
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransport mechanisms of water molecules and ions in sub-nano channels of nanostructured water treatment liquid-crystalline membranes: a molecular dynamics simulation study†
Hiroki
Nada
 *a,
Takeshi
Sakamoto
b,
Masahiro
Henmi
c,
Takafumi
Ogawa
d,
Masahiro
Kimura
d and
Takashi
Kato
*b
*a,
Takeshi
Sakamoto
b,
Masahiro
Henmi
c,
Takafumi
Ogawa
d,
Masahiro
Kimura
d and
Takashi
Kato
*b
aNational Institute of Advanced Industrial Science and Technology (AIST), 16-1 Onogawa, Tsukuba, Ibaraki 305-8569, Japan. E-mail: hiroki.nada@aist.go.jp
bDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kato@chiral.t.u-tokyo.ac.jp
cTechnology Center, Toray Industries, Inc., 1-1, Nihonbashi-Muromachi 2-chome, Chuo-ku, Tokyo 103-8666, Japan
dGlobal Environment Research Laboratories, Toray Industries, Inc., Sonoyama, Otsu, Shiga 520-0842, Japan
First published on 12th December 2019
Abstract
Membranes with sub-nano channels formed by self-organization of ionic liquid-crystalline (LC) compounds have great potential as water treatment membranes. In this study, the transport mechanisms of water molecules and ions in the sub-nano channels of the LC membranes are investigated by molecular dynamics simulations for NaCl and NaNO3 solutions. The simulation results suggest that there are different transport mechanisms for water molecules and ions; the transport of water molecules occurs by Brownian diffusion, whereas that of ions occurs by jump diffusion between particular sites in the sub-nano channel. A free-energy landscape of an ion in the channel is analyzed using a metadynamics method, which indicates distinct local minima at particular sites and supports the jump diffusion mechanism. The effects of the LC compounds' structural flexibility and the electrostatic interaction with the wall of the sub-nano channels on the permeability of water molecules and ions are also investigated by molecular dynamics simulations. Simulation results suggest both the structural flexibility and the electrostatic interaction, which are characteristics of the LC membranes, are important factors in determining the water treatment performance. The structural flexibility affects the permeability of the membrane to water molecules and the electrostatic interaction reduces the permeability to ions.
Water impactRecently, we developed liquid crystalline (LC) membranes with sub-nano channels formed by self-organization of thermotropic ionic LC compounds. The LC membranes have great potential as excellent water treat membranes. In this work, we elucidated the transport mechanisms of water molecules and ions in the LC membranes by molecular dynamics simulations. We also elucidated the effects of the structural flexibility and the electrostatic interaction, which are characteristics of the LC membranes, on the water treatment performance. The knowledge obtained in this work has great potential to realize the development of future LC membranes of which the performances to water treatment are significantly improved. |
1. Introduction
Membrane technologies for water treatment are essential to supply high-quality water at low cost and with low energy consumption.1,2 In particular, membranes removing small ions, such as nanofiltration (NF) membranes and reverse osmosis (RO) membranes, are widely used in seawater and brackish water desalination.1–11Conventional membranes contain films of cross-linked polyamides or cellulose acetates as separation functional layers. However, these films contain a disordered structure and the nanopores or sub-nanopores formed in those membranes for the transportation of water molecules and ions are not uniform.3–12 Hence, materials that contain nanopores or sub-nanopores with uniform sizes, such as carbon nanotubes (CNTs)13–20 and polymerized liquid crystals,21–29 have attracted a great deal of interest as candidates for efficient water treatment membranes. It is expected that the alignment of pores, which is required for high water permeability, will be easier for polymerized liquid crystals than for CNTs.
Previously, we reported the development of liquid-crystalline (LC) membranes with sub-nano channels formed by self-organization of thermotropic ionic LC compounds.23,25 These membranes showed high performance for water treatment, including excellent water permeability, salt rejection, and ion selectivity. We also investigated the mobility of water molecules and ions in the channel of the LC membranes by molecular dynamics (MD) simulations for a NaCl solution.25 The simulation results qualitatively explained the experimental results for the permeability of water molecules and ions. Like our LC membranes, some LC membranes developed by other groups have also exhibited selectivity for ions and molecules.22,28,29
The performance of LC membranes can be improved further, for example, by changing the chemical structure of the LC compounds. To develop better LC membranes, it is important to resolve factors determining the water treatment performance. It is essential to understand the transport mechanisms for water molecules and ions in sub-nano channels of LC membranes. Because it is difficult to analyze the transport mechanisms experimentally, MD simulations must be used. Although previous MD simulations have suggested that the mobility of water molecules and ions in sub-nano channels depends on the channel size,25 more detailed studies are needed to elucidate the factors that determine the water treatment performance.
The structures of the ionic LC compounds that form the sub-nano channels are flexible and are involved in strong electrostatic interactions with water molecules. These features of LC membranes are not seen in hard materials that have been considered for use as water treatment materials, such as zeolite30–32 and CNTs.13–20 Elucidating how each of these features affects the permeability of water molecules and ions in sub-nano channels will contribute greatly to elucidating the factors that determine the water treatment performance.
In this study, the transport mechanisms of water molecules and ions in sub-nano channels of LC membranes were examined by using MD simulations of NaCl and NaNO3 solutions. We examined a NaNO3 solution because in addition to the practical importance of removing Na+ and Cl− ions from natural water in desalination, removing NO3− ions is also important for environmental and human health reasons.33–35 Furthermore, our understanding of the transport mechanisms can be improved by examining different solutions for which the water treatment performance has been investigated experimentally.23 In addition to the transport mechanisms, the effects of the structural flexibility of LC compounds and the electrostatic interactions on the mobility of water molecules and ions were also investigated by performing MD simulations for both solutions.
2. Simulation methods
Wedge-shaped LC compounds 1 and 2 were used in this study. The cationic moiety for compound 1 was triethyl ammonium, and that for compound 2 was trimethyl ammonium (Fig. 1a).25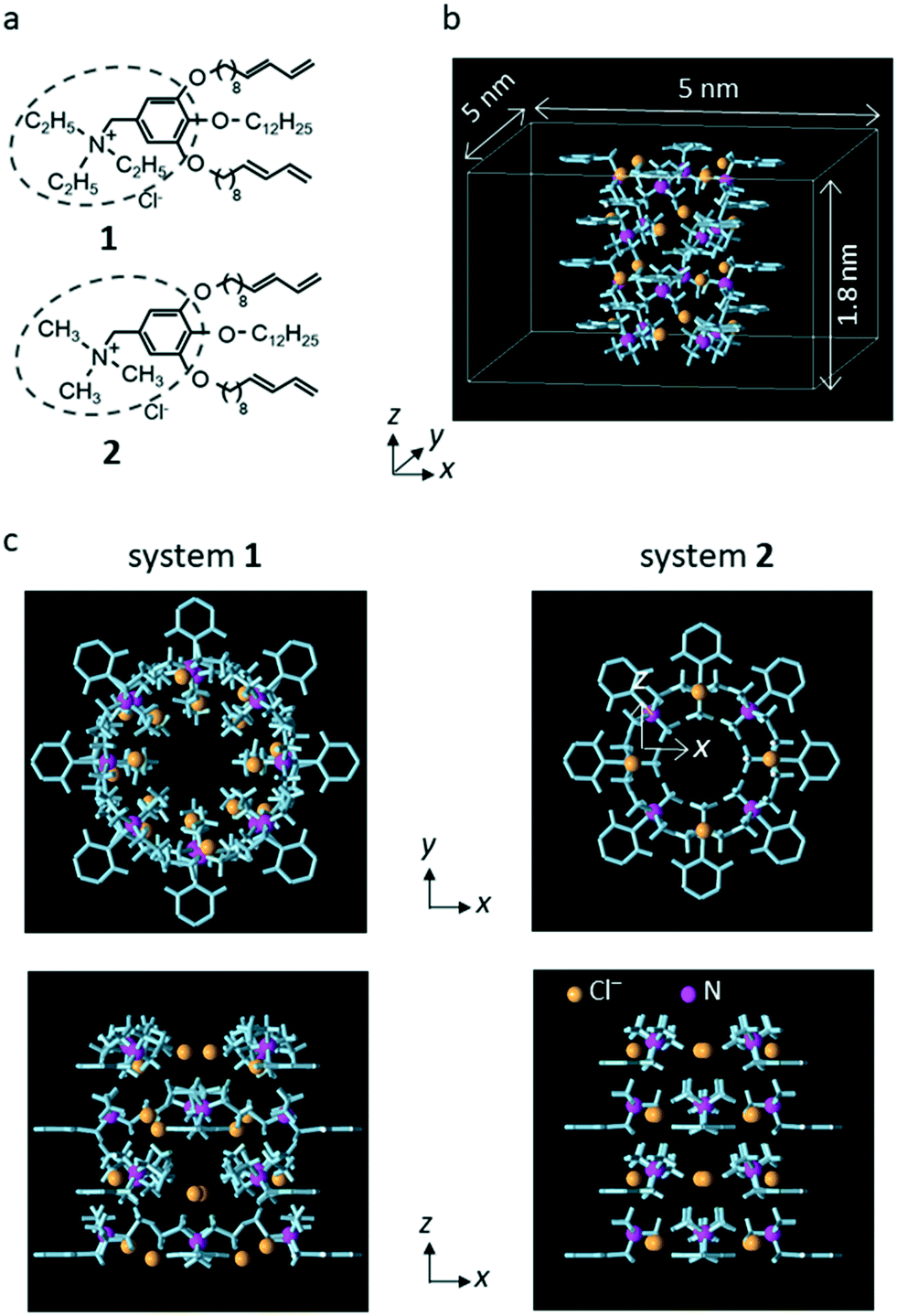 | ||
| Fig. 1 (a) Chemical structures of 1 and 2. (b) Unit system for system 2. Periodic boundary conditions were imposed in all x, y, and z directions of the unit system. Only the interatomic bonds (light blue columns) and the N atom (magenta spheres) of the LC monomers and the counter Cl− ions (chrome yellow spheres) are shown. The length of the simulation system in the z direction (3.6 nm) was twice that in unit system (1.8 nm). (c) The top view and side view of unit system for systems 1 and 2. The distance between the center of the system and the position of the N atom in the x–y plane was 0.67 nm for system 1 and 0.45 nm for system 2. (a) is reproduced from Fig. 1 of our previous paper.25 (b) is reproduced from Fig. S13 in the supporting information of our previous paper.25 (c) is reproduced from Fig. 5 of our previous paper.25 | ||
Following the previous MD simulation study,25 the simulation system including a sub-nano channel formed by the LC compounds was constructed using a simplified LC compound model from which the alkoxy chains of the nonionic moiety were eliminated (hereafter, the model is referred to as the LC monomer). For each LC compound, a rectangular parallelepiped system in which a sub-nano channel was created with 16 LC monomers and 16 counter Cl− ions (unit system, Fig. 1b) was prepared for a grand canonical Monte Carlo (GCMC) simulation to determine the number of water molecules in the channel. Then, using the final configuration generated by the GCMC simulation, the system for the MD simulations (simulation system) was constructed by putting a copy of the unit system filled with water molecules onto the original system in the z-axis direction for each LC compound. In the simulation systems, two water molecules in the channel were replaced with single Na+ and Cl− or NO3− ions. Hereafter, the simulation systems for 1 and 2 are referred to as system 1 and system 2, respectively.
The interactions for each LC compound were estimated using the potential model created in the previous MD simulation study.25 The interactions for water molecules were estimated using the TIP3P model,36 those for Na+ and Cl− ions were estimated using a model proposed by Joung and Cheatham,37 and those for NO3− ions were estimated using a model proposed by Banerjee et al.38
A standard MD simulation for the NVT ensemble at 298 K was performed for each simulation system. To investigate the effects of the LC monomer's structural flexibility and the electrostatic interaction with the LC monomers on the mobility of water molecules and ions in the channel, the MD simulation for each simulation system was performed under the following conditions: (1) the atoms constituting the LC monomers were free to move according to the force field, except for the benzene ring atoms, which were fixed at their equilibrium positions; (2) all atoms in the LC monomers were fixed at their equilibrium positions; and (3) all atoms in the LC monomers and the counter Cl− ions were fixed at their equilibrium positions, and the charges of the LC monomers' atoms and the counter Cl− ions were removed. Hereafter, conditions (1)–(3) are referred to as free, fixed, and chargeless conditions, respectively. Notably, the purpose of investigating the chargeless condition was qualitative understanding of the effect of the electrostatic interaction with the channel wall on the transport properties of water molecules and ions, but not reproducing the transport properties of water molecules and ions in the case that the charges were removed from real LC membranes. MD simulations of a NaCl solution for the free conditions were performed in our previous study.25 In the present study, MD simulations of a NaCl solution for the fixed and chargeless conditions, and MD simulations of a NaNO3 solution for all three conditions were performed. A free-energy landscape for the Na+ ion in the channel was also analyzed with a metadynamics (MTD) method39–41 to understand the transport mechanism of ions in the channel. Details of the simulation method are given in ESI.†
3. Results and discussion
3.1 Mobility of water molecules and ions in the NaNO3 solution in the channel under the free conditions
Fig. 2 shows the mean square displacements, 〈dr2〉, as a function of time, t, for the NaNO3 solution in systems 1 and 2 under the free conditions. Each 〈dr2〉 function was obtained by averaging 180 independent 〈dr2〉 functions, which were created using the simulation data for different periods in the three simulations. For both systems 1 and 2, the slope of the 〈dr2〉 function was much larger for water molecules than for the Na+ and NO3− ions. The slope of the 〈dr2〉 function for a species was proportional to the self-diffusion coefficient of the species. The larger the self-diffusion coefficient of a species, the higher is the permeability of the species. Therefore, the results of the 〈dr2〉 functions were qualitatively consistent with the experimental finding that the permeability is high for water molecules and low for NO3− ions.23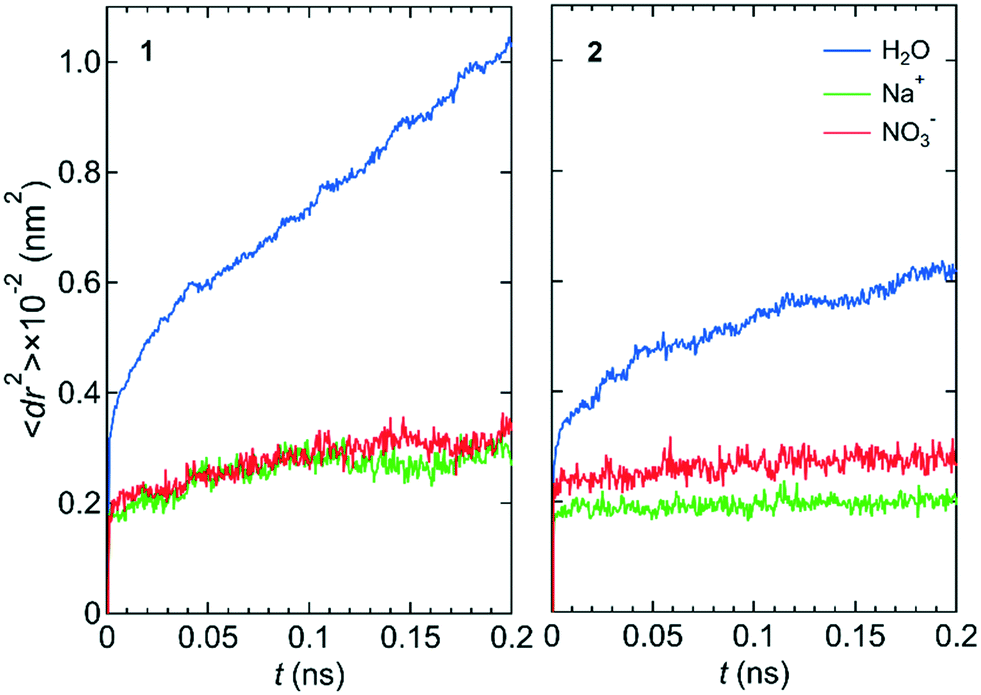 | ||
| Fig. 2 〈dr2〉 as a function of t for the center-of-mass of water molecules, the Na+ ion, and the center-of-mass of NO3− ions in NaNO3 solution for systems 1 and 2 under the free conditions. | ||
The lower permeability for the NO3− ion than for water molecules originated mainly from the stronger interaction with the wall of the channel for the NO3− ion than for water molecules. We confirmed it by comparing the potential energy, U, between the LC monomer and the NO3− ion (−48.77 kJ mol−1) and U between the LC monomer and a water molecule (the averaged value over all water molecules in the channel was −0.34 kJ mol−1). During the simulation, the NO3− ion was placed near the ammonium moiety of the LC monomer, which was positively charged. Therefore, the permeability of the NO3− ion might depend on the structure of the ammonium moiety.
Fig. 3 shows the number density profiles, ρ, for the O atom of the water molecule (Ow), the Na+ ion, the N atom in the NO3− ion (Nn), and the N atom in the LC monomer (N1) along the z-axis direction for system 1 under the free conditions. Each ρ was created using the data for all three simulations. For comparison, ρ for the Na+ and Cl− ions in the NaCl solution in the channel, which were analyzed using the previous MD simulation data,25 were also shown. ρ for both solutions indicate that water molecules were distributed over the channel. However, ions were localized at particular sites in the channel. This difference in ρ between water molecules and ions suggests that the transport mechanism is different between water molecules and ions; the transport of water molecules occurs by Brownian diffusion, whereas that of ions occurs by jump diffusion between particular sites.42 An example of the jump diffusion mechanism for Na+ observed during the simulation is shown in Fig. S3 (ESI†).
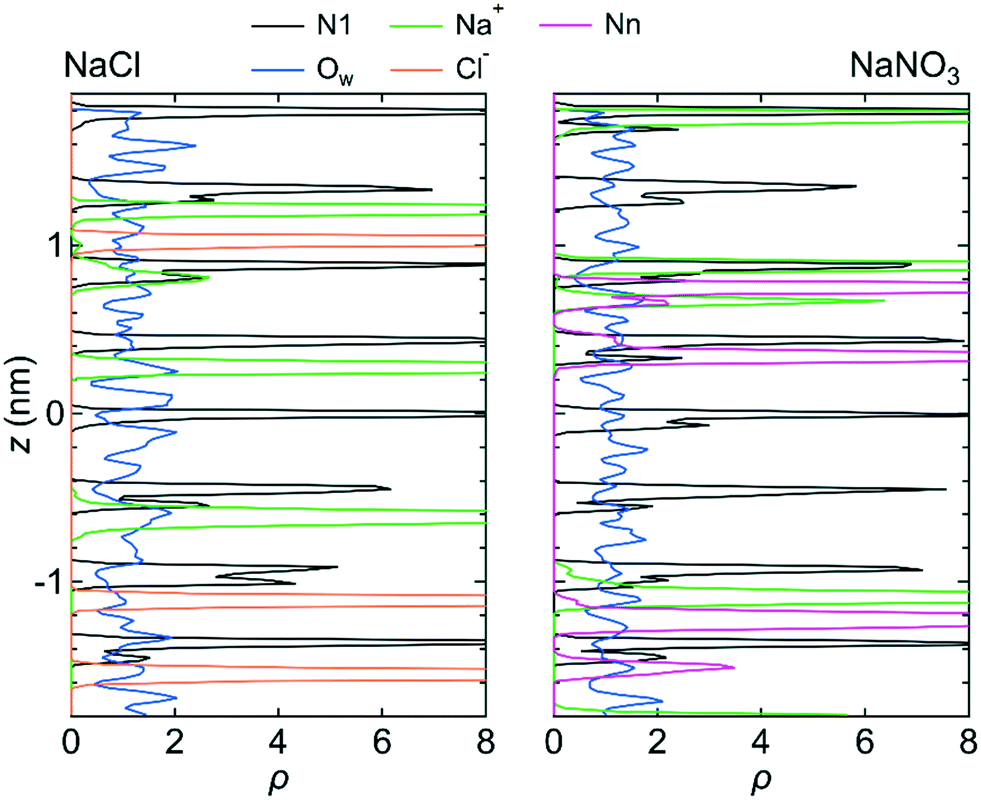 | ||
| Fig. 3 ρ of the N1 atom, Na+ ion, Ow atom, Cl− ion, and Nn atom along the z-axis direction for system 1. ρ was created so that ρ = 1 for all z components if the distribution of water molecules or ions was ideally random. The origin of the z-axis is the center of the system. | ||
In a bulk solution, each ion forms a hydration structure with the surrounding water molecules, and thus the Brownian diffusion of ions is accompanied by that of water molecules. In the channel, however, ions were stably located at particular positions, which were determined by the interaction with water molecules and the channel wall (i.e., with the LC monomers and the counter Cl− ions). Therefore, the ions in the channel did not undergo Brownian diffusion, even when the surrounding water molecules did. The positions at which ions were stably located in the channel were confirmed by creating a free-energy landscape of the Na+ ion using the MTD method (Fig. 4).
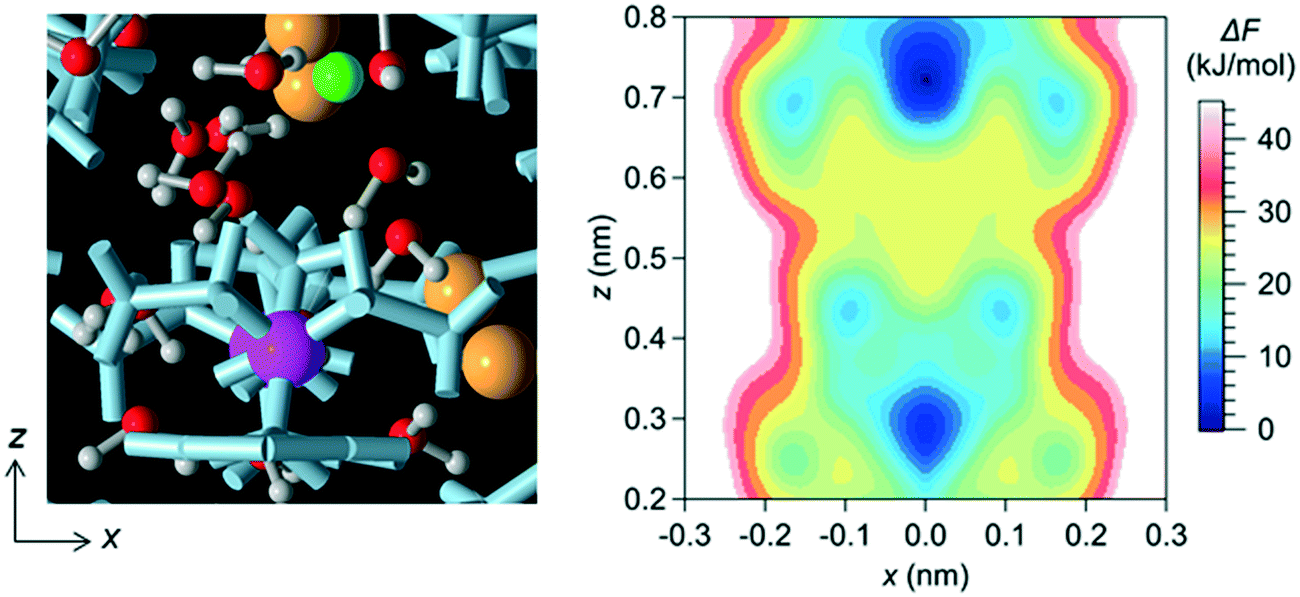 | ||
| Fig. 4 Free-energy landscape projected onto the x–z plane for the Na+ ion in the NaCl solution in the channel for system 1. The positions at which the Na+ ion could be stably located in the channel are the positions of the local minima, which appeared at (x, z) = (0.0, 0.28), (0.0, 0.72), (±0.09, 0.44), (±0.16, 0.25) and (±0.16, 0.69). ΔF is the free energy difference from the lowest value, which appeared at (x, z) = (0.0, 0.72). The origin of each axis is the center of the system. In the left-hand panel, the green sphere shows the Na+ ion, and the small red and white spheres show the O and H atoms of water molecules, respectively. The analysis of the free energy with the MTD method was done only for the region of x ≥ 0, and the free energy landscape was created by assuming that the landscapes for x ≥ 0 and x ≤ 0 were symmetric with respect to the z-axis. | ||
3.2 Effect of LC monomer's structural flexibility on the mobility of water molecules and ions in the channel
Because the mobilities of water molecules and ions were qualitatively the same for both systems 1 and 2, we hereafter discuss the relationship between each feature of the LC membranes and the mobility of water molecules and ions using the results for system 1 only. Fig. 5 shows the 〈dr2〉 as a function of t for both NaCl and NaNO3 solutions in system 1 for the fixed conditions. For comparison, the 〈dr2〉 functions for the free conditions are also shown. The mobility of water molecules was much lower for the fixed conditions than for the free conditions. The low mobility of water molecules for the fixed conditions was also seen in ρ of Ow along the z-axis direction (Fig. 6), which showed a periodic arrangement of distinct peaks, suggesting that the structure of water in the channel was solid-like.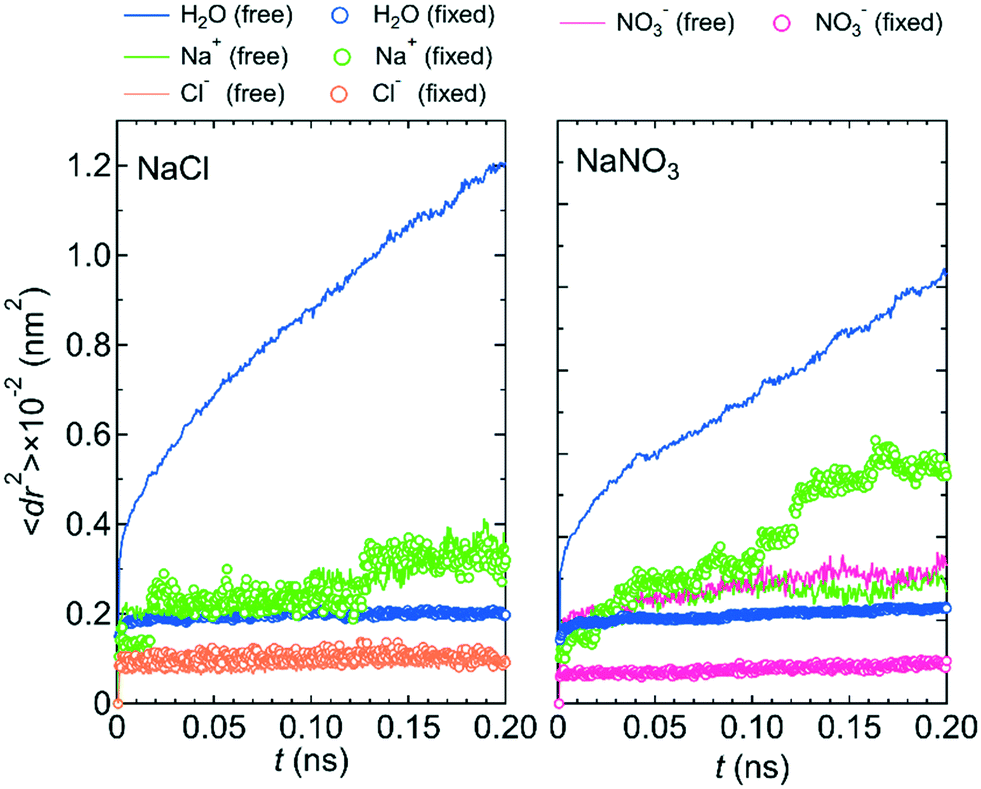 | ||
| Fig. 5 〈dr2〉 as a function of t for the NaCl and NaNO3 solutions in system 1 under the free (solid lines) and fixed (open circles) conditions. | ||
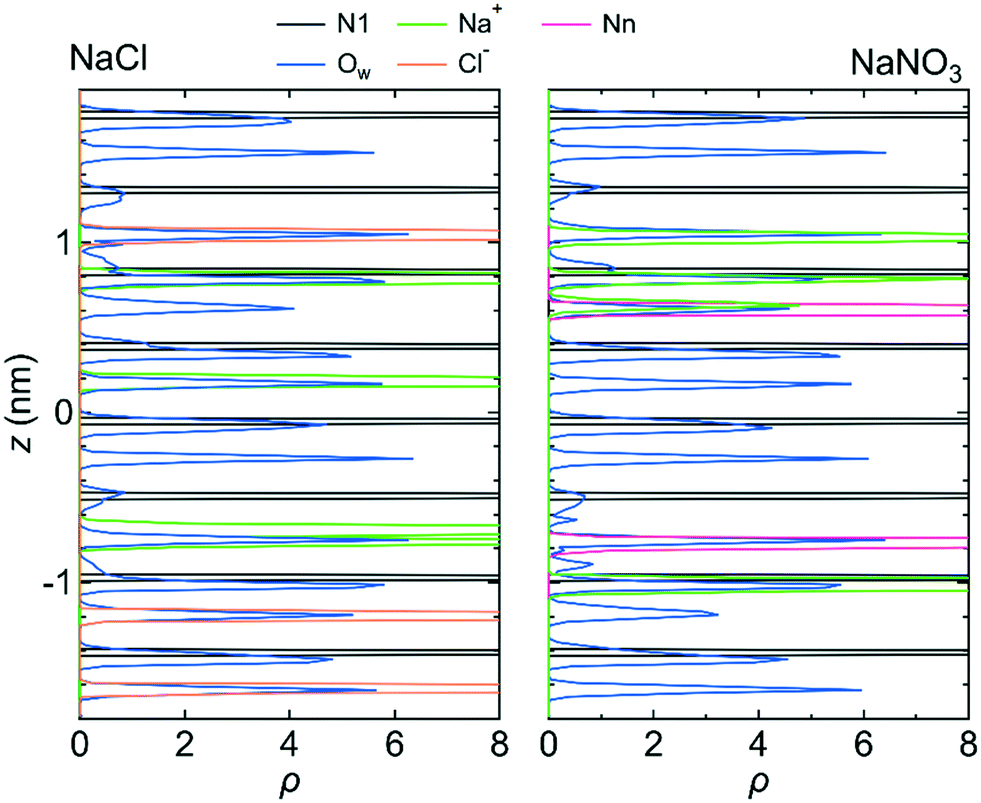 | ||
| Fig. 6 ρ of the N1 atom, Na+ ion, Ow atom, Cl− ion, and Nn atom along the z-axis direction for system 1 under the fixed conditions. The origin of the z-axis is the center of the system. | ||
However, the mobilities of ions for the NaCl solution for the free and fixed conditions were similar. For the NaNO3 solution, the mobility of the NO3− ion was slightly lower for the fixed conditions than for the free conditions, whereas the mobility of the Na+ ion was apparently higher for the fixed conditions than for the free conditions. For both solutions, the jump diffusion of ions occurred. The occurrence of the jump diffusion irrespective of the mobility of the surrounding water molecules suggests that the sites for the stable localization of ions in the channel were mainly determined by the interaction of ions with the channel wall. The substantial lowering of the mobility of water molecules just by fixing the structure of the LC monomers suggests that the LC monomer's structural flexibility is an important factor in increasing the permeability of water molecules in the channel. Future MD simulations for a sub-nano channel created with a realistic model of the LC compound without fixing the position of the benzene ring will provide detailed quantitative information on the relationship between the LC compound's structural flexibility and the water molecule permeability. Strictly, since the mobility of the Na+ ion for the NaNO3 solution increased by fixing the structure of the LC monomers, the LC monomer's structural flexibility would also influence somehow the permeability of ions. Performing a much larger number of the simulations with different initial positions of ions in the channel than in this study is needed to evaluate the degree of the influence.
3.3 Effect of the electrostatic interaction with the channel wall on the mobilities of water molecules and ions in the channel
Fig. 7 shows 〈dr2〉 as a function of t for both NaCl and NaNO3 solutions in system 1 under the chargeless conditions. For both solutions, the mobility of water molecules for the chargeless conditions was low. The mobility of the Na+ ion was also low. No jump diffusion of the Na+ ion was observed in the simulation. This result supports the hypothesis that the interaction with the channel wall mainly determines the mobility of the Na+ ion in the channel. The low mobility of water molecules and ions for the chargeless conditions can also be seen in ρ of the Ow atom, Na+ ion, Nn atom, and Cl− ion along the z-axis direction (Fig. 8), which all show a periodic arrangement of distinct peaks. These results for the chargeless conditions strongly suggest that the electrostatic interaction with the channel wall is an important factor in determining the mobility of water molecules and ions in the channel, in addition to the channel size and the flexibility of the LC monomer's structure.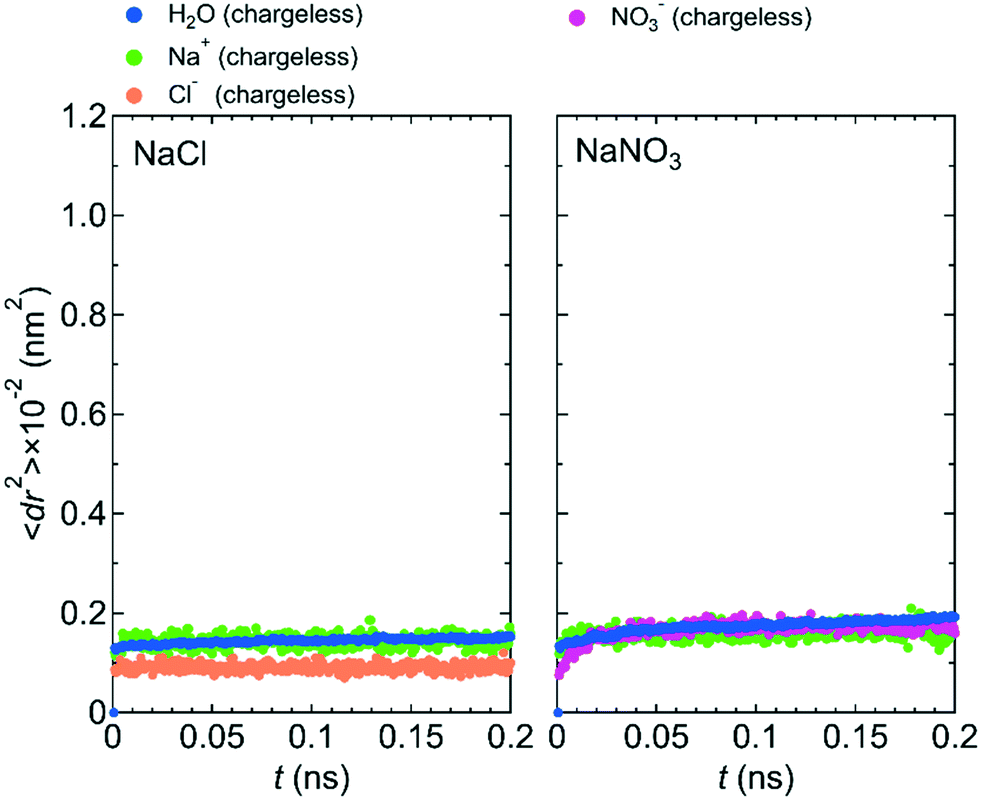 | ||
| Fig. 7 〈dr2〉 as a function of t for NaCl and NaNO3 solutions in system 1 for the chargeless condition. | ||
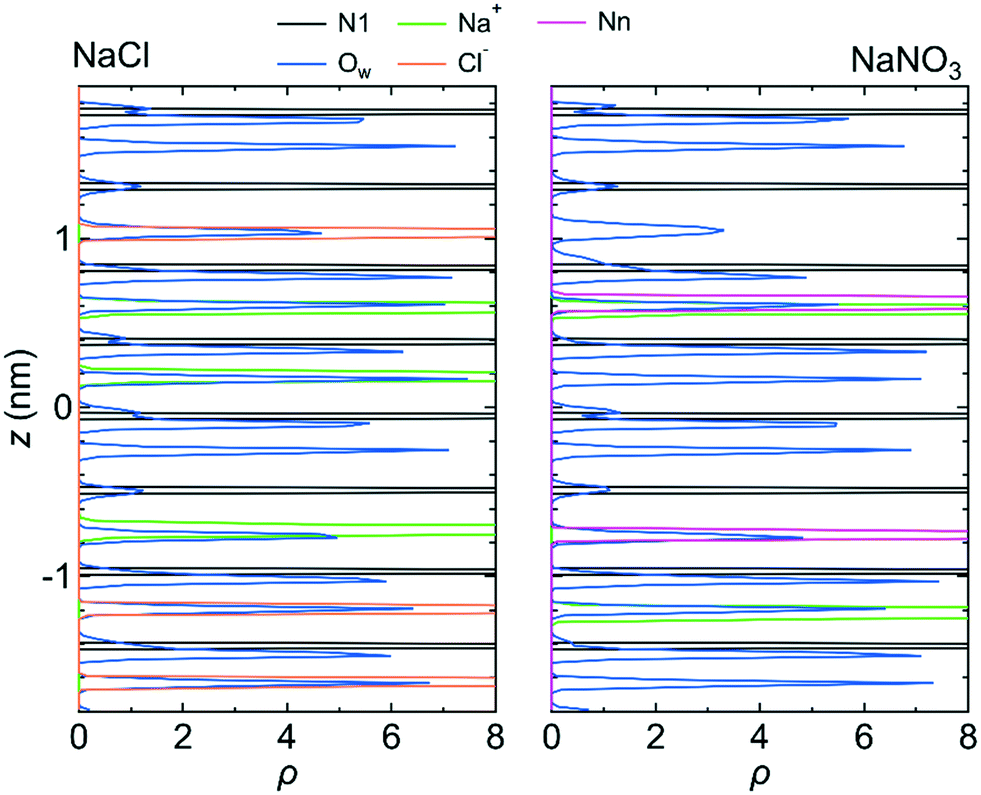 | ||
| Fig. 8 ρ of the N1 atom, Na+ ion, Ow atom, Cl− ion, and Nn atom along the z-axis direction for system 1 for the chargeless conditions. The origin of the z-axis is the center of the system. | ||
The low mobility of water molecules and ions in the channel under the chargeless conditions does not mean that the permeability of water molecules and ions is low. Because the interaction between the channel wall and each of the water molecules and ions under the chargeless conditions is weak, the interface between the channel wall and the solution is slippery as compared with the case that the electrostatic interaction acts on the water molecules and ions. Therefore, the transport of water molecules and ions along the channel should occur readily under external pressure. We confirmed it by performing a nonequilibrium MD simulation for the NaCl solution of system 1 under the fixed and chargeless conditions (Fig. S4†). The electrostatic interaction with the channel wall, which is required for the stable localization of ions at particular sites in the channel, is an important factor in reducing the permeability of ions. Analogously, the importance of the electrostatic interaction with the wall of CNTs and boron nitride nanotubes for the dynamics of water molecules in those nanotubes was discussed by Wei and Luo.20 The structure of the channel wall, which was geometrically not flat, might also influence the permeability of water molecules. Detailed studies about the effect of the channel wall structure on the permeability of water molecules and ions should be done in the future.
3.4 Water structure in the channel for the free, fixed, and chargeless conditions
The structure of the water in the channel was different under the free, fixed, and chargeless conditions. Fig. 9 shows the Ow–Ow pair distribution function, gOwOw, for the three conditions. Peaks appearing in gOwOw were more distinct for the chargeless conditions than for the free and fixed conditions, indicating that the structure of water was more highly ordered for the chargeless conditions than for the free and fixed conditions. The much lower U between water molecules, Uww, for the chargeless conditions than for the free and fixed conditions (Table 1) supports the formation of a highly ordered water structure under the chargeless conditions. The typical structure of water in the channel for each set of conditions is shown in Fig. 10.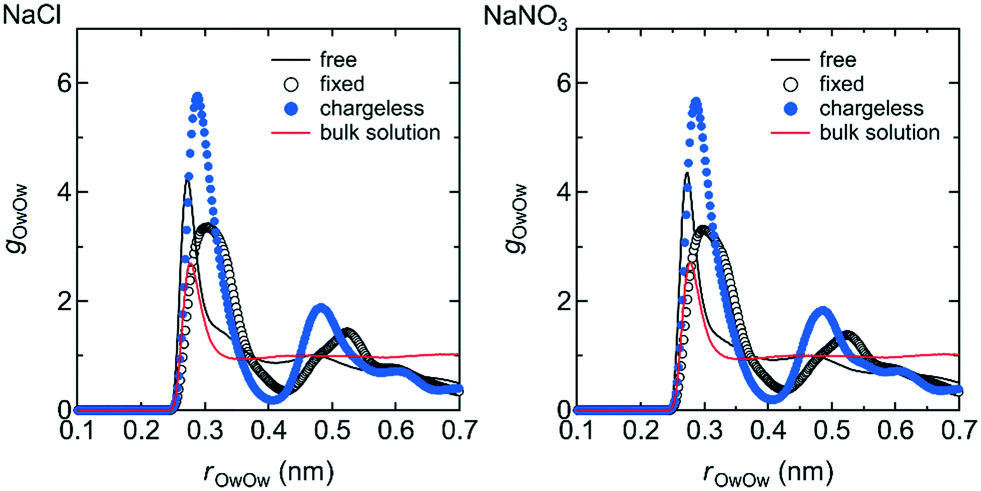 | ||
| Fig. 9 g OwOw in the channel for the free, fixed, and chargeless conditions as functions of the Ow–Ow distance, rOwOw. For reference, gOwOw for the bulk NaCl and NaNO3 solutions obtained by separate MD simulations are also shown. Details of the MD simulations for the bulk solutions are given in ESI† (S1-2). | ||
| Salt | Condition | U ww (kJ mol−1) |
|---|---|---|
| NaCl | Free | −16.78 |
| Fixed | −8.09 | |
| Chargeless | −23.23 | |
| NaNO3 | Free | −17.05 |
| Fixed | −9.08 | |
| Chargeless | −24.45 |
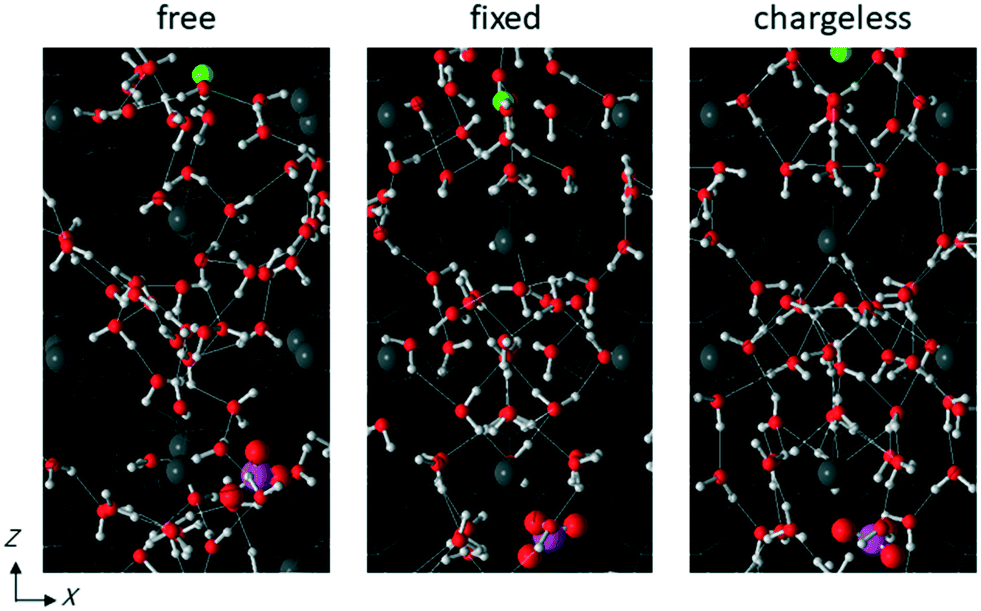 | ||
| Fig. 10 Typical structures of water in the channel in system 1 with NaNO3 solution for free, fixed, and chargeless conditions. Only water molecules and ions are shown in color. The green spheres are the Na+ ions. The large magenta and red spheres are the N and O atoms of the NO3− ion, respectively. The small red and white spheres show the O and H atoms of water molecules, respectively. The thin white lines connecting water molecules represent hydrogen bonds. The structure of water is more highly ordered for the fixed and chargeless conditions than for the free conditions. | ||
The formation of a highly ordered water structure in the channel for the chargeless conditions is analogous to the formation of an ice-like water structure in a restricted sub-nano space confined by hydrophobic walls, such as in a CNT.43,44Uww was much higher for the fixed conditions than for the free conditions (Table 1) because the arrangement of water molecules in the channel under the fixed conditions was dominated by the interaction with the channel wall. rOwOw at which the first peak of gOwOw appeared was longer for the fixed conditions than for the free and chargeless conditions for the same reason. Water molecules in the channels of polymer membranes are present as free and bound water.4 Our results suggest that the structural flexibility of the LC monomers is a factor in increasing the ratio of free water in the channels.
4. Conclusions
Following our previous MD simulation study of a NaCl solution in the channels of LC membranes,25 MD simulations were performed for a NaNO3 solution in the channel of membranes consisting of LC compounds 1 and 2. Similar to the previous MD simulation results for the NaCl solution,25 the present simulation results for the NaNO3 solution were qualitatively consistent with the experimentally measured permeability of water molecules and ions for LC membranes.23The transport mechanisms of water molecules and ions in the channel were analyzed for the NaCl and NaNO3 solutions. The results suggested that for both solutions, the water molecule and ion transport mechanisms were different. The transport of water molecules occurred by Brownian diffusion, whereas that of ions occurred by jump diffusion among sites where the ions were stably localized. Jump diffusion of ions occurred because the ions could only be stably located at particular positions in the channel, which were mainly determined by the interactions with the channel wall. The existence of particular positions at which ions were stably located in the channel was confirmed by analyzing the free-energy landscape for the Na+ ion in the channel using the MTD method.
The effects of the LC monomer's structural flexibility and the electrostatic interaction with the channel wall on the mobility of water molecules and ions were also investigated by performing MD simulations for fixed and chargeless conditions. The channel size, structural flexibility, and electrostatic interactions were important factors in determining the permeability of water molecules and ions in the channel. The simulations suggested that the structural flexibility increases the permeability of water molecules, and the electrostatic interactions decrease the permeability of ions.
The transport mechanisms of water molecules and ions, and the effects of the LC monomer's structural flexibility and the electrostatic interactions on the permeability of water molecules and ions in the channel clarified in this study will contribute to the development of superior water treatment LC membranes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST, CREST, JPMJCR1422 and JSPS KAKENHI Grant Number 19H05715. Some of the computations in this work were done using the facilities of the Super Computer Center, Institute of Solid State Physics, The University of Tokyo. H. N. is very grateful to Miharu Shimizu, Yukiyo Sakamoto, Atsuko Suzuki, Akiko Matsumoto, Miwako Takano, and Hiroko Kanno for their helpful assistance with computation, data analysis, creating figures, and creating tables in this paper.References
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Science and technology for water purification in the coming decades, Nature, 2008, 452, 301–310 CrossRef CAS.
- M. Henmi, Y. Fusaoka, H. Tomioka and M. Kurihara, High performance ro membranes for desalination and wastewater reclamation and their operation results, Water Sci. Technol., 2010, 62, 2134–2140 CrossRef CAS PubMed.
- T. Humplik, J. Lee, S. C. O'Hern, B. A. Fellman, M. A. Baig, S. F. Hassan, M. A. Atieh, F. Rahman, T. Laoui, R. Karnik and E. N. Wang, Nanostructured materials for water desalination, Nanotechnology, 2011, 22, 19 CrossRef PubMed.
- T. Kawakami, M. Nakada, H. Shimura, K. Okada and M. Kimura, Hydration structure of reverse osmosis membranes studied via neutron scattering and atomistic molecular simulation, Polym. J., 2018, 50, 327–336 CrossRef CAS.
- T. Wei, L. Zhang, H. Y. Zhao, H. Ma, M. S. J. Sajib, H. Jiang and S. Murad, Aromatic polyamide reverse-osmosis membrane: An atomistic molecular dynamics simulation, J. Phys. Chem. B, 2016, 120, 10311–10318 CrossRef CAS PubMed.
- W. M. Gao, F. H. She, J. Zhang, L. F. Dumee, L. He, P. D. Hodgson and L. X. Kong, Understanding water and ion transport behaviour and permeability through poly(amide) thin film composite membrane, J. Membr. Sci., 2015, 487, 32–39 CrossRef CAS.
- M. X. Ding, A. Szymczyk and A. Ghoufi, On the structure and rejection of ions by a polyamide membrane in pressure-driven molecular dynamics simulations, Desalination, 2015, 368, 76–80 CrossRef CAS.
- A. Ghoufi, E. Drazevic and A. Szymczyk, Interactions of organics within hydrated selective layer of reverse osmosis desalination membrane: A combined experimental and computational study, Environ. Sci. Technol., 2017, 51, 2714–2719 CrossRef CAS PubMed.
- Y. L. Liu, K. Xiao, A. Q. Zhang, X. M. Wang, H. W. Yang, X. Huang and Y. F. F. Xie, Exploring the interactions of organic micropollutants with polyamide nanofiltration membranes: A molecular docking study, J. Membr. Sci., 2019, 577, 285–293 CrossRef CAS.
- Y. J. Yao, M. Li, X. Z. Cao, P. Zhang, W. Zhang, J. F. Zheng, X. Zhang and L. J. Wang, A novel sulfonated reverse osmosis membrane for seawater desalination: Experimental and molecular dynamics studies, J. Membr. Sci., 2018, 550, 470–479 CrossRef CAS.
- M. Shen, S. Keten and R. M. Lueptow, Dynamics of water and solute transport in polymeric reverse osmosis membranes via molecular dynamics simulations, J. Membr. Sci., 2016, 506, 95–108 CrossRef CAS.
- J. Liu, X. Kong and J. W. Jiang, Solvent nanofiltration through polybenzimidazole membranes: Unravelling the role of pore size from molecular simulations, J. Membr. Sci., 2018, 564, 782–787 CrossRef CAS.
- J. K. Holt, H. G. Park, Y. M. Wang, M. Stadermann, A. B. Artyukhin, C. P. Grigoropoulos, A. Noy and O. Bakajin, Fast mass transport through sub-2-nanometer carbon nanotubes, Science, 2006, 312, 1034–1037 CrossRef CAS PubMed.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Water conduction through the hydrophobic channel of a carbon nanotube, Nature, 2001, 414, 188–190 CrossRef CAS.
- M. Thomas, B. Corry and T. A. Hilder, What have we learnt about the mechanisms of rapid water transport, ion rejection and selectivity in nanopores from molecular simulation?, Small, 2014, 10, 1453–1465 CrossRef CAS PubMed.
- Y. Hong, J. C. Zhang, C. Q. Zhu, X. C. Zeng and J. S. Francisco, Water desalination through rim functionalized carbon nanotubes, J. Mater. Chem. A, 2019, 7, 3583–3591 RSC.
- L. Y. Wang, R. S. Dumont and J. M. Dickson, Nonequilibrium molecular dynamics simulation of water transport through carbon nanotube membranes at low pressure, J. Chem. Phys., 2012, 137, 14 Search PubMed.
- T. B. Sisan and S. Lichter, Solitons transport water through narrow carbon nanotubes, Phys. Rev. Lett., 2014, 112, 5 CrossRef PubMed.
- J. Y. Su and D. C. Huang, Coupling transport of water and ions through a carbon nanotube: The role of ionic condition, J. Phys. Chem. C, 2016, 120, 11245–11252 CrossRef CAS.
- X. F. Wei and T. F. Luo, Effects of electrostatic interaction and chirality on the friction coefficient of water flow inside single-walled carbon nanotubes and boron nitride nanotubes, J. Phys. Chem. C, 2018, 122, 5131–5140 CrossRef CAS.
- D. L. Gin and R. D. Noble, Designing the next generation of chemical separation membranes, Science, 2011, 332, 674–676 CrossRef CAS PubMed.
- B. M. Carter, B. R. Wiesenauer, E. S. Hatakeyama, J. L. Barton, R. D. Noble and D. L. Gin, Glycerol-based bicontinuous cubic lyotropic liquid crystal monomer system for the fabrication of thin-film membranes with uniform nanopores, Chem. Mater., 2012, 24, 4005–4007 CrossRef CAS.
- M. Henmi, K. Nakatsuji, T. Ichikawa, H. Tomioka, T. Sakamoto, M. Yoshio and T. Kato, Self-organized liquid-crystalline nanostructured membranes for water treatment: Selective permeation of ions, Adv. Mater., 2012, 24, 2238–2241 CrossRef CAS PubMed.
- N. Marets, D. Kuo, J. R. Torrey, T. Sakamoto, M. Henmi, H. Katayama and T. Kato, Highly efficient virus rejection with self-organized membranes based on a crosslinked bicontinuous cubic liquid crystal, Adv. Healthcare Mater., 2017, 6, 6 Search PubMed.
- T. Sakamoto, T. Ogawa, H. Nada, K. Nakatsuji, M. Mitani, B. Soberats, K. Kawata, M. Yoshio, H. Tomioka, T. Sasaki, M. Kimura, M. Henmi and T. Kato, Development of nanostructured water treatment membranes based on thermotropic liquid crystals: Molecular design of sub-nanoporous materials, Adv. Sci., 2018, 5, 9 Search PubMed.
- K. Hamaguchi, D. Kuo, M. Liu, T. Sakamoto, M. Yoshio, H. Katayama and T. Kato, Nanostructured virus filtration membranes based on two-component columnar liquid crystals, ACS Macro Lett., 2019, 8, 24–30 CrossRef CAS.
- M. Gupta, Y. Suzuki, T. Sakamoto, M. Yoshio, S. Torii, H. Katayama and T. Kato, Polymerizable photocleavable columnar liquid crystals for nanoporous water treatment membranes, ACS Macro Lett., 2019, 8, 1303–1308 CrossRef CAS.
- M. J. Zhou, T. J. Kidd, R. D. Noble and D. L. Gin, Supported lyotropic liquid-crystal polymer membranes: Promising materials for molecular-size-selective aqueous nanofiltration, Adv. Mater., 2005, 17, 1850–1853 CrossRef CAS.
- M. J. Zhou, P. R. Nemade, X. Y. Lu, X. H. Zeng, E. S. Hatakeyama, R. D. Noble and D. L. Gin, New type of membrane material for water desalination based on a cross-linked bicontinuous cubic lyotropic liquid crystal assembly, J. Am. Chem. Soc., 2007, 129, 9574–9575 CrossRef CAS PubMed.
- Y. L. Liu and X. Chen, High permeability and salt rejection reverse osmosis by a zeolite nano-membrane, Phys. Chem. Chem. Phys., 2013, 15, 6817–6824 RSC.
- M. Fasano, D. Borri, E. Chiavazzo and P. Asinari, Protocols for atomistic modeling of water uptake into zeolite crystals for thermal storage and other applications, Appl. Therm. Eng., 2016, 101, 762–769 CrossRef CAS.
- S. H. Jamali, T. J. H. Vlugt and L. C. Lin, Atomistic understanding of zeolite nanosheets for water desalination, J. Phys. Chem. C, 2017, 121, 11273–11280 CrossRef CAS.
- S. Lee and R. M. Lueptow, Membrane rejection of nitrogen compounds, Environ. Sci. Technol., 2001, 35, 3008–3018 CrossRef CAS.
- A. Santafe-Moros, J. M. Gozalvez-Zafrilla and J. Lora-Garcia, Nitrate removal from ternary ionic solutions by a tight nanofiltration membrane, Desalination, 2007, 204, 63–71 CrossRef CAS.
- R. Epsztein, O. Nir, O. Lahav and M. Green, Selective nitrate removal from groundwater using a hybrid nanofiltration-reverse osmosis filtration scheme, Chem. Eng. J., 2015, 279, 372–378 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- I. S. Joung and T. E. Cheatham, Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef CAS PubMed.
- P. Banerjee, S. Yashonath and B. Bagchi, Coupled jump rotational dynamics in aqueous nitrate solutions, J. Chem. Phys., 2016, 145, 10 Search PubMed.
- A. Laio and F. L. Gervasio, Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science, Rep. Prog. Phys., 2008, 71, 22 CrossRef.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Plumed: A portable plugin for free-energy calculations with molecular dynamics, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- H. Nada, M. Kobayashi and M. Kakihana, Anisotropy in stable conformations of hydroxylate ions between the {001} and {110} planes of TiO2 rutile crystals for glycolate, lactate, and 2-hydroxybutyrate ions studied by metadynamics method, ACS Omega, 2019, 4, 11014–11024 CrossRef CAS PubMed.
- P. L. Hall and D. K. Ross, Incoherent neutron-scattering functions for random jump diffusion in bounded and infinite media, Mol. Phys., 1981, 42, 673–682 CrossRef CAS.
- Y. Maniwa, H. Kataura, M. Abe, A. Udaka, S. Suzuki, Y. Achiba, H. Kira, K. Matsuda, H. Kadowaki and Y. Okabe, Ordered water inside carbon nanotubes: Formation of pentagonal to octagonal ice-nanotubes, Chem. Phys. Lett., 2005, 401, 534–538 CrossRef CAS.
- K. Mochizuki and K. Koga, Solid-liquid critical behavior of water in nanopores, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8221–8226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the simulation method, MD simulations of the bulk solutions, an example of the jump diffusion mechanism for Na+ observed during the simulation, and the mobility of water molecules in a nonequilibrium MD simulation. See DOI: 10.1039/c9ew00842j |
| This journal is © The Royal Society of Chemistry 2020 |