
Coupled morphological and structural evolution of δ-MnO2 to α-MnO2 through multistage oriented assembly processes: the role of Mn(III)†
Xinran
Liang
 ab,
Jeffrey E.
Post
c,
Bruno
Lanson
d,
Xiaoming
Wang
a,
Mengqiang
Zhu
e,
Fan
Liu
a,
Wenfeng
Tan
a,
Xionghan
Feng
*a,
Guomin
Zhu
b,
Xin
Zhang
b and
James. J.
De Yoreo
b
ab,
Jeffrey E.
Post
c,
Bruno
Lanson
d,
Xiaoming
Wang
a,
Mengqiang
Zhu
e,
Fan
Liu
a,
Wenfeng
Tan
a,
Xionghan
Feng
*a,
Guomin
Zhu
b,
Xin
Zhang
b and
James. J.
De Yoreo
b
aKey Laboratory of Arable Land Conservation (Middle and Lower Reaches of Yangtze River), Ministry of Agriculture, College of Resources and Environment, Huazhong Agricultural University, Wuhan 430070, China. E-mail: fxh73@mail.hzau.edu.cn
bPhysical Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99352, USA
cDepartment of Mineral Sciences, Smithsonian Institution, Washington, DC 20013, USA
dUniversity of Grenoble Alpes, CNRS, ISTerre, F-38000 Grenoble, France
eDepartment of Ecosystem Science and Management, University of Wyoming, Laramie, WY 82071, USA
First published on 21st November 2019
Abstract
α-MnO2 is a typical tunneled Mn oxide (TMO) that is frequently associated with δ-MnO2 in the environment and exhibits strong adsorption and oxidation activity. The morphology of α-MnO2, which is controlled by an oriented attachment (OA) process, is one of the key factors affecting its reactivity. However, the detailed crystal growth process and coupling between morphology and structure of α-MnO2 during OA processes remain poorly understood. We propose that the transformation of layer-based δ-MnO2 to tunnel-based α-MnO2 occurs via a multistage OA process. In the initial stage, the produced δ-MnO2 nanoflakes are found to spontaneously self-assemble into a nanoribbon with a large number of lattice defects via edge-to-edge OA. The presence of lattice defects promotes the generation of oxygen vacancies, and the Mn(IV) ions in the [MnO6] octahedral layers of δ-MnO2 are reduced to Mn(III)/Mn(II). The reduced ions subsequently migrate from the [MnO6] octahedral layers to the interlayers during this process. The hydroxide which acts in coordination with the interlayer Mn(III)/Mn(II) and oxygen atoms coordination with adjacent nanoribbons attach to each other driven by hydrogen bonding and form primary nanorods through a face-to-face OA mechanism along the c-axis. Concomitantly, the bonding of [Mn(III)O6] octahedra in the interlayer of the nanoribbons with adjacent [MnO6] octahedral layers leads to the fabrication of a new α-MnO2 tunnel structure from the original δ-MnO2. These findings provide insights into both the transformation mechanisms of the layer-based to the tunnel-based nanoparticles and methods for the efficient and controlled synthesis of nanomaterials.
Environmental significanceδ-MnO2 and α-MnO2 are widespread near and on the earth's surface and play an important role in elemental cycles and pollutant dynamics. These behaviors are closely related to the structure and morphology of the minerals. Although documented in the literature, α-MnO2 nanorod crystal growth processes and the relationship between the structural transformation and morphological evolution are not clearly understood. In this study, the transformation of the layer-based δ-MnO2 to tunnel-based α-MnO2 is found to occur via a multistage OA process. The disordered structure produced by δ-MnO2 nanoflakes via the OA process facilitates the reduction of Mn(IV) in the layer to Mn(III), and Mn(III) migrates out to the interlayer. Interlayer Mn(III) promotes the face-to-face OA of adjacent nanoribbons to form a nanorod and to form a 2 × 2 α-MnO2 tunnel structure as tunnel “walls”. These findings provide insights into not only the transformation mechanisms of layer-based to tunnel-based nanoparticles in the environment but also the efficient and controlled synthesis of environmentally friendly materials. |
Introduction
Oriented attachment (OA) is a nonclassical mechanism of crystal growth, and it proceeds by repeated attachment events of crystalline particles on specific crystal faces that are lattice-matched with true crystallographic alignment.1 The growth of many crystalline substances, including engineered nanomaterials and natural nanoparticles, has been reported to occur via the OA mechanism.2 The properties of these nanoparticles are closely related to their morphologies and structures. The OA process only leads to morphological evolution, as indicated in previous reports, while the processes of structural changes associated with morphological evolutions have been explored to a lesser extent.1,2 Revealing the relationship between morphological evolution and structural changes during the OA process is not only important for controlling the growth of synthetic materials but also for providing novel ideas for the study of crystal growth.3Recent studies have reported that poor crystallinity layer manganese oxide (LMO) nanoparticles assemble as LMO nanoflowers and tunneled manganese oxide (TMO) nanorods via the OA mechanism.4–8 LMO-to-TMO reaction mechanisms have been extensively investigated because LMO association with TMOs occurs frequently in the environment.5,8–10 In addition, LMOs and TMOs have also been extensively applied as materials in catalysis, ion exchange, energy storage, and octahedral molecular sieves.9,10 There are some detailed studies on their structural evolution and a few studies on the morphological evolution during LMO-to-TMO reactions.8,11 The two separate studies on the structural and morphological evolution indicate that there are some difficulties in understanding their relationship and the reaction mechanism of the LMO-to-TMO process in the environment.
In the natural environment, cryptomelane (α-MnO2) represents an important family of TMOs with 2 × 2 and 1 × 1 (intergrowth little 2 × 3, 2 × 4 and “T” junction structure, etc.) tunnel structures, which are formed when double chains of edge-sharing [MnO6] octahedra share corners with neighboring chains (Fig. S1A†).12,13 The larger tunnels are generally stabilized by K+, Ba+, and Na+.14 Cryptomelane is the major manganese oxide in the supergene oxidation zones of Mn-bearing crusts and manganese deposits and lateritic weathered profiles.15–17 The unique physicochemical properties of α-MnO2 have provided a wide range of potential applications in removing environment pollutions.18–20 These applications are considerably affected by the structure and morphology of α-MnO2.20
δ-MnO2 (Fig. S1B†) is the most common LMO in the environment, with strong adsorption and oxidation activity frequently associated with α-MnO2.11 It was proposed that the formation and migration of Mn(III) occurred while δ-MnO2 transformed to α-MnO2.21 Yuan et al. also reported the transforming process of highly crystalline buserite to todorokite (2 × 3 tunnel structure) through Mn(III) migration by the combination of morphology, structure and theoretical calculation studies.20 Additionally, a detailed OA process based on the formation of long α-MnO2 nanorods from α-MnO2 primary nanorods was also reported.20,22–24 However, the formation process of the primary α-MnO2 nanorods from poor crystallinity δ-MnO2 nanoparticles is still unknown due to the too fast or slow conversion rates under different reaction conditions, which are detrimental to the capture of the transition state.12,13 In addition, it is unclear how the new tunnel structure formation corresponds with the morphological evolution by the OA process. A basic knowledge of the morphological evolution across the entire synthetic process and the relationship with the structural transformation is also critical for α-MnO2 material synthesis in various applications, and the understanding of the minerology and behavior of different types of MnO2 minerals in nature.
In this study, Na+/K+-stabilized α-MnO2, similar to environmental cryptomelane nanorods, was synthesized by co-precipitation of Na/KMnO4 with MnSO4. The dynamic process of the formation of the Na+-stabilized α-MnO2 nanorods was carefully explored. Given that the Na+-stabilized α-MnO2 forms at a slower rate compared to that of K+, as shown in previous experiments, it provides a more suitable system for the investigation of the process of α-MnO2 formation.14 During the synthesis, intermediate products were sampled and quenched in liquid N2 at different time intervals to stop crystal growth and to capture in situ morphologies and structures. These intermediate samples were characterized by using the pair distribution function (PDF), and X-ray photoelectron spectroscopy (XPS), to illustrate the structural changes at the atomic level. Meanwhile, morphological changes during the formation process were revealed with the use of high-resolution transmission electron microscopy (HRTEM) and field-emission scanning electron microscopy (FESEM). Possible mechanisms for the observed relationship between morphological and structural evolutions are proposed.
Experimental section
Synthesis of different types of cations of α-MnO2
In a typical experiment, 8.45 g of MnSO4·H2O (0.5 M) in 100 mL of acetic acid (2 M) and 80 mL of an aqueous solution of AMnO4 (0.4375 M, A = K or Na) were mixed together under vigorous stirring at 60 °C. This suspension was heated to 100 °C for 20 min and was then cooled to 80 °C in open air. After cooling to 80 °C, the suspension was frozen by liquid nitrogen and washed until the conductivity of the supernatant was less than 20.0 μS cm−1; the suspension was then freeze-dried.25After the two solutions were mixed and the suspension was heated to 100 °C, 10 mL of the suspension was extracted into a 50 mL centrifuge tube filled with 20 mL of liquid nitrogen at 0, 2, 4, 6, 8, 10, 14, 16 and 20 min. The extracted samples were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 8 min and the precipitates were washed with 50 mL of DDW multiple times until the conductivity of the supernatant was less than 20.0 μS cm−1.The precipitates were then freeze-dried for characterization. Samples were labelled AMix, A100-X and A80, where X is the heating time at 100 °C and A is the type of cation.
000 rpm for 8 min and the precipitates were washed with 50 mL of DDW multiple times until the conductivity of the supernatant was less than 20.0 μS cm−1.The precipitates were then freeze-dried for characterization. Samples were labelled AMix, A100-X and A80, where X is the heating time at 100 °C and A is the type of cation.
Powder X-ray diffraction (PXRD)
PXRD patterns were collected from the dried powders using a cavity mount on a Bruker D8 Advance diffractometer equipped with a LynxEye detector, using Ni-filtered Cu Kα radiation (λ = 0.15418 nm). The diffractometer was operated at a tube voltage of 40 kV and a current of 40 mA, with a scanning rate of 1° min−1 and a step size of 0.02°.Elemental analysis and Mn AOS
The chemical composition of the samples was determined by dissolving ∼0.1 g of each powder sample in 25 mL of 0.25 mol L−1 NH2OH·HCl. The concentrations of dissolved Mn and K were measured using inductively coupled plasma (ICP) and flame spectrometry, respectively. Mn average oxidation state (AOS) was measured by a back-titration method using a KMnO4 standard solution.26X-ray photoelectron spectroscopy
X-ray photoelectron spectra were collected using a VG Multilab2000 X-ray photoelectron spectrometer with an Al K X-ray source (1486 eV) and a base pressure of 3 × 10−9 Torr in the analytical chamber. The scans were recorded using the large area mode. The survey scans were collected using a fixed pass energy of 100 eV and an energy step size of 1.0 eV, whereas the narrow scans have a pass energy of 25 eV and an energy step size of 0.1 eV. The charge effect was corrected by adjusting the binding energy (BE) of C (1 s) to 284.62 eV. The spectra were analyzed using the Avantage software. The Shirley-type background was subtracted before deconvolution and fitting. The parameters used by Iton et al. for the multiple peaks of Mn (2p3/2) and Mn 3s for spectral fitting were adopted.27 A 20:80 ratio of the Lorentzian:Gaussian mix-sum function was used for all the fittings.
Transmission electron microscopy
The particle size and morphology of the samples were further examined on a JEM-2100F transmission electron microscope and by using FEI Titan 80/300 environmental transmission electron microscopy. Prior to observation, samples were embedded in epoxy resin, left to polymerize for 48 h in the dark and cut with an ultramicrotome (Leica EM UC6) equipped with a diamond knife. The ∼50 nm thick sections were picked up on lacey carbon films loaded on Cu grids. In addition, to assess possible preparation-induced artefacts (use of an ultramicrotome), all samples were prepared according to the above described protocol, filtered and re-suspended in ethanol. A drop of the obtained suspension was deposited on a copper microscope grid covered with perforated carbon.Atomic force microscopy
All of the images were recorded in air using tapping mode on a MultiMode VIII AFM (Bruker, CA). Rfespa-190 probes (Bruker, CA) were used in the experiment. The raw data were further analyzed using NanoScope Analysis v 1.50 offline software (Bruker, CA).Field-emission scanning electron microscopy
Detailed three-dimensional morphologies of particles were observed by using a field emission scanning electron microscope (SU8010, Hitachi) with a maximum resolution of 1 nm. For high-resolution SEM, the microscope was operated at 15 kV and the working distance was 0.5–30 mm; an in-lens secondary electron detector was used. Prior to SEM analysis, each sample was gold-coated.High-energy X-ray total scattering
Synchrotron-based X-ray total scattering data were collected using an X-ray energy of 58.6491 keV (λ = 0.2114 Å) at beamline 11-ID-B of the Advanced Photon Source (APS), Argonne National Laboratory (APS). The measurement was performed using the rapid acquisition PDF method by employing a Perkin Elmer amorphous silicon detector. The image plate was exposed for 1.2 s and the measurement was repeated 75 times for a total collection time of 90 s for each sample. The software Fit2D was used to integrate and convert the 2-D raw data to 1-D intensity versus wave vector (Q) data. The PDF, G(r), was obtained from the raw 1-D data using the program PDFgetX2.Results and discussion
During the synthesis of δ-MnO2, the amount of added MnO4− was in excess relative to the stoichiometric ratio of Mn(II), as shown in eqn (1).| 2MnO4− + 3Mn2+ + 2H2O → 5δ-MnO2 + 4H+ | (1) |
Therefore, the supernatant color changed to purple after the two solutions were mixed. After the solution was heated to 100 °C for 14 min, the color of the supernatant changed from purple to colorless owing to the complete reduction of the MnO4− ions under acidic conditions (pH = 0.86) according to eqn (2).22,28
| 4MnO4− + 4H+ → 4δ-MnO2 + 3O2 + 2H2O | (2) |
Morphological evolution
HRTEM, FESEM, and electron diffraction were used to elucidate the kinetics of the morphological evolution during the phase transformation. After NaMnO4 and MnSO4 were mixed, a three-dimensional morphology of aggregation of approximately 100 nm in width was observed (Fig. S2A†), and primary nanoflakes were observed using HRTEM after ultrasonic dispersion (Fig. 1A and B). This indicates that the primary nanoflakes aggregated loosely driven by the surface energy. The edges of the primary nanoflakes were approximately 3 to 4 nm (Fig. 1B and S4†). The lattice cannot be observed in Fig. 1B owing to the poor crystallinity of the nanoflakes. Furthermore, the selected area diffraction (SAED) pattern (inset of Fig. 1B) shows two diffuse diffraction rings at ∼0.24 nm and ∼0.14 nm, which is consistent with the d(100) = 0.24 nm and d(110) = 0.14 nm spacings of δ-MnO2, respectively. Additionally, the absence of the (001) and (002) diffraction rings in the SAED pattern indicates that at this stage, the poorly crystallized nanoflakes consist of no more than a few [MnO6] octahedral layers. The atomic force microscopy (AFM) results in Fig. 1C show two-dimensional nanoflakes with an average height of approximately 0.35 nm (Fig. 1D), which is consistent with the thickness of a single [MnO6] octahedral layer.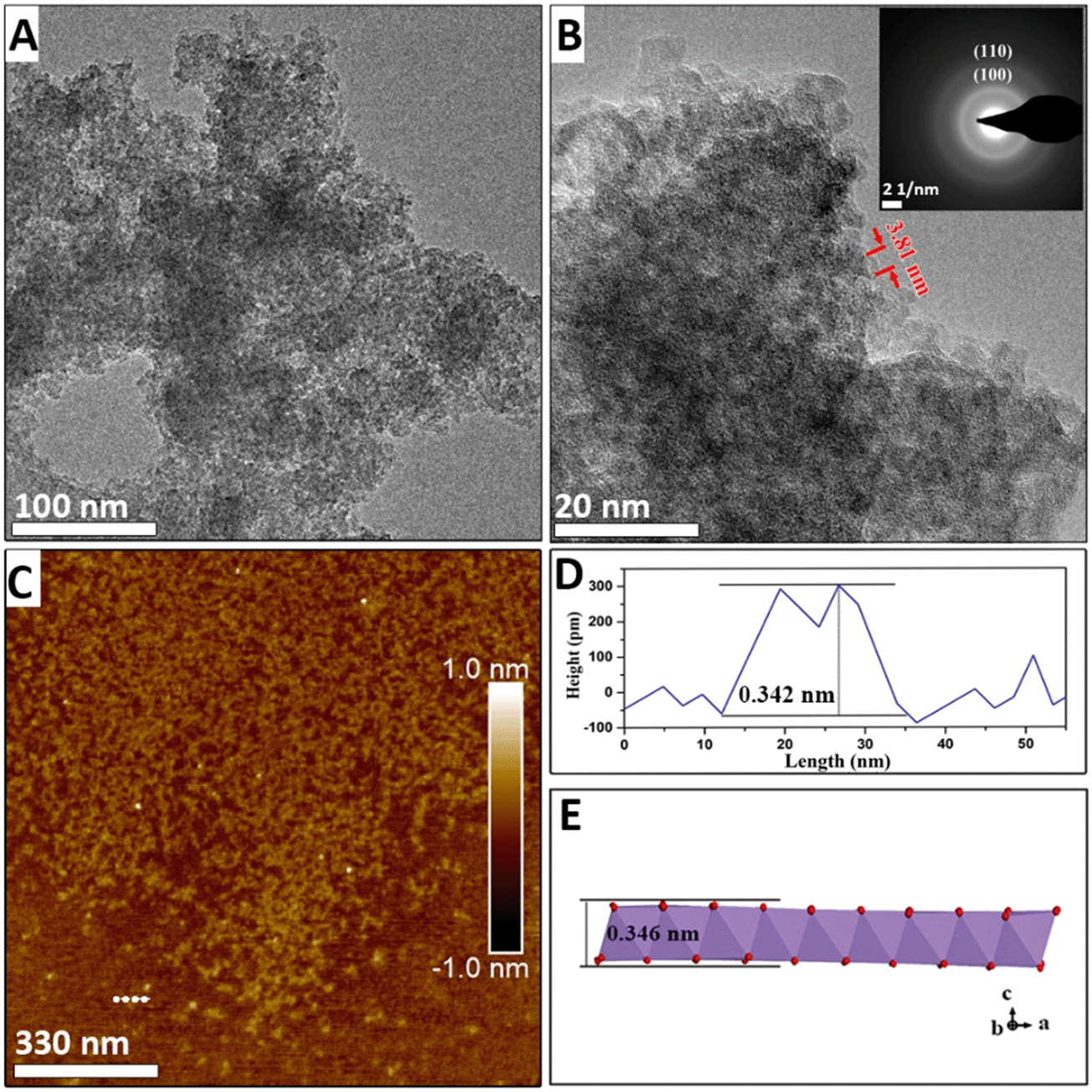 | ||
| Fig. 1 TEM images of intermediate product of NaMix (A and B) and the SAED pattern (B inset) recorded by focusing the electron beam in the area of image B. AFM image (C) of the intermediate product of NaMix. Profile image (D) corresponds to the trajectory shown in Fig. 1C. (E) Schematic of single layer δ-MnO2. | ||
When the suspension is heated for 6 min, nanoribbons appear (with lengths ranging from 20 to 60 nm and widths from 3 to 7 nm, as shown in Fig. 2A and B). In addition, the nanoribbon is aggregated from many nanoflakes, as shown in Fig. 2B and C. A diffraction ring is also observed in the SAED pattern at ∼0.14 nm and ∼0.24 nm and it originates from the (110) and (100) reflections of δ-MnO2 (inset of Fig. 2B). Although the morphology changes, no other diffraction peaks appear. The thickness of the nanoribbon in Fig. 2C is only 0.356 nm, which confirms that the nanoribbons maintain a single-layer δ-MnO2 structure. The assembly of δ-MnO2 at low pH is energetically favored because the primary nanoflakes have high-surface energy, low-Na+ content, and hydroxyls on edge sites that can generate hydrogen bonding with the adjacent nanoflakes.4
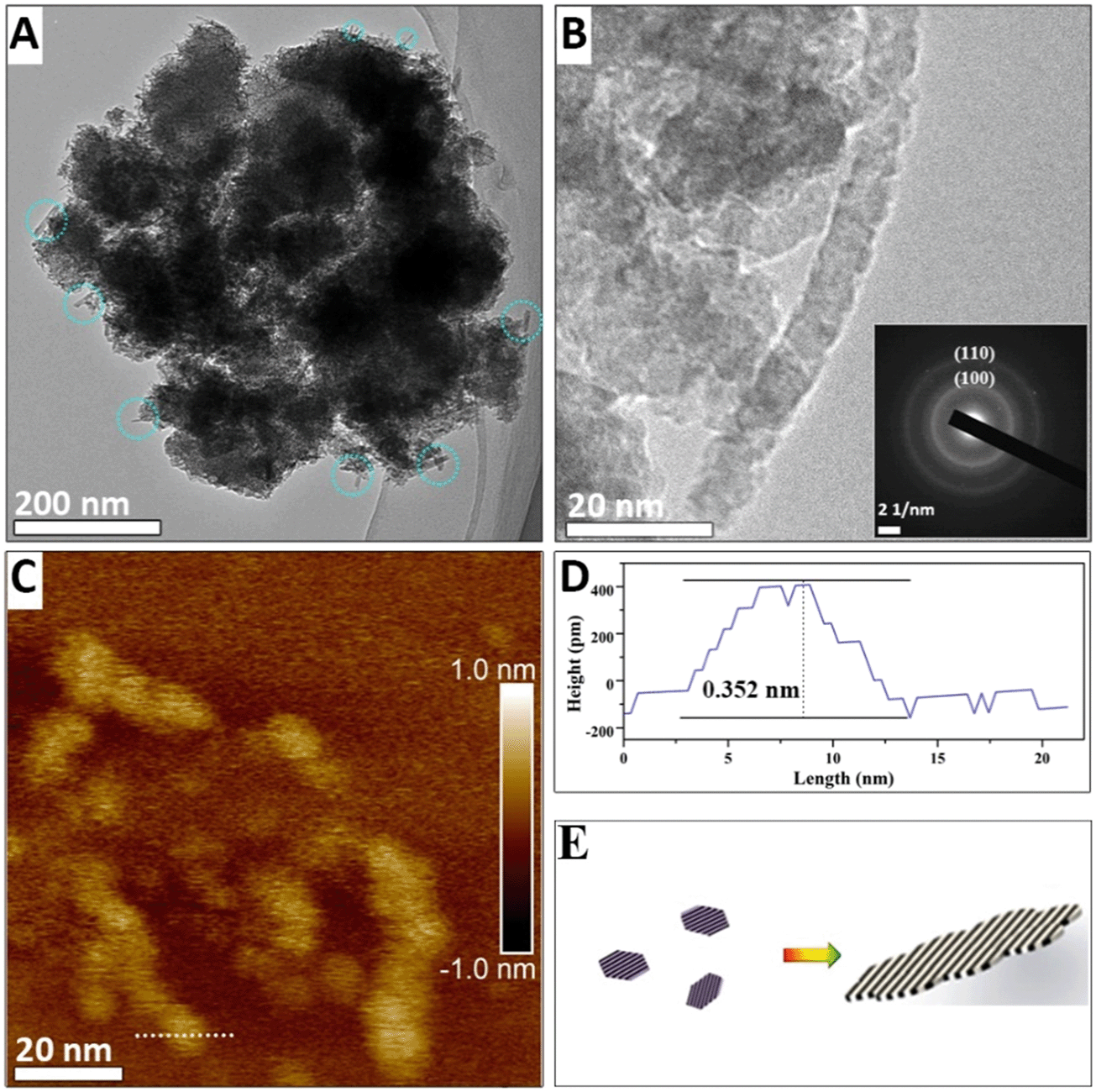 | ||
| Fig. 2 TEM images of intermediate product of Na100-6 (A and B) and the SAED pattern (B inset). The blue circle in image (A) indicates there are some nanoribbons. AFM image of the intermediate product of Na100-6 (C) and height profile of Na100-6. Profile image (D) corresponds to the trajectory shown in Fig. 2C. (E) Schematic of δ-MnO2 assembly process. | ||
When the suspension is heated for 10 min, the width and length of the nanoribbons increase to ∼30 nm and ∼100 nm, respectively (Fig. 3A). HRTEM images show in the sectioning of the nanosheets along the (001) plane in Fig. 3B that nanoribbons are thickened by the assembly and form nanorods with a thickness of ∼7 nm (the sectioning schematic along the (001) plane is shown in Fig. S3A†). The measured dhkl spacing of 0.69 nm corresponds to the (110) plane of α-MnO2 in Fig. 3D. The observed nanorods exhibit different sizes and crystallographic orientations owing to the assembly of multiple primary nanorods (Fig. 3B). The nanoribbons are cut off in the [110] direction (as shown in Fig. 3C, and the cutting schematic along the (001) plane is shown in Fig. S3B†). It is shown that the length of the nanoribbons is ∼40 nm. However, the observed thickness of the nanoribbons (2.24 nm) corresponds to only three [MnO6] octahedral layers. The top [MnO6] octahedral layer assembled on the nanosheet has a d spacing of 1.00 nm, which is larger than the d(110) value of the lower layer (Fig. 3C). Furthermore, the size of this layer shown in blue squares in Fig. 3C is 6.7 nm, which corresponds to the typical lateral dimensions of the nanoflakes, and indicates that the nanoribbons thicken by the face-to-face assembling of the nanoflakes. The AFM image in Fig. S5B† also reveals the assembly of the nanoflakes on the nanoribbons. The blue circles in Fig. 3C mark the dislocations along the [001] and [110] directions. Because the dislocations are a common remnant of attachment events, these may indicate the attachment of two small nanorods assembled by both end-to-end (along the (001) plane) and side-to-side OA (along the (110) plane).20 HRTEM shows that the nanorods have serrated sides that may form during the assembly of the nanoflakes (Fig. 3D).
 | ||
| Fig. 3 TEM (A) image of the intermediate Na100-10. (B) High-resolution transmission electron microscopy (HRTEM) image of a primary α-MnO2 nanorod cross-section viewed along the [001] zone axis. (C) HRTEM image of the δ-MnO2 nanoribbon cross-section viewed along the [110] zone axis. The blue circle indicates nanoflakes assembled along the (100) plane. (D) HRTEM image of nanorods. (E and F) Schematics of HRTEM images in (B) and (C), respectively. | ||
After the suspension is heated for 14 min, large numbers of nanorods appear, coincident with the disappearance of the δ-MnO2 nanoflakes (Fig. 4A). The widths of the α-MnO2 nanorods with better crystallinity, as shown in Fig. 4B, range from 8 to 10 nm. There are two nanorods that assemble at one end to form a continuous lattice along the (110) plane (Fig. 4C). The number of lattice dislocations observed at the attached interface (inset I in Fig. 4C) is higher than that of a single nanorod (inset II in Fig. 4C). The blue square in Fig. 4C indicates that two nanorods separate to form a gap at one end. This process has been extensively reported for the growth of α-MnO2 nanorods based on the side-to-side OA process.2,11,20,29,30
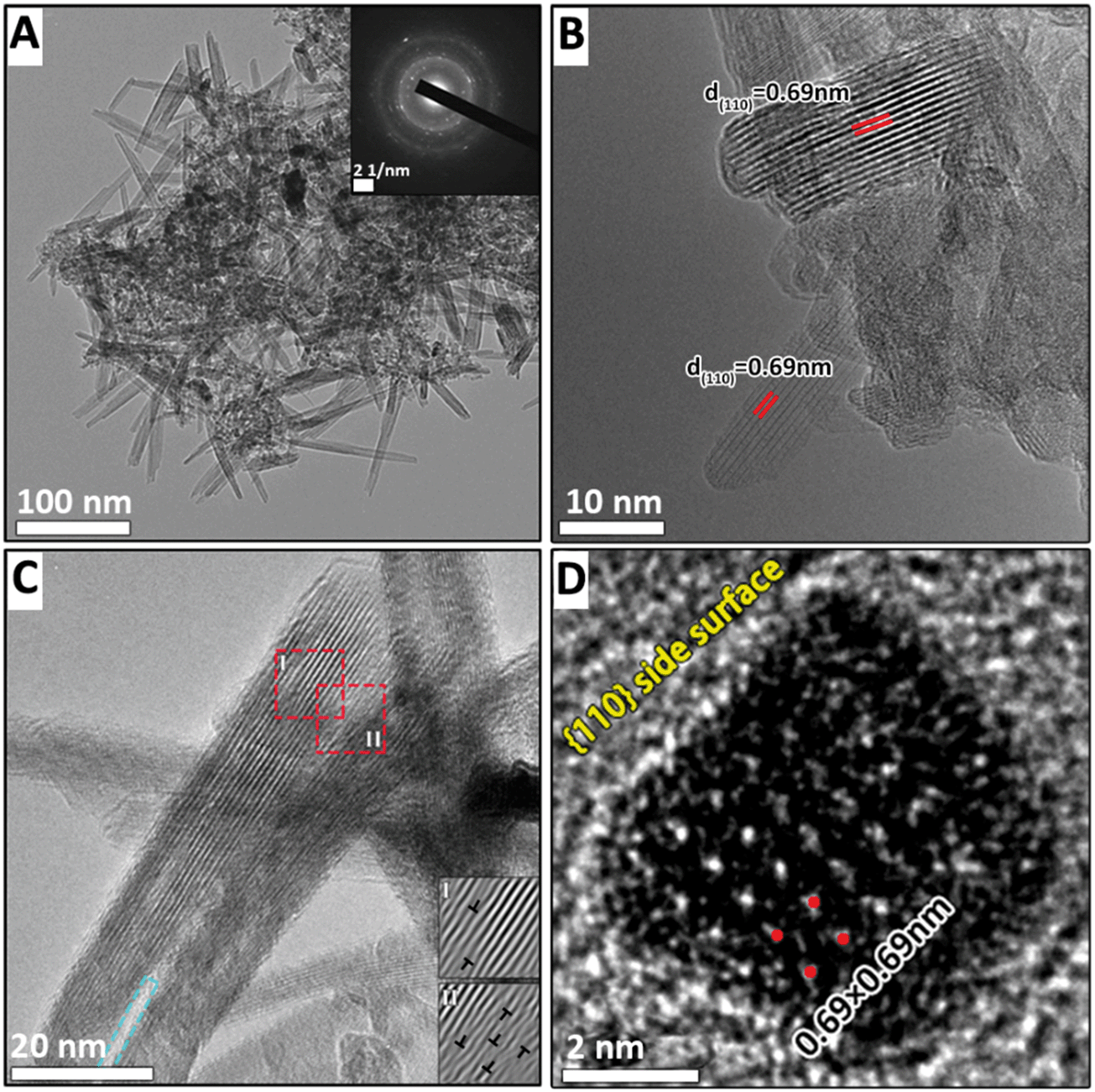 | ||
| Fig. 4 TEM (A) image of the intermediate Na100-14. (B) HRTEM image of nanorods. (C) HRTEM images show nanorods assemble along the (110) plane. The blue rectangle indicates that there is a gap between two nanorods. Fourier filtered images of zone I and zone II using red area outlined in (C), highlighting the defects. (D) An image of a nanorod cross section viewed along the [001] zone axis. | ||
After the final heating stage, the widths and lengths of nanorods increase to ∼40 nm and ∼400 nm, respectively (Fig. 5A and B). The serrated sides and internal lattice dislocations of nanorods shown in Fig. 5C have almost disappeared owing to the aging process. However, the overlap of individual sheets is still visible at the [001] ends of the rods (Fig. 5C).
 | ||
| Fig. 5 FESEM image of product of Na80 (A), TEM image (B) and HRTEM image (C) showing α-MnO2 nanoparticles. Fourier filtered image (C inset) using red area outlined in (C), highlighting the defects. (D) An image of a nanorod cross section viewed along the [001] zone axis. | ||
Structural evolution
TEM can only observe morphological changes. However, the mechanism based on which δ-MnO2 transformed to α-MnO2 at 100 °C for 10 min, and the exact structural changes experienced before transformation were studied using detailed structural characterization. The powder PXRD pattern of the initial δ-MnO2 shows two broad diffraction peaks at 37° (d(100) = 2.46 Å) and 65° (d(110) = 1.43 Å) (Fig. 6A), which can be attributed to δ-MnO2 with poor crystallinity, small-sized and randomly stacked [MnO6] octahedral layers, in good agreement with the SAED pattern shown in the inset of Fig. 1B.7,23,31 The d-spacing ratio of d(100) to d(110) is 1.73, which indicates a hexagonal layer symmetry.32–34 The absence of basal (001) and (002) peaks (d(001) = 7.2 Å, d(002) = 3.6 Å) is consistent with TEM and AFM images and further proves that δ-MnO2 possesses a single [MnO6] octahedral layer structure (Fig. 1).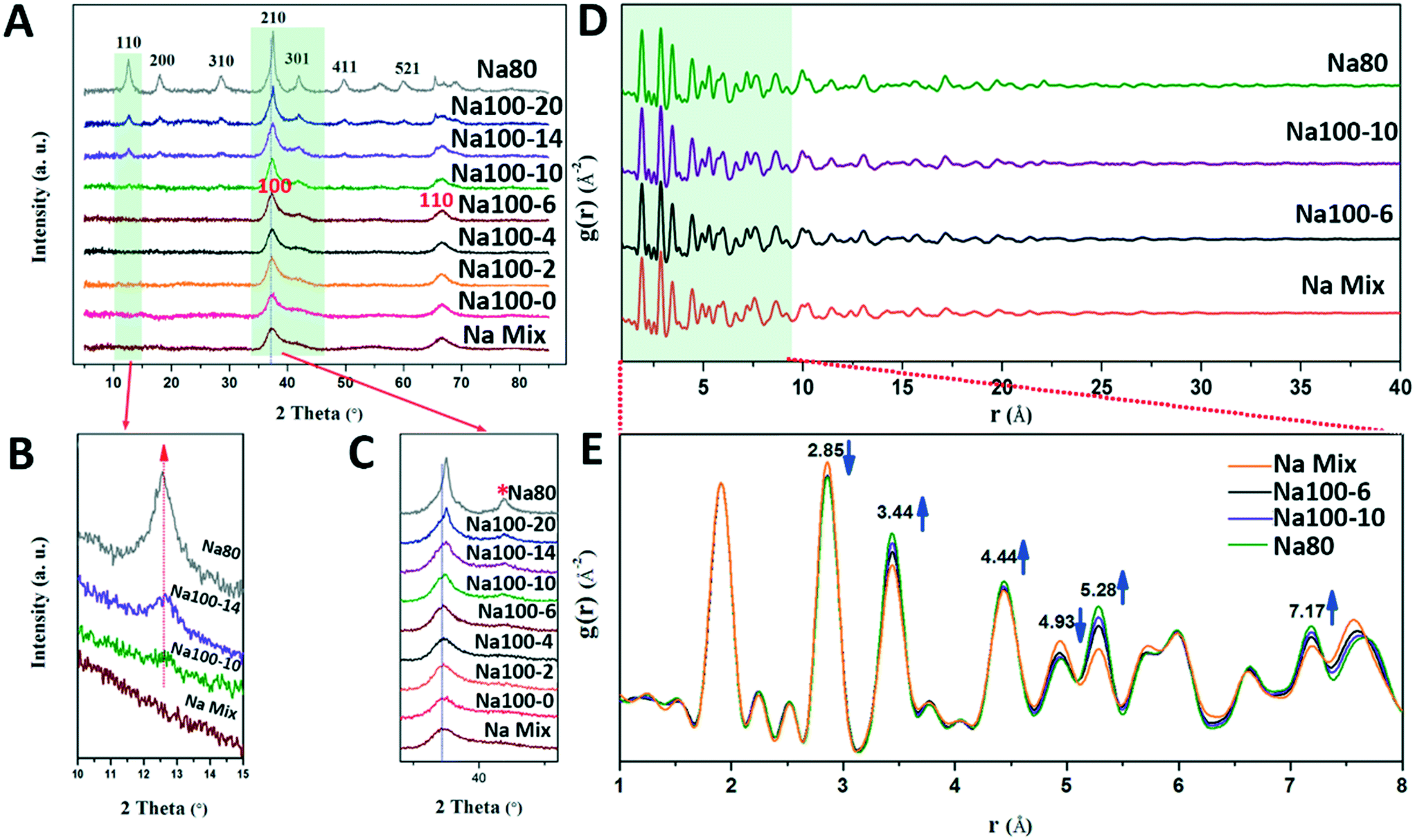 | ||
| Fig. 6 (A) PXRD patterns of intermediate products at different intervals of Na+ stabilized α-MnO2 formation and crystal growth. Red indicates the crystal surface of δ-MnO2, black indicates the crystal surface of α-MnO2 (B and C). The green zone is shown in a magnified view. The asterisk in (C) is a sign of shoulder peaks. (E and D) Pair distribution functions [G(r)s] of intermediate products at different time intervals of Na+ stabilized α-MnO2 formation and crystal growth. | ||
Before the suspension was heated at 100 °C for 10 min, only δ-MnO2 appeared in the PXRD pattern (Fig. 6A–C). The (100) and (110) diffraction peaks of δ-MnO2 are not sharper than those of the initially formed sample, thus indicating that the sizes of the crystallites did not increase within the a–b planes (Fig. 6C).18 A detailed inspection of these (100) and (110) peaks suggests a slight structural alteration of δ-MnO2 evidenced by the appearance of a small shoulder on the high angle side of the (100) peak (Fig. 6C, asterisk symbol). A previous study proposed that this feature could be related to an increased amount of Mn(III) capped on vacancies in the [MnO6] octahedral layers.32 These results suggest that Mn(IV) is being reduced to Mn(III)/Mn(II) and adsorbed on the vacancy sites with increasing reaction times.
After the suspension was heated for 14 min, (110), (200), and (310) α-MnO2 peaks with low intensity appeared (ICDD No. 00-29-1020, d(110) = 0.69 nm, d(200) = 0.48 nm, and d(310) = 0.31 nm), thus indicating that some of the δ-MnO2 was transformed to α-MnO2 during heating in the time interval from 10 min to 14 min (Fig. 6A).20 The lattice spacing of 0.69 nm (Fig. 3B) for the (110) plane of α-MnO2 can be observed in the PXRD pattern of Na100-10 (Fig. 6B). Therefore, the period along the α-MnO2 (110) plane was formed gradually and is consistent with the formation of α-MnO2 by the addition of the single-layer nanoflakes or the nanoribbons of δ-MnO2. Thickening of the nanoribbons and transformation of structures occurred simultaneously during the 10 min heating period.
Finally, the PXRD half peak width narrowed and the intensities increased gradually during the heating period from 14 to 20 min. This indicated that the crystallite size and crystallinity of α-MnO2 increased (Fig. 6).
During the structural transformation, subtle changes in the PXRD peak may occur owing to the reduction and migration of Mn. Therefore, we speculate that the production and adsorption of Mn(III)/Mn(II) play an important role in the transformation of δ-MnO2 to α-MnO2. The phenomenon based on which Mn(IV) was reduced to Mn(III)/Mn(II) and adsorbed on the vacancy sites was further confirmed by the results of the pair distribution functions (PDFs). From the peak intensity examination of the PDF, a systematic structural change was observed during the synthetic process (Fig. 6E). From Na100-6 to Na80, the peaks at 2.85 Å and 4.93 Å decreased in intensity. These peaks are attributed to atomic pairs involving the first and second Mn shells within the octahedral layer around specific Mn (MnL–MnL1 and MnL–MnL2) shells (Fig. S6†).31,35 By contrast, the correlations at 3.44 Å and 5.28 Å increased in intensity. These increases are attributed to the atomic pairs formed by MnL and the Mn interlayer (MnIL) at vacancies belonging to the first (Mn–MnIL1) and second (Mn–MnIL2) shells, respectively. The PDF results also suggest that MnL migrates from the layer to the interlayer, above or below the vacancies, to form mono-μ-oxo bridge (with an MnL–MnIL1 distance of approximately 3.44 Å) [MnO6] octahedra.35,36 It is likely that these interlayer Mn(III)/Mn(II) octahedra act as templates for the tunnel “walls” during the transformation to α-MnO2.11,12
Evolution of the chemical composition
The Mn average oxidation states (AOSs) of the intermediate products provide further quantification of the Mn(III)/Mn(II) production and structural changes of δ-MnO2. The detailed Mn(IV), Mn(III), and Mn(II) contents, which were obtained by fitting the XPS narrow scans of the Mn 3s spectra, are listed in Table 1 and Fig. S7.† The AOS of the samples decreased from 3.96 to 3.70 (Table 2, titration data) during the heating period of the suspension (100 °C for 10 min). XPS Mn 3s fitting data show that the percentages of Mn(IV) decreased from 92.5% to 84.1%; the percentage of Mn(III) increased from 6.7% to 12% during this stage. The values of AOS obtained by fitting of XPS Mn2p3/2 spectra are systematically lower than those obtained by the titration method, probably owing to the higher content of the lower valence Mn present at the surface.37–39 Thus, the XPS-derived AOS represents the “surface” Mn AOS, and it is possible that Mn(IV) is reduced to Mn(III)/Mn(II) and is adsorbed on the surface of the δ-MnO2 nanoparticles. Despite the discrepancy between the absolute AOS values, we obtained similar trends in the evolution of AOS with the reaction derived from the two methods.| Samples | Mn | O | ||||
|---|---|---|---|---|---|---|
| Mn(III) (±0.003) | Mn(II) (±0.001) | Mn(IV) (±0.003) | O2− | OH− | H2O | |
| NaMix | 0.067 | 0.008 | 0.925 | 0.699 | 0.181 | 0.120 |
| Na100-4 | 0.100 | 0.001 | 0.889 | 0.621 | 0.224 | 0.155 |
| Na100-10 | 0.120 | 0.039 | 0.841 | 0.562 | 0.263 | 0.175 |
| Na100-14 | 0.091 | 0.029 | 0.880 | 0.610 | 0.229 | 0.161 |
| Na80 | 0.048 | 0.00 | 0.931 | 0.685 | 0.192 | 0.123 |
| Samples | XPS (Mn 2p3/2) | XPS (Mn 3s) | Titration | Chemical composition |
|---|---|---|---|---|
| NaMix | 3.81 | 3.92 | 3.96 ± 0.01 | Na0.029MnO1.994·0.54H2O |
| Na100-0 | — | — | 3.96 ± 0.02 | Na0.039MnO1.999·0.63H2O |
| Na100-4 | 3.78 | 3.87 | 3.86 ± 0.02 | Na0.055MnO1.957·0.77H2O |
| Na100-6 | — | — | 3.75 ± 0.03 | — |
| Na100-10 | 3.76 | 3.79 | 3.70 ± 0.01 | Na0.090MnO1.895·0.86H2O |
| Na100-14 | 3.78 | 3.85 | 3.78 ± 0.04 | Na0.067MnO1.923·0.57H2O |
| Na100-16 | — | — | 3.95 ± 0.02 | Na0.061MnO2.006·0.67H2O |
| Na100-18 | — | — | 3.98 ± 0.03 | Na0.057MnO2.018·0.62H2O |
| Na100-20 | — | — | 3.96 ± 0.01 | Na0.048MnO2.004·0.53H2O |
| Na80 | 3.83 | 3.91 | 3.92 ± 0.02 | Na0.032MnO1.976·0.67H2O |
Previous studies have shown that structural Mn(III) is critical to the transformation of layer-structured Mn oxides into tunneled ones.40 However, the mechanism by which Mn(III) was reduced from Mn(IV) in the initial δ-MnO2 is still an open question. As described above, some of the octahedral Mn(IV) was initially reduced to Mn(II), and part of it was subsequently transformed to Mn(III). Given that apart from O2–H2O (H+), no other reductant existed during the transformation of δ-MnO2 to α-MnO2, there are two possible pathways to describe the reduction of Mn(IV).
One way is that the MnO2 is reduced by H2O at ∼100 °C and pH < 1, according to eqn (3).41
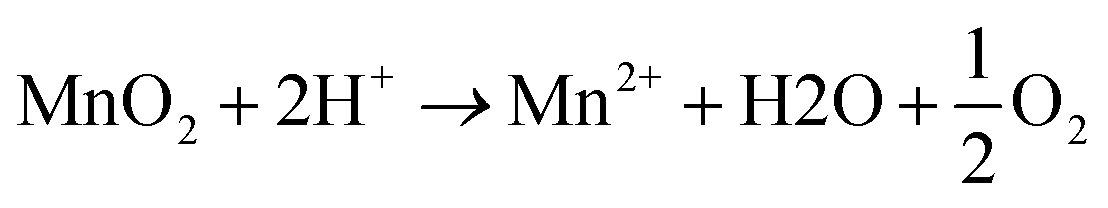 | (3) |
Depending on the pH, the Mn(II) cations originating from oxidation of lattice O2− under acidic conditions may attach above/below the layer vacancies.11,41,42 The PXRD pattern of NaMix in Fig. 6 shows that the crystallinity of δ-MnO2 was poor (i.e., a small crystallite size), thus indicating a large surface area, which favors eqn (3).
A second possible pathway for the reduction of Mn(IV) involves the poorly crystalline δ-MnO2 with mixed-Mn valences (II, III, and IV) and the release of structural oxygen, according to eqn (4).43,44
 | (4) |
The XPS fitting results of O1s are listed in Table 1. There are three species of oxygen in Mn oxides, lattice oxygen (O2−), hydroxide oxygen (–OH), and oxygen in molecular water. Among the three species of oxides, O2− decreased from 69.9% to 56.2%, and –OH increased from 18.1% to 26.3% during the heating of the suspension during the first 10 min. The large amount of lost O2− was not proportional to the formed –OH. It is interesting that this great loss of O2− is consistent with the creation of O vacancies in δ-MnO2, as proposed above (eqn (4)). The observed increase of the O1s peak at 532.4 eV in the XPS spectra is induced by the formation of oxygen vacancies (Fig. S8†).45–48 Furthermore, the increase in the content of Na+ and Mn(III) (longer Mn–O bond), and the poor crystallinity could facilitate the formation of oxygen vacancies (Tables 1 and 2), which promote the production of Mn(III) and the transformation of a 2D layer structure into a one-dimensional (1D) tunnel structure.41,49,50
After forming the tunnel structure, the intensities of the O1s XPS peaks at 532.4 eV were maximized, and the AOS of δ-MnO2 reached a minimum during the 10 min heating period. The reactions described in eqn (3) and (4) became less favorable owing to the formation of a more stable structure of α-MnO2, and the percentages of Mn(III) and Mn(II) decreased gradually (Table 1). Additionally, the decrease of the O1s XPS peak at 532.4 eV was induced by the reduction of oxygen vacancies (Fig. S8†). This indicated that dissolved oxygen generated by the reactions of eqn (3) and (4) could refill the oxygen vacancies and oxidize Mn(III)/Mn(II) back to Mn(IV) after the formation of α-MnO2, which caused the increase of AOS from 3.70 to 3.96 (Table 2).
Coupled morphological and structural evolution
Based on the above analyses, the coupled evolutions of structure (Fig. 6) and morphology (Fig. S9†) during the formation of α-MnO2 are illustrated in Fig. 7. Firstly, MnO4− is reduced by Mn(II) to produce primary nanoflakes of δ-MnO2 with poor crystallinity and with sizes in the range of 2–4 nm. Upon increasing the heating time, nanoflakes aggregate serially to form nanoribbons. Secondly, the nanoribbons stack with each other to form primary nanorods, which initiate the conversion of δ-MnO2 into α-MnO2. Finally, the primary nanorods assemble along the (110) and (001) planes by side-to-side and end-to-end OA, respectively.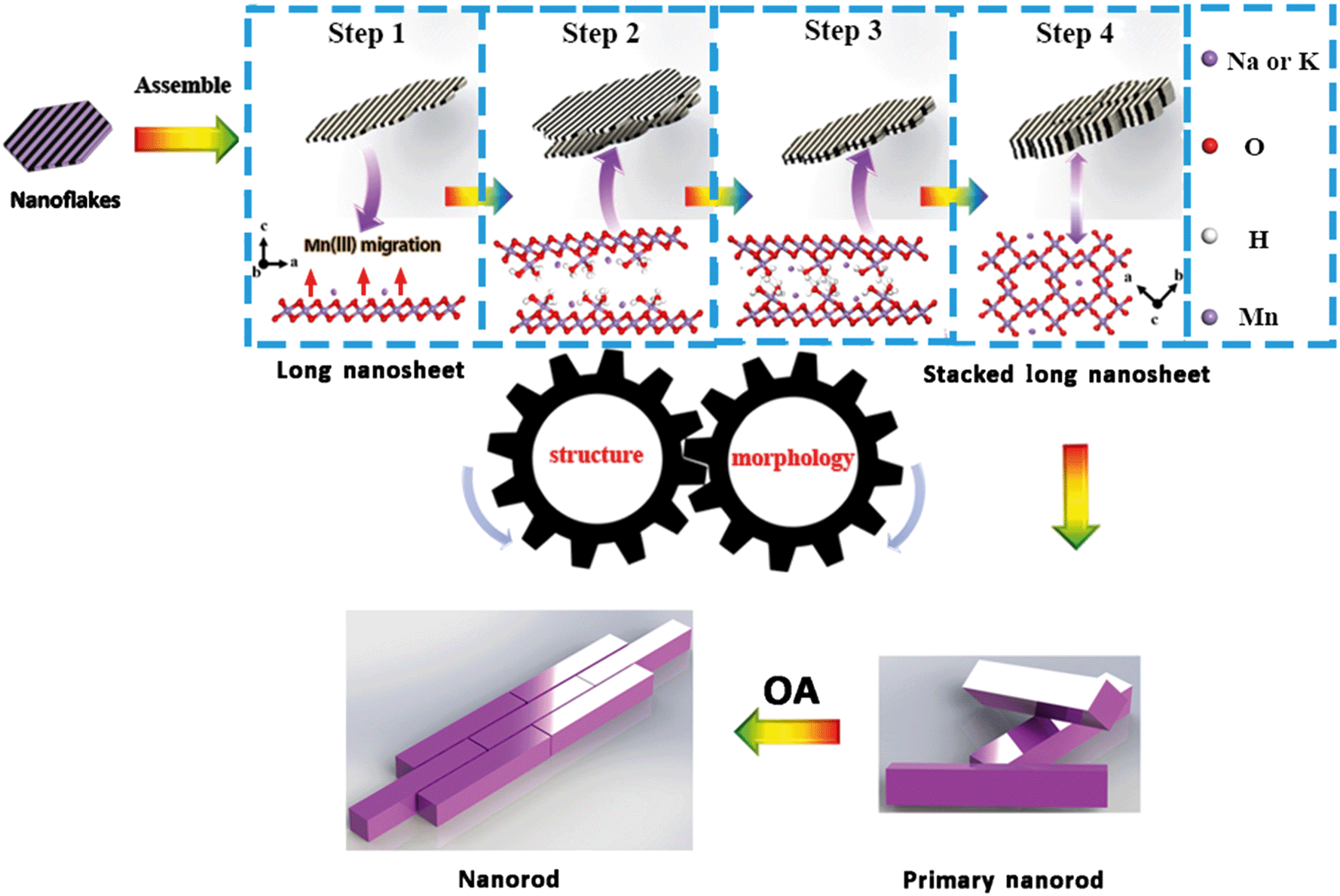 | ||
| Fig. 7 Proposed processes of assembly of nanoparticles and morphological evolution with time during the crystal growth of α-MnO2via a staged OA process. The schematic in the blue square illustrates the structural changes and morphological evolutions during the δ-MnO2 transformation to α-MnO2. | ||
The structure of δ-MnO2 produced in the initial crystallization stage does not meet the necessary conditions, i.e., increased proportion of Mn(III)/Mn(II) above or below the layer vacancies, for its conversion to α-MnO2. Although δ-MnO2 did not convert to α-MnO2 before it was heated (for a period of 10 min), the lower pH of 0.86 was favorable for edge-to-edge aggregation along the (100) plane.7 The initially formed nanoribbons that were formed by the nanoflakes and were attached to each other have many lattice dislocations at the grain boundaries (Fig. 2B and 3C) and promote the generation of oxygen vacancies.51 The Mn(III) moieties were generated based on the generation of oxygen.
Subsequently, structural adjustments occur through the production and migration of Mn(III) in the δ-MnO2 nanoflakes and nanoribbons. Recently, Ling et al. showed that the amount of Mn(III) has an impact on the structure of δ-MnO2.52 Furthermore, Cui et al. and Yuan et al. revealed that the interlayer Mn(III) plays an important role in building a tunnel structure during the conversion of LMOs (buserite) to TMOs (3 × 3 todorokite).21,40 The amount of Mn(IV)/(III) for the ideal transition state structure can be obtained, as shown in Fig. S10;†i.e., the required interlayer Mn(III) content is 30% of the total Mn to assemble into the α-MnO2 structure. This agrees very well with our experimental observations. The AOS of the production of onset of transformation into Na+-stabilized α-MnO2 and K+-stabilized α-MnO2 were 3.70 and 3.74 with Mn(III) concentration about 30% and 26%, respectively. Sufficient Mn(III) was required for the formation of the tunnel “walls” to support the transformation, and for the provision of sufficient hydrogen bonding. Indeed, [Mn(III)O6] octahedra are distorted by the Jahn–Teller effect, and bonding distortion within the octahedral layer is relieved when Mn(III) migrates from layers to locations above and below the vacancies (Fig. 7, step 1).11 These [Mn(III)O6] octahedra include three unsaturated oxygen molecules that were combined with H+ to form –OH. When the amount of –OH was large enough, a network of hydrogen bonds formed between the [Mn(III)O6] octahedra of adjacent nanosheets and nanoflakes (Fig. 7, step 2, 3). These [Mn(III)O6] octahedra dehydrated via a condensation reaction and formed the tunnel “walls.” Therefore, the number of –OH moieties is expected to be maximized at the initial stage of the transformation, and to decrease during the subsequent transformation stages. These nanoribbons and nanoflakes attach along the (001) plane of δ-MnO2 to form primary α-MnO2 nanorods based on face-to-face OA (Fig. 7, step 4). This is the process of structural evolution used to promote the transformation of morphological and crystal phase changes.
To confirm this formation mechanism in α-MnO2 with different tunnel cations, K+-stabilized α-MnO2 was synthesized with similar procedures, whereby only NaMnO4 was replaced by KMnO4. During the synthesis of K+-stabilized α-MnO2, all the trends of the structural evolution were consistent with those of Na+-stabilized α-MnO2 (Fig. S11 and Tables S1 and S2†). It is suggested that the different tunnel cations of α-MnO2 experienced a similar OA formation process. The δ-MnO2 molecules were converted to α-MnO2 using heating at 100 °C for a 2 min period. The PXRD analysis (Fig. S11†) indicated that the conversion rate of K+-stabilized δ-MnO2 was much faster than that of Na+-stabilized δ-MnO2. The structure of K+-stabilized α-MnO2 was stable when the suspension was aged for 24 h, while previous reports indicated that the stable (100) surface of nanorods could be formed during this stage.20 However, when the Na+-stabilized α-MnO2 was continually aged for 24 h, the PXRD patterns of the samples yielded a new peak, dhkl = 0.4 nm, of γ-MnO2 with a 1 × 2 tunnel structure (Fig. S12†) because of the small amount of Na+ used (3.24 ± 0.09% in Table 2) in the tunnel to stabilize the Na+-stabilized α-MnO2.50 These results indicate that the K+ adsorbed on δ-MnO2 not only accelerated the production and migration of Mn(III), but also promoted the structural conversion and stability of the produced α-MnO2. This finding agrees well with previous reports that indicated that the K+ ions, whose size (∼0.138 nm) facilitated trapping of the 2 × 2 tunnel size (∼0.46 nm), played important roles in the templating and stabilization of the tunneled framework.53
Another difference between the K+-stabilized α-MnO2 and Na+-stabilized α-MnO2 is the dominant side crystallographic face of the products. In this study, the cutting of the nanorods of Na+-stabilized α-MnO2 along the (001) plane at different stages shows that the side planes of the nanorods still correspond with the (110) plane (Fig. 5D and S13†). This is in part attributed to the fact that the open tunnel structure is formed more easily during the assembly process, and the morphology can be better captured during a slower Na synthesis. The nanorods of Na+ stabilized α-MnO2 with the exposed (110) lateral facets may be the intermediate products that can be stably present over a period of time due to a slow reaction rate. In the presence of K+, the side faces of the nanorods become four (100) faces with a lower energy of 0.44 J m−2 compared to the (110) surface energy of 0.74 J m−2 through a dissolution–recrystallization reaction (Fig. S13†).19 K+ stabilized α-MnO2 is formed more than 5 times faster than Na+ stabilized α-MnO2 during the same synthesis period, and it has a longer aging time, so that the K+ stabilized α-MnO2 nanorods tend to expose stable (100) edge surfaces by the dissolution–recrystallization driven by the surface energy difference. Therefore, α-MnO2 nanorods with different exposed lattice faces can be obtained during the synthesis according to the specific application. The Na+-stabilized α-MnO2 is more suitable for producing nanorods with four (110) lateral faces, which likely reduce the distance and energy barrier for ion diffusion, and improve the rate performance of α-MnO2 in various applications, such as rechargeable battery electrodes, supercapacitors, and Li–O2 battery catalysts.17,54–59 Conversely, the K+-stabilized α-MnO2 is more suitable for producing long nanowires because of its stable tunnel structure with the support of K+.
Although this study is not exhaustive, it does highlight an important area for further studies on the morphological and structural interactions. Indeed, future studies could evaluate K+ ion template effects on the migration of Mn(III) into tunnel “walls” and the changes in the reactivities of the products that are formed in different stages.
Conclusion
Na+-Stabilized tunnel structured α-MnO2 nanorods were found to assemble via a staged OA growth process. The coupled evolution of structure and morphology during the entire process has been explored.Firstly, the δ-MnO2 nanoflakes, which possessed poorly crystalline forms, aggregated along the (110) surface plane to form δ-MnO2 nanoribbons via the edge-to-edge aggregation mechanism. Meanwhile, the Mn(IV) ions in the [MnO6] octahedral layer of δ-MnO2 were reduced to Mn(III) by water, and then migrated above and below the vacancies. Secondly, the morphology and structure of δ-MnO2 evolved simultaneously. Mn(III), whose amount increased gradually, built up tunnel walls and triggered the conversion of the 2D layer structure to the 1D tunnel structure. A dynamic network of hydrogen bonds between the –OH groups, which were combined with Mn(III) of adjacent nanoribbons, was present as a mode of bonding to fabricate the tunnel structure. Therefore, adjacent nanoribbons aggregated and formed α-MnO2 primary nanorods based on the face-to-face OA mechanism. Thirdly, primary nanorods assembled with each other to form longer and wider nanorods based on respective end-to-end and side-to-side OA mechanisms. The defects that were formed by the assembly process were constantly smoothed via dissolution–recrystallization processes throughout the entire reaction. This work has filled the gap regarding the initial stage of 1D tunnel-structured α-MnO2 formation under natural conditions. Meanwhile, it also provided greater fundamental understanding of the relationship between the structure and morphological transformation during the crystal growth process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 41471194 & 41171197) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB15020402) for financial support of this research. Use of the Advanced Photon Source, Argonne National Laboratory, was supported by the U.S. DOE-BES under Contract DE-AC02-06CH11357.References
- L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef PubMed.
- J. J. De Yoreo, P. U. Gilbert, N. A. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky and J. F. Banfield, Science, 2015, 349, aaa6760 CrossRef PubMed.
- J. Zhang, F. Huang and Z. Lin, Nanoscale, 2010, 2, 18–34 RSC.
- X. Liang, Z. Zhao, M. Zhu, F. Liu, L. Wang, H. Yin, G. Qiu, F. Cao, X. Liu and X. Feng, Environ. Sci.: Nano, 2017, 4, 1656–1669 RSC.
- H. Zhao, M. Zhu, W. Li, E. J. Elzinga, M. Villalobos, F. Liu, J. Zhang, X. Feng and D. L. Sparks, Environ. Sci. Technol., 2016, 50, 1750–1758 CrossRef CAS PubMed.
- Q. Wang, X. Liao, W. Xu, R. Yang, K. J. Livi and M. Zhu, Inorg. Chem., 2016, 55, 10248–10258 CrossRef CAS PubMed.
- F. F. Marafatto, B. Lanson and J. Peña, Environ. Sci.: Nano, 2018, 5, 497–508 RSC.
- A. L. Atkins, S. Shaw and C. L. Peacock, Geochim. Cosmochim. Acta, 2014, 144, 109–125 CrossRef CAS.
- S. L. Suib, Acc. Chem. Res., 2008, 41, 479–487 CrossRef CAS PubMed.
- S. Dharmarathna, C. K. King'Ondu, W. Pedrick, L. Pahalagedara and S. L. Suib, Chem. Mater., 2012, 24, 705–712 CrossRef CAS.
- S. Grangeon, A. Fernandezmartinez, F. Warmont, A. Gloter, N. Marty, A. Poulain and B. Lanson, Geochem. Trans., 2015, 16, 12 CrossRef PubMed.
- S. Grangeon, B. Lanson and M. Lanson, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 828–838 CrossRef CAS PubMed.
- Y. F. Yuan, C. Liu, B. W. Byles, W. Yao, B. Song, M. Cheng, Z. Huang, K. Amine, E. Pomerantseva, R. Shahbazian-Yassar and J. Lu, Joule, 2019, 3, 1–14 CrossRef.
- J. Liu, V. Makwana, J. Cai, S. L. Suib and M. Aindow, J. Phys. Chem. B, 2003, 107, 9185–9194 CrossRef CAS.
- S. Parc, D. Nahon, Y. Tardy and P. Bieillard, Am. Mineral., 1989, 74, 466–475 CAS.
- J. Ostwald, Econ. Geol., 1992, 87, 1237–1252 CrossRef CAS.
- P. M. Vasconcelos, P. R. Renne, G. H. Brimhall and T. A. Becker, Geochim. Cosmochim. Acta, 1994, 58, 1635–1665 CrossRef CAS.
- T. T. Truong, Y. Liu, Y. Ren, L. Trahey and Y. Sun, ACS Nano, 2012, 6, 8067–8077 CrossRef CAS PubMed.
- K. Li, J. Chen, Y. Peng, W. Lin, T. Yan and J. Li, J. Mater. Chem. A, 2017, 5, 20911–20921 RSC.
- Y. Yuan, S. M. Wood, K. He, W. Yao, D. Tompsett, J. Lu, A. Nie, M. S. Islam and S. Santhanagopalan, ACS Nano, 2016, 10, 539–548 CrossRef CAS PubMed.
- Y. F. Yuan, K. He, B. W. Byles, C. Liu, K. Amine, J. Lu, E. Pomerantseva and R. Shahbazian-Yassar, Chem, 2019, 5, 1793–1805 CAS.
- S. Grangeon, B. Lanson and M. Lanson, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 828–838 CrossRef CAS PubMed.
- D. Portehault, S. Cassaignon, E. Baudrin and J. P. Jolivet, J. Mater. Chem., 2009, 19, 2407–2416 RSC.
- D. Portehault, S. Cassaignon, E. Baudrin and J. Jolivet, Chem. Mater., 2007, 19, 5410–5417 CrossRef CAS.
- D. Zhai, B. Li, C. Xu, H. Du, Y. He, C. Wei and F. Kang, J. Power Sources, 2011, 196, 7860–7867 CrossRef CAS.
- N. Kijima, H. Yasuda, T. Sato and Y. Yoshimura, J. Solid State Chem., 2001, 159, 94–102 CrossRef CAS.
- E. S. Iton, J. E. Post, P. J. Heavey, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef.
- R. M. Mckenzie, Mineral. Mag., 1971, 38, 493–502 CrossRef CAS.
- H. Zhang, J. J. De Yoreo and J. F. Banfield, ACS Nano, 2014, 8, 6526–6530 CrossRef CAS PubMed.
- X. Zhang, Y. He, M. L. Sushko, J. Liu, L. Luo, J. J. De Yoreo, S. X. Mao, C. M. Wang and K. M. Rosso, Science, 2017, 356, 434–437 CrossRef CAS PubMed.
- M. Villalobos, B. Toner, J. Bargar and G. Sposito, Geochim. Cosmochim. Acta, 2003, 67, 2649–2662 CrossRef CAS.
- V. A. Drits, B. Lanson and A. C. Gaillot, Am. Mineral., 2007, 92, 771–788 CrossRef CAS.
- S. Grangeon, B. Lanson, N. Miyata, Y. Tani and A. Manceau, Am. Mineral., 2010, 95, 1608–1616 CrossRef CAS.
- H. Yin, W. Tan, L. Zheng, H. Cui, G. Qiu, F. Liu and X. Feng, Geochim. Cosmochim. Acta, 2012, 93, 47–62 CrossRef CAS.
- A. Manceau, M. A. Marcus, S. Grangeon, M. Lanson, B. Lanson, A. C. Gaillot, S. Skanthakumar and L. Soderholm, J. Appl. Crystallogr., 2013, 46, 193–209 CrossRef CAS.
- V. Petkov, Y. Ren, I. Saratovsky, P. Pastén, S. J. Gurr, M. A. Hayward, K. R. Poeppelmeier and J. F. Gaillard, ACS Nano, 2009, 3, 441–445 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- H. Yin, F. Liu, X. Feng, T. Hu, L. Zheng, G. Qiu, L. K. Koopal and W. Tan, Geochim. Cosmochim. Acta, 2013, 117, 1–15 CrossRef CAS.
- H. Yin, X. Feng, G. Qiu, W. Tan and F. Liu, J. Hazard. Mater., 2011, 188, 341–349 CrossRef CAS PubMed.
- H. Cui, X. Liu, W. Tan, X. Feng, F. Liu and H. Daniel Ruan, Clays Clay Miner., 2008, 56, 397–403 CrossRef CAS.
- A. T. Stone, Environ. Sci. Technol., 1987, 2, 979–988 CrossRef PubMed.
- E. B. Godunov, A. D. Izotov and I. G. Gorichev, Inorg. Mater., 2018, 54, 66–71 CrossRef CAS.
- E. B. Godunov, I. V. Artamonova, I. G. Gorichev and Y. A. Lainer, Russ. Metall., 2012, 1, 39–44 CrossRef.
- J. Hou, Y. Li, L. Liu, L. Ren and X. Zhao, J. Mater. Chem. A, 2013, 1, 6736–6741 RSC.
- M. Xing, J. Zhang, F. Chen and B. Tian, Chem. Commun., 2011, 47, 4947–4949 RSC.
- K. Wang, Y. Chang, L. Lv and Y. Long, Appl. Surf. Sci., 2015, 351, 164–168 CrossRef CAS.
- Y. Sun, X. Yan, X. Zheng, Y. Liu, Y. Shen and Y. Zhang, Nano Res., 2016, 9, 1116–1124 CrossRef CAS.
- J. P. Holgado, G. Munuera, J. P. Espinós and A. R. González-Elipe, Appl. Surf. Sci., 2000, 158, 164–171 CrossRef CAS.
- X. Li, J. Ma, L. Yang, G. He, C. Zhang, R. Zhang and H. He, Environ. Sci. Technol., 2018, 52, 12685–12696 CrossRef CAS PubMed.
- G. Zhang, W. Dong, X. Huang and J. Zou, Catal. Commun., 2017, 89, 117–120 CrossRef CAS.
- J. A. Dawson and I. Tanaka, ACS Appl. Mater. Interfaces, 2014, 6, 17776–17784 CrossRef CAS PubMed.
- F. T. Ling, J. E. Post, P. J. Heaney and E. S. Ilton, Chem. Geol., 2018, 479, 216–227 CrossRef CAS.
- J. A. Dawson and I. Tanaka, ACS Appl. Mater. Interfaces, 2014, 6, 17776–17784 CrossRef CAS PubMed.
- C. Wei, C. Xu, B. Li, H. Du, D. Nan and F. Kang, J. Power Sources, 2013, 225, 226–230 CrossRef CAS.
- L. Li, C. Nan, J. Lu, Q. Peng and Y. Li, Chem. Commun., 2012, 48, 6945–6947 RSC.
- T. S. Arthur, R. Zhang, L. Chen, P. A. Glans, X. Fan, J. Guo and F. Mizuno, ACS Appl. Mater. Interfaces, 2014, 6, 7004–7008 CrossRef CAS PubMed.
- G. S. Hutchings, J. Rosen, D. Smiley, G. R. Goward, P. G. Bruce and F. Jiao, J. Phys. Chem. C, 2014, 118, 12617–12624 CrossRef CAS.
- Y. Qin, J. Lu, P. Du, Z. Chen, Y. Ren, T. Wu, J. T. Miller, J. Wen, D. J. Miller and Z. Zhang, Energy Environ. Sci., 2013, 6, 519–531 RSC.
- W. Xiao, H. Xia, J. Y. H. Fuh and L. Lu, J. Power Sources, 2009, 193, 935–938 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9en01000a |
| This journal is © The Royal Society of Chemistry 2020 |