
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of NOM on copper sulfide nanoparticle growth, stability, and oxidative dissolution†
Kevin
Hoffmann
 a,
Sylvain
Bouchet
a,
Iso
Christl
a,
Ralf
Kaegi
b and
Ruben
Kretzschmar
*a
a,
Sylvain
Bouchet
a,
Iso
Christl
a,
Ralf
Kaegi
b and
Ruben
Kretzschmar
*a
aInstitute of Biogeochemistry and Pollutant Dynamics, CHN, ETH Zürich, 8092 Zürich, Switzerland. E-mail: ruben.kretzschmar@env.ethz.ch
bEawag, Swiss Federal Institute of Aquatic Science and Technology, 8060 Dübendorf, Switzerland
First published on 25th March 2020
Abstract
Metal sulfide nanoparticles have recently been discovered in natural environments subject to fluctuating redox conditions such as wetlands and river floodplains, which are often very rich in natural organic matter (NOM). Strong binding of chalcophile metals like copper by NOM is expected to interfere with sulfide precipitation and is likely to affect the growth of these nanoparticles. We therefore conducted experiments on the formation of copper sulfide nanoparticles (CuxS NPs) in anoxic solutions with varying Cu (50, 500 μmol L−1) and sulfide (100, 1000 μmol L−1) concentrations in the absence and presence of Suwannee River fulvic acid (SRFA, 0, 5, 50 mg C L−1). The size development of the CuxS nanoparticles and their stability were tracked over 4 weeks using transmission electron microscopy (TEM) and size-exclusion chromatography coupled to ICP-MS (SEC-ICP-MS). Additional dissolution experiments were performed in closed containers in the absence and presence of O2 and fulvic acid over several months providing insights into the oxidative dissolution behavior of CuxS. Our results highlight the high colloidal stability of CuxS nanoparticles in anoxic environments irrespective of the NOM concentration. Median particle diameters ranged between a few and a few tens of nanometers with larger particles forming in more concentrated suspensions. At low Cu and S concentrations, fulvic acid restricted particle growth by up to 25% compared to SRFA-free suspensions and metal sulfide clusters even smaller than 1 nm were detected. The CuxS nanoparticles exhibited a remarkable chemical stability against oxidative dissolution and were only dissolved when both fulvic acid and O2 were present.
Environmental significanceRedox-dynamic environments such as wetlands and river floodplains, known as typical hotspots for natural organic matter (NOM) production, have recently been recognized as sources of natural metal sulfide nanoparticles. These sulfide particles are expected to have a profound impact on trace metal cycling in these environments. We show that NOM hampers the growth of CuxS nanoparticles and slows down their morphological development. Further, we demonstrate the importance of NOM in facilitating the oxidative dissolution of CuxS nanoparticles. Our findings suggest an increased mobility of the CuxS nanoparticles in NOM-rich environments, but also indicate enhanced oxidation of CuxS and release of Cu2+ ions in the presence of NOM upon aeration of anoxic waters. |
1. Introduction
The release of trace metals from enriched parent rock material by weathering as well as from mining, ore processing, industrial activities and agri- or vinicultural practices can lead to trace metal accumulation in natural environments. Many floodplain and wetland areas around the world are contaminated with metals due to upland mine waste discharge or tailings dam failures.1–7 Among those metals, copper (Cu) is an essential nutrient for all living organisms since it is required for the synthesis of numerous enzymes and proteins involved in redox-related biological functions. But its uptake is merely required in trace amounts.8 Exposure to elevated concentrations of Cu is highly toxic to a wide range of organisms with fish, insects, algae, and microorganisms being among the most susceptible.8,9 Therefore, excessive releases of Cu into natural environments pose a threat to ecosystem health, particularly when high biological diversity areas such as wetlands and marshes are affected. In this respect, a detailed understanding of trace metal behavior in these redox-variable systems is crucial for a risk assessment. Unlike oxic upland soils or permanently anoxic sediments, soils in these areas are subject to periodical flooding, leading to intense redox-dynamics.10 In periods of prolonged waterlogging, anoxic conditions can establish within a few days as a consequence of microbial respiration quickly consuming molecular dioxygen. The absence of molecular dioxygen favors the use of alternative electron acceptors (e.g., FeIII, MnIII/IV, and SVI) in biotic and abiotic reactions.10–12 Bisulfides (HS−) resulting from sulfate reduction have been shown to form sparingly soluble nanoscale precipitates with chalcophile trace metals (e.g., CuxS, CdS) in the pore water of floodplain soils.13–15 Likewise, other micro- and nanosized metal sulfides (mostly FeS, ZnS and HgS) have been observed in various natural environments, such as treated anaerobic wetlands,16 anoxic lakes,17 estuarine and marine waters and sediments,18–21 biofilms,22–24 and even in oxic surface waters,25–28 thus proving their persistence in oxic environments over several months. Considering the general stability of metal sulfide nanoparticles under various redox conditions, natural copper sulfide nanoparticles (CuxS NPs) may contribute to long-range transport of Cu in aquatic environments. However, in-depth knowledge about key factors controlling their distribution over various environmental compartments is still limited.Particle characteristics of CuxS such as size, morphology, surface charge, and aggregation behavior are decisive for their colloidal transport,29–32 control their reactivity towards the surrounding medium,31,33,34 the availability to cells and organisms35 and determine whether they can pass through a narrow soil pore.32,36–38 Key environmental factors such as ionic strength, pH, as well as copper, sulfide, and natural organic matter (NOM) concentrations control particle properties and may vary over a wide range depending on soil pore water composition. Large variations in the pore water concentrations of Cu(II) and S(-II) in floodplain soils are due to legacy contamination, regional parent rock composition, inflow of sulfur-rich waters, the flooding regime of the catchment or agricultural practices among others. In this context, the effect of elevated Cu(II) and S(-II) concentrations on the nucleation and growth of CuxS nanoparticles has not been addressed yet.
The poor aeration of soils, being typical for wetlands, was shown to slow down organic matter decomposition and favor the accumulation of humic substances.11,39 Since Cu exhibits a remarkable binding affinity towards NOM,40–42 this affects Cu cycling in these systems. Furthermore, Cu can take part in redox interactions with NOM, especially under anoxic conditions, which are known to induce high affinity proton binding groups in humic substances.39 However, inorganic HS− formed in anoxic pore waters is a much stronger ligand and competes for Cu binding so that Cu availability is limited by the formation of Cu-bearing sulfide (nano)phases.13,43 The relevance of NOM as key modulator for particle growth and stability has been shown for various engineered nanoparticles44–50 and other metal sulfides, e.g., ZnS and HgS.51–53 Moreover, several studies have demonstrated the ability of NOM coatings to protect various nanoparticles from aggregation via electrostatic or electrosteric stabilization.44–54 Up to now, knowledge on NOM effects on CuxS nanoparticle formation and growth is, however, still lacking.
The fate of nano-CuxS in variable redox environments can only be predicted reasonably if dissolution is taken into account. Even though bulk CuxS solubility is extremely low at circumneutral pH and under anoxic conditions,43 dissolution has to be considered as it can be triggered upon re-aeration of wetland soils in case of poorly crystalline nanoscale precipitates. When present in their nanoparticulate forms, the solubility of mineral phases can even be increased by several orders of magnitude.55–57 It has been suggested that partial oxidative dissolution of metal sulfides only occurs in the presence of NOM, e.g. HgS dissolution by various DOM isolates.58,59 Contrarily, dissolved Cu(II) has been detected after exposing sulfidized CuO nanoparticles to oxic solutions in the absence of NOM.60 Based on these, to some extent contradictory findings, together with the observations of metal sulfides in oxic natural waters, warrants further studies on the role of NOM in nano-CuxS dissolution.
Given the limited knowledge on CuxS NP properties and behavior under relevant environmental conditions, the aim of our study was to track particle growth of CuxS and to investigate the effects of absolute metal and sulfide concentration as well as NOM concentration on particle size, particle morphology, and colloidal stability of CuxS over four weeks. For this purpose, we used transmission electron microscopy (TEM) and adapted a hyphenated size-exclusion chromatography (SEC)-ICP-MS technique, which allowed the detection and sizing of CuxS nanoparticles between 2–50 nm in diameter. Additionally, oxidative dissolution experiments were conducted to study the influence of fulvic acid and dioxygen on CuxS NP solubility.
2. Materials and methods
Materials
Filtered (0.22 μm, sterile Millipak® 40, Millipore) ultrapure deionized water (Milli-Q®, 18 MΩ cm, Millipore) was used to prepare all solutions. All chemicals used were of at least analytical grade. For the preparation of anoxic solutions, ultrapure water was purged with N2 for at least 2 h under vigorous stirring. Anoxic work was performed in a glovebox (Labmaster 130, MBraun, Germany) under N2 atmosphere (O2 < 1 ppm). All glassware was acid-washed by soaking overnight in 1 mol L−1 HCl/1.5 mol L−1 HNO3 followed by three rinses with ultrapure water. All stock solutions were prepared in the glovebox and filtered (0.22 μm mixed cellulose ester membrane filters, BGB Analytik AG, Switzerland).Stock solutions of dissolved Cu(II) were prepared with CuCl2·2H2O (Merck). Using 0.1 mol L−1 HCl and 0.1 mol L−1 NaOH, the stock of 3-(N-morpholino)propanesulfonic acid (MOPS, Amresco) buffer was adjusted to pH 7.5. Likewise, the stock of Suwannee River fulvic acid II (SRFA, Cat. No. 2S101F, IHSS), serving as the representative for NOM, was adjusted to pH 6.1. Closed air-tight, the SRFA stock was stored outside the glovebox in the dark at 4 °C. Before use, the concentrations of Cu(II) and SRFA stocks were determined by inductively coupled plasma optical emission spectroscopy (5100 ICP-OES, Agilent) and by TOC analysis (DIMATOC® 2000, DIMATEC, Germany), respectively. Prior to the preparation of sulfide stock solutions, crystals of Na2S·9H2O (Sigma-Aldrich) were thoroughly rinsed with ultrapure water in order to remove potentially oxidized or polysulfidic surface coatings61 and dried for at least 24 h within the glovebox. The concentration of the sulfide stock was determined via iodometric titration, which was standardized against a thiosulfate solution (Titrisol®, Merck). Stability of the sulfide stock over 4 weeks within the glovebox atmosphere was verified using the same method. Sulfide working solutions were diluted from this stock. Ammonium nitrate (NH4NO3, Merck) and FL-70 (Fisher Scientific, USA) were used to prepare the mobile phase for SEC-ICP-MS analyses. Citrate-capped gold nanoparticles (Au NPs) with nominal sizes of 2, 5, 10, 20, 30, 40, and 50 nm as given by the manufacturer (BBI solutions, UK) served as the size calibration analogs for the CuxS NPs during the growth experiment. A quality assurance report of these well-characterized Au NPs is provided by the manufacturer.62 For the quantification of sulfur contents in the nanoparticles, sodium sulfate (Na2SO4, Merck) was used as the calibration standard. Calcium chloride (CaCl2·2H2O) required for the coagulation procedure in the dissolution experiments was obtained from Merck (Germany).
Preparation of CuxS suspensions for growth experiments
Transmission electron microscopy (TEM) and size-exclusion chromatography (SEC) coupled with inductively coupled plasma mass spectrometry (ICP-MS) were used to follow the size of the CuxS nanoparticles over time. For this purpose, we conducted experiments on CuxS nanoparticle formation from anoxic solutions with low copper (50 μmol L−1) and sulfide (100 μmol L−1) concentrations at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (henceforth denoted ‘dilute suspensions’), in absence and presence of SRFA (0, 5, 50 mg C L−1). Additionally, the influence of higher copper (500 μmol L−1) and sulfide (1000 μmol L−1) concentrations (henceforth called ‘concentrated suspensions’) on particle characteristics was investigated. All experiments were conducted at pH 7.5 (1 mmol L−1 MOPS) in a 10 mmol L−1 NaCl electrolyte at 25 ± 2 °C. Starting solutions were prepared in 100 mL serum glass bottles by mixing NaCl, MOPS buffer, SRFA, and Cu(II) at given concentrations. Subsequently, S(-II) was added in one spike while stirring vigorously (with a magnetic stirrer) to induce particle formation. After that, serum bottles were closed with a butyl rubber stopper (Rubberbv, Netherlands) and an aluminium cap (Chromacol 20-ACB, Thermo Scientific, Germany). The bottle headspace volume after addition of all aqueous reagents was 16 mL N2. To a large part of this study, the analyses (scanning transmission electron microscopy (STEM), energy dispersive X-ray spectroscopy (EDX)) were conducted with samples equilibrated for 24 h. Apart from these, growth experiments for selected treatments were conducted to investigate nanoparticle growth over time. For this, aliquots were withdrawn at selected time points (after 45 and 100 min, then 3, 5, 8, 24 and 48 h, then 5, 7, 14 and 28 d from physical triplicate samples for SEC-ICP-MS analysis and after 100 min, 24 h, 7 d and 28 d from one of the same triplicate samples for TEM analyses) for subsequent analyses using a plastic syringe (Amefa, Braun, Germany) equipped with a stainless steel needle (Sterican, Braun, Germany). For SEC-ICP-MS analysis, aliquots were diluted to ∼100 μg L−1 CuxS (assuming a Cu:S ratio around 1.0) in 10 mmol L−1 NaCl and 1% FL-70 into 2 mL HPLC glass vials and closed with a plastic screw cap within the glovebox before they were transferred to the instrument. The chemical speciation was calculated for all prepared solutions with Visual MINTEQ 3.163 to compare experimental conditions with thermodynamic equilibria (Tables S6a–e and S7a–e†).
:2 ratio (henceforth denoted ‘dilute suspensions’), in absence and presence of SRFA (0, 5, 50 mg C L−1). Additionally, the influence of higher copper (500 μmol L−1) and sulfide (1000 μmol L−1) concentrations (henceforth called ‘concentrated suspensions’) on particle characteristics was investigated. All experiments were conducted at pH 7.5 (1 mmol L−1 MOPS) in a 10 mmol L−1 NaCl electrolyte at 25 ± 2 °C. Starting solutions were prepared in 100 mL serum glass bottles by mixing NaCl, MOPS buffer, SRFA, and Cu(II) at given concentrations. Subsequently, S(-II) was added in one spike while stirring vigorously (with a magnetic stirrer) to induce particle formation. After that, serum bottles were closed with a butyl rubber stopper (Rubberbv, Netherlands) and an aluminium cap (Chromacol 20-ACB, Thermo Scientific, Germany). The bottle headspace volume after addition of all aqueous reagents was 16 mL N2. To a large part of this study, the analyses (scanning transmission electron microscopy (STEM), energy dispersive X-ray spectroscopy (EDX)) were conducted with samples equilibrated for 24 h. Apart from these, growth experiments for selected treatments were conducted to investigate nanoparticle growth over time. For this, aliquots were withdrawn at selected time points (after 45 and 100 min, then 3, 5, 8, 24 and 48 h, then 5, 7, 14 and 28 d from physical triplicate samples for SEC-ICP-MS analysis and after 100 min, 24 h, 7 d and 28 d from one of the same triplicate samples for TEM analyses) for subsequent analyses using a plastic syringe (Amefa, Braun, Germany) equipped with a stainless steel needle (Sterican, Braun, Germany). For SEC-ICP-MS analysis, aliquots were diluted to ∼100 μg L−1 CuxS (assuming a Cu:S ratio around 1.0) in 10 mmol L−1 NaCl and 1% FL-70 into 2 mL HPLC glass vials and closed with a plastic screw cap within the glovebox before they were transferred to the instrument. The chemical speciation was calculated for all prepared solutions with Visual MINTEQ 3.163 to compare experimental conditions with thermodynamic equilibria (Tables S6a–e and S7a–e†).
SEC-ICP-MS analysis
For size determination and quantification of the CuxS NPs, we adapted a SEC-ICP-MS method from Zhou et al. (2017),64 using Nucleosil-SiOH columns (250 × 4.6 mm) with pore sizes of 1000 Å or 4000 Å (Macherey-Nagel). The mobile phase was composed of 10 mmol L−1 NH4NO3 and 0.5% FL-70 at a flow rate of 0.5 mL min−1 and the injection volume was set to 50 μL. The setup consisted of a quaternary solvent pump (1200 series, Agilent Technologies), housing one of the above mentioned columns, coupled via a regular PEEK tubing to a triple quadrupole ICP-MS/MS instrument (ICP-MS 8800, Agilent Technologies) operated in tandem mode with either H2 or a mixture of O2 and H2 as reaction gas. The instrument was tuned daily before analysis with a multi-elemental solution and a solution containing Sc and Y (1000 μg L−1 and 50 μg L−1, respectively) was continuously admitted post-column through a T-piece to account for sensitivity variations during each session. The same solution was used over 28 d to correct for CuxS quantification. A summary of instrumental parameters is given in Table S1.† A series of 10 μg L−1 Au NPs was used for size calibration (R2 > 0.95, Fig. S7†), while Cu(II) and SO42− served as standards for quantification (range 0–500 μg L−1, R2 > 0.99). Matrices of all standards were matched with the sample matrix and prepared in 10 mmol L−1 NaCl and 1% FL-70, accordingly. The calibration series for quantification was measured once before every sample batch, whereas Au NP size calibration series was always measured at least once before and after each sample batch in order to check for minor retention time variations. In order to achieve conversion of peak retention times (tR) into particle diameters by means of established calibrations using Au nanoparticles, chromatographic data were first extracted from the Agilent Mass Hunter software. Then, Cu and S signals were corrected with the internal standards. After that, peak fitting of all Au NP and CuxS NP peaks was carried out with Origin 2018 (version b9.5.0.193) to determine peak properties such as peak maxima, center of gravity (COG) of the peaks and their full width at half maximum (FWHM). Finally, retention times at peak maxima for the internal standard corrected Cu signals were used to determine SEC diameters.TEM analysis
CuxS particles were investigated by scanning transmission electron microscopy (STEM, HD2700-Cs operated at 200 kV, Hitachi, Japan) using a high-angle annular dark-field (HAADF) detector for image formation. Elemental composition of individual particles was determined by energy dispersive X-ray spectroscopy (EDX, EDAX, USA). Samples for TEM analyses were prepared in the glovebox. Nickel grids with a holey carbon support film (S147N4, 400 mesh, Plano) were functionalized with 0.1% (w/v) poly-L-lysine (Sigma-Aldrich) to establish a positively charged collector surface. A drop of 25 μL of CuxS suspension collected from the growth experiments was placed on the grid, either undiluted (for the lower concentrated samples, final concentration in the droplet: ∼50 μmol L−1 CuxS) or up to 10 times diluted (for the higher concentrated samples, final concentration in the droplet: ∼50–250 μmol L−1 CuxS). Particles were allowed to attach to the grid surface for 10 min before the grid was washed with one drop of ultrapure water. Excess liquid from the grid was wicked away from underneath with a lint-free tissue after each step. Prepared grids were placed into plastic grid racks (Plano, Germany) wrapped in aluminum foil to prevent light exposure and stored under N2 atmosphere in the glovebox until analysis. Recorded TEM images were processed using different image analysis software packages (Digital Micrograph v3.01, ImageJ Fiji v1.51n, Adobe Illustrator CC v2015.0.0, Adobe Photoshop CC v2005.1.2). Particle sizing was performed either by using the Nanodefine Particle Sizer plugin65 incorporated into the ImageJ Fiji software or by manual size determination and counting using the Digital Micrograph software. A forward Fourier transformation was applied to high-resolution TEM images that showed crystal lattice fringes of CuxS particles to obtain corresponding selected area diffraction (SAED) patterns. Calculations on particle size distributions and statistical significance tests were performed with Origin 2018. To test for significant differences in particle sizes among treatments, pairwise Wilcoxon–Mann–Whitney-U-tests were performed with the TEM particle size distributions acquired during the growth experiments (Table S8–S10†).XRD analysis
Identification of mineralogical phases was accomplished by X-ray diffraction (XRD, D8 Advance, Bruker) analysis. For this, 10 mL of the concentrated sample suspensions were filled into polycarbonate tubes and ultracentrifuged (40000 rpm, 193000g, 3 h, 15 °C, fixed-angle rotor, Centrikon T-1080, Kontron). The supernatant was carefully discarded and the fragile pellet at the bottom of the tubes was resuspended in 1 mL of ultrapure water. The dispersed suspension was then centrifuged again for 30 min at otherwise same conditions. The supernatant was removed and the new pellet was dispersed in 0.2 mL ethanol. As a final step, 10 μL of the ethanolic suspension was repetitively (5–10 times) deposited and dried onto a zero background silicon wafer (orientation (510), Siltronix, France) until a thin CuxS layer was visible. For the measurement, the loaded silicon wafers were analyzed in Bragg–Brentano geometry employing Cu Kα radiation (λ = 1.5418 Å, 40 kV, 40 mA) and a high-resolution energy dispersive 1D detector (LynxEye XE). X-ray diffractograms were recorded from 4–90° 2θ in 0.02° steps with 10 s acquisition time per step. Qualitative analysis of the measured diffractograms was done via peak matching of the observed reflections with the ICDD PDF2 database used with the Bruker EVA software.
Dissolution experiments
To study the stability of CuxS against oxidative dissolution, a set of CuxS nanoparticle suspensions was prepared in triplicates in the absence and presence of fulvic acid (0, 5, 50 mg C L−1) and at lower (50 μmol L−1 Cu(II), 100 μmol L−1 S(-II)) and higher concentrations (500 μmol L−1 Cu(II), 1000 μmol L−1 S(-II)) in the same way as already described for growth experiments. The only two deviations from this procedure were the removal of the magnetic stirrer from the serum bottles (in order to prevent any potential adsorption of CuxS on the stirrer surface) together with the 1:10 dilution of the concentrated samples to 50 μmol L−1 CuxS, both after one hour to ensure comparable conditions. After 90 min from particle formation point, the N2 atmosphere in the headspace of the closed serum sample bottles was changed to 21% (v/v) O2 (synthetic air) by replacing equivalent volumes of N2 with O2 gas using a gas-tight syringe. The amounts of spiked O2 exceeded the amounts required to theoretically oxidize all CuxS in the samples 7.5-fold. Additionally, controls without oxygen gas spikes were run. Subsequently, suspensions were incubated in the dark under constant shaking at 25 °C. Aliquots of suspensions were collected with a gas-tight glass syringe at different time steps over several weeks (after 1, 4, 14, 22, 28, 42, 63, 87 d) and immediately processed by (i) an induced coagulation of the precipitates in 1 mmol L−1 CaCl2 solution followed by (ii) a filtration using nylon membrane filters (0.22 μm), thus effectively separating the initial solid from the liquid phase. Subsequently, Cu concentrations in the filtrate were determined with ICP-OES (method detection limit: 0.2 μmol L−1 Cu or 0.4% of total Cu). Preliminary tests with chemically stable, anoxic CuxS suspensions verified the suitability of this coagulation and filtration method in retaining all nanoparticulate CuxS (>1 nm) and even clusters between 0.5–1.0 nm as no Cu was found in the filtrates.
3. Results and discussion
Effects of reactant and NOM concentrations on CuxS nanoparticles
![[d with combining tilde]](https://www.rsc.org/images/entities/i_char_0064_0303.gif) TEM = 3.9 nm with very few larger particles between 14 and 20 nm as compared to TEM = 15.8 nm in concentrated suspensions with about 5% of the particles being larger than 45 nm (Fig. S1b and S4b†). In both cases, the CuxS nanoparticles appeared well-dispersed and no evidence for particle aggregation was observed at near-neutral pH. The comparison of the TEM images recorded for dilute and concentrated suspensions revealed clear differences in the speed at which particle morphology evolved, and also evidenced pronounced differences in particle sizes.
TEM = 3.9 nm with very few larger particles between 14 and 20 nm as compared to TEM = 15.8 nm in concentrated suspensions with about 5% of the particles being larger than 45 nm (Fig. S1b and S4b†). In both cases, the CuxS nanoparticles appeared well-dispersed and no evidence for particle aggregation was observed at near-neutral pH. The comparison of the TEM images recorded for dilute and concentrated suspensions revealed clear differences in the speed at which particle morphology evolved, and also evidenced pronounced differences in particle sizes.
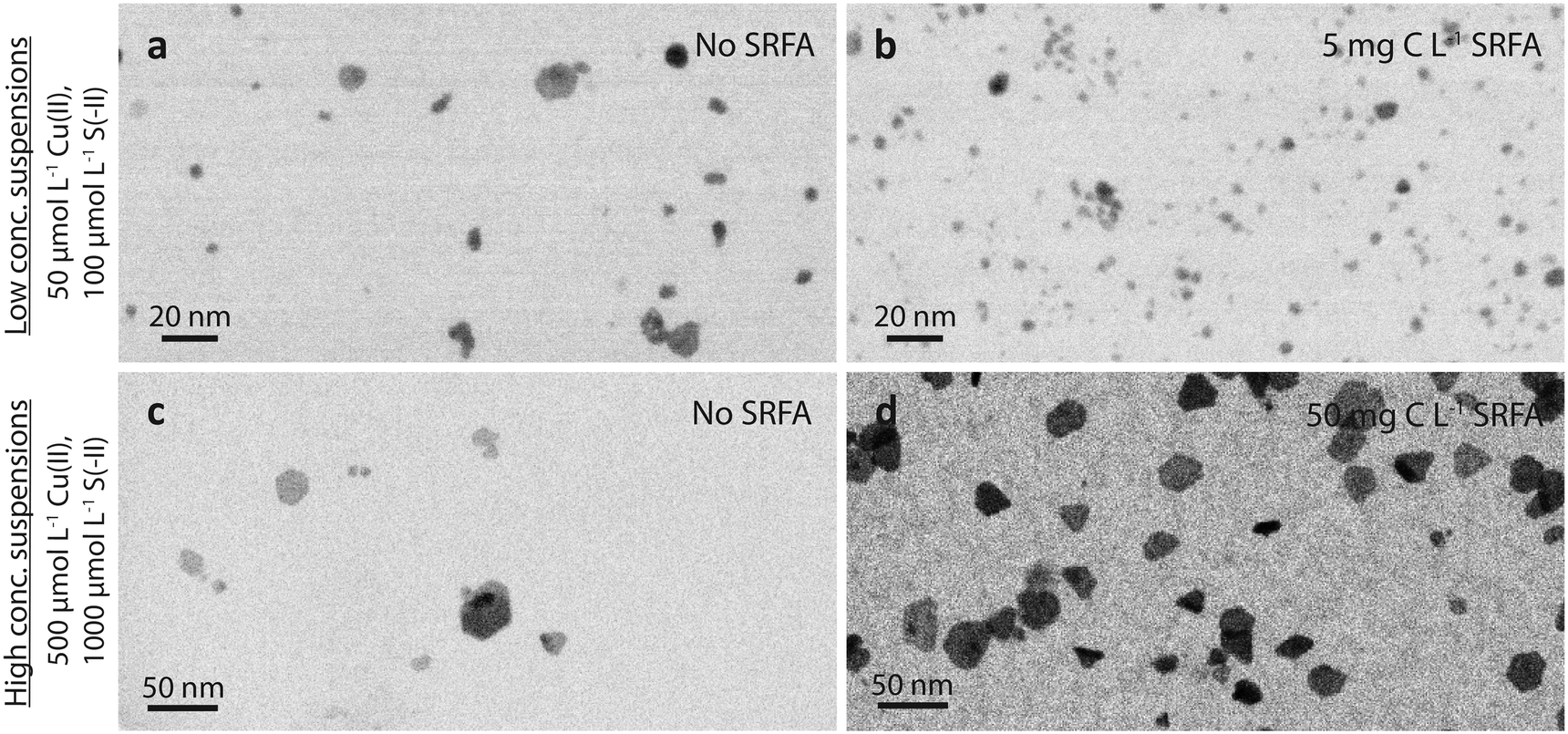 | ||
| Fig. 1 High-angle annular dark-field (HAADF) TEM images (shown as inverted images) of dilute (a and b, 50 μmol L−1 Cu and 100 μmol L−1 S) and concentrated (×10 reactants) CuxS suspensions (c and d) in the absence (a and c) and presence of 5 mg C L−1 fulvic acid (b) and 50 mg C L−1 fulvic acid (d), respectively, after 24 h equilibration time. Background solutions consisted of 10 mmol L−1 NaCl at pH 7.5 (MOPS buffer). | ||
TEM = 3.9 nm without vs. TEM = 2.9 nm with 5 mg C L−1 SRFA). Similar to these dilute SRFA-free suspensions, a 10-fold increase in reactant and SRFA concentrations of the SRFA-containing suspensions led to the formation of hexagonal platelets and larger particle sizes (Fig. 1b and d). Median TEM diameters were much larger in concentrated (TEM = 15.8 nm without SRFA, TEM = 20.8 nm with 50 mg C L−1 SRFA) than in dilute (TEM = 3.9 nm without SRFA, TEM = 2.9 nm with 5 mg C L−1 SRFA) suspensions (Fig. 1c, d, a and b). In all 24 h experiments with dilute suspensions, the fulvic acid hampered the development of the CuxS in particle size and morphology. This effect was much less pronounced in concentrated suspensions. However, in these concentrated suspensions, the fulvic acid hindered the growth of large particles exceeding sizes of 50 nm, which were only found in SRFA-free suspensions (Fig. S4b and S5b†).
A common observation for all investigated conditions was the extraordinary colloidal stability of the CuxS precipitates. Even without NOM, the suspensions remained stable under anoxic conditions for several months, likely due to the strongly negatively charged surfaces of the CuxS particles (zeta potentials around −50 mV, Table S5†) under the experimental conditions (pH 7.5, 10 mmol L−1 NaCl). This is consistent with a reported point of zero charge (PZC) for CuxS ranging between pH 1 and 3.68–72 Even a decrease in pH to 6 did not cause notable aggregation and only occasional aggregates ∼100 nm were observed in an otherwise well-dispersed suspension after 24 h (Fig. S10†).
Structure and composition of the CuxS nanoparticles
:S ratios were determined via SEC-ICP-MS measurements in a later phase (Fig. S9,† see also section below) and were found to be close to 1.0 (std. dev. = 0.1, min = 0.8, max = 1.4). High-resolution STEM images of CuxS nanoparticles (∼10 nm, 24 h) featured clear lattice fringes (Fig. 2). The identified lattice spacing of 0.19 nm corresponds to the crystallographic (110) plane d-spacing in the crystal structure of covellite (CuS) as well as to the (111) plane d-spacing in the crystal structure of yarrowite (Cu1.12S). Metastable CuxS mineral phases such as djurleite (Cu1.96S), digenite (Cu1.8S), anilite (Cu1.75S), geerite (Cu1.6S), spionkopite (Cu1.4S) and yarrowite (Cu1.12S) have been reported in earlier studies.73,74 Peak matching of the X-ray diffraction patterns of our samples suggested that the particles mainly consisted of covellite (CuS) but it cannot be excluded that metastable mineral phases coexisted during the first 24 h, since the theoretical positions of their XRD reflections partly overlapped with the observed broad diffraction peaks. The X-ray diffractograms depicted in Fig. S19† show the structural evolution of the CuxS particles over time and suggest a growth of crystalline domains (e.g., sharpening of (110) reflection peak with aging). This result together with STEM images confirmed that the larger nanoparticles developed crystalline domains.
 | ||
| Fig. 2 (High-resolution) STEM images (a (inverted HAADF), b (bright field)) and the corresponding forward Fourier transformation (c) of a 24 h aged dilute CuxS suspension (50 μmol L−1 Cu(II), 100 μmol L−1 S(-II)) formed in the presence of 5 mg C L−1 SRFA at pH 7.5 and an ionic strength (IS) of 10 mmol L−1 NaCl exhibiting the lattice fringes of covellite/yarrowite. | ||
Sizing of the CuxS nanoparticles by SEC-ICP-MS and comparison with TEM
The results presented above covered CuxS particle properties at 24 h aging. Particle properties also varied over time during particle growth and aging as suggested by the change in colors as discussed above. Therefore, we used SEC-ICP-MS to investigate the particle growth and composition (molar Cu:S ratios, Fig. S9†) over time in more detail. Typical chromatograms of the Cu and S signals recorded for a growth experiment at 100 min and 24 h are shown in Fig. 3a. Notice that the very large S peak starting at 5.75 min (Fig. 3a, inset) originates from the sulfonic acid group of the MOPS buffer. The S signal related to the CuxS nanoparticles could always be unambiguously distinguished from these large S background peaks given the excellent separation efficiency of the SEC columns with respect to dissolved species vs. nanoparticles. However, the Cu signal overall and especially the background signal was less noisy than the S signal, which made the Cu signal best suited for further use in data analysis of SEC diameters. The recoveries of Au nanoparticles ranged from 82 to 110% (ionic Au: 93%) and were even better for CuxS nanoparticles, i.e. 95–104% (ionic Cu: 109%; Table S2†), demonstrating the preservation and absence of loss during their transport through the size-exclusion column.
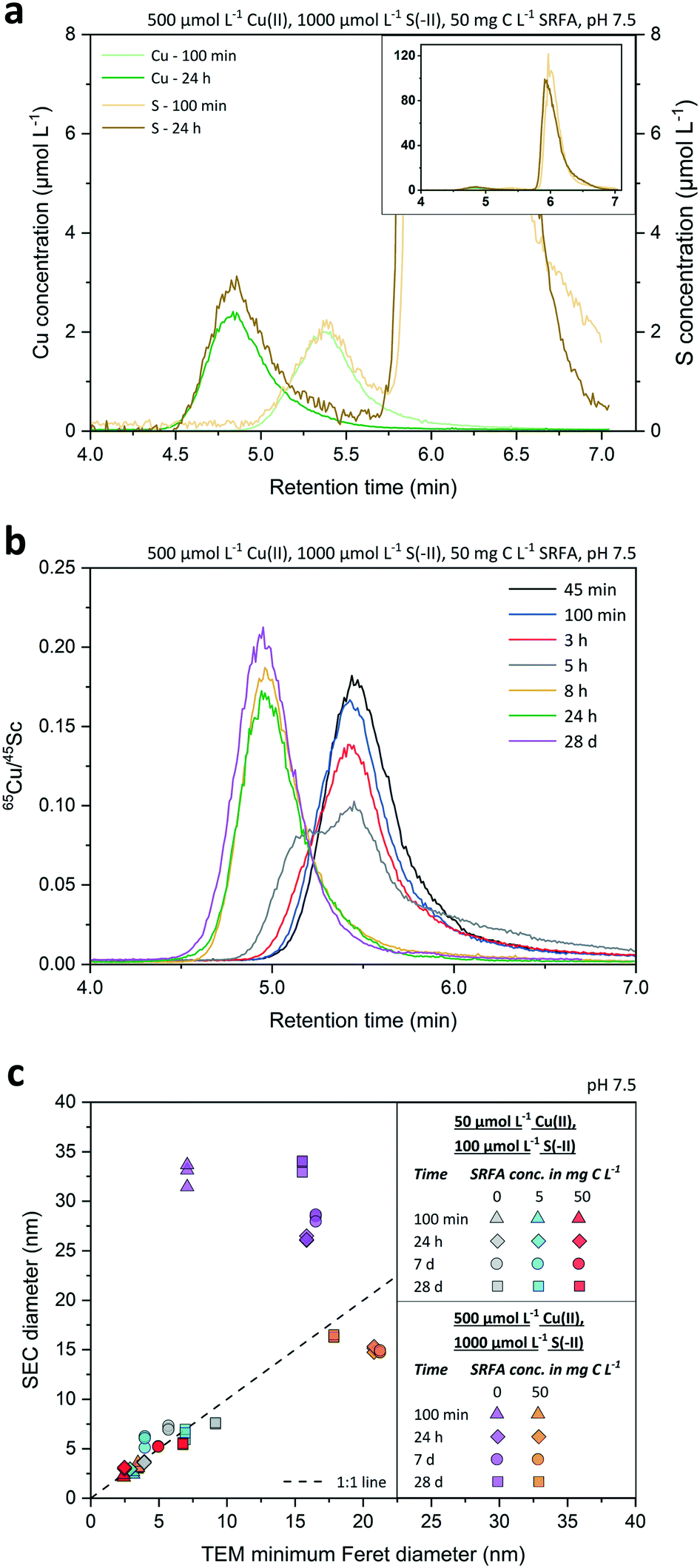 | ||
| Fig. 3 Panel (a) shows the SEC chromatograms of concentrated, SRFA-containing CuxS suspensions at two different growth stages (100 min, 24 h). Recorded elemental signal intensities for Cu and S were converted into Cu (green lines) and S (brownish lines) concentrations (in μmol L−1). The huge S peak shown in the inset of (a) is due to the sulfonic acid groups of the dissolved MOPS buffer. Panel (b) exemplifies the retention time shift towards earlier times upon growth of CuxS nanoparticles from a concentrated, SRFA-containing suspension during 28 d. Peaks of particle suspensions are represented as internal standard corrected 65Cu signal intensity ratios and are color-coded at different time steps. Panel (c) shows a regression plot of SEC diameter values measured during the CuxS growth experiment (after 100 min, 1 d, 7 d and 28 d) from various suspensions and the corresponding median minimum Feret TEM diameters of the same samples. Dashed line within (c) signifies the 1:1 line indicating 100% conformity of SEC and TEM sizes. In all cases, the ionic strength (IS) was 10 mmol L−1 NaCl. | ||
Fig. 3b presents the Cu signals as corrected with the 45Sc signals (using the same Sc solution over 28 d) at various time points along a growth experiment of CuxS particles formed in a concentrated, fulvic acid rich suspension. Overall, differences between peak retention time among the growing particles were very distinct, gradually decreasing from 5.45 min after 45 minutes to 4.95 min after 28 d, which corresponded to an increase in average diameter (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) SEC ± std. dev.) from 3.0 ± 0.2 nm to 16.4 ± 0.1 nm when applying the size calibration (Fig. S7†) using the Au nanoparticles. Interestingly, the peak shape markedly changed between 3 and 8 h with a clear bimodal distribution at 5 h, highlighting an important growing stage over that time period. Retention time differences assessed by either the peak maxima (PM) or peak center of gravity (COG) varied by less than 1.3% (n = 42) between same-sized Au nanoparticles, thus allowing a precise estimation of the median diameter of the nanoparticle population. Even small retention time shifts of the peaks ≥0.06 min were detectable over the whole course of the experiment, suggesting that changes in SEC diameters in the sub-nanometer range were detected. The applied technique exhibited a very high repeatability. Based on the retention time of the dissolved Au and Cu analyzed at the same concentration than the Au and CuxS nanoparticles, the size detection limit (lower size cutoff) was estimated to be 1 nm for Au and 1.6 nm for CuxS nanoparticles. The upper size cutoff of the method was not determined precisely but 50 nm Au NPs were still well detectable, even though peaks started to broaden above 40 nm (Fig. S8†), and recoveries were still excellent (Table S2†).
SEC ± std. dev.) from 3.0 ± 0.2 nm to 16.4 ± 0.1 nm when applying the size calibration (Fig. S7†) using the Au nanoparticles. Interestingly, the peak shape markedly changed between 3 and 8 h with a clear bimodal distribution at 5 h, highlighting an important growing stage over that time period. Retention time differences assessed by either the peak maxima (PM) or peak center of gravity (COG) varied by less than 1.3% (n = 42) between same-sized Au nanoparticles, thus allowing a precise estimation of the median diameter of the nanoparticle population. Even small retention time shifts of the peaks ≥0.06 min were detectable over the whole course of the experiment, suggesting that changes in SEC diameters in the sub-nanometer range were detected. The applied technique exhibited a very high repeatability. Based on the retention time of the dissolved Au and Cu analyzed at the same concentration than the Au and CuxS nanoparticles, the size detection limit (lower size cutoff) was estimated to be 1 nm for Au and 1.6 nm for CuxS nanoparticles. The upper size cutoff of the method was not determined precisely but 50 nm Au NPs were still well detectable, even though peaks started to broaden above 40 nm (Fig. S8†), and recoveries were still excellent (Table S2†).
We found a good agreement between the diameters derived from TEM and SEC analyses (Fig. 3c) for small CuxS nanoparticles (2.5–10 nm) that showed a rather narrow distribution (Fig. S1–S3†). In contrast, SEC diameters of samples containing few very large particles in an otherwise small particle size population led to mismatches with TEM diameters (cf.Fig. 3c purple dataset and Fig. S4†). The reason is that the TEM particle size corresponds to the number based average particle size whereas SEC-ICP-MS gives a mass based average particle size.31 As the mass of a particle scales with the third power of its size (diameter), the mass based average size of a particle population is shifted towards larger sizes compared to the number based average size. Hence, the large particles measured in SEC-ICP-MS increased the mass based average nanoparticle diameter compared to the number based average diameter. This effect becomes more pronounced with increasing polydispersity of the particle populations. Consequently, a larger discrepancy in diameters derived from SEC and TEM was found in CuxS samples that were formed in concentrated suspensions. In an attempt to explain this discrepancy in the SEC and TEM central tendency values in more detail, we converted the number based TEM particle size distributions of the CuxS suspensions into mass based TEM PSDs by assuming a spherical shape for all particles in the conversion (Fig. S11–S15†). While this procedure reduced the discrepancy between SEC and TEM average diameters in the concentrated SRFA-free suspensions, it led to considerable disagreement among the calculated mass based TEM average diameters and the SEC diameters in all other suspensions (Fig. S11–S16†). As particle shapes were different among the suspensions and changed during particle development (cf.Fig. 1, and S1–S5† addressed in later sections), the applied conversion procedure assuming a spherical particle model was prone to errors in the calculation of mass based diameters, particularly with suspensions that contained particles whose shape deviated the most from spheres. This is reflected by a larger shift from the 1:1 line in the comparison of converted mass TEM diameters and SEC diameters (Fig. S16†). In addition, recorded particle population statistics need to be considered with a few dozens to hundreds of particles in TEM datasets used for the conversion compared to millions of particles in SEC datasets. In this regard, much weight is given to a few large particles when applying this conversion, especially in low particle number TEM datasets, which then leads to a distortion in the calculated mass based TEM diameter. This clearly shows that such rather simple conversion procedures for particle size distributions should be applied and interpreted with great caution as respecting the correct particle geometry in x-, y- and z-direction as well as counting statistics are crucial.
The discrepancies were also slightly larger when peak maxima were used for the identification of peak retention times and the subsequent conversion into diameters. If instead the center of gravity (COG) obtained from peak fitting was used for the assignment of retention times and subsequent size conversion, average diameters of more polydisperse samples were in closer agreement with median TEM diameters (Fig. S17, Tables S3 and S4†). For all other samples, the SEC diameters derived from peak maxima agreed with the median TEM diameters. Therefore, peak maxima were preferably used to convert SEC retention times to equivalent diameters for the tracking of the nanoparticle size development in the growth experiments. Future studies could make use of such a diameter determination from size-exclusion chromatograms based on a central parameter of a Gaussian curve (used to fit the elution peak), in order to characterize different nanoparticle types with different coatings.
Nucleation and growth of CuxS nanoparticles in the absence of NOM
SEC = 3.0 nm. The corresponding median TEM minimum Feret diameter was only slightly larger (TEM = 3.4 nm). Thereafter, the growth of the CuxS particles proceeded slowly, with an average SEC diameter increase of less than 1 nm over the next 24 h. The major growth phase occurred between day 1 and day 7 with nearly a doubling in the average SEC diameter (from SEC = 3.7 nm to SEC = 7.1 nm). After 7 d, nanoparticle growth leveled off in all dilute suspensions, indicating that their size development was completed. Median TEM diameters for the selected dilute samples were very close to the SEC ones (max. difference <2 nm, Fig. 4a). TEM particle size distributions of all dilute suspensions widened in the course of the experiment without developing a clear bimodal size distribution (Fig. S1†).
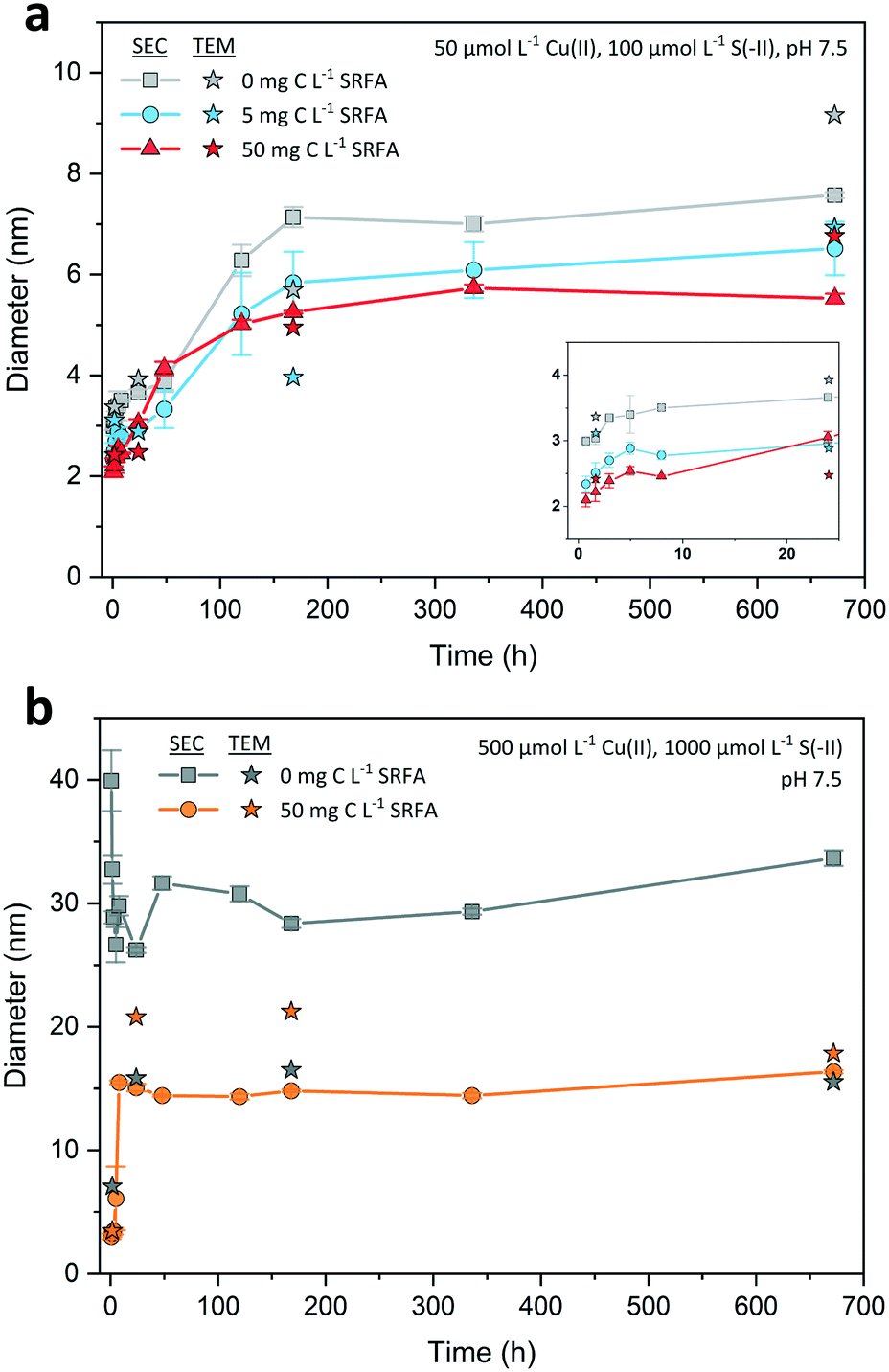 | ||
| Fig. 4 Development of average SEC and median minimum Feret TEM diameters of dilute (a) and concentrated (×10 reactants) CuxS suspensions (b) over the course of 4 weeks in the absence and presence of different Suwannee River fulvic acid (SRFA) concentrations. Background solutions consisted of 10 mmol L−1 NaCl at pH 7.5 (MOPS buffer). For reasons of visualization, the first 24 h of nanoparticle growth from dilute suspensions is exemplified in the inset of (a). Filled symbols connected with lines denote average SEC diameters, filled stars designate minimum Feret TEM diameters. Error bars of SEC diameters indicate the standard deviations derived from physical triplicate samples (n = 3). | ||
Thermodynamic calculations for the precursor solution (before sulfide addition) suggested the possible precipitation of 98.4% of the Cu as atacamite (Table S6†), a copper(II) chloride hydroxide mineral, and we actually found and isolated Cu(II)-hydroxide-like phases in the precursor solution after 30 min of equilibration time (Fig. S20†). Due to kinetic constraints on (par)atacamite precipitation,78 it is more likely that a simple Cu(II) hydroxide phase would form under these conditions. For nucleation, this means that the initial situation for metal sulfide formation is different than direct nucleation from solution as (par)atacamite seeds were already present in the system. Since there were no visible precipitates in the precursor solution and sodium sulfide was added within a maximum timespan of one minute, the growth of the potential nucleation seeds for CuxS was interrupted at a very early stage. We, therefore, assume that the formation of (par)atacamite was limited to nanoclusters or very small nanoparticles. In this case, growth initiation of CuxS occurs via Cu(II) that is released from the (par)atacamite structure by dissolution, before the sulfide forms CuxS complexes. Consequently, CuxS growth would also depend on the actual size and number of the (par)atacamite seeds. In this scenario, however, the supply of Cu might be slower compared to the situation where Cu is present as an aqueous Cu(II) complex, thereby resulting in a reduced nucleation rate. Generally, if the number of nucleation sites is low, particle size at the end of the main growth phase will be larger, and vice versa.79 Since reaction rates for metal sulfide cluster formation can be faster than 1 s−1,75 this primary growth phase was not resolved by our analytical methodology. According to Luther III (2016),75 cluster size definition ranges from 0.4–1.0 nm. In the SRFA-free suspensions, the smallest CuxS nanoparticles in the first TEM sample (after 100 min) showed minimum Feret diameters between 1.1–1.5 nm, indicating that cluster formation was already finished at this stage.
With increasing reaction time the particle size distributions of CuxS in SRFA-free suspensions considerably broadened, which we believe is due to an Ostwald ripening process that favors the growth of larger particles at the expense of smaller particles, which dissolve, reform clusters and contribute to the growth of the larger particles.80–84 The growth phase eventually ends due to the termination of the Ostwald ripening process,84–86 when the supply of small particles is exhausted and the dissolution of the bigger particles becomes negligible (Gibbs–Thomson effect86).
The relative proportions of precipitated (par)atacamite predicted by thermodynamic calculations were identical to dilute suspensions (Table S6†) and can therefore not explain the observed differences in particle sizes. However, the supersaturation with respect to (par)atacamite was higher in concentrated suspensions. Consequently, more (par)atacamite seeds may have been formed at the beginning in concentrated suspensions.87 When compared to the dilute suspensions, the (par)atacamite seeds may take longer to dissolve when the sulfide is added to the system to form CuxS particles. Such a retarded nucleus supply at initial stage in higher concentrated systems has occasionally been observed to induce the formation of condensed nucleus clusters at the cost of ‘true’ nucleus abundance.87,88 With less nuclei at growth initiation, the terminal particle size is expected to be larger, as was observed in our experiments with concentrated SRFA-free suspensions.79 With an increase in concentrations of the main reactants, a much larger reservoir for the reaction of copper and sulfide is available, resulting in a larger and prolonged supply of reactants during growth. On this account, some effects already observed in the SRFA-free experiments in dilute suspensions are magnified due to the extreme affinity of Cu(II) for S(-II), which drives crystal development and more rapidly leads to higher crystalline structures than in dilute CuxS variants (cf. hexagonal platelets already after 100 min in Fig. S4†vs. the still more spherical particles after 100 min in Fig. S1†). In concentrated suspensions, the color change from brownish to greenish was much more rapid (<1 h in concentrated suspensions vs. ≥24 h in dilute suspensions) indicating a strongly accelerated restructuring process from soluble copper bisulfide complexes over tetrameric clusters to the Cu3S–CuS3–S2–Cu3S–CuS3 layer structure of covellite. The increased supply of Cu assures a more continuous and sustained delivery of new reactants onto the surfaces of the precipitating clusters and nanocrystals promoting the fast structural and size-related development as observed in our experiments.89 Hence, the aforementioned plateau and thus also the final particle size is reached much faster for all concentrated suspensions.
Effect of NOM concentrations on CuxS nucleation and growth
The relative proportion of Cu initially precipitated as metal hydroxide depends on the concentration of SRFA. According to our thermodynamic calculations for the precursor solution before adding sulfide, 81% of the Cu(II) may have been bound in (par)atacamite nano-seeds in the presence of 5 mg C L−1 SRFA whereas the remaining fraction (19%) may have been bound by SRFA (Table S6†). Cu(II) exhibits a high affinity to major carboxylic and phenolic functional groups, but also to minor N-containing and reduced-sulfur (thiol) functional groups in fulvic acids.42,90,91 Accordingly, the precipitation of (par)atacamite was almost completely suppressed at the high SRFA concentrations (50 mg C L−1) as 99.8% of the Cu was predicted to be present as Cu(II)–SRFA-complexes (Table S6†). Therefore, the formation of CuxS nanoparticles from (par)atacamite nano-templates was only expected in the low SRFA case (5 mg C L−1) while CuxS particle growth at high SRFA concentration (50 mg C L−1) was primarily initiated from SRFA–Cu(II)–S(-II) complexes. An excess in SRFA was available in both cases for surface capping of the nanoparticles as only a small fraction of the fulvic acid was needed to bind the Cu in each case at initial formation stages (29% for low SRFA, 17% for high SRFA). The growth of CuxS in dilute suspensions was effectively slowed down by the fulvic acid as the first measured diameters were significantly lower compared to the ones found in the corresponding SRFA-free suspensions. CuxS nanoparticles with minimum diameters between 0.5–1.0 nm were occasionally observed in the first TEM sample (after 100 min), only in the presence of SRFA. This indicates that the cluster formation was not yet finished and that the length of the nucleation phase was longer when SRFA was present, as also discussed by Luther III (2016).75 If we consider nucleation from the different initial Cu(II) species, a nucleation of CuxS from SRFA–Cu(II) complexes would not only proceed faster than from (par)atacamite templates, it would also yield more nuclei in the same period of time. As SRFA concentration influenced the concentration level of (par)atacamite template seeds in the suspensions at initial stage, only 19% of the Cu(II) was readily available as SRFA–Cu(II) for cluster formation in suspensions containing 5 mg C L−1 SRFA, whereas most Cu(II) (99.8%) was quickly available as SRFA–Cu(II) for cluster formation in the high SRFA variants (50 mg C L−1 SRFA). Consequently, the number of nuclei at t = 0 was greater under the latter conditions, thus leading to smaller particle sizes after 4 weeks. This is consistent with the results of our experiments, where the final particle sizes of CuxS formed in dilute, SRFA-free suspensions were larger than in all NOM spiked suspensions that exhibited lower supersaturation levels.
SRFA may have interacted with early CuxS polynuclear complexes and clusters through chelation with (surface) copper cations or through sorption of hydrophobic fulvic acid sites to the newly formed clusters.92 Even though termination of CuxS surfaces by sulfur is expected due to the sulfide excess in the reactors, these early SRFA–Cu(II)–S(-II) associations may delay the process of further cluster formation especially in the beginning by steric hindrance as also observed by Poulin et al. (2017) in experiments involving DOM-HgS associations.93 If so, electrostatic repulsion of new clusters (negatively charged) and already existing SRFA–Cu(II)–S(-II) associations (negatively charged) additionally retard the reaction of Cu(II) and S(-II). In this way, SRFA inherently caps newly formed CuxS surfaces and protects the clusters and nanoparticles from extensive growth. Furthermore, it protects the nanoparticles from attachment to other CuxS particles. As a consequence, the presence of fulvic acid limited the broadening of the particle size distribution and hampered the development of large particles. Since CuxS particles were significantly smaller in dilute suspensions containing SRFA, a more pronounced Gibbs–Thomson effect may have facilitated Ostwald ripening. Additionally, SRFA functional groups, known to be strong Cu ligands, could have stimulated dissolution–reprecipitation reactions. Moreover, we observed a distinct deceleration of structural and morphological development of CuxS particles with the increase in fulvic acid concentration. A similar effect has been recently discovered in aged HgS-DOM suspensions.93 In accordance with Lee et al. (2005),83 the lack of hexagonally shaped particles with a higher structural order at the highest NOM concentration may be a hint for a strong contribution of Ostwald ripening in these suspensions. Surface ligands such as fulvic acid can also selectively bind to certain facets of growing nanocrystals to minimize their surface site energy.94 Like this, reactant or cluster delivery to these surfaces can be diminished or even be completely blocked, leading to a slower and restricted structural development with altered particle shapes compared to ligand-free suspensions89 as seen in our dilute CuxS suspensions.
SEC = 3.3 nm and TEM = 3.5 nm and increased to SEC = 15.1 nm and TEM = 20.8 nm within the first 8 to 24 h. At this time point, nanoparticle growth leveled off and the average SEC diameter remained nearly constant. Only the median TEM diameter of these suspensions decreased to 17.9 nm after 4 weeks, a value close to the corresponding SEC diameter (SEC = 16.4 nm, 28 d). A temporary bimodal phase in the development of the particles was detected with SEC as the 5 h chromatogram (Fig. 3b) constituted the rapid shift from a particle population that was mostly consisting of small particles <10 nm to a population that was dominated by larger particles ≥20 nm (cf. Fig. S4a and b†). This considerable change in the particle population coincided with median TEM and average SEC diameters recorded in the abovementioned intensive growth phase (Fig. 4b).
A comparison of the concentrated CuxS suspensions showed that after 4 weeks both the SRFA-free and SRFA-spiked samples were in a similar size range (TEM = 15.5 nm, no SRFA vs. TEM = 17.9 nm and SEC = 16.4 nm, with SRFA). When comparing the dilute SRFA-spiked suspensions to their concentrated counterparts, a 10-fold increase in reactant concentrations increased the particle size of CuxS in the most pronounced case by a factor of about 2.5 after 4 weeks irrespective of which measure was used (TEM = 6.9 nm and SEC = 6.5 nm in dilute, 5 mg C L−1 SRFA vs. TEM = 17.9 nm and SEC = 16.4 nm in concentrated, 50 mg C L−1 SRFA). With regard to the rapid transition in the particle populations from the initial to the plateau phase, we suggest that our SEC chromatograms show the ongoing process of Ostwald ripening as we observed the decline in contribution of smaller particles <5 nm which almost disappeared in later chromatograms and TEM particle size distributions.
Based on the capability of the fulvic acid to restrict particle growth in dilute suspensions, it is surprising that this does not seem to apply to the concentrated suspensions in the same way. In high concentrated suspensions, the fulvic acid may stabilize the CuxS particles only after crystal development into larger particles (TEM_high_28d = 17.9 nm vs. TEM_low_28d = 6.9 nm). In this case, the comparably large reactional driving force in the concentrated CuxS suspensions would favor the direct reaction of Cu(II) and S(-II) at the expense of early stage SRFA–Cu(II) complexation to functional groups that are responsible for effectively decreasing the size and structural order of the crystals. At Cu(II) concentrations this high, these high affinity Cu(II) binding sites can be saturated so that their size- and structure-restricting activity is considerably impeded.93 Poulin and coworkers have found similar decoupling of such size limiting effects of NOM when reactant concentrations exceeded a critical level at which saturation of strong binding sites prevailed.93 Additionally, the fast reaction kinetics in concentrated SRFA-containing suspensions provoke collisions that are more effective in removing surface bound fulvic acid. This also provides a reasonable explanation for the indifference in final CuxS particle size among concentrated suspensions in absence and presence of SRFA. Interestingly, even though the effectiveness in decreasing particle size is largely impaired, the occurrence of extraordinary large particles was still suppressed in the presence of SRFA, suggesting that other than high affinity functional groups are responsible for the limitation of maximum size or that surface bound fulvic acid possesses sufficient capabilities to prevent further growth, e.g. by providing additional electrostatic and steric stabilization.54
Dissolution of CuxS nanoparticles
The oxidative dissolution of CuxS nanoparticles in the absence and presence of fulvic acid and with CuxS particles of different morphologies is illustrated in Fig. 5. Under anoxic conditions, no dissolved Cu was detected (<0.2 μmol L−1 or <0.4% of total Cu) over several months at circumneutral pH, irrespective of the presence of fulvic acid. CuxS particles remained stable when exposed to O2 in the absence of fulvic acid and only the concurrent presence of O2 and SRFA led to their dissolution. In this case, the dissolution proceeded quickly (20 days) and levelled off between 22 μmol L−1 and 26 μmol L−1, values close to the Cu–SRFA complexation limit of the system (22.6 μmol L−1 Cu). The remaining solid fraction was determined by XRD and consisted exclusively of covellite (Fig. S21†). No significant difference was found among differently treated SRFA-containing samples. Accordingly, it was irrelevant whether fulvic acid was added before or 1 hour after particle formation. Likewise, the reaction was independent of the particle aging since the original dilute suspensions (containing more roundish-platy or nearly spherical shapes) behaved similar to samples from 10-fold diluted high concentrated suspensions (containing larger amounts of particles with hexagonal symmetries).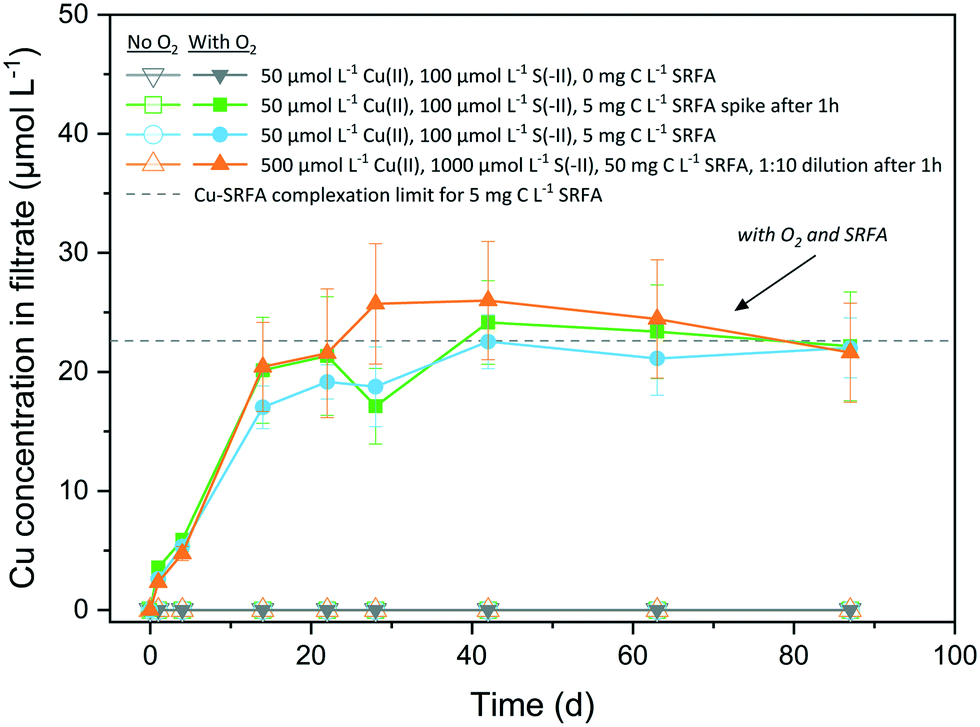 | ||
| Fig. 5 Copper ions released from CuxS nanoparticles under anoxic and oxic conditions over the course of 12.5 weeks. Open symbols and lines denote controls in absence of O2 (n = 3), filled symbols and lines indicate samples with O2 (n = 3–4). Dashed horizontal line marks the Cu–SRFA complexation limit of the system determined via Visual MINTEQ calculations. | ||
In the oxic, SRFA-free media, the particles maintained their integrity owing to the generally very low solubility of CuxS phases, that prevents dissolved O2 to successfully disintegrate the particle surface. The excess of reduced dissolved sulfur species in the reaction medium was most likely oxidized first, before S-terminating surface sites of the CuxS particles underwent oxidation, thus hindering dissolution. Our finding is opposed to the experiments of Sukola et al. (2005), who observed partial oxidative dissolution of CuS in synthetic suspensions that did not contain DOM.95 However, CuS samples were not kept strictly in the dark in their experiments, which could have provoked photochemical reactions facilitating dissolution and would be an explanation for the observed differences. An oxidation of the particle surface would lead to the transformation into a copper hydroxide phase on the surface, which would possess a much higher solubility than any CuxS phase. This may facilitate the dissolution promoted by strong ligands of the fulvic acid that help disintegrate the particle by liberating Cu at defect sites via complexation. Samples that did not contain fulvic acid were lacking this trigger for a ligand-promoted dissolution of the oxidized particle surfaces. The dissolution process continues until the maximum binding capacity of the fulvic acid for Cu(II) is reached, which corresponds to the Cu level of the plateau in the dissolution data. Depending on the batch variant taken for calculation, the half-life of CuxS was determined to be 27–32 d, which agreed well with the half-life of 22 d found for CuS in natural river water containing NOM, too.25 In natural waters, dissolution of CuxS could be influenced by its spatial residence as UV-light absorption by abundant aquatic species (DOM, NO3− and Fe(III)-complexes) leads to the formation of reactive oxygen species (ROS) in the upper layers of surface waters, which can take part in the oxidative dissolution process.96 In addition, several studies have identified oxic–anoxic interfaces in natural environments as hotspots for dark HO˙ formation when DOM and metal-based reductants (such as CuS) are present.97,98 A recent study even observed the formation of ROS in the dark in oxic solutions containing Cl− ions, SRFA and Cu(II), thus interfering in the redox cycle of such systems.99 Furthermore, the presence of different NOM types that exhibit increased binding affinities towards Cu(II), such as humic acids,40,42,100 could enhance CuxS dissolution.
4. Environmental implications
In this study, we demonstrated that fulvic acid as a NOM representative influences the formation of CuxS colloids by altering their nanoparticle size and structural development. Fulvic acid hampered the growth of the CuxS particles so that their sizes remained smaller than in fulvic acid free suspensions. Herein, the absolute concentrations of Cu and S had a substantial effect on the temporal evolution of CuxS particle development, resulting in morphologically more mature particles at higher reactant concentrations. Additionally, we observed a remarkable colloidal stability of CuxS suspensions at near neutral pH values relevant to natural or farmed wetland systems.10 Concerning oxic conditions, NOM acted as a key driver in the oxidative dissolution of CuxS nanoparticles.The growth-suppressing properties of NOM impact the potential mobility of small (<10 nm) CuxS nanoparticles forming at concentrations typical to soil pore waters, and also restrict size of particles precipitating from heavily contaminated sites with elevated Cu concentrations. This results in a high potential mobility of CuxS nanoparticles. Our findings suggest that CuxS will not grow into aggregated microparticles in natural environments, even at elevated Cu and bisulfide concentrations. Due to the strongly negatively charged surfaces resulting in an extraordinary long-term colloidal stability, CuxS is expected to contribute to the long-range transport of Cu, especially in NOM-rich, oxygen-depleted waters. In addition, our results provide strong evidence that Cu(II) is released from CuxS nanoparticles in NOM-rich oxic aquatic environments or upon aeration of (fertilized) wetland soils.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the Scientific Center for Optical and Electron Microscopy (Scope M) at ETH Zurich for providing access to their microscopes. Further, we thank Kurt Barmettler and Brian Sinnet for their support in the laboratories at ETH Zurich and EAWAG. This research was financially supported by the Swiss National Science Foundation under the grant No. 200021_156392.References
- K. A. Hudson-Edwards, M. G. Macklin, C. D. Curtis and D. J. Vaughan, Processes of formation and distribution of Pb-, Zn-, Cd-, and Cu-bearing minerals in the Tyne Basin, Northeast England: Implications for metal-contaminated river systems, Environ. Sci. Technol., 1996, 30, 72–80 CrossRef CAS.
- D. Wallschläger, M. V. M. Desai, M. Spengler and R.-D. Wilken, Mercury speciation in floodplain soils and sediments along a contaminated river transect, J. Environ. Qual., 1998, 27, 1034–1044 CrossRef.
- A. J. P. Smolders, R. A. C. Lock, G. Van der Velde, R. I. Medina Hoyos and J. G. M. Roelofs, Effects of mining activities on heavy metal concentrations in water, sediment, and macroinvertebrates in different reaches of the Pilcomayo River, South America, Arch. Environ. Contam. Toxicol., 2003, 44, 0314–0323 CrossRef CAS PubMed.
- R. W. Simmons, P. Pongsakul, D. Saiyasitpanich and S. Klinphoklap, Elevated levels of cadmium and zinc in paddy soils and elevated levels of cadmium in rice grain downstream of a zinc mineralized area in Thailand: Implications for public health, Environ. Geochem. Health, 2005, 27, 501–511 CrossRef CAS PubMed.
- P. Byrne, P. Wood and I. Reid, The impairment of river systems by metal mine contamination: A review including remediation options, Crit. Rev. Environ. Sci. Technol., 2012, 42, 2017–2077 CrossRef CAS.
- D. Kossoff, W. E. Dubbin, M. Alfredsson, S. J. Edwards, M. G. Macklin and K. A. Hudson-Edwards, Mine tailings dams: Characteristics, failure, environmental impacts, and remediation, Appl. Geochem., 2014, 51, 229–245 CrossRef CAS.
- M. Simmler, E. Suess, I. Christl, T. Kotsev and R. Kretzschmar, Soil-to-plant transfer of arsenic and phosphorus along a contamination gradient in the mining-impacted Ogosta River floodplain, Sci. Total Environ., 2016, 572, 742–754 CrossRef CAS PubMed.
- J. T. Trevors and C. M. Cotter, Copper toxicity and uptake in microorganisms, J. Ind. Microbiol., 1990, 6, 77–84 CrossRef CAS.
- D. G. Ellingsen, L. B. Møller and J. Aaseth, Chapter 35 - Copper in Handbook on the Toxicology of Metals, ed. G. F. Nordberg, B. A. Fowler and M. Nordberg, Academic Press, San Diego, 4th edn, 2015, pp. 765–786 Search PubMed.
- G. Kirk, The biogeochemistry of submerged soils, John Wiley & Sons, 2004 Search PubMed.
- K. R. Reddy and R. D. DeLaune, Biogeochemistry of wetlands: science and applications, CRC Press, 1st edn, 2008 Search PubMed.
- T. Borch, R. Kretzschmar, A. Kappler, P. Van Cappellen, M. Ginder-Vogel, A. Voegelin and K. Campbell, Biogeochemical redox processes and their impact on contaminant dynamics, Environ. Sci. Technol., 2010, 44, 15–23 CrossRef CAS PubMed.
- F.-A. Weber, A. Voegelin, R. Kaegi and R. Kretzschmar, Contaminant mobilization by metallic copper and metal sulphide colloids in flooded soil, Nat. Geosci., 2009, 2, 267–271 CrossRef CAS.
- A. F. Hofacker, A. Voegelin, R. Kaegi, F.-A. Weber and R. Kretzschmar, Temperature-dependent formation of metallic copper and metal sulfide nanoparticles during flooding of a contaminated soil, Geochim. Cosmochim. Acta, 2013, 103, 316–332 CrossRef CAS.
- A. F. Hofacker, A. Voegelin, R. Kaegi and R. Kretzschmar, Mercury mobilization in a flooded soil by incorporation into metallic copper and metal sulfide nanoparticles, Environ. Sci. Technol., 2013, 47, 7739–7746 CrossRef CAS PubMed.
- C. H. Gammons and A. K. Frandsen, Fate and transport of metals in H2S-rich waters at a treatment wetland, Geochem. Trans., 2001, 2, 1–15 CrossRef.
- G. W. Luther III, B. Glazer, S. Ma, R. Trouwborst, B. R. Shultz, G. Druschel and C. Kraiya, Iron and sulfur chemistry in a stratified lake: Evidence for iron-rich sulfide complexes, Aquat. Geochem., 2003, 9, 87–110 CrossRef.
- M. O. Barnett, L. A. Harris, R. R. Turner, R. J. Stevenson, T. J. Henson, R. C. Melton and D. P. Hoffman, Formation of mercuric sulfide in soil, Environ. Sci. Technol., 1997, 31, 3037–3043 CrossRef CAS.
- G. Du Laing, J. Rinklebe, B. Vandecasteele, E. Meers and F. M. Tack, Trace metal behaviour in estuarine and riverine floodplain soils and sediments: A review, Sci. Total Environ., 2009, 407, 3972–3985 CrossRef CAS PubMed.
- S. M. Theberge and G. W. Luther III, Determination of the electrochemical properties of a soluble aqueous FeS species present in sulfidic solutions, Aquat. Geochem., 1997, 3, 191–211 CrossRef CAS.
- G. W. Luther III, S. M. Theberge and D. T. Rickard, Evidence for aqueous clusters as intermediates during zinc sulfide formation, Geochim. Cosmochim. Acta, 1999, 63, 3159–3169 CrossRef.
- M. Labrenz, G. K. Druschel, T. Thomsen-Ebert, B. Gilbert, S. A. Welch, K. M. Kemner, G. A. Logan, R. E. Summons, G. D. Stasio, P. L. Bond, B. Lai, S. D. Kelly and J. F. Banfield, Formation of sphalerite (ZnS) deposits in natural biofilms of sulfate-reducing bacteria, Science, 2000, 290, 1744–1747 CrossRef CAS PubMed.
- G. Druschel, M. Labrenz, T. Thomsen-Ebert, D. Fowle and J. Banfield, Geochemical modeling of ZnS in biofilms: An example of ore depositional processes, Econ. Geol., 2002, 97, 1319–1329 CrossRef CAS.
- J. W. Moreau, R. I. Webb and J. F. Banfield, Ultrastructure, aggregation-state, and crystal growth of biogenic nanocrystalline sphalerite and wurtzite, Am. Mineral., 2004, 89, 950–960 CrossRef CAS.
- T. F. Rozan, G. Benoit and G. W. Luther III, Measuring metal sulfide complexes in oxic river waters with square wave voltammetry, Environ. Sci. Technol., 1999, 33, 3021–3026 CrossRef CAS.
- G. W. Luther III and D. T. Rickard, Metal sulfide cluster complexes and their biogeochemical importance in the environment, J. Nano Res., 2005, 7, 389–407 CrossRef.
- T. F. Rozan, M. E. Lassman, D. P. Ridge and G. W. Luther III, Evidence for iron, copper and zinc complexation as multinuclear sulphide clusters in oxic rivers, Nature, 2000, 406, 879–882 CrossRef CAS PubMed.
- T. F. Rozan and G. Benoit, Geochemical factors controlling free Cu ion concentrations in river water, Geochim. Cosmochim. Acta, 1999, 63, 3311–3319 CrossRef CAS.
- M. Elimelech and C. R. O'Melia, Kinetics of deposition of colloidal particles in porous media, Environ. Sci. Technol., 1990, 24, 1528–1536 CrossRef CAS.
- C. L. Tiller and C. R. O'Melia, Natural organic matter and colloidal stability: Models and measurements, Colloids Surf., A, 1993, 73, 89–102 CrossRef CAS.
- M. Hassellöv and R. Kaegi, Analysis and characterization of manufactured nanoparticles in aquatic environments in Environmental and Human Health Impacts of Nanotechnology, John Wiley & Sons, Ltd., 2009, pp. 211–266 Search PubMed.
- S. Wagner, A. Gondikas, E. Neubauer, T. Hofmann and F. von der Kammer, Spot the difference: engineered and natural nanoparticles in the environment—release, behavior, and fate, Angew. Chem., Int. Ed., 2014, 53, 12398–12419 CAS.
- P. Christian, F. von der Kammer, M. Baalousha and T. Hofmann, Nanoparticles: structure, properties, preparation and behaviour in environmental media, Ecotoxicology, 2008, 17, 326–343 CrossRef CAS PubMed.
- M. F. Hochella, S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Nanominerals, mineral nanoparticles, and earth systems, Science, 2008, 319, 1631–1635 CrossRef CAS PubMed.
- K. Donaldson, V. Stone, C. L. Tran, W. Kreyling and P. J. A. Borm, Nanotoxicology, Occup. Environ. Med., 2004, 61, 727–728 CrossRef CAS PubMed.
- C. Gustafsson and P. M. Gschwend, Aquatic colloids: Concepts, definitions, and current challenges, Limnol. Oceanogr., 1997, 42, 519–528 CrossRef.
- J. R. Lead and K. J. Wilkinson, Aquatic colloids and nanoparticles: Current knowledge and future trends, Environ. Chem., 2006, 3, 159–171 CrossRef CAS.
- G. Cornelis, L. Pang, C. Doolette, J. K. Kirby and M. J. McLaughlin, Transport of silver nanoparticles in saturated columns of natural soils, Sci. Total Environ., 2013, 463–464, 120–130 CrossRef CAS PubMed.
- F. Maurer, I. Christl and R. Kretzschmar, Reduction and reoxidation of humic acid: Influence on spectroscopic properties and proton binding, Environ. Sci. Technol., 2010, 44, 5787–5792 CrossRef CAS PubMed.
- K. Murray and P. W. Linder, Fulvic acids: structure and metal binding. II. Predominant metal binding sites, J. Soil Sci., 1984, 35, 217–222 CrossRef CAS.
- F. J. Stevenson and Y. Chen, Stability constants of copper(II)-humate complexes determined by modified potentiometric titration, Soil Sci. Soc. Am. J., 1991, 55, 1586–1591 CrossRef CAS.
- I. Christl, C. J. Milne, D. G. Kinniburgh and R. Kretzschmar, Relating ion binding by fulvic and humic acids to chemical composition and molecular size. 2. Metal binding, Environ. Sci. Technol., 2001, 35, 2512–2517 CrossRef CAS PubMed.
- F.-A. Weber, A. Voegelin and R. Kretzschmar, Multi-metal contaminant dynamics in temporarily flooded soil under sulfate limitation, Geochim. Cosmochim. Acta, 2009, 73, 5513–5527 CrossRef CAS.
- Y. Zhang, Y. Chen, P. Westerhoff and J. Crittenden, Impact of natural organic matter and divalent cations on the stability of aqueous nanoparticles, Water Res., 2009, 43, 4249–4257 CrossRef CAS PubMed.
- J. Labille, J. Feng, C. Botta, D. Borschneck, M. Sammut, M. Cabie, M. Auffan, J. Rose and J.-Y. Bottero, Aging of TiO2 nanocomposites used in sunscreen. Dispersion and fate of the degradation products in aqueous environment, Environ. Pollut., 2010, 158, 3482–3489 CrossRef CAS PubMed.
- J. T. K. Quik, I. Lynch, K. V. Hoecke, C. J. H. Miermans, K. A. C. D. Schamphelaere, C. R. Janssen, K. A. Dawson, M. A. C. Stuart and D. V. D. Meent, Effect of natural organic matter on cerium dioxide nanoparticles settling in model fresh water, Chemosphere, 2010, 81, 711–715 CrossRef CAS PubMed.
- H. Hyung, J. D. Fortner, J. B. Hughes and J.-H. Kim, Natural organic matter stabilizes carbon nanotubes in the aqueous phase, Environ. Sci. Technol., 2007, 41, 179–184 CrossRef CAS PubMed.
- A. A. Keller, H. Wang, D. Zhou, H. S. Lenihan, G. Cherr, B. J. Cardinale, R. Miller and Z. Ji, Stability and aggregation of metal oxide nanoparticles in natural aqueous matrices, Environ. Sci. Technol., 2010, 44, 1962–1967 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Interaction of fullerene (C60) nanoparticles with humic acid and alginate coated silica surfaces: Measurements, mechanisms, and environmental implications, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS PubMed.
- R. F. Domingos, N. Tufenkji and K. J. Wilkinson, Aggregation of titanium dioxide nanoparticles: Role of a fulvic acid, Environ. Sci. Technol., 2009, 43, 1282–1286 CrossRef CAS PubMed.
- A. Deonarine and H. Hsu-Kim, Precipitation of mercuric sulfide nanoparticles in NOM-containing water: Implications for the natural environment, Environ. Sci. Technol., 2009, 43, 2368–2373 CrossRef CAS PubMed.
- A. J. Slowey, Rate of formation and dissolution of mercury sulfide nanoparticles: The dual role of natural organic matter, Geochim. Cosmochim. Acta, 2010, 74, 4693–4708 CrossRef CAS.
- A. Deonarine, B. L. Lau, G. R. Aiken, J. N. Ryan and H. Hsu-Kim, Effects of humic substances on precipitation and aggregation of zinc sulfide nanoparticles, Environ. Sci. Technol., 2011, 45, 3217–3223 CrossRef CAS PubMed.
- A. Philippe and G. E. Schaumann, Interactions of dissolved organic matter with natural and engineered inorganic colloids: A review, Environ. Sci. Technol., 2014, 48, 8946–8962 CrossRef CAS PubMed.
- G. Kaptay, On the size and shape dependence of the solubility of nano-particles in solutions, Int. J. Pharm., 2012, 430, 253–257 CrossRef CAS PubMed.
- J. Sun, F. Wang, Y. Sui, Z. She, W. Zhai, C. Wang and Y. Deng, Effect of particle size on solubility, dissolution rate, and oral bioavailability: Evaluation using coenzyme Q10 as naked nanocrystals, Int. J. Nanomed., 2012, 7, 5733–5744 CAS.
- M. E. Essington, Soil and water chemistry: an integrative approach, CRC press, 2015 Search PubMed.
- J. S. Waples, K. L. Nagy, G. R. Aiken and J. N. Ryan, Dissolution of cinnabar (HgS) in the presence of natural organic matter, Geochim. Cosmochim. Acta, 2005, 69, 1575–1588 CrossRef CAS.
- M. Ravichandran, G. R. Aiken, M. M. Reddy and J. N. Ryan, Enhanced dissolution of cinnabar (mercuric sulfide) by dissolved organic matter isolated from the Florida Everglades, Environ. Sci. Technol., 1998, 32, 3305–3311 CrossRef CAS.
- R. Ma, J. Stegemeier, C. Levard, J. G. Dale, C. W. Noack, T. Yang, G. E. Brown and G. V. Lowry, Sulfidation of copper oxide nanoparticles and properties of resulting copper sulfide, Environ. Sci.: Nano, 2014, 1, 347–357 RSC.
- K. Y. Chen and J. C. Morris, Kinetics of oxidation of aqueous sulfide by oxygen, Environ. Sci. Technol., 1972, 6, 529–537 CrossRef CAS.
- BBI solutions, U. K., Gold nanoparticle specifications, https://www.bbisolutions.com/technical-support/gold/nanoparticles/gold-nanoparticle-specifications.html, (accessed 19.11. 2019).
- J. P. Gustafsson, Visual MINTEQ ver. 3.1, 2013, https://vminteq.lwr.kth.se/, (accessed 25.11.2019) Search PubMed.
- X.-X. Zhou, J.-F. Liu and G.-B. Jiang, Elemental mass size distribution for characterization, quantification and identification of trace nanoparticles in serum and environmental waters, Environ. Sci. Technol., 2017, 51, 3892–3901 CrossRef CAS PubMed.
- T. Wagner, ij-particlesizer: Ver. 1.0.7, 2016, DOI:10.5281/zenodo.163568.
- W. Lou, M. Chen, X. Wang and W. Liu, Size Control of Monodisperse Copper Sulfide Faceted Nanocrystals and Triangular Nanoplates, J. Phys. Chem. C, 2007, 111, 9658–9663 CrossRef CAS.
- Y. Liu, D. Qin, L. Wang and Y. Cao, A facile solution route to CuS hexagonal nanoplatelets, Mater. Chem. Phys., 2007, 102, 201–206 CrossRef CAS.
- J. Bebie, M. A. Schoonen, M. Fuhrmann and D. R. Strongin, Surface charge development on transition metal sulfides: An electrokinetic study, Geochim. Cosmochim. Acta, 1998, 62, 633–642 CrossRef CAS.
- M. J. Dekkers and M. A. Schoonen, An electrokinetic study of synthetic greigite and pyrrhotite, Geochim. Cosmochim. Acta, 1994, 58, 4147–4153 CrossRef CAS.
- Y. Nicolau and J. Menard, An electrokinetic study of ZnS and CdS surface chemistry, J. Colloid Interface Sci., 1992, 148, 551–570 CrossRef CAS.
- J. D. G. Durán, M. C. Guindo, A. V. Delgado and F. González-Caballero, Surface chemical analysis and electrokinetic properties of synthetic spherical mixed zinc–cadmium sulfides, J. Colloid Interface Sci., 1997, 193, 223–233 CrossRef PubMed.
- J. Liu and C. Huang, Electrokinetic characteristics of some metal sulfide-water interfaces, Langmuir, 1992, 8, 1851–1856 CrossRef CAS.
- R. A. D. Pattrick, J. F. W. Mosselmans, J. M. Charnock, K. E. R. England, G. R. Helz, C. D. Garner and D. J. Vaughan, The structure of amorphous copper sulfide precipitates: An X-ray absorption study, Geochim. Cosmochim. Acta, 1997, 61, 2023–2036 CrossRef CAS.
- R. J. Goble, The relationship between crystal structure, bonding and cell dimensions in the copper sulfides, Can. Mineral., 1985, 23, 61–76 CAS.
- G. W. Luther III, Metal sulfides in the environment and in bioinorganic chemistry in Inorganic Chemistry for Geochemistry and Environmental Sciences, John Wiley & Sons, Ltd, 2016, pp. 390–405 Search PubMed.
- G. W. Luther III, S. M. Theberge, T. F. Rozan, D. Rickard, C. Rowlands and A. Oldroyd, Aqueous copper sulfide clusters as intermediates during copper sulfide formation, Environ. Sci. Technol., 2002, 36, 394–402 CrossRef PubMed.
- D. J. Vaughan and J. A. Tossel, The chemical bond and the properties of sulfide minerals; I, Zn, Fe and Cu in tetrahedral and triangular coordinations with sulfur, Can. Mineral., 1980, 18, 157–163 CAS.
- A. Pollard, R. Thomas and P. Williams, Synthesis and stabilities of the basic copper (II) chlorides atacamite, paratacamite and botallackite, Min. Mag., 1989, 53, 557–563 CrossRef CAS.
- J. W. Mullin, Crystallization, Elsevier, 2001 Search PubMed.
- X. Peng, J. Wickham and A. P. Alivisatos, Kinetics of II-VI and III-V colloidal semiconductor nanocrystal growth: “Focusing” of size distributions, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS.
- Z. A. Peng and X. Peng, Mechanisms of the shape evolution of CdSe nanocrystals, J. Am. Chem. Soc., 2001, 123, 1389–1395 CrossRef CAS.
- Y. Du, Z. Yin, J. Zhu, X. Huang, X.-J. Wu, Z. Zeng, Q. Yan and H. Zhang, A general method for the large-scale synthesis of uniform ultrathin metal sulphide nanocrystals, Nat. Commun., 2012, 3, 1177 CrossRef PubMed.
- E. J. H. Lee, C. Ribeiro, E. Longo and E. R. Leite, Oriented attachment: An effective mechanism in the formation of anisotropic nanocrystals, J. Phys. Chem. B, 2005, 109, 20842–20846 CrossRef CAS PubMed.
- K. Ding, J. Zeng, L. Jing, R. Qiao, C. Liu, M. Jiao, Z. Li and M. Gao, Aqueous synthesis of PEGylated copper sulfide nanoparticles for photoacoustic imaging of tumors, Nanoscale, 2015, 7, 11075–11081 RSC.
- A. L. Brazeau and N. D. Jones, Growth mechanisms in nanocrystalline lead sulfide by stopped-flow kinetic analysis, J. Phys. Chem. C, 2009, 113, 20246–20251 CrossRef CAS.
- C. A. Johnson, Generalization of the Gibbs-Thomson equation, Surf. Sci., 1965, 3, 429–444 CrossRef.
- P. G. Vekilov, Nucleation, Cryst. Growth Des., 2010, 10, 5007–5019 CrossRef CAS PubMed.
- D. Kashchiev, P. G. Vekilov and A. B. Kolomeisky, Kinetics of two-step nucleation of crystals, J. Chem. Phys., 2005, 122, 244706 CrossRef PubMed.
- J. J. De Yoreo and P. G. Vekilov, Principles of Crystal Nucleation and Growth, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS.
- J. C. Masini, G. Abate, E. C. Lima, L. C. Hahn, M. S. Nakamura, J. Lichtig and H. R. Nagatomy, Comparison of methodologies for determination of carboxylic and phenolic groups in humic acids, Anal. Chim. Acta, 1998, 364, 223–233 CrossRef CAS.
- T. Karlsson, P. Persson and U. Skyllberg, Complexation of copper(II) in organic soils and in dissolved organic matter − EXAFS evidence for chelate ring structures, Environ. Sci. Technol., 2006, 40, 2623–2628 CrossRef CAS PubMed.
- J. Yu, Q. Xu, Z. Liu, X. Guo, S. Han, S. Yuan and L. Tong, Morphological characteristics of fulvic acid fractions observed by atomic force microscopy, J. Microsc., 2013, 252, 71–78 CrossRef CAS PubMed.
- B. A. Poulin, C. A. Gerbig, C. S. Kim, J. P. Stegemeier, J. N. Ryan and G. R. Aiken, Effects of sulfide concentration and dissolved organic matter characteristics on the structure of nanocolloidal metacinnabar, Environ. Sci. Technol., 2017, 51, 13133–13142 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, The surface science of nanocrystals, Nat. Mater., 2016, 15, 141 CrossRef CAS PubMed.
- K. Sukola, F. Wang and A. Tessier, Metal-sulfide species in oxic waters, Anal. Chim. Acta, 2005, 528, 183–195 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic chemistry: chemical equilibria and rates in natural waters, John Wiley & Sons, 1996 Search PubMed.
- X. Yuan, P. S. Nico, X. Huang, T. Liu, C. Ulrich, K. H. Williams and J. A. Davis, Production of Hydrogen Peroxide in Groundwater at Rifle, Colorado, Environ. Sci. Technol., 2017, 51, 7881–7891 CrossRef CAS PubMed.
- P. Liao, Y. Liang and Z. Shi, Impact of Divalent Cations on Dark Production of Hydroxyl Radicals from Oxygenation of Reduced Humic Acids at Anoxic–Oxic Interfaces, ACS Earth Space Chem., 2019, 3, 484–494 CrossRef CAS.
- G. Xing, S. Garg, C. J. Miller, A. N. Pham and T. D. Waite, Effect of Chloride and Suwannee River Fulvic Acid on Cu Speciation: Implications to Cu Redox Transformations in Simulated Natural Waters, Environ. Sci. Technol., 2020, 54, 2334–2343 CrossRef CAS PubMed.
- K. Murray and P. W. Linder, Fulvic acids: structure and metal binding. I. A random molecular model, J. Soil Sci., 1983, 34, 511–523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9en01448a |
| This journal is © The Royal Society of Chemistry 2020 |