 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The persistence of a proxy for cooking emissions in megacities: a kinetic study of the ozonolysis of self-assembled films by simultaneous small and wide angle X-ray scattering (SAXS/WAXS) and Raman microscopy†
Adam
Milsom
 a,
Adam M.
Squires
b,
Ben
Woden
c,
Nicholas J.
Terrill
d,
Andrew D.
Ward
e and
Christian
Pfrang
*af
a,
Adam M.
Squires
b,
Ben
Woden
c,
Nicholas J.
Terrill
d,
Andrew D.
Ward
e and
Christian
Pfrang
*af
aUniversity of Birmingham, School of Geography, Earth and Environmental Sciences, Edgbaston, Birmingham, B15 2TT, UK. E-mail: c.pfrang@bham.ac.uk
bUniversity of Bath, Department of Chemistry, South Building, Soldier Down Ln, Claverton Down, Bath, BA2 7AX, UK
cDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK
dDiamond Light Source, Diamond House, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
eCentral Laser Facility, STFC Rutherford Appleton Laboratory, Didcot OX11 0FA, UK
fDepartment of Meteorology, University of Reading, Whiteknights, Earley Gate, Reading, RG6 6BB, UK
First published on 7th September 2020
Abstract
Cooking emissions account for a significant proportion of the organic aerosols emitted into the urban environment and high pollution events have been linked to an increased organic content on urban particulate matter surfaces. We present a kinetic study on surface coatings of self-assembled (semi-solid) oleic acid–sodium oleate cooking aerosol proxies undergoing ozonolysis. We found clear film thickness-dependent kinetic behaviour and measured the effect of the organic phase on the kinetics for this system. In addition to the thickness-dependent kinetics, we show that significant fractions of unreacted proxy remain after extensive ozone exposure and that this effect scales approximately linearly with film thickness, suggesting that a late-stage inert reaction product may form and inhibit reaction progress – effectively building up an inert crust. We determine this by using a range of simultaneous analytical techniques; most notably Small-Angle X-ray Scattering (SAXS) has been used for the first time to measure the reaction kinetics of films of a wide range of thicknesses from ca. 0.59 to 73 μm with films <10 μm thick being of potential atmospheric relevance. These observations have implications for the evolution of particulate matter in the urban environment, potentially extending the atmospheric lifetimes of harmful aerosol components and affecting the local urban air quality and climate.
1 Introduction
Particulate matter is a major contributor to urban air pollution affecting both human health and air quality.1–3 Organic matter represents a large proportion of urban aerosols.4 Organic cooking aerosols have been estimated to contribute an additional ca. 10% to PM2.5 emissions in the UK,5 and cooking emissions were recently identified as a major contributor to the organic PM1 flux in aerosol flux measurements in a Chinese megacity.6 Unsaturated fatty acids, such as oleic acid, have been identified as key components of urban cooking emissions up to PM10.7–10 For this reason, oleic acid has been used as a proxy for the reactive fraction of cooking aerosols and as a representative of unsaturated fatty acids in the atmosphere, which makes it a well-studied model compound.11–20Solid and semi-solid phases have been observed for secondary organic aerosols.21 In addition to the effects of organic–aqueous phase separation,22 viscous phases are identified as a key factor in influencing fundamental aerosol processes such as uptake of water and reactive gasses.23 In particular, glassy and semi-solid phases have been studied and show drastic differences in their physical properties.24–27 Therefore, organic aerosol lifetime can vary significantly depending on temperature and humidity-induced phase changes, which simultaneously alter the viscosity of the aerosol and the diffusion coefficients of key atmospheric species within it.28,29
Oleic acid is a surfactant and, upon mixing with its salt (sodium oleate) and water, is able to form a range of lyotropic liquid crystalline (LLC) phases.30 Oleic acid and its LLC phase behaviour have been studied extensively in biological and cosmetic contexts, exhibiting a range of different 3-D molecular assemblies.31–33 More recently, this phase behaviour has been studied in an atmospheric context in levitated droplets.17 All of these phases exhibit unique physical properties such as viscosity,34 and the rate of diffusion of small molecules through them can vary significantly; these are important factors exploited in drug delivery applications that are also applicable to the diffusion of molecules through atmospherically-relevant particles.35 In the present study we focus on the lamellar phase formed by the oleic acid–sodium oleate cooking aerosol proxy and explore the impact of this phase on the kinetic behaviour.
Coatings of organic material have been found and characterised on aerosol surfaces, with fatty acids identified as one of the components on sulfate aerosols.36,37 Recently, high pollution events have been linked to greater organic content at the surface of urban PM2.5 emissions in Beijing, China.38 Urban particulate matter coatings have also been shown to contain harmful species such as Polycyclic Aromatic Hydrocarbons (PAHs) and heavy metals, making these surfaces a potential health hazard in the urban environment.39 PAHs and oleic acid have been identified in marine aerosols influenced by urban emissions, suggesting a degree of long-range transport from the urban environment.40 Long-range transport of PAHs and the link to lung cancer risk have been associated with shielding by organic aerosol coatings, and the extent of this effect is attributed to the phase of the PAH-containing aerosol, which is dependent on the surrounding environment.41,42
In this work we investigate the effect of film thickness on the reactivity of a self-assembled (semi-solid) form of oleic acid–sodium oleate using simultaneous Small-Angle X-ray Scattering (SAXS – kinetics and self-assembled structure), Raman microscopy (kinetics) and Wide-Angle X-ray Scattering (WAXS – fatty acid tail packing). These techniques are complemented by Grazing-Incidence SAXS (GI-SAXS – surface orientation). Most notably, we present a novel method of measuring reaction kinetics using SAXS data, taking advantage of the high X-ray flux available at a synchrotron source to enable sub-micron-sized films to be probed – increasing the atmospheric relevance of this proxy compared to the large levitated particles in our previous study.17 We compare the reactivity of oleic acid in the solid, semi-solid and liquid forms under the same conditions, highlighting drastic differences linked to the phase state. Reaction stagnation is observed for the thickest films and we link this to the formation of an inert crust, which has previously been postulated.43 We associate these findings with the persistence of organic coatings in the atmosphere and potential effects on the urban environment.
2 Experimental
2.1 Preparation of self-assembled fatty acid coatings inside quartz capillaries
Oleic acid (Sigma-Aldrich, 90%) and sodium oleate (Sigma-Aldrich, 99%) were dissolved separately in methanol as 10 wt% solutions. The sodium oleate mixture required gentle heating to ∼50 °C for 10 min in order to fully dissolve. These solutions were then mixed together in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio to afford the oleic acid–sodium oleate coating solution.
:1 weight ratio to afford the oleic acid–sodium oleate coating solution.
A 70 μL aliquot of the coating solution was injected inside a quartz capillary tube (Capillary Tube Supplies Ltd., UK, 1.5 ± 0.25 mm diameter, wall thickness 0.010 mm) which was embedded inside a metal holder. The sample solution was passed up and down the tube. Simultaneously, compressed air was passed gently through the tube to facilitate evaporation of the methanol. Room-temperature compressed air was chosen in order not to risk the breakdown of the possibly temperature-sensitive self-assembled phase. After evaporation, a translucent film of the oleic acid–sodium oleate lamellar phase sample was observed. This method of film coating left a film of unequal thickness along the capillary, enabling us to probe sections of the film of different thicknesses under the same conditions (see Results and discussion). This coated tube was placed into a sample holder on the I22 beamline at the Diamond Light Source (UK). Dry oxygen was passed through the capillary for ∼10 min before the kinetic experiments began to ensure that all of the methanol had evaporated.
2.2 SAXS/WAXS/Raman microscopy during ozonolysis of films coated inside quartz capillaries
SAXS/WAXS is a scattering technique which probes aggregate materials at the nanometre scale (i.e. interactions between aggregate structures rather than atoms within molecules).44,45 The scattered intensity is measured against a scattering parameter (q), which is inversely proportional to the spacing between equivalent scattering planes (Fig. 1). | ||
| Fig. 1 The initial SAXS pattern of a 73 ± 2 μm thick film. Numbers indicate the lamellar phase Bragg peaks. The scattering peak from Kapton film on the X-ray detector is also labelled and was used as an internal standard. | ||
SAXS/WAXS patterns were collected as a series of 1 s frames along the length of the coated quartz capillary. We introduced a delay period of 75 s between each set of scans to avoid any possible X-ray beam damage since the signal-to-noise ratios were sufficient at 1 s exposure times. The size of the collimated X-ray beam was approximately 320 μm × 400 μm (FWHM). It should be noted that the film thickness was independent of the size of the X-ray beam: a large X-ray footprint was chosen specifically in order to maximise the signal-to-noise ratios and thus be able to detect sub-micron-sized films, making this a key advantage of the experimental setup. The beam size was determined by attenuation of the X-rays by the metal capillary holder used during the experiments. On allowing the X-ray beam to hit the metal, a diode close to the detector did not detect the X-ray beam. Moving the beam in small 20 μm steps until the diode detected the full X-ray flux allowed the beam size to be determined (i.e. the difference between the stage position at half and full X-ray flux is the radius of the beam in the scanning direction). This was done for the horizontal and vertical directions. Scattering patterns were acquired in the q-range 0.008–0.6 Å−1 by the SAXS detector (Pilatus P3-2M) and simultaneously from 0.48 to 5.28 Å−1 by the WAXS detector (Pilatus P3-2M-L).
Ozone was produced by exposing oxygen (BOC, 99.5%) to UV radiation through a pen-ray ozoniser (Ultraviolet Products Ltd, Cambridge, UK). The ozone concentration was controlled by exposing the oxygen flow to the desired amount of UV light. The ozone concentration was calibrated offline using UV spectroscopy, monitoring the 254 nm absorption band of ozone using the absorption cross-section for ozone at this wavelength ((1.14 ± 0.07) × 10−17 cm2).46 The concentration of ozone used in these experiments was 77 ± 5 ppm. Such a high concentration of ozone was employed as past experience with self-assembled phases suggests that the ozonolysis reaction is very slow.17 In the timeframe of a synchrotron beamtime experiment, exposing the samples to more atmospherically-relevant concentrations of ozone (ppb levels) would have resulted in only very few experiments being feasible, thus making a rigorous kinetic study impossible.
A 60 mL min−1 flow of the oxygen/ozone mixture was passed through the fatty acid-coated capillaries. The sample was exposed to X-rays from the synchrotron. Scans across the capillary were carried out, with each scan consisting of 11 scattering patterns along the length of the tube, i.e. 11 different sections of the capillary tube.
As the X-ray beam passes through two film surfaces, one surface on the ‘beam’ side and the other on the ‘detector’ side of the capillary, the resulting X-ray scattering pattern is two-fold more intense for a particular thickness than it would be if the X-ray beam were to pass through just one surface. This is an advantage of using capillary tubes as surfaces for the deposition of films.
For these chosen experimental conditions, the lamellar phase was expected in our fatty acid proxy (Fig. 1). Bragg reflections for the lamellar phase were evident in the SAXS patterns. The first Bragg peak at ∼0.15 Å−1 was chosen as the peak of interest for the kinetic study. Kapton is a material used in the detector setup at the synchrotron and has a scattering peak at ∼0.37 Å−1. This Kapton peak is not affected by the experiment and was used as an internal standard for the measurement of the lamellar phase peak area. Normalising to the Kapton peak area has the advantage of accounting for fluctuations in the synchrotron beam intensity and other possible variations in the sample exposure.
The film thickness was determined by measuring the first lamellar peak area (peak “1” in Fig. 1) and normalising to the Kapton peak for a quartz capillary of known internal diameter (1.45 mm – measured by attenuation of the X-ray beam while scanning across an empty capillary) entirely filled with the sample. The lamellar peak/Kapton peak ratio of each film was then used to determine the film thickness, taking into account the X-ray beam passing through two films on either side of the capillary. A wide range of film thicknesses (0.59–73 μm) were measured. Although organic coatings are known to be present in atmospheric aerosols, to our knowledge there are no explicit studies to date directly characterising organic film thickness in urban particulate matter. However, the morphology of sea spray aerosols with an organic coating has been reported recently, demonstrating that organic film thicknesses in the order of microns are possible.47 With particulate matter up to PM10 being the most common size observed in the urban environment, we assume that films up to 10 μm thick are potentially relevant to the urban environment. The thicker films investigated here together with the atmospherically-relevant <10 μm films are however very useful to demonstrate that the kinetic trends are valid and consistent across more than two orders of magnitude of film thicknesses accessible with our experimental set-up in otherwise identical conditions. The uncertainties associated with these thickness measurements were determined from the error in the intensities recorded over the q-range of the lamellar peak. This was done for every peak measurement in order to quantify the uncertainty for each point in the decay plots (see Fig. 3(b) below). These numbers are small due to the better statistics associated with high-flux synchrotron radiation compared to lab-based X-ray sources. The uncertainties, along with all kinetic data, are listed in Table S1.†
The additional scattering peak at a lower q (∼0.11 Å−1) than the first lamellar scattering peak is likely to be a result of excess oleic acid in the mixture and is observed in the thickest films (Fig. 1). This phase is most likely another arrangement of oleic acid, although phase assignment was not possible as more than one peak is required for this.
The peak area of the first lamellar phase reflection was measured as a function of ozone exposure. Measurements of the SAXS patterns were started ∼3 min after the ozone production was initiated due to the safety procedures necessary at the synchrotron beamline.
As ozone attacked the unsaturated fatty acid double bond, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C Raman scattering peak area at ∼1650 cm−1 (normalised to the –CH2 deformation band at ∼1442 cm−1) was followed during ozonolysis in order to provide complementary kinetic data to the more sensitive SAXS measurements. The reactive decay of disordered oleic acid cannot be measured by SAXS as it does not have a strong SAXS peak due to the lack of self-assembly. For this reason, Raman microscopy is essential for measuring the reaction kinetics for comparison with the self-assembled samples.
C Raman scattering peak area at ∼1650 cm−1 (normalised to the –CH2 deformation band at ∼1442 cm−1) was followed during ozonolysis in order to provide complementary kinetic data to the more sensitive SAXS measurements. The reactive decay of disordered oleic acid cannot be measured by SAXS as it does not have a strong SAXS peak due to the lack of self-assembly. For this reason, Raman microscopy is essential for measuring the reaction kinetics for comparison with the self-assembled samples.
Simultaneous Raman microscopy was possible on the I22 beamline. A 532 nm Raman laser probe with a long working distance objective (numerical aperture: 0.42) and a minimum spot diameter of ∼1.5 μm was focussed onto the coated film. The laser power emitted from the objective end was between 20 and 50 mW. The focal depth of the Raman probe beam only permitted measurement of the Raman spectrum of the film on one side of the capillary.
2.3 Challenges associated with the novel SAXS–kinetics method
Fig. 2 illustrates the correction applied to the raw SAXS data for derivation of meaningful kinetic information. At the beginning of the ozonolysis experiment a broad peak arising from a minor co-existent phase (ca. 0.20–0.25 Å−1) decays, accompanied by an increase in the lamellar peak intensity (Fig. 2(b)). The measured peak area for the thickest films consequently showed an increase during the first ∼10 min of the reaction (Fig. 2(a)). This was accompanied by a small shift in the peak position (q), corresponding to a decrease in spacing (d) between lamellar sheets (q ∝ 1/d). It is likely that this minor phase was an inverse micellar arrangement of oleic acid due to the 7.8% molar excess of oleic acid in the mixture and the dryness of the sample.31 The effect was most prominent for the thickest films. To account for this effect, the raw peak area decays were adjusted so that time zero was set as the point at which the maximum lamellar peak area was measured, with all other peak areas being normalised to this new initial value (Fig. 2(a)). The adjustments were of the order of ca. 0–15 min depending on the magnitude of this effect for the different film thicknesses. The observed kinetic decay parameters were calculated from this corrected data. This effect is not dissimilar to that of overlapping peaks for spectroscopic methods, but if a co-existing phase leading to an interfering peak is not well defined or characterised, corrections can be challenging.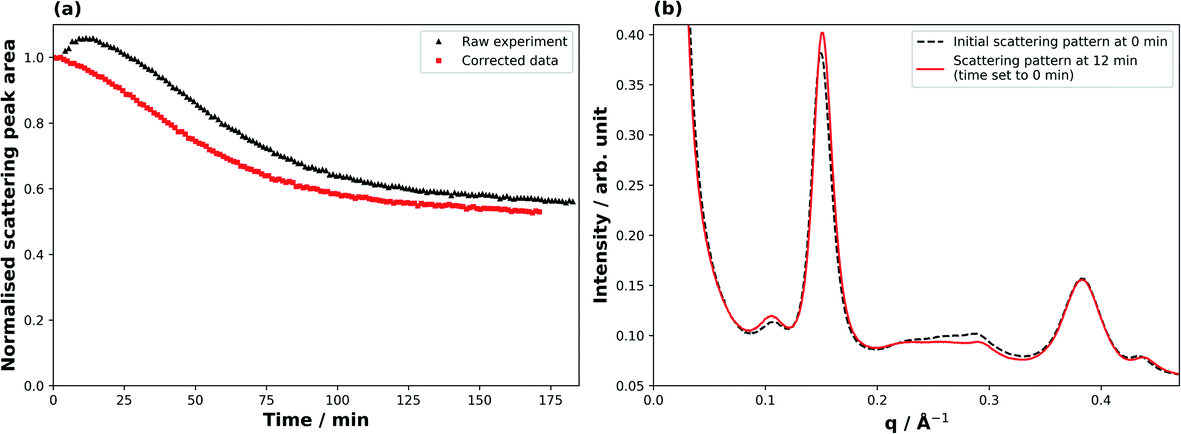 | ||
| Fig. 2 Illustration of data correction (film thickness: 73 ± 2 μm). (a) Scattering peak area vs. time exposed to ozone; both raw and corrected data are plotted. (b) SAXS patterns of the sample at the beginning of the experiment (black dashed line) and at the time (after 12 min) at which the peak scattering area was measured, which was defined as t = 0 for the decay of the lamellar phase (red solid line). | ||
3 Results and discussion
3.1 Film thickness-dependent kinetics
We were able to take advantage of the smooth decay plots obtained using the synchrotron-SAXS technique and derive observed pseudo-first order decay constants (kobs), measured during the fastest period of the reaction (see Fig. 3(b)), for the range of film thicknesses probed in this study (Fig. 3).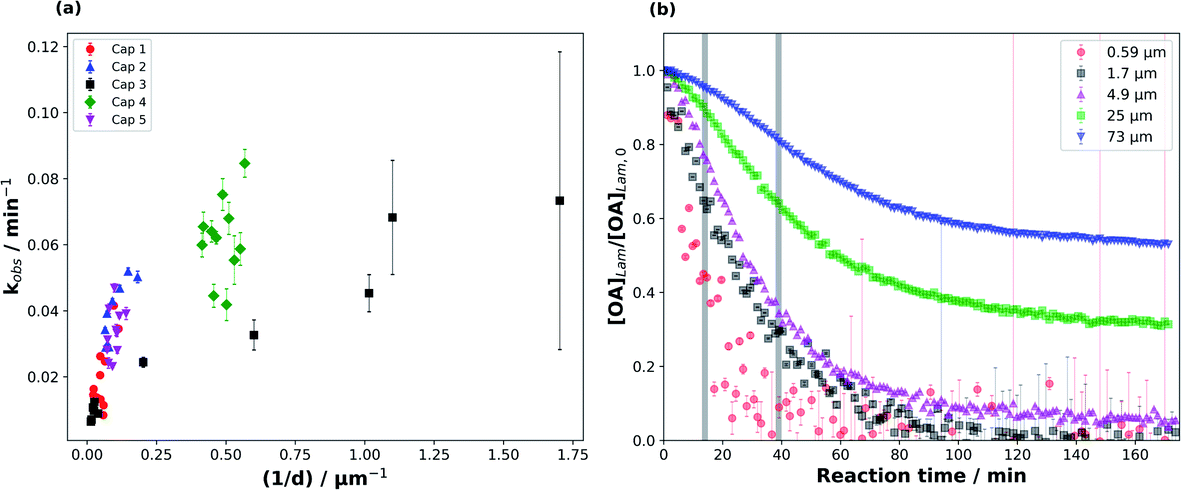 | ||
| Fig. 3 (a) The observed pseudo-first order decay constant (kobs) as a function of inverse film thickness (1/d), demonstrating the film thickness-dependence of the reaction kinetics. Different capillary (“Cap”) experiments are distinguished by different colours and symbols. (b) Decay plots of the normalised amount of lamellar oleic acid ([OA]Lam/[OA]Lam,0) vs. reaction time (t) for different thicknesses – the data are from one capillary experiment (“Cap 3”) and the grey bars indicate the reaction times between which kobs was measured for films with d > 2.5 μm. [O3] is 77 ± 5 ppm. | ||
The data presented in this study come from five different capillary coatings exposed to ozone under the same controlled conditions. Kinetic parameters from different capillaries are highlighted by different colours and symbols in Fig. 3. The higher uncertainty in the measured kobs for the thinnest films is derived from fitting fewer points to the decay function and the proximity of the 1st order lamellar peak to the baseline (Fig. S1†).
The range of film thicknesses for each coated capillary is generally small (Fig. 3(a)), with one exception (“Cap 3”) showing a range of thicknesses from the thinnest (0.59 ± 0.02 μm) to the thickest (73 ± 2 μm) films (Table S1†).
Generally, kobs scales with 1/d for the thickest films (Fig. 3(a)). However, this trend does not hold as well for the thinner films, though the increase in the uncertainty of kobs at thinner film thicknesses complicates this judgement. This deviation implies that the limiting factors governing kobs change with film thickness.48 The shape of the decay plots changes between the thinnest and thickest films (Fig. 3(b)). The decay plot for the thickest film (d = 73 ± 2 μm) reveals an initial slow decay in self-assembled oleic acid–sodium oleate concentration followed by a faster period of decay. Finally, the reaction stagnates with a significant portion of self-assembled oleic acid remaining (this stagnation is discussed in Section 3.3). A thinner film from the same capillary experiment (d = 0.59 ± 0.02 μm) exhibits a decay profile that has a more “classical” shape, following pseudo-first order kinetics with one clear reaction regime, similar to experiments performed on liquid oleic acid.13,48–50 The reaction does slow down for the thinnest films, but the low signal-to-noise ratio close to the baseline means that there is no clear evidence of stagnation.
It is known that self-assembled phases, such as the lamellar phase studied here, are viscous,34 and certainly more viscous than pure oleic acid, which is a liquid at room temperature. This means that diffusion through a particle of this composition is likely to be hindered for both the reactive gas (ozone) and oleic acid/sodium oleate. The observation that kobs scales with 1/d for the thickest films suggests that the reaction is limited to the surface of these particles and/or limited by the diffusion of O3 through the particle.48,50
The oleic acid–ozone heterogeneous reaction has been shown to be surface-dominated.48 It has been suggested that oleic acid (in the liquid form) can form ordered structures such as dimers.51 Indeed, under certain conditions, oleic acid can even form helices and vesicles.52,53 Hearn et al. suggested that on the particle surface, the alkyl chains in oleic acid are more ordered and that there is an increase in double bond density at the surface, decreasing the rate of ozone diffusion into the bulk and implying that the reaction is surface-dominated.48 Furthermore, they stated that the short spacing between alkyl chains results in reduced diffusion of ozone past them, further limiting bulk uptake of ozone. WAXS patterns obtained simultaneously during our experiments show that the short spacing between oleic acid molecules in the liquid crystalline lamellar phase is 4.41 Å (Fig. S2†). This is shorter than the 4.57 Å measured by Iwahashi et al.51 and confirmed by our WAXS experiments on liquid oleic acid, indicating that diffusion of ozone past the ordered alkyl chains in our self-assembled case is even slower. Complementary GI-SAXS patterns demonstrate that our lamellar phase sample, deposited on a silicon wafer, shows significant alignment parallel to the silicon surface, i.e. the alkyl chains are oriented perpendicular to the surface (Fig. S3†). Our experiments suggest therefore that the reaction is most likely surface-dominated at the beginning. This is clearly visible for the films thicker than ∼2 μm, which exhibit a slow initial decay (Fig. 3(b)).
There is evidence to suggest that the kinetic regime for the reaction changes after the initial surface-dominated period for the thickest films. The increase in reaction rate after the slow initial reaction period may be due to the initial surface layers of highly ordered oleic acid–sodium oleate having reacted, leaving behind disordered products (and possibly some unreacted oleic acid) which would allow for increased bulk ozone uptake due to a reduction in steric and diffusional hindrance. We suggest that after the initial surface-limited reaction, ozone starts to diffuse further into the bulk layers, forming a concentration gradient along the way. The bulk layers are still composed of the highly viscous lamellar phase with its impaired oleic acid and ozone diffusion. Noting that kobs (calculated from this region) does not scale with 1/d2, we conclude that in this reaction period the reaction is limited by ozone diffusion through the bulk and not by oleic acid diffusion. The third part of the decay profile, exhibiting reaction stagnation, will be discussed in Section 3.3.
The kinetic values presented here are comparable to those reported in previous kinetic studies on particles or films of oleic acid.13,54,55 In particular, an ozonolysis dataset reported by He et al.,49 studying deposited oleic acid particles in the size range of 50 to 200 μm using an IR-based technique, is the most comparable to our study (Table 1). Note that the deposited particles observed by He et al. will have a curvature that may affect ozone uptake.56 Our films are assumed to have a flat geometry on the μm scale (there may also be a slight curvature of the films, given the mm-scale round capillaries, but this is not considered significant on the μm scale).
Comparing our film with thickness d = 46 ± 2 μm to the ∼50 μm diameter oleic acid particle deposited by He et al., we find that kobs is 0.0107 ± 0.0002 min−1 and ∼0.135 min−1 for our film and the deposited particle in He et al., respectively. This suggests that our film reacts ca. one order of magnitude more slowly than a deposited oleic acid particle of similar size, even though we used a nearly 8-fold greater ozone concentration. Despite there being a difference in geometry, this large difference in reactivity is further evidence of the slowing effect our viscous self-assembled phase has on the reaction kinetics.
3.2 Quantification of the effect of self-assembly on reaction kinetics
The ability to coat capillaries with oleic acid in various states of solidity under the same laboratory conditions allows us to quantify the effect of phase and, more specifically, of self-assembly on the reaction kinetics of the oleic acid–ozone system.A comparison of kobs between this self-assembled system and its constituent parts shows a marked difference in reactivity as the system moves from a liquid (oleic acid) via a semi-solid (self-assembled oleic acid–sodium oleate) to a solid (sodium oleate) phase (Fig. 4 – note the log scale). There is approximately an order of magnitude difference in reactivity when going from liquid (∼10−1 min−1) via semi-solid (∼10−2 min−1) to solid (∼10−3 min−1).
 | ||
| Fig. 4 Comparison of kobs between a capillary coating of oleic acid (liquid), two coatings of self-assembled oleic acid–sodium oleate proxy (semi-solid: 0.59 and 73 μm thick) and a coating of sodium oleate (solid). *The oleic acid and sodium oleate decays were followed by Raman microscopy, using the CC band at 1650 cm−1. The thickness of the oleic acid and sodium oleate coatings was measured to be ∼50 μm based on the X-ray attenuation of these samples compared to self-assembled coatings of known thickness (OA: oleic acid; SO: sodium oleate). [O3] = 77 ± 5 ppm. | ||
To our knowledge, this is the first time the effect of self-assembly on reaction kinetics has been quantified. The data also highlight the significance of phase state and viscosity in determining the reactivity of an organic aerosol, which has been the subject of recent debate in the atmospheric literature.23,24,29,57 As discussed previously, the same reacto-diffusive arguments related to the viscosity of the lamellar phase provide a reasonable explanation for the much reduced kobs as the system’s solidity is increased.
3.3 Reaction stagnation as a function of film thickness
The decay profiles presented in this study show that as film thickness increases, the lamellar SAXS peak area decay slows down significantly by the end of the ∼3 h experiment (Fig. 3(b)), with an increasing fraction of self-assembled material retained, demonstrating an approximately linear relationship (Fig. 5).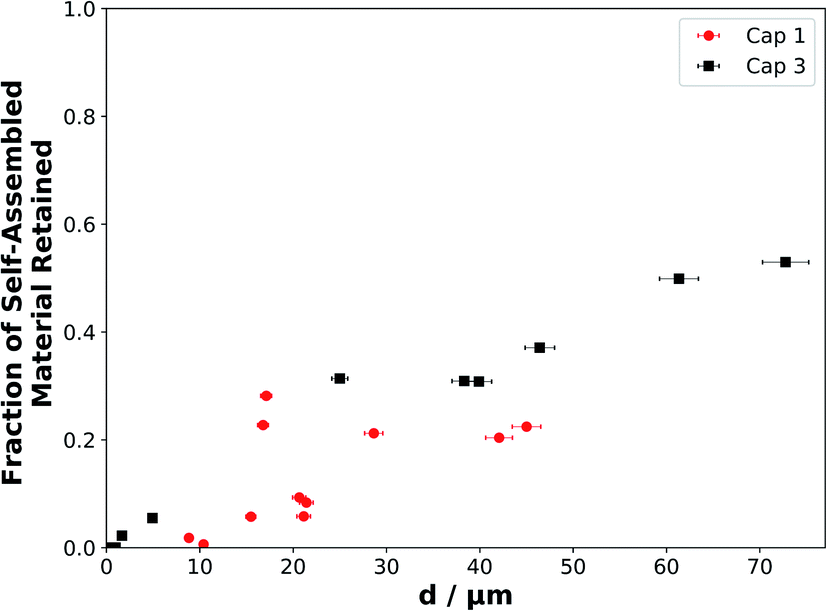 | ||
| Fig. 5 The fraction of self-assembled material retained at the end of each experiment (after ∼175 min of ozonolysis), when the reaction was deemed to have stopped or slowed significantly, as a function of film thickness. Symbols, colours and labels correspond to the same experiments presented in Fig. 3. | ||
There is a strong correlation between the fraction of self-assembled material left and the initial film thickness (Fig. 5). We assume that the thinnest films react to completion as no SAXS signal remains and simultaneous Raman microscopy shows that there is no CC (∼1650 cm−1) peak remaining by the end of the experiment. Note that the Raman spectra of these coatings were considerably noisier than the SAXS patterns, so that the CC signal became indistinguishable from the background signal earlier than the SAXS peak.
In this final reaction stage, the reaction has stagnated (Fig. 3(b)). The diffusion of ozone and oleic acid becomes hindered and this must be a result of the ozonolysis reaction. We suggest that an inert, highly viscous product is formed. This is likely because whatever is causing this stagnation has to have resulted from the reaction as this is the only process occurring. As the reaction proceeds mainly in the surface layers of the film, with a reacto-diffusive length of the order of ∼10 nm for oleic acid,15,50,58 it is likely that the outer layers of the film start to form an inert crust of these viscous products. Our results support the idea of inert crust formation, postulated by Pfrang et al.43 and suggested by others more recently,59 and are consistent with the notion that viscous phases suppress reactive gas uptake.24,29,57 High-molecular-weight oligomeric products have been observed in the oleic acid–ozone heterogeneous reaction system.60–64 They are formed by the reaction of stabilised Criegee intermediates with other reaction products. Wang et al. used a simplified model of this reaction to follow the formation of products as a function of the percentage of oleic acid conversion.62 Their output suggests that at higher oleic acid conversion, there is an increased fraction of high-molecular-weight product of up to 10%. This is a result of high-molecular-weight products forming from first-, second- and multi-generational ozonolysis products,60 suggesting a time delay before an appreciable amount of these products is formed. Our data show that there is indeed a time delay before an appreciable stagnation is observed. Considering the literature and these results, the thickest films would form a greater mass of higher molecular weight products. Therefore a thicker crust with a higher concentration of oligomeric products would be formed compared to the thinnest films, which would not have had the time to form significant concentrations of oligomeric products.
These results are in line with the recent observations of Alpert et al. who reported unreacted material remaining in the cores of viscous particles after ozonolysis at low relative humidity (RH, 0–20%).65 In particular, similarities may be drawn between our viscous system and that of Berkemeier et al. who also observed changes in the kinetic regime and rationalised this using the “kinetic cube”,66 demonstrating a transformation from a surface-dominated regime to a bulk diffusion-limited regime for viscous particles during ozonolysis at low RH (∼20%).24 We suggest that a similar reaction regime evolution prevails in our system.
The evolution of the kinetic regime is being explored in an ongoing kinetic modelling study based on the PRA framework18,20,56 and suggests that the additional stagnation is not fully accounted for when evolving particle diffusion using either obstruction theory (e.g. Pfrang et al.)43 or a Vignes-type regime (e.g. Zhou et al.).59
3.4 Implications for the urban atmosphere
Oleic acid has been measured as a component of cooking emissions in urban areas of the UK and China,7,10 and cooking aerosols have been estimated to account for an additional 10% of officially reported PM2.5 emissions in the UK.5 Our observations show that the reactive lifetime of oleic acid can vary by orders of magnitude when moving from a solid form via a self-assembled (semi-solid) form to a liquid form. In addition, there is a thickness-dependent reactivity associated with these self-assembled films, adding to the increased oleic acid lifetime. This effect suggests that the phase state of organic films formed by cooking emissions, controlled by both the urban environment and aerosol composition, could play a significant part in determining the lifetime of urban particulate matter. Moreover, assuming the thickness of an organic film is related to the amount of cooking activity, the organic film thickness could also vary – implying that the reactivity and persistence of the aerosol could be impacted by the amount of organic emissions. This is likely as high pollution events have been linked to a greater surface organic content in PM2.5 emissions in Beijing, China.38Heterogeneous reactions of oleic acid will also occur with other key initiators of atmospheric oxidation. Of particular importance at night time are nitrate radicals, NO3.67 It has been shown that the reactivity of unsaturated fatty acids towards NO3 is approximately two orders of magnitude higher than that towards ozone.68 We suggest that the phase and thickness kinetic effects reported here could also be observed for other heterogeneous reactions of this and related systems initiated by NO3. The oleic acid–ozone reaction is severely impeded by the semi-solid nature of this proxy even at a high ozone concentration. This suggests that a heterogeneous reaction with a more reactive species, such as NO3, is likely to also be impeded by the semi-solid nature of this proxy.
Urban air pollution has been linked to health conditions such as asthma,3 and recently a large range of microorganisms have been identified in urban particulate matter emitted in a megacity, implying potential consequences for human health.69 A wide range of compounds emitted into the urban atmosphere are known to have adverse health effects. These include phthalates, which affect male reproductive development70 and are emitted from plastics,71 as well as polycyclic aromatic hydrocarbons (PAHs), which have been linked to cancer risk,72 are emitted by combustion and vehicles73 and have been observed on the surface of urban particulate matter.39 Both of these classes of toxic compounds, along with oleic acid, have been found in the same marine aerosols which have been influenced by continental urban emissions.40 These compounds therefore survived the journey from the urban to the marine environment. The transport of PAHs in particular has been shown to be highly dependent on aerosol phase, which in turn is dependent on atmospheric conditions.42 Coatings of semi-solid organic aerosols are thought to shield PAHs, increasing their global atmospheric transport potential and thus increasing lung cancer risks – again, the effect is highly phase-dependent.41 The increased reactive lifetime of our self-assembled (semi-solid) films of oleic acid–sodium oleate suggests that the aerosol would travel further, take part in aerosol processes such as cloud nucleation and contribute to the persistence of pollutants in the urban environment and beyond. The fact that all three of these pollutants have been observed in the same urban-influenced marine aerosol samples indicates that viscous forms of oleic acid, such as the form studied here, could contribute to the extended transport distances of the aerosol and the potentially harmful components within it. Additionally, we have demonstrated that there is a film thickness-dependent reactivity, and as film thickness increases there is a significant reaction stagnation due to the formation of inert products – this effect is observed down to d = 4.9 ± 0.2 μm (Table S1†). Unreacted oleic acid, and reaction products, would therefore be protected from further oxidative ageing. This protection is analogous to what has been postulated about the shielding of PAHs by semi-solid organic coatings.41 As particulate organic content in urban PM2.5 emissions is linked with air quality38 and cooking aerosols contribute to PM2.5 (ref. 10) and PM10 (ref. 7 and 74) emissions, the observations for d < 10 μm are most atmospherically-relevant and demonstrate that (i) reactivity slows down over the whole film thickness range studied here, and (ii) reaction stagnation is observed only for the thicker films (from d = 4.9 ± 0.2 μm) and is therefore most relevant to PM10 emissions – though the effect is not clear for the thinnest films within the limitations of experimental signal-to-noise ratios (Fig. 3(b)).
Oleic acid is surface active, along with its primary low-volatility ozonolysis products azelaic acid75 and nonanoic acid.19 We suggest that surface active material is retained after extensive oxidation. This is because low volatility primary products are likely to remain in the surface layers of the film. Surface active organic material affects aerosol hygroscopicity by reducing aqueous droplet surface tension.76–78 Our findings suggest that the surface active components of an organic aerosol could have an increased residence time in the atmosphere, increasing the potential of the aerosol to affect cloud and fog formation in the local urban environment.79 Our results are consistent with the organic residues observed in unsaturated organic films at the air–water interface, and have similar implications.19,68,80,81
The e-folding time (τe) can be calculated as the inverse of kobs. These values are valid for the fixed ozone concentration used here, however the general trend is thought to be comparable with what would be observed at ambient ozone concentrations. τe ranges from ∼13 min (d = 0.59 ± 0.02 μm) and ∼41 min (d = 4.9 ± 0.2 μm) at the lower end of the thickness range to ∼156 min (d = 73 ± 2 μm) for the thickest film (see Table S1†). Note that kobs was measured at the fastest point of the reaction, meaning that these τe values do not take into account the effect of reaction stagnation and the evolution of the kinetic regime observed and discussed here. All τe values are longer than that estimated for a film coated with oleic acid, ∼4 min (d ∼ 50 μm, kinetics followed by Raman microscopy). This large range of τe values suggests that film thickness and phase state have a significant effect on oleic acid reactivity, implying that organic aerosols containing oleic acid in a semi-solid state such as this are likely to persist for longer times in the atmosphere. These experiments provide some explanation for the apparent extended atmospheric lifetime of oleic acid in the atmosphere compared with laboratory observations.82,83
4 Conclusions
We have conducted a series of ozonolysis experiments on capillaries coated with self-assembled oleic acid–sodium oleate cooking emission proxies. By taking advantage of the self-assembled nature of these proxies and the high intensity radiation available at a synchrotron X-ray source, we present a novel way of measuring reaction kinetics from time-resolved SAXS data; specifically we measure the rate at which self-assembly disappears during ozonolysis. There is a clear film thickness-dependent reactivity with a significant difference in reactivity between the thickest and thinnest films. We have quantified the effect of phase state on reactivity under the same reaction conditions by utilising simultaneous Raman microscopy to follow the reaction kinetics of non-self-assembled oleic acid and its salt sodium oleate. Importantly, we have found that the reaction stagnates and there remains a significant amount of unreacted proxy at the end of the experiment. This effect was most pronounced for the thickest films and the fraction of reactant remaining at the end of the experiment exhibited an approximately linear relationship with film thickness.Our work provides new evidence on the effect of particle phase on atmospheric oxidation processes. We have shown that reactant remains in the particle after ozonolysis down to a film thickness of d = 4.9 ± 0.2 μm and that generally particle phase can alter particle reactivity, which is in line with recent literature highlighting the impact of particle phase on the kinetic regime observed for heterogeneous reaction systems of atmospheric relevance.
We have presented a large dataset of film thickness-dependent kinetic decay parameters for oleic acid in a semi-solid form and have shown how the phase state of this proxy accounts for the reduction in reactivity. The thickness-dependent kinetics trend is consistent with particle size/film thickness studies in the literature and suggests that the reaction is diffusion limited.49,84,85 This diffusion limitation is likely to be enhanced by the formation of inert, viscous oligomeric products which hinder the transport of other molecules in the film. These findings are valid for the range of film thicknesses likely to be found in urban particulate matter (d < 10 μm).
In terms of atmospheric implications, our findings demonstrate that both phase state and organic film thickness significantly affect the reactive lifetime of an aerosol particle. There is clear evidence for reaction stagnation as a result of the formation of an inert crust, contributing to a longer film lifetime. These observed effects would contribute to the protection not only of urban cooking emission surfactant molecules (with implications for cloud/fog droplet formation) but also of potentially harmful compounds associated with aerosols from similar urban sources, enabling extended transport distances and persistence in the atmosphere with associated adverse health implications.
Data access statement
Kinetic data relating to this study are available in the ESI and from the corresponding author upon request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was carried out with the support of the Diamond Light Source (DLS), instrument I22 (proposals SM21663 and NT23096). AM wishes to acknowledge funding from NERC SCENARIO DTP award number NE/L002566/1 and CENTA DTP; Eleonore Mason (University of Bath) is thanked for inviting AM/CP to her GI-SAXS beamtime. The authors would like to thank Andy Smith (DLS), Tim Snow (DLS) and Lee Davidson (DLS) for technical support during beamtime experiments; Jacob Boswell is acknowledged for help during beamtimes. The authors are grateful to the Central Laser Facility for access to key equipment for the Raman work carried out simultaneously with the DLS beamtime experiments.References
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef.
- C. K. Chan and X. Yao, Atmos. Environ., 2008, 42, 1–42 CrossRef CAS.
- M. Guarnieri and J. R. Balmes, Lancet, 2014, 383, 1581–1592 CrossRef CAS.
- A. J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, P. F. Decarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, A. S. H. Prevot, Q. Zhang and A. P. Grieshop, Science, 2009, 326, 1525–1529 CrossRef.
- R. Ots, M. Vieno, J. D. Allan, S. Reis, E. Nemitz, D. E. Young, H. Coe, C. Di Marco, A. Detournay, I. A. Mackenzie, D. C. Green and M. R. Heal, Atmos. Chem. Phys., 2016, 16, 13773–13789 CrossRef CAS.
- F. Squires, E. Nemitz, B. Langford, O. Wild, W. Drysdale, P. Fu, J. Hamilton, M. Hollaway, S. Kotthaus, J. Lee, S. Metzger, N. Pingintha-Durden, M. Shaw, A. Vaughan, X. Wang, R. Wu, Q. Zhang and Y. Zhang, Atmos. Chem. Phys., 2020, 1–33 Search PubMed.
- J. D. Allan, P. I. Williams, W. T. Morgan, C. L. Martin, M. J. Flynn, J. Lee, E. Nemitz and G. J. Phillips, Atmos. Chem. Phys., 2010, 10, 647–668 CrossRef CAS.
- C. A. Alves, E. D. Vicente, M. Evtyugina, A. M. Vicente, T. Nunes, F. Lucarelli, G. Calzolai, S. Nava, A. I. Calvo, C. d. B. Alegre, F. Oduber, A. Castro and R. Fraile, Atmos. Pollut. Res., 2020, 11, 531–544 CrossRef CAS.
- E. D. Vicente, A. Vicente, M. Evtyugina, R. Carvalho, L. A. C. Tarelho, F. I. Oduber and C. Alves, Fuel Process. Technol., 2018, 176, 296–306 CrossRef CAS.
- X. Zhao, Q. Hu, X. Wang, X. Ding, Q. He, Z. Zhang, R. Shen, S. Lü, T. Liu, X. Fu and L. Chen, J. Atmos. Chem., 2015, 72, 1–18 CrossRef CAS.
- J. Zahardis and G. A. Petrucci, Atmos. Chem. Phys., 2007, 7, 1237–1274 CrossRef CAS.
- M. D. King, A. R. Rennie, C. Pfrang, A. V. Hughes and K. C. Thompson, Atmos. Environ., 2010, 44, 1822–1825 CrossRef CAS.
- P. J. Gallimore, P. T. Griffiths, F. D. Pope, J. P. Reid and M. Kalberer, J. Geophys. Res.: Atmos., 2017, 122, 4364–4377 CAS.
- M. D. King, K. C. Thompson and A. D. Ward, J. Am. Chem. Soc., 2004, 126, 16710–16711 CrossRef CAS.
- J. W. Morris, P. Davidovits, J. T. Jayne, J. L. Jimenez, Q. Shi, C. E. Kolb, D. R. Worsnop, W. S. Barney and G. Cass, Geophys. Res. Lett., 2002, 29, 71-1–71-4 CrossRef.
- A. N. Schwier, N. Sareen, T. L. Lathem, A. Nenes and V. F. McNeill, J. Geophys. Res.: Atmos., 2011, 116, D16202 CrossRef.
- C. Pfrang, K. Rastogi, E. R. Cabrera-Martinez, A. M. Seddon, C. Dicko, A. Labrador, T. S. Plivelic, N. Cowieson and A. M. Squires, Nat. Commun., 2017, 8, 1724 CrossRef CAS.
- M. Shiraiwa, C. Pfrang and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- M. Shiraiwa, C. Pfrang, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2012, 12, 2777–2794 CrossRef CAS.
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirilä, J. Leskinen, J. M. Mäkelä, J. K. Holopainen, U. Pöschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, Nature, 2010, 467, 824–827 CrossRef CAS.
- M. A. Freedman, Acc. Chem. Res., 2020, 53, 1102–1110 CrossRef CAS.
- J. P. Reid, A. K. Bertram, D. O. Topping, A. Laskin, S. T. Martin, M. D. Petters, F. D. Pope and G. Rovelli, Nat. Commun., 2018, 9, 1–14 CrossRef CAS.
- T. Berkemeier, S. S. Steimer, U. K. Krieger, T. Peter, U. Pöschl, M. Ammann and M. Shiraiwa, Phys. Chem. Chem. Phys., 2016, 18, 12662–12674 RSC.
- B. Zobrist, V. Soonsin, B. P. Luo, U. K. Krieger, C. Marcolli, T. Peter and T. Koop, Phys. Chem. Chem. Phys., 2011, 13, 3514–3526 RSC.
- T. Koop, J. Bookhold, M. Shiraiwa and U. Pöschl, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 RSC.
- E. Mikhailov, S. Vlasenko, S. T. Martin, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2009, 9, 9491–9522 CrossRef CAS.
- L. Renbaum-Wolff, J. W. Grayson, A. P. Bateman, M. Kuwata, M. Sellier, B. J. Murray, J. E. Shilling, S. T. Martin and A. K. Bertram, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8014–8019 CrossRef CAS.
- M. Shiraiwa, M. Ammann, T. Koop and U. Poschl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS.
- G. J. T. Tiddy, Phys. Rep., 1980, 57, 1–46 CrossRef CAS.
- S. Mele, O. Söderman, H. Ljusberg-Wahrén, K. Thuresson, M. Monduzzi and T. Nylander, Chem. Phys. Lipids, 2018, 211, 30–36 CrossRef CAS.
- J. M. Seddon, E. A. Bartle and J. Mingins, J. Phys.: Condens. Matter, 1990, 2, SA285–SA290 CrossRef CAS.
- J. Engblom, S. Engström and K. Fontell, J. Controlled Release, 1995, 33, 299–305 CrossRef CAS.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21, 3322–3333 CrossRef CAS.
- A. Zabara and R. Mezzenga, J. Controlled Release, 2014, 188, 31–43 CrossRef CAS.
- H. Tervahattu, J. Juhanoja and K. Kupiainen, J. Geophys. Res.: Atmos., 2002, 107, 1–7 Search PubMed.
- H. Tervahattu, J. Juhanoja, V. Vaida, A. F. Tuck, J. V. Niemi, K. Kupiainen, M. Kulmala and H. Vehkamäki, J. Geophys. Res., D: Atmos., 2005, 110, 1–9 CrossRef.
- Y. Zhao, H. Huang, Y. Zhang, K. Wu, F. Zeng, J. Wang, X. Yu, Z. Zhu, X.-Y. Yu and F. Wang, Atmos. Environ., 2020, 220, 117090 CrossRef CAS.
- W. Li, H. Li, J. Li, X. Cheng, Z. Zhang, F. Chai, H. Zhang, T. Yang, P. Duan, D. Lu and Y. Chen, J. Environ. Sci., 2018, 69, 61–76 CrossRef.
- M. Kang, F. Yang, H. Ren, W. Zhao, Y. Zhao, L. Li, Y. Yan, Y. Zhang, S. Lai, Y. Zhang, Y. Yang, Z. Wang, Y. Sun and P. Fu, Sci. Total Environ., 2017, 607–608, 339–350 CrossRef CAS.
- M. Shrivastava, S. Lou, A. Zelenyuk, R. C. Easter, R. A. Corley, B. D. Thrall, P. J. Rasch, J. D. Fast, S. L. M. Simonich, H. Shen and S. Tao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1246–1251 CrossRef CAS.
- Q. Mu, M. Shiraiwa, M. Octaviani, N. Ma, A. Ding, H. Su, G. Lammel, U. Pöschl and Y. Cheng, Sci. Adv., 2018, 4, eaap7314 CrossRef.
- C. Pfrang, M. Shiraiwa and U. Pöschl, Atmos. Chem. Phys., 2011, 11, 7343–7354 CrossRef CAS.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS.
- B. R. Pauw, J. Phys.: Condens. Matter, 2013, 25, 383201 CrossRef.
- K. Mauersberger, J. Barnes, D. Hanson and J. Morton, Geophys. Res. Lett., 1986, 13, 671–673 CrossRef CAS.
- R. M. Kirpes, D. Bonanno, N. W. May, M. Fraund, A. J. Barget, R. C. Moffet, A. P. Ault and K. A. Pratt, ACS Cent. Sci., 2019, 5, 1760–1767 CrossRef CAS.
- J. D. Hearn, G. D. Smith and A. J. Lovett, Phys. Chem. Chem. Phys., 2005, 7, 501–511 RSC.
- X. He, C. Leng, S. Pang and Y. Zhang, RSC Adv., 2017, 7, 3204–3213 RSC.
- G. D. Smith, E. Woods, C. L. DeForest, T. Baer and R. E. Miller, J. Phys. Chem. A, 2002, 106, 8085–8095 CrossRef CAS.
- M. Iwahashi, Y. Yamaguchi, T. Kato, T. Horiuchi, I. Sakurai and M. Suzuki, J. Phys. Chem., 1991, 95, 445–451 CrossRef CAS.
- M. Ishimaru, T. Toyota, K. Takakura, T. Sugawara and Y. Sugawara, Chem. Lett., 2005, 34, 46–47 CrossRef CAS.
- E. Blöchliger, M. Blocher, P. Walde and P. L. Luisi, J. Phys. Chem. B, 1998, 102, 10383–10390 CrossRef.
- E. P. Rosen, E. R. Garland and T. Baer, J. Phys. Chem. A, 2008, 112, 10315–10324 CrossRef CAS.
- E. González-Labrada, R. Schmidt and C. E. DeWolf, Phys. Chem. Chem. Phys., 2007, 9, 5814–5821 RSC.
- U. Pöschl, Y. Rudich and M. Ammann, Atmos. Chem. Phys., 2007, 7, 5989–6023 CrossRef.
- F. H. Marshall, R. E. H. Miles, Y. Song, P. B. Ohm, R. M. Power, J. P. Reid and C. S. Dutcher, Chem. Sci., 2016, 7, 1298–1308 RSC.
- T. Moise and Y. Rudich, J. Phys. Chem. A, 2002, 106, 6469–6476 CrossRef CAS.
- S. Zhou, B. C. H. Hwang, P. S. J. Lakey, A. Zuend, J. P. D. Abbatt and M. Shiraiwa, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11658–11663 CAS.
- J. Zahardis, B. W. LaFranchi and G. A. Petrucci, Atmos. Environ., 2006, 40, 1661–1670 CrossRef CAS.
- J. C. Reynolds, D. J. Last, M. McGillen, A. Nijs, A. B. Horn, C. Percival, L. J. Carpenter and A. C. Lewis, Environ. Sci. Technol., 2006, 40, 6674–6681 CrossRef CAS.
- M. Wang, L. Yao, J. Zheng, X. Wang, J. Chen, X. Yang, D. R. Worsnop, N. M. Donahue and L. Wang, Environ. Sci. Technol., 2016, 50, 5702–5710 CrossRef CAS.
- J. Zahardis, B. W. LaFranchi and G. A. Petrucci, J. Geophys. Res., D: Atmos., 2005, 110, 1–10 CrossRef.
- J. W. L. Lee, V. Carrascón, P. J. Gallimore, S. J. Fuller, A. Björkegren, D. R. Spring, F. D. Pope and M. Kalberer, Phys. Chem. Chem. Phys., 2012, 14, 8023–8031 RSC.
- P. A. Alpert, P. C. Arroyo, J. Dou, U. K. Krieger, S. S. Steimer, J. D. Förster, F. Ditas, C. Pöhlker, S. Rossignol, M. Passananti, S. Perrier, C. George, M. Shiraiwa, T. Berkemeier, B. Watts and M. Ammann, Phys. Chem. Chem. Phys., 2019, 21, 20613–20627 RSC.
- T. Berkemeier, A. J. Huisman, M. Ammann, M. Shiraiwa, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2013, 13, 6663–6686 CrossRef.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. Le Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, Atmos. Environ., Part A, 1991, 25, 1–203 CrossRef.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Atmos. Chem. Phys., 2018, 18, 3249–3268 CrossRef CAS.
- N. Qin, P. Liang, C. Wu, G. Wang, Q. Xu, X. Xiong, T. Wang, M. Zolfo, N. Segata, H. Qin, R. Knight, J. A. Gilbert and T. F. Zhu, Genome Biol., 2020, 21, 1–11 CrossRef.
- S. H. Swan, K. M. Main, F. Liu, S. L. Stewart, R. L. Kruse, A. M. Calafat, C. S. Mao, J. B. Redmon, C. L. Ternand, S. Sullivan, J. L. Teague, E. Z. Drobnis, B. S. Carter, D. Kelly, T. M. Simmons, C. Wang, L. Lumbreras, S. Villanueva, M. Diaz-Romero, M. B. Lomeli, E. Otero-Salazar, C. Hobel, B. Brock, C. Kwong, A. Muehlen, A. Sparks, A. Wolf, J. Whitham, M. Hatterman-Zogg and M. Maifeld, Environ. Health Perspect., 2005, 113, 1056–1061 CrossRef CAS.
- G. Wang, K. Kawamura, L. Shuncheng, K. Ho and J. Cao, Environ. Sci. Technol., 2006, 40, 4619–4625 CrossRef CAS.
- P. Boffetta, N. Jourenkova and P. Gustavsson, Cancer, Causes Control, 1997, 8, 444–472 CrossRef CAS.
- R. M. Harrison, D. I. T. Smith and L. Luhana, Environ. Sci. Technol., 1996, 30, 825–832 CrossRef CAS.
- K. R. Daellenbach, G. Stefenelli, C. Bozzetti, A. Vlachou, P. Fermo, R. Gonzalez, A. Piazzalunga, C. Colombi, F. Canonaco, C. Hueglin, A. Kasper-Giebl, J. L. Jaffrezo, F. Bianchi, J. G. Slowik, U. Baltensperger, I. El-Haddad and A. S. H. Prévôt, Atmos. Chem. Phys., 2017, 17, 13265–13282 CrossRef CAS.
- R. Tuckermann, Atmos. Environ., 2007, 41, 6265–6275 CrossRef CAS.
- M. C. Facchini, M. Mircea and S. Fuzzi, Nature, 1999, 401, 257–260 CrossRef CAS.
- J. Ovadnevaite, A. Zuend, A. Laaksonen, K. J. Sanchez, G. Roberts, D. Ceburnis, S. Decesari, M. Rinaldi, N. Hodas, M. C. Facchini, J. H. Seinfeld and C. O’Dowd, Nature, 2017, 546, 637–641 CrossRef CAS.
- B. R. Bzdek, J. P. Reid, J. Malila and N. L. Prisle, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8335–8343 CrossRef CAS.
- M. C. Facchini, S. Decesari, M. Mircea, S. Fuzzi and G. Loglio, Atmos. Environ., 2000, 34, 4853–4857 CrossRef CAS.
- B. Woden, M. W. A. Skoda, M. Hagreen and C. Pfrang, Atmosphere, 2018, 9, 471 CrossRef CAS.
- C. Pfrang, F. Sebastiani, C. O. M. Lucas, M. D. King, I. D. Hoare, D. Chang and R. A. Campbell, Phys. Chem. Chem. Phys., 2014, 16, 13220–13228 RSC.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS.
- A. L. Robinson, N. M. Donahue and W. F. Rogge, J. Geophys. Res.: Atmos., 2006, 111, 1–15 Search PubMed.
- S. S. Al-Kindi, F. D. Pope, D. C. Beddows, W. J. Bloss and R. M. Harrison, Atmos. Chem. Phys., 2016, 16, 15561–15579 CrossRef.
- S. S. Steimer, M. Lampimäki, E. Coz, G. Grzinic and M. Ammann, Atmos. Chem. Phys., 2014, 14, 10761–10772 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00088d |
| This journal is © The Royal Society of Chemistry 2021 |