
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface precipitation of Mn2+ on clay minerals enhances Cd2+ sorption under anoxic conditions†
Natacha
Van Groeningen
 ,
Blanche
Glück
,
Iso
Christl
* and
Ruben
Kretzschmar
,
Blanche
Glück
,
Iso
Christl
* and
Ruben
Kretzschmar
Institute of Biogeochemistry and Pollutant Dynamics, Department of Environmental Systems Science, ETH Zurich, Universitätstrasse 16, CHN, 8092 Zurich, Switzerland. E-mail: iso.christl@env.ethz.ch; Tel: +41 44 633 60 01
First published on 6th July 2020
Abstract
The influence of Mn2+ on the sorption of metal(loid)s onto clay minerals is still unclear despite its relevance in suboxic and anoxic environments which often exhibit elevated dissolved Mn2+ concentrations. In this study, the effects of Mn2+ on Cd2+ sorption to two types of clay minerals, a well-crystalline natural kaolinite (KGa-1b) and a synthetic montmorillonite (Syn-1), were investigated. Batch experiments on Mn2+ and Cd2+ sorption to Ca-saturated KGa-1b and Syn-1 were conducted under anoxic conditions. At low Mn2+ and Cd2+ concentrations (1 and 5 µM), both metals exhibited similar affinity for sorption to the clays, suggesting that elevated Mn2+ concentrations might effectively decrease Cd2+ sorption as predicted using a three-plane surface complexation model. However, competitive Mn–Cd experiments at higher concentrations (≥50 µM) revealed that for both clay minerals, the presence of Mn2+ increased Cd2+ sorption to the solid phases. Although solutions were undersaturated with respect to known Mn(II) solid phases, analysis using X-ray absorption spectroscopy (XAS) evidenced the formation of Mn(II)-containing solid phases which can specifically adsorb or incorporate Cd2+. This process, which was mediated by the presence of clay minerals, overcompensated the decrease in Cd2+ adsorption to clay surfaces due to competition with Mn2+. We conclude that, contrary to predictions based on a competitive surface complexation model, elevated Mn2+ concentrations can contribute to decrease dissolved Cd2+ concentrations in anoxic clay-containing environments, such as contaminated sediments or flooded paddy soils.
Environmental significanceClay minerals represent major sorbents in soils and sediments for nutrients and contaminants. In suboxic and anoxic environments, elevated concentrations of Mn2+ can establish as the result of reductive dissolution of Mn(III/IV) solid phases, e.g., during water saturation of paddy soils. The impact of elevated Mn2+ concentrations on trace metal interactions with clay minerals is largely unknown, but is highly relevant for trace metal availability to organisms. In this study on Mn–Cd interactions with clay minerals under anoxic conditions, we demonstrate that elevated Mn2+ concentrations do not cause a net decrease in Cd2+ sorption due to competition, but lead to an overall increase of Cd2+ sorption due to the formation of Mn(II)-containing surface precipitates that adsorb or incorporate Cd2+. |
Introduction
Elevated concentrations of cadmium (Cd) in agricultural soils are of widespread concern due to its high toxicity, long half-life in the human body, and high bioavailability in many soils. Cadmium has no known biological function and is toxic to humans,1 causing liver and kidney damage, low bone density and increasing the risk of cancer.2 It was identified by the World Health Organization (WHO) as one of the “ten leading chemicals of concern”. Elevated dissolved concentrations of Cd can be found in the environment due to release from both natural sources (e.g., weathering of trace metal-enriched minerals) and anthropogenic sources (e.g., agriculture, industrial and mining activities). Cd concentrations in soils have been reported to range from geologic background concentrations, being typically below 0.10 mg kg−1, up to about 10 mg kg−1 indicating geogenically or anthropogenically elevated concentrations.3 Accumulation in plants can occur as the result of uptake from contaminated soils and contaminated water used for irrigation. Some plants like, e.g., rice, cacao, tobacco, and mushrooms are known to bioaccumulate Cd and thus are an important pathway for human exposure to Cd.4 Approximately 90% of the exposure to Cd for non-smokers is through diet.5 Herein, rice represents one of the most important pathways for Cd uptake, especially in many parts of Asia.The retention of Cd2+ in soils and sediments is, as for most divalent cations, governed by sorption processes which include adsorption, polymerization, and surface precipitation.6 Herein, clay minerals, carbonates, as well as Fe(III)- and Mn(IV,III)-(oxyhydr)oxides are most important due to their abundance, large specific surface areas, and specific sorption properties. In addition, Cd can be retained in the solid phase when precipitating as carbonate, hydroxide, phosphate, or sulfide. In soils low in Cd, adsorption is considered the dominant mechanism controlling Cd mobility,7 whereas precipitation gains importance in soils with higher Cd activity, for example, in highly contaminated soils. In soils where exchangeable Cd represents a major fraction of total Cd,8 Cd is expected to be easily re-mobilized from mineral surfaces by competing divalent ions such as e.g., Ca2+ and Zn2+ and thus to become bioavailable to plants.9
Agricultural soils commonly exhibit weakly acidic to neutral pH values, at which clay minerals and Fe(III) (oxyhydr)oxides are the most important mineral sorbent phases for Cd2+. Several studies have investigated the adsorption of Cd to Fe(III)-(oxyhydr)oxides, showing that Cd can form inner-sphere complexes at the mineral–water interface10 and ternary mineral–organic–Cd complexes.11 Similar sorption processes also occur on edge surfaces of clay minerals.12–15 To date, most studies on Cd adsorption in soils were conducted under oxic conditions applicable to upland soils, while the adsorption of Cd in suboxic to anoxic soils, such as rice paddy soils, is much less understood.
Compared to well-aerated soils, the adsorption of Cd2+ to mineral phases may be markedly altered in periodically anoxic soils of riparian floodplains, wetlands, and rice paddies. Even though Cd is not directly subject to redox reactions, changes in soil pore water conditions as well as reductive dissolution processes affect Cd adsorption during periods with suboxic to anoxic conditions. Oxides and oxyhydroxides of Mn(III/IV) become thermodynamically favorable electron acceptors for microbial respiration already at low oxygen levels still preventing Fe(III) and sulfate reduction to predominate. As a result, dissolved Mn2+ concentrations in soil solutions increase before Fe2+ and sulfide. In a study by Kirk,16 flooding of acidic soils containing high contents of manganese and organic matter led to dissolved Mn2+ concentrations up to 1.7 mM after one week of submergence. For flooded alkaline soils and soils low in manganese, dissolved Mn2+ concentrations up to 0.2 mM were reported.16 These findings indicate that in submerged soils, Mn2+ becomes a major cation in soil solution and is therefore expected to compete with trace element cations (e.g., Zn2+, Cd2+) for adsorption to mineral surfaces influencing their fate in the environment. During soil aeration, Fe2+ is rapidly oxidized by molecular O2, contrary to Mn2+, for which oxidation kinetics by dioxygen is much slower.17 Therefore, Mn2+ may persist under oxic conditions for prolonged periods and continue to act as a cation competing with trace metals for adsorption.
In a competitive situation, trace metal retention by solids may, however, be altered by metal cation-specific interactions on mineral surfaces as evidenced by Soltermann et al.18 for Fe2+ and Zn2+ adsorption to a synthetic montmorillonite. Their results indicated the existence of specific sites which were not accessible to Zn2+, but were able to induce oxidation of Fe2+ and enhance Fe sorption.18 These results show that ion-specific sorption can occur, leading to metal retention deviating from simple competitive adsorption. This is likely to hold true for many other systems, including Mn2+ and Cd2+ sorption to clay minerals. In addition, metal ion-specific formation of new mineral phases may be induced by the presence of clay minerals and contribute to metal sorption. If newly formed surface precipitates exhibit a lower affinity for the trace metal of interest than the clay mineral surfaces and concomitantly block the reactive clay sites, the formation of surface precipitates might lead to a decrease in trace metal sorption. However, if the newly formed phases have a similar or even higher affinity than the clay minerals, overall sorption may be increased markedly, especially in case the new phases add a significant amount of surfaces, i.e., sorption sites. Hence, surface precipitation may considerably modify sorption patterns of trace metals.
At elevated concentrations, many divalent metal cations (Me2+, first-row transition series) have been shown to precipitate as Me2+Al3+-layered double hydroxides (LDH) under alkaline conditions in presence of Al-containing minerals.19–25 Starcher et al. demonstrated that the FeAl-LDH phase formed during Fe2+ sorption to an Al-bearing sorbent can sorb or incorporate Zn2+ when both metals were co-sorbed to the Al-bearing mineral.26 However, Bhattacharya and Elzinga demonstrated that dissolved Mn2+ concentrations significantly higher than normally encountered in anoxic soils (i.e., [Mn2+] > 10−2 M) are required to precipitate Mn2+Al3+-LDH, making the precipitation of this phase unlikely.27 Despite the relevance of such processes affecting the mobility of metals in the environment, to date the above mentioned sorption processes have not been investigated in detail and it is difficult to predict which process would predominantly control Cd2+ sorption at elevated Mn2+ concentrations in clay-rich environments.
Therefore, the objective of this research was to assess the interactions between Mn2+ and Cd2+ during sorption to clay minerals. Kaolinite and montmorillonite are two very widespread clay minerals in the environment and were selected as the clay sorbents representing 1:1- and 2:1-type phyllosilicate minerals, respectively. CaCl2 was chosen as the background electrolyte, as calcium is often the predominant divalent cation in soils.28 Acid–base titrations and batch experiments on metal sorption of Mn2+ and Cd2+ onto the Ca-clays were carried out. The data were used to optimize parameters of a two-site Three-Plane Model (TPM)29 to best describe H+, Ca2+, Mn2+, and Cd2+ adsorption to the clay surfaces. Predictions of competitive Mn2+–Cd2+ adsorption as calculated with model parameters based on binary adsorption data (Mn–Ca and Cd–Ca clay systems) were compared to experimental results in ternary Mn–Cd–Ca clay systems. Additionally, solid phase speciation was carried out for Mn and Cd using synchrotron X-ray absorption spectroscopy (XAS) to identify the dominant sorption mechanisms.
Materials and methods
Clay minerals
A well-ordered kaolinite (KGa-1b) and a synthetic montmorillonite (Barasym SSM-100), referred to as KGa-1b and Syn-1 in the following, were used in this study. KGa-1b and Syn-1 were selected as the solids, as both are low in Fe in order to minimize potential oxidation of Mn2+ by structural FeIII. Both materials were purchased from the Source Clays Repository of the Clay Minerals Society (Purdue University, West Lafayette, IN).Particles with a hydrodynamic diameter of less than 2 µm were isolated by repeated centrifugation and dispersion steps. Briefly, clay particles were dispersed by adjusting the suspension pH to 9.0 with NaOH and subsequent ultrasonication for 3 min prior to centrifugation at 100g (600 rpm) for 5 min (Mistral 6000). The supernatant containing the particles smaller than 2 µm was collected and the procedure was repeated until the supernatant was clear. A yield of 62% for KGa-1b and 95% for Syn-1 was obtained after size fractionation. Size-fractionated particles were saturated with calcium by adding CaCl2 solution to achieve a concentration of 0.1 M. The suspensions were subsequently placed on an overhead shaker overnight and then centrifuged at 200g (1200 rpm) for 5 min for both KGa-1b and Syn-1. To ensure complete Ca saturation, the Ca saturation step was repeated two more times by adding 0.1 M CaCl2 solution and shaking the suspensions for 3–4 hours before centrifugation. After Ca saturation, excess salt was removed by repeated washing with doubly deionized (DDI) water (≥18.2 MΩ cm, Milli-Q, Millipore, Merck KGaA, Darmstadt, Germany) until the supernatants were found to be free of chloride using AgNO3. The final products were freeze-dried and stored as dry powders.
Characterization of clay minerals
The size-fractionated and Ca-saturated KGa-1b and Syn-1 were analyzed as random-oriented powder samples by X-ray diffraction (XRD, D8 Advance, Bruker) to test for mineralogical purity. Diffractograms were recorded in Bragg–Brentano geometry from 3 to 80° 2θ with a step size of 0.02° 2θ and 4 s acquisition time per step, using Cu Kα1,2 radiation. Energy-dispersive X-ray fluorescence analysis (XRF, XEPOS, Spectro) using pressed pellets was performed for elemental analysis of the clay minerals. The specific surface areas of the clay minerals were measured by multi-point N2-BET analyses (ASiQwin, Quantachrome).The measured diffractogram of KGa-1b indicated trace impurities of TiO2 and for Syn-1 trace amounts of γ-AlOOH which is in agreement with published results for KGa-1b and Syn-1 (see Fig. S1†).30 Total iron contents as determined using XRF amounted to 1.41 g kg−1 for KGa-1b and 0.26 g kg−1 for Syn-1 (see also Table S1†). We determined a N2-BET surface area of 12.8 ± 0.1 m2 g−1 for KGa-1b and 95 ± 2 m2 g−1 for Syn-1. Mermut and Cano31 reported iron contents of 2.5 ± 0.3 g kg−1 and 0.9 ± 0.2 g kg−1 Fe for KGa-1b and Syn-1, respectively. N2-BET surface areas as published by Dogan et al.32 amounted to 13.1 ± 0.1 m2 g−1 and 118 ± 1 m2 g−1, for KGa-1b and Syn-1, respectively. The minor differences in Fe contents and surface areas between our measurements and literature are likely due to the sample pretreatment applied in this study, whereas literature values given above refer to untreated source clays.
Potentiometric titrations of KGa-1b and Syn-1 suspensions were carried out at three CaCl2 concentrations (0.01, 0.03, and 0.1 M) with an automated titration setup33 located in a thermostated room at 25 ± 1 °C. The setup was equipped with four burettes (Dosimat 605, Metrohm) containing 0.05 M HCl, ∼0.02 M Ca(OH)2, 0.6 M CaCl2, and CO2-free DDI water, respectively. The four burettes were connected to a computer by a Microlink MF18 interface (Biodata, Manchester, UK). All solutions were prepared with CO2-free DDI water. For the acid–base titrations, 2.5 g KGa-1b and 0.75 g Syn-1, respectively, were suspended in 40 mL DDI water in a Teflon vessel, which was continuously purged with CO2-free, water saturated N2-gas during the whole experiment. At the beginning, the starting ionic strength (0.01 M) was adjusted by adding CaCl2 solution. As a preconditioning prior to the acid–base titration, the pH of the suspension was first increased to pH 9 to disperse the particles, then adjusted to pH 5 and kept at this pH for one hour to remove CO2. During the subsequent acid–base titration, the clay suspensions were titrated in the potential range of 200 mV to −180 mV (about pH 3.8–10) with increments of 15 mV as the target step size for acid and base additions, respectively. After each titrant addition, the suspension was first stirred for 2 minutes before monitoring the pH value. The ionic strength was automatically readjusted after each acid or base addition using either CaCl2 solution or DDI water. Reported data correspond to pH values recorded once pH drift was below 0.05 mV per minute or after a maximum equilibration time of 30 minutes. After completion of a full acid–base titration cycle, the ionic strength was increased to the next higher ionic strength (0.03 and 0.1 M). This way a set of acid–base titration data at three different ionic strengths was recorded in one experiment ensuring that there was no experimental offset bias between the data measured at the different ionic strengths.
The proton binding by the clay surfaces was calculated as a H+–OH− mass balance. The additions of acid and bases, the concentrations of H+ and OH− in the electrolyte solutions, as well as the consumption of OH− by the formation of aqueous Ca hydroxide complexes and proton consumption by dissolved silicate were considered in the calculations (see ESI† for species). The concentration of dissolved species was calculated for each data point with the geochemical speciation program PHREEQC (version 3) (ref. 34) using measured pH and the known total Ca and Cl concentrations as input. For silicate, the total dissolved Si concentration as determined at the end of the titration experiment was used. Hydrolysis of dissolved Al was not considered as including a correction for Al hydrolysis as based on total dissolved Al determined at the end of the experiment led to an obvious overcorrection of proton consumption (see Fig. S5†).
Metal sorption experiments
The sorption of Mn2+ and Cd2+ on Ca-saturated KGa-1b and Syn-1 was studied in batch experiments as a function of pH (4–10) and CaCl2 concentration (0.1, 1, 3, 10, and 30 mM) at fixed total sorbate concentrations (10−3 to 10−8 M) at 25 ± 1 °C. Based on kinetic pre-experiments (see Fig. S2 and S3†), an equilibration time of 24 h was chosen for all experiments. Clay suspensions were prepared with a solid-to-solution ratio of ∼1 g L−1 in 0.1, 1.0, 3.0, 10, and 30 mM CaCl2, respectively. All solutions were prepared with CO2-free DDI water to avoid the formation of carbonate solids at alkaline pH values contributing to the removal of Mn2+ and Cd2+ from solution. Before starting the sorption experiments, suspensions were equilibrated overnight at pH 5 to remove CO2. Subsequently, CdCl2 or MnCl2 solutions were added to the clay suspensions. The pH was adjusted by adding a pre-determined amount of base (0.01 or 0.1 M NaOH) to reach the target pH value. After 24 hours of equilibration on an end-over-end shaker, samples were passed through 0.22 µm Nylon filters (VWR International), acidified, and analyzed for Ca, Al, Si, Cd, Mn, Zr, V and Fe by inductively coupled plasma-optical emission spectrometry (ICP-OES, Vista MPX, Varian) or mass spectrometry (ICP-MS, 8800 QQQ-ICP-MS, Agilent). The pH of the clay suspensions was measured for all samples at the end of the 24 hours equilibration period. Competitive sorption experiments were performed at a CaCl2 concentration of 10 mM, which was chosen to saturate non-specific cation exchange sites with calcium and more selectively observe specific sorption of Mn2+ and Cd2+ on edge-sites of the clays. The influence of various Mn2+ concentrations (0.05 mM and 0.5 mM MnCl2) on the sorption of Cd2+ at trace concentrations (0.001 mM and 0.05 mM) was studied as function of pH (4–10) for both KGa-1b and Syn-1.Batches for binary Mn–Ca and ternary Mn–Cd–Ca sorption experiments were prepared in an anoxic glovebox (<10 ppm O2) using O2-free solutions to prevent catalyzed oxidation of Mn2+ by the clay minerals.35 For equilibration, the anoxic Mn2+-containing samples were sealed in two layers of gas-tight aluminum foil inside the anoxic glovebox but equilibrated on an end-over-end shaker outside the glovebox for 24 hours, similarly as for the samples containing Cd2+, only.
Modeling
Clay minerals exhibit permanent negative charge due to isomorphic substitution in the crystal structure,36,37 giving rise to cation exchange sites on face surfaces of the clay platelets. In addition, reactive surface hydroxyl groups located at edge surfaces can have pH-dependent charge. Both types of surface sites contribute to total cation binding. In the modeling approach, one type of permanently charged sites, assigned to face surface areas and denoted as Ex−, and one type of pH-dependent charged sites, assigned to edges surface areas and denoted as![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SOH−0.5, were considered. An edge-to-face surface area ratio of 1:4 was assumed and corresponding fractions of the total N2-BET surface area were assigned to edge and face surfaces, respectively. This percentage of edge surface area is in line with published aspect ratio measurements for KGa-1b.38–40 As the permanent charge of KGa-1b was reported to be located predominantly in the tetrahedral sheet, 50 percent of the face surface were assumed to carry small amounts of permanent negative charge. Correspondingly, reactive surface areas of edge and face sites were set to 2.56 m2 g−1 and 5.12 m2 g−1 for KGa-1b and to 18.71 m2 g−1 and 74.84 m2 g−1 for Syn-1, respectively (see Tables S2 and S3†). Ion sorption on both face and edge surfaces was described with a 1 pK Three-Plane surface complexation model (TPM)29 (see Fig. S4†). More detailed information about the modeling parameters can be found in the ESI.†
SOH−0.5, were considered. An edge-to-face surface area ratio of 1:4 was assumed and corresponding fractions of the total N2-BET surface area were assigned to edge and face surfaces, respectively. This percentage of edge surface area is in line with published aspect ratio measurements for KGa-1b.38–40 As the permanent charge of KGa-1b was reported to be located predominantly in the tetrahedral sheet, 50 percent of the face surface were assumed to carry small amounts of permanent negative charge. Correspondingly, reactive surface areas of edge and face sites were set to 2.56 m2 g−1 and 5.12 m2 g−1 for KGa-1b and to 18.71 m2 g−1 and 74.84 m2 g−1 for Syn-1, respectively (see Tables S2 and S3†). Ion sorption on both face and edge surfaces was described with a 1 pK Three-Plane surface complexation model (TPM)29 (see Fig. S4†). More detailed information about the modeling parameters can be found in the ESI.†
PHREEQC coupled with the parameter optimization code PEST41 was used for data fitting. The stability constants for the formation of aqueous complexes as well as solubility product constants of possible solid phases were taken from Smith and Martell.42 The acid–base titration data recorded in CaCl2 background electrolyte were modeled in order to obtain the surface parameters of the clay minerals, namely the site densities of SOH−0.5 on edge and Ex− on face surfaces and the protonation and surface complex formation constants for Ca2+ and Cl−. Capacitances of both the 1- and 2-plane were fixed to 1 and 5 F m−2, respectively.10 Surface complex formation of Mn2+ and Cd2+ on the clay minerals were optimized using the binary metal adsorption data, while fitted surface parameters were fixed according to the best fit of acid–base titration data. Finally, the competitive effect of Mn2+ on Cd2+ adsorption was predicted by running forward calculations with PHREEQC using all optimized surface complex formation parameters. Model predictions were compared to experimental data on competitive sorption.
X-ray absorption spectroscopy
A second set of samples was prepared to investigate the solid phase speciation of Mn and Cd and to analyze the effects of an elevated Mn2+ concentration (2.5 mM) on the sorption of Cd2+ (0.25 mM) to clay minerals at pH ∼8 using XAS. The samples were prepared with a solid-to-solution ratio of ∼5 g clay per L and a CaCl2 concentration of 50 mM in order to maintain similar conditions to the batch experiments described above. The suspensions were equilibrated in 250 mL Nalgene® bottles for 1 day under anoxic conditions while stirred at 800 rpm. After equilibration in the anoxic glovebox, 10 mL aliquots were taken and filtered for liquid analysis using ICP-OES or ICP-MS. For XAS analysis an aliquot of the suspension was filtered through a 0.22 µm cellulose nitrate filter. The collected solid was washed with deoxygenated CO2-free DDI and subsequently dried in the glovebox in the dark.Solid samples from these batch experiments were analyzed by Mn K-edge (6539 eV) and Cd K-edge (26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 711 eV) X-ray absorption spectroscopy (XAS) at the SAMBA beamline of SOLEIL (Saint-Aubin, France). Dried solids were homogenized and pressed into 13 mm pellets and sealed between Kapton® tape. For transport to the synchrotron, samples were sealed in two layers of gas-tight aluminum foil under N2 gas. Two samples were additionally prepared as oriented clay films and measured with polarized-XAS (α = 10°, 35°, 55°, and 80°). The oriented samples were prepared by collecting the clay on a cellulose nitrate filter, cutting and stacking the filters, and sealing them between Kapton® tape. All measurements were conducted at 25 K to avoid beam damage and oxidation of O2-sensitive samples. Sample spectra were evaluated by linear combination fitting (LCF) to obtain information about the speciation and the (trans)formation of solid-phases. Shell-fit analyses of the extended X-ray absorption fine structure (EXAFS) spectra were performed in order to gain information about the short range local coordination environment of Mn and Cd. The nature of the backscattering atoms in the Mn sorption samples was determined using Morlet wavelet transforms (WT) of k3-weighted EXAFS data using the Fortran version of the HAMA software developed by Funke et al.43 Details on all measurements, data reduction, and analyses are provided in the ESI.†
711 eV) X-ray absorption spectroscopy (XAS) at the SAMBA beamline of SOLEIL (Saint-Aubin, France). Dried solids were homogenized and pressed into 13 mm pellets and sealed between Kapton® tape. For transport to the synchrotron, samples were sealed in two layers of gas-tight aluminum foil under N2 gas. Two samples were additionally prepared as oriented clay films and measured with polarized-XAS (α = 10°, 35°, 55°, and 80°). The oriented samples were prepared by collecting the clay on a cellulose nitrate filter, cutting and stacking the filters, and sealing them between Kapton® tape. All measurements were conducted at 25 K to avoid beam damage and oxidation of O2-sensitive samples. Sample spectra were evaluated by linear combination fitting (LCF) to obtain information about the speciation and the (trans)formation of solid-phases. Shell-fit analyses of the extended X-ray absorption fine structure (EXAFS) spectra were performed in order to gain information about the short range local coordination environment of Mn and Cd. The nature of the backscattering atoms in the Mn sorption samples was determined using Morlet wavelet transforms (WT) of k3-weighted EXAFS data using the Fortran version of the HAMA software developed by Funke et al.43 Details on all measurements, data reduction, and analyses are provided in the ESI.†
Results and discussion
Sorption of Mn2+ and Cd2+ to Ca-clays
The pH-dependent sorption of Mn2+ and Cd2+ to KGa-1b and Syn-1 is shown in Fig. 1, S7, and S8† along with the best surface complexation model fits. For both clay minerals, an increase of Mn2+ and Cd2+ sorption was observed with increasing pH (Fig. 1, S7 and S8†). Mn2+ and Cd2+ sorption isotherms (Fig. S9 and S10†) did not exhibit an apparent sorption maximum for either cation when plotted on a log–log scale. This may indicate that the sorption sites had not been saturated with respect to Mn2+ or Cd2+ within the studied range of metal concentration in solution or that additional sorption processes aside of adsorption (e.g., surface precipitation) contributed to the overall uptake (sorption) of Mn2+ and Cd2+ (see isotherms in Fig. S9 and S10†). Furthermore, the sorption of both metal cations decreased with increasing Ca2+ concentration, especially in the acidic pH range. This observation points towards the presence of two distinct types of surface sites on the clay mineral surfaces, one site dominating sorption in the acidic pH range and a second site dominating sorption under neutral to alkaline conditions. In the acidic pH range, elevated Ca2+ concentrations competed for cation binding indicating that cation sorption was dominated by cation exchange. Under neutral to alkaline conditions, cation sorption was governed by sorption on pH-dependent edge sites, for which Cd2+ and Mn2+ showed a higher sorption affinity than Ca2+. A closer look at the effects of Ca2+ concentration furthermore reveals that the sorption of Cd2+ was decreased to a larger extent by increasing Ca2+ levels than was the sorption of Mn2+. This finding implies that below pH 7, Ca2+ more effectively competed with Cd2+ than with Mn2+. Fitted affinities of each divalent cation to permanently charged sites (logKEx2Me) were similar for both clay minerals and followed the order Mn2+ > Cd2+ > Ca2+ (Tables S2 and S3†), which is in line with the above mentioned observations.
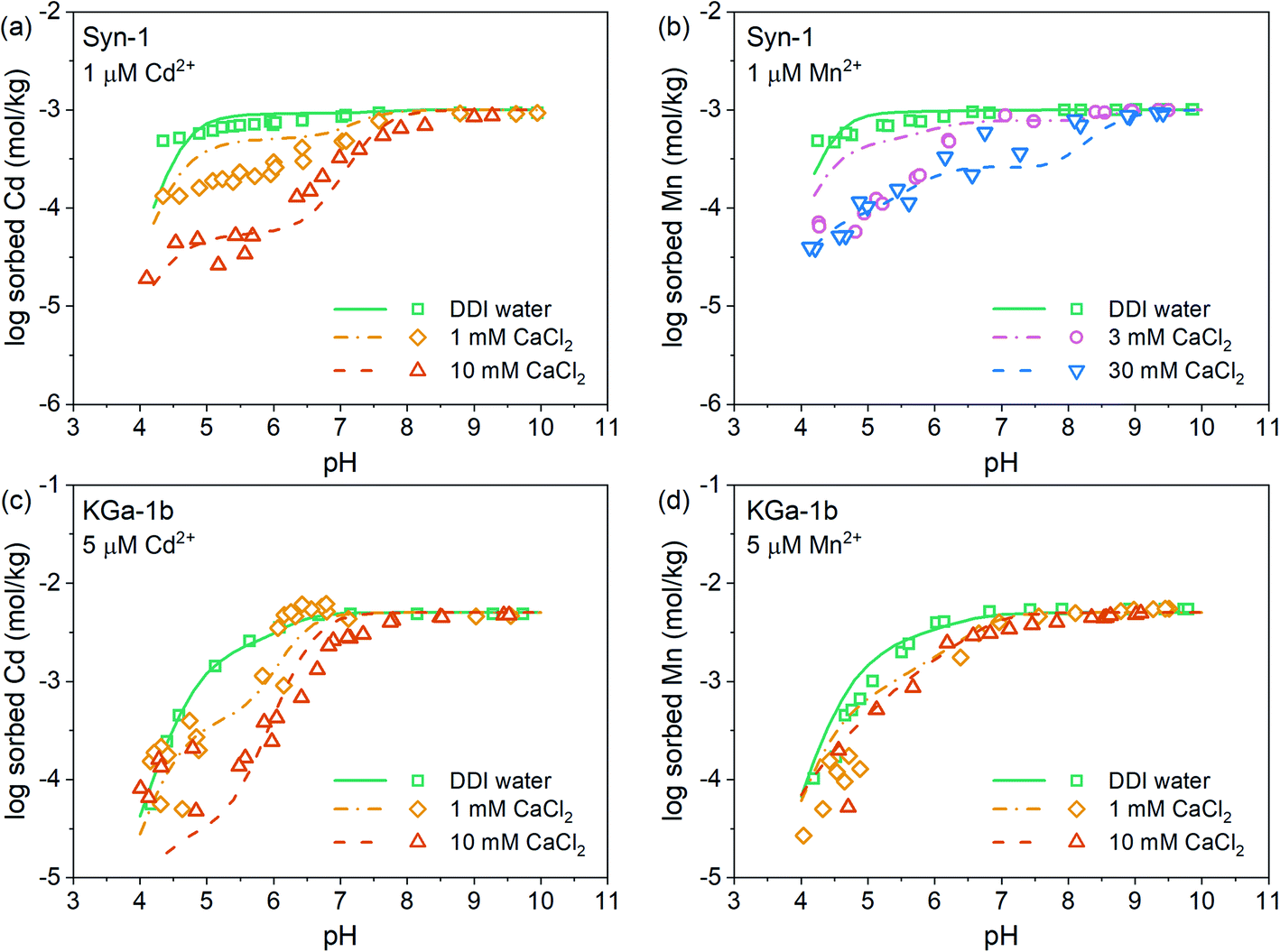 | ||
| Fig. 1 Sorption of Cd2+ and Mn2+ to (a, b) Syn-1 and (c, d) KGa-1b as a function of pH using Mn2+ and Cd2+ concentration of 1 and 5 µM at various CaCl2 concentrations (from 1 mM to 30 mM CaCl2, for DDI water samples CaCl2 was of ∼0.1 mM and ∼0.001 mM for Syn-1 and KGa-1b respectively). The solid concentration was ∼1 g L−1. Lines represent fits for Cd2+ and Mn2+ sorption described with the two-site TPM, calibrated with the protonation data. | ||
From neutral to alkaline pH, the sorption of cations is governed by sorption to edge sites. For both clay minerals, similar sorption of Cd2+ and Mn2+ to edge sites was observed (Fig. 1, S7 and S8†). This is in line with the fitted affinities of Cd and Mn hydroxide surface complexes (Tables S2 and S3†), which differed negligibly. Additionally, this observation is in good agreement with the known minor differences in metal first hydrolysis between these two metal cations (Me2+ + H2O ⇌ MeOH+ + H+ with logKMnOH+ = 3.4 and logKCdOH+ = 3.9) as affinities of cation adsorption to reactive surface hydroxyl groups was reported to be related to the first cation hydrolysis constant.44–46
Competitive sorption between Mn2+ and Cd2+ on Ca-clays
Experimental data and modeling of the Mn2+ and Cd2+ sorption data revealed that Mn2+ had a slightly higher affinity than Cd2+ for sorption to face sites, being the dominant sorption site on both clay minerals in the acidic pH range, and that the affinities for surface complexation on edge sites were similar for both metal cations (see ESI, Tables S2 and S3 and Fig. S13†). In natural systems, Mn2+ concentrations usually exceed Cd2+ concentrations in solution during anoxic conditions. Especially in submerged soils, Mn2+ concentrations can be highly elevated, reaching up to millimolar concentrations. A high Mn2+ to Cd2+ ratio in solution is therefore expected to decrease Cd adsorption to clay minerals as based on the findings on the binary Mn–Ca and Cd–Ca sorption experiments presented above. Predictions based on the calibrated Three-Plane Model, which considers Mn2+–Cd2+ competition during adsorption, illustrate this effect which becomes prominent at total Mn concentrations exceeding 50 µM (see lines in Fig. 2). However, the data of the competitive sorption experiments revealed that Cd2+ adsorption increased with increasing total Mn2+ concentration over the entire pH range (see Fig. 2). The clear difference between model predictions and experimental data indicates that the presence of Mn2+ triggered a process that counteracted competitive metal adsorption and led to a net increase in Cd2+ sorption. We therefore hypothesized that the observed increase of Cd2+ sorption in the presence of Mn2+ was caused by a newly formed, Mn-containing solid phases. This newly formed Mn-containing solid phases may serve as an additional sorbent for Cd2+ or incorporate Cd2+ in its structure. Ainsworth et al. found that Cd2+ could be incorporated in hydrous ferric oxide (HFO) by recrystallization.47 Similarly, Cd2+ could be sorbed by a newly formed Mn-containing solid phases through more than just adsorption. Thermodynamically, elevated Mn2+ concentrations are expected to form pyrochroite (Mn(OH)2) at alkaline pH (see Fig. S14†). But the studied suspensions were undersaturated with respect to the solubility of known Mn- and Cd-phases (Table S5†). Alternatively, Me(II)Al-LDH phases, which have already been evidenced in literature for alkaline conditions,19–25 may have formed. It has been demonstrated that LDH phases are able to sorb or incorporate contaminants.26 However, it is unknown to what extent Mn-containing LDH phases are able to sorb or incorporate Cd2+.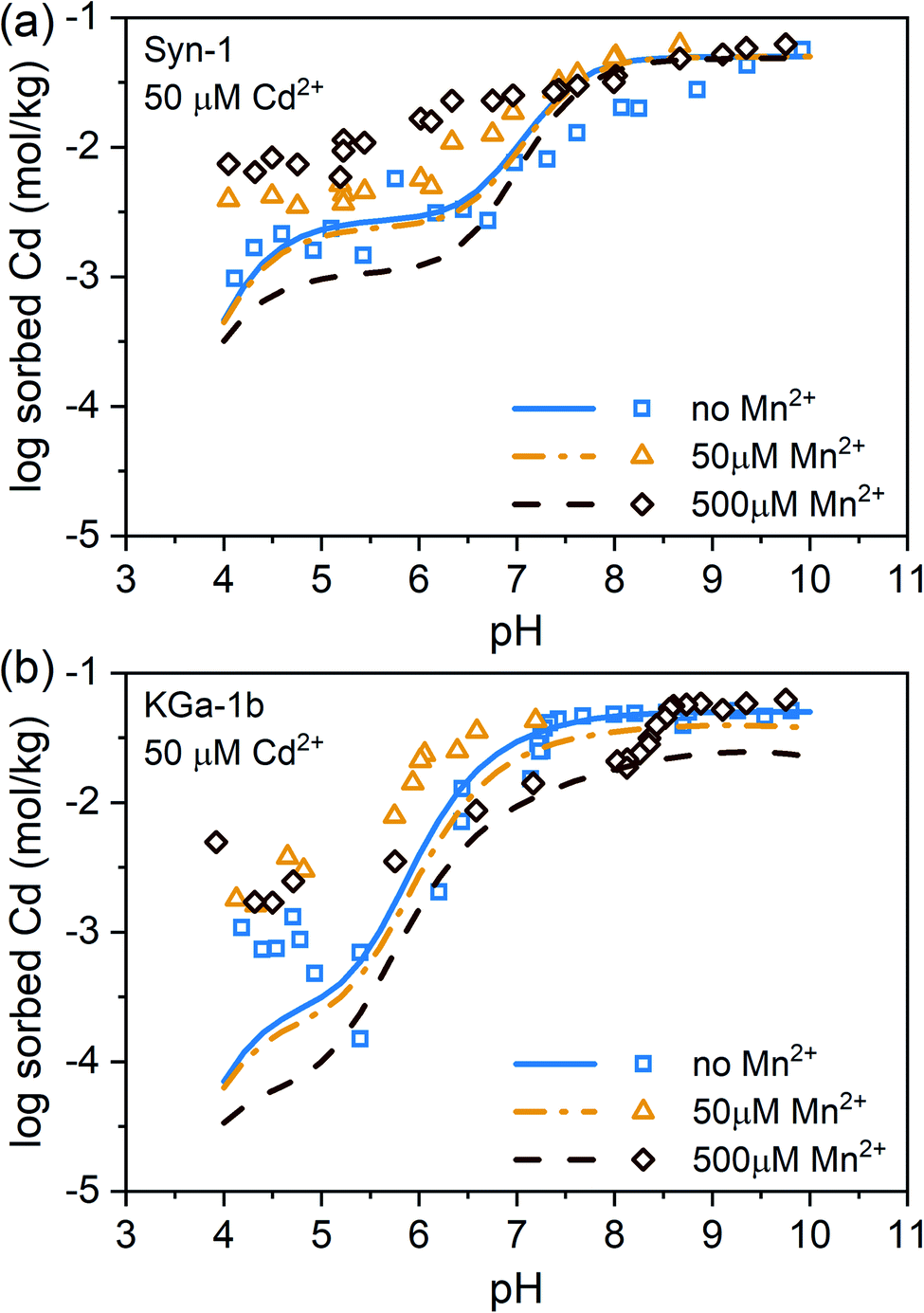 | ||
| Fig. 2 Sorption of Cd2+ (50 µM) to (a) Syn-1 and (b) KGa-1b in 10 mM CaCl2 with various Mn2+ concentrations (0 mM, 50 µM and 500 µM) as a function of pH. The solid concentration was ∼1 g L−1. Lines show the predicted effect of Mn2+ on Cd2+ sorption assuming that competition for adsorption sites is the dominant type of interaction in a competitive system. | ||
X-ray absorption spectroscopy
To clarify the predominant mechanisms leading to the observed deviations from the model predictions in competitive systems, the solid phase speciation of both Cd and Mn was investigated by synchrotron XAS. As observed for the competitive sorption experiments, the XAS samples displayed an increase of Cd2+ sorption upon addition of Mn2+ to the clay suspension at pH ∼8 (see Fig. 3). The marginally higher suspension pH for samples with Mn2+ is insufficient to explain the pronounced increase of Cd2+ sorption in KGa-1b samples. Thus, XAS samples furthermore point towards the formation of a newly formed Mn-containing solid phases, which adsorbed or incorporated Cd2+.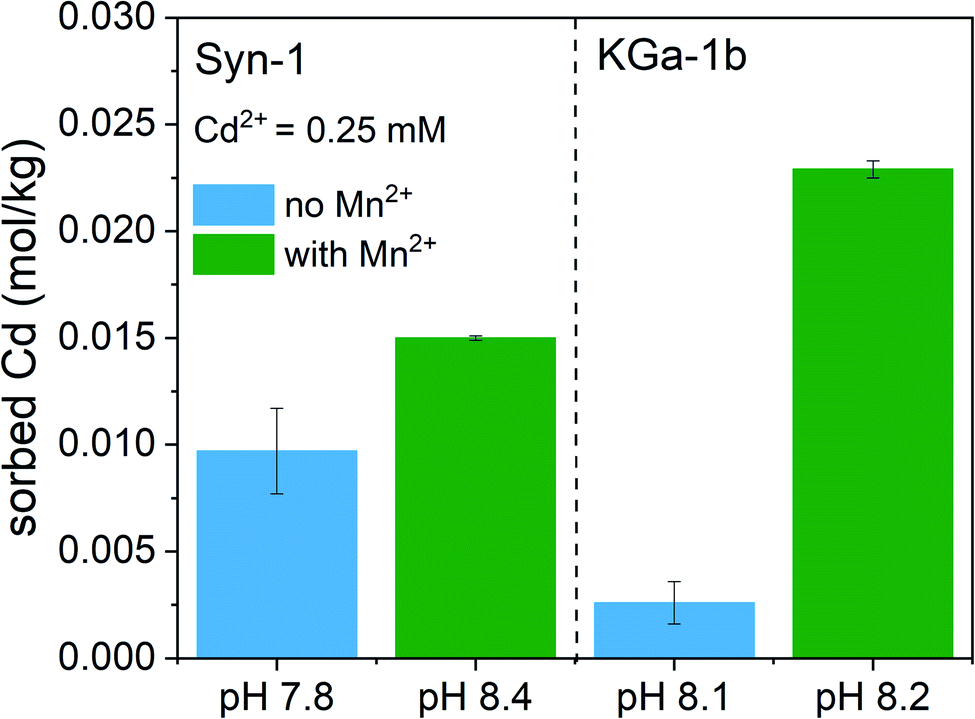 | ||
| Fig. 3 Sorption of Cd2+ (0.25 mM) to Syn-1 and KGa-1b in 50 mM CaCl2 in the absence and presence of Mn2+ (2.5 mM) at pH ∼8. The solid concentration was ∼5 g L−1. Samples were equilibrated for 1 day under anoxic conditions. Displayed pH values correspond to the pH measured at the end of the equilibration period for each sample. The error bars indicate the standard deviation of duplicates. | ||
The Mn K-edge XANES spectra for selected Mn2+ sorption samples are shown in Fig. 4 together with relevant Mn reference compounds. The position of the white line between 6553 and 6557 eV in the Mn XANES spectra for all the sorption sample suggests a valence state close to 2+ for the sorbed Mn.
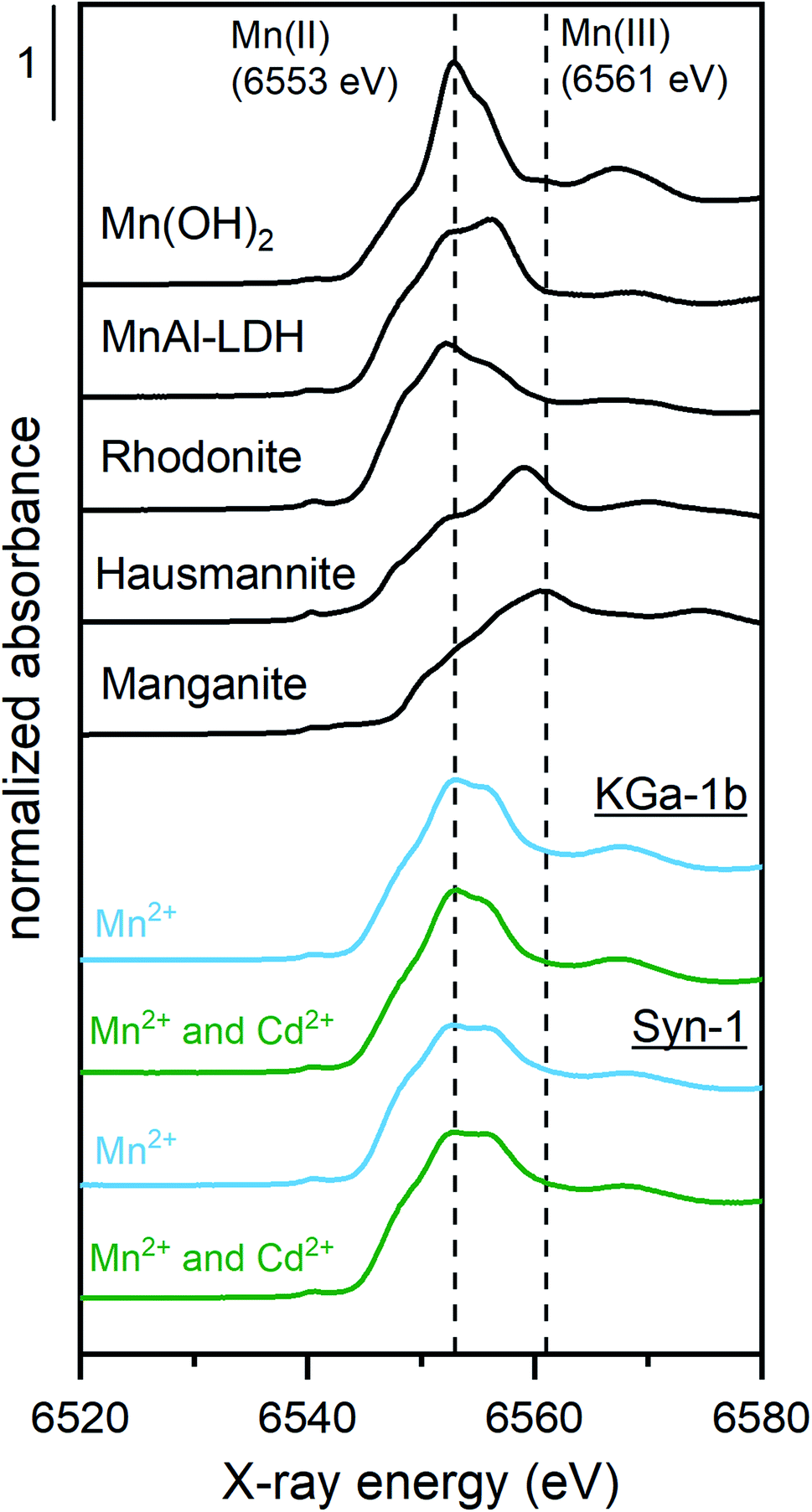 | ||
| Fig. 4 Normalized XANES spectra of the Mn K-edge of reference materials (black lines) and clay mineral samples reacted with 2.5 mM Mn2+ in absence (blue line) or presence (green line) of Cd2+ (0.25 mM Cd2+). Clay mineral samples were equilibrated for 1 day under anoxic conditions at ∼pH 8 with a solid concentration of ∼5 g L−1 KGa-1b or Syn-1. Vertical dashed lines correspond to peak maxima of Mn(OH)2 and manganite representing Mn(II)- and Mn(III)-containing solid phases, respectively. | ||
Dissolved Si released from Syn-1 and KGa-1b during 1 day equilibration between pH 6 and 10 amounted on average to ∼0.016 mmol Si per g clay and 0.0016 mmol Si per g clay for Syn-1 and KGa-1b (see Fig. S16†), respectively, when Mn was absent. In the presence of Mn, up to 2 times lower dissolved Si concentrations were observed (see Fig. S17†). Likewise, the clay mineral samples equilibrated with Cd displayed a decrease in dissolved Si concentration, but to a smaller extent. These findings suggest that the released Si was adsorbed or incorporated into newly formed phases. Alternatively, the addition of Mn and Cd to the clay suspension may have limited the dissolution of the clay minerals and thereby decreased the Si release. It has been shown by Soltermann et al.48 that Fe2+ can be taken up by the clay minerals, leading to the formation of Fe-bearing phyllosilicate structures. Similarly, the observed decrease of the concentration of dissolved Si upon addition of Mn2+ may be due to the incorporation of Mn and Si into a newly crystallized phyllosilicate phases.
To investigate the possible solid phases Mn species formed, linear combination fits (LCF) of Mn K-edge XANES spectra were performed (Fig. S19 and Table S7†). A two-component LCF model was selected as the addition of more components did not decrease the reduced χ2-values by at least a factor two.49 LCF results showed that the sorbed Mn remained as Mn2+ (Table S7†), as was already indicated by the white line position. The XANES spectra of sorption samples were adequately fitted by including Mn(II)Al-layered double hydroxide (LDH) (with generic composition [M1−x2+Mx3+(OH)2](Cl−)x/n·yH2O, 40–66%) and Mn(II)-hydroxide (Mn(OH)2, 34–38%) or rhodonite (MnSiO3, 42–60%). To fit the Mn spectra of the Syn-1 samples, the reference compound rhodonite (42–60%) was required, which suggests the incorporation of the released Si into a newly formed Mn-silicate phases. This is in line with the observed higher dissolved Si concentration for Syn-1 compared KGa-1b, which would promote the formation of Mn-phyllosilicates upon Mn2+ addition to Syn-1 compared to KGa-1b were Mn(OH)2 formation was promoted.
It has been shown in many studies that at elevated concentrations of divalent metal cations (Me2+, first-row transition series), Me(II)Al(III)-LDH can form under alkaline conditions in the presence of Al-containing minerals.19–22,24–27,50–52 The formation of Mn(II)Al-LDH formation is favored over Mn(OH)2 in environments where Al3+ is present, because MnAl-LDH phases are by two orders of magnitude less soluble than is Mn(OH)2.27 Based on solubility calculations by Bhattacharya and Elzinga, the formation of Mn(II)Al-LDH did not appear likely to occur in our samples, as very high Mn2+ concentrations (>10−2 M) are required to induce the precipitation of such phases at near-neutral pH.27 However, LCF of XANES spectra suggested that a large fraction of the sorbed Mn2+ was present as MnAl-LDH. Potentially, this may be explained by the ability of Si to be incorporated in LDH phases, which increases the stability of LDH precipitates.53 Because including a reference spectrum for Mn2+ sorbed to illite (IMt from the Source Clay Repository of the Clay Minerals Society) did not improve the LCF significantly, we concluded that surface adsorption of Mn2+ to the clay mineral, under these conditions, was a minor contribution and the majority of Mn2+ was sorbed by formation of secondary phases.
In order to get more detailed information about the binding environment of Mn and Cd on Syn-1 and KGa-1b, shell-by-shell fitting of the Fourier transforms (FT) of k3-weighted Mn and Cd K-edge EXAFS spectra (see Fig. 5) were performed. For all Mn-containing samples, a first shell was observed at ∼1.7 Å (uncorrected for phase shift), representing the first oxygen shell surrounding the Mn atoms. A second shell with R + ΔR at 2.8 Å was observed, which can be attributed to Mn and Al or Si backscatterers. Due to the similar or even identical crystallographic distances of Mn2+ and Al3+ in LDH phases (3.318 Å),54 it is difficult to unequivocally differentiate these two atoms. Additionally, constructive interferences from Si4+ atoms could occur when present at a slightly larger distance of 3.393 Å like e.g., in rhodonite.55
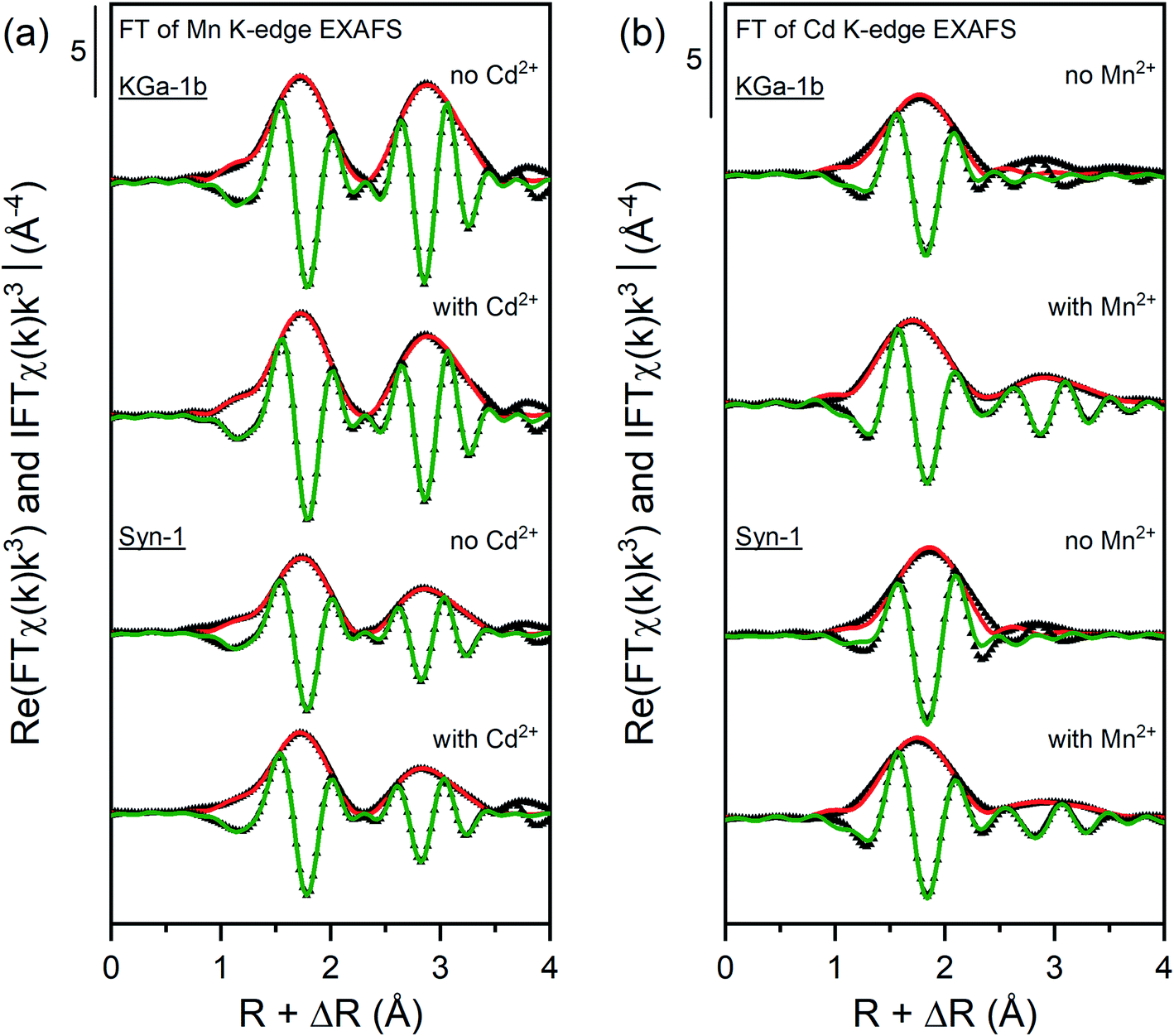 | ||
| Fig. 5 Fourier transform real parts and magnitudes of the Mn and Cd K-edge EXAFS spectra of selected samples with corresponding shell fit. Points indicate experimental data and solid lines show the model fits. The fits were performed over a R-range of 1.2–3.5 Å (k-weight = 3) except for the samples equilibrated with Cd2+ which were fitted over the R-range of 1.2–2.5 Å. The corresponding fitting parameters are reported in Tables 1 and 2. | ||
For the Mn-containing samples (see Table 1 and Fig. 5a), the first shell was fitted with 4.0–5.5 O atoms at a distance of 2.18 Å, which is characteristic for an octahedral arrangement of O around Mn(II).54 Fitting of the FT of the k3χ(k) EXAFS spectra was unsatisfactory when including a Mn–Si or Mn–Al backscattering path for the second shell. Hence, the second shell was fitted with a Mn atom located at radial distance of 3.27–3.29 Å and with a coordination number between 2.4–5.0. The fitted distance of the second shell corresponds to a Mn atom around Mn(II), confirming that a Mn-containing solid phases formed under anoxic conditions.54 The higher second-shell CN found for KGa-1b samples indicate that the formation of a Mn(II)-containing solid phases was more favored in presence of KGa-1b than in presence of Syn-1. This difference may be due to sorption of Mn2+ to KGa-1b compared to Syn-1 (Table S7†), which resulted in higher dissolved Mn2+ concentration and thus enhanced the formation of Mn(OH)2 in the KGa-1b suspensions.
| Syn-1 | pH | Mn–O | Mn–Mn | ΔE0e | Red. χ2f | R-Factorg | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CNb | R (Å) | σ 2 (Å2) | CNb | R (Å) | σ 2 (Å2) | |||||
a The amplitude reduction factor S02, was set to 0.7 based on first shell optimization for all sorption samples (R-range 1.1–2.5 Å).
b Path degeneracy (coordination number).
c Mean half path length.
d Debye–Waller parameter.
e Energy-shift parameter.
f Fit accuracy; 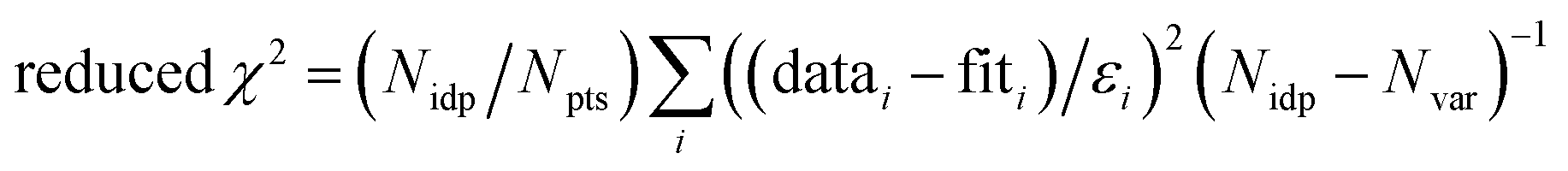 . Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (7), the total number of data points (141), and the number of fit variables (7). εi is the uncertainty of the ith data point.
g
R-Factor; normalized sum of squared residuals . Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (7), the total number of data points (141), and the number of fit variables (7). εi is the uncertainty of the ith data point.
g
R-Factor; normalized sum of squared residuals 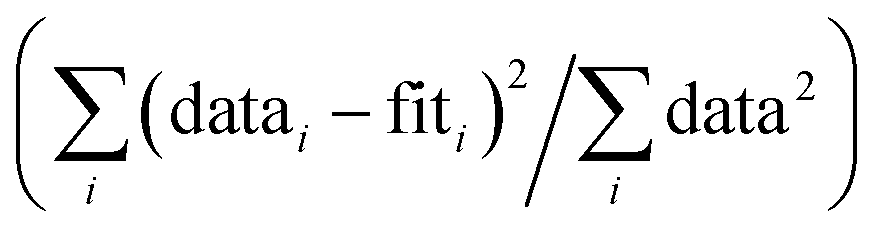 . Note: displayed pH values correspond to the pH measured at the end of the equilibration period for each sample. . Note: displayed pH values correspond to the pH measured at the end of the equilibration period for each sample.
|
||||||||||
| No Cd2+ | 8.1 | 4.0 (0.2) | 2.18 (0.01) | 0.006 (0.006) | 2.4 (0.3) | 3.29 (0.01) | 0.006 (0.001) | 3.13 (0.50) | 10 | 0.0014 |
| With Cd2+ | 8.4 | 4.6 (0.3) | 2.18 (0.01) | 0.007 (0.001) | 2.8 (0.5) | 3.27 (0.01) | 0.008 (0.001) | 2.13 (0.68) | 7 | 0.0025 |
The k3-weighted EXAFS spectra of the Syn-1 samples showed a truncated oscillation at 7–8 Å−1 similar to that of the MnAl-LDH reference (Fig. S20†). This feature has been identified as a signature feature for the presence of Al in the octahedral MeII(OH)2 sheets formed by divalent cations such as Ni, Zn, Co, and Fe.52 To gain more information about the possible incorporation of Al into the Mn-containing solid phases, wavelet transforms (WT) of EXAFS spectra were performed for the samples and compared to the references Mn(OH)2, MnAl-LDH, and rhodonite. WT complements the FT by resolving the k-dependence of the absorption signal, allowing better distinction between heavier and lighter backscattering atoms. The WT of the second coordination shell of the Mn-containing samples and references are shown in Fig. S22 and S23.† The WT plots of the samples exhibited two overlapping maxima at R = 2.8 Å. The observed WT displayed as an intermediate between MnAl-LDH and Mn(OH)2 which may suggest that Al released from the clay minerals was incorporated into the newly formed Mn-bearing solid phases. The shape of the WT of the Syn-1 samples resembled more the MnAl-LDH reference than Mn(OH)2, suggesting that more Al was incorporated into the Mn-containing solid phases in these samples. As more dissolution of the clay mineral was observed for Syn-1 samples compared to KGa-1b, it is likely that also more Al was released and available for incorporation.
The FT of the Cd K-edge EXAFS spectra for Cd-containing samples (see Table 2 and Fig. 5b) showed a first shell at ∼1.7 Å (uncorrected for phase shift). A second shell was observed in the FT of the Cd K-edge EXAFS spectra for samples containing both Cd and Mn (at ∼2.8 Å, uncorrected for phase shift). The presence of a minor second shell in Cd samples suggests that Cd was mainly adsorbed to the clay mineral through outer-sphere complexation. In contrast to that, the clear appearance of a second shell for samples containing both Cd and Mn pointed towards the sorption of Cd onto high-affinity sites on the newly formed Mn-containing solid phases or the incorporation of Cd in the formed Mn-containing solid phases.
| Syn-1 | pH | Cd–O | Cd–Mn | ΔE0e | Red. χ2f | R-Factorg | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CNb | R (Å) | σ 2 (Å2) | CNb | R (Å) | σ 2 (Å2) | |||||
a The amplitude reduction factor S02, was set to 0.7 based on first shell optimization for all sorption samples (R-range 1.1–2.5 Å).
b Path degeneracy (coordination number).
c Mean half path length.
d Debye–Waller parameter. Debye–Waller parameter was fixed to 0.006 Å2 for Cd–O based on the σ2 obtained for first shell fitting of all sorption samples. σ2 (Mn) was fixed to 0.004 Å2 based on σ2 fitted for Mn(OH)2 reference.
e Energy-shift parameter.
f Fit accuracy; 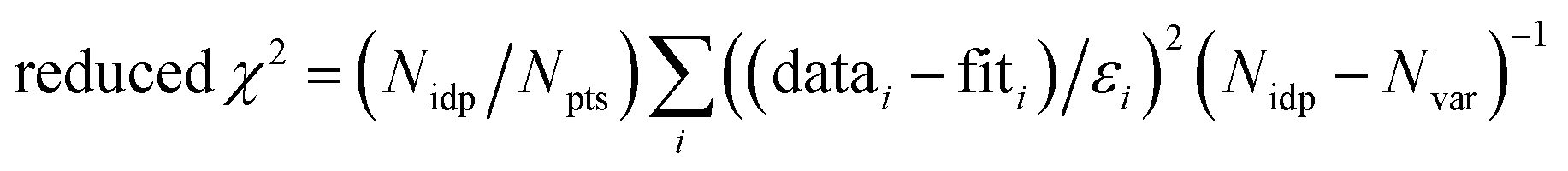 . Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (4–7), the total number of data points (101), and the number of fit variables (3–5). εi is the uncertainty of the ith data point.
g
R-Factor; normalized sum of squared residuals . Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (4–7), the total number of data points (101), and the number of fit variables (3–5). εi is the uncertainty of the ith data point.
g
R-Factor; normalized sum of squared residuals 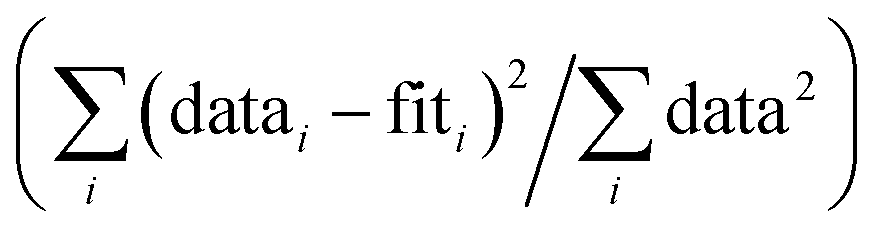 . Note: displayed pH values correspond to the pH measured at the end of the equilibration period for each sample. . Note: displayed pH values correspond to the pH measured at the end of the equilibration period for each sample.
|
||||||||||
| No Mn2+ | 7.8 | 3.9 (0.7) | 2.33 (0.03) | 0.006 | 11.89 (3.65) | 1318 | 0.0271 | |||
| With Mn2+ | 8.4 | 3.9 (0.2) | 2.28 (0.01) | 0.006 | 1.0 (0.2) | 3.33 (0.02) | 0.004 | 6.03 (1.39) | 80 | 0.0067 |
Fitting the first shell of Cd-containing samples required a Cd–O backscattering path at a radial distance between 2.26–2.33 Å and coordination number (CN) between 3.7–4.2. The minor changes in CN or radial distance of the first shell suggest that the type of surface complex Cd formed did not change substantially upon the addition of Mn2+. Shell fitting of the minor second shell for the Cd2+-containing samples in absence of Mn2+ was not possible, which may indicate that only small amounts of Cd precipitates were formed. The second shell observed in the FT of the Cd K-edge EXAFS spectra of Mn and Cd-containing samples was successfully fitted with a Cd–Mn backscattering path. This backscattering path was constructed by replacing one Mn atom in a pyrochroite structure by a Cd atom.54 Correspondingly, the second shell was fitted with a radial distance of 3.33–3.34 Å and a CN between 1.0 and 1.5. This radial distance is in agreement with the crystallographic distance of Mn in pyrochroite (3.318 Å).54
The Mn K-edge P-EXAFS spectra of the oriented sample recorded at α = 10°, 35°, 55° and 80° are presented in Fig. S24.† The presence of isosbestic points in the spectra provided evidence that differences in the measured spectra were due to orientation effects alone.56–59 The pronounced dependence upon orientation in the regions between the isosbestic points confirmed the successful preparation of a clay film with preferred orientation.57–60 The angle dependence of the spectra also confirmed the anisotropic formation of the secondary Mn-containing solid phases in KGa-1b samples. However, no clear angle dependency was observed in the Cd K-edge P-EXAFS spectra (Fig. S25†).
Conclusion
Modeling metal sorption data showed that both Cd2+ and Mn2+ had similar affinities for surface complexation on edge sites, suggesting that high Mn2+ concentration would lead to less sorption of Cd2+ at alkaline pH as indicated by the model prediction for competitive systems. Yet, in competitive experiments containing both Mn2+ and Cd2+, Mn2+ enhanced Cd2+ sorption, demonstrating that Mn2+ induced a process that led to a pronounced net increase of Cd2+ sorption. Mn K-edge XAS data analysis revealed that Mn2+ sorption was partly through the formation of Mn(II)-containing solid phases, although sorption experiments were performed under anoxic conditions and the solution were undersaturated with respect to common Mn-phases. Shell fitting of the FT of Cd K-edge EXAFS spectra indicated that Cd was sorbed by forming inner sphere surface complexes or by being incorporated into the formed Mn(II)-containing solid phases. This way, the Mn2+ addition increased the retention of Cd in the presence of clay minerals. Our findings imply that in anoxic submerged soils, where pH values typically are circumneutral, elevated Mn2+ concentration may contribute to increased retention of Cd by adsorption or incorporation into newly formed Mn(II)-containing solid phases on clay minerals. Consequently, the dynamics of dissolved Mn2+ may play an important role in the cycling of Cd in redox variable environments such as e.g., contaminated paddy fields used for rice production.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank M. Fischer and K. Barmettler (Soil Chemistry Group, ETH Zurich) for assistance in the laboratory, K. Hoffmann (Soil Chemistry Group, ETH Zurich) for his help during XAS measurements and Dr M. Plötze (ETH Zurich) for support with the N2-BET measurements. We would like to gratefully acknowledge Dr C. A. J. Appelo for his support with PHREEQC modelling. We acknowledge SOLEIL for the provision of synchrotron radiation facilities and thank G. Landrot for his support during the synchrotron measurements. This research was funded by the Swiss National Science Foundation (grant no. 200021_156392).References
- J. O. Nriagu and J. M. Pacyna, Quantitative Assessment of Worldwide Contamination of Air, Water and Soils by Trace-Metals, Nature, 1988, 333, 134–139 CrossRef CAS PubMed.
- J. L. Pan, J. A. Plant, N. Voulvoulis, C. J. Oates and C. Ihlenfeld, Cadmium levels in Europe: implications for human health, Environ. Geochem. Health, 2010, 32, 1–12 CrossRef CAS PubMed.
- D. C. Adriano, Trace elements in the terrestrial environments, Springer-Verlag, New York, 1986 Search PubMed.
- H. Kupper and B. Leitenmaier, Metal Ions in Life Science, ed. S. H. Sigel, A. Sigel and R. K. O. Sigel, Springer, Dordrecht, 2013, vol. 11 Search PubMed.
- WHO, Cadmium, 2000 Search PubMed.
- D. L. Sparks, Environmental soil chemistry, Academic Press, San Diego, 1995 Search PubMed.
- T. H. Christensen, Cadmium soil sorption at low concentrations. I. Effect of time, cadmium load, pH, and calcium, Water, Air, Soil, Pollut., 1984, 21, 105–114 CrossRef CAS.
- M. G. Hickey and J. A. Kittrick, Chemical partitioning of cadmium, copper, nickel and zinc in soils and sediments containing high levels of heavy metals, J. Environ. Qual., 1984, 13, 372–376 CrossRef CAS.
- H. B. Bradl, Adsorption of heavy metal ions on soils and soils constituents, J. Colloid Interface Sci., 2004, 277, 1–18 CrossRef CAS PubMed.
- P. Venema, T. Hiemstra and W. H. van Riemsdijk, Multisite adsorption of cadmium on goethite, J. Colloid Interface Sci., 1996, 183, 515–527 CrossRef CAS PubMed.
- A. Tessier, D. Fortin, N. Belzile, R. R. DeVitre and G. G. Leppard, Metal sorption to diagenetic iron and manganese oxyhydroxides and associated organic matter: Narrowing the gap between field and laboratory measurements, Geochim. Cosmochim. Acta, 1996, 60, 387–404 CrossRef CAS.
- K. G. Tiller, V. K. Nayyar and P. M. Clayton, Specific and nonspecific sorption of cadmium by soil clays as influenced by zinc and calcium, Aust. J. Soil Res., 1979, 17, 17–28 CrossRef CAS.
- M. H. Bradbury and B. Baeyens, Modelling the sorption of Mn(II), Co(II), Ni(II), Zn(II), Cd(II), Eu(III), Am(III), Sn(IV),Th(IV), Np(V) and U(VI) on montmorillonite: Linear free energy relationships and estimates of surface binding constants for some selected heavy metals and actinides, Geochim. Cosmochim. Acta, 2005, 69, 875–892 CrossRef CAS.
- J. M. Zachara, S. C. Smith, J. P. Mckinley and C. T. Resch, Cadmium sorption on specimen and soil smectites in sodium and calcium electrolytes, Soil Sci. Soc. Am. J., 1993, 57, 1491–1501 CrossRef CAS.
- C. Tournassat, S. Grangeon, P. Leroy and E. Giffaut, Modeling specific pH dependent sorption of divalent metals on montmorillonite surfaces. A review of pitfalls, recent achievement and current challenges, Am. J. Sci., 2013, 313, 395–451 CrossRef CAS.
- G. Kirk, The Biogeochemistry of Submerged Soils, John Wiley & Sons, Ltd, Chichester, UK, 2004 Search PubMed.
- D. Diem and W. Stumm, Is dissolved Mn2+ being oxidized by O2 in absence of Mn-bacteria or surface catalysts?, Geochim. Cosmochim. Acta, 1984, 48, 1571–1573 CrossRef CAS.
- D. Soltermann, M. Marques Fernandes, B. Baeyens, J. Miehé-Brendlé and R. Dähn, Competitive Fe(II)–Zn(II) uptake on a synthetic montmorillonite, Environ. Sci. Technol., 2014, 48, 190–198 CrossRef CAS PubMed.
- O. Jacquat, A. Voegelin, A. Villard, M. A. Marcus and R. Kretzschmar, Formation of Zn-rich phyllosilicate, Zn-layered double hydroxide and hydrozincite in contaminated calcareous soils, Geochim. Cosmochim. Acta, 2008, 72, 5037–5054 CrossRef CAS.
- A. C. Scheinost and D. L. Sparks, Formation of layered single- and double-metal hydroxide precipitates at the mineral/water interface: A multiple-scattering XAFS analysis, J. Colloid Interface Sci., 2000, 223, 167–178 CrossRef CAS PubMed.
- W. Li, K. J. T. Livi, W. Q. Xu, M. G. Siebecker, Y. J. Wang, B. L. Phillips and D. L. Sparks, Formation of crystalline Zn-Al layered double hydroxide precipitates on gamma-alumina: The role of mineral dissolution, Environ. Sci. Technol., 2012, 46, 11670–11677 CrossRef CAS PubMed.
- A. N. Starcher, W. Li, R. K. Kukkadapu, E. J. Elzinga and D. L. Sparks, Fe(II) sorption on pyrophyllite: Effect of structural Fe(III) (impurity) in pyrophyllite on nature of layered double hydroxide (LDH) secondary mineral formation, Chem. Geol., 2016, 439, 152–160 CrossRef CAS.
- Y. Zhu, J. J. Liu, O. Goswami, A. A. Rouff and E. J. Elzinga, Effects of humic substances on Fe(II) sorption onto aluminum oxide and clay, Geochem. Trans., 2018, 19, 3 CrossRef PubMed.
- A. Voegelin, A. C. Scheinost, K. Buhlmann, K. Barmettler and R. Kretzschmar, Slow formation and dissolution of Zn precipitates in soil – A combined column-transport and XAFS study, Environ. Sci. Technol., 2002, 36, 3749–3754 CrossRef CAS PubMed.
- Y. Zhu and E. J. Elzinga, Formation of layered Fe(II)-hydroxides during Fe(II) sorption onto clay and metal-oxide substrates, Environ. Sci. Technol., 2014, 48, 4937–4945 CrossRef CAS PubMed.
- A. N. Starcher, E. J. Elzinga and D. L. Sparks, Formation of a mixed Fe(II)-Zn-Al layered hydroxide: Effects of Zn co-sorption on Fe(II) layered hydroxide formation and kinetics, Chem. Geol., 2017, 464, 46–56 CrossRef CAS.
- L. Bhattacharya and E. J. Elzinga, A comparison of the solubility products of layered Me(II)-Al(III) hydroxides based on sorption studies with Ni(II), Zn(II), Co(II), Fe(II), and Mn(II), Soil Syst., 2018, 2, 20 CrossRef CAS.
- K. H. Tan, Principles of Soil Chemistry, CRC Press, Boca Raton, Florida, 4th edn, 2011 Search PubMed.
- T. Hiemstra and W. H. van Riemsdijk, A surface structural approach to ion adsorption: The charge distribution (CD) model, J. Colloid Interface Sci., 1996, 179, 488–508 CrossRef CAS.
- S. J. Chipera and D. L. Bish, Baseline studies of the clay minerals society source clays: Powder X-ray diffraction analyses, Clays Clay Miner., 2001, 49, 398–409 CrossRef CAS.
- A. R. Mermut and A. F. Cano, Baseline studies of The Clay Minerals Society Source Clays: Chemical analyses of major elements, Clays Clay Miner., 2001, 49, 381–386 CrossRef CAS.
- A. U. Dogan, M. Dogan, M. Onal, Y. Sarikaya, A. Aburub and D. E. Wurster, Baseline studies of The Clay Minerals Society Source Clays: specific surface area by the Brunauer Emmett Teller (BET) method, Clays Clay Miner., 2006, 54, 62–66 CrossRef CAS.
- D. G. Kinniburgh, C. J. Milne and P. Venema, Design and construction of a personal-computer-based automatic titrator, Soil Sci. Soc. Am. J., 1996, 59, 417–422 CrossRef.
- D. L. Parkhurst and C. A. J. Appelo, User's guide to PHREEQC (version 2)–A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, 1999 Search PubMed.
- S. H. R. Davies and J. J. Morgan, Manganese(II) oxidation-kinetics on metal-oxide surfaces, J. Colloid Interface Sci., 1989, 129, 63–77 CrossRef CAS.
- P. V. Brady, R. T. Cygan and K. L. Nagy, Molecular controls on kaolinite surface charge, J. Colloid Interface Sci., 1996, 183, 356–364 CrossRef CAS PubMed.
- G. Sposito and A. L. Page, in Metal ions in biological systems, ed. H. Sigel, Marcel Dekker, Inc, New York and Basel, 1984, vol. 18 Search PubMed.
- I. Heidmann, I. Christl, C. Leu and R. Kretzschmar, Competitive sorption of protons and metal cations onto kaolinite: experiments and modeling, J. Colloid Interface Sci., 2005, 282, 270–282 CrossRef CAS PubMed.
- S. H. Sutheimer, P. A. Maurice and Q. H. Zhou, Dissolution of well and poorly crystallized kaolinites: Al speciation and effects of surface characteristics, Am. Mineral., 1999, 84, 620–628 CrossRef CAS.
- B. R. Bickmore, K. L. Nagy, P. E. Sandlin and T. S. Crater, Quantifying surface areas of clays by atomic force microscopy, Am. Mineral., 2002, 87, 780–783 CrossRef CAS.
- J. Doherty, PEST model-independent parameter estimation, User Manual, Institution of Engineers, Australia, 5th edn, 1994 Search PubMed.
- R. M. Smith and A. E. Martell, Critical stability-constants, enthalpies and entropies for the formation of metal-complexes of aminopolycarboxylic acids and carboxylic-acids, Sci. Total Environ., 1987, 64, 125–147 CrossRef CAS.
- H. Funke, A. C. Scheinost and M. Chukalina, Wavelet analysis of extended x-ray absorption fine structure data, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71 Search PubMed.
- E. R. Nightingale Jr, Phenomenological theory of ion solvation. Effective radii of hydrated ions, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef.
- C. L. Chi, W. W. Emerson and D. G. Lewis, Exchangeable calcium, magnesium and sodium and dispersion of illites in water. I. Characterization of illites and exchange-reactions, Aust. J. Soil Res., 1977, 15, 243–253 CrossRef CAS.
- A. G. Volkov, S. Paula and D. W. Deamer, Two mechanisms of permeation of small neutral molecules and hydrated ions across phospholipid bilayers, Bioelectrochem. Bioenerg., 1997, 42, 153–160 CrossRef CAS.
- C. C. Ainsworth, J. L. Pilon, P. L. Gassman and W. G. Vandersluys, Cobalt, cadmium, and lead sorption to hydrous iron-oxide-residence time effect, Soil Sci. Soc. Am. J., 1994, 58, 1615–1623 CrossRef CAS.
- D. Soltermann, M. Marques Fernandes, B. Baeyens, R. Dähn, P. A. Joshi, A. C. Scheinost and C. A. Gorski, Fe(II) uptake on natural montmorillonites. I. Macroscopic and spectroscopic characterization, Environ. Sci. Technol., 2014, 48, 8688–8697 CrossRef CAS PubMed.
- S. D. Kelly, B. Hesterdberg and B. Ravel, in Methods of Soil Analysis Part 5—Mineralogical Methods, SSSA Book Ser. 5.5. SSSA, ed. L. R. Drees and A. L. Ulery, Madison, WI, 2008, pp. 387–463, DOI:10.2136/sssabookser5.5.c14.
- A. M. Scheidegger, G. M. Lamble and D. L. Sparks, Spectroscopic evidence for the formation of mixed-cation hydroxide phases upon metal sorption on clays and aluminum oxides, J. Colloid Interface Sci., 1997, 186, 118–128 CrossRef CAS PubMed.
- A. M. Scheidegger, D. G. Strawn, G. M. Lamble and D. L. Sparks, The kinetics of mixed Ni-Al hydroxide formation on clay and aluminum oxide minerals: A time-resolved XAFS study, Geochim. Cosmochim. Acta, 1998, 62, 2233–2245 CrossRef CAS.
- A. C. Scheinost, R. G. Ford and D. L. Sparks, The role of Al in the formation of secondary Ni precipitates on pyrophyllite, gibbsite, talc, and amorphous silica: A DRS study, Geochim. Cosmochim. Acta, 1999, 63, 3193–3203 CrossRef CAS.
- E. Peltier, R. Allada, A. Navrotsky and D. L. Sparks, Nickel solubility and precipitation in soils: A thermodynamic study, Clays Clay Miner., 2006, 54, 153–164 CrossRef CAS.
- J. B. Parise, B. Theroux, R. Li, J. S. Loveday, W. G. Marshall and S. Klotz, Pressure dependence of hydrogen bonding in metal deuteroxides: a neutron powder diffraction study of Mn(OD)2 and beta-Co(OD)2, Phys. Chem. Miner., 1998, 25, 130–137 CrossRef CAS.
- P. Leverett, P. A. Williams and D. E. Hibbs, Ca-Mg-Fe-rich rhodonite from the Morro da Mina mine, Conselheiro Lafaiete, Minas Gerais, Brazil, Mineral. Rec., 2008, 39, 125–130 CAS.
- R. Dähn, B. Baeyens and M. H. Bradbury, Investigation of the different binding edge sites for Zn on montmorillonite using P-EXAFS – The strong/weak site concept in the 2SPNE SC/CE sorption model, Geochim. Cosmochim. Acta, 2011, 75, 5154–5168 CrossRef.
- R. Dähn, A. M. Scheidegger, A. Manceau, M. L. Schlegel, B. Baeyens, M. H. Bradbury and M. Morales, Neoformation of Ni phyllosilicate upon Ni uptake on montmorillonite: A kinetics study by powder and polarized extended X-ray absorption fine structure spectroscopy, Geochim. Cosmochim. Acta, 2002, 66, 2335–2347 CrossRef.
- A. Manceau, D. Chateigner and W. P. Gates, Polarized EXAFS, distance-valence least-squares modeling (DVLS), and quantitative texture analysis approaches to the structural refinement of Garfield nontronite, Phys. Chem. Miner., 1998, 25, 347–365 CrossRef CAS.
- A. Manceau and M. L. Schlegel, Texture effect on polarized EXAFS amplitude, Phys. Chem. Miner., 2001, 28, 52–56 CrossRef CAS.
- A. Manceau, D. Bonnin, P. Kaiser and C. Fretigny, Polarized EXAFS of Biotite and Chlorite, Phys. Chem. Miner., 1988, 16, 180–185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data for the characterization of the clay minerals, information about the kinetics of metal sorption, acid–base titration of the clay minerals, additional data for Cd2+ and Mn2+ adsorption on KGa-1b and Syn-1, detailed information about the model and the used fitting parameters, a second set of competition sorption experiments, calculation of saturation indices, protocol to ensure anoxic conditions, percentage of Cd2+ and Mn2+ sorbed in samples for solid phase analysis, concentration of Si release during sorption experiments, details on XAS data collection and reduction and additional XAS results. See DOI: 10.1039/d0em00155d |
| This journal is © The Royal Society of Chemistry 2020 |