 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCritical assessment of wet-chemical oxidation synthesis of silicon quantum dots†
Jonathan L.
Wilbrink
abc,
Chia-Ching
Huang
b,
Katerina
Dohnalova
 bc and
Jos M. J.
Paulusse
*ac
bc and
Jos M. J.
Paulusse
*ac
aDepartment of Biomolecular Nanotechnology, MESA+ Institute for Nanotechnology, TechMed Institute for Health and Biomedical Technologies, Faculty of Science and Technology, University of Twente, P.O. Box 217, Enschede, 7500 AE, The Netherlands. E-mail: J.M.J.Paulusse@utwente.nl
bInstitute of Physics, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cSpectriS-dot b.v., Wilsonstraat 34, 2131 PT Hoofddorp, The Netherlands
First published on 1st November 2019
Abstract
Wet-chemical synthetic procedures are powerful strategies to afford fluorescent silicon quantum dots (Si QDs) in a versatile and scalable manner. However, development of Si QDs is still hampered by a lack of control over photoluminescence emission, in addition to synthesis and characterization complexities. The wet-chemical Si QD synthesis by oxidation of magnesium silicide (Mg2Si) with bromine (Br2) was revisited and a control reaction was carried out where the silicon source was omitted. Both reaction conditions resulted in substantial quantities of fluorescent material. Moreover, a comparative analysis of their optical properties (UV-vis/fluorescence) revealed no apparent differences. Other characterization techniques also confirmed the resemblance of the two materials as 1H NMR, FTIR and XPS spectra were nearly identical for both samples. Elemental analysis revealed the presence of only 2 wt% silicon in the Si QD sample. No evidence was found for the formation of significant amounts of Si QDs via this wet-chemical procedure.
Introduction
Quantum dots (QDs), also coined semiconductor nanocrystals, are nanoparticles that possess unique properties on the quantum level that differ from bulk properties.1,2 Depending on the particle size and shape, QDs display photoluminescence, following the quantum confinement model.1 As a consequence, by varying QD dimensions, emission can be tuned over a broad spectral range. Owing to their superior optical properties, such as size-dependent fluorescence,3–5 color definition6 and resistance against photobleaching,7,8 QDs are promising materials for a wide variety of applications, including photovoltaics,9–11 lighting and displays,6,12 and biomedical imaging.13–19 This application potential is, however, severely limited by the fact that conventional QDs typically contain toxic and/or scarce elements (e.g. Cd, In, Pb, Se or Te). Silicon quantum dots (Si QDs) signify an appealing, non-toxic and highly sustainable alternative.20–22Synthesis strategies for Si QDs are typically divided into top-down and bottom-up approaches (and combinations).16 In top-down approaches, Si QDs are prepared from larger macroscopic materials, e.g. silicon wafers, to construct nanoparticles, which can be done mechanically,23 as well as by etching with strong acids, such as HF.24 In addition, the group of Veinot and coworkers developed an effective route based on the thermal decomposition of hydrogen-terminated silsesquioxane to produce Si QDs within a silica matrix, followed by liberation of the Si QDs by HF etching.4,25,26
In bottom-up approaches, Si QDs are prepared from molecular precursors. This includes syntheses by physical means, such as condensation of silane gas into Si QDs via laser pyrolysis27,28 or plasma synthesis.3,29,30 Other strategies that have been reported to yield Si QDs include decomposition of precursors in supercritical fluids31 and by microwave irradiation.32–34 Bottom-up approaches by chemical means are typically wet-chemical syntheses, where reactions are carried out in solution-phase.16,21,35,36 Strategies include reduction of SiCl4 (or SiBr4, and sometimes co-reaction with alkyltrichlorosilanes) using reducing agents, such as alkali metals (e.g. Na, K),37–39 alkaline earth metals (e.g. Mg),40 sodium naphthalide41,42 or LiAlH4,43–49 and in molten salt syntheses using active Al species.50,51 Synthetic routes towards Si QDs have also been based on the oxidation of silicon species, typically Zintl salts (XSin, where X is an alkali metal or alkaline earth metal). Common methods are the oxidation of these Zintl salts by Br2,52–56 NH4Br,34,57–59 or NH4Cl.60,61 Furthermore, Zintl salts have been reacted with SiCl4 to yield Si QDs in a metathesis type synthesis.62–64 Regarding synthesis strategies, several reviews are recommended for a more complete and detailed overview.16,21,36,65
Wet-chemical synthesis is generally more versatile, less laborious and highly scalable, while elevated temperatures can be avoided, although limited control over fluorescence emission remains a major challenge.16 Moreover, the number of reliable wet-chemical synthetic strategies towards Si QDs is still limited, with complications concerning synthesis and characterization hampering research into Si QDs, as well as their commercialization.
Fluorescence emission generally correlates well with Si QD size,66 although discrepancies between Si QD size and emission have been reported.25 In particular for wet-chemical syntheses, primarily blue or green fluorescence emission is observed, regardless of the reported size. There is still debate as to whether quantum confinement is in fact the origin of photoluminescence in Si QDs.67–69 Bulk silicon is an indirect bandgap material,70 and even on the nanoscale it is often implicated that fluorescence emission is either inefficient or mostly governed by surface defects rather than quantum confinement effects.9,71 Moreover, such defects are frequently reported,9,69,71,72e.g. due to nitrogen-containing species on Si QD surfaces73 or particle oxidation.72,74 Interestingly, even two seemingly comparable Si QD samples, albeit synthesized via different procedures, displayed very different optical properties, i.e. blue and red emission.75 These differences were explained by the occurrence of nitrogen-containing impurities in the blue-emitting Si QDs. Emission of Si QDs was demonstrated by Wiggers and coworkers to change from red to blue upon exposure to air, confirming that blue fluorescence can be induced by oxidation.74 Surface-related fluorescence is typically undesirable, as the emission is no longer tunable by adjusting the particle size, and such particles are more susceptible to photobleaching.7 Although photoluminescence is frequently assigned to surface defects when the quantum confinement model cannot be applied,71,75 this is not necessarily true in certain cases, as discussed below.
Interestingly, side reactions occurring during Si QD synthesis may also induce the (majority of) fluorescence and obscure results. Recently, Oliinyk et al. criticized76 the preparation of Si QDs via a one-pot microwave synthesis employing 3-aminopropyltrimethoxysilane (APTMS) as the silicon source, as reported earlier by He and coworkers.32 Their criticism focused on discrepancies concerning the XRD data when comparing with other Si QD literature,77 the absence of XPS data, TEM images showing crystalline material that is inconsistent with silicon, and most strikingly, very comparable optical properties for materials prepared in the presence, as well as in the absence of the silicon source.
Purification of fluorescent nanomaterials has also been pointed out to be critical.78–80 For instance, fluorescent by-products (e.g. small molecules) were frequently insufficiently removed.78–82 Crystalline nanoparticles of molybdenum sulfide, as observed by TEM, were reported to display strong fluorescence emission,83 although more recently, smaller impurities were demonstrated to be the actual source of fluorescence.84 Hence, connecting the optical properties to a nanomaterial remains a considerable challenge, as also pointed out earlier by Nandi and coworkers,80 who particularly stressed the importance of careful interpretation of TEM data.79,85
Our investigations into the preparation of Si QDs via the oxidation of Mg2Si with Br2, a well-established, facile reaction generally resulting in high product yields,52,53 revealed that attempts to alter the optical properties by changing the reaction parameters, such as temperature, reaction time, concentration and reagents, remained unsuccessful. A lack of size-control (and thus control over the emission) was noted earlier, but remained unexplained.52 This prompted us to investigate this reaction in more detail. An in-depth comparative study on these Si QDs is presented here, and a comparison is made with material resulting from a silicon-free synthesis, where the Zintl salt was omitted. Both materials were characterized using a number of different techniques, including 1H-NMR spectroscopy, FTIR, TEM, elemental analysis, XPS, fluorescence spectroscopy, UV-vis spectroscopy, and SEC. Both products possessed highly comparable properties, hinting towards a mechanism that presumably does not (exclusively) produce Si QDs, but primarily fluorescent by-products.
Experimental
Materials and methods
Syntheses were carried out under a nitrogen atmosphere, unless stated otherwise. Glassware was dried before use. Solvents were dried over 4 Å molecular sieves for at least 24 h before use.n-Butyllithium (n-BuLi, 2.5 M, in hexanes), chloroform-d (99.8 atom% D) and n-octane (≥97%) were obtained from Acros, bromine (Br2, ≥99.99% trace metals basis), 9,10-diphenylanthracene, hydrochloric acid (HCl, 37%), magnesium silicide (Mg2Si, ≥99% trace metals basis, −20 mesh), molecular sieves (0.3 nm, rods), sodium sulfate (Na2SO4, anhydrous), and sodium thiosulfate (Na2S2O3, ≥98%, anhydrous) were obtained from Sigma Aldrich, chloroform, ethanol, ethyl acetate and methanol were obtained from VWR, TEM grids were obtained from EM Resolutions (Sheffield, United Kingdom), and XPS substrates from SSENS (Hengelo, The Netherlands).
Characterization
Nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker 400 MHz or Bruker 600 MHz spectrometer. Samples were measured in chloroform-d. Attenuated total reflectance (ATR) Fourier-transform infrared (FTIR) spectra were obtained by drop-casting samples from chloroform solution on a Bruker Alpha-p. Elemental analysis for carbon was performed on a Vario Micro Cube CHNS-Analyser from Elementar, for bromine on an 883 Plus IC from Metrohm, and for silicon on a Specord 50 Plus UV/vis spectrophotometer from Analytik Jena. Transmission electron microscopy (TEM) was performed on a Philips CM300ST-FEG transmission electron microscope operated at 300 kV. Samples were drop-cast from a dilute chloroform solution on holey carbon films on a 200 mesh copper substrate. X-ray photoelectron spectroscopy (XPS) was measured on a PHI Quantera SXM with a monochromatic Al Kα source (1486.6 eV). The carbon C 1s signal at 284.8 eV was taken as the reference binding energy. Samples were drop-cast from a chloroform solution on gold-coated glass substrates (20 nm gold). UV-vis spectra were recorded on a Shimadzu UV-2401PC UV-vis spectrophotometer. Absorbance was kept below 0.05 for quantum yield determinations to avoid inner filter effects. Ethyl acetate was employed as the solvent for UV-vis and fluorescence spectroscopy. Fluorescence was measured on a Varian Cary Eclipse fluorescence spectrophotometer. Size exclusion chromatography (SEC) measurements were performed on a Waters Alliance e2696, equipped with a 2475 fluorescence detector, and a 2998 photodiode array detector to determine absorbance. Samples were run on an Agilent PLgel 5 μm MIXED-D column, using HPLC-grade chloroform as the eluent. Before measurement, samples were filtered on 0.2 μm Whatman Spartan syringe filters.Synthesis of Si QDs
Si QDs were synthesized, with only minor changes, according to earlier literature procedures.52,53 Degassed n-octane (100 mL, 0.615 mol) was added to Mg2Si (100 mg, 1.30 mmol), followed by the addition of Br2 (0.54 mL, 10.5 mmol). Subsequently, the dispersion was stirred for 2 h, whereupon the color changed from deep orange to pale orange. The dispersion was heated to reflux for 18 h (or 60 h, isolated yield 37.8 mg). Then, the solvent was removed under reduced pressure, under an inert atmosphere. Fresh, degassed n-octane (100 mL, 0.615 mol) was added and the mixture was cooled on ice. n-Butyllithium in hexane (2.5 M, 2.09 mL, 5.22 mmol) was slowly added and run overnight for complete reaction, while the dispersion was allowed to warm up to room temperature. Unreacted n-butyllithium was quenched with excess methanol (10 mL), and left to react for 1 h. Thereafter, the dispersion was filtered and extracted against aqueous HCl (1 M, 100 mL, 1×) and distilled water (100 mL, 3×). During extraction, the organic phase was deep orange, with some insoluble materials, especially at the solvent–solvent interface, which were discarded. The organic phase was dried over Na2SO4 and filtered, and the solvent was removed under reduced pressure. The obtained product was a dark brown material (24.4 mg).Control synthesis
The control synthesis was performed similarly to the procedure described above, with the only change being that the silicon source, Mg2Si, was omitted during the synthesis. Initially, more coloration was observed during the reaction, due to the absence of the dark purple Mg2Si. The obtained product was a dark brown material (24.5 mg).Reaction between n-octane and Br2
Br2 (0.54 mL, 10.5 mmol) was added to degassed n-octane (100 mL, 0.615 mol) and reacted for 2 h at room temperature. Upon reaction, the orange color of the Br2 slowly fades. Subsequently, samples were drawn and analyzed with 1H-NMR spectroscopy.Results and discussion
Synthesis of Si QDs by oxidation of Mg2Si by Br2
To investigate the structural and optical properties of the nanomaterials prepared via the oxidation of Mg2Si by Br2,52–56 as depicted in Scheme 1, this synthesis was carried out with only minor changes: most importantly, commercial Mg2Si was used instead of that synthesized from Mg and Si, solvent was removed at reflux temperature, rather than at room temperature, and surface passivation with n-butyllithium was carried out for 1 day, instead of for 2 days. The products of these reactions here are denoted as Si QDs (in the presence of Mg2Si) and control (in the absence of Mg2Si).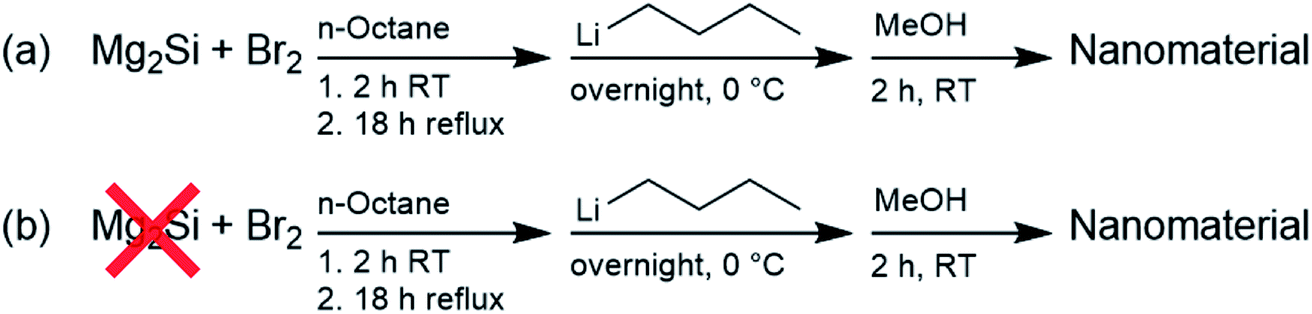 | ||
| Scheme 1 Synthesis of Si QDs (a) and the control (b). | ||
The products were first characterized using 1H NMR spectroscopy, as shown in Fig. 1. The spectrum for Si QDs is in accordance with spectra published before. Signals at 0.88 ppm and 1.25 ppm were observed and have been assigned previously to –CH3 and –CH2– protons of the butyl surface passivation, respectively.86 Similar peak broadening was observed as previously reported.53 However, no proof for silicon-bound –CH2– protons around 0.6 ppm (ref. 87) was found, although whether such signals can be observed for these kinds of material is still the subject of debate.88 In comparison with the control, no obvious differences between the spectra are apparent. Signal intensity was low for both samples, even at high concentrations (>20 mg mL−1), while no precipitation was observed.
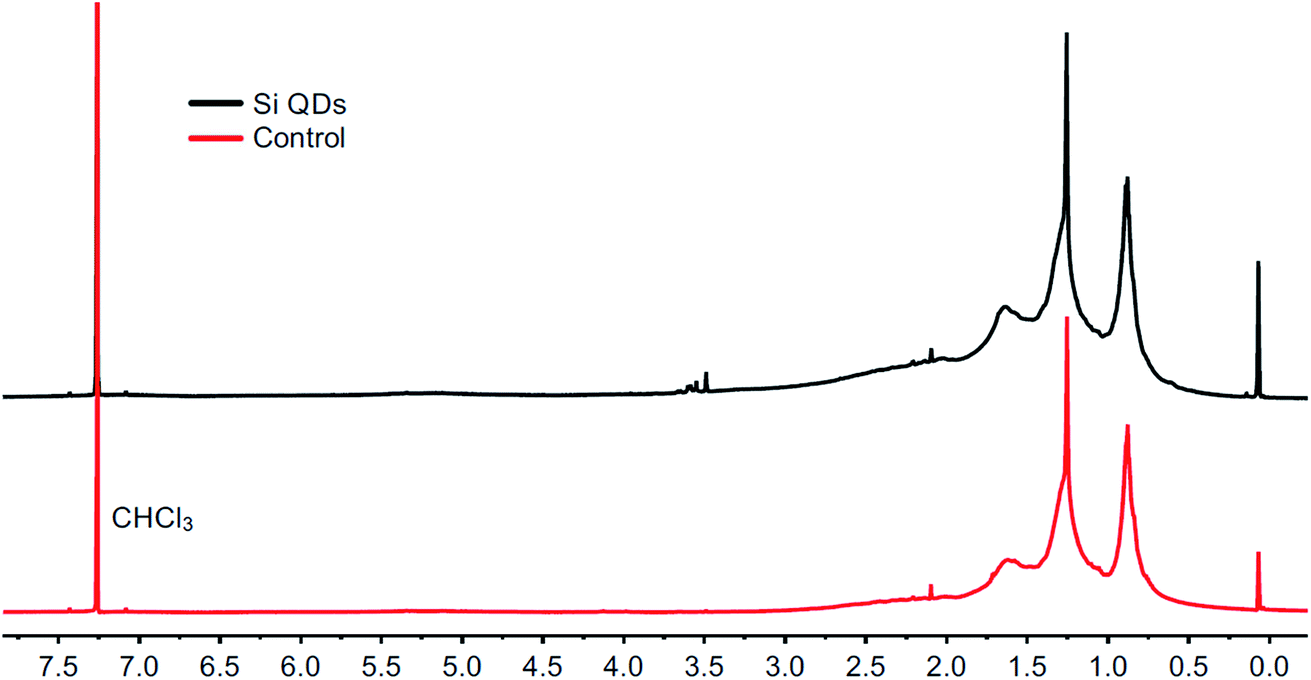 | ||
| Fig. 1 1H NMR spectra from the Si QD synthesis (top) and the control synthesis (bottom) in chloroform-d. | ||
The FTIR spectrum of the Si QDs (see Fig. 2) corresponds well with those from previous literature, although OH vibrations around 3425 cm−1 were not observed in previous reports,52 or only to a minor extent.53 Vibrations around 2927 cm−1 correspond to CH2 and CH3 groups, vibrations at 1456 cm−1 to CH2 and CH3 groups, and vibrations at 1377 cm−1 to CH3 groups; all of them are characteristic for alkyl functionalities. Vibrations corresponding to Si–CH2 bonds have been reported before to appear at approximately 1260 cm−1 and 1460 cm−1,49,64,89,90 but are generally weak. Small vibrations were indeed observed at 1261 cm−1, which might be related to Si–CH2 vibrations, although it is not strong evidence for Si QDs, as it may also be assigned to other species. Vibrations at 1705 cm−1 are also present, which has been observed earlier.52,53 However, the nature of these species remains unknown, as these vibrations were not assigned. The distinctive vibrations at 1073 cm−1 are attributed to Si–O bonds, as a result of (partial) oxidation of the Si QDs.4,74 Notably, this signal was also observed in the control, which makes the assignment of these vibrations as unique for Si QDs uncertain. Moreover, no discernible differences between the Si QDs and the control were observed.
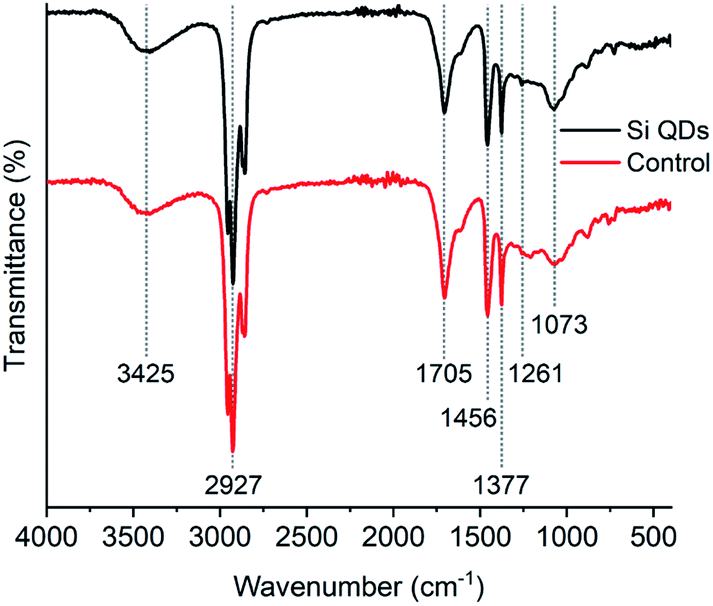 | ||
| Fig. 2 FTIR spectra of Si QDs and the control. | ||
The samples were further analyzed with TEM, as shown in Fig. 3. It can be observed that non-crystalline nanosized particles are present (size not quantified) in both cases. It should be mentioned that both TEM grids were evaluated until areas were found with high contrast, given that these images do not represent the full sample. However, this was performed in order to show the notable resemblance with Si QDs reported previously.53 For the control, it can be excluded that these particles on TEM are Si QDs, indicating that mere particle observations using TEM do not infer the presence of Si QDs by default. Particles might be aggregates of organic matter, carbon dots, or a result of TEM sample preparation.79,80,85 Fluorescence by quantum confinement effects needs to be confirmed, regardless of whether the Si QDs are amorphous91 or crystalline.92
 | ||
| Fig. 3 TEM images of Si QDs (a) and the control (b). | ||
Elemental analysis was performed in order to examine the sample composition, and the presence of silicon in particular, as shown in Table S1.† A Si amount of 2.2 wt% was found for the Si QDs, while for the control a Si amount of 0.4 wt% was determined. In both samples, carbon (>60 wt%) and bromine (>3 wt%) were observed. In combination with the product yield, the reaction efficiency for silicon can be derived. At best, less than 1 mg of elemental Si from the starting material (Mg2Si) can be traced back to the Si QDs, which is equal to 2.2% conversion of Mg2Si. Considering the (slightly) higher Si content in Si QDs, conversion of Mg2Si may have occurred, but is only marginal.
XPS analysis was performed to obtain more knowledge on the type of Si bonding in particular, and further confirm the presence of silicon (see Fig. 4 and S1–S4†). After deconvolution, a binding energy of 102.2 eV was observed for silicon in the Si QDs, and a binding energy of 102.4 eV was determined for the control. In the case of the control, Si arose from minor impurities, and therefore will be neglected. Binding energies around 102.2 eV were found for the Si QDs, indicating that no fully oxidized SiO2 species were present, as binding energies would be observed around ∼103.5 eV.25,56 However, XPS data for siloxanes shows similar binding energies to that for Si QDs,93 whereas such materials consist primarily of Si–O bonds. In both the control and the Si QDs, it is not unlikely that similar oxygen-bound silicon material is present. However, most importantly, binding energies at ∼99.5 eV (depending on the oxidation state), corresponding to Si–Si bonds, are absent, though expected in the case of Si QDs.25,77 Hence, the presence of appreciable amounts of Si QDs cannot be confirmed.
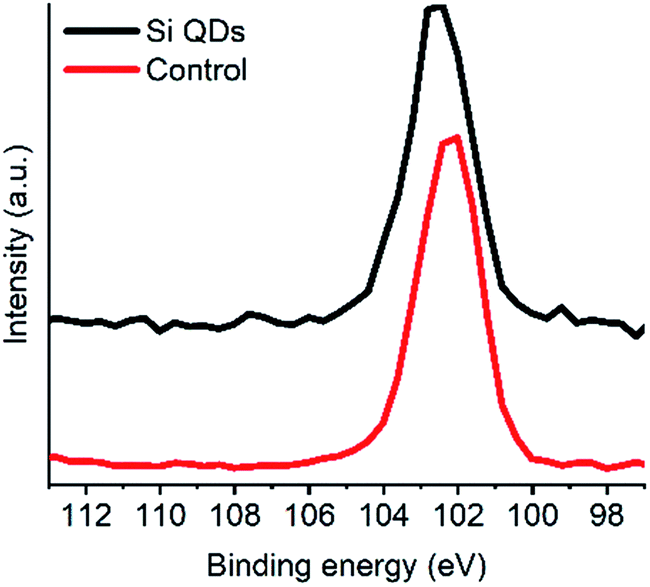 | ||
| Fig. 4 XPS spectra Si QDs and the control (Si 2p scan). | ||
The absorbance and fluorescence of Si QDs and the control are depicted in Fig. 5. The Si QDs revealed no characteristic features in the absorbance, as described earlier.52,53 While it is known that QDs show features that correlate to their size, proposed for Si QDs as well,94 the absence of features may also be an effect of a heterogeneous particle size distribution. However, the similarity with the control is of more concern, indicating no discernable differences.
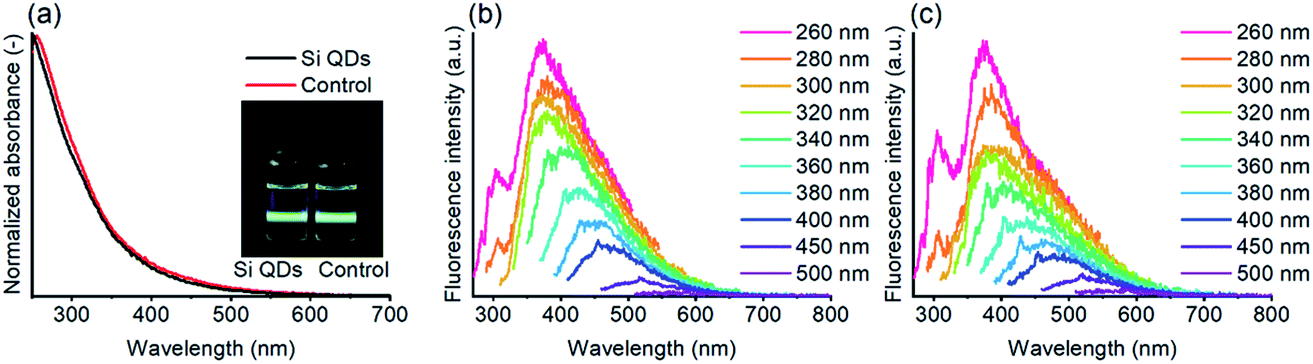 | ||
| Fig. 5 Absorbance of Si QDs and the control, with an insert showing both samples under UV (excitation 405 nm) illumination (a), and excitation plots of Si QDs (b) and the control (c) in ethyl acetate, with the excitation wavelength ranging from 260 to 500 nm (see also Fig. S5†). | ||
The fluorescence properties of the samples were subsequently assessed, as shown in Fig. 5. Under visible light, both samples are colorless to pale yellow, while under UV illumination bright blue/green fluorescence can be observed, consistent with the emission observed in the excitation plots. For the Si QDs, the fluorescence spectra are comparable to those reported before,52 although the emission observed for the Si QDs at lower excitation wavelengths is somewhat stronger. For excitation wavelengths between ∼260–320 nm, the emission wavelength is centered around ∼380 nm, while for higher excitation wavelengths, the emission was red-shifted and rapidly decreased in intensity. Thus, below excitation wavelengths of 320 nm, excitation-independent emission dominates, while for higher excitation wavelengths, excitation-dependent emission is stronger. The reported emission peak maximum (383 nm) in the literature is consistent with the maximum in our spectra. However, the most effective excitation wavelength was reported to be at ∼320 nm, whilst in our case, the lowest excitation wavelength at 260 nm was the most efficient. The most plausible explanation would be that in the current work, more blue-emitting matter was produced due to slight experimental differences. For instance, shorter reaction times or slightly lower temperatures might possibly result in more blue-emitting material.80 In Fig. S5,† the strongly overlapping emission of the Si QDs and the control can be observed, regardless of excitation wavelength. The fluorescence emission of the control cannot originate from Si QDs, yet still the spectra from Si QDs show the same identical emission spectra as the control, confirming that fluorescence emission in both the Si QDs and control samples is (mostly) determined by species other than Si QDs.
The quantum yield (QY) of the Si QDs, at an excitation wavelength of 350 nm, was determined via a relative method.95 The QY was found to be 2.2%, which is comparable with previously published data.53 For the control, the QY was somewhat lower at 1.1%.
By means of size exclusion chromatography (SEC) it was possible to separate the materials based on their size, as shown in Fig. 6, and evaluate the optical properties accordingly. For the Si QDs, it was observed that two populations are present, while only the smaller material showed significant fluorescence. Complete SEC traces of absorbance and fluorescence can be found in Fig. S6.† For fluorescence emission, it can be observed that for the Si QDs and the control, the fluorescence spectra were very comparable. In addition, both materials showed size-dependent fluorescence, which before was thought to be a result of quantum confinement.52 Emission at 400 nm was strongest at 8.87 min, while emission at 550 nm was strongest at an elution time of 8.60 min. Therefore, it can be concluded that solely size-dependent emission is not proof of Si QD emission.
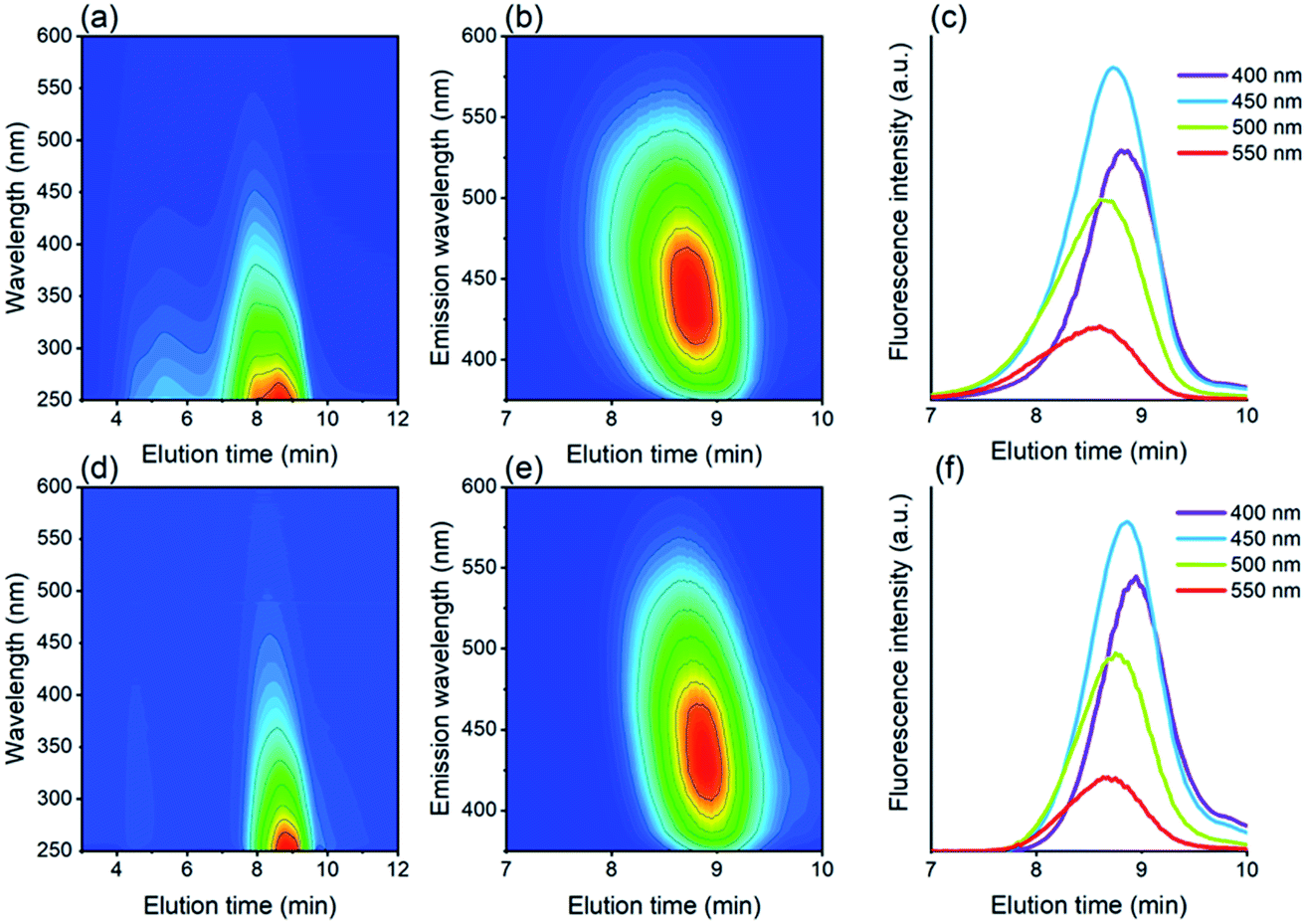 | ||
| Fig. 6 SEC traces of absorbance (a) and fluorescence (b and c) of Si QDs, and absorbance (d) and fluorescence (e and f) of the control. The intensity of absorbance and fluorescence increases from blue to red in the contour plots. The emission intensities in (c) and (f) were measured at four different emission wavelengths. | ||
There are, however, more notable deviations in the case of the absorbance spectra. It can be observed in Fig. 6 that for both samples, absorbance is observed over a much broader range (∼3.5–10 min) than fluorescence (∼8–9.5 min), indicating the presence of an emissive fraction at longer elution times, and a less- or non-emissive fraction at shorter elution times. The absorbance for Si QDs is much stronger than for the control at short elution times (∼3.5–7 min), although also in the control absorbance was observed to a minor extent (see also Fig. S7†). Size fractionation of this material has been carried out, revealing enhanced silicon content (0.6 wt% vs. 0.2 wt%),96 however, this species does not display strong fluorescence. Remarkably, a similar effect was also found in carbon dot synthesis, where size-separation was performed via dialysis.79 Smaller materials (<1 kDa) showed strong fluorescence, while the larger materials (≥50 kDa) showed much weaker fluorescence. This again suggests that in the samples investigated here, the fluorescence emission is likely caused by small fluorescent materials other than Si QDs.
The findings that the reactions carried out in the presence and absence of Mg2Si result in products with almost identical properties raises questions concerning the actual nature of the material formed. Therefore, determining the origin of these by-products was attempted. Direct exposure of Mg2Si to Br2 in the absence of a solvent with overnight stirring did not result in any noticeable reaction. Alternatively, elemental (insoluble) silicon may have been formed, as presumed for the Si QDs synthesis carried out in glyme.52 In addition, it is known that reactions between Mg2Si and aqueous HCl yield (higher) silanes,97,98 assuming that aqueous HBr reacts in a similar fashion. Hereunder, it is shown that HBr is indeed formed during Si QD synthesis, although it remains unclear whether other species (besides octyl bromides and HBr) would form and how the reaction might proceed further.
After addition of Br2 to the reaction mixture containing Mg2Si and n-octane, the color of the Br2 fades. This is also the case in the absence of Mg2Si. Bromination of alkanes has been demonstrated before,99–102 and results in the formation of alkyl bromides and HBr gas.1021H NMR spectroscopic analysis indeed confirms the formation of octyl bromides, as evidenced by signals at 3.99, 4.04 and 4.13 ppm, which were assigned (primarily) to three isomers of secondary octyl bromides (ratios not quantified, see Fig. S8†). Formation of considerable amounts of HBr gas was visually confirmed, as well as through the use of a pH indicator. Despite considerable evolution of HBr gas, it should be noted that the solubility of HBr in n-octane is also significant.103 Formation of octyl bromides was observed before, but was then not considered to be the origin of strongly fluorescent material.53 Experimentally, products of the reaction between n-octane and Br2 were collected, possible residual Br2 and HBr were removed by extraction against water, and the organic phase, containing n-octane and octyl bromides, was dried over Na2SO4. Subsequently, this mixture of n-octane and octyl bromides was heated to reflux, and fluorescent material was again obtained. This indicates that HBr and Br2 are likely not essential for formation of fluorescent by-products upon heating, but rather that other, earlier formed species are, e.g. octyl bromides.
In addition to our critical assessment of Si QD synthesis by means of wet-chemical oxidation, we stress that related work on Si QDs should be critically assessed as well, and bottom-up syntheses in particular.
In our view, most essential in Si QD synthesis are two aspects. Firstly, it should be shown convincingly that (crystalline) Si QDs have been synthesized successfully, preferably using an appropriate combination of methods discussed below.26,76 Secondly, it should be confirmed that these Si QDs are indeed the true origin of displayed fluorescence. Careful purification and characterization are therefore essential. Frequently, successful fabrication of (size-tunable) fluorescent Si QDs is claimed, almost entirely based on particles observed by TEM in combination with fluorescence spectra. However, as indicated, the presence of (crystalline) particles alone is not direct proof of the formation of fluorescing Si QDs.79,85 Furthermore, it is not improbable that side products formed during synthesis might attach to the surface of the true QDs, further complicating observations.80
As long as the quantum confinement model is applicable, evidence of a direct relation between determined size and fluorescence emission would be of great value in this regard, for instance achieved by fluorescence spectroscopy on size-separated samples. Alternatively, differently sized particles may be prepared and their respective sizes and fluorescence emissions can be assessed.4,104 Unfortunately, surface effects influencing fluorescence, and thus reducing or eliminating the effect of size on emission, especially towards smaller sizes, may complicate such characterization methods.105
Veinot and coworkers provided guidelines for the evaluation of Si QDs,26 which are in line with a more general approach published on inorganic colloidal materials.106 In fact, characterization can be performed almost inexhaustibly, and a combination of techniques will always be required due to the complex nature of Si QDs.26 Most conveniently, information regarding crystallinity can be obtained using TEM107,108 and XRD,77 size is readily assessed by TEM and SAXS,25,66 while the nature of Si-bonding can be determined using Raman spectroscopy,109 FTIR108,110 and XPS.25 The optical properties can be ascertained through fluorescence and UV-vis spectroscopic analysis, while chemical composition can be determined by elemental analysis and, trivially but importantly, by reporting product yields. Further insights can be gained from techniques such as dynamic light scattering,111 atomic force microscopy,112 thermogravimetric analysis,113 NMR spectroscopy,77 fluorescence lifetime experiments,114 ultracentrifugation,115 size-selective precipitation,105 and size exclusion chromatography.28,116
For purification, chromatographic separation based on size (e.g. SEC) or polarity (e.g. HPLC) are powerful tools for removing (fluorescent) by-products,80,117 while dialysis may be convenient as well.79 It has been shown that purification in the field of nanotechnology has been frequently performed only to a limited extent, if at all, leading to misinterpretations.78 For instance, centrifugation or filtration alone is typically insufficient.79 Dialysis, especially when using a molecular weight cut-off below 20 kDa, is not recommended, due to its limited ability to remove organic residues.79
Our studies indicate that the reaction solvents may not be as inert as generally considered, and can induce fluorescence (possibly in combination with other reactants), especially when heat or microwave radiation is applied. For instance, it has been shown that refluxing ethylene glycol readily results in fluorescent material,84 as reported for poly(ethylene glycol) as well.118 The combination of sodium and refluxing diglyme has also been demonstrated to lead to fluorescent by-products (suggested to be polymerized diglyme).119 These by-products could also form during a Si QD synthesis where sodium naphthalide is reacted in refluxing glyme.41 Gu et al. obtained fluorescent materials when heating a series of solvents (DMF, DMAc, xylene, n-hexane and cyclohexane) to 260 °C in an autoclave.120 Generally, reaction temperatures in wet-chemical Si QD syntheses (e.g. glyme at 85 °C,52,64n-octane at 126 °C,52,53 DMF at 153 °C (ref. 57 and 58)) and microwave-assisted Si QD syntheses (e.g. DMSO, DMF, acetonitrile at 160 °C,33 glycerol at 180 °C121 and DMF at 275 °C (ref. 34)) are slightly lower. However, it is important to rule out the formation and/or insufficient removal of such fluorescent materials when comparable conditions are employed. For example, microwave-assisted synthesis in DMF at 275 °C was confirmed to result in the formation of fluorescent by-products.34 Hence the requirement for effective purification. Straightforward control experiments (without a source of silicon, in the case of Si QDs) would help to confirm that the observed fluorescence does not originate from by-products.76,84
Conclusions
In the assessment of the wet-chemical oxidation of Mg2Si with Br2 we observed the formation of fluorescent nanomaterials, even in the absence of a silicon precursor. We demonstrated that the materials formed in the presence and absence of Mg2Si display very comparable optical properties. Not only were the optical properties similar, but other physicochemical properties, as determined using other characterization techniques, were found to be similar. In fact, only SEC characterization and elemental analysis revealed minute differences. However, no evidence was found supporting the formation of Si QDs in substantial amounts. No crystallinity corresponding to silicon was observed, no size-dependent emission different from the control without Mg2Si was shown, and no proof of Si–Si bonds was observed. We suggest that wet-chemical syntheses, especially those involving heating, can lead to the formation of fluorescent by-products with similar optical properties to those of Si QDs. The presence of fluorescent impurities might not easily be distinguished or detected due to a generally high content of organic material in the samples and there being no need for the carbon material to be crystalline to show size-dependent properties.78The optical properties observed in Si QD synthesis should be convincingly linked to the actual nanomaterial. This may be achieved by appropriate purification by chromatography and a combination of characterization techniques to determine Si QD crystallinity, size, bonds, optical properties, composition and product yields. Considerations regarding Si QDs may also be extended to other types of fluorescent nanoparticles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Rico Keim for performing TEM measurements and Mr Gerard Kip for performing XPS measurements.References
- A. D. Yoffe, Adv. Phys., 2001, 50, 1–208 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- A. Gupta, M. T. Swihart and H. Wiggers, Adv. Funct. Mater., 2009, 19, 696–703 CrossRef CAS.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, Chem. Mater., 2006, 18, 6139–6146 CrossRef CAS.
- D. Norris and M. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16338–16346 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- M. Dasog, G. B. De Los Reyes, L. V. Titova, F. A. Hegmann and J. G. C. Veinot, ACS Nano, 2014, 8, 9636–9648 CrossRef CAS PubMed.
- X. Gao, L. Yang, J. A. Petros, F. F. Marshall, J. W. Simons and S. Nie, Curr. Opin. Biotechnol., 2005, 16, 63–72 CrossRef CAS PubMed.
- Z. Ni, S. Zhou, S. Zhao, W. Peng, D. Yang and X. Pi, Mater. Sci. Eng., R, 2019, 138, 85–117 CrossRef.
- F. Priolo, T. Gregorkiewicz, M. Galli and T. F. Krauss, Nat. Nanotechnol., 2014, 9, 19–32 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CrossRef CAS.
- F. Maier-Flaig, J. Rinck, M. Stephan, T. Bocksrocker, M. Bruns, C. Kübel, A. K. Powell, G. A. Ozin and U. Lemmer, Nano Lett., 2013, 13, 475–480 CrossRef CAS PubMed.
- E. J. Henderson, A. J. Shuhendler, P. Prasad, V. Baumann, F. Maier-Flaig, D. O. Faulkner, U. Lemmer, X. Y. Wu and G. A. Ozin, Small, 2011, 7, 2507–2516 CAS.
- F. Erogbogbo, K. T. Yong, I. Roy, G. X. Xu, P. N. Prasad and M. T. Swihart, ACS Nano, 2008, 2, 873–878 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- X. Cheng, S. B. Lowe, P. J. Reece and J. J. Gooding, Chem. Soc. Rev., 2014, 43, 2680–2700 RSC.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- L. M. Kustov, P. V. Mashkin, V. N. Zakharov, N. B. Abramenko, E. Y. Krysanov, L. A. Aslanov and W. Peijnenburg, Environ. Sci.: Nano, 2018, 5, 2945–2951 RSC.
- M. Dasog, J. Kehrle, B. Rieger and J. G. C. Veinot, Angew. Chem., Int. Ed., 2016, 55, 2322–2339 CrossRef CAS PubMed.
- P. Reiss, M. Carrière, C. Lincheneau, L. Vaure and S. Tamang, Chem. Rev., 2016, 116, 10731–10819 CrossRef CAS PubMed.
- A. S. Heintz, M. J. Fink and B. S. Mitchell, Adv. Mater., 2007, 19, 3984–3988 CrossRef CAS.
- K. Sato, H. Tsuji, K. Hirakuri, N. Fukata and Y. Yamauchi, Chem. Commun., 2009, 3759–3761 RSC.
- C. M. Hessel, D. Reid, M. G. Panthani, M. R. Rasch, B. W. Goodfellow, J. Wei, H. Fujii, V. Akhavan and B. A. Korgel, Chem. Mater., 2012, 24, 393–401 CrossRef CAS.
- R. J. Clark, M. Aghajamali, C. M. Gonzalez, L. Hadidi, M. A. Islam, M. Javadi, M. H. Mobarok, T. K. Purkait, C. J. T. Robidillo, R. Sinelnikov, A. N. Thiessen, J. Washington, H. Yu and J. G. C. Veinot, Chem. Mater., 2017, 29, 80–89 CrossRef CAS.
- X. Li, Y. He, S. S. Talukdar and M. T. Swihart, Langmuir, 2003, 19, 8490–8496 CrossRef CAS.
- G. Ledoux, J. Gong, F. Huisken, O. Guillois and C. Reynaud, Appl. Phys. Lett., 2002, 80, 4834–4836 CrossRef CAS.
- L. Mangolini, E. Thimsen and U. Kortshagen, Nano Lett., 2005, 5, 655–659 CrossRef CAS PubMed.
- F. Kunze, S. Kuns, M. Spree, T. Hülser, C. Schulz, H. Wiggers and S. M. Schnurre, Powder Technol., 2019, 342, 880–886 CrossRef CAS.
- J. D. Holmes, K. J. Ziegler, R. C. Doty, L. E. Pell, K. P. Johnston and B. A. Korgel, J. Am. Chem. Soc., 2001, 123, 3743–3748 CrossRef CAS.
- Y. Zhong, F. Peng, F. Bao, S. Wang, X. Ji, L. Yang, Y. Su, S. T. Lee and Y. He, J. Am. Chem. Soc., 2013, 135, 8350–8356 CrossRef CAS PubMed.
- S. P. Pujari, H. Driss, F. Bannani, B. Van Lagen and H. Zuilhof, Chem. Mater., 2018, 30, 6503–6512 CrossRef CAS PubMed.
- T. M. Atkins, A. Thibert, D. S. Larsen, S. Dey, N. D. Browning and S. M. Kauzlarich, J. Am. Chem. Soc., 2011, 133, 20664–20667 CrossRef CAS PubMed.
- B. Song and Y. He, Nano Today, 2019, 26, 149–163 CrossRef CAS.
- B. F. P. McVey, S. Prabakar, J. J. Gooding and R. D. Tilley, ChemPlusChem, 2017, 82, 60–73 CrossRef CAS PubMed.
- J. R. Heath, Science, 1992, 258, 1131–1133 CrossRef CAS PubMed.
- A. Kamyshny, V. Zakharov, M. Zakharov, A. Yatsenko, S. Savilov, L. Aslanov and S. Magdassi, J. Nanopart. Res., 2011, 13, 1971–1978 CrossRef CAS.
- L. A. Aslanov, V. N. Zakharov, A. V. Pavlikov, S. V. Savilov, V. Y. Timoshenko and A. V. Yatsenko, Russ. J. Coord. Chem., 2013, 39, 427–431 CrossRef CAS.
- S. Semlali, B. Cormary, M. L. De Marco, J. Majimel, A. Saquet, Y. Coppel, M. Gonidec, P. Rosa and G. L. Drisko, Nanoscale, 2019, 11, 4696–4700 RSC.
- R. K. Baldwin, K. A. Pettigrew, E. Ratai, M. P. Augustine and S. M. Kauzlarich, Chem. Commun., 2002, 17, 1822–1823 RSC.
- Q. Li, T. Y. Luo, M. Zhou, H. Abroshan, J. Huang, H. J. Kim, N. L. Rosi, Z. Shao and R. Jin, ACS Nano, 2016, 10, 8385–8393 CrossRef CAS PubMed.
- R. D. Tilley and K. Yamamoto, Adv. Mater., 2006, 18, 2053–2056 CrossRef CAS.
- R. D. Tilley, J. H. Warner, K. Yamamoto, I. Matsui and H. Fujimori, Chem. Commun., 2005, 1833–1835 RSC.
- J. Wang, S. Sun, F. Peng, L. Cao and L. Sun, Chem. Commun., 2011, 47, 4941–4943 RSC.
- X. Cheng, R. Gondosiswanto, S. Ciampi, P. J. Reece and J. J. Gooding, Chem. Commun., 2012, 48, 11874–11876 RSC.
- J. P. Wilcoxon, G. A. Samara and P. N. Provencio, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 2704–2714 CrossRef CAS.
- K. Linehan and H. Doyle, Small, 2014, 10, 584–590 CrossRef CAS PubMed.
- M. Rosso-Vasic, E. Spruijt, B. Van Lagen, L. De Cola and H. Zuilhof, Small, 2008, 4, 1835–1841 CrossRef CAS PubMed.
- N. Lin, Y. Han, L. Wang, J. Zhou, J. Zhou, Y. Zhu and Y. Qian, Angew. Chem., Int. Ed., 2015, 54, 3822–3825 CrossRef CAS PubMed.
- A. Shavel, L. Guerrini and R. A. Alvarez-Puebla, Nanoscale, 2017, 9, 8157–8163 RSC.
- K. A. Pettigrew, Q. Liu, P. P. Power and S. M. Kauzlarich, Chem. Mater., 2003, 15, 4005–4011 CrossRef CAS.
- L. Ruizendaal, S. P. Pujari, V. Gevaerts, J. M. J. Paulusse and H. Zuilhof, Chem.–Asian J., 2011, 6, 2776–2786 CrossRef CAS.
- Q. Liu and S. M. Kauzlarich, Mater. Sci. Eng., B, 2002, 96, 72–75 CrossRef.
- M. Imamura, J. Nakamura, S. Fujimasa, H. Yasuda, H. Kobayashi and Y. Negishi, Eur. Phys. J. D, 2011, 63, 289–292 CrossRef CAS.
- C. W. Hsu, D. Septiadi, C. H. Lai, P. Chen, P. H. Seeberger and L. De Cola, ChemPlusChem, 2017, 82, 660–667 CrossRef CAS.
- X. Zhang, D. Neiner, S. Wang, A. Y. Louie and S. M. Kauzlarich, Nanotechnology, 2007, 18, 095601 CrossRef.
- B. M. Nolan, T. Henneberger, M. Waibel, T. F. Fässler and S. M. Kauzlarich, Inorg. Chem., 2015, 54, 396–401 CrossRef CAS PubMed.
- D. Neiner, H. W. Chiu and S. M. Kauzlarich, J. Am. Chem. Soc., 2006, 128, 11016–11017 CrossRef CAS PubMed.
- B. Cho, S. Baek, H. G. Woo and H. Sohn, J. Nanosci. Nanotechnol., 2014, 14, 5868–5872 CrossRef CAS PubMed.
- D. Rodríguez Sartori, C. R. Lillo, J. J. Romero, M. Laura Dellarciprete, A. Miñán, M. Fernández Lorenzo De Mele and M. C. Gonzalez, Nanotechnology, 2016, 27, 475704 CrossRef PubMed.
- D. Mayeri, B. L. Phillips, M. P. Augustine and S. M. Kauzlarich, Chem. Mater., 2001, 13, 765–770 CrossRef CAS.
- R. A. Bley and S. M. Kauzlarich, J. Am. Chem. Soc., 1996, 118, 12461–12462 CrossRef CAS.
- C. S. Yang, R. A. Bley, S. M. Kauzlarich, H. W. H. Lee and G. R. Delgado, J. Am. Chem. Soc., 1999, 121, 5191–5195 CrossRef CAS.
- B. Ghosh and N. Shirahata, Sci. Technol. Adv. Mater., 2014, 15, 014207 CrossRef PubMed.
- Y. Yu, G. Fan, A. Fermi, R. Mazzaro, V. Morandi, P. Ceroni, D. M. Smilgies and B. A. Korgel, J. Phys. Chem. C, 2017, 121, 23240–23248 CrossRef CAS.
- K. Dohnalová, A. Fučíková, C. P. Umesh, J. Humpolíčková, J. M. J. Paulusse, J. Valenta, H. Zuilhof, M. Hof and T. Gregorkiewicz, Small, 2012, 8, 3185–3191 CrossRef PubMed.
- X. Wen, P. Zhang, T. A. Smith, R. J. Anthony, U. R. Kortshagen, P. Yu, Y. Feng, S. Shrestha, G. Coniber and S. Huang, Sci. Rep., 2015, 5, 12469 CrossRef PubMed.
- R. Sinelnikov, M. Dasog, J. Beamish, A. Meldrum and J. G. C. Veinot, ACS Photonics, 2017, 4, 1920–1929 CrossRef CAS.
- T. Takagahara and K. Takeda, Phys. Rev. B, 1992, 46, 15578–15581 CrossRef CAS PubMed.
- D. C. Hannah, J. Yang, P. Podsiadlo, M. K. Y. Chan, A. Demortière, D. J. Gosztola, V. B. Prakapenka, G. C. Schatz, U. Kortshagen and R. D. Schaller, Nano Lett., 2012, 12, 4200–4205 CrossRef CAS PubMed.
- K. Dohnalová, A. N. Poddubny, A. A. Prokofiev, W. D. De Boer, C. P. Umesh, J. M. Paulusse, H. Zuilhof and T. Gregorkiewicz, Light: Sci. Appl., 2013, 2, e47 CrossRef.
- M. Dasog, Z. Yang, S. Regli, T. M. Atkins, A. Faramus, M. P. Singh, E. Muthuswamy, S. M. Kauzlarich, R. D. Tilley and J. G. C. Veinot, ACS Nano, 2013, 7, 2676–2685 CrossRef CAS PubMed.
- A. Gupta and H. Wiggers, Nanotechnology, 2011, 22, 055707 CrossRef PubMed.
- J. Fuzell, A. Thibert, T. M. Atkins, M. Dasog, E. Busby, J. G. C. Veinot, S. M. Kauzlarich and D. S. Larsen, J. Phys. Chem. Lett., 2013, 4, 3806–3812 CrossRef CAS.
- B. V. Oliinyk, D. Korytko, V. Lysenko and S. Alekseev, Chem. Mater., 2019, 31, 7167 CrossRef CAS.
- A. N. Thiessen, M. Ha, R. W. Hooper, H. Yu, A. O. Oliynyk, J. G. C. Veinot and V. K. Michaelis, Chem. Mater., 2019, 31, 678–688 CrossRef CAS.
- J. B. Essner and G. A. Baker, Environ. Sci.: Nano, 2017, 4, 1216–1263 RSC.
- J. B. Essner, J. A. Kist, L. Polo-Parada and G. A. Baker, Chem. Mater., 2018, 30, 1878–1887 CrossRef CAS.
- N. C. Verma, A. Yadav and C. K. Nandi, Nat. Commun., 2019, 10, 2391 CrossRef PubMed.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Häusler, P. Kasák, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef CAS.
- V. Štengl and J. Henych, Nanoscale, 2013, 5, 3387–3394 RSC.
- L. Jiang and H. Zeng, Nanoscale, 2015, 7, 4580–4583 RSC.
- S. Khan, A. Sharma, S. Ghoshal, S. Jain, M. K. Hazra and C. K. Nandi, Chem. Sci., 2017, 9, 175–180 RSC.
- L. Ruizendaal, Functional Silicon Nanoparticles, Wageningen University, 2011 Search PubMed.
- W. Biesta, B. Van Lagen, V. S. Gevaert, A. T. M. Marcelis, J. M. J. Paulusse, M. W. F. Nielen and H. Zuilhof, Chem. Mater., 2012, 24, 4311–4318 CrossRef CAS.
- I. M. D. Höhlein, J. Kehrle, T. K. Purkait, J. G. C. Veinot and B. Rieger, Nanoscale, 2015, 7, 914–918 RSC.
- J. H. Warner, A. Hoshino, K. Yamamoto and R. D. Tilley, Angew. Chem., Int. Ed., 2005, 44, 4550–4554 CrossRef CAS PubMed.
- A. Shiohara, S. Hanada, S. Prabakar, K. Fujioka, T. H. Lim, K. Yamamoto, P. T. Northcote and R. D. Tilley, J. Am. Chem. Soc., 2010, 132, 248–253 CrossRef CAS PubMed.
- N. M. Park, C. J. Choi, T. Y. Seong and S. J. Park, Phys. Rev. Lett., 2001, 86, 1355–1357 CrossRef CAS PubMed.
- W. L. Wilson, P. F. Szajowski and L. E. Brus, Science, 1993, 262, 1242–1244 CrossRef CAS PubMed.
- M. Ouyang, C. Yuan, R. J. Muisener, A. Boulares and J. T. Koberstein, Chem. Mater., 2000, 12, 1591–1596 CrossRef CAS.
- R. Intartaglia, K. Bagga, F. Brandi, G. Das, A. Genovese, E. Di Fabrizio and A. Diaspro, J. Phys. Chem. C, 2011, 115, 5102–5107 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- Reaction conditions differed from the syntheses presented in this article and concern higher Mg2Si concentrations.
- A. Stock and C. Somieski, Ber. Dtsch. Chem. Ges., 1916, 49, 111–157 CrossRef CAS.
- H. J. Emeléus and A. G. Maddock, J. Chem. Soc., 1946, 1131–1134 RSC.
- Y. Manabe, Y. Kitawaki, M. Nagasaki, K. Fukase, H. Matsubara, Y. Hino, T. Fukuyama and I. Ryu, Chem.–Eur. J., 2014, 20, 12750–12753 CrossRef CAS PubMed.
- M. S. Kharasch, W. Hered and F. R. Mayo, J. Org. Chem., 1941, 6, 818–829 CrossRef CAS.
- G. Egloff, R. E. Schaad and C. D. Lowry, Chem. Rev., 1931, 8, 1–80 CrossRef CAS.
- X. Wang, R. F. Liu, J. Cork, Y. Gu, D. C. Upham, B. Laycock and E. W. McFarland, Ind. Eng. Chem. Res., 2017, 56, 9411–9418 CrossRef CAS.
- E. R. Boedeker and C. C. Lynch, J. Am. Chem. Soc., 1950, 72, 3234–3236 CrossRef CAS.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, J. Phys. Chem. C, 2007, 111, 6956–6961 CrossRef CAS.
- M. L. Mastronardi, F. Maier-Flaig, D. Faulkner, E. J. Henderson, C. Kübel, U. Lemmer and G. A. Ozin, Nano Lett., 2012, 12, 337–342 CrossRef CAS PubMed.
- C. J. Murphy and J. M. Buriak, Chem. Mater., 2015, 27, 4911–4913 CrossRef CAS.
- M. G. Panthani, C. M. Hessel, D. Reid, G. Casillas, M. José-Yacamán and B. A. Korgel, J. Phys. Chem. C, 2012, 116, 22463–22468 CrossRef CAS.
- Y. Yu, C. M. Hessel, T. D. Bogart, M. G. Panthani, M. R. Rasch and B. A. Korgel, Langmuir, 2013, 29, 1533–1540 CrossRef CAS PubMed.
- C. M. Hessel, J. Wei, D. Reid, H. Fujii, M. C. Downer and B. A. Korgel, J. Phys. Chem. Lett., 2012, 3, 1089–1093 CrossRef CAS PubMed.
- L. Mangolini and U. Kortshagen, Adv. Mater., 2007, 19, 2513–2519 CrossRef CAS.
- K. Kůsová, O. Cibulka, K. Dohnalová, I. Pelant, J. Valenta, A. Fǔcíková, K. Žídek, J. Lang, J. Englich, P. Matějka, P. Štěpánek and S. Bakardjieva, ACS Nano, 2010, 4, 4495–4504 CrossRef PubMed.
- M. Rosso-Vasic, E. Spruijt, Z. Popović, K. Overgaag, B. Van Lagen, B. Grandidier, D. Vanmaekelbergh, D. Domínguez-Gutiérrez, L. De Cola and H. Zuilhof, J. Mater. Chem., 2009, 19, 5926–5933 RSC.
- F. Hua, M. T. Swihart and E. Ruckenstein, Langmuir, 2005, 21, 6054–6062 CrossRef CAS PubMed.
- M. Jakob, A. Aissiou, W. Morrish, F. Marsiglio, M. Islam, A. Kartouzian and A. Meldrum, Nanoscale Res. Lett., 2018, 13, 338 CrossRef PubMed.
- J. B. Miller, A. R. Van Sickle, R. J. Anthony, D. M. Kroll, U. R. Kortshagen and E. K. Hobbie, ACS Nano, 2012, 6, 7389–7396 CrossRef CAS.
- L. Pitkänen and A. M. Striegel, TrAC, Trends Anal. Chem., 2016, 80, 311–320 CrossRef PubMed.
- Y. Shen, M. Y. Gee, R. Tan, P. J. Pellechia and A. B. Greytak, Chem. Mater., 2013, 25, 2838–2848 CrossRef CAS.
- M. Chen, W. Wang and X. Wu, J. Mater. Chem. B, 2014, 2, 3937–3945 RSC.
- D. E. Harwell, J. C. Croney, W. Qin, J. T. Thornton, J. H. Day, E. K. Hajime and D. M. Jameson, Chem. Lett., 2003, 32, 1194–1195 CrossRef CAS.
- J. Gu, X. Li, D. Hu, Y. Liu, G. Zhang, X. Jia, W. Huang and K. Xi, RSC Adv., 2018, 8, 12556–12561 RSC.
- H. L. Ye, S. J. Cai, S. Li, X. W. He, W. Y. Li, Y. H. Li and Y. K. Zhang, Anal. Chem., 2016, 88, 11631–11638 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fd00099b |
| This journal is © The Royal Society of Chemistry 2020 |