on the non-coated dishes, the cells on col I-coated dishes showed actin patches underneath the cell membrane (Fig. 1B). The results of cell migration assay showed that collagen I enhanced the migration of C2C12 cells in a concentration-dependent manner (Fig. 1C and D). Myogenic differentiation of C2C12 cells was induced by serum deprivation using differentiation medium. As shown in Fig. 1E and F, col I increases the portion of muscle-specific protein MyHC-positive cells in a dose-dependent manner, suggesting a promotion of myogenic differentiation. The expressions of myogenesis-related proteins, myogenin and MyoD and MyHC, increase in the cells cultured on col I-coated dishes (Fig. 1G). The mRNA levels of myogenin, MyoD and MyHC are also up-regulated in the cells cultured on col I-coated dishes (Fig. 1H). These results demonstrate that col I promotes the migration and differentiation of C2C12 cells, but does not influence their proliferation.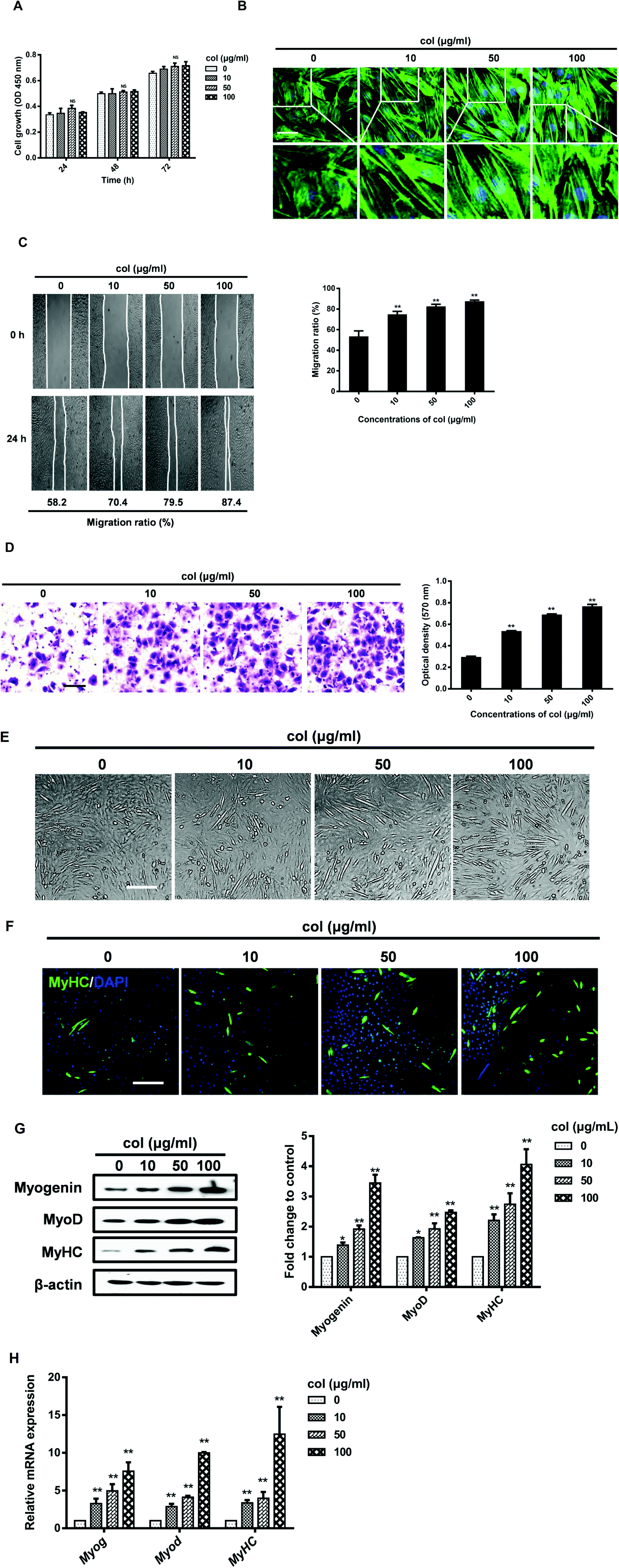 | ||
| Fig. 1 Effects of col I on the proliferation, migration and differentiation of C2C12 cells. (A) The cells were cultured on col I-coated dishes (0, 10, 50, and 100 μg mL−1) for 24, 48 and 72 h, and the cell growth was examined by using the CCK8 kits. NS, not significant compared with the 0 μg mL−1 group (n = 3, mean ± SD). (B) The cells were stained with Alexa Fluor 488 phalloidin and captured using confocal microscopy. Scale bar, 30 μm. (C) The cells were cultured for 48 h, reaching about 90% confluence, and were scratched off using 200 μl tips. Then the cells were observed under a phase contrast microscope at 24 h. The cell migration is presented as the percentage closure of the scratch area. The scratch gap was measured using Image Pro Plus 6.0 software. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (D) An in vitro transwell plate assay was conducted using Costar transwell membranes. Scale bar, 50 μm. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (E and F) The cells were induced to myogenic differentiation as described in Materials and Methods. The differentiated cells were observed using a phase contrast microscope and confirmed by immunostaining of MyHC. Scale bar, 100 μm. (G) Western blot analyses of myogenesis-related proteins. β-Actin was used as an equal loading control. Band density of the myogenesis-related proteins was analyzed with Image Pro Plus software and the results are expressed as the average density to β-actin. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (H) The mRNA level of myogenin, MyoD and MyHC was examined. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). | ||
Col I promotes the production and release of IL-6 followed by up-regulation of the migration and myogenic differentiation of cells
Our previous study has shown that the behaviors of 3T3-L1 cells are affected by the release of pro-inflammatory cytokines in the cells cultured on col I-coated dishes.29 In the current study, we found that the level of IL-6 both in cell culture supernatants and in cells increased when cultured on col I-coated dishes (Fig. 2A and B). The mRNA level of IL-6 was also up-regulated in cells cultured on col I-coated dishes (Fig. 2C). To directly examine the effect of IL-6 on cell differentiation, we exogenously added mouse recombinant IL-6 to C2C12 cells. The results showed that IL-6 increased the expression of myogenic regulatory factors, myogenin and MyoD, as well as the muscle-specific marker, myosin heavy chain (Fig. 2D). In the subsequent experiments, col I (50 μg mL−1) was used for coating dishes. In order to investigate whether IL-6 is involved in col I-enhanced migration and myogenic differentiation of C2C12 cells, an anti-mouse IL-6 neutralizing monoclonal antibody (IL-6 NA) was used, expected for blockade of IL-6 signaling in cells cultured on col I-coated dishes (50 μg mL−1). The results reveal that IL-6 NA effectively down-regulated the protein and mRNA expression levels of myogenin, MyoD and MyHC in C2C12 cells cultured on col I-coated dishes (Fig. 2E and F). Consistently, in the transwell cell migration assays, IL-6 NA reversed the enhancement effect of col I on cell migration (Fig. 2G).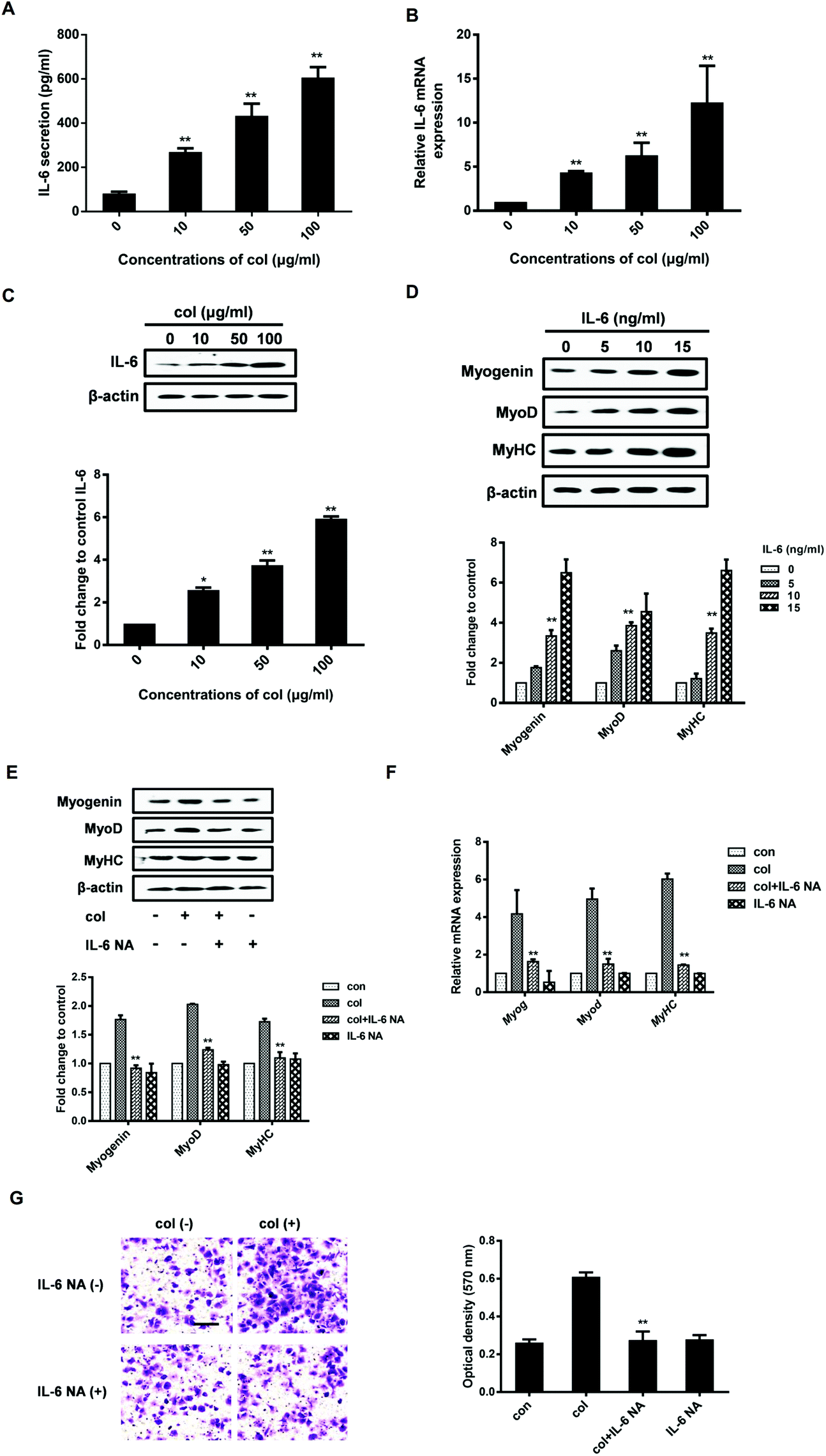 | ||
| Fig. 2 Col I promotion of the production and release of IL-6 up-regulates the migration and myogenic differentiation of cells. (A) The cells were cultured on col I-coated dishes (0, 10, 50, and 100 μg mL−1) for 90% confluence and the secretion level of IL-6 was detected by ELISA. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (B) Western blot analyses of IL-6 protein. β-Actin was used as an equal loading control. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (C) The mRNA levels of IL-6 are shown. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (D) The cells were cultured with 90% confluence and then the medium was replaced by differentiation medium containing IL-6 protein (5, 10, and 15 ng ml−1). The expressions of myogenin, MyoD and MyHC are examined by western blot. β-Actin is used as an equal loading control. **Compared with the col group, P < 0.01 (n = 3, mean ± SD). (E) The cells were cultured on col I-coated dishes (50 μg mL−1) for 24 h and then were treated with IL-6 NA (neutralization antibody) for 24 h (90% confluence). Then the cells were induced to differentiation for 5 days. The expressions of myogenin, MyoD and MyHC are examined by western blot. β-Actin is used as an equal loading control. **Compared with the col group, P < 0.01 (n = 3, mean ± SD). (F) The mRNA levels of myogenin, MyoD and MyHC are shown. **Compared with the col group, P < 0.01. (n = 3, mean ± SD). (G) The cell migration with transwell migration assay. **Compared with the col group, P < 0.01 (n = 3, mean ± SD). Scale bar, 50 μm. | ||
The nuclear translocation of NF-κB p65 enhances the production and release of IL-6
It is known that IL-6 signaling is dependent upon activation of the NF-κB pathway.32 NF-κB p65 is an important transcription factor, participating in various physiological and pathological processes via inducing the expression of a variety of target genes.33 Once activated, the NF-κB p65 dimers translocate to the nucleus, bind with specific DNA sequences, and promote the transcription of some pro-inflammatory genes including IL-6.34 NF-κB p65 induces IL-6 expression, leading to STAT3 activation and tumor progression in glioblastoma.35 NF-κB p65 is also involved in the production of IL-6 stimulated by IL-1β in skeletal muscle cells.36 Our previous study has shown that col I increases the nuclear translocation of NF-κB p65, leading to the enhancement of 3T3-L1 cell migration.29,37 However, whether NF-κB p65 participates in the myogenic differentiation of C2C12 cells is unclear. By the analysis of extraction of nuclear fraction, we found that col I increases the nuclear concentration of NF-κB p65 in C2C12 cells (Fig. 3A). The results of immunofluorescence analysis demonstrate enhanced nuclear translocation of NF-κB p65 in the cells cultured on col I-coated dishes (Fig. 3B). Then the cells were transfected with NF-κB p65 small interfering RNA (siRNA). Transfection with p65 siRNA resulted in decreased release of IL-6 from the cells cultured on col I-coated dishes (Fig. 3C). The expression of myogenic proteins and col I-enhanced migration were also down-regulated (Fig. 3D and E). The results indicate that the nuclear translocation of NF-κB p65 plays an important role in inducing IL-6-enhanced migration and myogenic differentiation of C2C12 cells.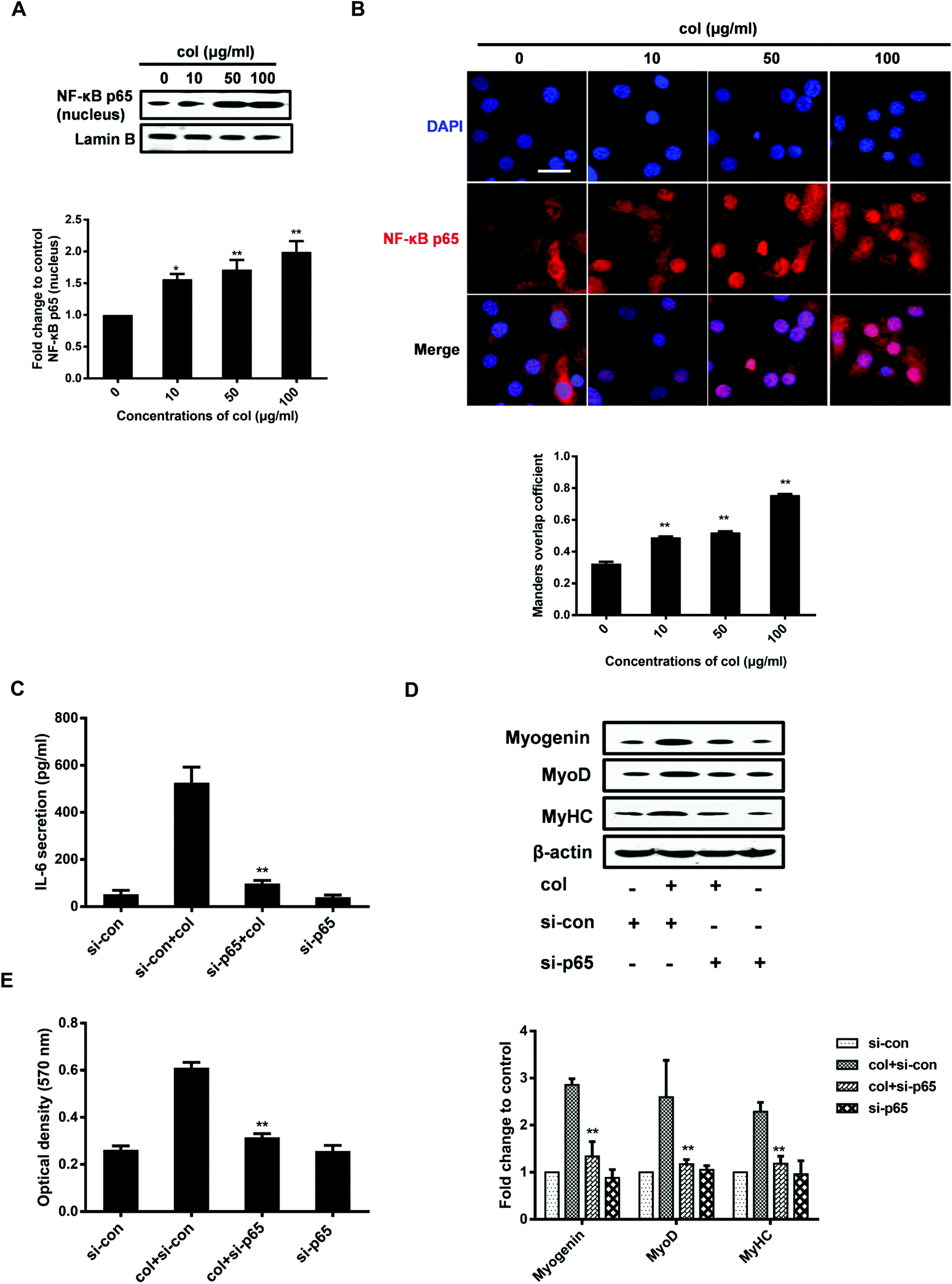 | ||
| Fig. 3 The increased nuclear translocation of NF-κB p65 plays an important role in IL-6 production and release. (A) The cells were cultured on col I-coated dishes (0, 10, 50, and 100 μg mL−1) for 90% confluence and the cells were harvested. Then the expression of NF-κB p65 in the nuclei was detected by western blot. Lamin B is used as an equal loading control. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (B) Representative immunofluorescence images of NF-κB p65 (red) and DAPI (blue) in cells. Scale bar, 30 μm. Its nuclear translocation is quantified using Image Pro Plus software. Mander's overlap coefficient refers to the spatial overlap between red (NF-κB p65) and blue (DAPI), indicating the nuclear translocation ratio of NF-κB p65. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (C) Cells were cultured on col I-coated dishes (50 μg mL−1) for 70% confluence and were transfected with control or NF-κB p65-targeting siRNA for 24 h. Then the medium was replaced by fresh DMEM for another 24 h (90% confluence). The secretion levels of IL-6 in the media were determined by ELISA. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). (D) The expression levels of myogenin, MyoD and MyHC were examined by western blot. β-Actin is used as an equal loading control. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). (E) The cell migration by transwell migration assay. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). | ||
Col I enhances the nuclear translocation of NF-κB p65 via increased phosphorylation of FAK
As an extracellular factor, how does col I transduce the signal from extra- to intra-cellular space is a critical question. FAK situates at the site of cell attachment to the ECM, sensing the extracellular stimuli. Activation of FAK by autophosphorylation initiates the downstream signaling cascades that regulate the cell behaviors.38 The ECM, including col I, is able to induce the activation of FAK.30,39 FAK also plays an important role in the regulation of NF-κB p65. It is reported that oxidized LDL drives NF-κB p65 activation through the FAK pathway in endothelial cells.40 FAK promotes the activation of NF-κB p65 signaling in human embryonic kidney cells (HEK293T).41 Therefore, we investigated whether the nuclear translocation of NF-κB p65 induced by culture on col I involves FAK signaling in C2C12 cells. The results show that col I increases the phosphorylation of FAK in a dose-dependent manner (Fig. 4A). Then we tested the effect of FAK silencing with small interfering RNA (siRNA) on the cells. In the cells cultured on col I-coated dishes, transfection with FAK siRNA results in a decrease of nuclear NF-κB p65 (Fig. 4B and C), as well as the release of IL-6 (Fig. 4D). The col I-enhanced migration and the expression of myogenic proteins were also down-regulated in the cells transfected with FAK siRNA (Fig. 4E and F). The results indicate that col I promotes the migration and myogenic differentiation of C2C12 myoblasts via an FAK/NF-κB p65/IL-6 pathway.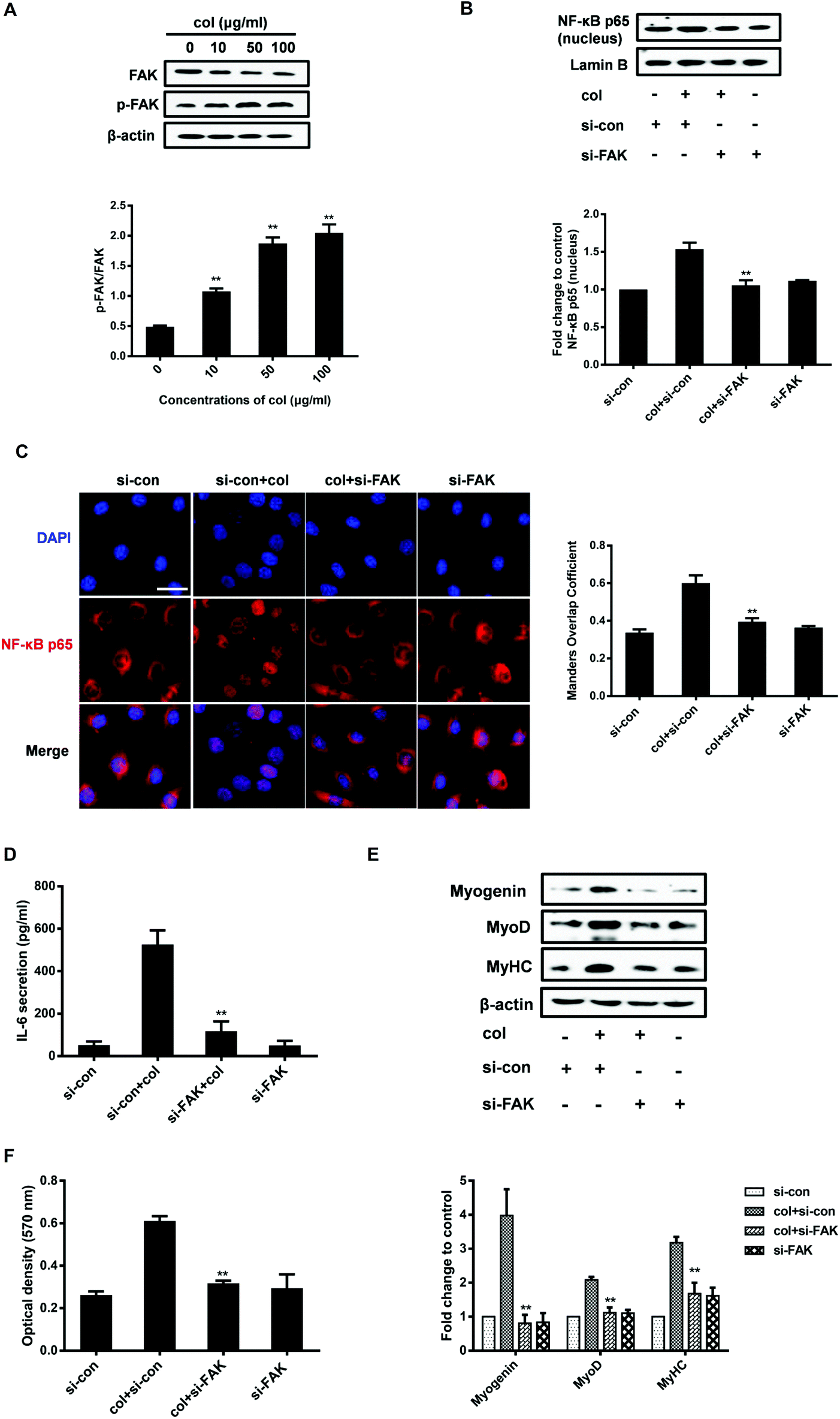 | ||
| Fig. 4 Col I enhances the nuclear translocation of NF-κB p65 via increasing the phosphorylation of FAK. (A) The expression levels of FAK and p-FAK were detected by western blot. β-Actin was used as an equal loading control. The ratios of p-FAK/FAK are analyzed with Image Pro Plus software. **Compared with the 0 μg mL−1 group, P < 0.01 (n = 3, mean ± SD). (B) Cells were transfected with control or FAK-targeting siRNA, and the expression levels of NF-κB p65 in the nuclei were determined by western blot. Lamin B is used as an equal loading control. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). (C) Representative immunofluorescence images of NF-κB p65 (red) and DAPI (blue) in the cells. Scale bar, 30 μm. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). (D) The secretion levels of IL-6 in the media were determined by ELISA. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). (D) The expression levels of myogenin, MyoD and MyHC were examined by western blot. β-Actin is used as an equal loading control. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). (E) The cell migration by transwell migration assay. **Compared with the si-con + col group, P < 0.01 (n = 3, mean ± SD). | ||
Discussion
Proliferation, migration and differentiation of myoblasts are necessary for skeletal muscle regeneration in vivo. The enhanced activities of migration and differentiation of myoblasts potentially contribute to the therapies for muscular dystrophies by transplantation of myoblasts. In this study, mouse C2C12 myoblasts cultured on collagen I-coated dishes show the enhanced activities of migration and myogenic differentiation regulated by the increased release and production of IL-6 that is up-regulated by FAK-induced augmentation of nuclear translocation of NF-κB p65.As the main component in animal connective tissue, collagen has been widely used as food and as a bioactive functional material. The fibrillar structure of collagen contributes to enhancing the elasticity and tenacity properties of collagen-based biomaterials, which supports the application of biomaterials, pharmaceutical materials, and food-packaging materials.42 Furthermore, there is increasing evidence indicating that collagen and its hydrolysates can be used as nutraceutical agents, influencing both physiological and pathological processes.43 It was reported that collagen peptides have the ability to attenuate skin aging and prevent cardiovascular diseases.44 Our results showed that collagen has a close relationship with muscle regeneration processes by influencing myoblast migration and differentiation. We suspect that the poor cell migration and differentiation capacity in myogenic cell transplantation might be ameliorated by collagen-based materials or nutraceutical agents in muscular dystrophic pathologies.
Various components of the ECM participate in muscle regeneration, not only serving as the structure support, but also affecting the myoblast cell function and myofiber formation.45 Collagen is the major structural protein in the muscle ECM, accounting for 1–10% muscle mass dry weight. Type I, III, IV and V collagens are the main types of collagen in the muscle ECM, existing in different parts of muscle tissue, and type I collagen is the most abundant protein.14 Type I collagen is predominantly expressed in the perimysium and epimysium in fibrillar and molecular states.17 There is a small amount of fibrillar type III collagen distributed between the endomysium and epimysium. Type V collagen, another fibrillar collagen, co-exists with type I and III collagen and constitutes a core for type I collagen fibrils in the perimysium and endomysium. The muscle basement membrane consists of type IV collagen that mainly localizes in the endomysium.46
There is increasing evidence that ECM molecules play an important role in the regulation of pro-inflammatory cytokines, influencing various cell behaviors. Our previous study shows that col I promotes the release of IL-6, IL-1β and TNFα in the migration of 3T3-L1 cells.29 In this study, we found that col I increases the production and release of IL-6 via the nuclear translocation of NF-κB p65, enhancing the migration and myogenic differentiation of C2C12 cells. Another report has shown that jellyfish collagen stimulates the production of TNFα and IL-6 in J774.1 cells via the activation of NF-κB p65.47 Colombo et al. also reported that col I increases the release of IL-6 and IL-8 to support the isolation and expansion of endothelial progenitor cells from human adult peripheral blood.48
In vivo, NF-κB p65 is involved in the regulation of a wide array of cellular processes. A previous study has shown that the activation of NF-κB p65 enhances the proliferation and migration of 3T3-L1 cells cultured on collagen I-coated dishes. In this study, we found that the increased nuclear translocation of NF-κB p65 promotes the migration and myogenic differentiation of C2C12 cells through the release of IL-6. Guoping Ji et al. reported a p38-dependent elevation of NF-κB activity in p65 DNA binding during the myogenic differentiation of C2C12 cells, which is required for the production of IL-6 to promote myogenic differentiation.49 However, a growing body of evidence has shown that NF-κB p65 plays a dual role in skeletal myogenesis, playing both positive and negative regulatory roles in myogenesis. Inhibition of the inhibitor of the kappa B kinase (IKK)/NF-κB p65 pathway enhances muscle regeneration in injured and diseased skeletal muscle.50 Glutathione depletion impairs myogenic differentiation through sustained activation of NF-κB.51 NF-κB p65 is activated by a variety of upstream signals and induces transcription of different genes. TNFα inhibits myogenic differentiation of C2C12 cells through NF-κB p65 activation.52 IL-6 promotes the myogenic differentiation of C2C12 cells through the activation of NF-κB p65 in IL-1β-stimulated C2C12 cells.36 This study also shows that col I promotes the myogenic differentiation of C2C12 cells through NF-κB p65-induced release of IL-6. Therefore, we suppose that the difference in upstream activators and downstream targets of NF-κB p65 might account for its dual roles in myogenesis.
Skeletal muscle differentiation requires rearrangement of intensive actin cytoskeleton to maintain cell shape, coordinate cell locomotion, adhesion and fusion.53 It is reported that the actin-associated protein paladin is required for the migration behavior and myogenic differentiation potential of C2C12 myoblast cells.54 In this study, the staining of the cytoskeleton using Alexa Fluor 488 Phalloidin showed that col I changes the cell morphology from a large and flattened one to an elongated, bipolar, fibroblast-like one. Some actin bundles are extended into pseudopodia and actin patches close to the cell membrane. We suppose that the col I-induced morphological change might contribute to the enhanced migration and myogenic differentiation of C2C12 cells.
Taken together, col I promotes the migration and myogenic differentiation of C2C12 cells via the release of IL-6 through activation of the FAK/NF-κB p65 pathway. Understanding the biological function of col I on myogenesis could potentially lead to a novel therapeutic strategy to combat muscular dystrophic pathologies.
Conflicts of interest
There are no conflicts to declare.References
- D. J. Watt, J. Karasinski, J. Moss and M. A. England, Nature, 1994, 368, 406–407 CrossRef CAS PubMed.
- L. Leloup, L. Daury, G. Mazeres, P. Cottin and J. J. Brustis, Int. J. Biochem. Cell Biol., 2007, 39, 1177–1189 CrossRef CAS PubMed.
- J. G. Tidball, Compr. Physiol., 2011, 1, 2029–2062 Search PubMed.
- N. A. Dumont, C. F. Bentzinger, M. C. Sincennes and M. A. Rudnicki, Compr. Physiol., 2015, 5, 1027–1059 Search PubMed.
- A. E. Emery, Lancet, 2002, 359, 687–695 CrossRef CAS.
- P. M. Van Ry, T. M. Fontelonga, P. Barraza-Flores, A. Sarathy, A. M. Nunes and D. J. Burkin, Compr. Physiol., 2017, 7, 1519–1536 Search PubMed.
- D. Briggs and J. E. Morgan, FEBS J., 2013, 280, 4281–4293 CrossRef CAS PubMed.
- F. D. Price, K. Kuroda and M. A. Rudnicki, Biochim. Biophys. Acta, 2007, 1772, 272–283 CrossRef CAS.
- W. P. Daley and K. M. Yamada, Curr. Opin. Genet. Dev., 2013, 23, 408–414 CrossRef CAS.
- K. A. Clark, A. S. McElhinny, M. C. Beckerle and C. C. Gregorio, Annu. Rev. Cell Dev. Biol., 2002, 18, 637–706 CrossRef CAS PubMed.
- N. Osses and E. Brandan, Am. J. Physiol.: Cell Physiol., 2002, 282, C383–C394 CrossRef CAS PubMed.
- H. Lei, D. Leong, L. R. Smith and E. R. Barton, Am. J. Physiol.: Cell Physiol., 2013, 305, C529–C538 CrossRef CAS PubMed.
- I. Jun, S. J. Kim, E. Choi, K. M. Park, T. Rhim, J. Park, K. D. Park and H. Shin, Macromol. Biosci., 2012, 12, 1502–1513 CrossRef CAS PubMed.
- A. R. Gillies and R. L. Lieber, Muscle Nerve, 2011, 44, 318–331 CAS.
- T. Nishimura, Anim. Sci. J., 2010, 81, 21–27 CrossRef.
- N. Light and A. E. Champion, Biochem. J., 1984, 219, 1017–1026 CrossRef CAS.
- K. Mukund and S. Subramaniam, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2019, e1462 Search PubMed.
- D. Nandan, E. P. Clarke, E. H. Ball and B. D. Sanwal, J. Cell Biol., 1990, 110, 1673–1679 CrossRef CAS.
- T. Kitakaze, T. Sakamoto, T. Kitano, N. Inoue, F. Sugihara, N. Harada and R. Yamaji, Biochem. Biophys. Res. Commun., 2016, 478, 1292–1297 CrossRef CAS PubMed.
- T. Tanaka, M. Narazaki and T. Kishimoto, Cold Spring Harbor Perspect. Biol., 2014, 6, a016295 CrossRef.
- B. K. Pedersen, T. C. Akerstrom, A. R. Nielsen and C. P. Fischer, J. Appl. Physiol., 2007, 103, 1093–1098 CrossRef CAS PubMed.
- A. Steensberg, G. van Hall, T. Osada, M. Sacchetti, B. Saltin and B. Klarlund Pedersen, J. Physiol., 2000, 529(Pt 1), 237–242 CrossRef CAS PubMed.
- R. L. Starkie, M. J. Arkinstall, I. Koukoulas, J. A. Hawley and M. A. Febbraio, J. Physiol., 2001, 533, 585–591 CrossRef CAS.
- J. B. Kurek, L. Austin, S. S. Cheema, P. F. Bartlett and M. Murphy, Neuromuscular Disord., 1996, 6, 105–114 CrossRef CAS.
- J. B. Kurek, S. Nouri, G. Kannourakis, M. Murphy and L. Austin, Muscle Nerve, 1996, 19, 1291–1301 CrossRef CAS PubMed.
- M. Hoene, H. Runge, H. U. Haring, E. D. Schleicher and C. Weigert, Am. J. Physiol.: Cell Physiol., 2013, 304, C128–C136 CrossRef CAS PubMed.
- D. Yaffe and O. Saxel, Nature, 1977, 270, 725–727 CrossRef CAS PubMed.
- R. Vaz, G. G. Martins, S. Thorsteinsdottir and G. Rodrigues, Cell Tissue Res., 2012, 348, 569–578 CrossRef CAS PubMed.
- X. Liu, Q. Xu, W. Liu, G. Yao, Y. Zhao, F. Xu, T. Hayashi, H. Fujisaki, S. Hattori, S. I. Tashiro, S. Onodera, M. Yamato and T. Ikejima, Mol. Cell. Biochem., 2018, 441, 35–62 CrossRef CAS PubMed.
- X. Liu, X. Long, W. Liu, Y. Zhao, T. Hayashi, M. Yamato, K. Mizuno, H. Fujisaki, S. Hattori, S. I. Tashiro, T. Ogura, Y. Atsuzawa and T. Ikejima, Biochimie, 2018, 150, 110–130 CrossRef CAS.
- X. Liu, Q. Xu, X. Long, W. Liu, Y. Zhao, T. Hayashi, S. Hattori, H. Fujisaki, T. Ogura, S. I. Tashiro, S. Onodera, M. Yamato and T. Ikejima, Mol. Cell. Biochem., 2018, 450(1–2), 1–23 CrossRef PubMed.
- H. Yu, W. Jiang, H. Du, Y. Xing, G. Bai, Y. Zhang, Y. Li, H. Jiang, Y. Zhang, J. Wang, P. Wang and X. Bai, PLoS One, 2014, 9, e93810 CrossRef PubMed.
- G. Diamant and R. Dikstein, Biochim. Biophys. Acta, 2013, 1829, 937–945 CrossRef CAS PubMed.
- T. Lawrence, Cold Spring Harbor Perspect. Biol., 2009, 1, a001651 Search PubMed.
- B. C. McFarland, S. W. Hong, R. Rajbhandari, G. B. Twitty Jr., G. K. Gray, H. Yu, E. N. Benveniste and S. E. Nozell, PLoS One, 2013, 8, e78728 CrossRef CAS PubMed.
- G. Luo, D. D. Hershko, B. W. Robb, C. J. Wray and P. O. Hasselgren, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2003, 284, R1249–R1254 CrossRef CAS.
- X. Liu, X. Long, W. Liu, G. Yao, Y. Zhao, T. Hayashi, S. Hattori, H. Fujisaki, T. Ogura, S. I. Tashiro, S. Onodera, M. Yamato and T. Ikejima, Free Radical Res., 2018, 1–261 Search PubMed.
- X. K. Zhao, Y. Cheng, M. Liang Cheng, L. Yu, M. Mu, H. Li, Y. Liu, B. Zhang, Y. Yao, H. Guo, R. Wang and Q. Zhang, Sci. Rep., 2016, 6, 19276 CrossRef CAS.
- J. B. Heim, C. A. McDonald, S. P. Wyles, S. Sominidi-Damodaran, E. J. Squirewell, M. Li, C. Motsonelidze, R. T. Bottcher, J. van Deursen and A. Meves, PLoS One, 2018, 13, e0200558 CrossRef.
- A. Yurdagul Jr., F. J. Sulzmaier, X. L. Chen, C. B. Pattillo, D. D. Schlaepfer and A. W. Orr, J. Cell Sci., 2016, 129, 1580–1591 CrossRef PubMed.
- S. F. Dwyer, L. Gao and I. H. Gelman, Int. J. Biol. Sci., 2015, 11, 404–410 CrossRef CAS PubMed.
- Z. Bazrafshan and G. K. Stylios, Int. J. Biol. Macromol., 2019, 129, 693–705 CrossRef CAS PubMed.
- M. I. Avila Rodriguez, L. G. Rodriguez Barroso and M. L. Sanchez, J. Cosmet. Dermatol., 2018, 17, 20–26 CrossRef PubMed.
- E. Proksch, D. Segger, J. Degwert, M. Schunck, V. Zague and S. Oesser, Skin Pharmacol. Physiol., 2014, 27, 47–55 CrossRef CAS PubMed.
- K. M. Stearns-Reider, A. D'Amore, K. Beezhold, B. Rothrauff, L. Cavalli, W. R. Wagner, D. A. Vorp, A. Tsamis, S. Shinde, C. Zhang, A. Barchowsky, T. A. Rando, R. S. Tuan and F. Ambrosio, Aging Cell, 2017, 16, 518–528 CrossRef CAS PubMed.
- A. Urciuolo, M. Quarta, V. Morbidoni, F. Gattazzo, S. Molon, P. Grumati, F. Montemurro, F. S. Tedesco, B. Blaauw, G. Cossu, G. Vozzi, T. A. Rando and P. Bonaldo, Nat. Commun., 2013, 4, 1964 CrossRef PubMed.
- A. B. Putra, K. Nishi, R. Shiraishi, M. Doi and T. Sugahara, Mol. Immunol., 2014, 58, 32–37 CrossRef CAS PubMed.
- E. Colombo, F. Calcaterra, M. Cappelletti, D. Mavilio and S. Della Bella, PLoS One, 2013, 8, e66734 CrossRef CAS.
- G. Ji, D. Liu, J. Liu, H. Gao, X. Yuan and G. Shen, Biochem. Biophys. Res. Commun., 2010, 391, 547–551 CrossRef CAS PubMed.
- A. Lu, J. D. Proto, L. Guo, Y. Tang, M. Lavasani, J. S. Tilstra, L. J. Niedernhofer, B. Wang, D. C. Guttridge, P. D. Robbins and J. Huard, Mol. Ther., 2012, 20, 661–668 CrossRef CAS PubMed.
- E. Ardite, J. A. Barbera, J. Roca and J. C. Fernandez-Checa, Am. J. Pathol., 2004, 165, 719–728 CrossRef CAS.
- Q. Zhao, S. T. Yang, J. J. Wang, J. Zhou, S. S. Xing, C. C. Shen, X. X. Wang, Y. X. Yue, J. Song, M. Chen, Y. Y. Wei, Q. P. Zhou, T. Dai and Y. H. Song, Biochem. Biophys. Res. Commun., 2015, 458, 790–795 CrossRef CAS PubMed.
- M. A. Geeves and K. C. Holmes, Annu. Rev. Biochem., 1999, 68, 687–728 CrossRef CAS PubMed.
- N. U. Nguyen, V. R. Liang and H. V. Wang, Biochem. Biophys. Res. Commun., 2014, 452, 728–733 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |