
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComposition and photo-reactivity of organic matter from permafrost soils and surface waters in interior Alaska†
Kristin R.
Gagné
a,
Sara C.
Ewers
a,
Carl J.
Murphy
b,
Ronald
Daanen
c,
Katey
Walter Anthony
de and
Jennifer J.
Guerard
 *a
*a
aDepartment of Chemistry & Biochemistry, University of Alaska Fairbanks, Fairbanks, Alaska 99775, USA. E-mail: jguerard@alaska.edu
bInstitute of Arctic Biology, University of Alaska Fairbanks, Fairbanks, Alaska 99775, USA
cDivision of Geological and Geophysical Surveys, Alaska Department of Natural Resources, Fairbanks, Alaska 99709, USA
dWater and Environmental Research Center, University of Alaska Fairbanks, Fairbanks, Alaska 99775, USA
eInternational Arctic Research Center, University of Alaska Fairbanks, Fairbanks, Alaska 99775, USA
First published on 22nd June 2020
Abstract
Yedoma permafrost soils are especially susceptible to abrupt thaw due to their exceptional thickness and high ice content. Compared to other mineral soils, yedoma has a high organic carbon content, which has shown to be particularly biolabile. The organic carbon in these deposits needs to be characterised to provide an identification toolkit for detecting and monitoring the thaw, mobilisation and mineralisation of yedoma permafrost. This study characterised organic carbon isolates from thermokarst lakes (either receiving inputs from thaw of original yedoma or refrozen-thermokarst deposits, or lacking recent thaw) during winter and summer seasons within the Goldstream Creek watershed, a discontinuous permafrost watershed in interior Alaska, to identify the extent to which thermokarst-lake environments are impacted by degradation of yedoma permafrost. Waters from lakes of varied age and thermokarst activity, as well as active layer and undisturbed yedoma permafrost soils were isolated and characterised by functional group abundance (multiCP-MAS 13C and SPR-W5-WATERGATE 1H NMR), absorbance and fluorescence, and photobleaching ability. DOM isolated from winter and summer seasons revealed differing composition and photoreactivity, suggesting varied active layer and permafrost influence under differing ground water flow regimes. Water extractable organic matter isolates from permafrost leachates revealed variation in terms of photoreactivity and photolability, with the youngest sampled permafrost isolate being the most photoreactive and photolabile. As temperatures increase, release of permafrost organic matter is inevitable. Obtaining a holistic understanding of DOM composition and photoreactivity will allow for a better prediction of permafrost thaw impacts in the coming decades.
Environmental significanceYedoma permafrost deposits are rapidly thawing in high latitudes, causing mobilisation and transformation of surface and sub-surface organic carbon pools. This study characterised organic carbon isolates from thermokarst-lakes (either receiving inputs from thaw of original yedoma or refrozen-thermokarst deposits, or lacking recent thaw) during winter and summer seasons within the Goldstream Creek watershed, a discontinuous permafrost watershed in interior Alaska, to identify the extent to which thermokarst-lake environments are impacted by degradation of yedoma permafrost. |
Introduction
Northern permafrost soils hold the largest pool of terrestrial organic carbon on Earth, estimated at 1307 Gt C.1 Of this, 450 Pg (25–35%) occurs in yedoma, yet comprises only about 7% of the permafrost area.1–5 This means that yedoma has a disproportionately large concentration of permafrost soil organic carbon. Yedoma deposits formed in unglaciated regions of Eurasia and North America during the late Pleistocene when syngenetic sediment, peat, and ice accumulated.3,6,7 Yedoma tends to be much older than non-yedoma permafrost soils, so there is arguably a more significant permafrost C feedback since the C has been locked away from the atmosphere for tens of thousands of years compared to some younger (e.g. Little Ice Age) permafrost soils.1,8,9These deposits, covering 1.8 million square kilometres in North Siberia and Alaska, are especially susceptible to abrupt thaw (thermokarst) due to their exceptional thickness and high ice content.3,6,10–12 Ice melt leads to ground subsidence, ponding of water, and rapid talik development beneath lakes.6,13–15 80% of the original late Pleistocene yedoma carbon pool has already thawed under anaerobic conditions beneath thermokarst lakes and gullies during the Holocene.5,8 The majority of these thermokarst deposits refroze after lakes drained, sequestering organic carbon of a different and unknown quality in less ice-rich epigenetic permafrost sediments. The remaining original yedoma is thus highly dissected across the landscape.
Thaw may be exacerbated by the infiltration of supra- or sub-permafrost ground water into unfrozen soils, further mobilizing previously held organic carbon. Release of materials from thawed permafrost soils also impacts surface water composition. For example, Toohey et al.16 attributed increased ions, nutrients, and observed positive monthly dissolved organic carbon (DOC) flux trends to active layer expansion and permafrost thaw in the Yukon and Tanana Rivers. In thermokarst lakes in Western Siberia, DOC concentration has been shown to be negatively correlated with lake stage development,17 with younger lakes containing more total DOC than more mature lakes, which the authors suggested was due to the progressive consumption and photodegradation of organic matter over time. However, thermokarst features on lake shores can remain active for centuries, allowing them to be a driving mechanism for the delivery of permafrost organic matter to surface waters.18
The organic carbon in original and refrozen yedoma deposits needs to be characterised in order to provide an identification toolkit for detecting and monitoring the thaw, mobilisation and mineralisation of yedoma permafrost soils. While permafrost-derived organic matter is largely comprised of smaller molecular weight, increased aliphatic moieties, and relatively less aromaticity compared to active layer carbon,19–22 fewer studies have characterised yedoma organic carbon.19,22 Organic matter in yedoma deposits in lake sediments in western Alaska were identified as largely terrestrially derived, based on C/N ratios and lipid biomarkers,23 implying a heterogeneity in permafrost carbon composition.
Biolability of permafrost derived DOM has been observed to be dependent upon the composition of organic matter6,18,24,25 and lake ontogeny,26 and yedoma organic matter may be especially labile.6,11,27 For example, Heslop et al.6 observed that increased biolability along four depths of yedoma lake sediment was due to a higher abundance of reduced and saturated organic compounds. Permafrost organic matter is less degraded, which may stimulate microbial processing of permafrost carbon upon thaw,23 contributing significant radiative forcing from CO2 and CH4 emissions over the next century.28
Permafrost organic carbon is also photoreactive. The extent of photo-oxidation influences its composition and thus propensity for biodegradation.29,30 For example, exposure to sunlight primes Alaskan Arctic soil organic carbon for microbial processing upon release and impacts its ability to produce hydroxyl radical,31,32 but surface waters from the Bolshezemelskaya Tundra peatlands were found to be resistant to both bio- and photodegradation over a 1 month period.33 Fewer studies though have focused on the photoreactivity of yedoma permafrost carbon. Stubbins et al.22 identified transformation but little photomineralisation of ancient yedoma-derived DOM from north-eastern Siberia near the Kolyma River. Photoreactivity can transform DOM to other products besides mineralised carbon. DOM photo-oxidation in permafrost underlain surface waters can occur via the formation of reactive oxygen species (ROS) such as hydroxyl radical30,32,34,35 or triplet excited state DOM.36
DOM has been characterised in several thermokarst-impacted watersheds in Alaska, however the relationships between permafrost and surface water composition of thermokarst lakes remain poorly understood. Also, while the number of studies characterising permafrost and biogeochemical impacts upon thaw have expanded greatly over recent decades, the composition of yedoma permafrost carbon pools remains relatively unexplored. Further, even fewer studies have characterised the photoreactivity of DOM from permafrost and thermokarst-influenced waters. Given yedoma permafrost's relatively high carbon and susceptibility to form thermokarst, it is necessary to better qualify the content and behaviour of this carbon pool in order to gain understanding of its influence on the biogeochemical function of permafrost organic carbon and ultimately the ecological impacts upon its release in a warming climate.
This study characterised the composition and photoreactivity of DOM isolated from thermokarst lakes, including thaw inputs from original yedoma, thaw of refrozen-thermokarst, and lacking recent thaw, as well as surface soils, undisturbed permafrost soils, and groundwater beneath permafrost within the Goldstream Creek watershed in interior Alaska. Our objective was to identify the extent to which organic carbon pools in thermokarst-lake environments are impacted by degradation of yedoma permafrost, leaching of surface soils, and mixing with groundwater. Isolates were characterised by organic functional group abundance and optical properties, as well as susceptibility to photobleaching in simulated sunlight.
Materials and methods
Study sites
The Goldstream Creek watershed37 (Fig. 1) is a sub-Arctic residential watershed underlain by discontinuous yedoma permafrost approximately 11 km northeast of Fairbanks, Alaska that drains into the Tanana River within the broader Yukon River Basin.38 This study area has a mean annual temperature of −2.4 °C and a mean annual precipitation of 274 mm (Fairbanks Int. Airport, 1981–2010, U.S. National Climatic Data Center) and is dominated by boreal forest vegetation. This watershed contains several thermokarst lakes, with some lakes having formed from abrupt thaw within the past 60 years.28 Within this watershed, waters from multiple lakes of varying talik development and thermokarst activity (Table 1), a high carbon well, active layer soils, and permafrost soils (Table 2) were sampled. The four study lakes are located in the bottom of the watershed, which is dominated by discontinuous permafrost consisting of Pleistocene organic and ice-rich deposits of the Goldstream Loess formation (e.g., yedoma), as well as its refrozen thermokarst deposits.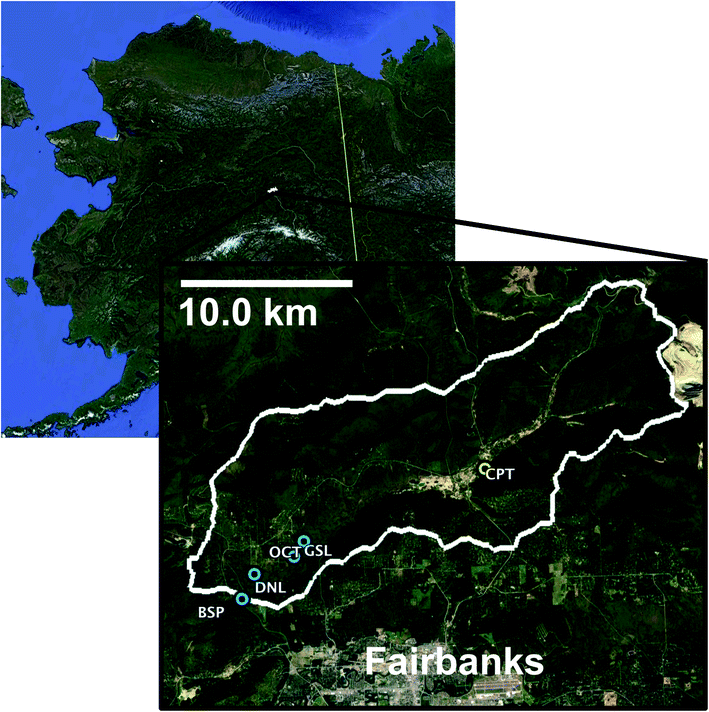 | ||
| Fig. 1 Location and outline of Goldstream Watershed and sample sites. Blue locations represent surface waters. Yellow location represents CRREL Permafrost Tunnel (CPT). Active layer samples were taken at GSL site. | ||
| Study sitea | Goldstream Lake (GSL) | Octopus Lake (OCT) | Doughnut Lake (DNL) | Blacksheep Pond (BSP) | Residential well (GSRW) |
|---|---|---|---|---|---|
| a Unofficial names. b Data from Elder et al.38 c TOC/TN data unavailable for this sample, reported values from a prior Aug 2016 sampling. d Corrections to SUVA for iron content are described in the ESI. | |||||
| GPS (°N, °W) | 64.916, 147.847 | 64.907, 147.860 | 64.899, 147.908 | 64.888, 147.920 | Undisclosed |
| Surface area (m2) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 030b 030b |
22000 |
34000b |
540 | N/A |
| Lake max depth (m) | 4.7b | 2 | 3.8a | <2 | N/A |
| Permafrost type | Original yedoma | Original yedoma | Refrozen thermokarst | Refrozen thermokarst | Unknown |
| Recent thermokarst | Yes | Some | No | Yes | Unknown |
| Est. age (years) | >120b | — | 1000a | <60 | N/A |
| Date(s) sampled | Jan 17 | Jun 17 | Aug 18 | Jan 17 | Aug 18 | Mar 17 | Jul 16 |
| Sampled depth (m) | 0 | 0 | 0 | 0 | 0 | 0 | 72.5 |
| pH | 6.30 | 8.20 | 8.46 | 5.09 | 8.43 | — | 6.93 |
| Sp. conductivity (mS cm−1) | 1.21 | 0.47 | 0.31 | 0.63 | 0.41 | — | 0.99 |
| DOC (mg C L−1) | 125 ± 1 | 556 ± 1 | 36.3 ± 0.7 | 60.2 ± 0.2 | 58.0 ± 0.3 | 216 ± 4 | 11.52 ± 0.04c |
| TDN (mg N L−1) | 5.99 ± 0.08 | 2.73 ± 0.03 | 0.16 ± <0.01 | 2.90 ± 0.05 | 0.17 ± 0.01 | 16.6 ± 0.3 | 4.33 ± 0.04c |
| SUVA (L mg C−1 m−1) | 2.53 ± 0.03 | 5.4 ± 0.1 | 6.3 ± 0.1 | 2.48 ± 0.01 | 3.94 ± 0.02 | 2.97 ± 0.05 | 3.78 ± 0.02c |
| SUVAcorr (L mg C−1 m−1)d | 1.99 | 5.16 | 5.56 | 2.23 | 3.90 | 2.95 | 3.64c |
| S R | 0.82 | 0.86 | 0.87 | 0.89 | 1.01 | 0.93 | 0.93 |
| E2:E3 |
8.55 | 7.90 | 5.87 | 8.39 | 9.82 | 8.12 | 6.55 |
| FI | 1.70 | 1.62 | 1.54 | 1.56 | 1.52 | 1.63 | 1.66 |
| BIX | 0.74 | 0.70 | 0.60 | 0.70 | 0.72 | 0.70 | 0.69 |
| Freshness | 0.71 | 0.69 | 0.58 | 0.66 | 0.69 | 0.68 | 0.68 |
| HIX | 0.95 | 0.94 | 0.92 | 1.03 | 0.89 | 0.95 | 0.92 |
| Dissolved Fe (μg L−1) | 2960 ± 20 | 503 ± 2 | 1470 ± 30 | 1243 ± 2 | 116 ± 1 | 72 ± 3 | 430 ± 40 |
| Goldstream active layer (GSAL) | CRREL permafrost tunnel (CPT) | ||||
|---|---|---|---|---|---|
| a Data from Mackelprang et al.47 | |||||
| Date(s) sampled | Apr 18 | Apr 18 | |||
| GPS (°N, °W) | 64.92, −147.830 | 64.951, −147.621 | |||
| Soil depth (m) | 0–0.19 | 0–0.26 | 20 | 54 | 81 |
| Soil 14C age (years) | — | — | 19000a |
27000a |
33000a |
| Gravimetric % water/ice | 90% | 88% | 17% | 20% | 22% |
| % C | 3 ± 2 | 3.1 ± 0.3 | 3.1 ± 0.4 | ||
|
|||||
| Soil leachate properties | |||||
| pH | 5.20 | 5.15 | 8.05 | 7.72 | 8.30 |
| TOC (mg C L−1) | 22.0 ± 0.4 | 26.1 ± 0.8 | 2.9 ± 0.2 | 11.67 ± 0.1 | 23.7 ± 0.3 |
| TDN (mg N L−1) | 0.58 ± 0.03 | BDL | 0.49 ± 0.01 | 1.28 ± 0.05 | 1.87 ± 0.04 |
| SUVA (L mg C−1 m−1) | 2.57 ± 0.04 | 1.87 ± 0.06 | 2.0 ± 0.1 | 1.72 ± 0.02 | 2.28 ± 0.03 |
| SUVAcorr (L mg C−1 m−1) | 2.49 | 1.85 | 2.0 | 1.67 | 2.20 |
| S R | 0.59 | 0.59 | 0.94 | 0.89 | 0.91 |
| E2:E3 |
4.69 | 4.66 | 5.24 | 6.26 | 6.18 |
| FI | 1.34 | 1.32 | 1.51 | 1.53 | — |
| BIX | 0.42 | 0.42 | 0.58 | 0.58 | — |
| Freshness | 0.40 | 0.39 | 0.57 | 0.57 | — |
| HIX | 0.88 | 0.90 | 0.95 | 0.93 | — |
| Dissolved Fe (μg L−1) | 188 ± 4 | 112 ± 2 | 13 ± 2 | 348 ± 3 | 377 ± 3 |
Goldstream Lake (informal name; GSL) has been previously described.38–40 The eastern margin of Goldstream Lake is expanding from active thermokarst into original yedoma.40 Octopus Lake (informal name; OCT) appears to have minor thermokarst activity suspected to also be into original yedoma as determined by 14C radiodating of methane and geophysical analysis.41 Blacksheep Pond (informal name; BSP) is a very small closed talik lake, less than 60 years old and resulting from abrupt thaw of refrozen thermokarst deposits. Doughnut Lake (informal name; DNL) may have also formed from thermokarst into refrozen deposits based on methane radiodating,41 but its currently insignificant thermokarst activity37 allows it to serve as a negative control for thermokarst influence.
Sampling
Epilimnion surface waters were sampled for DOM isolation during summer for three lakes (DNL, GSL, OCT), and additionally in winter prior to melt for three lakes (BSP, DNL, GSL) and were transferred in a cooler to the laboratory where the waters were then analysed and processed for DOM isolation. Ground water was obtained from a high dissolved methane, high carbon residential well near Blacksheep Pond (GSRW). This water was sampled via an outdoor tap before filtration and softener systems in July 2016, and stored and transported as described above.Two replicate active layer samples were obtained using a SIPRE corer during April 2018 near Goldstream Lake (GSAL1, GSAL2). The active layer depth of the cores was no more than 60 cm, however exact active layer depth was unable to be determined as supra-permafrost waters mixed with unfrozen portions of active layer while coring. Soil samples were obtained from the Cold Regions Research and Engineering Laboratory (CRREL) permafrost tunnel (CPT) with a round key hole saw attached to a power drill at 20 m, 54 m, and 81 m from the tunnel opening. The CRREL permafrost tunnel has been well characterised in terms of its geocryology and stratigraphy,42–48 and is comprised of syngenetic ice-rich permafrost that has been characterised as largely containing Pleistocene aged loess and gravels of the Goldstream Formation.42,45,46 These depths were reported by Mackelprang et al.47 as radiocarbon dated to 19000 years, 27000 years, and 33000 years, respectively (Table 2). Immediately after collection, cores were wrapped in plastic and then stored at −80 °C. Soil cores were then freeze-dried to constant mass followed by storage in acid washed plastic bags in the dark.
DOM isolation and characterisation
Freeze-dried soils were leached with 18.2 MΩ water at a ratio of 1:200 soil:water (w/w) for a period of 7 d in the dark at 4 °C with daily agitation. Soil leachate and sampled surface waters were filtered to 0.45 μm (Pall, Port Washington, New York). Following filtering, small aliquots were sampled for total organic carbon (TOC), total nitrogen, absorbance, and fluorescence analysis. DOM was then isolated as described in Dittmar et al.49 Briefly, filtered water was acidified to pH 2 with concentrated hydrochloric acid and pumped onto Bond-Elut PPL solid phase extraction cartridges (Agilent; Santa Clara, CA) at 18 mL min−1. DOM was extracted from PPL cartridges with methanol, whereby eluent was rotovapped and diluted back up to 90% water before freeze-drying to constant mass. Chemicals and analytical instrumentation methods (absorbance, fluorescence, nuclear magnetic resonance; NMR) are provided in the ESI.†
Photochemical experiments
DOM isolates were reconstituted to 10 mg C L−1 in aqueous solution and pH adjusted to 7.61–7.68 with NaOH or HCl and placed in 10 mL quartz test tubes with a 1 cm inner diameter (Robson Scientific; Hertfordshire, England). Solutions were run as either pH adjusted reconstituted DOM, or with additions of 20 μM Fe(III) or 12.5 μM methanol.50,51 Solutions were irradiated for 24 h in an Atlas CPS+ Suntest (ATLAS; Mount Prospect, Illinois) solar simulator with a 1500 W Xe lamp set to 15 °C with duplicate samples sacrificed at each time point. Dark controls were wrapped in aluminium foil. Irradiance was monitored for consistency by a PMA2100 radiometer (Solar Light Co. Inc, Glenside, PA). Samples were stored in the dark until analysed for absorbance and fluorescence within 2 h of irradiation.Solutions of 10 μM 2,4,6-trimethylphenol (TMP) in the presence of 10 mg C L−1 DOM solutions were used to probe triplet excited DOM reactivity.36 Photolysis experiments were set up as described above, and run in duplicate over 6 h with dark controls. TMP was quantified on an Agilent 1100 Series reverse-phase HPLC (Agilent Technologies, Santa Clara, CA) using a Restek Ultra C18 5 μm column (Restek Corporation, Centre County, PA) and a 60:40 acetonitrile:water (v/v) mobile phase at 1 mL min−1, detected by fluorescence at λex = 230 nm and λem = 305 nm.
Light screening was determined from the absorbance data by eqn (1),52
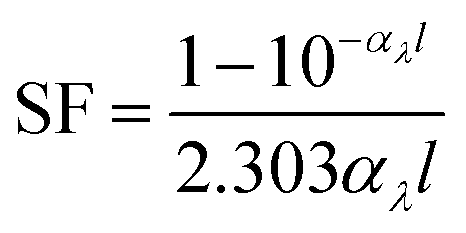 | (1) |
Apparent quantum yields (AQY) for DOC loss were quantified using the p-nitroanisole (PNA) chemical actinometer, in order to compare extent of DOC loss between DOM and water extractable organic matter (WEOM) isolates, assuming all DOC was lost as CO2.53 No pyridine was added, so that PNA losses could be measured over 24 h to match the DOM photoirradiation periods. Because the solar simulator is a polychromatic light source, AQYs were calculated from 290–400 nm according to a similar method as Page et al.32 shown in eqn (2).
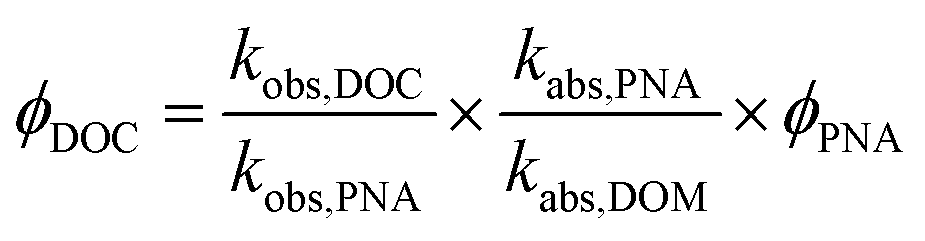 | (2) |
290–400 nm was chosen because of the cut-off of the daylight filter, and beyond 400 nm absorbances were very small. ϕDOC is the AQY for photochemical loss of DOC, kobs,DOC and kobs,PNA are the fitted first order rate constants of DOC and PNA loss respectively, kabs,PNA and kabs,DOM represent the integrated light absorption over 290–400 nm for PNA and DOM respectively, and ϕPNA is the quantum yield for PNA. Uncertainty in AQY derives from standard deviations in rates, which averaged ∼8% for PNA and ≤ 4% for measured DOC losses (Table 3).
| SUVA | ΔSUVA24 h | % DOCphoto | AQYDOC × 10−5 | k obs,254 nm L mg C −1 h −1 × 10−3 | Δkobs,254, + Fe L mg C−1 h−1 × 10−3 | Δkobs,254, + MeOH L mg C−1 h−1 × 10−3 | TMP kobs L mg C −1 h −1 | |
|---|---|---|---|---|---|---|---|---|
| a Rates calculated to only 6 h photolysis due minimal absorbance loss and non-first order kinetics after 6 h. b Errors listed are standard deviations of fitted kobs from a single experiment. | ||||||||
| DOM isolates | ||||||||
| GSL S (Jun 17) | 2.64 ± 0.03 | −0.63 ± 0.05 | 9 ± 3 | 32 | 1.15 ± <0.01 | 2.07 ± 0.05 | −0.31 ± 0.01 | 0.119 ± 0.007 |
| OCT S (Aug 18) | 3.11 ± 0.02 | −0.95 ± 0.03 | 7 ± 1 | 16 | 2.2 ± 0.4 | −0.6 ± 0.4 | −1.0 ± 0.4 | 0.129 ± 0.005 |
| DNL S (Aug 18) | 2.49 ± 0.02 | −0.66 ± 0.03 | Neg. | — | 1.25 ± <0.01 | 0.18 ± <0.01 | −0.11 ± 0.04 | 0.105 ± 0.007 |
| GSL W (Jan 17) | 2.77 ± 0.05 | −0.76 ± 0.06 | 4 ± 3 | 11 | 1.8 ± 0.2 | 4.5 ± 0.2 | −0.4 ± 0.3 | 0.11 ± 0.01 |
| DNL W (Jan 17) | 2.79 ± 0.04 | −0.78 ± 0.06 | 6 ± 3 | 17 | 1.45 ± 0.03 | 2.9 ± 0.3 | −0.2 ± 0.2 | 0.09 ± 0.02 |
| BSP W (mar 17) | 3.02 ± 0.02 | −0.69 ± 0.03 | 7 ± 2 | 20 | 1.70 ± 0.04 | 7 ± 1 | −0.2 ± 0.1 | 0.12 ± 0.03 |
| GSRW (Jul 16) | 3.82 ± 0.03 | −0.69 ± 0.03 | 14 ± 3 | 29 | 2.0 ± 0.2 | 2.5 ± 0.8 | −0.3 ± 0.5 | 0.11 + 0.02 |
|
||||||||
| WEOM isolates | ||||||||
| GSAL1 (Apr 18) | 3.73 ± 0.06 | −0.68 ± 0.07 | 4 ± 2 | 8.1 | 1.1 ± 0.1 | 0.1 ± 0.1 | −0.2 ± 0.2 | 0.050 ± 0.008 |
| GSAL2 (Apr 18) | 3.45 ± 0.06 | −0.46 ± 0.07 | 7 ± 3 | 9.8 | 1.16 ± 0.08 | 0.3 ± 0.1 | −0.2 ± 0.1 | 0.047 ± 0.003 |
| CPT1 (Apr 18) | 2.16 ± 0.02 | −0.09 ± 0.04 | 22 ± 1 | 72 | 5.19 ± 0.04a | −0.5 ± 0.3 | −1.2 ± 0.6a | 0.14 ± 0.01 |
| CPT2 (Apr 18) | 2.73 ± 0.03 | −0.80 ± 0.04 | 8 ± 3 | 23 | 1.3 ± 0.1 | 0.4 ± 0.2 | −0.2 ± 0.2 | 0.09 ± 0.02 |
| CPT3 (Apr 18) | 2.81 ± 0.05 | −0.76 ± 0.06 | 7 ± 3 | 17 | 1.68 ± 0.06 | 0.6 ± 0.1 | −0.1 ± 0.1 | 0.09 + 0.02 |
|
||||||||
| IHSS reference | ||||||||
| SRFA | 3.86 ± 0.05 | −0.50 ± 0.06 | 9 ± 4 | 15 | 1.1 ± 0.2b | 0.6 ± 0.3b | −0.0 ± 0.4b | 0.065 ± 0.005 |
| PLFA | 2.68 ± 0.03 | −0.61 ± 0.03 | 7 ± 3 | 15 | 0.9 ± 0.2b | 0.1 ± 0.3b | −0.02 ± 0.3b | 0.066 ± 0.004 |
Results
Surface water characterisation
For all of the sampled waters, DOC concentration was largely comparable to other boreal lakes,54–56 though in some cases over 100 mg C L−1 in the winter months of the active thermokarst lakes (GSL, BSP). GSL's carbon content varied by over a factor of two between summer and winter, whereas DNL's carbon content was more similar (Table 1).Specific UV absorbance at 254 nm (SUVA) values are similar to that of other lakes in interior Alaska,54 except for GSL summer and OCT, which had SUVA > 5.0 (Table 1). Despite the similar DOC concentrations, SUVA values for GSL and DNL differ between the summer and winter. For example, GSL's SUVA values are over twice as high in the summer compared to the winter (Table 1 and Fig. S3†). Even accounting for the higher iron content in the winter (described in ESI†), compared to the summer sample, the summer sample had the higher SUVA value, indicating differences in DOM composition. Other optical indices are less variable across lakes and sampling events, further detailed in the ESI.†
Soil and leachate characterisation
All three permafrost soils have similar carbon content (∼3% C, Table 2), consistent with previously reported measurements in the CRREL tunnel46 and indicative of yedoma permafrost,57 although samples were separated by over 10000 years according to soil 14C age.47 Ice content in permafrost soils is considerably lower than active layer moisture content (17–22% vs. 88–90%, respectively), though ice content could be underestimated due to sublimation occurring within the tunnel.48 Soil leachate DOC differs between the sampled soils (oldest permafrost leachate had the highest DOC at 23.65 ± 0.30 mg C L−1), however, optical indices are relatively stable across leachates. Replicate active layer leachates from nearby GSL display near identical optical qualities (Table 2), further detailed in the ESI.†
DOM and WEOM isolate optical characterisation
In contrast to the sampled whole waters, the DOM isolates display more similar SUVA values between samples, ranging from 2.49–3.82 (Table 3). The GSL winter isolate SUVA is higher than that of the sampled filtered water (Table 1), however the summer isolate's SUVA is much lower. This could be due to the differences in iron content, except all the isolate solutions contain less than 5 μM Fe by ferrozine (data not shown) and these differences are still present for iron corrected SUVA values (Table 1). For DNL, a similar trend occurs, where the isolate from the winter sample has a higher SUVA than the filtered water, but the summer isolate's SUVA value is lower than the sampled water, though not as dramatically as GSL. BSP and GSRW isolates display nearly the same SUVA values as their sampled waters, within the margin of error. The other optical indices are less variable between samples than their respective sampled waters and are further detailed in the ESI.†SUVA values for WEOM isolates from soil leachates (Table 3) are higher compared to their requisite leachate solutions (Table 2), often a value of more than 1 L mg C−1 m−1 greater. Increased SUVA values have been correlated with increased aromaticity,58 suggesting a preferential capture of aromatic groups during isolation, which may bias the carbon fraction studied relative to the entire mobilisable carbon pool. However, optical indices from soil WEOM isolates are much closer to their leachate solution optical properties than the DOM isolates are to their requisite sampled waters (Table 2 and Fig. S10†). This suggests a more uniform capture of the chromophoric and fluorescent carbon WEOM pool occurred during the isolation process compared to the sampled waters, but that non-chromophoric DOM was less retained, giving rise to the higher SUVA value in the WEOM isolates compared to their leachates.
NMR spectral features
Overall, bulk functional group composition and distinct features from NMR spectra are similar across surface water DOM isolates, for both 13C and 1H spectra (Fig. 2 and S1†). More detailed results of these features are described in the ESI.† However, the WEOM isolates from sampled soils are more variable (Fig. 2). The active layer (GSAL) samples are near identical, but compared to the surface water 13C spectra display a much sharper O-alkyl peak (70 ppm), a more defined anomeric C peak (110 ppm), a less pronounced carbonyl signal, and in some cases more defined features in the aromatic region (Fig. 2). The three permafrost tunnel isolates vary considerably, with the youngest WEOM isolate (CPT1) presenting sharper peaks in the aliphatic regions, as well as overall more defined peaks across the chemical shift range, consistent with less humification extent.59 CPT1 contains spectral features present in some of CPT2 and CPT3 WEOM isolates. For example, CPT1 contains a less defined carbonyl peak than CPT2 or CPT3. CPT1 and CPT 3have two defined aromatic peaks at 125 and 145 ppm, but CPT2 has nearly no defined aromatic peak at 145 ppm. CPT1 also has a much less distinct O-alkyl peak at 75 ppm compared to CPT2 and CPT3.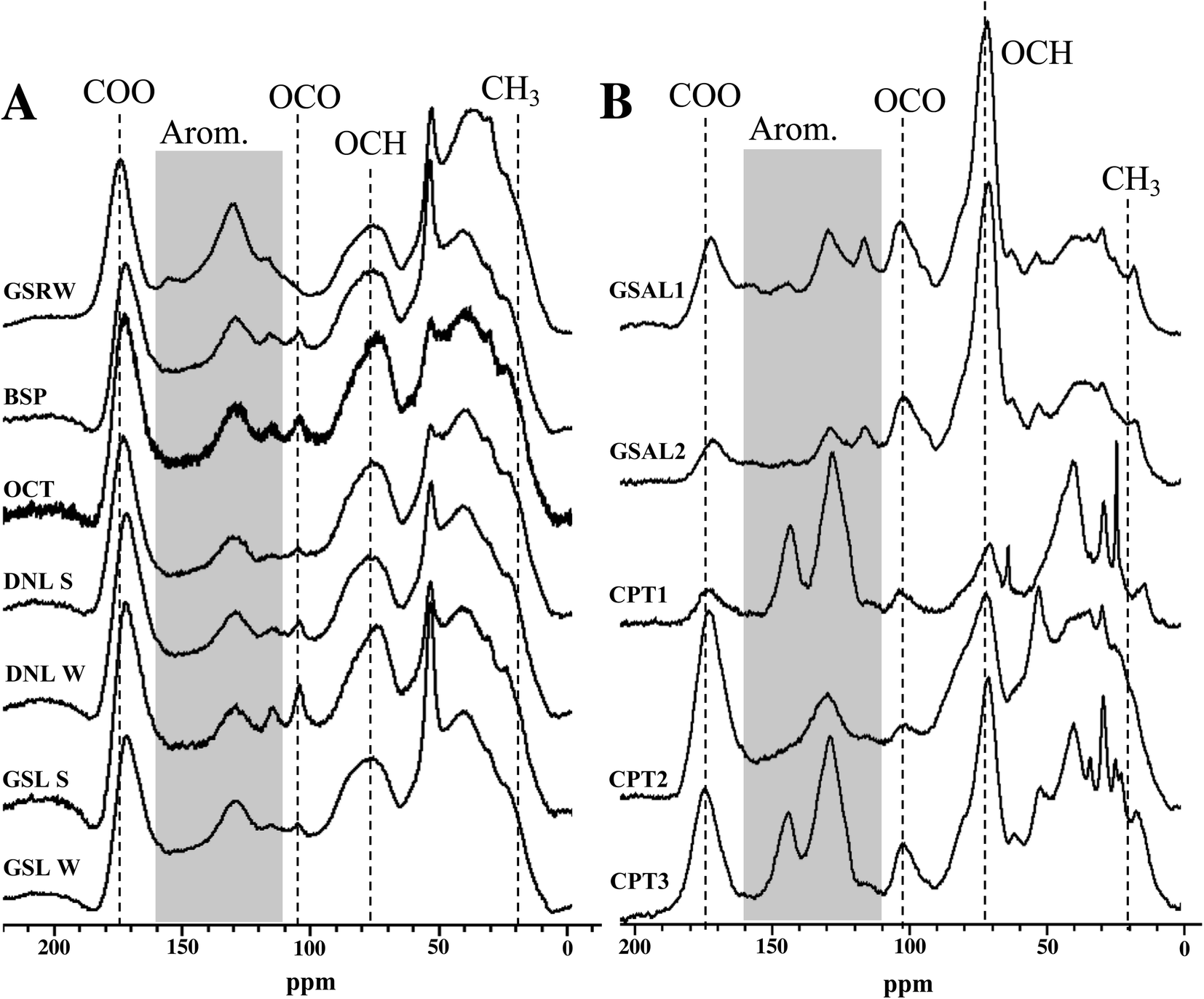 | ||
| Fig. 2 13C multiCP-MAS NMR spectra for PPL isolates. (A) Surface water isolates. (B) WEOM isolates. | ||
Functional group distribution of DOM and WEOM isolates
Most of the DOM isolates from lake waters yield relative integrations similar to each other, with standard deviation of signal ≤3.5% for all regions. The ability of 13C multiCP-MAS to standardise signal sensitivity across chemical shift similar to DP methods60 means that quaternary and carbonyl containing groups are likely to be elevated compared to traditional CP-MAS spectra.61,62 Aliphatics (0–45 ppm) and O-alkyl carbons (60–110 ppm) are the most abundant functional groups, averaging 26.0 ± 3.5% and 27.5 ± 3.4% of 13C NMR spectra, respectively, followed by aromatic regions (110–160 ppm, 19.1 ± 1.9%; Table S1†). Anomeric/acetal carbon (90–110 ppm) comprised 5.3–9.3% of the DOM isolates, with GSRW containing the smallest proportion of 13C signal from this region. However, the anomeric region may actually reflect a mixture of both anomeric and aromatics found in DOM,63 and are difficult to resolve from each other without other spectral editing techniques. Comparison to International Humic Substances Society (IHSS) isolates are presented in the ESI.†Summer and winter DOM isolates from the same lakes (GSL, DNL) have similar 13C NMR integrations (Table S1†), despite their differences in SUVA (Table 3). The winter DNL isolate has 3.8% more aromaticity and 3.9% less aliphatic content than its summer counterpart (Table S1†). However, in the 1H spectra, DNL summer isolate has more aromatic signal than the winter isolate, suggesting that the increased aromaticity observed in the winter isolate 13C spectrum consists of more substituted aromatics. In contrast, GSL's 13C spectra from both seasons integrate to near identical relative contributions for each region, within 2.1% for any given region.
WEOM isolates exhibit variation in both active layer and permafrost isolates, dominated by O-alkyl (32.8 ± 10.4%), followed by alkyl (24.2 ± 4.4%), and aromatic regions (22.3 ± 6.8%). The high average O-alkyl content is largely derived from the active layer and not the permafrost isolates, whose O-alkyl content was in some cases less than that of the DOM isolates (Table S1†). A more detailed description of WEOM isolate NMR spectra is presented in the ESI.†
The permafrost isolates differ from each other, in some cases more dramatically than compared to other isolate types. Aromaticity spread over a range from 17.7–32.9%, with no trend associated with radiocarbon age of the sampled permafrost. The isolate from the youngest permafrost sampled (CPT1) contains the highest aromaticity, followed by the isolate from the oldest (CPT3) and then the second oldest (CPT2) permafrost soil. CPT1 also contains the lowest carbonyl and O-alkyl content, but the highest aliphatic content of the permafrost isolates.
Photolability of DOM and WEOM isolates
Total organic carbon, absorbance and fluorescence losses upon photoirradiation were used as a probe for composition and reactivity. Several isolates saw DOC losses of over 5% from just 24 h irradiation in a solar simulator (Table 3). However, GSRW lost 13.5 ± 2.7% of initial DOC upon photoirradiation, and the WEOM isolate from the youngest permafrost soil (CPT1) lost 21.6 ± 1.4% of initial DOC. The only isolates to see DOC losses less than 5% were GSL winter, one of the active layer isolates, and negligible DOC losses were observed for only one DOM isolate, DNL summer. According to PNA actinometer degradation (kobs,PNA = 0.24 ± 0.02 h−1), actinometry showed about 4.1× intensity compared to summer noon sunlight at 40° N, or 9.7× intensity compared to at 70° N,64 so these DOC losses are likely considerably higher than what would be expected to be observed in natural sunlight. It is also possible that since samples were not sterilised or sterile-filtered before experimentation, some biomineralisation could have occurred, but under 24 h of continuous photoirradiation, it is unlikely to have occurred to an extent that could account for the drastic DOC losses. Overall, significant photolability was observed.Apparent quantum yields were calculated over 290–400 nm against PNA actinometry to account for differences in light absorption properties between the different DOM and WEOM isolates. AQYs span an order of magnitude, ranging from 8.05 × 10−5 to 7.23 × 10−4. The highest AQYs are CPT1, followed by GSL summer, GSRW, CPT2, and BSP. The active layer WEOM isolates show the smallest AQYs of the isolates, both having an AQY < 0.0001.
Photobleaching of DOM and WEOM isolates
Absorbance curves decay upon increased photoexposure (Fig. 3), with losses at 254 nm on average 32 ± 10% and 24 ± 3% over 24 h for DOM and WEOM isolates, respectively. Absorbance degradation follows pseudo-first order kinetics, with R2 of first order fits >0.92 for chromophores absorbing <400 nm. For several cases, fitted absorbance loss rates at higher wavelengths (>400 nm) are less first order, with R2 < 0.90, likely due to detection limits arising from low absorbance at those wavelengths. In general, absorbance loss rates are slowest at lower wavelengths (<260 nm), and steadily increase or level off with increasing wavelength (Fig. S5 and S6†). For several DOM isolates, a local absorbance loss rate maximum was observed ∼320 nm (BSP, DNL summer, GSL, OCT; Fig. S5†), while for the WEOM isolates a local maximum in absorbance loss appears at a slightly higher wavelength, ∼340 nm (Fig. S6†). | ||
| Fig. 3 Absorbance loss of 10 mg C L−1 GSL summer DOM isolate and amended solutions over 24 h irradiation. (A) Log linearised pseudo first-order kinetics of absorbance loss at 254 nm. (B) Fitted observed pseudo first order rates of absorbance loss across wavelengths. | ||
Absorbance loss rates at 254 nm range from 1.15–2.2 × 10−3 L mg−1 C−1 h−1 for DOM isolates, and 1.1–5.19 × 10−3 L mg−1 C−1 h−1 for WEOM isolates (Table 3). Of the DOM isolates, GSRW and OCT photobleach the fastest (Table 3 and Fig. 4). Though CPT1's absorbance loss could only be measured over 6 h, its absorbance loss rate is over 3–4 times faster than the other WEOM isolates (Table 3). Six-hour loss rates of the other isolates average only 1.3 ± 0.3 times faster than their 24 h loss rates (data not shown). Both DOM winter isolates photobleach faster than their summer counterparts (254 nm absorbance losses are 0.2 and 0.65 L mg−1 C−1 h−1 faster, respectively, for DNL and GSL winter isolates). Only the summer DOMs, active layer WEOMs, and CPT2 photobleaching rates fall within the range of IHSS reference absorbance losses, as all the other isolates' absorbance decay rates are faster than the IHSS references (Table 3).
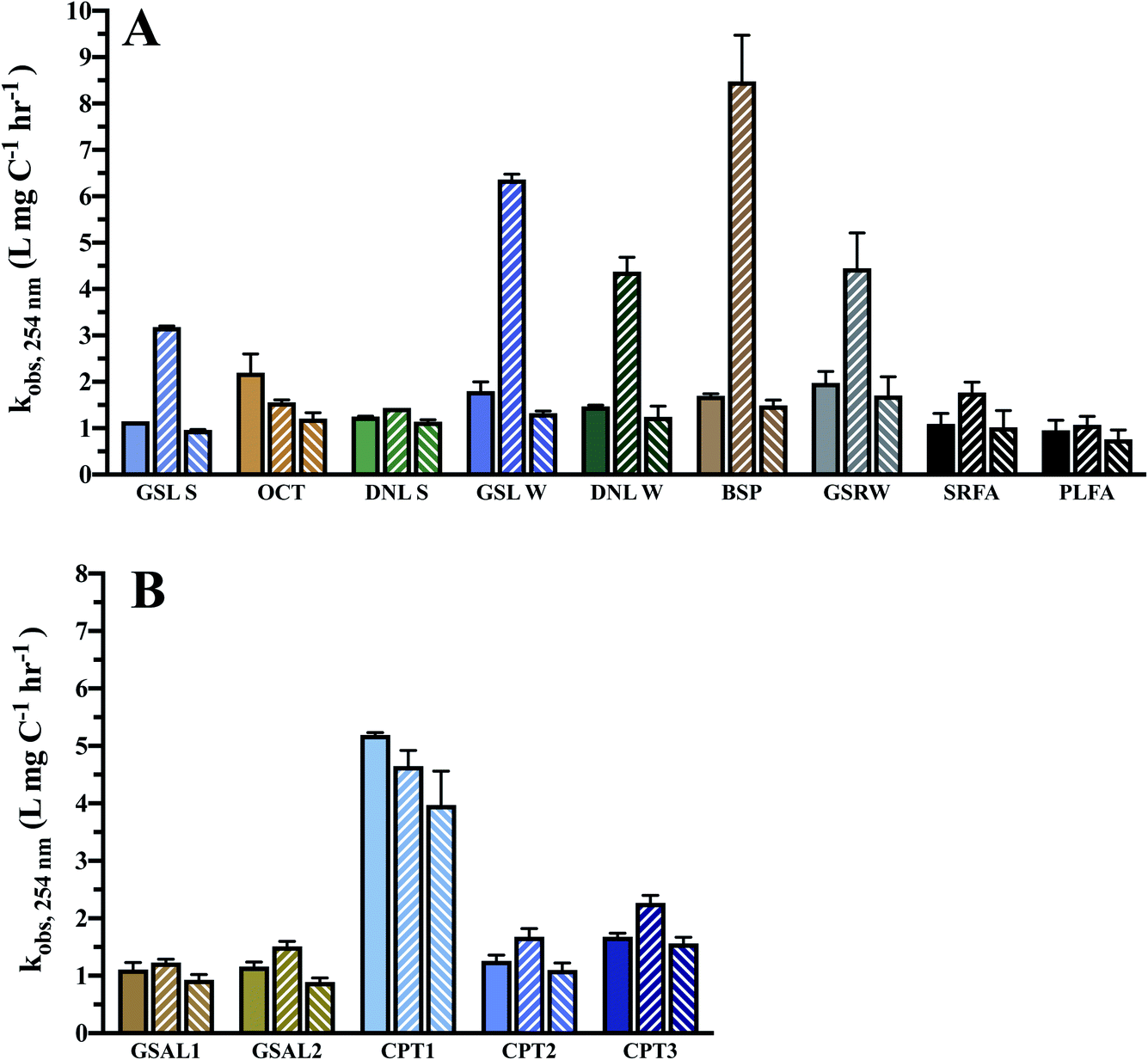 | ||
| Fig. 4 Observed pseudo first order absorbance loss rates at 254 nm in 10 mg C L−1 reconstituted PPL and reference isolate solutions, corrected for light screening and normalised to DOC concentration. Solid fill = DOM only. Upward slanted fill = DOM + 20 μm Fe. Downward slanted fill = DOM + 12.5 μm MeOH. (A) Surface water DOM isolates. (B) WEOM isolates. | ||
Optical indices also changed during irradiation, indicating non-uniform absorbance and fluorescence losses and a transformation in overall DOM composition (Fig. S9 and S10†). SUVA values decreased on average by 0.6 ± 0.3 L mg C−1 m−1 over 24 h photolysis, implying a loss of aromaticity and chromophoric DOM. SUVA of the OCT isolate decreased most dramatically, by 0.95 L mg C−1 m−1. While a positive relationship between SUVA and kobs,254 occurred for DOM isolates (R2 = 0.53), this was not the case for the WEOM isolates and neither set of isolates had a relationship between SUVA or Al:Ar and kobs,254 that was significantly non-zero.
Influence of sensitisers and quenchers on photoreactivity
Iron(III) was added to reconstituted isolate solutions to probe for the ability of the DOM and WEOM isolates to promote photo-Fenton processes,65–67 whereby production of ˙OH, a non-selective radical, may enhance photobleaching of DOM isolates in the presence of sunlight. Photoreduction of Fe(III) to Fe(II) reacts with photoproduced hydrogen peroxide to form ˙OH (eqn (3)):| Fe(II) + H2O2 → Fe(III) + OH− + ˙OH | (3) |
Given the high total dissolved iron present in sampled waters (Table 1), photo-Fenton processes may be extremely relevant for the phototransformation of DOM in surface waters. Most of the DOM and WEOM isolates exhibit enhanced rates of absorbance loss at 254 nm upon the addition of 20 μM Fe(III) (1116.9 μg L−1), except for OCT and CPT1 (Table 3 and Fig. 4), consistent with findings of hydroxyl radical production in Arctic surface waters.32 The WEOM isolates were not especially vulnerable to photo-Fenton processes, which may indicate compositional differences between mobilised thermokarst and permafrost DOM.
Enhanced chromophore decay upon iron addition is consistent across 240–450 nm (Fig. S5†) for the DOM isolates, except for DNL summer and OCT. The winter DOM isolates and GSRW show the most enhancement of absorbance loss at 254 nm, consistent across all wavelengths. In contrast, most of the soil WEOM isolates only exhibit enhancement of absorbance loss at lower wavelengths (Fig. S6†), but at wavelengths above 275–290 nm, iron addition slows absorbance loss. Slower loss rates could be indicative of precipitation of Fe(III), as experiments were not anoxic and occurred at circumneutral pH, as observed in studied lakes (Table 1); however, no precipitate formation was observed. Differing levels of enhancement or retardation of absorbance loss could then be attributed to either efficiency of H2O2 photoproduction, and/or the ability to stabilise iron in solution.
Methanol was utilised to broadly scavenge radicals formed during photobleaching.68 In most cases, the rate of absorbance loss at 254 nm upon addition of methanol ranged from 0.1–1.2 × 10−3 L mg C−1 h−1 slower (Table 3 and Fig. 4), indicating that a portion of observed photobleaching was due to radical production. In general, decreased rates of absorbance loss were consistent across wavelengths (Fig. S5 and S6†). No significant difference in absorbance loss rates were observed for several isolates (Table 3), suggesting either minimal photochemical radical production or the prevalence of a quenching mechanism that did not lead to absorbance losses.
2,4,6-Trimethylphenol (TMP) degradation rates may then be indicative of either triplet excited state reactivity or singlet oxygen production.36,68,69 Triplet excited state DOM (3DOM*) has been shown to react efficiently with phenolic, anilinic, and sulfidic groups.69 TMP's one-electron oxidation potential (1.22 V) renders it susceptible to single electron transfer with several quinoid moieties, or proton coupled electron transfer through phenoxy radical formation for weaker sensitisers.69 DOM isolates facilitated the photodegradation of TMP relatively similarly, with pseudo-first order degradation rates ranging from 0.09–0.129 L mg C−1 h−1 (Table 3). More variability was exhibited by WEOM isolates, where active layer WEOM isolates facilitated TMP photodegradation the least (0.048 L mg C−1 h−1 on average), and CPT1 facilitating TMP photodegradation the most. Except for the active layer WEOM isolates, all isolates facilitated TMP photodegradation faster than the IHSS isolates.
Discussion
This study compared several lakes with differing thermokarst activity within a single watershed underlain by discontinuous original yedoma or refrozen permafrost, along with WEOM isolates from permafrost and active layer soils within the watershed. Goldstream Valley is classified as yedoma permafrost. This is the same classification as yedoma permafrost in northeast Siberia, as it is defined as ice-supersaturated loess deposits that formed syngenetically in unglaciated regions of Eurasia and North America during the Late Pleistocene.3,5 The vast majority of yedoma permafrost occurs in northeast Siberia; however, there are pockets of it in Alaska.3,5 In both regions, organic carbon contents are usually less than 3%. In both regions, yedoma can be tens of meters thick.3,5 Unlike Siberia, where most of the yedoma occurs in the zone of continuous permafrost, the Goldstream Valley study sites are characterised by discontinuous permafrost. This means that there is a higher likelihood of surface and groundwater exchange. DOM isolates from winter sampling of active thermokarst lakes, where active layer inputs are minimal, revealed elevated photobleaching ability compared to summer isolates from the same lakes, more similar to the youngest sampled permafrost isolate than older or active layer isolates, suggesting that some permafrost thaw may enhance photoreactivity of DOM in lakes underlain by thawing yedoma. Permafrost WEOM isolates and several DOM isolates from this watershed were found to be both photolabile and photoreactive, even though reactivity could not necessarily be directly tied to permafrost input.Composition of DOM and WEOM isolates
NMR spectral features of permafrost WEOM isolates appear distinct from both the active layer and surface water isolates. The carbohydrate signal (O-alkyl region, 60–90 ppm, and anomeric C peak, 110 ppm)63 in the permafrost WEOM isolates is increasingly intense with older soil 14C age. Pautler et al.70 associated increased carbohydrate abundance with undisturbed permafrost in the Canadian High Arctic, suggesting that depleted carbohydrate levels in exposed permafrost through active layer detachments, even recent detachments, stimulate microbial activity and degradation through release of organic material. While biolability was not measured on the WEOM isolates in this study, the permafrost WEOM isolates may have differing ability to stimulate microbial activity due to their varied carbohydrate content.Permafrost WEOM isolates were not necessarily less aromatic than active layer isolates (Table S1†), contrasting reports elsewhere characterizing permafrost thaw waters.21,22,71 This elevated aromaticity may be a feature of yedoma specifically, bias of the isolation method, or a result of active layer being sampled at a different location within the watershed. The PPL isolation method likely biases toward capturing the most light-absorbing fraction of the carbon pool, preferentially extracting the chromophoric DOM and a more efficient capture of aromatic functional groups compared to natural abundance.49 Nearly all isolation methods contain some selectivity bias, however, PPL sorbents are less biased toward aromatics than for example, XAD-8 resins.72 Carbon recoveries for PPL extractions are within typical ranges,49 capturing a majority but not all of the carbon pool (>60%), which somewhat limits the representativity compared to what may be present in natural interstitial thaw waters. Other differences from the leaching process compared to a natural thaw are potentially temperature, time of water contact, and the volumes that were required in order to capture DOM by PPL isolation. However, all soil leachates were isolated according to the same conditions and method, and thus any bias would be expected to be systematic, and relative differences between samples would still be observed. A supplemental leaching study was performed on a different permafrost soil core sampled from just a few miles outside this watershed, in the Bonanza Creek Experimental Forest, to identify how representative the leached organic matter is relative to the entire carbon pool from those soils, and is detailed further in the ESI.† In general, PPL extraction from leached soils as described above captured a representative fraction of the soil organic carbon pool with the exception of aliphatics, which were largely retained on the soils even after leaching (Fig. S12†).
NMR characterisation of relative aromaticity (Al:Ar)73 identified permafrost WEOM isolates as having composition intermediate of that of the DOM and active layer WEOM isolates. However, optical indices such as SUVA, SR, and FI, which have been observed to correlate with aromaticity of DOM,74–77 revealed few trends (Fig. S11†). For example, CPT1's relative O-alkyl and aromatic contributions more closely resemble that of GSRW than surface water DOM isolates. GSRW was sampled well below the active layer, so it may be possible that some of the carbon pool of this well is from deeper thawing permafrost, though more extensive sampling and characterisation is needed to determine if that is actually the case. In some cases, trends could be observed amongst various optical properties and Al:Ar, with significantly non-zero slopes for HIX, BIX, and freshness (β:α), all fluorescent measurements. HIX trended negatively with Al:Ar, which is consistent with the idea that higher aromaticity indicates more humification.77 BIX and β:α both trended positively with Al:Ar, associating less processed material with higher aliphatic content, and largely placing the permafrost WEOM isolates between the DOM isolates and the active layer WEOM isolates in terms of composition. While SUVA has been reported to correlate with aromaticity,76 the slope in this dataset could not be determined to be non-zero.
Several of these measurements revealed a disparate pattern of trends, despite sharing commonalities in their methods. For example, FI revealed no significant trend with Al:Ar, even though BIX and freshness did, and they share similar excitation wavelength (λ = 370 or 380 nm), and are separated by as little as 10 nm in emission wavelengths. A relationship was found with Al:Ar and HIX, which shares the same excitation wavelength (λ = 254 nm) as the reported SUVA measurement, but no such trend was found with SUVA. Fundamentally the optical properties represent but a subset of organic pool (and fluorescence an even smaller subset) compared to the nearly quantitative NMR method. This could indicate a more significant presence of overlapping anomeric carbon signal such as that from carbohydrates, convoluting the determination of total aromaticity by NMR,63 and that reported relative aromaticity values in this study may be higher than the actual aromatic content of the isolates. A lack of trends could also be due to the fact that the dataset is relatively small.
Permafrost WEOM is photolabile and photoreactive
Permafrost WEOM isolates are both photoreactive and photolabile. However, while the permafrost WEOM isolates were on average more photolabile than the active layer WEOM or the DOM isolates, the difference between DOC losses or AQYs was not statistically different by a student t-test (p > 0.05), likely influenced by the large variance and small sample sizes. Most strikingly, the youngest permafrost WEOM isolate, CPT1, exhibits the highest aromaticity, photolability, absorbance loss rate, and TMP photodegradation rate (Table 3). Significant photoreactivity has been observed in DOM from the Alaskan Arctic, where in headwater streams carrying DOM with less prior light exposure, sunlight was responsible for over 90% of carbon processing.29 However, in other Arctic systems little photodegradability has been observed,30 and limited data exists on yedoma DOM photoreactivity. Stubbins et al.22 found that permafrost thaw waters from near the Kolyma River, Russia was not very photolabile, observing DOC losses over 30 d in a solar simulator <5%. The increased photolability observed in this study could be due to the PPL isolation method preferentially extracting the chromophoric pool of DOM.49 Though, even if chromophores were preferentially extracted through the isolation process, the observed results still suggest that their sampled waters and leachates are both photoreactive and photolabile.Integrating from 290–400 nm, AQYs are on the order of 10−4 to 10−5, showing that these carbon pools are indeed photoreactive. As AQYs were calculated over a large range of wavelengths, it is possible that DOC photomineralisation also occurred from chromophores absorbing light outside these wavelength bounds, and thus the AQYs presented may represent a high estimate. However, these values are a similar order of magnitude as those reported by Cory et al. for thermokarst soils in the Arctic over a slightly narrower wavelength range (305–395 nm).31 Apparent quantum yields were also highest for CPT1, indicating that the high DOC losses were not simply from higher rates of light absorption for this particular isolate, which is corroborated by the lower SUVA value for CPT1 compared to the other permafrost WEOM isolates.
The small change in SUVA CPT1 suggests DOC losses included organic carbon not absorbing at 254 nm. However, there was faster absorbance loss at this wavelength for CPT1 than the other isolates. Some of the DOC loss thus could have also resulted after transformation of chromophores absorbing at 254 nm. Stubbins et al.22 similarly observed absorbance losses from photoirradiation of yedoma permafrost thaw waters that were considered to be associated with coloured aromatic moieties. Discrepancies between absorbance loss rates and SUVA decreases may reflect the degree of chromophore transformation, as differing chromophores will exhibit differing molar absorptivities, which may alter both absorbance loss rates and SUVA.
Both absorbance and fluorescence for all isolates decreased upon exposure to photoirradation, however CPT1 did not stand out from the other WEOM isolates with respect to other optical indices, convoluting this observation somewhat. SR and E2:E3 increased with photoirradiation, corroborating a loss of aromaticity consistent with other reported observations upon photoexposure,77,78 while FI, BIX, and β:α decreased, indicating compositional shifts in both the chromophoric and fluorophoric pools (Fig. S10†). However, FI decreases upon photoirradiation for both DOM and WEOM isolates instead of increasing with a loss of aromaticity (Fig. S9 and S10†), contrasting with the SUVA results above, and similar to previous studies by Cory et al.31,79 While FI has been negatively correlated with aromaticity, multiple fluorescent components may give rise to detected signal in the FI region of an excitation-emission matrix (EEM).75,77,80 In addition, a change in fluorescence or absorbance at any single wavelength could include the formation of new chromophores or fluorophores, partly offsetting the loss of others. While net decay of fluorescence (Fig. S7 and S8†) and absorbance (Fig. S4†) occurs, optical index changes (Fig. S9 and S10†) indicate these losses are not uniform across spectra. At least some selective transformation occurs upon photoexposure, despite the fact that no significant trends (slope significantly non-zero by linear regression) between photobleaching rates, photolability, and Al:Ar could be identified. More studies would need to be done to ascertain which part of the chromophoric pool is undergoing transformation vs. mineralisation.
All permafrost WEOM isolates facilitate the photodegradation of TMP more dramatically than the active layer WEOM isolates (Table 3), corroborated by their lower SUVA values and despite not necessarily having a lower relative aromaticity. Differences in TMP reactivity may be due to differences in 3DOM* species or self-quenching by the higher antioxidant capacity of quinoid groups.81 For example, a variety of anilinic and phenolic compounds react with 3DOM*, and are reported to have more inhibition efficiency by more aliphatic Pony Lake fulvic acid (PLFA) than Suwannee River fulvic acid (SRFA).82 The slower observed TMP photodegradation rates in the presence of the active layer WEOM isolates (GSAL1, GSAL2) are thus consistent with these observations. TMP degradation with isolated DOM from a North Slope river in Alaska reported TMP degradation rates ranging from 0.075 to 0.135 L mg C−1 h−1,36 encompassing similar rates as for the DOM isolates in this study (Table 3). Quantifying the antioxidant capacity of the permafrost WEOM isolates may also help to parse out the full nature of triplet excited state reactivity of these DOM pools.
Thermokarst influence on DOM characterisation
Like the WEOM isolates, nearly all DOM isolates were photolabile, with the exception of the summer DNL isolate, spanning AQYs on the order of 1.1–2.9 × 10−4 over 290–400 nm (Table 3). AQYs are not statistically different from that measured from the IHSS isolates. Photoreactivity was broadly similar in terms of observed absorbance loss rates at 254 nm, facilitation of TMP photodegradation, and shifts in SUVA upon photoirradiation, making it difficult to discern potential influence of thermokarst activity or original vs. refrozen yedoma on DOM behaviour.In DOM isolate solutions sourced from waters with either thermokarst activity and/or minimal active layer influence (winter DOM isolates and GSRW), the addition of Fe(III) resulted in increased absorbance losses from 250–275 nm, where small aromatics absorb. This enhancement of absorbance loss upon Fe(III) addition suggests that thermokarst-influenced DOM may be more efficient at promoting photo-Fenton reactions, as iron has been correlated to aromatics in other permafrost containing systems.83 However, CPT1, the most aromatic WEOM isolate by 13C multiCP-MAS NMR, did not increase in photobleaching rate upon iron addition, and instead its absorbance loss slowed. This may reflect its ability for peroxide production or to associate with iron in solution, though neither of these were measured in this study. The winter BSP isolate was most responsive to iron addition, suggesting it may better stabilise iron to promote photo-Fenton pathways, potentially through better association with DOM ligands, indicating some distinct compositional differences, even if those compositional differences were not directly measured in this study.
Few 13C NMR data from other Arctic or sub-Arctic lakes are reported to compare the representativity of these samples. Toolik Lake (northern Alaska, continuous non-yedoma permafrost) had a reported aliphatic (0–60 ppm) to aromatic ratio (90–160 ppm) (Al:Ar)73 of 1.94 for XAD-8 isolate,34 which is slightly lower than the average Al:Ar for the PPL isolates from the lakes in this study (2.09 ± 0.32), indicating slightly more aliphatic content relative to aromatic content than Toolik Lake. However, it should be noted that the isolation method, ecosystem, permafrost type, and NMR methods reported for Toolik Lake are all slightly different from this study,36 and so a direct comparison may not be completely equivalent. GSL, OCT, and BSP had the lowest Al:Ar ratios, indicating the highest proportion of aromatic content relative to aliphatic groups, and also represent surface waters with active thermokarst activity.
Winter DOM isolates present an opportunity to control for active layer inputs into lake waters, as the winter DOM is theoretically more cut off from direct active layer inputs, and thus would be receive inputs dominated by thawed sediments. However, as temperatures increase, the active layer may not completely freeze in winter, allowing lateral flow year-round.84–87 Thus, these relatively shallow lakes are not necessarily devoid of active layer source material in the winter, from either unfrozen active layer or recalcitrant materials remaining in the water column. The two winter isolates comprise similar relative contributions of functional groups, varying within just 2% of each other for any given region. This decrease in variability during wintertime could be due to restricted lateral flow through active layers, limiting input into the lakes. This also suggests that the similarity between GSL's summer and winter isolate composition, including aromaticity, may be from significant contributions other than the active layer, such as thermokarst activity.
DNL, in contrast, does not have active thermokarst activity,37 and seasonal variation may instead reflect differences solely from surface vs. ground water inputs. This is supported by the fact that the DOM isolates from active thermokarst systems were more similar in composition and rates of absorbance losses to winter DOM than summer non-thermokarst DOM isolates, and also exhibited more similarity to the permafrost WEOM isolates than the active layer WEOM isolates. For example, the GSL isolates appear to have similar composition between the summer and winter samples in terms of both optical and NMR composition, despite large differences in whole water carbon and iron concentration. The GSL summer isolate was less susceptible to absorbance decay than its winter isolate, but otherwise, characterisation did not yield strong differences between DOM isolates from either winter or summer (Fig. S11†), with the exception of DNL summer standing out with a higher Al:Ar ratio.
Permafrost underlain soils and sediments around and under GSL may have the potential to enhance lateral contributions through the active layer by limiting subsurface drainage to ground waters. In contrast, winter DOM isolates may have more compositional similarities to the permafrost WEOM isolates due to being more isolated from active layer inputs, regardless of thermokarst activity. However, such a relationship cannot be conclusive, as DNL's winter isolate also had increased photobleaching ability, yet has no observed thermokarst activity.
Increased photoreactivity in winter isolates may also reflect a lack of in situ photodegradation during dark winter months compared to summer, where DOM may have already undergone some photobleaching at time of collection. This could be reflected in the fact that photobleaching of the winter isolates was significantly enhanced upon addition of iron, presumably through reaction with hydroxyl radical, which has been shown to correlate with photo exposure.32 However, there is no requisite observed trend in photolability that one would expect to be consistent with this hypothesis, which may be complicated by transformation (biotic and abiotic) of permafrost material prior to transport to the water column or other mechanisms. For example, several studies have reported biolability and characterised the biotransformation of permafrost material.6,19,70,88–90 Morgalev et al.91 found that the active layer/permafrost interface displayed enhanced metabolic activity in regions of discontinuous permafrost peatlands of western Siberia. Thus, any contribution of permafrost derived material to the studied thermokarst lakes may in fact be quite different in composition compared to what was characterised from the permafrost tunnel WEOM isolates. Quantifying the flux of ground waters in contact with permafrost thaw to examine the extent of thermokarst contribution to these lakes may help shed to some light on this process.
Conclusions
Overall, there were few observations that clearly separated waters with thermokarst activity from original yedoma permafrost and permafrost soils from waters of refrozen deposits or active layer. It should be noted that NMR, optical properties, and to some extent photobleaching ability are by definition bulk properties of a particular DOM pool. However, all isolates were photoreactive, and several of the permafrost influenced DOM and permafrost WEOM isolates were especially photolabile, with relatively high AQYs for DOC loss. More specific compositional analyses or broader sampling may shed light on any compositional dependence of DOM reactivity from active thermokarst inputs. Expanding the data set to develop more statistical robustness between lakes of varied thermokarst activity and exploring inter- and intra-annual variability92 and the impact of differing hydrological regimes will also help to resolve the extent of thermokarst influence on surface water DOM. In addition, sampling sediment pore waters in locations of active thaw rather than solely lake waters, which represent a combination of all the DOM sources to the water column, may help to identify and quantify the specific compositional influences of thermokarst activity on the surface water carbon pool. As temperatures increase, the release of permafrost organic matter is inevitable, and thus, obtaining a holistic understanding of the variability in functional group composition and photoreactivity will allow for a better understanding of how DOM's ecological role will be impacted by permafrost thaw in the coming decades.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this project was supplied by the Alaska Space Grant Graduate Research Fellowship, National Institutes for Water Resources Graduate Student-Lead Proposal through the UAF Water & Environmental Research Center and the U.S. Department of Interior – Geological Survey, and the University of Alaska Fairbanks Undergraduate Research and Scholarly Activity program. Access to residential lakes were obtained through permissions supplied by NSF ARCSS-1500931. Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103395. The content is solely the responsibility of the authors and does not necessarily reflect the views of the NIH. A sincere thank you to A. Liljedahl, D. Barnes, and V. Romanovsky, for allowing access and providing intellectual support. Thank you to A. Kholodov for assistance in collecting and obtaining active layer samples and T. Douglas at Cold Regions Research and Engineering Laboratory with the U.S.A. Army for access and sampling of the CRREL Permafrost Tunnel. Thank you to A. Simpson for providing the SPR-W5-WATERGATE pulse sequence for the Bruker NMR. Further thanks to B. Eckhardt, N. Ramos, D. Knight, N. Ruckhaus, R. Noratuk, J. Sterle, A. Mutschlecner, R. Voight, R. Davey, M. Ross and T. Harms for instrumentation and sampling assistance, and G. Quesada for actinometry assistance.References
- E. A. G. Schuur, A. D. McGuire, C. Schädel, G. Grosse, J. W. Harden, D. J. Hayes, G. Hugelius, C. D. Koven, P. Kuhry, D. M. Lawrence, S. M. Natali, D. Olefeldt, V. E. Romanovsky, K. Schaefer, M. R. Turetsky, C. C. Treat and J. E. Vonk, Nature, 2015, 520, 171–179 CrossRef CAS PubMed.
- L. Schirrmeister, D. Froese, V. Tumskoy, G. Grosse and S. Wetterich, in Encyclopedia of Quaternary Science, 2nd edn, 2013, pp. 542–552 Search PubMed.
- J. Strauss, L. Schirrmeister, G. Grosse, D. Fortier, G. Hugelius, C. Knoblauch, V. Romanovsky, C. Schädel, T. Schneider von Deimling, E. A. G. Schuur, D. Shmelev, M. Ulrich and A. Veremeeva, Earth-Sci. Rev., 2017, 172, 75–86 CrossRef CAS.
- G. Hugelius, J. Strauss, S. Zubrzycki, J. W. Harden, E. A. G. Schuur, C.-L. Ping, L. Schirrmeister, G. Grosse, G. J. Michaelson, C. D. Koven, J. A. O'Donnell, B. Elberling, U. Mishra, P. Camill, Z. Yu, J. Palmtag and P. Kuhry, Biogeosciences, 2014, 11, 6573–6593 CrossRef.
- J. Strauss, L. Schirrmeister, G. Grosse, S. Wetterich, M. Ulrich, U. Herzschuh and H. W. Hubberten, Geophys. Res. Lett., 2013, 40, 6165–6170 CrossRef CAS PubMed.
- J. K. Heslop, M. Winkel, K. M. Walter Anthony, R. G. M. Spencer, D. C. Podgorski, P. Zito, A. Kholodov, M. Zhang and S. Liebner, J. Geophys. Res.: Biogeosci., 2019, 124, 1–18 Search PubMed.
- L. Schirrmeister, G. Grosse, S. Wetterich, P. P. Overduin, J. Strauss, E. A. G. Schuur and H. W. Hubberten, J. Geophys. Res.: Biogeosci., 2011, 116, G00M02 Search PubMed.
- K. M. Walter Anthony, S. A. Zimov, G. Grosse, M. C. Jones, P. M. Anthony, F. S. C. Iii, J. C. Finlay, M. C. Mack, S. Davydov, P. Frenzel and S. Frolking, Nature, 2014, 511, 452–456 CrossRef PubMed.
- E. A. G. Schuur, A. D. McGuire, V. Romanovsky, C. Schädel, and M. Mack, Chapter 11: Arctic and boreal carbon, in Second State of the Carbon Cycle Report (SOCCR2): A Sustained Assessment Report, ed. N. Cavallaro, G. Shrestha, R. Birdsey, M. A. Mayes, R. G. Najjar, S. C. Reed, P. Romero-Lankao and Z. Zhu, U.S. Global Change Research Program, Washington, DC, USA, pp. 428–468 Search PubMed.
- S. A. Zimov, S. P. Davydov, G. M. Zimova, A. I. Davydova, E. A. G. Schuur, K. Dutta and I. S. Chapin, Geophys. Res. Lett., 2006, 33, L20502 CrossRef.
- J. E. Vonk, P. J. Mann, K. L. Dowdy, A. Davydova, S. P. Davydov, N. Zimov, R. G. M. Spencer, E. B. Bulygina, T. I. Eglinton and R. M. Holmes, Environ. Res. Lett., 2011, 40, 2689–2693 CrossRef.
- D. Olefeldt, S. Goswami, G. Grosse, D. Hayes, G. Hugelius, P. Kuhry, A. D. Mcguire, V. E. Romanovsky, A. B. K. Sannel, E. A. G. Schuur and M. R. Turetsky, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- J. K. Heslop, K. M. Walter Anthony, A. Sepulveda-Jauregui, K. Martinez-Cruz, A. Bondurant, G. Grosse and M. C. Jones, Biogeosciences, 2015, 12, 4317–4331 CrossRef.
- M. A. Kessler, L. J. Plug and K. M. Walter Anthony, J. Geophys. Res.: Biogeosci., 2012, 117, G00M06 Search PubMed.
- T. Schneider Von Deimling, G. Grosse, J. Strauss, L. Schirrmeister, A. Morgenstern, S. Schaphoff, M. Meinshausen and J. Boike, Biogeosciences, 2015, 12, 3469–3488 CrossRef.
- R. C. Toohey, N. M. Herman-Mercer, P. F. Schuster, E. A. Mutter and J. C. Koch, Geophys. Res. Lett., 2016, 43, 12120–12130 CrossRef.
- O. S. Pokrovsky, L. S. Shirokova, S. N. Kirpotin, S. Audry, J. Viers and B. Dupré, Biogeosciences, 2011, 8, 565–583 CrossRef CAS.
- B. W. Abbott, J. R. Larouche, J. B. Jones, W. B. Bowden and A. W. Balser, J. Geophys. Res.: Biogeosci., 2014, 119, 2049–2063 CrossRef CAS.
- T. W. Drake, K. P. Wickland, R. G. M. Spencer, D. M. McKnight and R. G. Striegl, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13946–13951 CrossRef CAS PubMed.
- R. G. M. Spencer, P. J. Mann, T. Dittmar, T. I. Eglinton, C. McIntyre, R. M. Holmes, N. Zimov and A. Stubbins, Geophys. Res. Lett., 2015, 42, 2830–2835 CrossRef.
- C. P. Ward and R. M. Cory, Geochim. Cosmochim. Acta, 2015, 167, 63–79 CrossRef CAS.
- A. Stubbins, P. J. Mann, L. Powers, T. B. Bittar, T. Dittmar, C. P. McIntyre, T. I. Eglinton, N. Zimov and R. G. M. Spencer, J. Geophys. Res.: Biogeosci., 2017, 122, 200–211 CrossRef CAS.
- L. L. Jongejans, J. Strauss, J. Lenz, F. Peterse, K. Mangelsdorf, M. Fuchs and G. Grosse, Biogeosciences, 2018, 15, 6033–6048 CrossRef CAS.
- N. Weiss, D. Blok, B. Elberling, G. Hugelius, C. J. Jørgensen, M. B. Siewert and P. Kuhry, Sediment. Geol., 2016, 340, 38–48 CrossRef CAS.
- J. K. Heslop, S. Chandra, W. V. Sobzcak, S. P. Davydov, A. I. Davydova, V. V. Spektor and K. M. Walter Anthony, Polar Res., 2017, 36, 1305157 CrossRef.
- S. Peura, M. Wauthy, D. Simone, A. Eiler, K. Einarsdóttir, M. Rautio and S. Bertilsson, Limnol. Oceanogr., 2020, 65, S248–S263 CrossRef CAS.
- C. Knoblauch, C. Beer, S. Liebner, M. N. Grigoriev and E. M. Pfeiffer, Nat. Clim. Change, 2018, 8, 309–312 CrossRef CAS.
- K. Walter Anthony, T. Schneider Von Deimling, I. Nitze, S. Frolking, A. Emond, R. Daanen, P. Anthony, P. Lindgren, B. Jones, G. Grosse, K. Walter Anthony, T. Schneider Von Deimling, I. Nitze, S. Frolking, A. Emond, R. Daanen, P. Anthony, P. Lindgren, B. Jones and G. Grosse, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- R. M. Cory, C. P. Ward, B. C. Crump and G. W. Kling, Science, 2014, 345, 925–928 CrossRef CAS PubMed.
- C. P. Ward, S. G. Nalven, B. C. Crump, G. W. Kling and R. M. Cory, Nat. Commun., 2017, 8, 772 CrossRef PubMed.
- R. M. Cory, B. C. Crump, J. A. Dobkowski and G. W. Kling, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3429–3434 CrossRef CAS PubMed.
- S. E. Page, J. R. Logan, R. M. Cory and K. McNeill, Environ. Sci.: Processes Impacts, 2014, 16, 807–822 RSC.
- L. S. Shirokova, A. V. Chupakov, S. A. Zabelina, N. V. Neverova, D. Payandi-Rolland, C. Causserand, J. Karlsson and O. S. Pokrovsky, Biogeosci, 2019, 16, 2155–2526 Search PubMed.
- A. M. Grannas, C. B. Martin, Y. P. Chin and M. Platz, Biogeochem, 2006, 78, 51–66 CrossRef CAS.
- C. P. Ward and R. M. Cory, Environ. Sci. Technol., 2016, 50, 3545–3553 CrossRef CAS PubMed.
- K. M. Cawley, J. A. Hakala and Y. P. Chin, Limnol. Oceanogr.: Methods, 2009, 7, 391–398 CrossRef CAS.
- A. M. Emond, R. P. Daanen, G. R. C. Graham, K. W. Anthony, A. K. Liljedahl, B. J. Minsley, D. L. Barnes and V. E. Romanovsky, Airborne electromagnetic and magnetic survey, Goldstream Creek watershed, interior Alaska, Fairbanks, 2018 Search PubMed.
- C. D. Elder, M. Schweiger, B. Lam, E. D. Crook, X. Xu, J. Walker, K. M. Walter Anthony and C. I. Czimczik, J. Geophys. Res.: Biogeosci., 2019, 124, 1209–1229 CrossRef CAS.
- S. Greene, K. M. Walter Anthony, D. Archer, A. Sepulveda-Jauregui and K. Martinez-Cruz, Biogeosciences, 2014, 11, 6791–6811 CrossRef.
- P. R. Lindgren, G. Grosse, K. M. Walter Anthony and F. J. Meyer, Biogeosciences, 2016, 13, 27–44 CrossRef CAS.
- K. M. Walter Anthony, personal communication, Feb 15, 2020.
- T. L. Péwé, Quaternary Geology of Alaska, 1975, USGS Series, vol. 835, p. 145 Search PubMed.
- T. D. Hamilton, J. L. Craig and P. V. Sellmann, Geol. Soc. Am. Bull., 1988, 100, 948–969 CrossRef.
- M. T. Bray, H. M. French and Y. Shur, Permafr. Periglac. Process., 2006, 17, 233–243 CrossRef.
- Y. Shur, H. M. French, M. T. Bray and D. A. Anderson, Permafr. Periglac. Process., 2004, 15, 339–347 CrossRef.
- T. A. Douglas, D. Fortier, Y. L. Shur, M. Z. Kanevskiy, L. Guo, Y. Cai and M. T. Bray, Permafr. Periglac. Process., 2011, 22, 120–128 CrossRef.
- R. Mackelprang, A. Burkert, M. Haw, T. Mahendrarajah, C. H. Conaway, T. A. Douglas and M. P. Waldrop, ISME J., 2017, 11, 2305–2318 CrossRef CAS PubMed.
- T. A. Douglas and M. T. Mellon, Nat. Commun., 2019, 10, 1716 CrossRef PubMed.
- T. Dittmar, B. Koch, N. Hertkorn and G. Kattner, Limnol. Oceanogr.: Methods, 2008, 6, 230–235 CrossRef CAS.
- S. E. Page, W. A. Arnold and K. McNeill, Environ. Sci. Technol., 2011, 45, 2818–2825 CrossRef CAS PubMed.
- Y. P. Chin, P. L. Miller, L. Zeng, K. M. Cawley and L. K. Weavers, Environ. Sci. Technol., 2004, 38, 5888–5894 CrossRef CAS PubMed.
- P. L. Miller and Y. P. Chin, J. Agric. Food Chem., 2002, 50, 6758–6765 CrossRef CAS.
- D. Dulin and T. Mill, Environ. Sci. Technol., 1982, 16, 815–820 CrossRef CAS PubMed.
- D. R. Halm and D. B. Griffith, Water-Quality Data from Lakes in the Yukon Flats, Alaska, 2010–2011, 2014–1181, p. 6 Search PubMed.
- S. E. Johnston, R. G. Striegl, M. J. Bogard, M. M. Dornblaser, D. E. Butman, A. M. Kellerman, K. P. Wickland, D. C. Podgorski and R. G. M. Spencer, Limnol. Oceanogr., 2020, 9999, 1–17 Search PubMed.
- S. E. Johnston, M. J. Bogard, J. A. Rogers, D. Butman, R. G. Striegl, M. Dornblaser and R. G. M. Spencer, Biogeochemistry, 2019, 146, 271–292 CrossRef CAS.
- S. A. Zimov, E. A. G. Schuur and F. Stuart Chapin, Science, 2006, 312, 1612–1613 CrossRef CAS PubMed.
- Y. P. Chin, G. R. Aiken and K. M. Danielsen, Environ. Sci. Technol., 1997, 31, 1630–1635 CrossRef CAS.
- J. Mao, N. Chen, X. Cao, R. M. Cory, D. M. McKnight and K. Schmidt-Rohr, Org. Geochem., 2007, 38, 1277–1292 CrossRef CAS.
- R. L. Johnson and K. Schmidt-Rohr, J. Magn. Reson., 2014, 239, 44–49 CrossRef CAS PubMed.
- K. M. Cawley, D. M. McKnight, P. Miller, R. M. Cory, R. L. Fimmen, J. J. Guerard, M. Dieser, C. Jaros, Y. P. Chin and C. Foreman, Environ. Res. Lett., 2013, 8, 045015 CrossRef.
- R. L. Fimmen, R. M. Cory, Y. P. Chin, T. D. Trouts and D. M. McKnight, Geochim. Cosmochim. Acta, 2007, 71, 3003–3015 CrossRef CAS.
- J. Mao, X. Cao, D. C. Olk, W. Chu and K. Schmidt-Rohr, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 100, 17–51 CrossRef CAS PubMed.
- A. Leifer. The Kinetics of Environmental Aquatic Chemistry: Theory and Practice, 1988, American Chemical Society, Washingtong, p. 304 Search PubMed.
- J. J. Guerard, P. L. Miller, T. D. Trouts and Y. P. Chin, Aquat. Sci., 2009, 71, 160–169 CrossRef CAS.
- R. G. Zepp, B. C. Faust, H. Jürg and J. Holgné, Environ. Sci. Technol., 1992, 26, 313–319 CrossRef CAS.
- B. A. Southworth and B. M. Voelker, Environ. Sci. Technol., 2003, 37, 1130–1136 CrossRef CAS PubMed.
- S. Canonica and M. Freiburghaus, Environ. Sci. Technol., 2001, 35, 690–559 CrossRef CAS PubMed; A. E. Mutschlecner, J. J. Guerard, J. B. Jones and T. K. Harms, Limnol. Oceanogr., 2018, 63, 1605–1621 CrossRef.
- K. McNeill and S. Canonica, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- B. G. Pautler, A. J. Simpson, D. J. McNally, S. F. Lamoureux and M. J. Simpson, Environ. Sci. Technol., 2010, 44, 4076–4082 CrossRef CAS PubMed.
- S. R. Textor, K. P. Wickland, D. C. Podgorski, S. E. Johnston and R. G. M. Spencer, Front. Earth Sci., 2019, 7, 1–17 CrossRef.
- I. V. Perminova, I. V. Dubinenkov, A. S. Kononikhin, A. I. Konstantinov, A. Y. Zherebker, M. A. Andzhushev, V. A. Lebedev, E. Bulvgina, R. M. Holmes, Y. I. Kostyukevich, I. A. Popov and E. N. Nikolaev, Environ. Sci. Technol., 2014, 38, 7461–7468 CrossRef PubMed.
- S. B. Schwede-Thomas, Y. P. Chin, K. J. Dria, P. Hatcher, E. Kaiser and B. Sulzberger, Aquat. Sci., 2005, 67, 61–71 CrossRef CAS.
- Y. P. Chin, G. Alken and E. O'Loughlin, Environ. Sci. Technol., 1994, 28, 1853–1858 CrossRef CAS PubMed.
- D. M. McKnight, E. W. Boyer, P. K. Westerhoff, P. T. Doran, T. Kulbe and D. T. Andersen, Limnol. Oceanogr., 2001, 46, 38–48 CrossRef CAS.
- J. L. Weishaar, G. R. Aiken, B. A. Bergamaschi, M. S. Fram, R. Fujii and K. Mopper, Environ. Sci. Technol., 2003, 37, 4702–4708 CrossRef CAS PubMed.
- A. M. Hansen, T. E. C. Kraus, B. A. Pellerin, J. A. Fleck, B. D. Downing and B. A. Bergamaschi, Limnol. Oceanogr., 2016, 61, 1015–1032 CrossRef CAS.
- J. R. Helms, A. Stubbins, J. D. Ritchie, E. C. Minor, D. J. Kieber and K. Mopper, Limnol. Oceanogr., 2008, 53, 955–969 CrossRef.
- R. M. Cory, D. M. McKnight, Y.-P. Chin, P. Miller and C. L. Jaros, J. Geophys. Res.: Biogeosci., 2007, 112, G04S51 Search PubMed.
- R. M. Cory and D. M. McKnight, Environ. Sci. Technol., 2005, 39, 8142–8149 CrossRef CAS PubMed.
- J. Wenk, S. N. Eustis, K. McNeill and S. Canonica, Environ. Sci. Technol., 2013, 47, 12802–12810 CrossRef CAS PubMed.
- J. Wenk and S. Canonica, Environ. Sci. Technol., 2012, 46, 5455–5462 CrossRef CAS PubMed.
- V. Mangal, S. DeGasparro, D. V. Beresford and C. Guéguen, Sci. Total Environ., 2020, 204, 135415 CrossRef PubMed.
- V. F. Bense, G. Ferguson and H. Kooi, Geophys. Res. Lett., 2009, 36, L22401 CrossRef.
- S. Ge, J. McKenzie, C. Voss and Q. Wu, Geophys. Res. Lett., 2011, 38, L14402 CrossRef.
- Y. Shur, K. M. Hinkel and F. E. Nelson, Permafr. Periglac. Process., 2005, 16, 5–17 CrossRef.
- J. A. O'Donnell, G. R. Aiken, M. A. Walvoord and K. D. Butler, Global Biogeochem. Cycles, 2012, 26, GB0E06 CrossRef.
- B. P. Selvam, J. F. Lapierre, F. Guillemette, C. Voigt, R. E. Lamprecht, C. Biasi, T. R. Christensen, P. J. Martikainen and M. Berggren, Sci. Rep., 2017, 7, 45811 CrossRef.
- K. Dutta, E. A. G. Schuur, J. C. Neff and S. A. Zimov, Glob. Change Biol., 2006, 12, 2336–2351 CrossRef.
- E. A. G. Schuur, J. G. Vogel, K. G. Crummer, H. Lee, J. O. Sickman and T. E. Osterkamp, Nature, 2009, 459, 556–559 CrossRef CAS PubMed.
- S. Y. Morgalev, I. V. Lushchaeva, T. G. Morgaleva, L. G. Kolesnichenko, S. V. Loiko, I. V. Kirckov, A. Lim, I. I. Volkova, L. S. Shirokova, S. Y. Morgalev, S. N. Vorobyev, S. N. Kirpotin and O. S. Pokrovsky, Polar Biol., 2017, 40, 1645–1659 CrossRef.
- N. J. Shatilla and S. Carey, Hydrol. Earth Syst. Sci., 2019, 23, 3571–3591 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detail on chemicals utilised, elemental characterisation, NMR, and optical characterisation methods, 1H NMR results and NMR results from IHSS isolates, as well as absorbance loss and optical index plots and representative fluorescence spectra. See DOI: 10.1039/d0em00097c |
| This journal is © The Royal Society of Chemistry 2020 |