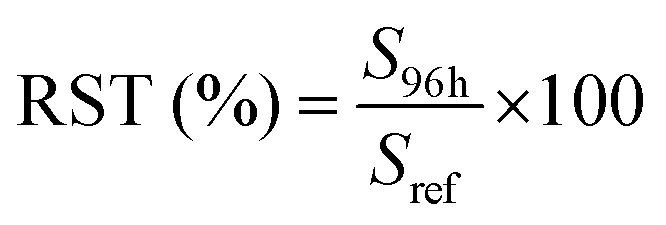Highly sensitive multi-residue analysis of veterinary drugs including coccidiostats and anthelmintics in pond water using UHPLC-MS/MS: application to freshwater ponds in Flanders, Belgium†
Received
12th May 2020
, Accepted 3rd September 2020
First published on 18th September 2020
Abstract
Veterinary drugs, such as coccidiostats and anthelmintics are routinely administered in extensive animal husbandry, finding their way into the aquatic environment through urine and/or feces of treated animals kept outdoors or by the application of contaminated liquid manure on agricultural fields and subsequent mechanisms of surface run-off, leaching and drift. Several of these compounds are known to exert acute and chronic toxicity effects on aquatic organisms, and can lead to changes in biodiversity and ecosystem functioning. The overall objective of this research was to develop, validate and apply a highly sensitive, multi-residue SPE-UHPLC-MS/MS method for the determination of 12 coccidiostats, registered as a feed supplement or veterinary medicine in Europe and three regularly used anthelmintics, in pond water, often functioning as amphibian habitat. Sample extraction was optimized using a fractional factorial resolution design. Pond water filtration efficiency (i.e. 80–118%, ≤25% RSD) and matrix effects (i.e. 72–119%, ≤39% RSD) were evaluated using water from respectively 3 and 20 different ponds in Flanders. By incorporating internal standards, overall results improved and adequate precision values (i.e. ≤15%) were obtained according to the EMA guidelines. Acceptable within-run and between-run apparent recoveries, satisfactory precision as well as good linearity were demonstrated according to the CD 2002/657/EC, SANTE/12682/2019 and VICH 49 guidelines, except for robenidine for which the between-day precision was between 21.0 and 34.5%. Sample storage stability studies indicated that storage at 4 °C and analysis performed within 96 hours after sampling was sufficient to avoid loss by degradation for all compounds, excluding robenidine. Values for the limit of detection (LOD) and quantification (LOQ) were in nanograms per liter, which was essential for the environmental application of this novel method. The method was successfully applied on grab water samples from the water surface of 18 different ponds across Flanders, Belgium, detecting amprolium and levamisole at concentrations below the LOQ of 2.5 ng L−1 and at 250.0 ng L−1 or below the LOQ of 250.0 ng L−1, respectively. In conclusion, our newly developed method may provide insights about the contamination status of amphibian breeding ponds.
Environmental significance
This study provides the first SPE-UHPLC-MS/MS method for the simultaneous quantification of 12 coccidiostats registered as feed additives or veterinary medicine in Europe and three frequently used anthelmintics, in pond water. Our newly developed method provides information about pond water health, i.e. contamination status and potential toxicity to aquatic organisms including aquatic microorganisms and amphibians. The method will be applied in the establishment of a mechanistic model of food-web and infectious disease dynamics in natural ponds, in which the relationship between contaminants, aquatic microorganisms and amphibians (i.e. food-web) and the relationship between all the latter and chytrid infections (i.e. infectious disease dynamics) will be studied. As such, the method may contribute to highly demanded mitigating strategies, preventing further loss of biodiversity worldwide.
|
1. Introduction
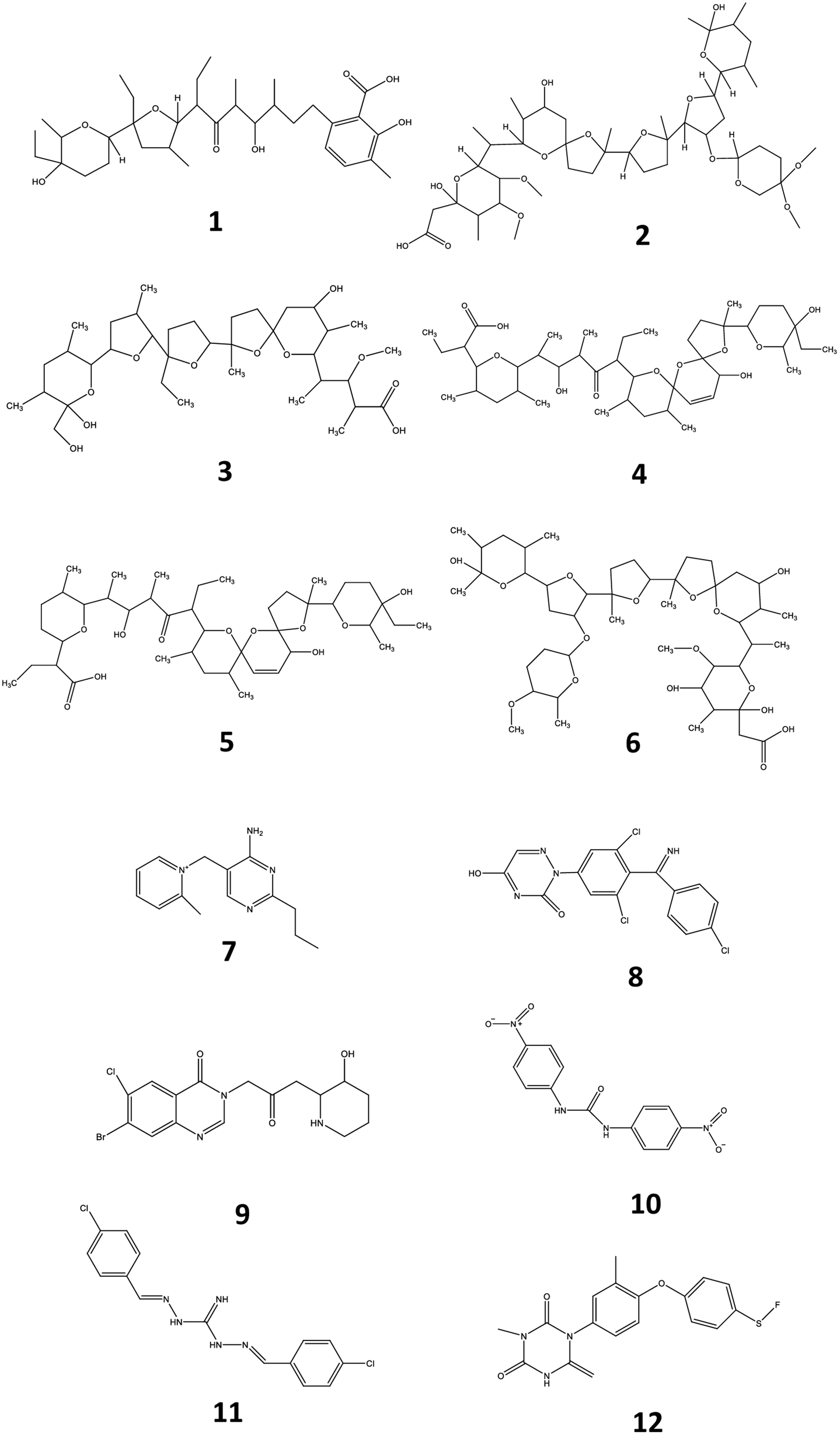
Veterinary drugs and feed additives are widely used in animal farming for the prevention and treatment of disease and the overall protection of animal health. Many of these compounds, such as coccidiostats and anthelmintics are routinely administered to avoid economic losses related to disease in extensive animal husbandry.1 Coccidiostats are a group of compounds that are regularly added in feed and used for the prevention and treatment of coccidiosis, an infectious disease caused by protozoa of the genus Eimeria and responsible for gastroenteritis in poultry and livestock.2 Currently, 12 coccidiostats are registered in Europe, either as a feed supplement (n = 11)3 or as a veterinary medicine (i.e. toltrazuril).4 These compounds can be categorized in non-ionophoric coccidiostats (amprolium, diclazuril, halofuginone, nicarbazin, robendine and toltrazuril) and ionophoric coccidiostats (lasalocid, maduramicin, monensin, narasin, salinomycin and semduramicin), the latter naturally produced by certain strains of Streptomyces. Nicarbazin is the generic name for the complex of 4,4′-dinitrocarbanilide (4,4′-DNC) and 2-hydroxy-4,6-dimethylpyrimidine (HDP). However, 4,4′-DNC is mostly monitored for its persistence in the environment.2 The chemical structure of coccidiostats consists of multiple cyclic ethers and is shown in Fig. 1. Because ionophoric coccidiostats form complexes with cations such as K+, Na+, Ca2+ and Mg2+ and are lipid soluble, they are able to conduct cation transport across the cell membrane which results in osmotic imbalance. In this way these compounds induce parasitic cell death.5 Non-ionophoric coccidiostats act by interfering with either the mitochondrial metabolism, oxidative phosphorylation or cell division.6
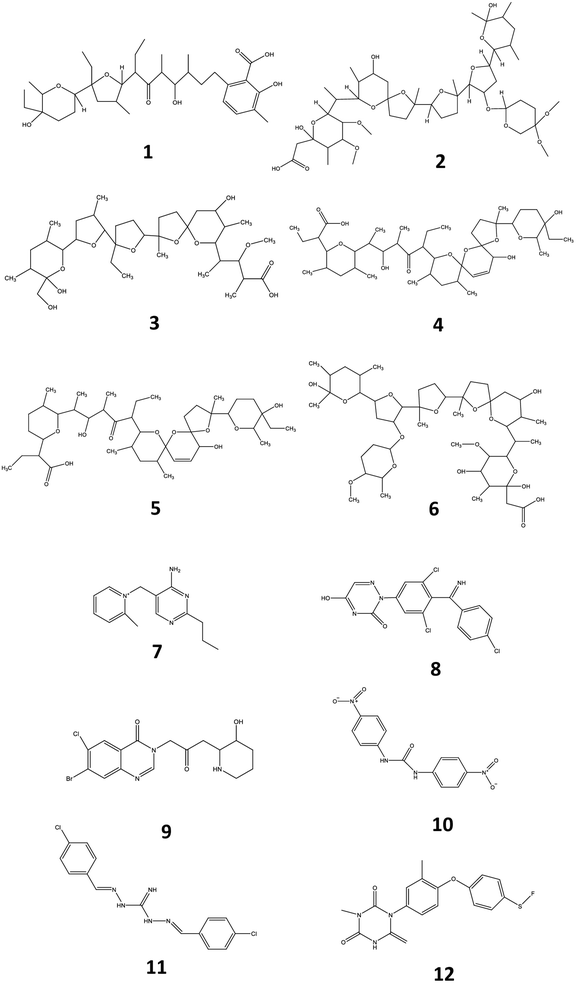 |
| | Fig. 1 Chemical structures of lasalocid (1), maduramicin (2), monensin (3), narasin (4), salinomycin (5), semduramicin (6) (ionophoric coccidiostats) and amprolium (7), diclazuril (8), halofuginone (9), 4,4′-dinitrocarbanilide (10), robenidine (11) and toltrazuril (12) (non-ionophoric coccidiostats). | |
An important group of veterinary drugs are anthelmintics, which are used for treating parasitic worm (helminths) infections, very often as group medication in cattle, pig and poultry industry.7,8 Levamisole belongs to the group of imidazothiazoles which is characterized by a chemical structure containing an imidazole and thiazole ring (Fig. 2). It works as a nicotine receptor agonist that causes continued parasitic muscle contractions, leading to paralysis.9 Flubendazole is a major representative of the group of benzimidazoles which contain a benzene and imidazole ring structure. These drugs exert their function by inhibiting the synthesis of microtubules in the cell.10 Ivermectin is an important macrocyclic lactone which induces paralysis in nematodes by the overstimulation of gamma-aminobutyric acid receptors.11 The chemical structures of levamisole, flubendazole and ivermectin are presented in Fig. 2.
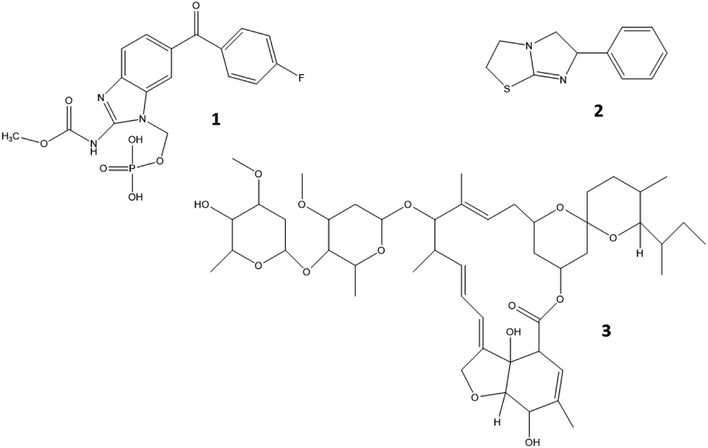 |
| | Fig. 2 Chemical structures of flubendazole (1), levamisole (2) and ivermectin (3). | |
Most of these compounds find their way into the aquatic environment by direct application in aquaculture, through urine and/or feces of animals kept outdoors or by the application of liquid manure on agricultural fields and subsequent mechanisms of surface run-off, leaching to groundwater and drift, the latter being displacement of aerosolized manure droplets after certain manure spraying techniques.12 Once in the environment, these veterinary drugs can be subjected to transport processes between different environmental compartments (e.g. sorption–desorption) as well as removal processes such as hydrolysis and photolysis, depending on the environmental conditions and the specific physicochemical properties of each compound (Table S1†).13 Even after certain degrees of metabolization in the animal body, degradation and different sorption kinetics to sludge and soil, several coccidiostats and anthelmintics have been found in river waters adjacent to agricultural fields at concentrations of 15 ng L−1 (monensin), 17 ng L−1 (salinomycin) and 23 ng L−1 (maduramicin).14 Several of these compounds such as monensin, narasin, salinomycin, lasalocid and flubendazole are known to exert acute (i.e. mostly immobility) and/or chronic toxic effects (i.e. reproduction and growth impairment) to aquatic organisms with reported half maximal concentrations (EC50, 48h) for immobility ranging between 0.045 and 403 mg L−1 and lowest-observed-effect-concentrations (LOEC21d) for reproduction or growth impairment ranging between 0.005 and 0.09 mg L−1 in daphnids and rotifers. Furthermore, ivermectin has shown to be very toxic to daphnids, with EC50, 48h for immobility ranging from 0.006 to 0.59 μg L−1 and a reported LOEC21d of 0.001 ng L−1.15–20 As these aquatic organisms are crucial elements in the aquatic foodweb, their loss induces drastic changes in ecosystem functioning and structure21 and even in aquatic vertebrate disease dynamics.22,23 Up to now, research has mainly focused on the detection of coccidiostats and anthelmintics in rivers, large lakes, ground and tap water.14,15,24,25 Only three papers have been published on the determination of these veterinary products (n ≤ 3) in natural ponds.12,26,27 Comparing biodiversity of macrophytes and macroinvertebrates, the European Pond Conservation Network28 concluded that ponds at the regional level contribute most to biodiversity by supporting more unique species as do rivers, streams and ditches. Consequently, as research has shown that several of the compounds mentioned above (e.g. ivermectin, monensin, salinomycin) pose significant treat to aquatic organisms,16–20,29 it is highly recommended to determine the presence and concentration of these pharmaceuticals in pond water as well.
A major challenge concerning environmental water (e.g. river and ground water) analysis is the determination of low concentrations (ng L−1 range) which implies the need for sensitive methods able to analyze a wide variety of environmental pollutants. Analyzing pond water poses additional challenges such as pH variability, the presence of organic matter and the overall pond matrix complexity,30 which implies the need for appropriate filters and internal standards. Furthermore, a wide concentration range needs to be considered, ranging from low to high nanograms per liter, as detected concentrations can depend on variable nearby agricultural activities and livestock production,31 as well as pond size (i.e. lower concentrations can be detected in larger ponds due to dilution and vice versa).32 Furthermore, pond size may vary between ponds but also within ponds over time: ponds may be smaller in size during prolonged warm periods for example in spring and summer.32
This study provides the first full-scale screening analysis of toltrazuril and robenidine, and quantification of the other 10 coccidiostats registered as feed additive or veterinary medicine in Europe3,4 and three regularly used anthelmintics33 in pond water using solid phase extraction (SPE) combined with UHPLC-MS/MS.
2. Material and methods
2.1 Chemicals and reagents
Concerning anthelmintics, one representative from three major anthelmintic groups (i.e. macrocyclic lactones, benzimidazoles and imidazothiazoles)33 was included in the method. Within each group, compounds were selected based upon previous detection in surface water in Belgium (i.e. flubendazole),34 sales data provided by Elanco Animal Health (Antwerp, Belgium) showing high sales percentages for ivermectin, and the fact that compounds are registered as feed and/or drinking water medication (i.e. mass medication) in Belgium.35 All analytical standards including the selected internal standards (ISs) (i.e. DNC-d8, diclazuril-methyl, nigericin and toltrazuril-d3) were purchased from Sigma-Aldrich (Overijse, Belgium). A broad polarity range of compounds was covered with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values ranging from 1.04 to 7.51 (Table 1). Stock solutions of amprolium, flubendazole, ivermectin, levamisole, maduramicin, monensin, narasin, nigericin, salinomycin, semduramicin, toltrazuril and toltrazuril-d3 were prepared in methanol (MeOH) at a concentration of 1 mg mL−1. Stock solutions of lasalocid (0.1 mg mL−1) and halofuginone (0.1 mg mL−1) were prepared in acetonitrile (ACN). Stock solutions of diclazuril and diclazuril-methyl (0.1 mg mL−1), 4,4′-DNC and DNC-d8 (1 mg mL−1) and robenidine (1 mg mL−1) were prepared in MeOH/n,n-dimethylformamide (DMF) (90/10, v/v%), ACN/dimethylsulfoxide (DMSO) (75/25, v/v%) and MeOH/DMSO (90/10, v/v%), respectively. From the individual stock solutions, a mixed standard working solution (WO1) of 0.5 μg mL−1 (i.e. amprolium, flubendazole and halofuginone), 4 μg mL−1 (i.e. lasalocid), 5 μg mL−1 (i.e. diclazuril, 4,4′-DNC, maduramicin, monensin, narasin, robenidine, salinomycin, semduramicin and toltrazuril) and 25 μg mL−1 (i.e. ivermectin and levamisole) was prepared in ACN. Serial dilution resulted in mixed standard working solutions of 0.05, 0.4, 0.5 and 2.5 μg mL−1 (WO2) and 0.005, 0.04, 0.05 and 0.25 μg mL−1 (WO3) in ACN, respectively. Additionally, a mixed standard working solution (WOIS) (1 μg mL−1) containing the selected ISs was prepared in ACN. Stock and working solutions were stored at ≤−15 °C. A Milli-Q Advantage A10® System (Merck Millipore, Darmstadt, Germany) was used to obtain ultrapure water. The organic solvents were of UPLC grade, purchased from Biosolve BV (Valkenswaard, The Netherlands). Formic acid (99.5%) and ammonia solution (25.0%) was of UPLC grade, purchased from Merck (Darmstadt). DMSO (99.5%, LC-MS grade) and DMF (99.9%, HPLC grade) were provided by Sigma-Aldrich (Belgium). Ammonium formate (UPLC grade) was procured from Biosolve BV. The Oasis® hydrophilic–lipophilic-balanced (HLB) cartridges (500 mg, 6 mL) were purchased from Waters (Zellik, Belgium).
P values ranging from 1.04 to 7.51 (Table 1). Stock solutions of amprolium, flubendazole, ivermectin, levamisole, maduramicin, monensin, narasin, nigericin, salinomycin, semduramicin, toltrazuril and toltrazuril-d3 were prepared in methanol (MeOH) at a concentration of 1 mg mL−1. Stock solutions of lasalocid (0.1 mg mL−1) and halofuginone (0.1 mg mL−1) were prepared in acetonitrile (ACN). Stock solutions of diclazuril and diclazuril-methyl (0.1 mg mL−1), 4,4′-DNC and DNC-d8 (1 mg mL−1) and robenidine (1 mg mL−1) were prepared in MeOH/n,n-dimethylformamide (DMF) (90/10, v/v%), ACN/dimethylsulfoxide (DMSO) (75/25, v/v%) and MeOH/DMSO (90/10, v/v%), respectively. From the individual stock solutions, a mixed standard working solution (WO1) of 0.5 μg mL−1 (i.e. amprolium, flubendazole and halofuginone), 4 μg mL−1 (i.e. lasalocid), 5 μg mL−1 (i.e. diclazuril, 4,4′-DNC, maduramicin, monensin, narasin, robenidine, salinomycin, semduramicin and toltrazuril) and 25 μg mL−1 (i.e. ivermectin and levamisole) was prepared in ACN. Serial dilution resulted in mixed standard working solutions of 0.05, 0.4, 0.5 and 2.5 μg mL−1 (WO2) and 0.005, 0.04, 0.05 and 0.25 μg mL−1 (WO3) in ACN, respectively. Additionally, a mixed standard working solution (WOIS) (1 μg mL−1) containing the selected ISs was prepared in ACN. Stock and working solutions were stored at ≤−15 °C. A Milli-Q Advantage A10® System (Merck Millipore, Darmstadt, Germany) was used to obtain ultrapure water. The organic solvents were of UPLC grade, purchased from Biosolve BV (Valkenswaard, The Netherlands). Formic acid (99.5%) and ammonia solution (25.0%) was of UPLC grade, purchased from Merck (Darmstadt). DMSO (99.5%, LC-MS grade) and DMF (99.9%, HPLC grade) were provided by Sigma-Aldrich (Belgium). Ammonium formate (UPLC grade) was procured from Biosolve BV. The Oasis® hydrophilic–lipophilic-balanced (HLB) cartridges (500 mg, 6 mL) were purchased from Waters (Zellik, Belgium).
Table 1 Mass spectrometric parameters for the 12 registered coccidiostats, 3 anthelmintics and 4 internal standards included in the multi-residue UHPLC-MS/MS method. IS = internal standarda
| Compounds |
IS |
Elemental formula |
Theoretical mass (g mol−1) |
t
R (min) |
ΔtRb (min) |
ESI precursor ion |
Precursor ion (m/z) |
Quantifier ion (m/z) |
Qualifier ion (m/z) |
Cone voltage (V) |
Collison energy (eV) (A–B) |
|
m/z = mass-to-charge ratio; (A–B): collision energy for the quantifier (A) and qualifier ion (B), respectively.
ΔtR = standard deviation of the retention time originating from 3 samples in three different analytical batches using an Acquity HSS T3 UPLC column (1.8 μm particle size) and Vanguard. Acquity HSS T3 UPLC pre-column (1.8 μm particle size).
|
|
Coccidiostats
|
| 4,4′-Dinitrocarbanilide |
a |
C13H10N4O5 |
302.1 |
8.21 |
0.010 |
[M − H]− |
301.0 |
137.1 |
107.0 |
30 |
10–25 |
| Amprolium |
c |
C14H19ClN4 |
278.1 |
0.90 |
0.012 |
[M − Cl]+ |
243.0 |
150.0 |
94.0 |
20 |
15–10 |
| Diclazuril |
b |
C17H9Cl3N4O2 |
406.0 |
8.63 |
0.010 |
[M − H]− |
405.0 |
335.9 |
333.8 |
30 |
20–20 |
| Halofuginone |
c |
C16H17BrClN3O3 |
414.7 |
5.17 |
0.010 |
[M + H]+ |
416.0 |
120.0 |
100.0 |
50 |
20–20 |
| Lasalocid |
c |
C34H54O8 |
590.4 |
12.13 |
0.026 |
[M + Na]+ |
613.2 |
595.2 |
377.2 |
30 |
20–30 |
| Maduramicin |
c |
C47H80O17 |
916.5 |
11.30 |
0.006 |
[M + Na]+ |
939.5 |
877.3 |
859.3 |
30 |
30–30 |
| Monensin |
c |
C36H62O11 |
670.4 |
11.04 |
0.012 |
[M + Na]+ |
693.39 |
675.3 |
461.3 |
40 |
40–40 |
| Narasin |
c |
C43H72O11 |
765.0 |
11.68 |
0.015 |
[M + Na]+ |
787.5 |
531.0 |
431.0 |
20 |
40–45 |
| Robenidine |
c |
C15H13Cl2N5 |
333.1 |
7.04 |
0.012 |
[M + H]+ |
334.0 |
155.0 |
137.7 |
20 |
20–25 |
| Salinomycin |
c |
C42H70O11 |
750.5 |
11.43 |
0.015 |
[M + Na]+ |
773.1 |
431.3 |
531.4 |
30 |
50–50 |
| Semduramicin |
c |
C45H76O16 |
872.5 |
10.76 |
0.006 |
[M + Na]+ |
895.5 |
833.5 |
851.2 |
30 |
40–30 |
| Toltrazuril |
d |
C18H14F3N304S |
425.4 |
8.93 |
0.017 |
[M − H]− |
423.9 |
423.9 |
— |
20 |
5 |
|
|
Anthelmintics
|
| Flubendazole |
c |
C16H12FN3O3 |
313.1 |
6.75 |
0.006 |
[M + H]+ |
313.9 |
281.8 |
122.7 |
30 |
20–20 |
| Ivermectin |
c |
C48H74O14 |
875.1 |
11.18 |
0.006 |
[M + Na]+ |
897.3 |
752.9 |
609.0 |
30 |
40–40 |
| Levamisole |
c |
C11H12N2S |
204.1 |
0.91 |
0.012 |
[M + H]+ |
204.8 |
177.9 |
122.9 |
30 |
10–20 |
|
|
IS
|
| 4,4′-Dinitrocarbanilide-d8 (a) |
IS |
C13H2N4O5D8 |
310.3 |
8.21 |
0.023 |
[M − H]− |
309.1 |
141.0 |
111.0 |
30 |
10–20 |
| diclazuril-methyl (b) |
IS |
C18H11Cl3N4O2 |
421.7 |
8.84 |
0.006 |
[M − H]− |
420.9 |
348.0 |
321.0 |
40 |
30–30 |
| Nigericin (c) |
IS |
C40H68O11 |
725.5 |
11.71 |
0.010 |
[M + NH4]+ |
742.3 |
729.6 |
657.2 |
30 |
30–30 |
| Toltrazuril-d3 (d) |
IS |
C18H14F3N304SD3 |
428.4 |
8.94 |
0.010 |
[M − H]− |
426.9 |
426.9 |
— |
20 |
5 |
2.2 Instrumentation
Chromatographic separation was achieved using an Acquity H-Class ultra-high performance liquid chromatograph (UHPLC) system (Waters, Milford, USA) equipped with an Acquity HSS T3 100 × 2.1 mm UPLC column (1.8 μm particle size) and Acquity Vanguard HSS T3 10 × 2.1 mm UPLC pre-column (1.8 μm particle size), both from Waters. Column and autosampler temperature were set at 45 °C and 7 °C, respectively. The injection volume was 5 μL. Gradient elution was established with a mobile phase consisting of 0.01% formic acid in Milli-Q Ultrapure water (mobile phase A), MeOH/ACN (50/50, v/v%) (mobile phase B) and MeOH containing 1 mM ammonium formate (mobile phase C) at a flow rate of 0.3 mL min−1. The gradient program was as follows: 0.0–1.0 min, 85% A and 15% B; 1.0–8.2 min: linear gradient to 5% A and 95% B; 8.2–9.0 min: linear gradient to 100% C; 9.0–14.0 min: 100% C; 14.0–14.2 min: linear gradient to 85% A and 15% B; 14.2–20.0 min: 85% A and 15% B. Analytes were detected using a Waters Xevo® TQ-XS triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) interface. The settings on the Xevo® TQ-XS mass spectrometer were as follows: desolvation gas flow rate: 800 L h−1; desolvation temperature: 600 °C; cone gas flow rate: 150 L h−1; source temperature: 150 °C. The capillary voltage was optimized at 3.0 kV and 2.5 kV for ESI positive and ESI negative mode, respectively. Dwell times between 10 and 110 ms per transition were selected for each analyte separately. The Xevo® TQ-XS mass spectrometer was operated in the selected reaction monitoring (SRM) mode. For every compound, the most intense and second most intense product ions were selected for quantification (i.e. quantifier) and qualification (i.e. qualifier), respectively. The compound specific MS/MS parameters at the selected ionization mode (ESI negative or positive) are presented in Table 1.
2.3 Sample preparation and solid-phase extraction
2.3.1 Extraction optimization.
Five parameters that could affect the extraction efficiency were selected based on literature, i.e. the addition of ethylenediaminetetraacetic acid (EDTA) to prevent complexation of ionophores with Ca2+, Mg2+ and residual metal ions,12 the addition of sodium chloride (NaCl) to facilitate conversion of ionophores into single sodium adducts,36 acidification of the sample for a better interaction between the SPE sorbent and the drug14 and different SPE loading and elution volumes to improve sensitivity.15,37,38 Following a previously described approach,39 the software program JMP 12.0 (SAS Institute Inc, Cary, USA) was used to fit and model a statistical screening design (fractional factorial resolution or FRR) consisting of 18 experiments taking into account different options for the selected extraction parameters (Table S2†). Experiments were conducted using tap water spiked with each individual analyte to reach a concentration of 50 ng L−1. To this extent, stock solutions were diluted with ACN to establish individual working solutions with a concentration of 0.5 μg mL−1 and 500 mL water samples were spiked with 50 μL of the working solution. For each experiment, the normalized peak areas of the analytes was summed (AUCSUM), taking into account the number of analytes detected while ensuring equal compound distribution. Using the statistical FFR design, the optimized value for each of the selected extraction parameters was provided based on the highest AUCSUM. Obtained results were evaluated to ensure a signal-to-noise ratio (S/N) ≥ 3, adequate peak shape and baseline separation. One-way analysis of variance (ANOVA) at a confidence interval of 95% was used to statistically evaluate responses and retain parameters with a significant effect (p-value < 0.05).
2.3.2 Sample preparation and extraction.
Grab water samples were collected with plastic buckets and sieved on spot to eliminate large particles (Retsch® sieve, Novolab NV, Geraardsbergen, Belgium, 250 μm, 50 × 200 mm). Samples were transported in cooled glass amber bottles and stored for less than 96 hours at 4 °C. For each analyte, the appropriate volume of mixed standard working solution was added to blank pond water (500 mL) and carried throughout the extraction procedure to establish a total of 10 calibration points. A detailed protocol concerning the fortification of different calibrator samples was added to the ESI (Table S3†). Furthermore, 25 μL of the IS working solution (1 μg mL−1) was added to 500 mL samples to reach a concentration of 50 ng L−1. All 500 mL samples were filtered a second time through a 0.45 μm glass fiber filter (Whatman™, Buckinghamshire, UK) to remove solid particles. Prior to extraction, the pH of each sample was measured and if necessary, adjusted by acidification or alkalinization to achieve a pH of 7 using formic acid or ammonia 25% solution, respectively. An Oasis® hydrophilic–lipophilic balanced (HLB) cartridge (6 mL, 500 mg, Waters) was used to extract analytes from water and served as a cleanup and concentration step in the sample preparation. The cartridge was pre-conditioned with 6 mL of methanol and 6 mL of ultrapure water to remove any potential traces and liberate adsorption sites. Samples were loaded under vacuum at 15 mmHg Next, the cartridge was washed with 5 mL of ultrapure water and dried under vacuum for 5 min. Analytes were eluted subsequently with 5 mL of methanol and 5 mL of methanol acidified with 0.1% formic acid (i.e. concentration step). Finally, 200 μL of the combined eluate was transferred to an UHPLC autosampler vial containing 50 μL of ultrapure water followed by 10 seconds of vortex mixing. This procedure resulted in an overall concentration factor of 40. All vials were immediately subjected to UHPLC-MS/MS analysis.
2.4 Method validation
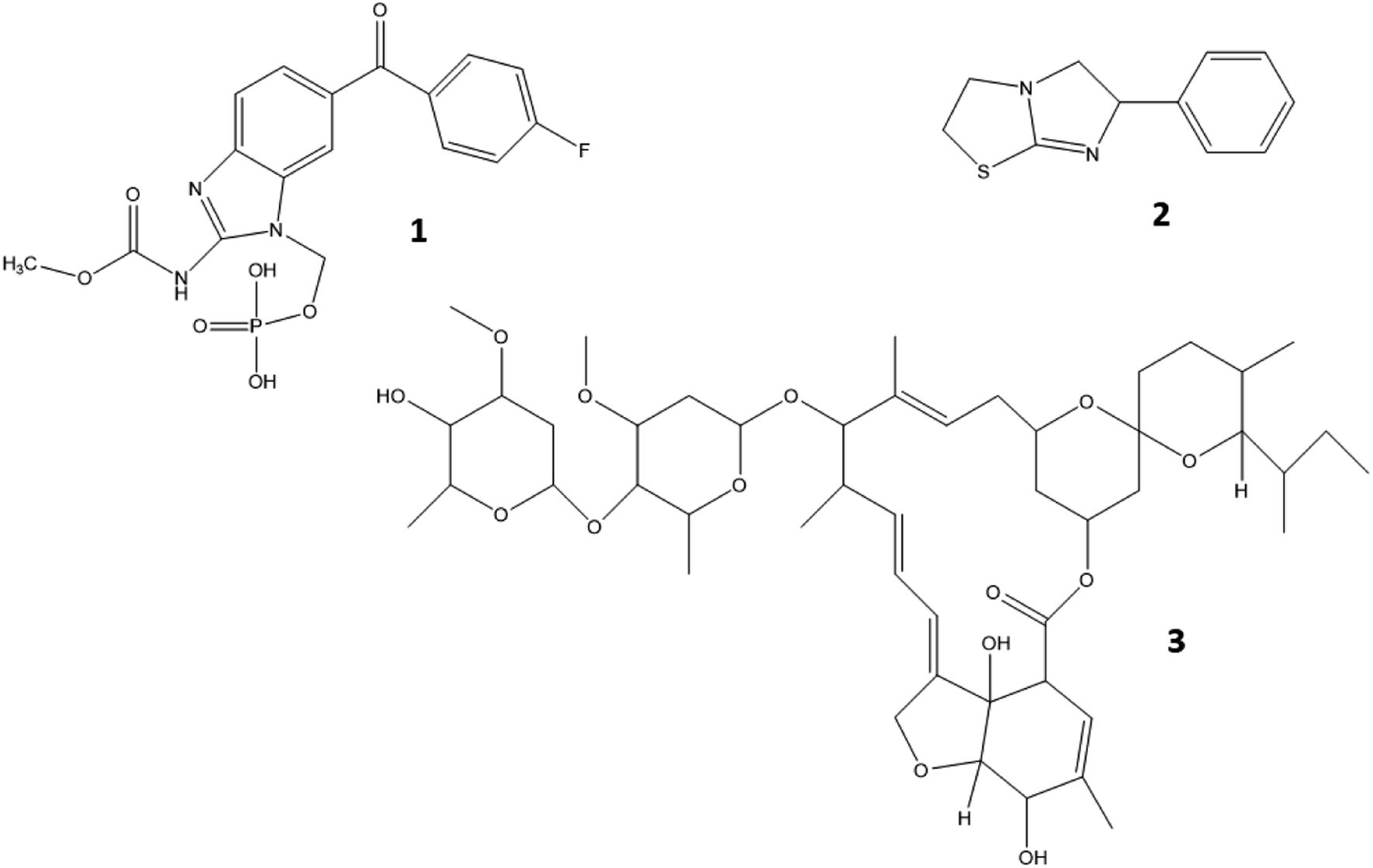
Guidelines concerning validation for the analysis of micro-pollutants in fresh water ecosystems are lacking. The only European guideline that is available is CD 2009/90/EC in which the water status is analytically evaluated by defining environmental quality standards (EQS).40,41 It is stated in the guideline that the measurement uncertainty must be below 50% and the detection limit (LOD) has to be 30% below the EQS, the latter defined as the concentration of a substance in water that should not be exceeded in order to sustain the environmental quality objective.41 In the directive, EQS are stated for several heavy metals, pesticides and organic compounds based upon their aquatic toxicity and bioaccumulating properties.42 However, no EQS are stated for any of the compounds included in the current study and other validation guidelines, i.e. CD 2002/657 of the European Commission,43 SANTE/12682/2019,44 EMA45 and VICH 49 guidelines46 were consulted to ensure complete analytical method validation. A set of parameters was used to evaluate method performance: specificity, linearity, LOD and limit of quantification (LOQ), pond water filtration efficiency, matrix effects, within-day and between-day apparent recovery (Rapp) (universally referred to as “trueness”), within-day and between-day precision and carry-over. According to the European guideline CD 2002/657, laying down the performance criteria of analytical methods, four identification criteria should be fulfilled to ensure identity confirmation of the detected analytes: one precursor and two product ions should be detected, the relative intensity of the detected product ions (i.e. ion ratio) should correspond to the product ions of the calibration curve (i.e. reference ion ratio) within accepted deviation margins, the relative retention time of the analytes must range within a margin of 2.5% and a signal-to-noise ratio (S/N) of at least 3 is obtained for each compound. The ion ratio is calculated as the intensity (or peak area) ratio of the less intense product ion (qualifier) to that of the more intense product ion (quantifier). The reference ion ratio is calculated as an average of ion ratios of calibration solutions. To assess compliance of ion ratios in samples, the following equation was used:| |  | (1) |
To ensure stringent method validation a maximum relative difference of 30% was accepted according to the recent SANTE/12682/2019 guideline, which is more strict than the European guideline CD 2002/657.43,44
2.4.1 Specificity, linearity, LOD and LOQ.
Specificity was determined by investigating the absence of possible interfering chromatographic peaks within the retention time margin of 2.5% for all analytes in three pond water samples, blank for the compounds of interest. Different matrix-matched calibration curves were obtained for different compounds with concentrations ranging between 8 and 10000 ng L−1 (Table 2). The coefficient of determination (R2) and goodness-of-fit (g) (%) were used to evaluate and confirm linearity, the latter being a better criterion to evaluate the calibration curve, since it considers the difference between the nominal values of the calibration samples and the calculated concentration (eqn (2)). According to the guidelines discussed above, acceptance criteria of R2 and g were set at ≥0.99 and ≤20%.43,47| | 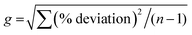 | (2) |
with  .
.
Table 2 Validation results for linearity (linear range, number of calibration points used to calculate linear range (n), coefficient of determination (R2) and goodness-of-fit coefficient (g)), limit of quantification (LOQ) and limit of detection (LOD) for the coccidiostats and anthelmintics included in the multi-residue UHPLC-MS/MS method
|
|
Linear range, ng L−1 |
Calibration points, n |
R
2
|
g, % |
LOD, ng L−1 |
LOQ, ng L−1 |
|
Coccidiostats
|
| 4,4′-Dinitrocarbanilide |
10–2000 |
9 |
0.9990 |
3.89 |
1.5 |
10 |
| Amprolium |
2.5–200 |
8 |
0.9943 |
8.91 |
1.3 |
2.5 |
| Diclazuril |
10–1000 |
8 |
0.9979 |
5.52 |
2.2 |
10 |
| Halofuginone |
2.5–200 |
8 |
0.9983 |
4.82 |
0.7 |
2.5 |
| Lasalocid |
8–1600 |
10 |
0.9990 |
3.95 |
1.5 |
8 |
| Maduramicin |
25–2000 |
7 |
0.9973 |
6.16 |
8.5 |
25 |
| Monensin |
10–2000 |
9 |
0.9983 |
5.19 |
4.0 |
10 |
| Narasin |
10–2000 |
8 |
0.9990 |
3.98 |
4.3 |
10 |
| Robenidin |
25–2000 |
7 |
0.9939 |
9.22 |
1.8 |
25 |
| Salinomycin |
10–2000 |
9 |
0.9988 |
4.39 |
2.7 |
10 |
| Semduramicin |
25–2000 |
7 |
0.9992 |
3.41 |
12.8 |
25 |
| Toltrazuril |
10–500 |
8 |
0.9977 |
5.63 |
4.3 |
10 |
|
|
Anthelmintics
|
| Flubendazole |
2.5–2000 |
10 |
0.9991 |
3.63 |
1.0 |
2.5 |
| Ivermectin |
125–10000 |
7 |
0.9963 |
7.83 |
32.3 |
125 |
| Levamisole |
250–10000 |
8 |
0.9980 |
5.08 |
40.0 |
250 |
For the determination of the LOQ, blank samples were spiked within the expected concentration range determined during method optimization (8–10000 ng L−1). The LOD was calculated as 3 times the standard deviation of the y intercept divided by the average slope of three independent calibration curves. Furthermore, detected peaks and relative retention time deviations (≤2.5%) were evaluated and a S/N ratio ≥ 3 was considered acceptable for the estimation of LOD. The LOQ was determined using spiked samples as the lowest concentration that could be analyzed within the limits of apparent recovery and precision according to the guidelines described above.43
2.4.2 Filtration efficiency and matrix effects.
Filtration efficiency was determined by comparing peak areas of analytes spiked before filtration (i.e. filtration step 1 and 2) to peak areas of analytes spiked after filtration at calibrator level no 8 (Table S3†). To this extent, pond water samples were used, originating from 3 different ponds in Flanders, situated in different geological regions (Table S6†). The corresponding precision was assessed by calculating the relative standard deviation (RSD, %) of the obtained results per compound.
Matrix effects (ME) were assessed in pond water samples, resulting from 20 different ponds in Flanders, Belgium (Table S5†) and at two concentration levels, i.e. one low (calibrator no 5) and one high level (calibrator no 8) (Table S3†). Following the approach described by Matuszweski et al. (2003),48 ME were calculated as follows (eqn (3)):
| | 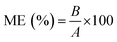 | (3) |
For which A denotes the peak area of the analyte in neat solution and B the peak area of the analyte spiked after extraction. The corresponding precision was assessed by calculating the RSD (%) of the obtained results per compound. Furthermore, filtration efficiency, ME and corresponding precision were recalculated using peak area ratios instead of absolute peak areas, to evaluate if selected ISs could adequately compensate for loss of analyte due to filtration and ME, and improve corresponding precision.
2.4.3 Apparent recovery, precision and carry-over.
Within-day and between-day apparent recovery (Rapp, %) is defined as the ratio between the measured concentration and the theoretical (spiked) concentration and was determined by analyzing respectively six and three blank samples spiked at three different concentration levels (high, medium and low concentration level) on three different days (Table S4†).46 The measured concentration was calculated from a matrix-matched calibration curve. According to the VICH 49 guideline, acceptance criteria for within-day and between-day Rapp are between 50 and 120%.46 However, to ensure rigorous validation, more stringent acceptance criteria were applied, i.e. between 70 and 120%, as proposed by the SANTE/12682/2019 guideline.44 Within-day precision (repeatability) and between-day precision (reproducibility) were determined by calculating the residual standard deviation (RSD, %) of respectively 6 and 3 spiked concentration levels during three subsequent days.46 According to the VICH 49 guideline46 for the validation of analytical methods, obtained RSD values must be ≤30% for the within-day precision and ≤45% for the between-day precision when dealing with analyte concentrations <1000 ng L−1. However, because the European guideline CD 2002/657 states that for concentrations below 100 μg L−1, values for precision should be as low as possible, a maximum value of 25% was considered acceptable for validation.43 Finally, carry-over was evaluated by injecting three solvent samples containing ultrapure water and MeOH (1/4, v/v%) directly after the highest calibrator in each curve. Ideally no peaks eluting at the same retention time as the analyte should be detected. If however signal detection is observed at equal retention time, the response of the peak should not exceed 10% of the mean peak area of the analyte in the LOQ samples.43,46
2.4.4 Sample storage stability study.
To fulfill the requirements of the EMA guidelines,45 stability was assessed for all compounds at a high (cal. 8) and low (cal. 5) concentration level in pond water samples originating from 3 different ponds. Grab samples were collected in amber glass bottles and filtered before dividing into 500 mL subsamples. Three subsamples per pond were analyzed immediately to ensure the absence of compounds and verify if the representative samples were truly blank. Subsequently, three subsamples per pond were spiked with a mixture of all 15 compounds to reach a high and low concentration of 50, 400, 500 or 2500 ng L−1, and 5, 40, 50 or 250 ng L−1, respectively, according to the measured analyte (Table S3†). Samples were stored at 4 °C in the dark until analysis 96 hours later. Prior to extraction, ISs were added and each 500 mL sample was filtered, extracted and analyzed as described before. Stability, expressed as recovery (RST), was evaluated according to the method of Kruve et al. (2015)49 by using the chromatographic peak areas, as follows (eqn (4)):| | 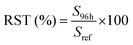 | (4) |
where Sref represents the peak area of the compound analyzed immediately after spiking and S96h represents the peak area of the compound analyzed after 96 hours of storage in glass amber bottles at 4 °C. By using pond water from 3 different ponds, situated in different geological regions, the applicability of the stability test was assessed. Coordinates and corresponding soil types of the 3 different ponds are presented in Table S6 of the ESI† data.
2.5 Application to environmental samples
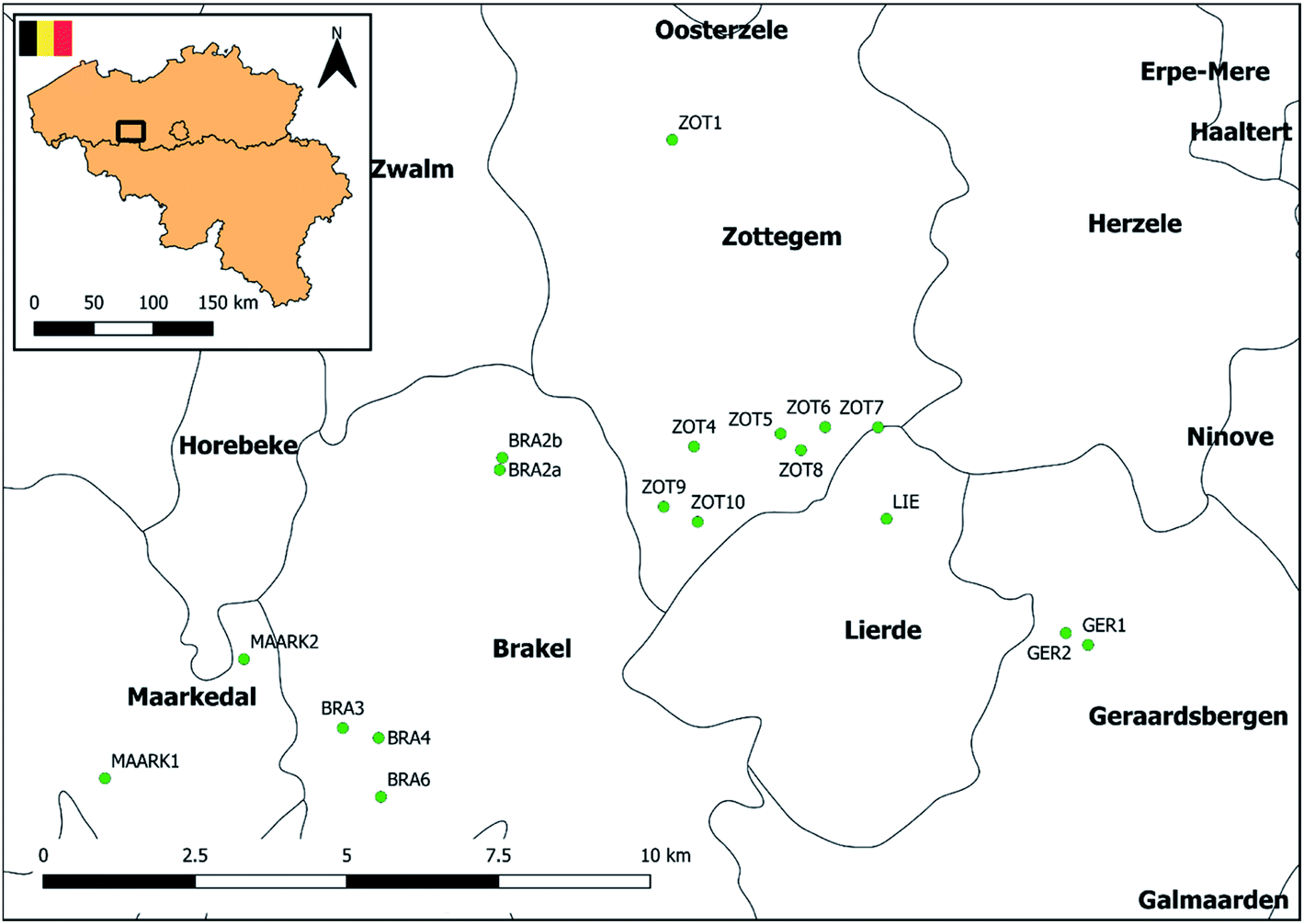
The applicability of the method was evaluated using grab water samples collected from 18 different ponds situated in agricultural areas across Flanders, Belgium (Fig. 3). Samples were taken between August and October 2018. Initial information on pond localization in Belgium was provided by the database of the Pondscape project,50 following a geographical selection to those located in Flanders using QGIS 2.14 software. Definitions of the term ‘pond’ may vary and there is no universal agreement of what a pond is.28 However, in this study, we defined ponds based on the study of Oertli et al. (2005)51 and Biggs et al. (2005)52 as permanent or seasonal waterbodies between 1 m2 and 20000 m2 in area, with a maximum depth of no more than 8 m. A field study was performed to exclude permanent dried up ponds or ponds with <1 m2 in area or >8 m in depth. Finally, information on surrounding agricultural land use was gathered through the Geopunt database (Land Cover Data 2018, LANDBGBRP, shapefile) and ponds without surrounding agricultural activities within 200 m were withdrawn from the study. Site locations, coordinates and surrounding agricultural land use of the 18 selected ponds are presented in Fig. 3, Tables S7 and S8,† respectively. Three types of pond shape were distinguished, i.e. rectangular, elliptic and circular. Three locations were identified along the shore of the ponds situated equidistantly from each other. At two of these locations, surface water was collected at <1 m from shore. At the third location surface water was collected at >1 m from shore (Fig. S4†). Finally, samples were sieved (Retch sieve®, 250 μm) and pooled per pond. Following cooled transport, samples were stored at 4 °C until analysis within 96 hours.
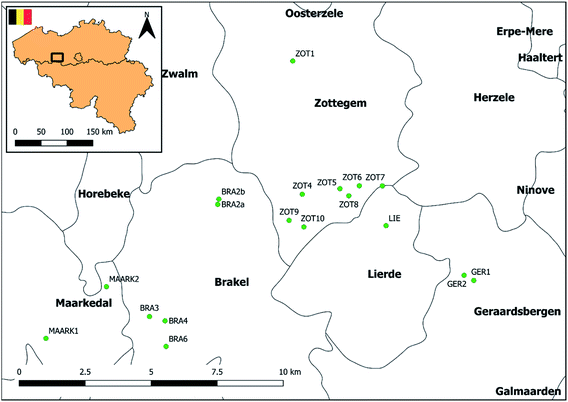 |
| | Fig. 3 Site locations of the sampled fresh water ponds in Flanders, Belgium. | |
2.5 Data analysis
LC-MS/MS data acquisition and data processing were performed with Masslynx software version 4.2 (Waters). Compound identification was realized by the relative intensity of the detected ions corresponding to the ions of the calibration curve within accepted deviation margins, S/N ratio (≥3) and the retention time relative to that of the IS (deviation ≤ 2.5%), all being investigated from the corresponding reference standard.
3. Results and discussion
3.1 Method development
3.1.1 UHPLC-MS/MS conditions.
Tuning was performed by direct infusion of a 10, 100 and 1000 ng L−1 standard mix working solution (flow: 10 μL min−1) in combination with the mobile phase (200 μL min−1, A/B: 50/50, v/v) in positive (ESI+) and negative ion (ESI−) mode. According to the optimal signal intensity for each analyte, adducts were selected as precursor ion (i.e. [M + NH4]+, [M + Na]+, [M + H]+ and [M − H]−) and cone voltages were optimized (Table 1). The ability of ionophoric coccidiostats to bind sodium frequently results in the formation of sodium adducts even with the use of UHPLC-MS/MS graded solvents.53 However, amprolium lacks such ability and exists as positively charged precursor ion in aqueous solution. For this reason the cationic fraction of the molecule (without chloride) was selected as precursor ion.54 Next, to determine the two most optimal product ions for the SRM transitions, different collision energies were applied. The most intense product ion was used for quantification while other product ions were used for identification (Table 1). In the case of toltrazuril, only the precursor ion was selected for further optimization since application of the collision energy yielded insufficient fragmentation patterns consisting of small fragments (<100 m/z) with low signal intensity (<35%). For this reason, the detection of this analyte is rather suitable for screening purposes, as it did not yield a sufficient fragmentation pattern to meet the CD 2002/657 identification criteria.
An Acquity HSS T3 UPLC column (1.8 μm particle size) and Vanguard Acquity HSS T3 UPLC pre-column (1.8 μm particle size) was tested, based upon literature,55 resulting in narrow and symmetric peaks. A selection of mobile phase compositions was based upon literature.12,56 Two different buffer additives (i.e. 0.1% formic acid and 0.1% ammonium formate) were evaluated for the aqueous (i.e. Milli-Q Ultrapure water) and organic solvents (i.e. MeOH and ACN). Optimization of the organic mobile phase demonstrated higher peak intensities for 7 compounds (i.e. ivermectin, lasalocid, maduramicin, monensin, narasin, salinomycin and toltrazuril) in the presence of MeOH containing 0.1% ammonium formate, whereas higher peak intensities were obtained in the presence of ACN/MeOH (50/50, v/v%) for amprolium, diclazuril, 4,4′-DNC, flubendazole, halofuginone, levamisole and robenidine. Subsequently, it was found that the combination of the Acquity HSS T3 column with one aqueous (mobile phase A) and two organic (mobile phase B and C) solvents gave the best retention, peak shape and separation for all compounds within an acceptable run time of 20 minutes. These findings are in line with literature using three mobile phases and an ammonium formate additive for the detection of various coccidiostats.57,58
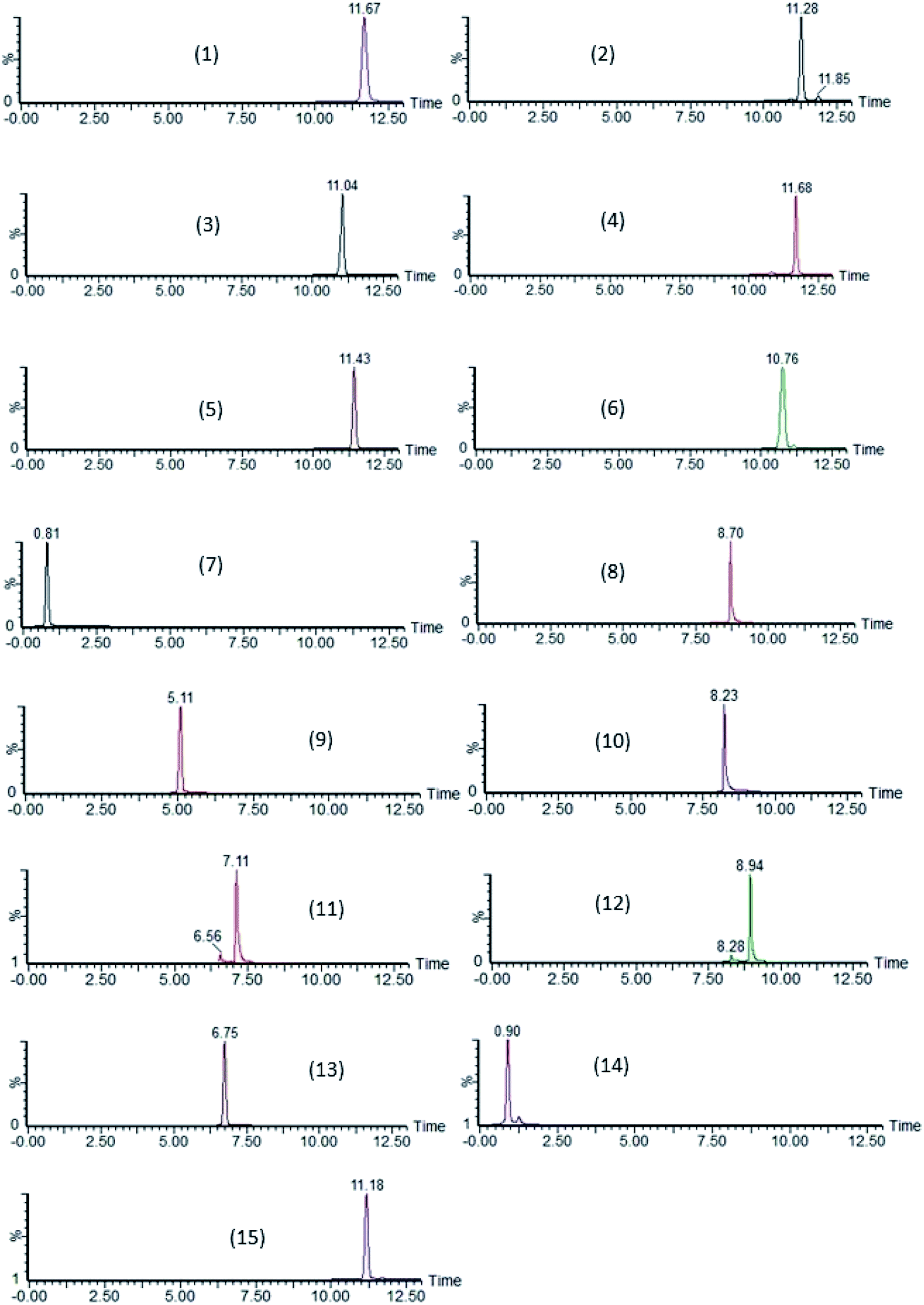
By further studying the symmetry of the peak shape (Asminimal = 1.50 and Asoptimal = 1.00) and resolution, the optimal conditions of the stationary phase, flow rate, solvent gradient, column temperature and injection volume were determined. Separation of the 15 compounds was achieved covering a broad polarity range (logP ranging from 1.04 to 7.51) with retention times ranging from 0.90 to 12.13 min (Table 1). Chromatograms of each compound are presented in Fig. 4.
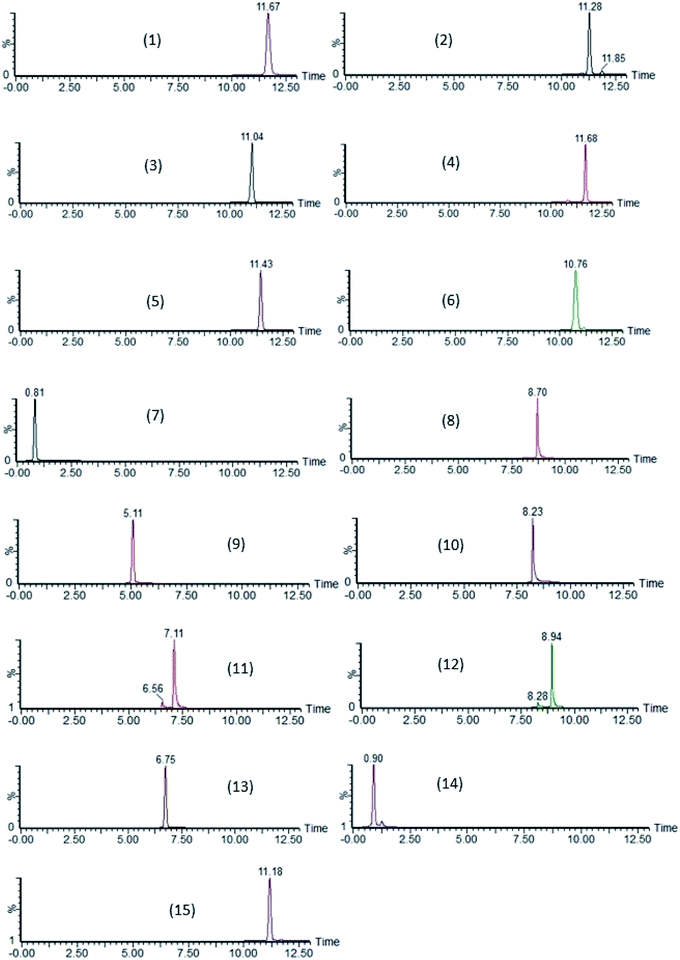 |
| | Fig. 4 SRM chromatograms of lasalocid (1), maduramicin (2), monensin (3), narasin (4), salinomycin (5), semduramicin (6) (ionophoric coccidiostats) and amprolium (7), diclazuril (8), halofuginone (9), 4,4′-DNC = 4,4′-dinitrocarbanilide (10), robenidine (11), toltrazuril (12) (non-ionophoric coccidiostats), flubendazole (13), levamisole (14) and ivermectin (15) (anthelmintics) using the validated method on water samples spiked at LOQ-concentration level. Quantification ions are shown. | |
3.1.2 Sample extraction.
Although solid-phase extraction is the most commonly used technique for extracting veterinary drugs from aquatic matrices, a direct and simple SPE-method for the determination of coccidiostats and anthelmintics remains challenging due to their diverse chemical structures and properties2,14 (Table 1 and Fig. 1 and 2). For this reason, an Oasis® HLB cartridge was selected to cover a broad polarity range of compounds. The lipophilic divinylbenzene and hydrophilic N-vinylpyrrolidone co-polymer in this cartridge provided good results for all polar and apolar analytes included in this study (logP = 1.04–7.51) and no other purification techniques were tested. Conditioning (i.e. MeOH and ultrapure water) and elution solvents (i.e. MeOH) were selected based upon previous literature,2,14,54,59 resulting in acceptable recoveries and sensitivity for all compounds, except for robenidine. However, by subsequently eluting cartridges with methanol acidified with formic acid, recoveries for robenidine (logP = 3.17) ameliorated. To avoid degradation of the acid labile ionophores,60 samples were subjected immediately to UHPLC-MS/MS analysis. Further optimization was performed using a fractional factorial resolution experimental design in which the effect of the selected parameters on the extraction efficiency of the 15 compounds was assessed (Fig. S1†). The addition of NaCl was not significant (p-value > 0.05; F1,1 = 4.7), whereas addition of EDTA, pH adjustment of the sample, loading and elution volume exerted a significant effect (p-value < 0.05 and F1,1 = 15.5, 5.5, 45.6 and 26.8, respectively) on AUCSUM of the compounds (Fig. S2†). More specific, a higher AUCSUM was observed in the absence of EDTA, higher elution volumes (>4 mL) and higher loading volumes (>300 mL) (Fig. S3†). For practical reasons, an optimal loading volume of 500 mL was selected since higher volumes tended to clog the sorbent phase, resulting in long loading times. Additionally, best results were obtained for samples at pH 7. In this respect, analyzing samples with a pH value that differs greatly from 7 is considered less optimal and sample pH should be adjusted accordingly (Fig. S3†). These findings are in line with previous research regarding feed additives in the aquatic environment and are related to the hemiacetal or ketal structures of the ionophores that tend to be acid labile.60
3.2 Method validation
3.2.1 Specificity, linearity, LOD and LOQ.
Good specificity implicates the ability of the method to distinguish between the target analyte and other substances present in the sample.43 No interfering peaks (S/N ratio > 3) were detected within the retention time margin of 2.5% for all analytes in any of the blank samples analyzed, confirming the specificity of the method. Linear calibration curves were constructed for each compound and good linearity was obtained for all target compounds with R2 ≥ 0.994 and g ≤ 9.22% (Table 2). A weighted least-squares regression (WLSLR) applying an optimal weighing factor of 1/x2 was used to compensate the observed heteroscedasticity. LOD values ranged from 0.7 to 12.8 ng L−1 for coccidiostats and from 1.0 to 40.0 ng L−1 for anthelmintics. LOQ values were between 2.5 and 25.0 ng L−1 (coccidiostats) and between 2.5 and 250.0 ng L−1 (anthelmintics). Regarding coccidiostats, obtained results are better than those reported in previous literature using HPLC-MS/MS for the detection of maximum 9 compounds in surface water.14,59,61 However, a recent study, comprising a total of 26 coccidiostats in surface and ground water showed lower values for LOD and LOQ for all compounds, except for toltrazuril for which obtained values were two times higher than those obtained in the current study.25 To the authors' knowledge, no studies have been published applying an UHPLC-MS/MS method for the detection of coccidiostats in pond water. In comparison to a recent study detecting multiple anthelmintics in surface water using UHPLC-MS/MS,62 values for LOD and LOQ are slightly lower than those obtained in the current study i.e. 0.1 and 1.0 ng L−1versus 1.0 and 2.5 ng L−1 for flubendazole, respectively. These findings illustrate the well-known limitation of multi-residue methodologies, where not the best conditions for all individual target analytes can be achieved but a compromise on final analytical conditions has to be made, compromising on method sensitivity. However, taking reported EC50, 48h and LOEC21d values for various aquatic organisms into account, the authors can state that the obtained LODs are sufficient to evaluate potential ecotoxicity for the majority of the organisms.16–20,29
3.2.2 Filtration efficiency and matrix effects.
Experiments showed good results for the filtration efficiency, with values ranging between 80 and 118% for all compounds. Furthermore adequate precision was demonstrated, with RSD values <20% for all compounds, except for robenidine and ivermectin, for which precision was 25%. However, by incorporating ISs, precision values for robenidine and ivermectin improved, resulting in RSD values of 9% and 3%, respectively. Results of filtration efficiency and corresponding precision with and without taking the ISs into account, are presented in Table 3.
Table 3 Filtration efficiency (%) and corresponding precision (RSD, %) of the 15 veterinary drugs included in the multi-residue UHPLC-MS/MS method, using samples from 3 different ponds in Flanders, Belgium. Samples were spiked at calibrator level no 8, i.e. 50, 400, 500 or 2500 ng L−1, according to the measured analyte
|
|
Filtration efficiency (%) |
RSD (%) |
| Mean (n = 3) |
n = 3 |
| No IS |
With IS |
No IS |
With IS |
|
Coccidiostats
|
| 4,4′-Dinitrocarbanilide |
89.6 |
112.9 |
12.4 |
6.7 |
| Amprolium |
102.7 |
104.3 |
2.7 |
8.6 |
| Diclazuril |
100.7 |
94.2 |
8.5 |
5.8 |
| Halofuginone |
107.8 |
112.0 |
5.1 |
0.1 |
| Lasalocid |
88.2 |
106.3 |
14.8 |
13.9 |
| Maduramicin |
80.8 |
96.1 |
12.7 |
12.8 |
| Monensin |
102.2 |
113.1 |
14.8 |
6.5 |
| Narasin |
90.4 |
109.1 |
14.2 |
15.1 |
| Robenidine |
79.5 |
95.0 |
25.0 |
9.1 |
| Salinomycin |
91.0 |
109.8 |
14.5 |
14.9 |
| Semduramicin |
91.7 |
93.6 |
0.1 |
4.5 |
| Toltrazuril |
101.8 |
101.5 |
1.4 |
11.6 |
|
|
Anthelmintics
|
| Flubendazole |
117.8 |
117.7 |
8.5 |
5.7 |
| Ivermectin |
105.7 |
94.0 |
24.5 |
10.1 |
| Levamisole |
80.2 |
106.9 |
5.9 |
3.2 |
Matrix effects are caused by competition between analyte and co-eluting matrix compounds with primary ions formed in the LC-MS/MS interface, resulting in an enhanced or suppressed ionization of the analyte.48 Analyte quantification can be strongly affected by matrix effects, which can be expected in a complex matrix such as pond water, implying the need to examine this further. According to the SANTE/12682/2019 criteria,44 obtained results for ME (n = 20) were acceptable for 14 out of 15 compounds with values ranging between 81.7 and 108.9% for analytes spiked at low concentration, and between 80.4 and 119.1% for analytes spiked at high concentration. Slightly lower values (i.e. 71.5 and 76.5%) were obtained for robenidine. Furthermore, precision was evaluated between the 20 different samples, with RSD values ranging between 7.0 and 38.6% for analytes spiked at low concentration and 7.4 and 22.6% for analytes spiked at high concentration. By incorporating ISs, overall results for ME improved and adequate precision values (i.e. ≤15%) were obtained according to the EMA guidelines.45 Results confirm the suitability of the selected ISs in our newly developed method. Details of ME are summarized in Table 4. The use of ISs and a matrix-matched calibration curve was employed to address any potential ME, further satisfying validation criteria.
Table 4 Results of the matrix effects (ME) and relative standard deviation (RSD) for each of the 15 veterinary drugs, using samples from 20 different ponds in Flanders, Belgium. Samples were spiked at calibrator level no 5 (i.e. low = 5, 40, 50 or 250 ng L−1) and calibrator level no 8 (i.e. high = 50, 400, 500 or 2500 ng L−1)
|
|
Mean ME (%), (n = 20) |
RSD (%), (n = 20) |
| Low |
High |
Low |
High |
| No IS |
With IS |
No IS |
With IS |
No IS |
With IS |
No IS |
With IS |
|
Calculated on 7 pond water samples as other samples contained traces of flubendazole (i.e. 9–16 ng L−1).
|
|
Coccidiostats
|
| 4,4′-Dinitrocarbanilide |
81.7 |
98.4 |
84.9 |
101.2 |
12.0 |
2.4 |
8.2 |
2.2 |
| Amprolium |
106.7 |
104.1 |
81.2 |
86.2 |
38.6 |
15.2 |
12.6 |
13.8 |
| Diclazuril |
94.2 |
99.3 |
101.0 |
103.5 |
13.6 |
2.9 |
8.3 |
3.4 |
| Halofuginone |
88.5 |
85.4 |
100.9 |
95.7 |
7.0 |
8.2 |
7.4 |
9.7 |
| Lasalocid |
84.9 |
84.2 |
93.1 |
93.7 |
12.1 |
3.7 |
8.9 |
8.5 |
| Maduramicin |
107.9 |
103.3 |
102.7 |
103.7 |
14.2 |
9.7 |
18.2 |
4.8 |
| Monensin |
108.9 |
103.2 |
102.7 |
104.2 |
20.1 |
6.7 |
14.8 |
8.6 |
| Narasin |
84.8 |
82.6 |
96.2 |
98.1 |
10.7 |
5.3 |
10.5 |
8.4 |
| Robenidine |
71.5 |
72.1 |
76.5 |
78.6 |
14.6 |
9.1 |
11.2 |
9.5 |
| Salinomycin |
88.3 |
85.3 |
89.3 |
90.5 |
12.9 |
3.6 |
15.7 |
2.7 |
| Semduramicin |
104.4 |
100.1 |
98.2 |
100.0 |
11.4 |
6.6 |
10.7 |
9.9 |
| Toltrazuril |
86.2 |
99.3 |
88.7 |
101.0 |
11.9 |
2.5 |
10.8 |
2.9 |
|
|
Anthelmintics
|
| Flubendazole |
99.6a |
88.9a |
119.1 |
115.0 |
11.8a |
5.1a |
22.6 |
15.3 |
| Ivermectin |
96.9 |
96.0 |
93.9 |
95.8 |
13.8 |
10.3 |
19.4 |
12.0 |
| Levamisole |
83.9 |
83.0 |
80.4 |
81.0 |
19.5 |
15.4 |
16.4 |
10.2 |
3.2.3 Apparent recovery, precision and carry-over.
The validation parameters with respect to apparent recovery and precision are presented in Table S4.† According to the previously described guidelines, Rapp results were within acceptable limits for both group of compounds. Values for within-day Rapp ranged between 73.4 and 112.8% and between 80.0 and 107.3% for all but one of the coccidiostats and anthelmintics, respectively, spiked at high, medium and low concentration level (Table S4†). Between-day Rapp ranged between 73.5 and 112.6% for the coccidiostats and between 81.5 and 115.8% for anthelmintics, implying good accuracy of the validated method. These results are comparable,2,14,15 more variable25 or better62 (i.e. levamisole) than those reported in previous studies detecting coccidiostats or anthelmintics in freshwater. Overall, the within-day and between-day precision RSDs were within acceptable limits, i.e. ≤25%, for all compounds except for robenidine, for which the between-day precision was between 21.0 and 34.5%. This finding could be attributed to the well-known limitation of multi-residue methodologies, where conditions are optimized for the group of compounds rather than the individual analyte, resulting in a compromise on final analytical performance. For this reason, the method may be less fit for quantification of robenidine. Finally, no carry-over was detected in any of the solvent samples containing ultrapure water and methanol (1/4, v/v%) injected directly after the highest calibrator of every compound.
3.2.4 Stability of coccidiostats and anthelmintics in spiked pond water samples.
The stability of the investigated compounds in a biologically active matrix such as pond water, often affected by anthropogenic and agricultural pollution, is a crucial factor affecting the reliability of published data. However, very little research has been performed on the stability of coccidiostats and anthelmintics in pond water27 and half-lives in other aquatic matrices vary between and 7 and >34 days (Table S1†).62 In this study, the stability of coccidiostats and anthelmintics in pond water was evaluated for all compounds after 96 hours of storage at 4 °C in amber glass bottles. By using pond water from 3 different ponds, situated in different geological regions, the applicability of the stability test was assessed, as well. Results indicated that 14 out of 15 compounds were stable under storage conditions for 96 hours, reaching recoveries between 86.6 and 115.6% for analytes spiked at high concentrations, and between 92.7 and 116.7% for analytes spiked at low concentrations (Table 5). Robenidine was not stable under storage conditions for 96 hours, showing a recovery of 18.2 and 29.0% respectively, at high and low spiking level. These findings are in line with previous research16,27,63 and indicate that analysis performed within 96 hours was sufficient to avoid loss by degradation for 14 out of 15 compounds included in this method. In the case of robenidine, no studies have been reported regarding the stability in environmental waters to compare or support these findings.
Table 5 Mean recovery (RST) (n = 3), standard deviation (SD) and stable storage time of coccidiostats and anthelmintics in pond water after storage for 96 hours at 4 °C in amber glass bottles
|
|
Recovery (RST) |
Stable storage time (hours) |
| Mean ± SD (%) |
| Low concentration |
High concentration |
|
Coccidiostats
|
| 4,4′-Dinitrocarbanilide |
97.6 ± 3.6 |
92.7 ± 1.0 |
96 |
| Amprolium |
86.6 ± 4.3 |
96.3 ± 10.2 |
96 |
| Diclazuril |
98.6 ± 3.9 |
104.6 ± 9.7 |
96 |
| Halofuginone |
99.5 ± 2.2 |
100.4 ± 6.4 |
96 |
| Lasalocid |
115.6 ± 8.8 |
116.7 ± 3.8 |
96 |
| Maduramicin |
113.2 ± 7.4 |
115.5 ± 15.5 |
96 |
| Monensin |
102.1 ± 11.9 |
111.8 ± 13.6 |
96 |
| Narasin |
109.3 ± 10.8 |
115.3 ± 10.8 |
96 |
| Robenidin |
29.0 ± 0.6 |
18.2 ± 5.8 |
<96 |
| Salinomycin |
100.8 ± 5.0 |
108.8 ± 17.2 |
96 |
| Semduramicin |
115.5 ± 8.7 |
112.8 ± 13.9 |
96 |
| Toltrazuril |
102.5 ± 2.6 |
111.7 ± 12.1 |
96 |
|
|
Anthelmintics
|
| Flubendazole |
103.1 ± 4.4 |
112.7 ± 11.5 |
96 |
| Ivermectin |
95.0 ± 5.4 |
108.3 ± 2.6 |
96 |
| Levamisole |
90.3 ± 1.5 |
97.4 ± 18.7 |
96 |
3.3 Environmental sample analysis
To demonstrate the applicability of the newly developed UHPLC-MS/MS method, analysis of freshwater samples was performed, originating from 18 different ponds in agricultural areas in Flanders, Belgium. As the pH of each of the pond samples did not differ greatly from 7 (Table S7†), no further pH adjustment was necessary during sample preparation. Except for amprolium, no coccidiostats were detected in any of the ponds. Amprolium was present in one pond (i.e. LIE) at concentrations below the LOQ (<2.5 ng L−1), comparable to the study of Song et al. (2007) in which the compound was detected in surface run-off water from a livestock farm.38 Although recent use as animal feed supplement and subsequent run-off, leaching or drift from fertilized fields could account for the presence in ponds situated in an agricultural landscape, the presence of residues must be related to the physicochemical properties as well. As an example, amprolium has a higher aqueous solubility (i.e. 540 mg L−1) and a lower logP (i.e. 1.04) than the other coccidiostats, which could be an additional explanation for its presence in the aquatic compartment.64 Furthermore, as robenidine turned out to be unstable in pond water under the used storage conditions, it is unlikely that it will be detected in pond waters which are subject to varying temperature and sunlight, unless it is recently used. Levamisole was present in one pond at a concentration of 250.0 ng L−1 (i.e. ZOT 8) and in all other ponds at concentrations below the LOQ (<250.0 ng L−1). Although levamisole is extensively metabolized in most species,65 it is not surprising that it has been detected in ponds that are receiving run-off water (or contaminated groundwater) from agricultural areas considering that levamisole is a frequently oral and intramuscular administered anthelmintic for pigs and cattle.33 Furthermore, levamisole exerts a high aqueous solubility (i.e. 1440 mg L−1) and has a lower logP value than flubendazole and ivermectin, i.e. 2.36 versus 3.40 and 5.83, respectively.66,67 Alternate use, low aqueous solubility (i.e. 4 mg L−1 for ivermectin), higher logP values and a high ability to bind to soil (Koc ivermectin = 3981 L kg−1) could account for the fact that ivermectin and flubendazole were not detected in any of the ponds, the latter drug being extensively metabolized in mammals as well.68 For this reason, future studies, determining anthelmintic transformation products such as amino-flubendazole and hydroxy-flubendazole metabolites are warranted. Obtained results are in line with previous research describing levamisole as most frequent and abundant detection among different classes of anthelmintics including macrocyclic lactones and benzimidazoles in river water originating from agricultural areas.62 Due to the hydrophobic character of many of the target compounds (logP ≥ 3), it is possible to have accumulation in other compartments of the aquatic environment, such as sediment or biota, potentially posing an environmental risk there, as well. This was, however, outside the scope of this study.
4. Conclusions
A novel, highly sensitive, multi-residue SPE-UHPLC-MS/MS method was successfully developed and validated for the simultaneous screening of toltrazuril and robenidine, and quantification of the other 10 coccidiostats registered as feed additives or veterinary medicine in Europe and three frequently used anthelmintics, in pond water, covering a broad polarity range of compounds (logP = 1.04–7.51). Filtration efficiency, matrix effects and corresponding precision were evaluated and obtained results overall improved by incorporating internal standards resulting in adequate values according to the EMA guideline. Acceptable within-run and between-run apparent recoveries, satisfactory precision as well as good linearity were demonstrated according to the guidelines discussed above, except for robenidine, for which the between-day precision was between 21.0 and 34.5%. Storage stability studies indicated that sample storage at 4 °C and analysis performed within 96 hours was sufficient to avoid loss by degradation for all compounds, except for robenidine which implied its improbability of detection in pond waters subject to varying temperature and sunlight, unless recently used. Values for LOD and LOQ were in nanograms per liter which was essential for the environmental application of this novel method. The method was successfully applied on water samples from 18 different ponds situated in agricultural areas across Flanders, Belgium. Amprolium was found in one pond at a concentration level below the LOQ of 2.5 ng L−1 and levamisole was found in every pond at concentrations ranging between 250.0 ng L−1 and below the LOQ of 250.0 ng L−1. The former probably resulting from surface run-off, leaching or drift after applying liquid manure from recent treated animals on fields in an agricultural landscape. In conclusion, our newly developed method may provide insights about the contamination status of freshwater ponds and the potential toxicity to aquatic organisms.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors wish to thank Jelle Lambrecht for the technical contribution to the manuscript. The research that yielded these results, was funded by the Special Research Fund of Ghent University, Belgium [BOF16-GOA-024.08].
References
- European Parliament, Off. J. Eur. Union, 2003, 268, 29–43 Search PubMed.
- A. Martínez-Villalba, E. Moyano and M. T. Galceran, Rapid Commun. Mass Spectrom., 2009, 23, 1255–1263 Search PubMed.
- European Commission, Health and Food Safety, 2019, 11, 1–277 Search PubMed.
-
European Medicines Agency, Toltrazuril, article 35 referral, annex I, II, III, 2008 Search PubMed.
-
M. N. Novilla, in Veterinary Toxicology: Basic and Clinical Principles, 3rd edn, Elsevier, 2018, pp. 1073–1092 Search PubMed.
- A.-C. Huet, L. Mortier, E. Daeseleire, T. Fodey, C. Elliott and P. Delahaut, Food Addit. Contam., 2005, 22, 128–134 Search PubMed.
- P. A. Belœil, C. Chauvin, C. Fablet, J. P. Jolly, E. Eveno, F. Madec and J. M. Reperant, Livest. Prod. Sci., 2003, 81, 99–104 Search PubMed.
- B. H. B. Baiak, C. R. Lehnen and R. A. da Rocha, Livest. Sci., 2018, 217, 127–135 Search PubMed.
- M. M. Ayaz and M. Hanif, Pak. J. Pharm. Sci., 2018, 31, 961–966 Search PubMed.
- P. A. Spagnuolo, J. Hu, R. Hurren, X. Wang, M. Gronda, M. A. Sukhai, A. Di Meo, J. Boss, I. Ashali, R. Beheshti Zavareh, N. Fine, C. D. Simpson, S. Sharmeen, R. Rottapel and A. D. Schimmer, Blood, 2010, 115, 4824–4833 Search PubMed.
- K. L. Goa, D. McTavish and S. P. Clissold, Drugs, 1991, 42, 640–658 Search PubMed.
- J. Cha and K. H. Carlson, Sci. Total Environ., 2018, 640–641, 1346–1353 Search PubMed.
- R. Moreno-González, S. Rodríguez-Mozaz, M. Gros, E. Pérez-Cánovas, D. Barceló and V. M. León, Sci. Total Environ., 2014, 490, 59–72 Search PubMed.
- A. Iglesias, C. Nebot, J. M. Miranda, B. I. Vázquez and A. Cepeda, Environ. Sci. Pollut. Res., 2012, 19, 3235–3249 Search PubMed.
- M. Wagil, J. Maszkowska, A. Białk-Bielińska, P. Stepnowski and J. Kumirska, Chemosphere, 2015, 119, S35–S41 Search PubMed.
- H. Yoshimura and Y. S. Endoh, Wiley Interscience, 2005, 10, 60–66 Search PubMed.
- S. J. Oh, J. Park, M. J. Lee, S. Y. Park, J.-H. Lee and K. Choi, Environ. Toxicol. Chem., 2006, 25, 2221 Search PubMed.
-
B. A. I. de Sousa, Toxicological evaluation of mixtures of veterinary drugs (Monensin, Sulfamethazine and Enrofloxacin) in Daphnia magna (Cladocera, Crustacea), Biblioteca Digital de Teses e Dissertações da Universidade de São Paulo, Piracicaba, 2013 Search PubMed.
- J. Garric, B. Vollat, K. Duis, A. Péry, T. Junker, M. Ramil, G. Fink and T. A. Ternes, Chemosphere, 2007, 69, 903–910 Search PubMed.
- M. Bundschuh, T. Hahn, B. Ehrlich, S. Höltge, R. Kreuzig and R. Schulz, Bull. Environ. Contam. Toxicol., 2016, 96, 139–143 Search PubMed.
- M. E. DeLorenzo, G. I. Scott and P. E. Ross, Environ. Toxicol. Chem., 2001, 20, 84–98 Search PubMed.
- D. S. Schmeller, M. Blooi, A. Martel, T. W. J. Garner, M. C. Fisher, F. Azemar, F.
C. Clare, C. Leclerc, L. Jäger, M. Guevara-Nieto, A. Loyau and F. Pasmans, Curr. Biol., 2014, 24, 176–180 Search PubMed.
-
A. Martel, F. Pasmans, M. C. Fisher, L. F. Grogan, L. F. Skerratt and L. Berger, in Emerging and Epizootic Fungal Infections in Animals, Springer International Publishing, 2018, pp. 309–335 Search PubMed.
- A. J. Watkinson, E. J. Murby, D. W. Kolpin and S. D. Costanzo, Sci. Total Environ., 2009, 407, 2711–2723 Search PubMed.
- D. Mooney, C. Coxon, K. G. Richards, L. W. Gill, P. E. Mellander and M. Danaher, J. Chromatogr. A, 2020, 1618, 460857 Search PubMed.
- J. Olsen, E. Björklund, K. A. Krogh and M. Hansen, Anal. Chim. Acta, 2012, 755, 69–76 Search PubMed.
- H. Sanderson, B. Laird, L. Pope, R. Brain, C. Wilson, D. Johnson, G. Bryning, A. S. Peregrine, A. Boxall and K. Solomon, Aquat. Toxicol., 2007, 85, 229–240 Search PubMed.
-
T. E. P. and C. Network, The Pond Manifesto, 2008 Search PubMed.
- M. Wagil, A. Białk-Bielińska, A. Puckowski, K. Wychodnik, J. Maszkowska, E. Mulkiewicz, J. Kumirska, P. Stepnowski and S. Stolte, Environ. Sci. Pollut. Res., 2015, 22, 2566–2573 Search PubMed.
- M. Søndergaard, E. Jeppesen and J. P. Jensen, Arch. Hydrobiol., 2005, 162, 143–165 Search PubMed.
- M. Akasaka, N. Takamura, H. Mitsuhashi and Y. Kadono, Freshw. Biol., 2010, 55, 909–922 Search PubMed.
- S. Hecnar and R. M'Closkey, Freshw. Biol., 1996, 36, 7–15 Search PubMed.
- A. El-Abdellati, J. Charlier, P. Geldhof, B. Levecke, J. Demeler, G. von Samson-Himmelstjerna, E. Claerebout and J. Vercruysse, Vet. Parasitol., 2010, 169, 352–357 Search PubMed.
- J. C. Van De Steene, C. P. Stove and W. E. Lambert, Sci. Total Environ., 2010, 408, 3448–3453 Search PubMed.
-
BCFI Vet (Belgian Centre for Farmacotherapeutic Information), Vetcompendium - Commented Veterinary Drug Overview, ed. P. Gustin, Liège, 2018 Search PubMed.
-
T. Christiaens, Commented Report For Human Medicine 2019, Belgian Centre for Farmacotherapeutic Information, Ghent, 2019 Search PubMed.
- F. Hernández, M. Ibáñez, R. Bade, L. Bijlsma and J. V. Sancho, Trac. Trends Anal. Chem., 2014, 63, 140–157 Search PubMed.
- W. Song, M. Huang, W. Rumbeiha and H. Li, Rapid Commun. Mass Spectrom., 2007, 21, 1944–1950 Search PubMed.
- S. Huysman, L. Van Meulebroek, F. Vanryckeghem, H. Van Langenhove, K. Demeestere and L. Vanhaecke, Anal. Chim. Acta, 2017, 984, 140–150 Search PubMed.
- European Commission, Off. J. Eur. Union, 2009, 201, 36–38 Search PubMed.
- European Parliament, Off. J. Eur. Communities: Legis., 2008, 348, 84–97 Search PubMed.
- European Parliament, Off. J. Eur. Communities: Legis., 2006, 327, 1–70 Search PubMed.
- European Commission, Off. J. Eur. Communities: Legis., 2002, 221, 8–36 Search PubMed.
-
T. Pihlström, A. R. Fernández-Alba, M. Gamón, C. F. Amate, A. De Kok, F. O'regan, P. Pelosi, H. Mol and M. Jezussek, Sante/12682/2019: analytical quality control and method validation procedures for pesticide residues analysis in food and feed, 2019 Search PubMed.
-
European Medicines Agency, Guideline on bioanalytical method validation, 2011 Search PubMed.
-
VICH GL 49, Studies to evaluate the metabolism and residue kinetics of veterinary drugs in food producing animals: validation of analytical methods used in residue depletion studies 2016 Search PubMed.
- J. Knecht and G. Stork, Fresenius Z. Anal. Chem., 1974, 270, 97–99 Search PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 18, 39 Search PubMed.
- A. Kruve, R. Rebane, K. Kipper, M.-L. Oldekop, H. Evard, K. Herodes, P. Ravio and I. Leito, Anal. Chim. Acta, 2015, 870, 8–28 Search PubMed.
- P. Lemmens, A. De Wever, N. Bonjean, A. Castiaux, L. Colson, T. De Bie, E. Decoster, C. Denis, D. Roeck, D. Ercken, B. Goddeeris, R. Goyvaerts, S. N. M. Mandiki, K. Morelle, E. Praca, I. Schön, J. Van Wichelen, K. Van Der Gucht, J. Vandekerckhoven, P. Vanormelingen, M. Villena Alvarez, D. Bauwens, L. Denys, M. Herremans, L. Vanhecke, B. Losson, Y. Caron, H.-M. Cauchie, P. Kestemont, W. Vyverman, L. De Meester, S. A. J. Declerck and K. Martens, Freshwater Metadata Journal, 2018, 31, 1–10 Search PubMed.
- B. Oertli, J. Biggs, G. Cé, R. É. Ghino, P. Grillas, P. Joly and J.-B. Lachavanne, Aquat. Conserv. Mar. Freshw. Ecosyst., 2005, 15, 535–540 Search PubMed.
- J. Biggs, P. Williams, M. Whitfield, P. Nicolet and A. Weatherby, Aquat. Conserv. Mar. Freshw. Ecosyst., 2005, 15, 693–714 Search PubMed.
- M. Alam, O. A. Arikan, E. Yuksel, M. Eyvaz, E. Gurbulak and O. Gunaydin, J. Liq. Chromatogr. Relat. Technol., 2019, 42, 555–562 Search PubMed.
- W. Song, M. Huang, W. Rumbeiha and H. Li, Rapid Commun. Mass Spectrom., 2007, 21, 1944–1950 Search PubMed.
- S. J. Lehotay and A. R. Lightfield, Anal. Bioanal. Chem., 2018, 410, 1095–1109 Search PubMed.
- E. De Paepe, J. Wauters, M. Van Der Borght, J. Claes, S. Huysman, S. Croubels and L. Vanhaecke, Food Chem., 2019, 293, 187–196 Search PubMed.
- M. Olejnik, T. Szprengier-Juszkiewicz and P. Jedziniak, J. Chromatogr. A, 2009, 1216, 8141–8148 Search PubMed.
- F. Buiarelli, P. Di Filippo, C. Riccardi, D. Pomata, L. Giannetti, B. Neri and D. Rago, Separations, 2017, 4, 15 Search PubMed.
- S.-C. Kim and K. Carlson, Water Res., 2006, 40, 2549–2560 Search PubMed.
- J. M. Cha, S. Yang and K. H. Carlson, J. Chromatogr. A, 2005, 1065, 187–198 Search PubMed.
- L. Capitan-Vallvey, A. Ariza, R. Checa and N. Navas, J. Chromatogr. A, 2002, 978, 243–248 Search PubMed.
- M. Zrnčić, M. Gros, S. Babić, M. Kaštelan-Macan, D. Barcelo and M. Petrović, Chemosphere, 2014, 99, 224–232 Search PubMed.
- M. Hansen, E. Björklund, K. A. Krogh and B. Halling-Sørensen, Trac. Trends Anal. Chem., 2009, 28, 521–533 Search PubMed.
- G. Rychen, G. Aquilina, G. Azimonti, V. Bampidis, M. de L. Bastos, G. Bories, A. Chesson, P. S. Cocconcelli, G. Flachowsky, B. Kolar, M. Kouba, M. López-Alonso, S. López Puente, A. Mantovani, B. Mayo, F. Ramos, M. Saarela, R. E. Villa, R. J. Wallace, P. Wester, P. Brantom, I. Halle, P. van Beelen, O. Holczknecht, M. V. Vettori and J. Gropp, EFSA J., 2018, 16, 1–28 Search PubMed.
-
P. Nielsen and F. Rasmussen, Pharmacokinetics of levamisole in goats and pigs, Springer, Dordrecht, 1983 Search PubMed.
- H. Yoshimura, Reprod. Toxicol., 2003, 17, 377–385 Search PubMed.
- M. Liebig, Á. A. Fernandez, E. Blübaum-Gronau, A. Boxall, M. Brinke, G. Carbonell, P. Egeler, K. Fenner, C. Fernandez, G. Fink, J. Garric, B. Halling-Sørensen, T. Knacker, K. A. Krogh, A. Küster, D. Löffler, M. Á. P. Cots, L. Pope, C. Prasse, J. Römbke, I. Rönnefahrt, M. K. Schneider, N. Schweitzer, J. V Tarazona, T. A. Ternes, W. Traunspurger, A. Wehrhan and K. Duis, Integrated Environ. Assess. Manag., 2010, 6, 567–587 Search PubMed.
- M. L. Maté, T. Geary, C. Mackenzie, C. Lanusse and G. Virkel, J. Vet. Pharmacol. Therapeut., 2017, 40, 493–499 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0em00215a |
|
| This journal is © The Royal Society of Chemistry 2020 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
S. D.
Baere
a,
N. D.
Troyer
b,
A.
Deknock
b,
P.
Goethals
b,
L.
Lens
c,
F.
Pasmans
d and
S.
Croubels
*a,
S. D.
Baere
a,
N. D.
Troyer
b,
A.
Deknock
b,
P.
Goethals
b,
L.
Lens
c,
F.
Pasmans
d and
S.
Croubels
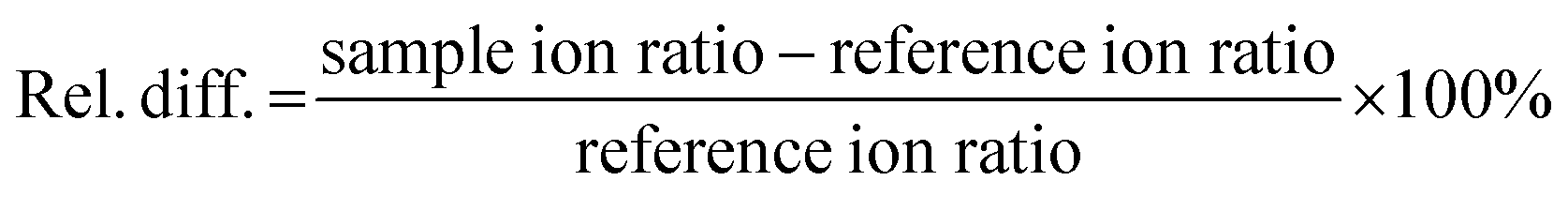

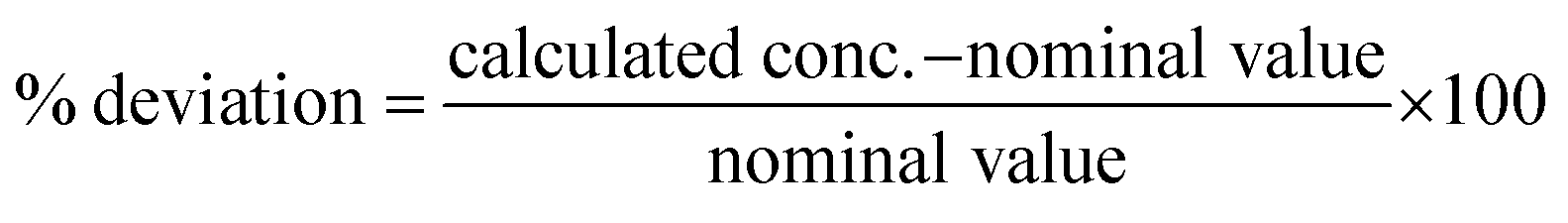 .
.