
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into origins and function of the unexplored majority of the reductive dehalogenase gene family as a result of genome assembly and ortholog group classification†
Olivia
Molenda
 a,
Luz A.
Puentes Jácome
a,
Xuan
Cao
b,
Camilla L.
Nesbø
ab,
Shuiquan
Tang
a,
Nadia
Morson
c,
Jonas
Patron
b,
Line
Lomheim
a,
David S.
Wishart
bd and
Elizabeth A.
Edwards
*ac
a,
Luz A.
Puentes Jácome
a,
Xuan
Cao
b,
Camilla L.
Nesbø
ab,
Shuiquan
Tang
a,
Nadia
Morson
c,
Jonas
Patron
b,
Line
Lomheim
a,
David S.
Wishart
bd and
Elizabeth A.
Edwards
*ac
aDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Ontario, Canada. E-mail: elizabeth.edwards@utoronto.ca
bDepartment of Biological Sciences, Faculty of Science, University of Alberta, Edmonton, Canada
cDepartment of Cell and Systems Biology, University of Toronto, Ontario, Canada
dDepartment of Computing Science, University of Alberta, Edmonton, Canada
First published on 11th March 2020
Abstract
Organohalide respiring bacteria (OHRB) express reductive dehalogenases for energy conservation and growth. Some of these enzymes catalyze the reductive dehalogenation of chlorinated and brominated pollutants in anaerobic subsurface environments, providing a valuable ecosystem service. Dehalococcoides mccartyi strains have been most extensively studied owing to their ability to dechlorinate all chlorinated ethenes – most notably carcinogenic vinyl chloride – to ethene. The genomes of OHRB, particularly obligate OHRB, often harbour multiple putative reductive dehalogenase genes (rdhA), most of which have yet to be characterized. We recently sequenced and closed the genomes of eight new strains, increasing the number of available D. mccartyi genomes in NCBI from 16 to 24. From all available OHRB genomes, we classified predicted translations of reductive dehalogenase genes using a previously established 90% amino acid pairwise identity cut-off to identify Ortholog Groups (OGs). Interestingly, the majority of D. mccartyi dehalogenase gene sequences, once classified into OGs, exhibited a remarkable degree of synteny (gene order) in all genomes sequenced to date. This organization was not apparent without the classification. A high degree of synteny indicates that differences arose from rdhA gene loss rather than recombination. Phylogenetic analysis suggests that most rdhA genes have a long evolutionary history in the Dehalococcoidia with origin prior to speciation of Dehalococcoides and Dehalogenimonas. We also looked for evidence of synteny in the genomes of other species of OHRB. Unfortunately, there are too few closed Dehalogenimonas genomes to compare at this time. There is some partial evidence for synteny in the Dehalobacter restrictus genomes, but here too more closed genomes are needed for confirmation. Interestingly, we found that the rdhA genes that encode enzymes that catalyze dehalogenation of industrial pollutants are the only rdhA genes with strong evidence of recent lateral transfer – at least in the genomes examined herein. Given the utility of the RdhA sequence classification to comparative analyses, we are building a public web server (http://reductivedehalogenasedb.ca) for the community to use, which allows users to add and classify new sequences, and download the entire curated database of reductive dehalogenases.
Environmental significanceOrganohalide-respiring bacteria (OHRB) including Dehalococcoides mccartyi, Dehalobacter restrictus and Dehalogenimonas are important biocatalysts of a wide variety of dehalogenation reactions. An OHRB genome can harbour over 30 different copies of reductive dehalogenase-encoding genes and public databases currently contain thousands of representatives, yet fewer than 1% of all sequences have a characterized function. We used an amino acid sequence similarity-based method to classify all publicly available reductive dehalogenase sequences from closed genomes or metagenome-assembled genomes. Comparative phylogenetics combined with classification gave us new insights into origins and function of the unexplored majority of the reductive dehalogenase gene family. Given the utility of the approach, we created a public web server (http://reductivedehalogenasedb.ca) with a curated database of reductive dehalogenases and a classification tool to easily situate newly identified sequences. |
Introduction
Exposure to toxic or carcinogenic halogenated organic compounds, particularly chlorinated aliphatic hydrocarbons (CAHs), via groundwater and soil is of public concern.1 While biological transformation processes proceed to some extent naturally, these processes can be very effectively accelerated.2 Natural attenuation, biostimulation, and bioaugmentation using mixed microbial consortia capable of reductive dechlorination are now commonly used to remediate sites contaminated with CAHs. Biologically-mediated reductive dechlorination of chlorinated pesticides was reported as early as 1967.3 Dechlorination of CAHs such as perchloroethene (PCE) to less chlorinated ethenes under methanogenic conditions was first reported in 1983.4 The idea to deploy specific bacteria to degrade CAHs became more viable after 1987, upon the discovery of anaerobic mixed cultures that could dechlorinate the last and most toxic daughter product, vinyl chloride, from PCE stepwise dechlorination5 and the subsequent isolation of Dehalococcoides as the responsible organism.6,7 At present, the efficacy of in situ bioaugmentation to transform contaminants such as PCE and trichloroethene (TCE), and daughter products cis-dichloroethene (cDCE) and vinyl chloride (VC) to ethene is well established.8,9Organohalide respiring bacteria (OHRB) conserve energy for growth from cleavage of carbon–halogen bonds. These bacteria use halogenated organic compounds as terminal electron acceptors in their metabolism. DCB-1,10 later named Desulfomonile tiedjei11 was the first characterized OHRB (∼1986) capable of growing with 3-chlorobenzoate as an electron acceptor. OHRB come from many different phyla including Deltaproteobacteria (Geobacter, Desulfuromonas, Anaeromyxobacter, Desulfoluna, Desulfomonile and Desulfovibrio), Epsilonproteobacteria (Sulfurospirillum), Betaproteobacteria (Shewanella and Comamonas), Firmicutes (Dehalobacter and Desulfitobacterium) and Chloroflexi (Dehalococcoides, Dehalogenimonas and Dehalobium)12 – see recent reviews.13–16 The success of bioremediation is largely due to the discovery of highly substrate-specific OHRB that link dehalogenation with growth, and thus whose populations increase as dehalogenation proceeds. These OHRB express respiratory reductive dehalogenases that can be categorized as part of a quinone-dependent or quinone-independent electron transport chain.14 The Dehalococcoidia, including Dehalococcoides and Dehalogenimonas, are quinone-independent, obligate organohalide respiring bacteria for which no other growth supporting electron acceptors are known. Characterized members of the Dehalococcoidia are capable of using more difficult-to-dechlorinate CAHs that only have one or two chlorine substituents. This ability of the Dehalococcoidia is key to complete detoxification. The enrichment culture studied herein (called KB-1) is routinely used for bioaugmentation and contains multiple organohalide-respiring bacteria (OHRB), including Dehalococcoides mccartyi, Dehalobacter restrictus and Geobacter lovleyi, which, in concert, are capable of complete dechlorination of many chlorinated ethenes, ethanes and benzenes.8,17 From the lens of geologic time, OHRB are phylogenetically diverse microbes of ancient origin that participate not only in detoxification of man-made contaminants, but also in the global halogen cycle, recycling a multitude of naturally-occurring organohalides.12,18,19
Many OHRB genomes contain multiple distinct genes predicted to be reductive dehalogenases, termed rdhA genes16 sometimes accounting for a significant proportion of their genomes. For example, D. mccartyi have remarkably small genomes (∼1.4 Mbp) yet some strains contain up to 37 rdhA genes. D. mccartyi contain a core syntenic region encoding for “housekeeping” genes such as biosynthesis of amino acids, cell components, transcription/translation, nutrient transport and energy conservation.20,21 The differences between strains in this region are no more than single nucleotide polymorphisms (SNPs) with average nucleotide identity (ANI) of 85–99%.22 This stable core genome is interrupted by two variable regions flanking the origin of replication commonly referred to as High Plasticity Regions (HPRs, individually HPR1 and HPR2).21 HPRs show signs of both lateral gene transfer and homologous recombination including repeats, duplication events, insertion sequences, genomic islands, and phage-related sequences. The majority of reductive dehalogenases in D. mccartyi are located on these HPRs. In addition, several characterized dehalogenases are within genomic islands with evidence of site-specific integration, such as the VC reductase genes vcrA and bvcA,23 and TCE reductase gene tceA.24 It is generally thought that recombination events have allowed D. mccartyi to adapt to naturally-occurring and anthropogenic halogenated compounds.21,23
Reductive dehalogenase genes occur in an operon containing the rdhA, rdhB, and sometimes, additional genes. The rdhA gene encodes the catalytically active enzyme (RdhA or RDase if characterized) and the B gene encodes a membrane anchor protein containing two or sometimes three transmembrane helices.13–15,25,26D. mccartyi RdhA and RdhB subunits are part of a unique protein complex which is currently considered to be a fully functional stand-alone respiratory chain without quinone or cytochrome involvement.27 Other than RdhAB, the protein complex includes the organohalide respiration molybdoenzyme (OmeA), formally called the complex iron–sulfur cluster molybdoenzyme (CISM),27 found to have hydrogenase activity,28 its putative membrane anchor (OmeB), hydrogen uptake hydrogenase (HupX) with a [NiFe] large subunit (HupL) and iron–sulfur containing small subunit (HupS).
RdhAs are identified based on the presence of two motifs: a twin-arginine TAT membrane export sequence (RRXFXK), and an eight-iron ferredoxin cluster binding motif (CXXCXXCXXXCP). RDases can have one or two iron–sulfur binding motifs. In certain RDases, such as Dehalogenimonas lykanthroporepellens BL-DC-9 DcpA, a cobalamin binding motif can be identified (DXHXXGSXLGG).29–31 A corrinoid molecule is a critical co-factor for folding and function of RdhA proteins.32–34 However, outside of motif regions, RdhAs can have as little as 30% full-length pairwise identity (PID) even when found in the same genome. While many putative dehalogenases sequences can be found in any particular genome, only a select few have been found expressed in response to different CAHs.14 As a result, without a universal functioning heterologous expression system, only a few dehalogenases have been characterized out of the thousands of sequences in the National Center for Biotechnology Information (NCBI) database. To help classify newly found putative dehalogenases with unknown functions, Hug et al. developed a classification system in 2013, grouping RdhAs with upwards of 90% amino acid PID into sets of orthologous sequences (i.e. Ortholog Groups, or OGs) which most likely share similar function.19,35 Since 2013, two crystal structures of RdhA proteins have been solved,36,37 confirming previous features and providing more resolution to the critical substrate-binding domain.
The first objective of this study was to identify and sequence the different strains of OHRB in a set of related KB-1 sub-cultures amended with different chlorinated electron acceptors. We had previously noted that different electron acceptors enriched for different strains of D. mccartyi.38 From three cultures, 8 new D. mccartyi genomes were closed, increasing the total number of publicly available D. mccartyi genomes to 24. As well, one new Dehalobacter restrictus genome was closed increasing the number publicly available to 4 (as of December 2019). The second objective was to compare these genomes to identify key trends in genome evolution, homologous recombination, and lateral gene transfer. We first specifically investigated the evolution of reductive dehalogenase genes in D. mccartyi since most genomes were available for comparison. We then tried to extend the analysis to Dehalogenimonas, a close relative of Dehalococcoides, and finally to Dehalobacter – a distinct genus in the Firmicutes. The Ortholog Group classification method was extremely useful in this analysis, which led us to the development of a public web server to make it easier for the community to compare and classify new sequences (http://RDaseDB.biozone.utoronto.ca). The user can also download part of or the entire manually curated reductive dehalogenase sequence database for further analyses.
Methods
Enrichment cultures analysed
The KB-1 cultures were originally enriched from aquifer materials at a TCE contaminated site in southern Ontario as previously described.8,9,39 A commercial version of the culture is maintained at SiREM (Guelph, ON) for bioaugmentation. The parent enrichment culture maintained at the University of Toronto, referred to as KB-1/TCE-MeOH, has been maintained since 1998 in a 2 L glass bottle (∼1.3 L culture with 0.7 L headspace) in anaerobic mineral medium amended with 100 mg L−1 TCE as electron acceptor and methanol (MeOH) as electron donor, added at 5× the electron equivalents (eeq.) required for complete dechlorination, as previously described.39–41 TCE is completely dechlorinated to ethene prior to refeeding, approximately every 2–4 weeks. Acetogens in the mixed culture ferment added methanol to hydrogen and acetate required by D. mccartyi. In 2001, a sub-culture was created with a 2% transfer into pre-reduced anaerobic mineral medium and maintained on VC and hydrogen as described in39 and is referred to as KB-1/VC-H2. This 200 mL VC enrichment was maintained in a 250 mL glass bottle sealed using a Teflon mininert cap and was amended with 55 mg L−1 VC (supplied as 5 mL of pure VC gas) and 5× eeq. hydrogen gas (supplied as 5 mL 80% H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20% CO2, Praxair). The KB-1/VC-H2 enrichment bottle was also amended with 0.5 mM sodium acetate as a carbon source every ten feedings. In 2003, a 1,2-dichloroethane (DCA)-fed enrichment culture was created (KB-1/1,2-DCA-MeOH culture) fed with 250 mg L−1 1,2-DCA and 5× eeq. MeOH, in a 2 L glass Pyrex bottle containing 1.3 L culture with 0.7 L headspace. Approximately every 6 months, half of each culture was removed and substituted with fresh pre-reduced anaerobic mineral medium to replenish vitamins and buffer. In all enrichment cultures, chlorinated ethenes/ethanes, methane and ethene concentrations were monitored using gas chromatography with an FID detector (Agilent 7890A GC system, G1888 auto-sampler, helium used as carrier gas, packed inlet, Agilent GSQ-Plot column 0.53 mm × 30 mm), calibrated with external standards.
:20% CO2, Praxair). The KB-1/VC-H2 enrichment bottle was also amended with 0.5 mM sodium acetate as a carbon source every ten feedings. In 2003, a 1,2-dichloroethane (DCA)-fed enrichment culture was created (KB-1/1,2-DCA-MeOH culture) fed with 250 mg L−1 1,2-DCA and 5× eeq. MeOH, in a 2 L glass Pyrex bottle containing 1.3 L culture with 0.7 L headspace. Approximately every 6 months, half of each culture was removed and substituted with fresh pre-reduced anaerobic mineral medium to replenish vitamins and buffer. In all enrichment cultures, chlorinated ethenes/ethanes, methane and ethene concentrations were monitored using gas chromatography with an FID detector (Agilent 7890A GC system, G1888 auto-sampler, helium used as carrier gas, packed inlet, Agilent GSQ-Plot column 0.53 mm × 30 mm), calibrated with external standards.
Metagenomic sequencing and genome assembly
DNA for metagenome sequencing was extracted from larger samples (40–615 mL) taken from the three functionally-stable enrichment cultures described above: KB-1/VC-H2 (40 mL sample), KB-1/TCE-MeOH (500 mL sample), KB-1/cDCE-MeOH (300 mL sample) and KB-1/1,2-DCA-MeOH (615 mL sample). Extractions were conducted between February and May 2013. Cultures were filtered using Sterivex™ filters (Millipore 0.2 μm) and the DNA was extracted using the CTAB method (JGI bacterial genomic DNA isolation using CTAB protocol v.3). DNA was sequenced at the Genome Quebec Innovation Sequencing Centre using Illumina HiSeq 2500 technology. Paired-end sequencing with an insert size of ∼400 bp and read length of ∼150 bp provided roughly 50 million reads per culture. Additional mate-pair sequencing with insert size of ∼8000 bp and read length of ∼100 bp was conducted for the KB-1/TCE-MeOH and KB-1/1,2-DCA-MeOH cultures where we had more DNA (>10 μg each). For metagenomic sequencing using short-read Next Generation Sequencing (NGS), we have demonstrated the utility of integrating long-insert mate-pair data to resolve challenges in the metagenomic assembly, especially those related to repeat elements and strain variation.42 Raw sequences were trimmed with Trimmomatic43 to remove bases of low quality and to remove adapters.The D. mccartyi genomes were assembled in six steps as described in supplemental methods. Quantitative PCR to quantify the abundance of specific rdhA genes and 16S rRNA amplicon sequencing for relative abundances of different organisms (methods provided in ESI†) were also performed to guide the metagenomic assembly (illustrated in Fig. S1†). In all cases, multiple genomes could be closed (assembled into a single circular scaffold without gaps) from a single enrichment culture because the different populations of D. mccartyi were present at different abundances (as inferred from read depth) at the time of sampling. Two complete genomes, each containing a vinyl chloride reductase gene (vcrA), were closed from the KB-1/VC-H2. The naming convention developed identifies culture (KB-1) electron acceptor (in this case vinyl chloride or VC) and relative abundance (number 1 for highest abundance, 2 for next and so on). Thus, the D. mccartyi strains from culture KB-1/VC-H2 are named strains KBVC1 and KBVC2. Three D. mccartyi genomes containing the vinyl chloride reductase bvcA were closed from the KB-1/1,2-DCA enrichment culture referred to as strains KBDCA1, KBDCA2 and KBDCA3 and one Dehalobacter restrictus genome named strain 12DCA. A D. mccartyi complete genome containing a vcrA gene was assembled from KB-1/TCE-MeOH culture, named strain KBTCE1. Additionally, two genomes containing trichloroethene reductase tceA genes were assembled from the KB-1/TCE-MeOH culture, strains KBTCE2 and KBTCE3. The genomes were annotated using the RAST44 and BASyS45 servers; results were manually inspected and corrected where required and automatically annotated after submission to NCBI by PGAP.46 Additional searches for conserved domains were conducted using NCBI conserved domain search (E-value threshold of 0.01). The origin of replication was identified using Oriloc in R.47
Alignments and phylogenetic trees
A concatenated gene alignment was created by aligning a set of 109 homologous genes found in genomes of Dehalococcoides mccartyi (22 strains), Dehalogenimonas lykanthroporepellens, D. alkenigignens, Dehalogenimonas sp. WBC-2, and a Chloroflexi out-group Sphaerobacter thermophilus. Outgroups were used to better visualize branching and to improve the estimate of the number of mutations along each branch. Outgroups were selected to be as closely related to in-group as reasonably possible without being part of the in-group. Homologous genes were identified using reciprocal BLASTp and OrthoMCL followed by manual inspection using the Get Homologues package.48 Each gene was aligned using MUSCLE v3.8.3.149 with default settings. The alignments were concatenated to create one long alignment (138334 bp long alignment, 26 sequences, 83% average pairwise identity, gaps included). A maximum likelihood (ML) tree was built using the RAxML50 plugin in Geneious 8.1.851 with the GTR substitution model and 100 bootstrap replicates. The best scoring ML tree was chosen as the final tree.
Five-hundred and fifty-one reductive dehalogenase sequences were selected from Dehalococcoidia to create an amino acid and a separate nucleotide phylogenetic tree using Geneious 8.1.8. These included all rdhA which have been assigned an Ortholog Group (OG) number from the RDase database, all rdhA from three species (D. lykanthroporepellens, D. alkenigignens, and Dehalogenimonas sp. WBC-2) and a reductive dehalogenase from Desulfoluna spongiiphila as the out-group. D. spongiiphila is a reductive dehalogenating, anaerobic, sulfate-reducing bacterium isolated from a marine sponge.52 RDases which cannot be classified into a group at this time have still been assigned a number in the database along with the letter “S” to indicate that this sequence is a singleton. A second tree was created using the same technique from all Dehalobacter reductive dehalogenase gene and protein sequences, and a third tree with a combination of previously functionally characterized RDases from Dehalococcoides, Dehalobacter and Dehalogenimonas. Alignments and tree-building were conducted using MUSCLE and RAxML as described above. FigTree 1.4.2 was used to visualize and further edit the tree to generate figures in this study (http://tree.bio.ed.ac.uk/software/figtree/).
Time since divergence analysis and detection of positive selection
The time since divergence was calculated using the number of mutations identified from phylogenetic concatenated gene alignments and OG rdhA gene alignments to date (in substitutions per site). Substitutions per site identified from rdhA OG alignments were compared to see whether rdhA could have originated before or after (1) the divergence of Dehalococcoidia into Dehalococcoides and Dehalogenimonas and (2) the divergence of Dehalococcoides into three clades: Pinellas, Cornell and Victoria.We compared the ratio of non-synonymous (Ka) to synonymous substitutions rates (Ks) in rdhA OG alignments because the Ka/Ks ratio can identify positive selection. If Ka/Ks < 1, then non-synonymous mutations are either neutral or deleterious, whereas if Ka/Ks > 1, then positive selection occurred.53Ka/Ks ratios were calculated using the PAML package.54,55 Only OGs with five or more rdhA were used for the analysis since using fewer than five sequences is not recommended.56 Two models were run: one which described neutral or nearly neutral evolution and one which additionally accounted for adaptive evolution. The χ2 test was used to determine whether the Ka/Ks gene ratios were significantly greater than 1, suggesting positive selection.
Nucleotide sequence accession numbers
The KB-1 sequencing bioproject can be accessed in the National Center for Biotechnology Information (NCBI) database using bioproject no. PRJNA376155. Dehalococcoides mccartyi KB-1 closed genome nucleotide accession numbers in NCBI are: strain KBDCA1 CP019867, strain KBDCA2 CP019868, strain KBDCA3 CP019946, strain KBVC1 CP019968, strain KBVC2 CP19969, strain KBTCE1 CP01999, strain KBTCE2 CP019865, and strain KBCTCE3 CP019866. KB-1 Dehalobacter restrictus 12DCA closed genome: CP046996. All metagenomic raw reads are available in NCBI short-read archive (SRA) accession no. SRR11085567-71. 16S rRNA amplicon sequences have been deposited in NCBI SRA accession no. SRP144609.Results & discussion
General features of Dehalococcoides and Dehalobacter genomes from KB-1 enrichment cultures
Eight complete genomes of D. mccartyi strains and one Dehalobacter restrictus strain were assembled (Fig. S1† for assembly methodology) and annotated from three different enrichment cultures (Table 1) using Illumina mate-pair and paired-end metagenomic sequencing in combination with 16S rRNA amplicon sequencing and qPCR of functional rdhA to guide assembly (Fig. S2†). In all KB-1 enrichments, we found lower abundance, fragmented Dehalococcoides and Dehalobacter contigs which could not be assembled into complete genomes indicating that each of the three KB-1 sub-cultures must have at least one additional strain which could not be assembled totalling at least eleven strains of Dehalococcoides mccartyi and at least two strains of Dehalobacter restrictus. The strains whose genomes were assembled and closed have been named based on contaminant/electron acceptor amended and a number to indicate rank abundance with respect to other strains found in that same enrichment culture (Table 1). D. mccartyi strains, overall, share at least 98% nucleotide sequence identity of the 16S rRNA gene57 (Fig. S3†) and fall into three clades known as the Pinellas, Victoria and Cornell groupings.58 Most KB-1 strains (KBVC1, KBVC2, KBDCA1, KBDCA2, KBDCA3 and KBTCE1) fall into the Pinellas clade (which also contains strains CBDB1,20 BTF08,59 DCMB5,59 11a5,60 WBC-2,35 GT,61 IBARAKI62 and BAV1), while only two KB-1 strains (KBTCE2 and KBTCE3) fall into the Cornell clade (containing strain 195, MB63 and CG464) (ESI Fig. S3†). All genomes have a clear GC skew with one origin of replication, and the majority of reductive dehalogenase genes are found flanking the origin of replication, primarily coded on the leading strands.
| Dehalococcoides | Dehalobacter | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| KBVC1 | KBVC2 | KBTCE1 | KBTCE2 | KBTCE3 | KBDCA1 | KBDCA2 | KBDCA3 | 12DCA | |
| a Identifiable rdhA indicates the presence of a rdhA gene whose protein product was characterized in a different study. It is not known whether these rdhA are expressed by these specific strains. | |||||||||
| Genome size (mbp) | 1.39 | 1.35 | 1.39 | 1.33 | 1.27 | 1.43 | 1.39 | 1.34 | 3.05 |
| G + C content (%) | 47.3 | 47.2 | 47.3 | 49.1 | 49.3 | 47.4 | 47.5 | 47.6 | 44.5 |
| Protein coding genes | 1468 | 1432 | 1451 | 1381 | 1319 | 1496 | 1462 | 1404 | 2906 |
| Hypothetical genes (%) | 31.1 | 30.2 | 30.3 | 29.1 | 26.8 | 32.8 | 31.9 | 29.1 | 19.1 |
| tRNA | 47 | 48 | 47 | 45 | 45 | 47 | 46 | 46 | 52 |
| CRISPR-Cas genes | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 |
| Nitrogen fixation genes | 0 | 0 | 0 | 9 | 9 | 0 | 0 | 0 | 0 |
| Sub-group/Clade | Pinellas | Pinellas | Pinellas | Cornell | Cornell | Pinellas | Pinellas | Pinellas | n/a |
| Electron acceptor provided to culture | VC | VC | TCE | TCE | TCE | 1,2-DCA | 1,2-DCA | 1,2-DCA | 1,2-DCA |
| rdhA genes | 22 | 16 | 16 | 5 | 5 | 7 | 7 | 9 | 22 |
| Identifiable rdhAa | vcrA, pceA | vcrA, pceA | vcrA, pceA | tceA | tceA | bvcA | bvcA | bvcA | Unknown |
| NCBI accession number | CP019968 | CP019969 | CP019999 | CP019865 | CP019866 | CP019867 | CP019868 | CP019946 | CP046996 |
Strains KBVC1 and KBDCA3 contain complete CRISPR-Cas systems65 that are similar to those in published strains 11a,66 CBDB1,20 DCBM559 and GT.61 Cornell-clade strains KBTCE2 and KBTCE3 contain nitrogen fixation genes similar to type strain 195.67 The number of putative reductive dehalogenase genes (rdhA) among all strains varies from five to twenty-two (Table 1). Strain KBTCE3 has the smallest genome of all known D. mccartyi (1.27 Mbp), with the fewest number of rdhA (only five in both KBTCE3 and KBTCE2) and has highest GC content (49.3% in KBTCE3) (Table 1). We consider all eight KB-1 D. mccartyi genomes to be representative of independent strains because the number of accumulated SNPs and genomic rearrangements has clearly resulted in functional changes and differential growth patterns as inferred from the observed different abundances in the same KB-1 sub-culture.
The KB-1 1,2-DCA enrichment culture contains a population of Dehalobacter, as determined by 16S rRNA amplicon analysis (Fig. S2†). We closed a genome from this population whose 16S rRNA gene sequence shares 99.9% nucleotide PID with Dehalobacter restrictus PER-K2368 and 99.8% average nucleotide identity (ANI). Several rearrangements and insertions have occurred which makes the whole genome alignment of the two strains 76.9% nucleotide PID. This KB-1 Dehalobacter genome will be further referred to as Dehalobacter restrictus strain 12DCA.
Reductive dehalogenase genes in Dehalococcoides mccartyi and conserved synteny
Hug et al. (2013)19 developed a classification system for reductive dehalogenases where sequences were assigned to Ortholog Groups (OGs) in an attempt to cluster rdhA sequences into groups with similar activity on specific halogenated electron acceptors.35 Previous studies identified 32 distinct rdhA genes in the KB-1/TCE-MeOH culture,38,69,70 with two being identical to genes found previously in D. mccartyi CBDB1. Although 104 RdhA were found in this study, only three new OGs could be described, and the remaining RdhA sequences fell into previously identified groupings (Table S2†). As more D. mccartyi genomes are closed, fewer new dehalogenases are discovered that do not belong to an existing OG (Fig. 2); as of December 2019, a total of 84 OGs from D. mccartyi have been identified. We further applied this classification to 144 Dehalogenimonas and 356 Dehalobacter reductive dehalogenases. A similar trend occurred with Dehalobacter restrictus RdhAs, but not with Dehalogenimonas, because too few Dehalogenimonas genomes are available, and those that are available, are not from the same species (Fig. 2). With the strict 90% amino acid full-length alignment identity cut off, resulting ortholog groups were found to be genus-specific for Dehalococcoides and Dehalogenimonas.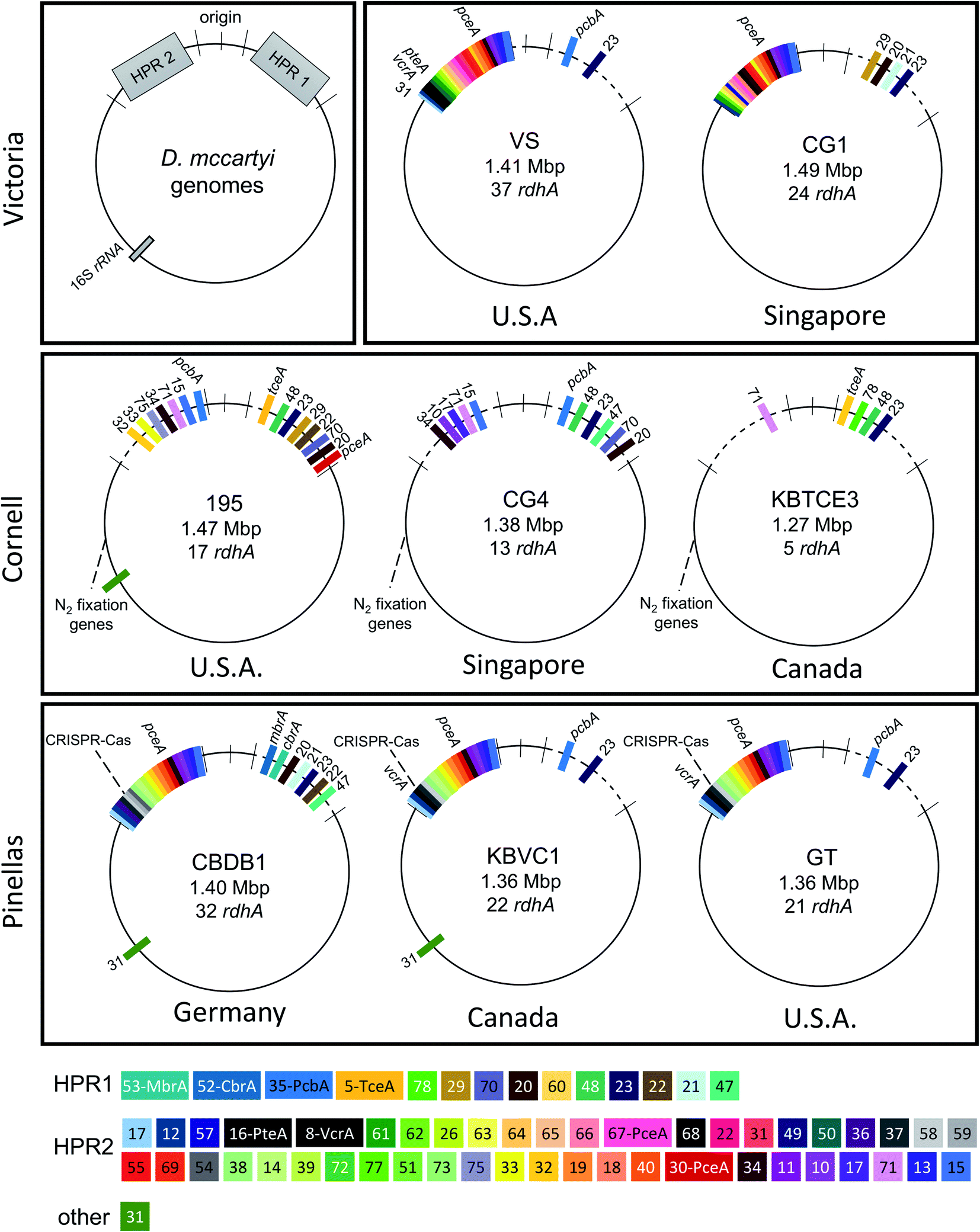 | ||
| Fig. 1 Overview of reductive dehalogenase genes found in eight D. mccartyi genomes originating from different geographical locations. Top left shows the general architecture of D. mccartyi genomes. Genomes are identified by strain name and country of origin indicated beneath. Reductive dehalogenase homologous genes (rdhA) are marked on each genome, coloured by Ortholog Group (OG). Genomes are aligned (dotted lines indicating gaps) to visualize OGs. Genome high plasticity region (HPR), individually HPR1 and HPR2. | ||
 | ||
| Fig. 2 The number of new Ortholog Groups (OGs) of reductive dehalogenases (RDases) found with each new Dehalococcoides mccartyi, Dehalobacter restrictus, or Dehalogenimonas (various species) genome or Metagenome Assembled Genome (MAG) available from NCBI or IMG. | ||
Whole genome alignments of D. mccartyi generally result in strong core-genome region alignments (∼90% nucleotide pairwise identity), with poor alignments of two regions flanking the origin, deemed the high plasticity regions (HPR) (<30% pairwise nucleotide identity) suggesting that HPRs are unique per strain. Analysing twenty-four closed genomes has revealed that most of the rdhA in HPRs are actually syntenous, and that poor alignments are a result of gene loss of specific rdhA genes (Fig. 1 and 3). Therefore, most strain HPRs are much more similar than previously thought.21 A conserved sequential order of rdhA exists in all D. mccartyi strains (Fig. 3a HPR1, Fig. 3b HPR2, with accession numbers Table S2†). For example, at the end of HPR2, the most common order of RdhA OGs is 5′-40:30:34:11:10:17-3′. Some strains have all of these OGs (CG5, BTF08, CBDB1, KBVC1, GT, KBVC2, KBTCE1, 11a5) while certain strains only have a select few, such as IBARAKI 5′-30:11-3′, CG4 5′-10:17-3′, or GY50, that have 5′-40:30:10:17-3’ (Fig. 3B). The visualization of this synteny would not have been possible without grouping of rdhA sequences, nor without the numerous complete genomes currently publicly available in NCBI.
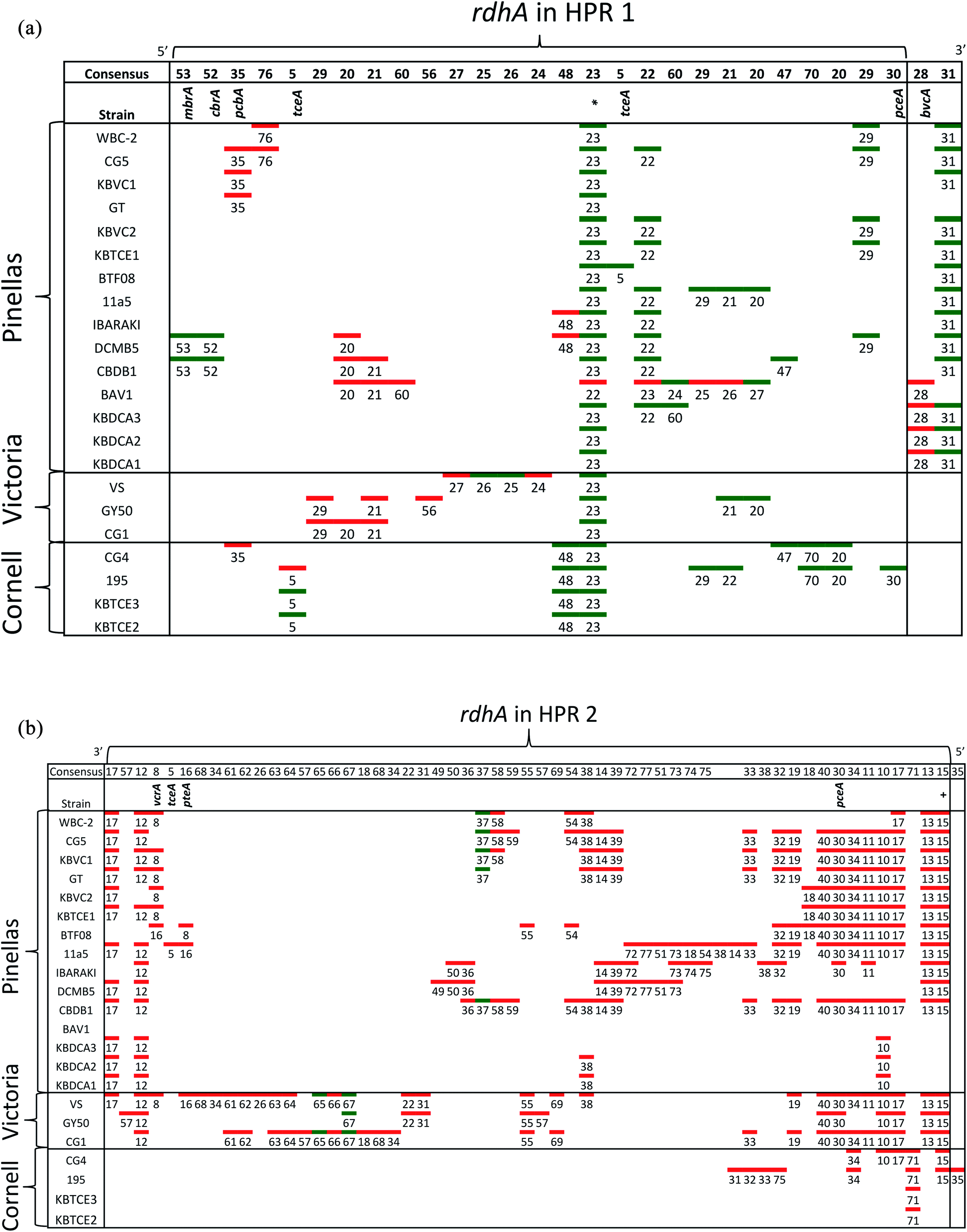 | ||
| Fig. 3 Synteny in Dehalococcoides mccartyi genomes. (a) Order of rdhA found in high plasticity region one (HPR1) in 22 D. mccartyi genomes labeled by strain name. rdhA are labeled by Ortholog Group (OG) number. rdhA of the same OG share >90% amino acid pairwise identity. Unpaired rdhA (i.e. singletons) are not included due to space limitations (see Table S2† for all genomes and rdhA). Green (leading strand) and red (lagging strand) accents indicate DNA strand clockwise from the oriC. The majority of rdhA are on the leading strand. * found in all D. mccartyi strains. (b) Order of rdhA found in high plasticity region two (HPR2) in 22 D. mccartyi genomes labeled by strain name. rdhA are labeled as in panel (a) except colors reflect different leading strands. Green (lagging strand) and red (leading strand) accents indicate DNA strand anticlockwise from the oriC. The majority of rdhA are on the leading strand. HPR2 starts at tRNA-Leu/Arg/Val at approximately 1.2 Mbp from the origin. OG 35 is only in strain 195 after tRNA-Ala, 1.3 Mbp from the origin. +OG known to be expressed during starvation. | ||
Trends in Dehalococcoides rdhA: acquisition, loss and evolution
There are two evolutionary time points which can be identified from the concatenated homologous gene alignment in the Dehalococcoides and Dehalogenimonas phylogenetic tree (Fig. 4). First, an estimate of the point of divergence of Dehalococcoides and Dehalogenimonas (Dehalococcoidia speciation) and secondly the separation of Dehalococcoides mccartyi into three distinct clades: the Pinellas, Cornell and Victoria (clade speciation). The clade separation in D. mccartyi is evident not only at the 16S rRNA level (Fig. S3†) but also in each homologous rdhA gene alignment (Fig. 5). D. mccartyi strains that are in the same clade typically have the same SNPs. These divergence time points could also be seen in rdhA alignments within OG groups. Each OG rdhA alignment includes sequences with varying numbers of nucleotide substitutions. These substitutions are sometimes clade-specific, meaning that the OG was present prior to clade speciation, and the divergences occurred after the clade speciation event. Other OGs include sequences with very few substitutions, which are not clade-specific, suggesting more recent acquisition (Fig. 4 and 5 with specific examples; all OG analyzed in Fig. S4 and S5†).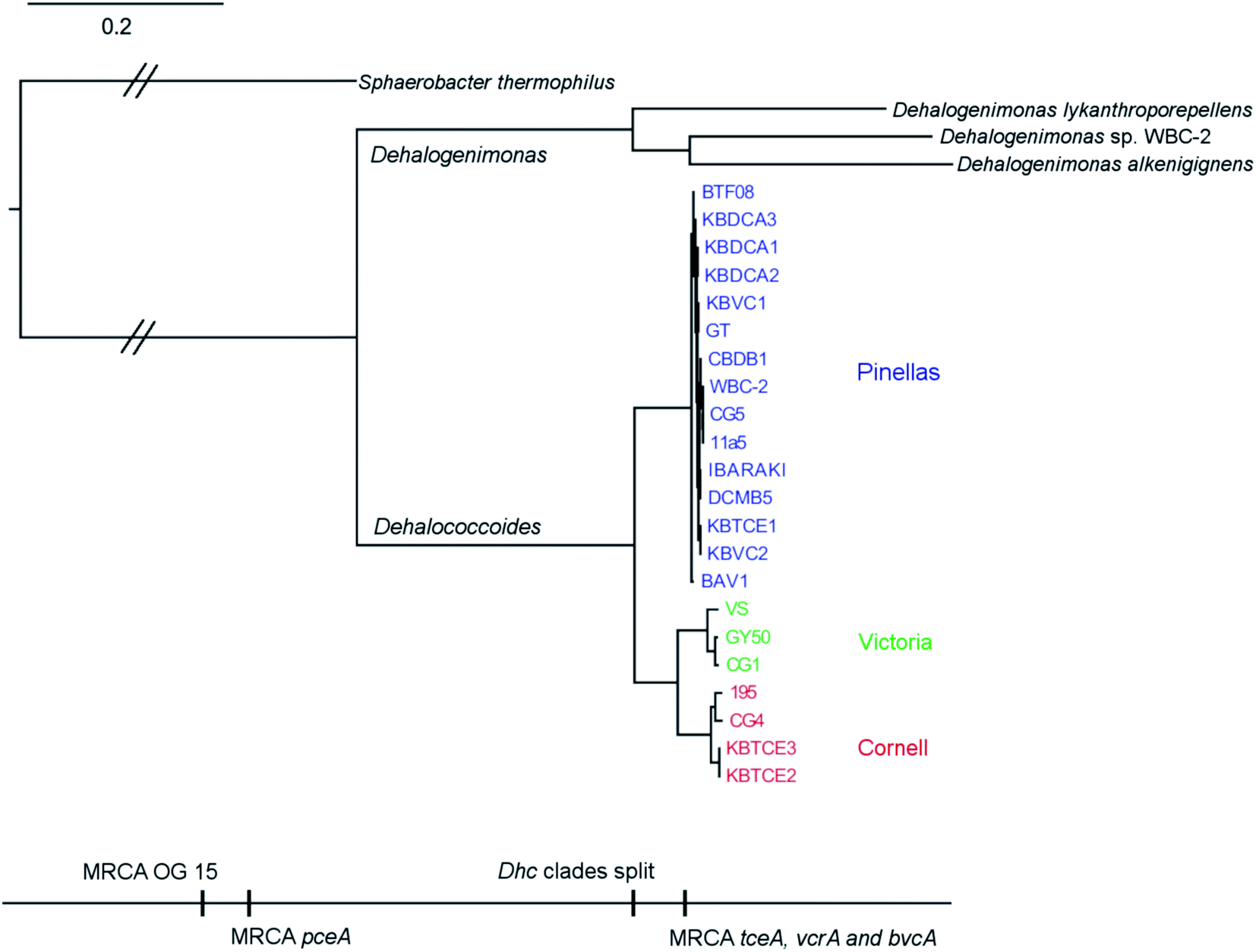 | ||
| Fig. 4 Phylogenetic tree created from an alignment of 109 concatenated core genes from Dehalococcoides mccartyi closed genomes and Dehalogenimonas closed genomes with chloroflexi Sphaerobacter thermophilus as out-group. Best scoring maximum likelihood tree is displayed. Bottom scale indicates timing of key events including clade separation in D. mccartyi and the Most Recent Common Ancestor (MRCA) of several dehalogenases listed by name or by Ortholog Group (OG) if uncharacterized. D. mccartyi clades are highlighted in common colour. Scale indicates number of substitutions per site. Double cross-hatching indicates this branch was reduced in length by half for visualization purposes. | ||
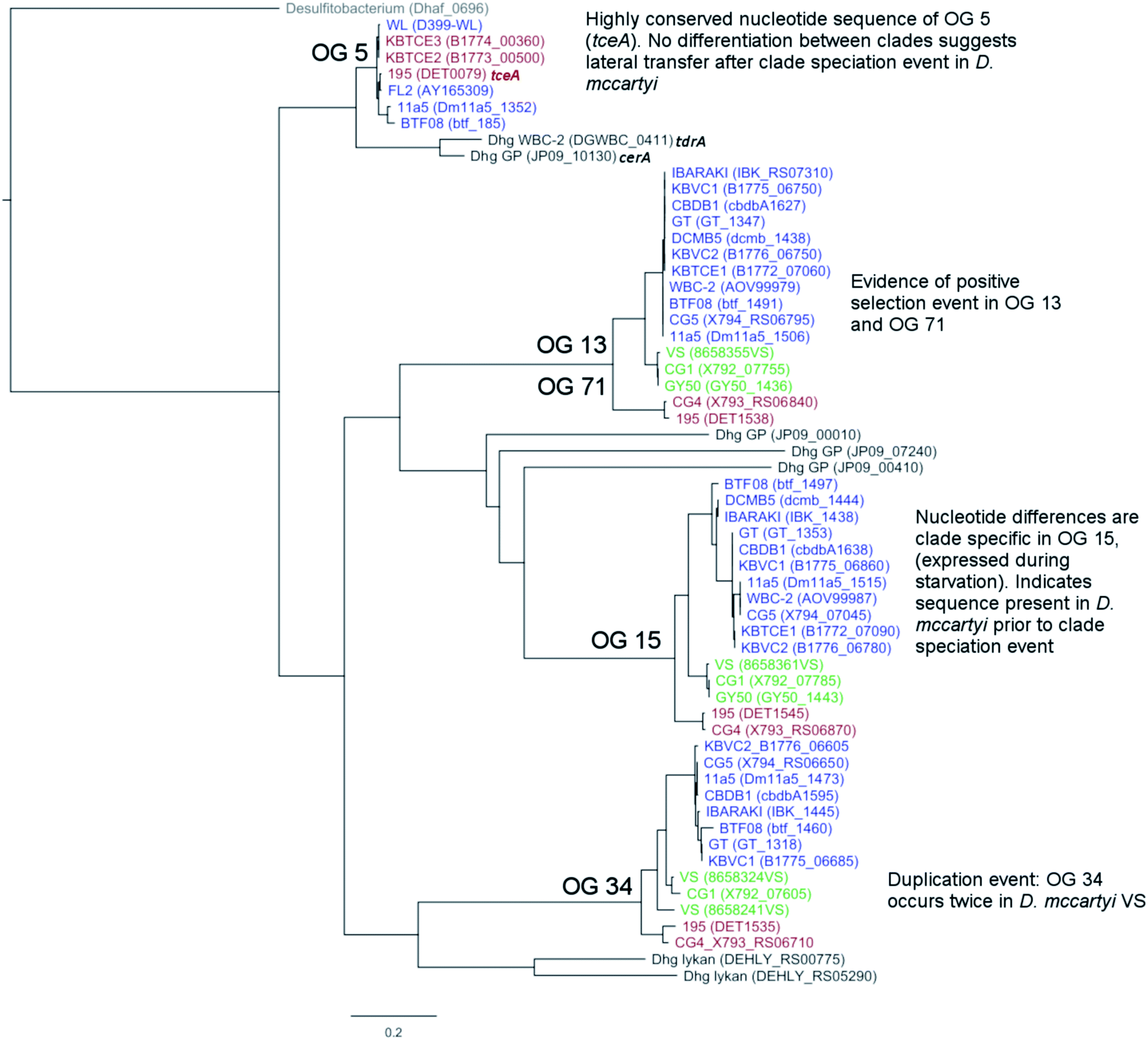 | ||
| Fig. 5 Phylogenetic tree of reductive dehalogenase genes which belong to Ortholog Group (OG) 5, 13, 71, 15 and 34. Best scoring maximum likelihood tree displayed. Scale shows number of substitutions per site. A trichloroethene dehalogenase from Desulfitobacterium (Dhaf_0696) used as out-group. The rdhA in the tree are coloured by clade: blue (Pinellas), green (Victoria), red (Cornell) and identified with strain name followed by locus tag of rdha in parentheses. Dehalogenimonas rdha similar to these OG groups are shown in black. | ||
Reductive dehalogenase sequences belonging to an OG with a long history with Dehalococcoides (before the clade speciation event, or even before the Dehalococcoidia speciation event, Tables S3 and S4†) occur syntenically and are typically preceded by MarR-type regulators, such as MarR-type regulator Rdh2R (cbdb1456), which was found to supress downstream rdhA expression in CBDB1.71 Syntenic OG alignments have small (∼0) Ka/Ks ratios meaning that syntenic OG rdhAs are not currently under selective pressure (Fig. 5). The first and most common trend observed from sequence classification and analysis is a consensus order of rdhA genes which are ancestral and not found expressed or synthesized at this time15 (summarized in Table 2). Dehalococcoides OGs whose level of divergence pre-dates the Dehalococcoidia speciation event, such as OG 15 and OG 34 have homologous representatives in Dehalogenimonas, including Dehalogenimonas sp. strains WBC-2, GP, D. alkenigignens and D. formicexedens.72
| Type | OG #s | General features | Protein information |
|---|---|---|---|
| a PID – pairwise identity. | |||
| 1 | 10, 11, 15, 17, 19, 21, 22, 23, 30, 48, 55, 60, 66 | Present in more than one clade | OG 15 known to be expressed by D. mccartyi during starvation101,102 |
| Found in D. mccartyi before clades separated. Synteny within genomes conserved. OG 15 illustrated in Fig. 5 | OG 23 found in all D. mccartyi genomes at time of analysis | ||
| 2 | 13/71, 18/40, 32/69, 33/38 | Pairs of OGs which have recently diverged from one another. Location of rdhA is syntenic across all genomes. OG 13 and 71 illustrated in Fig. 5 | No characterized members at time of study |
| 3 | 5, 8, 16, 28, 52, 53, 35 | Highly similar PID in different strains, regardless of clades. Often associated with mobile genes or on a genomic island. OG 5 illustrated in Fig. 5 | 5 – TceA103 |
| 8 – VcrA100 | |||
| 16 – PteA60 | |||
| 28 – BvcA104 | |||
| 52 – MbrA63 | |||
| 53 – CbrA105 | |||
| 35 –PcbA64 | |||
| 4 | 26, 34, 57 | Duplicated within the same genome. OG 34 illustrated in Fig. 5 | No characterized members at time of study |
For some OGs (Table 2), there is evidence for positive selection on RdhA according to Ka/Ks ratios. For example, we found that OGs 13 and 71 occur in the exactly the same position in different genomes, and that genomes contain either one or the other, but not both. OG 13 (Fig. 3) is found in both Pinellas and Victoria clades and is highly similar to OG 71 which is only in Cornell strains (Fig. 3). OG 13 dehalogenases have small Ka/Ks ratios while RdhAs in OG 71 have high ratios, suggesting positive selection. The conditions experienced by Cornell strains likely led to the use and specialization of this dehalogenase. Therefore, the second trend in dehalogenases in D. mccartyi is the evolution of new dehalogenases from existing ones (Summarized in Table 2).
Some rdhA are thought to have been acquired laterally by D. mccartyi, including tceA (OG 5, Fig. 4),24bvcA (OG 28),21 and vcrA (OG 8)21,23 due to their location on genomic islands, and very high nucleotide and amino acid sequence conservation among strains from distant geographical locations and sometimes different clades. Additionally, cbrA (OG 53), mbrA (OG 52) pcbA (OG 35) and pteA (OG 16) currently also appear to have very few mutations and occur in the vicinity of mobile elements. McMurdie et al. (2011)23 established that vcrA nucleotide polymorphisms indicate that it was acquired by Dehalococcoides approximately ∼1000 years ago, after the Dehalococcoides clade speciation event, meaning vcrA looks identical across all clades. All rdhA predicted to occur on mobile elements also have few mutations within OGs like vcrA, suggesting that they were acquired in a similarly recent time frame (Table S3†). Interestingly, all of these mobile OGs have members which have been biochemically characterized due to their association with dehalogenation of industrial pollutants and growth on these substrates in a laboratory setting.
Some OGs display movement within a genome, rather than between genomes. Victoria clade strains VS, CG1 and GY50 contain examples of an OG occurring twice in the same genome suggesting that a duplication event occurred. Duplicated OGs occur in different HPRs, and not in tandem (Fig. 3). In one case, pceA (OG 30) appears to have moved within a genome and without duplication. OG 30 is syntenic in HPR2 in 14 strains, with the exception of strain 195 where it occurs in HPR1 (Fig. 3). In strain 195, pceA is located near a serine recombinase which possibly mediated its movement. Additionally, the strain 195 pceA is present without its usual upstream transcriptional regulator, which was thought to be responsible for rdhA regulation.71,73 Transcriptomic studies show that strain 195 will continue to produce high transcript levels of pceA regardless of starvation or TCE amendment.74 Furthermore, strain 195 is the only strain to produce pceA in the presence of PCE, where other strains, such as CG5, transcribe pceA in the presence of multiple PCB congeners64 and CBDB1 was found to transcribe pceA in the presence of 2,3-dichlorophenol (DCP).75,76 Thus, the third trend we observed in dehalogenase evolution is mobility, either within or between strains (summarized as types 3 (lateral transfer) and 4 (within genome duplication or rearrangement) in Table 2).
Gene loss and genomic streamlining in Dehalococcoides mccartyi
D. mccartyi genomes are unique in that they are among the smallest genomes found in free-living bacteria (avg. 1.4 Mbp and 1451 protein-coding genes). A common theme among all small free-living prokaryotes is their high niche specialization and low-nutrient level environments.77D. mccartyi genomes probably required an extensive period of time to become as specialized and as small as they currently are. Wolf et al. (2013) proposed a theory that gene loss is equally or even more important than lateral gene transfer in shaping genomes.78 The high level of synteny and number of mutations found in Ortholog Groups of dehalogenases supports this theory in D. mccartyi. Given that all dehalogenases capable of dehalogenating industrial contaminants also appear to be mobile, it is possible that anthropogenic releases of organohalides have caused D. mccartyi genomes to enter a period of complexification involving sharing select rdhA. The exchange of key reductive dehalogenases among D. mccartyi can is akin to the recent dissemination of antibiotic resistance genes in the natural environment.79–81 Transfer mechanisms may even be related considering the homology observed between the integration module of the vcrA containing genomic island and the Staphylococcal cassette chromosome that carries the broad spectrum beta-lactam resistance gene.23,82Reductive dehalogenases in other obligate dechlorinating organisms: Dehalobacter and Dehalogenimonas
We analyzed the reductive dehalogenases from other OHRB including Dehalobacter and Dehalogenimonas to extend the OG classification system beyond Dehalococcoides. In both cases, there are very few publicly available closed genomes, which limited our analysis. Members of the genus Dehalobacter are also organohalide-respiring bacteria that use hydrogen or formate as electron donor, acetate as carbon source, and organohalides as electron acceptors.83,84Dehalobacter spp. have been shown to respire and/or be implicated in the reductive dechlorination of chlorinated methanes,85 ethanes,86–88 ethenes,68,89 cyclohexanes,90,91 benzenes,92,93 phenols,94 biphenyls,95 and phthalides.95 As of November of 2019, ten Dehalobacter genome assemblies, corresponding to strains PER-K23 (DSM 9455), CF, DCA, UNSWDHB, TeCB1, MCB1, 12DCB1, 14DCB1, E1, and FTH1 were publicly available NCBI, but only three of these genomes are closed (strains PER-K23, CF, DCA) (Table S6† for all genomes analyzed in this study). Four additional metagenome-assembled-genomes of Dehalobacter from KB-1 subcultures are found in the Integrated Microbial Genomes & Microbiomes (IMG/M) system, corresponding to strains TCB1 (2775506732), TCB2 (2786546105), TCB3 (2821530269), and HCH1 (2823892816). Like Dehalococcoides, many rdhA genes (∼10 to 30) can be found in each Dehalobacter genome, but only a subset of genes has been characterized (see Table S5†). As part of this study, a new closed Dehalobacter restrictus genome was submitted to NCBI, strain 12DCA (CP046996).The rdhA genes of the 15 publicly available Dehalobacter restrictus genome assemblies were translated and classified into OGs. Like Dehalococcoides mccartyi, fewer new Dehalobacter restrictus dehalogenases were classified into new OGs with an increasing number of distinct genome assemblies (Fig. 2). A total of 80 OGs resulted from the classification of 356 RdhA protein sequences from 15 genomes (or metagenome assembled genomes/binned genomes), approaching saturation, as with Dehalococcoides mccartyi sequences (Fig. 2). A full Dehalobacter RDase tree annotated by OG can be found in Fig. S6.† An initial examination of the distribution of OGs in closed Dehalobacter genomes as well as in some of the contigs from other Dehalobacter spp., (Fig. S7†) suggests that some Dehalobacter RdhA OGs also display synteny across genomes. As additional Dehalobacter complete and closed genomes become available, trends such as synteny and gene evolution will become easier to see.
With respect to Dehalogenimonas, to-date only six genomes have been closed or binned (D. lykanthroporepellans,96Dehalogenimonas sp. strain WBC-2,35D. alkenigignens strain IP3-3,97D. alkenigignens strain BRE15M, D. formicexedens98 and Dehalogenimonas sp. strain GP99) contributing 144 reductive dehalogenase sequences. These dehalogenases formed only 18 OGs, where 16 of the 18 come from two similar strains of Dehalogenimonas alkenigignens. Unlike Dehalococcoides mccartyi and Dehalobacter restrictus, Dehalogenimonas is a more divergent group at this time (Fig. 4 and 2) with multiple species identified. Much more sequencing is required to more clearly define the extent and organization of OGs in Dehalogenimonas.
We next considered only the reductive dehalogenases from Dehalococcoides, Dehalobacter and Dehalogenimonas that have been characterized, either biochemically or genetically, to reveal relationships between function and OG classification (Fig. 6). From Dehalococcoides dehalogenases, all characterized representatives in the same OG have activity on the same substrates. For example VcrA from different D. mccartyi (strain VS,100 WBC-235 or from the KB-1 consortium101) are active on the same tested substrates (see Table S5† for full list of characterized dehalogenases). We found the same to be true with BvcA (D. mccartyi BAV1 and KB-1) and TceA (D. mccartyi 195 and KB-1). It is important to note that the number of substrates tested was a limited list in all cases. From Dehalogenimonas there are no OGs with more than one characterized representative. From Dehalobacter, OG 97 can be described as a 1,1,1-trichloroethane/chloroform/1,1-dichloroethane group of dehalogenases with several overlapping functions, and OG 96 includes a perchloroethene dehalogenase and a tetrachlorobenzene/trichlorobenzene dehalogenase. At present, the members of these OGs have not been extensively tested on different substrates. Future work and new protein structures will refine the definition of OGs to better associate dehalogenase function to sequence.
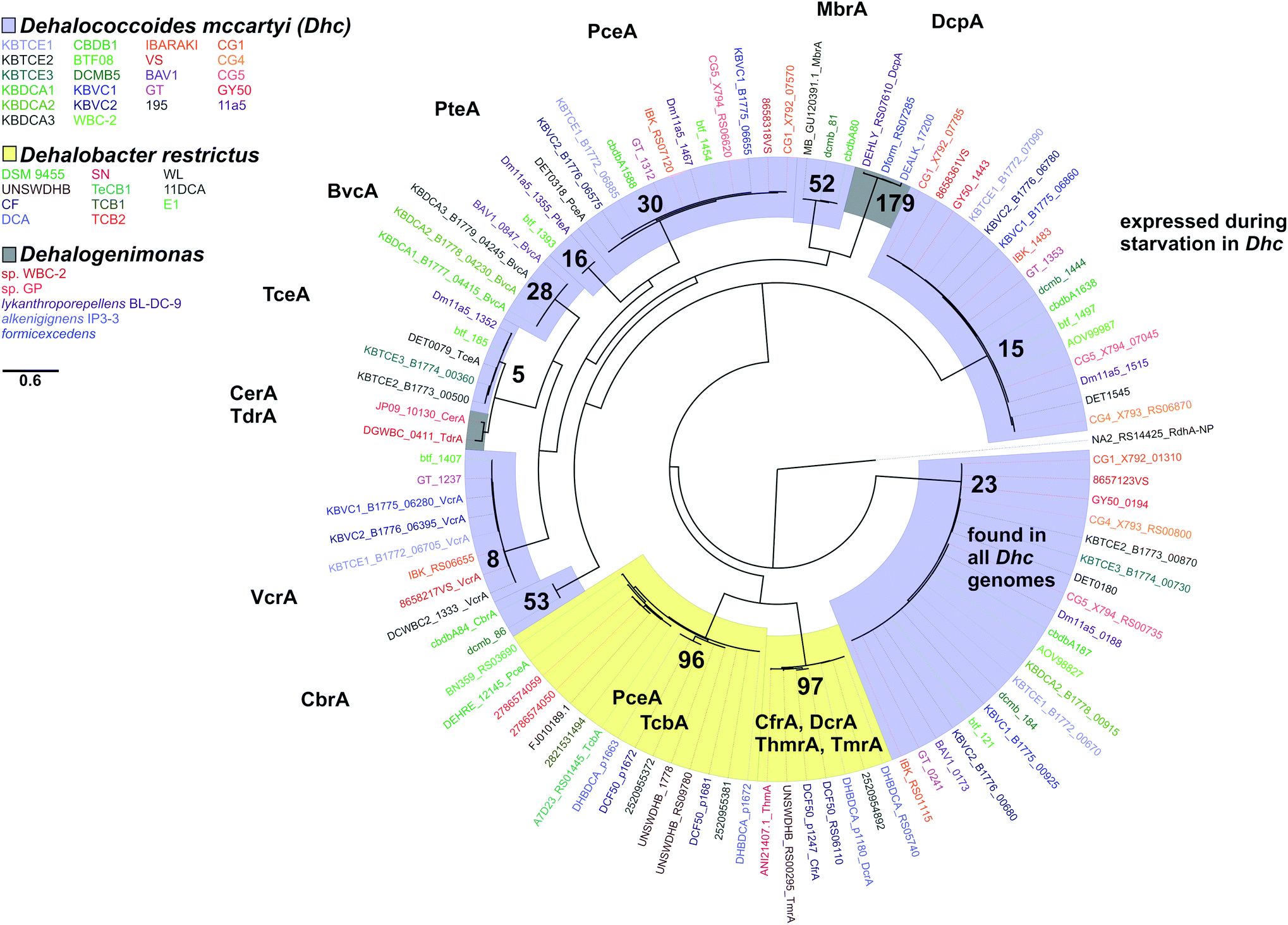 | ||
| Fig. 6 Phylogenetic amino acid tree of reductive dehalogenases which have been at least partially characterized from Dehalococcoides (Dhc, in blue), Dehalogenimonas (Dhg, in grey) and Dehalobacter (Dhb, in yellow). Best scoring maximum likelihood tree displayed. Scale indicates number of substitutions per site. Ortholog Groups (OGs) of dehalogenases with upwards of 90% amino acid identity are highlighted and identified by number. OGs containing a functionally characterized representative are annotated by dehalogenase name. rdhA sequences are coloured by genome they originated from. rdhA are named by NCBI locus tag. | ||
New community resource: the reductive dehalogenase database web server
A database of reductive dehalogenase gene sequences and protein sequences was previously located in a publicly accessible Google Doc.19 We have transformed this resource into a web-based database where the data can be interactively viewed and downloaded. Additionally, users can check and add new sequences, classify RdhA into Ortholog Groups and visualize sequences in a tree with other RdhA sequences. The webserver can be accessed at: http://reductivedehalogenasedb.ca.Conclusions
Sequencing and completely closing multiple genomes of D. mccartyi has provided new insights into the evolution of rdhA genes in this genus. The majority of rdhA found in D. mccartyi display a much higher degree of synteny between genomes than previously appreciated. Evolutionary analysis revealed that most rdhA sequences are old, and have likely been in D. mccartyi genomes since the time of a Dehalococcoidia common ancestor (Fig. 4 and Table S4-4†). It is possible that the relatively recent anthropogenic release of specific industrial chemicals has enabled the dissemination of specific reductive dehalogenases capable of solvent dehalogenation, initiating a period of adaptation and complexification of the D. mccartyi genome. These genome comparisons have provided specific examples of how the complement of D. mccartyi rdhA has been shaped, revealing (1) adaptation of existing rdhA to new substrates, (2) lateral gene transfer of rdhA from the environment and (3) duplication or movement within genomes. While too few genomes are currently available from other OHRB, it is anticipated that these trends will still prevail. The classification of reductive dehalogenase sequences into Ortholog Groups has enabled a better understanding of the evolution and function of rdhA sequences, clearly showing that they are of pre-industrial origin. The OG classification prevents over-reporting of “novel” sequences, but rather allows for rapid situation of newly sequenced members in the greater context. To assist future studies of organohalide-respiring bacteria and their reductive dehalogenases, we have begun a public resource to assist the community in the classification of reductive dehalogenase sequences by building a high-quality curated database of all reductive dehalogenases, and maintaining a simple classification system and nomenclature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support was provided by the Government of Canada through Genome Canada and the Ontario Genomics Institute (no. 2009-OGI-ABC-1405 to E. A. E.), the Natural Sciences and Engineering Research Council of Canada (NSERC PGS-D to O. M.), the NSERC CREATE RENEW program [180804567], and the Government of Ontario through the Ontario Graduate Scholarship program (OGS to L. A. P. J.). Support was also provided by the Government of Ontario through the ORF-GL2 program and the Unites States Department of Defense through the strategic Environmental Research and Development Program (SERDP) under contract (W912HQ-07-C-0036 project ER-1586). Special thanks to Laura Hug for initially coming up with the OG classification and her input on the reductive dehalogenase web server. Enormous thanks to Herbert Molenda for programming the website and database. Also special thanks to anonymous reviewers for their thorough comments and suggestions.References
- H. F. Stroo, A. Leeson and C. H. Ward, Bioaugmentation for Groundwater Remediation, Springer, New York, 2012 Search PubMed.
- S. S. Suthersan, M. Schnobrich, J. Martin, J. F. Horst and E. Gates, Three Decades of Solvent Bioremediation: The Evolution from Innovation to Conventional Practice, Groundwater Monit. Rem., 2017, 37, 14–23 CrossRef.
- D. W. Hill and P. L. McCarty, Anaerobic degradation of selected chlorinated hydrocarbon pesticides, J. – Water Pollut. Control Fed., 1967, 39, 1259–1277 CAS.
- E. J. Bouwer and P. L. McCarty, Transformations of 1- and 2-carbon halogenated aliphatic organic compounds under methanogenic conditions, Appl. Environ. Microbiol., 1983, 45, 1286–1294 CrossRef CAS PubMed.
- D. L. Freedman and J. M. Gossett, Biological reductive dechlorination of tetrachloroethylene and trichloroethylene to ethylene under methanogenic conditions, Appl. Environ. Microbiol., 1989, 55, 2144–2151 CrossRef CAS PubMed.
- R. Seshardi, L. Adrian, D. E. Fouts, J. A. Eisen, A. M. Phillippy, B. A. Methe, N. L. Ward, W. C. Nelson, R. J. Dobson, S. C. Daugherty, L. M. Brinkac, S. A. Sullivan, R. Madupu, K. E. Nelson, K. H. Kang, M. Impraim, K. Tran, J. M. Robinson, H. A. Forberger, C. M. Fraser, S. H. Zinder and J. F. Heidelberg, Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes, Science, 2005, 307, 105–108 CrossRef PubMed.
- X. Maymo-Gatell, Y. Chien, J. M. Gossett and S. H. Zinder, Isolation of a bacterium that reductively dechlorinates tetrachloroethene to ethene, Science, 1997, 276, 1568–1571 CrossRef CAS PubMed.
- D. J. Major, M. L. McMaster, E. E. Cox, E. A. Edwards, S. M. Dwortzek, E. R. Hendrickson, M. G. Starr, J. A. Payne and L. W. Buonamici, Field demonstration of sucessful bioaugmentation to achieve dechlorination of tetrachloroethene to ethene, Environ. Sci. Technol., 2000, 36, 5106–5116 CrossRef PubMed.
- M. Duhamel, S. D. Wehr, L. Yu, H. Rizvi, D. Seepersad, S. Dworatzek, E. E. Cox and E. A. Edwards, Comparison of anaerobic dechlorinating enrichment cultures maintained on tetrachloroethene, trichloroethene, cis-dichloroethene and vinyl chloride, Water Res., 2002, 36, 4193–4202 CrossRef CAS PubMed.
- J. Dolfing and J. M. Tiedje, Hydrogen cycling in a three-tiered food web growing on the methanogenic conversion of 3-chlorobenzoate, FEMS Microbiol. Ecol., 1986, 2, 293–298 CrossRef.
- K. A. DeWeerd, L. Mandelco, R. S. Tanner, C. R. Woese and J. M. Suflita, Desulfomonile tiedjei gen. nov. and sp. nov., a novel anaerobic, dehalogenating, sulfate-reducing bacterium, Arch. Microbiol., 1990, 154, 23–30 CrossRef CAS.
- F. E. L. Lorenz Adrian, Organohalide-Respiring Bacteria, ed. F. L. Lorenz Adrian, Heidelberg, Berlin, 2006 Search PubMed.
- T. Schubert, L. Adrian, R. G. Sawers and G. Diekert, Organohalide respiratory chains: composition, topology and key enzymes, FEMS Microbiol. Ecol., 2018, 94 Search PubMed.
- M. Fincker and A. M. Spormann, Biochemistry of Catabolic Reductive Dehalogenation, Annu. Rev. Biochem., 2017, 86, 357–386 CrossRef CAS PubMed.
- D. Türkowsky, N. Jehmlich, G. Diekert, L. Adrian, M. von Bergen and T. Goris, An integrative overview of genomic, transcriptomic and proteomic analyses in organohalide respiration research, FEMS Microbiol. Ecol., 2018, 94 Search PubMed.
- S. Atashgahi, Y. Lu and H. Smidt, Organohalide-Respiring Bacteria, ed. L. Adrian and F. E. Löffler, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 63–105, DOI:10.1007/978-3-662-49875-0_5.
- L. A. Puentes Jácome and E. A. Edwards, A switch of chlorinated substrate causes emergence of a previously undetected native Dehalobacter population in an established Dehalococcoides-dominated chloroethene-dechlorinating enrichment culture, FEMS Microbiol. Ecol., 2017, 93 Search PubMed.
- M. J. Krzmarzick, B. B. Crary, J. J. Harding, O. O. Oyerinde, A. C. Leri, S. C. B. Myneni and P. J. Novak, Natural Niche for Organohalide-Respiring Chloroflexi, Appl. Environ. Microbiol., 2012, 78, 393–401 CrossRef CAS PubMed.
- L. A. Hug, F. Maphosa, D. Leys, F. E. Löffler, H. Smidt, E. A. Edwards and L. Adrian, Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases, Philos. Trans. R. Soc., B, 2013, 368, 1–10 CrossRef PubMed.
- M. Kube, A. Beck, S. H. Zinder, H. Kuhl, R. Reinhardt and L. Adrian, Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1, Nat. Biotechnol., 2005, 23, 1269–1273 CrossRef CAS PubMed.
- P. J. McMurdie, S. F. Behrens, J. A. Müller, J. Goke, K. M. Ritalahti, R. Wagner, E. Goltsman, A. Lapidus, S. Holmes, F. E. Löffler and A. M. Spormann, Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides, PLoS Genet., 2009, 5, e1000714 CrossRef PubMed.
- C. Ding, M. J. Rogers, K.-L. Yang and J. He, Loss of the ssrA genome island led to partial debromination in the PBDE respiring Dehalococcoides mccartyi strain GY50, Environ. Microbiol., 2017, 19, 2906–2915 CrossRef CAS PubMed.
- P. J. McMurdie, L. A. Hug, E. A. Edwards, S. Holmes and A. A. Spormann, Site-specific mobilization of vinyl-chloride respiration islands by a mechanism common in Dehalococcoides, BMC Genomics, 2011, 12, 287–302 CrossRef CAS PubMed.
- R. Krajmalnik-Brown, Y. Sung, K. M. Ritalahti, F. M. Saunders and F. E. Löffler, Environmental distribution of the trichloroethene reductive dehalogenase gene (tceA) suggests lateral gene transfer among Dehalococcoides, FEMS Microbiol. Ecol., 2007, 59, 206–214 CrossRef CAS PubMed.
- H. Smidt, M. Van Leest, J. van der Oost and W. M. De Vos, Transcriptional regulation of the cpr gene cluster in ortho-chlorophenol respiring Desulfitobacterium dehalogenans, J. Bacteriol., 2000, 182, 5683–5691 CrossRef CAS PubMed.
- K. Chen, L. Huang, C. Xu, X. Liu, J. He, S. H. Zinder, S. Li and J. Jiang, Molecular characterization of the enzymes involved in degradation of a brominated aromatic herbicide, Mol. Microbiol., 2013, 89, 1121–1139 CrossRef CAS PubMed.
- A. Kublik, D. Deobald, S. Hartwig, C. L. Schiffmann, A. Andrades, M. von Bergen, R. G. Sawers and L. Adrian, Identification of a multi-protein reductive dehalogenase complex in Dehalococcoides mccartyi strain CBDB1 suggests a protein-dependent respiratory electron transport chain obviating quinone involvement, Environ. Microbiol., 2016, 18, 3044–3056 CrossRef CAS PubMed.
- S. Hartwig, N. Dragomirova, A. Kublik, D. Türkowsky, M. von Bergen, U. Lechner, L. Adrian and R. G. Sawers, A H2-oxidizing, 1,2,3-trichlorobenzene-reducing multienzyme complex isolated from the obligately organohalide-respiring bacterium Dehalococcoides mccartyi strain CBDB1, Environ. Microbiol. Rep., 2017, 9, 618–625 CrossRef CAS PubMed.
- R. Banerjee and S. W. Ragsdale, The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes, Annu. Rev. Biochem., 2003, 72, 209–247 CrossRef CAS PubMed.
- Y. T. Marcy, R. S. Ishoey, T. B. Laksen, B. P. Stockwell, A. L. Walenz, K. Y. Halpern, K. Y. Beeson, S. M. Goldberg and S. R. Quake, Nanoliter reactors improve multiple displacement amplification of genomes from single cells, PLoS Genet., 2007, 3, 1702–1708 CAS.
- Y. Morita, T. Futagami, M. Goto and K. Furukawa, Functional characterization of the trigger factor protein PceT of a tetrachloroethene-dechlorinating Desulfitobacterium hafniense Y51, Appl. Microbiol. Biotechnol., 2009, 83, 775–781 CrossRef CAS PubMed.
- A. Sunyama, M. Yamashita, S. Yoshino and K. Furukawa, Molecular characterization of the PceA dehalogenase of Desulfitobacterium sp. strain Y51, Bacteriol, 2002, 184, 3419–3425 CrossRef PubMed.
- J. Yan, K. M. Ritalahti, D. D. Wagner and F. L. Löffler, Unexpected specificity of interspecies cobamide transfer from Geobacter spp. to organohalide-respiring Dehalococcoides mccartyi strains, Appl. Environ. Microbiol., 2012, 78, 6630–6636 CrossRef CAS PubMed.
- A. Parthasarathy, T. A. Stich, S. T. Lohner, A. Lesnefsky, R. D. Britt and A. Spormann, Heterologously expressed vinyl chloride reductive dehalogenase (VcrA) from Dehalococcoides mccartyi strain VS, J. Am. Chem. Soc., 2015, 137, 3525–3532 CrossRef CAS PubMed.
- O. Molenda, A. T. Quaile and E. A. Edwards, Dehalogenimonas sp. strain WBC-2 genome and identification of its trans-dichloroethene reductive dehalogenase, TdrA, Appl. Environ. Microbiol., 2016, 82, 40–50 CrossRef CAS PubMed.
- K. A. Payne, C. P. Quezada, K. Fisher, M. S. Dunstan, F. A. Collins, H. Sjuts, C. Levy, S. Hay, S. E. Rigby and D. Leys, Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation, Nature, 2015, 517, 513–516 CrossRef CAS PubMed.
- M. Bommer, C. Kunze, J. Fesseler, T. Schubert, G. Diekert and H. Dobbek, Structural basis for organohalide respiration, Science, 2014, 346, 455–458 CrossRef CAS PubMed.
- A. Pérez-de-Mora, A. Lacourt, M. L. McMaster, X. Liang, S. M. Dworatzek and E. A. Edwards, Chlorinated electron acceptor abundance drives selection of Dehalococcoides mccartyi (D. mccartyi) strains in dechlorinating enrichment cultures and groundwater environments, Front. Microbiol., 2018, 9 Search PubMed.
- M. Duhamel, K. Mo and E. A. Edwards, Characterization of a highly enriched Dehalococcoides-containing culture that grows on vinyl chloride and trichloroethene, Appl. Environ. Microbiol., 2004, 70, 5538–5545 CrossRef CAS PubMed.
- M. Duhamel and E. A. Edwards, Growth and yields of dechlorinators, acetogens, and methanogens during reductive dechlorination of chlorinated ethenes and dihaloelimination of 1,2-dichloroethane, Environ. Sci. Technol., 2007, 41, 2303–2310 CrossRef CAS PubMed.
- M. Duhamel, S. D. Wehr, L. Yu, H. Rizvi, D. Seepersad, S. Dworatzek, E. E. Cox and E. A. Edwards, Comparison of anaerobic dechlorinating enrichment cultures maintained on tetrachloroethene, trichloroethene, cis-dichloroethene and vinyl chloride, Water Res., 2002, 36, 4193–4202 CrossRef CAS PubMed.
- S. Tang, Y. Gong and E. A. Edwards, Semi-automatic in silico gap closure enabled de novo assembly of two Dehalobacter genomes from metagenomic data, PLoS One, 2012, 7 CAS.
- A. M. Bolger, M. Lohse and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 2014, 30, 2114–2120 CrossRef CAS PubMed.
- R. Aziz, D. Bartels, A. D. Best, M. DeJongh, T. Disz, R. Edwards, K. Forsma, S. Gerdes, E. M. Glass, M. Kubal, F. Meyer, G. Olsen, R. Olson, A. Ostermn, R. Overbeek, L. McNeil, D. Paarmann, T. Paczian, B. Parello, G. Push, C. Reich, R. Stevens, O. Vassieva, V. Vonstein, A. Wilke and O. Zagnitko, The RAST server: rapid annotations using subsystems technology, BMC Genomics, 2008, 9 Search PubMed.
- G. H. Van Domselaar, P. Stothard, S. Shrivastava, J. A. Cruz, A. Guo, X. Dong, P. Lu, D. Szafron, R. Greiner and D. S. Wishart, BASys: a web server for automated bacterial genome annotation, Nucleic Acids Res., 2005, 33, W455–W459 CrossRef CAS PubMed.
- T. Tatusova, M. DiCuccio, A. Badretdin, V. Chetvernin, E. P. Nawrocki, L. Zaslavsky, A. Lomsadze, K. D. Pruitt, M. Borodovsky and J. Ostell, NCBI prokaryotic genome annotation pipeline, Nucleic Acids Res., 2016, 44, 6614–6624 CrossRef CAS PubMed.
- A. Frank and J. Lobry, Oriloc: prediction of replication boundaries in unannotated bacterial chromosomes, Bioinformatics, 2000, 16, 566–567 CrossRef PubMed.
- B. Contreras-Moreira and P. Vinuesa, GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis, Appl. Environ. Microbiol., 2013, 79, 7696–7701 CrossRef CAS PubMed.
- R. C. Edgar, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res., 2004, 32, 1792–1797 CrossRef CAS PubMed.
- A. Stamatakis, RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models, Bioinformatics, 2006, 22, 2688–2690 CrossRef CAS PubMed.
- M. Kearse, R. Moir, A. Wilson, S. Stones-Havas, M. Cheung, S. Sturrock, S. Buxton, A. Cooper, S. Markowitz, C. Duran, T. Thierer, B. Ashton, P. Meintjes and A. Drummond, Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data, Bioinformatics, 2012, 28, 1647–1649 CrossRef PubMed.
- Y. B. Ahn, L. J. Kerkhof and M. M. Haggblom, Desulfoluna spongiiphila sp. nov., a dehalogenating bacterium in the Desulfobacteraceae from the marine sponge Aplysina aerophoba, Int. J. Syst. Evol. Microbiol., 2009, 59, 2133–2139 CrossRef CAS PubMed.
- A. Nekrutenko, K. D. Makova and W.-H. Li, The K(A)/K(S) Ratio Test for Assessing the Protein-Coding Potential of Genomic Regions: An Empirical and Simulation Study, Genome Res., 2002, 12, 198–202 CrossRef CAS PubMed.
- B. Xu and Z. Yang, pamlX: A Graphical User Interface for PAML, Mol. Biol. Evol., 2013, 30, 2723–2724 CrossRef CAS PubMed.
- Z. Yang, PAML: a program package for phylogenetic analysis by maximum likelihood, CABIOS, Comput. Appl. Biosci., 1997, 13, 555–556 CAS.
- Z. Yang and J. P. Bielawski, Statistical methods for detecting molecular adaptation, Trends Ecol. Evol., 2000, 15, 496–503 CrossRef.
- F. E. Löffler, J. Yan, K. M. Ritalahti, L. Adrian, E. A. Edwards, K. T. Konstantinidis, J. A. Müller, H. Fullerton, S. H. Zinder and A. M. Spormann, Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi, Int. J. Syst. Evol. Microbiol., 2013, 63, 625–635 CrossRef PubMed.
- E. R. Hendrickson, J. A. Payne, R. M. Young, M. G. Starr, M. P. Perry, S. Fahnestock, D. E. Ellis and R. C. Ebersole, Molecular Analysis of Dehalococcoides 16S Ribosomal DNA from Chloroethene-Contaminated Sites throughout North America and Europe, Appl. Environ. Microbiol., 2002, 68, 485–495 CrossRef CAS PubMed.
- M. Pöritz, T. Goris, T. Wubet, M. T. Tarkka, F. Buscot, I. Nijenhuis, U. Lechner and L. Adrian, Genome sequences of two dehalogenation specialists – Dehalococcoides mccartyi strains BTF08 and DCMB5 enriched from the highly polluted Bitterfeld region, FEMS Microbiol. Lett., 2013, 343, 101–104 CrossRef PubMed.
- S. Zhao, C. Ding and J. He, Genomic characterization of Dehalococcoides mccartyi strain 11a5 reveals a circular extrachromosomal genetic element and a new tetrachloroethene reductive dehalogenase gene, FEMS Microbiol. Ecol., 2017, 93, fiw235 Search PubMed.
- Y. Sung, K. M. Ritalahti, R. P. Apkarian and F. E. Löffler, Quantitative PCR confirms purity of strain GT, a novel trichloroethene-to-ethene-respiring Dehalococcoides isolate, Appl. Environ. Microbiol., 2006, 72, 1980–1987 CrossRef CAS PubMed.
- M. Yohda, O. Yagi, A. Takechi, M. Kitajima, H. Matsuda, N. Miyamura, T. Aizawa, M. Nakajima, M. Sunairi, A. Daiba, T. Miyajima, M. Teruya, K. Teruya, A. Shiroma, M. Shimoji, H. Tamotsu, A. Juan, K. Nakano, M. Aoyama, Y. Terabayashi, K. Satou and T. Hirano, Genome sequence determination and metagenomic characterization of a Dehalococcoides mixed culture grown on cis-1,2-dichloroethene, J. Biosci. Bioeng., 2015, 120, 69–77 CrossRef CAS PubMed.
- D. Cheng and J. He, Isolation and characterization of “Dehalococcoides” sp. strain MB which dechlorinates tetrachloroethene to trans-1,2-dichloroethene, Appl. Environ. Microbiol., 2009, 75, 5910–5918 CrossRef CAS PubMed.
- S. Wang, K. R. Chng, A. Wilm, S. Zhao, K.-L. Yang, N. Nagarajan and J. He, Genomic characterization of three unique Dehalococcoides that respire on persistent polychlorinated biphenyls, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12103–12108 CrossRef CAS PubMed.
- O. Molenda, S. Tang, L. Lomheim, G. Vasu, S. Lemak, A. F. Yakunin and E. A. Edwards, Extrachromosomal circular elements targeted by CRISPR-Cas in Dehalococcoides mccartyi are linked to mobilization of reductive dehalogenase genes, ISME J., 2019, 13, 24–38 CrossRef CAS PubMed.
- A. Low, Z. Shen, D. Cheng, M. J. Rogers, P. K. H. Lee and J. He, A comparative genomics and reductive dehalogenase gene transcription study of two chloroethene-respiring bacteria, Dehalococcoides mccartyi strains MB and 11a, Sci. Rep., 2015, 5, 15204 CrossRef CAS PubMed.
- P. K. H. Lee, J. He, S. H. Zinder and L. Alvarez-Cohen, Evidence for nitrogen fixation by “Dehalococcoides ethenogenes” strain 195, Appl. Environ. Microbiol., 2009, 75, 7551–7555 CrossRef CAS PubMed.
- T. Kruse, J. Maillard, L. Goodwin, T. Woyke, H. Teshima, D. Bruce, C. Detter, R. Tapia, C. Han, M. Huntemann, C. L. Wei, J. Han, A. Chen, N. Kyrpides, E. Szeto, V. Markowitz, N. Ivanova, I. Pagani, A. Pati, S. Pitluck, M. Nolan, C. Holliger and H. Smidt, Complete genome sequence of Dehalobacter restrictus PER-K23(T), Stand. Genomic Sci., 2013, 8, 375–388 CrossRef PubMed.
- L. A. Hug, R. G. Beiko, A. R. Rowe, R. E. Richardson and E. A. Edwards, Comparative metagenomics of three Dehalococcoides-containing enrichment cultures: the role of the non-dechlorinating community, BMC Genomics, 2012, 13, 327 CrossRef CAS PubMed.
- A. S. Waller, R. Krajmalnik-Brown, F. E. Löffler and E. A. Edwards, Multiple reductive-dehalogenase-homologous genes are simultaneously transcribed during dechlorination by Dehalococcoides-containing cultures, Appl. Environ. Microbiol., 2005, 71, 8257–8264 CrossRef CAS PubMed.
- A. Wagner, L. Segler, S. Kleinsteuber, G. Sawers, H. Smidt and U. Lechner, Regulation of reductive dehalogenase gene transcription in Dehalococcoides mccartyi, Philos. Trans. R. Soc., B, 2013, 368, 20120317 CrossRef PubMed.
- O. Molenda, S. Tang, L. Lomheim and E. A. Edwards, Eight new genomes of organohalide-respiring Dehalococcoides mccartyi reveal evolutionary trends in reductive dehalogenase enzymes, bioRxiv, Bioinf., 2018, 345173 Search PubMed.
- L. Krasper, H. Lilie, A. Kublik, L. Adrian, R. Golbik and U. Lechner, The MarR-Type Regulator Rdh2R Regulates rdh Gene Transcription in Dehalococcoides mccartyi strain CBDB1, J. Bacteriol., 2016, 198, 3130–3141 CrossRef CAS PubMed.
- C. B. Mansfeldt, A. R. Rowe, G. L. W. Heavner, S. H. Zinder and R. E. Richardson, Meta-analyses of Dehalococcoides mccartyi strain 195 transcriptomic profiles identify a respiration rate-related gene expression transition point and interoperon recruitment of a key oxidoreductase subunit, Appl. Environ. Microbiol., 2014, 80, 6062–6072 CrossRef PubMed.
- R. M. Morris, J. M. Fung, B. G. Rahm, S. Zhang, D. L. Freedman, S. H. Zinder and R. E. Richardson, Comparative proteomics of Dehalococcoides spp. reveals strain-specific peptides associated with activity, Appl. Environ. Microbiol., 2007, 73, 320–326 CrossRef CAS PubMed.
- J. M. Fung, R. M. Morris, L. Adrian and S. H. Zinder, Expression of reductive dehalogenase genes in Dehalococcoides ethenogenes strain 195 growing on tetrachloroethene, trichloroethene, or 2,3-dichlorophenol, Appl. Environ. Microbiol., 2007, 73, 4439–4445 CrossRef CAS PubMed.
- D. J. Martínez-Cano, M. Reyes-Prieto, E. Martínez-Romero, L. P. Partida-Martínez, A. Latorre, A. Moya and L. Delaye, Evolution of small prokaryotic genomes, Front. Microbiol., 2015, 5 Search PubMed.
- Y. I. Wolf and E. V. Koonin, Genome reduction as the dominant mode of evolution, Bioessays, 2013, 35, 829–837 CrossRef PubMed.
- H. K. Allen, J. Donato, H. H. Wang, K. A. Cloud-Hansen, J. Davies and J. Handelsman, Call of the wild: antibiotic resistance genes in natural environments, Nat. Rev. Microbiol., 2010, 8, 251–259 CrossRef CAS PubMed.
- H. Ochman, J. G. Lawrence and E. A. Groisman, Lateral gene transfer and the nature of bacterial innovation, Nature, 2000, 405, 299–304 CrossRef CAS PubMed.
- J. Davies and D. Davies, Origins and evolution of antibiotic resistance, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- A.-M. Hanssen and J. U. Ericson Sollid, SCCmec in staphylococci: genes on the move, FEMS Immunol. Med. Microbiol., 2006, 46, 8–20 CrossRef CAS PubMed.
- J. Maillard and C. Holliger, in Organohalide-Respiring Bacteria, ed. L. Adrian and F. E. Löffler, Springer, Berlin, 2016, pp. 153–168 Search PubMed.
- P. H. Wang, S. Q. Tang, K. Nemr, R. Flick, J. Yan, R. Mahadevan, A. F. Yakunin, F. E. Löffler and E. A. Edwards, Refined experimental annotation reveals conserved corrinoid autotrophy in chloroform-respiring Dehalobacter isolates, ISME J., 2017, 11, 626–640 CrossRef PubMed.
- Y. K. Wong, S. I. Holland, H. Ertan, M. Manefield and M. Lee, Isolation and characterization of Dehalobacter sp. strain UNSWDHB capable of chloroform and chlorinated ethane respiration, Environ. Microbiol., 2016, 18, 3092–3105 CrossRef CAS PubMed.
- S. Tang and E. A. Edwards, Identification of Dehalobacter reductive dehalogenases that catalyse dechlorination of chloroform, 1,1,1-trichloroethane and 1,1-dichloroethane, Philos. Trans. R. Soc., B, 2013, 368, 20120318 CrossRef PubMed.
- A. Grostern, W. W. Chan and E. A. Edwards, 1,1,1-trichloroethane and 1,1-dichloroethane reductive dechlorination kinetics and co-contaminant effects in a Dehalobacter-containing mixed culture, Environ. Sci. Technol., 2009, 43, 6799–6807 CrossRef CAS PubMed.
- B. Sun, B. M. Griffin, L. A.-d.-R. Héctor, S. A. Hashsham and J. M. Tiedje, Microbial Dehalorespiration with 1, 1, 1-Trichloroethane, Science, 2002, 298, 1023–1025 CrossRef CAS PubMed.
- C. Holliger, D. Hahn, H. Harmsen, W. Ludwig, W. Schumacher, B. Tindall, F. Vazquez, N. Weiss and A. J. B. Zehnder, Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration, Arch. Microbiol., 1998, 169, 313–321 CrossRef CAS PubMed.
- F. Maphosa, M. van Passel, W. De Vos and H. Smidt, Metagenome analysis reveals yet unexplored reductive dechlorinating potential of Dehalobacter sp. E1 growing in co-culture with Sedimentibacter sp., Environ. Microbiol. Rep., 2012, 4, 604–616 CAS.
- W. Qiao, F. Luo, L. Lomheim, E. E. Mack, S. Ye, J. Wu and E. A. Edwards, A Dehalogenimonas population respires 1,2,4-trichlorobenzene and dichlorobenzenes, Environ. Sci. Technol., 2018, 52, 13391–13398 CrossRef CAS PubMed.
- J. L. Nelson, J. M. Fung, H. Cadillo-Quiroz, X. Cheng and S. H. Zinder, A Role for Dehalobacter spp. in the Reductive Dehalogenation of Dichlorobenzenes and Monochlorobenzene, Environ. Sci. Technol., 2011, 45, 6806–6813 CrossRef CAS PubMed.
- L. A. Puentes Jacome, P.-H. Wang, Y. X. Li, O. Molenda, K. Krivushin, M. Ahsanul Islam and E. A. Edwards, Sustained dechlorination of vinyl chloride to ethene in Dehalococcoides-enriched cultures grown without addition of exogenous vitamins at low pH, Environ. Sci. Technol., 2019, 53, 11364–11374 CrossRef CAS PubMed.
- S. Wang, W. Zhang, K. L. Yang and J. He, Isolation and characterization of a novel Dehalobacter species strain TCP1 that reductively dechlorinates 2,4,6-trichlorophenol, Biodegradation, 2014, 25, 313–323 CrossRef CAS PubMed.
- N. Yoshida, L. Ye, D. Baba and A. Katayama, A novel Dehalobacter species is involved in extensive 4,5,6,7-tetrachlorophthalide dechlorination, Appl. Environ. Microbiol., 2009, 75, 2400 CrossRef CAS PubMed.
- S. Siddaramappa, J. F. Challacombe, S. F. Delano, L. D. Green, H. Daligault, D. Bruce, C. Detter, R. Tapia, S. Han, L. Goodwin, J. Han, T. Woyke, S. Pitluck, L. Pennacchio, M. Nolan, M. Land, Y. J. Chang, N. C. Kyrpides, G. Ovchinnikova, L. Hauser, A. Lapidus, J. Yan, K. S. Bowman, M. S. da Costa, F. A. Rainey and W. M. Moe, Complete genome sequence of Dehalogenimonas lykanthroporepellens type strain (BL-DC-9(T)) and comparison to “Dehalococcoides” strains, Stand. Genomic Sci., 2012, 6, 251–264 CrossRef PubMed.
- T. A. Key, D. P. Richmond, K. S. Bowman, Y. J. Cho, J. Chun, M. S. da Costa, F. A. Rainey and W. M. Moe, Genome sequence of the organohalide-respiring Dehalogenimonas alkenigignens type strain (IP3-3(T)), Stand. Genomic Sci., 2016, 11, 44 CrossRef PubMed.
- T. A. Key, K. S. Bowman, I. Lee, J. Chun, L. Albuquerque, M. S. da Costa, F. A. Rainey and W. M. Moe, Dehalogenimonas formicexedens sp. nov., a chlorinated alkane-respiring bacterium isolated from contaminated groundwater, Int. J. Syst. Evol. Microbiol., 2017, 67, 1366–1373 CrossRef CAS PubMed.
- Y. Yang, S. A. Higgins, J. Yan, B. Simsir, K. Chourey, R. Iyer, R. L. Hettich, B. Baldwin, D. M. Ogles and F. E. Löffler, Grape pomace compost harbors organohalide-respiring Dehalogenimonas species with novel reductive dehalogenase genes, ISME J., 2017, 11, 2767–2780 CrossRef CAS PubMed.
- J. A. Müller, B. M. Rosner, G. Von Abendroth, G. Meshulam-Simon, P. L. McCarty and A. M. Spormann, Molecular identification of the catabolic vinyl chloride reductase from Dehalococcodies sp. strain VS and its environmental distribution, Appl. Environ. Microbiol., 2004, 70, 4880–4888 CrossRef PubMed.
- X. Liang, O. Molenda, S. Tang and E. A. Edwards, Identity and substrate-specificity of reductive dehalogenases expressed in Dehalococcoides-containing enrichment cultures maintained on different chlorinated ethenes, Appl. Environ. Microbiol., 2015, 81, 4626–4633 CrossRef CAS PubMed.
- D. R. Johnson, E. L. Brodie, A. E. Hubbard, G. L. Andersen, S. H. Zinder and L. Alvarez-Cohen, Temporal transcriptomic microarray analysis of “Dehalococcoides ethenogenes” strain 195 during the transition into stationary phase, Appl. Environ. Microbiol., 2008, 74, 2864–2872 CrossRef CAS PubMed.
- J. K. Magnuson, M. F. Romine, D. R. Burris and M. T. Kingsley, Trichloroethene reductive dehalogenase from Dehalococcoides ethenogenes: sequence of tceA and substrate range characterization, Appl. Environ. Microbiol., 2000, 66, 5141–5147 CrossRef CAS PubMed.
- S. Tang, W. M. W. Chan, K. E. Fletcher, J. Seifert, X. Liang, F. E. Löffler, E. A. Edwards and L. Adrian, Functional characterizayion of reductive dehalogenases by using blue native polyacrylamide gel electrophoresis, Appl. Environ. Microbiol., 2013, 79, 974–981 CrossRef CAS PubMed.
- L. Adrian, J. Rahnenführer, J. Gobom and T. Hölscher, Identification of a chlorobenzene reductive dehalogenase in Dehalococcoides sp. strain CBDB1, Appl. Environ. Microbiol., 2007, 73, 7717–7724 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00605b |
| This journal is © The Royal Society of Chemistry 2020 |