
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAging induced changes in ice nucleation activity of combustion aerosol as determined by near edge X-ray absorption fine structure (NEXAFS) spectroscopy†
Fabian
Mahrt‡
 *a,
Peter A.
Alpert
b,
Jing
Dou
a,
Philippe
Grönquist
cd,
Pablo Corral
Arroyo§
b,
Markus
Ammann
b,
Ulrike
Lohmann
a and
Zamin A.
Kanji
*a
*a,
Peter A.
Alpert
b,
Jing
Dou
a,
Philippe
Grönquist
cd,
Pablo Corral
Arroyo§
b,
Markus
Ammann
b,
Ulrike
Lohmann
a and
Zamin A.
Kanji
*a
aDepartment of Environmental System Science, Institute for Atmospheric and Climate Science, ETH Zurich, 8092 Zurich, Switzerland. E-mail: mahrt@chem.ubc.ca; zamin.kanji@env.ethz.ch; Tel: +41 44 633 61 61
bLaboratory of Environmental Chemistry, Paul Scherrer Institute, 5232 Villigen, Switzerland
cDepartment of Civil, Environmental and Geomatic Engineering, Institute for Building Materials, ETH Zurich, 8093 Zurich, Switzerland
dDepartment of Functional Materials, Cellulose & Wood Materials, EMPA, 8600 Dubendorf, Switzerland
First published on 18th March 2020
Abstract
Fresh soot particles are generally hydrophobic, however, particle hydrophilicity can be increased through atmospheric aging processes. At present little is known on how particle chemical composition and hydrophilicity change upon atmospheric aging and associated uncertainties governing the ice cloud formation potential of soot. Here we sampled two propane flame soots referred to as brown and black soot, characterized as organic carbon rich and poor, respectively. We investigated how the ice nucleation activity of these particles changed through aging in water and aqueous acidic solutions, using a continuous flow diffusion chamber operated at cirrus cloud temperatures (T ≤ 233 K). Single aggregates of both unaged and aged soot were chemically characterized by scanning transmission X-ray microscopy and near edge X-ray absorption fine structure (STXM/NEXAFS) measurements. Particle wettability was determined through water sorption measurements. Unaged black and brown soot particles exhibited significantly different ice nucleation activities. Our experiments revealed significantly enhanced ice nucleation activity of the aged soot particles compared to the fresh samples, lowering the required relative humidities at which ice formation can take place at T = 218 K by up to 15% with respect to water (ΔRHi ≈ 25%). We observed an enhanced water uptake capacity for the aged compared to the unaged samples, which was more pronounced for the black soot. From these measurements we concluded that there is a change in ice nucleation mechanism when aging brown soot. Comparison of the NEXAFS spectra of unaged soot samples revealed a unique spectral feature around 287.5 eV in the case of black soot that was absent for the brown soot, indicative of carbon with hydroxyl functionalities. Comparison of the NEXAFS spectra of unaged and aged soot particles indicates changes in organic functional groups, and the aged spectra were found to be largely similar across soot types, with the exception of the water aged brown soot. Overall, we conclude that atmospheric aging is important to representatively assess the ice cloud formation activity of soot particles.
Environmental significanceSoot is an important anthropogenic pollutant known to impact human health and climate. Nevertheless, the aerosol–cloud interactions of soot particles (indirect climate effect) remains poorly constrained. In particular, the impact of atmospheric aging mechanisms on the ice nucleating ability of soot remains a key uncertainty due to changes in particle physicochemical properties. We investigate changes in the ice nucleating activity of two soot types following aging in aqueous environments, mimicking acidic conditions found in cloud and haze droplets. Aged soot particles reveal enhanced ice nucleating abilities comparable to that of mineral dust particles. Aged particles also show distinct chemical changes and enhanced hydrophilicity. Such changes will have implications for the aerosol–ice cloud interactions of soot particles. |
1. Introduction
Soot particles influence climate directly by absorbing and scattering shortwave radiation, as well as indirectly through cloud formation.1–3 Soot is mainly composed of elemental and organic carbon (OC). Elemental carbon (EC) denotes water-insoluble, refractory graphitic black carbon which strongly absorbs light in the visible and near-visible spectral regions.4 The OC fraction encompasses saturated, unsaturated and polyaromatic hydrocarbons (PAHs) along with other organic compounds.5 In addition, soot can contain (inorganic) impurities, depending on the combustion condition and source.6 Here, we explicitly include these compounds in the term soot, given their importance for the lifetimes and cloud-forming effects of the combustion particles. For instance, the heterogeneous ice nucleation ability of soot has been observed to be closely linked to its physicochemical particle properties.7,8 The extent to which soot properties are affected by atmospheric aging is still largely unknown. Aging can result in processes that affect particle hydrophilicity, including changes in particle morphology and in chemical surface functionalities induced through e.g. oxidation processes. While freshly emitted soot particles are usually hydrophobic,9 both the physical and chemical properties of the soot particles can be altered during atmospheric aging.10 The breadth of atmospheric aging covers a large variety of processes, including oxidation by atmospheric trace gases,11 photochemical reactions12 as well as condensation (coating) of semi-volatile material such as secondary organic aerosol (SOA) species13 and sulfuric acid (H2SO4) on the soot particles,14 changing the mixing state of the aerosol particles15 and in turn their radiative properties.16,17 Some studies have investigated the various consequences of atmospheric aging of soot particles for their susceptibility to interact with atmospheric water and their ability to serve as nucleation sites for water vapor, which is critical to quantify their cloud formation potential.18 For instance, initially hydrophobic soot aggregates can be transformed into more hydrophilic19 or even hygroscopic particles,20 in case the aging leads to an increase in the particle water-soluble mass fraction.6,21Water uptake on soot particles is determined through an interplay of particle physical properties (morphology) and chemistry. For soot particles with water-soluble material, water uptake can take place through absorption. Water molecules can also adsorb on hydrophilic (polar) surface sites with e.g. oxygenated surface functional groups,22,23 followed by the formation of water clusters that can freeze and cause macroscopic ice formation. For instance, Koehler et al.7 found both an increased water uptake capacity and ice nucleation activity after oxidizing thermal soot in mixtures of nitric and sulfuric acid. They attributed the enhanced ice nucleation to so-called adsorption freezing, where an increased number of hydrophilic functional groups on the particle surface allows for water adsorption and subsequent freezing of the adsorbate. In the same study, another soot type derived from an aircraft engine was found to be more hydrophilic but at the same time less ice active than the oxidized thermal soot. Koehler et al.7 argued that the availability of water soluble material on the aircraft soot causes a change in the water uptake mechanism and ultimately impacts the ice nucleation activity, with the aircraft soot taking up water hygroscopically (absorption). Within classical nucleation theory, which is commonly used to describe the heterogeneous ice formation on a particle surface, the particle hydrophilicity is usually accounted for by the contact angle, denoting a macroscopic parameter to describe particle wettability.24,25 Aging processes have been reported to decrease the contact angle, i.e. enhance the wettability of soot samples26 with consequences for their ice cloud formation potential. Finally, soot particle morphology can affect their water uptake capacity and ultimately ice formation.27 If the soot aggregate hosts mesopores with typical sizes of about dpore = 2–50 nm, water can be taken up into such cavities by capillary condensation.28,29 Following classical nucleation theory, pore water in mesopores with sizes comparable to the critical ice embryo can freeze homogeneously at temperatures below the homogeneous nucleation temperatures (235 K), forming ice through the pore condensation and freezing (PCF) mechanism.30–32 In fact, soot particles have been reported to form ice through PCF at cirrus temperatures.8,33
Considerable efforts to link the ice nucleation activity of soot particles to their physical and chemical properties have been made.7,34 In this study we expose soot particles for long time scales on the order of months, to study the effect of water and dilute acid exposure on ice nucleation. Particle physicochemical properties were observed concurrently while quantifying the ability of aged soot particles to nucleate ice in controlled laboratory experiments. We use soot derived from a commercial propane flame burner and the ice nucleation experiments are performed using the horizontal ice nucleation chamber (HINC).33,35 Two soot types were investigated that differ in their OC content, derived from different combustion conditions, in order to mimic different particle properties and sources. Aging of the soot particles is achieved by submersing them in pure water and aqueous H2SO4 to address the acidic environment frequently found in cloud droplets.36,37 The compositional changes in soot before and after aging are probed with scanning transmission X-ray microscopy coupled with near edge absorption fine structure (STXM/NEXAFS) following previous studies.38–44 We complement these experiments by water vapor sorption measurements to quantify the particle hydrophilicity, as has been done in several previous studies on carbonaceous aerosol.28,45–49 Drawing from these different measurements, we provide a comprehensive discussion of soot composition and water sorption and its link with the ability of aged soot to nucleate ice at upper tropospheric conditions.
2. Methods
2.1 Soot particle generation and aging
Soot particles were generated using a miniature combustion aerosol standard (miniCAST, Model 4200, Jing Ltd., Zollikofen, Switzerland). The miniCAST is a propane flame diffusion burner that allows generation of soot particles of different sizes and OC fractions by changing the combustion conditions.50–52 Here we chose to use soot derived from the miniCAST because it allows generation of particles in a stable and reproducible way, a premise to analyse aging induced changes in the particle physicochemical and ice nucleation properties. MiniCAST soot is frequently used as proxy for aircraft exhaust, when operating the burner under fuel-lean combustion conditions,53–55 whereas soot with higher OC content is more representative of particles emitted from e.g. biomass burning. In general, the content of (semi-volatile) organic material decreases with decreasing fuel/oxygen ratio,56 due to the higher combustion efficiency of oxygen rich flames, allowing most of the PAHs and hydrocarbons formed during the combustion process to become oxidized or transformed into soot. Hence, the types of organics and overall properties of the soot particles strongly depend on the combustion condition.50,54 Combustion conditions have also been reported to alter soot particle hydrophilicity with increasing hydrophilicity exhibited with decreasing fuel/oxygen ratio.57–59 By using fuel-lean and fuel-rich flame conditions Mahrt et al.33 generated and characterized soots with low and high OC fraction, termed mCASTbl and mCASTbr for miniCASTblack and brown, respectively, and the soots used here for aging experiments were generated using identical combustion conditions as Mahrt et al.33 They reported fractal dimensions of 1.86 and 2.31 for miniCASTbl and mCASTbr, respectively. While the slightly higher fractal dimension for mCASTbr suggest more round, i.e. more compact aggregates, compared to mCASTbl, the transmission electron microscopy analysis revealed an overall comparable aggregate structure for both soot types. Samples of each soot type were collected at the miniCAST outlet on quartz fiber filters (Tissuequartz filters, type 2500QAT-UP, Pall Corporation). Acidic suspensions (pH = 4) of soot in Milli-Q water (18.2 MΩ cm, pH = 5.8) and H2SO4 (95%, Sigma-Aldrich) were prepared, by scraping the soot off the filters with a metal spatula. Similarly, suspensions with soot only in Milli-Q water were prepared for comparison. Prior to chemical and ice nucleation analysis, the solutions were stored for two months under dark conditions and at constant temperature, T, of 277 K. This aging time was chosen to estimate the upper limit of magnitude of soot particle aging in aqueous solutions on their ice nucleation ability. Suspension of the soot particles was ensured through regular agitation of the samples during aging and particle generation from solution as described below. Unaged soot directly from the miniCAST exhaust was also characterized and tested for its ice nucleation activity. For these experiments, the miniCAST exhaust was diluted by a factor 10 using synthetic, VOC-filtered air in a dilution stage (Model VKL10, Palas), and fed to a 0.125 m3 stainless steel mixing chamber, from which soot particles were sampled for ice nucleation measurements. In the mixing chamber aggregate size increases through coagulation and aggregation, achieving a sufficiently high and stable number concentration of 400 nm soot aggregates for our ice nucleation measurements.2.2 Ice nucleation experiments
Aged soot was generated with a nebulizer (Model 8026, TSI Inc.) mounted on a magnetic stirrer to ensure a uniform suspension from which the soot was being aerosolized and subsequently passed through a diffusion drier. Particles with an electrical mobility diameter of dm = 400 nm were selected using a differential mobility analyzer (DMA, classifier 3080, column 3081, TSI Inc., aerosol to sheath flow ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7). Similarly, a DMA was used to sample the fresh, unaged soot particles directly from the mixing volume described above. Ice nucleation experiments were performed using HINC, a continuous flow diffusion chamber, described in detail elsewhere.33,35 Briefly, two ice coated parallel plates are cooled to different temperatures, so that a linear temperature gradient between the two horizontally aligned plates is established. This results in a water vapor supersaturation profile across the vertical extent of HINC. The parametrization of Murphy and Koop60 is used to define the relative humidity (RH) across the so-called aerosol lamina, the soot particles are exposed to within HINC. Particle residence time for the ice nucleation experiments in HINC was τ ≈ 10.4 s. An ice nucleation experiment in HINC is performed by slowly scanning RH (RHi < 3% min−1) by increasing the temperature gradient between the top and the bottom wall of HINC, but keeping the centerline temperature constant. The ice nucleation activity was quantified by measuring the activated fraction (AF) given as:
:7). Similarly, a DMA was used to sample the fresh, unaged soot particles directly from the mixing volume described above. Ice nucleation experiments were performed using HINC, a continuous flow diffusion chamber, described in detail elsewhere.33,35 Briefly, two ice coated parallel plates are cooled to different temperatures, so that a linear temperature gradient between the two horizontally aligned plates is established. This results in a water vapor supersaturation profile across the vertical extent of HINC. The parametrization of Murphy and Koop60 is used to define the relative humidity (RH) across the so-called aerosol lamina, the soot particles are exposed to within HINC. Particle residence time for the ice nucleation experiments in HINC was τ ≈ 10.4 s. An ice nucleation experiment in HINC is performed by slowly scanning RH (RHi < 3% min−1) by increasing the temperature gradient between the top and the bottom wall of HINC, but keeping the centerline temperature constant. The ice nucleation activity was quantified by measuring the activated fraction (AF) given as: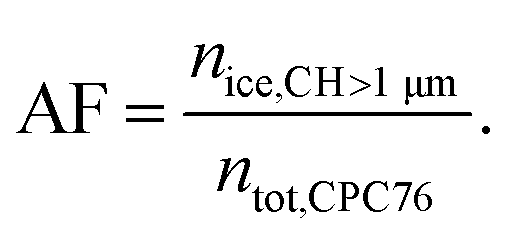 | (1) |
:10, and evaluated for a maximum RHw = 105% reached at the end of our RH-scans. The ice nucleation results presented here represent mean values over two independent RH-scans at each T and for each soot sample.
2.3 Soot particle collection
For each soot sample, size selected particles were collected on standard Cu grids with a single-coated continuous carbon film (Quantifoil Micro Tools GmbH, Grosslöbichau, Germany), using the Zurich electron microscope impactor (ZEMI). ZEMI is a rotating drum impactor that allows independent control of aerosol flow rate, sampling duration and impaction distance.33 Impacted particles were then subjected to chemical analysis as described next.2.4 Chemical characterization by STXM/NEXAFS
Chemical analysis of individual particles was achieved by STXM/NEXAFS performed at the PolLux facility of the Swiss Light Source, located at the Paul Scherrer Institute, Villigen, Switzerland.61 Descriptions of the STXM/NEXAFS technique are well documented41,62 and are only briefly described here. Monochromatic X-rays are focused on a fixed position on the sample. Scanning the sample position and measuring the transmitted photons through the particles on the sample generated an X-ray optical density, OD, image. According to Beer–Lambert's law: | (2) |
Using soft X-rays (λ ≈ 1–10 nm), absorption peaks arise from electronic transitions 1s → π* and 1s → σ*, where π* excitations denote transitions at relatively low energies into orbitals of the lowest unoccupied molecular orbitals (conduction band), and σ* transitions take place at higher energies denoting excitations of core electrons to energy levels above the Fermi level, i.e. above the ionization threshold, and take place at relatively high energies.65 X-ray absorption is sensitive to the chemical bonding environment of the absorbing atom according to the optical dipole selection rules resulting in characteristic spectral features.62,65 In case multiple electronic transitions can occur at a given energy, the resulting absorption denotes the total of all potential contributions. In this study, our energy resolution was E/dE = 1500, which translates to the ability to differentiate between peaks separated by 0.2 eV. We acquired NEXAFS spectra at the C K-edge (278–320 eV) and O K-edge (525–550 eV) to identify carbon and oxygen functionalities of our soot samples.41 An energy offset calibration was determined by matching the well defined transition of polystyrene in literature66–68 to measurement of polystyrene latex spheres. To compare STXM/NEXAFS results between different particles and samples, spectra were background subtracted (mean absorption between 278–282 eV in the case of C K-edge) and normalized to OD integrated between 305–320 eV following Takahama et al.69 We used the peak height ratio, r = Iπ*/Iσ* = OD285 eV/OD292 eV, from the normalized and background subtracted spectra to estimate the graphitic character of a soot in a semi quantitative manner, following previous work.39,64,70–72 Higher values of r indicate a greater degree of graphitization, with the highest value of r for highly oriented pyrolytic graphite.40 We carefully assessed beam damage which was successfully avoided as described in ESI.†
2.5 Characterization of particle hydrophilicity and OH accessibility
The hydrophilicity of the bulk soot samples over the RHw range of 0–98% was characterized by dynamic vapor sorption (DVS, Model Advantage ET 1, Surface Measurement Systems Ltd., London, UK). The amount of water taken up by the soot at a given RHw is equivalent to the relative mass change of the sample, Δm, given by the difference in weight of the sample at any RHw probed and the sample dry weight. DVS is a gravimetric technique with a mass sensitivity of 0.1 μg and an absolute RHw accuracy of ±0.5%. This RHw uncertainty dominates the uncertainties in Δm, when using the DVS to test sorption isotherms, i.e. determining the mass change when scanning RHw, resulting in maximal uncertainties in Δm of ±0.75% for our samples. In our experiments each soot sample was dried at ambient pressure and at 313 K for 400 min within the DVS cell in order to out-gas any pre-adsorbed water in a pure N2 atmosphere (5.0 grade), prior to the water uptake and loss measurements, while constantly monitoring the sample mass. RHw was increased and decreased in increments of 5 and 3% between 0–80 and 80–98%, respectively, for both adsorption and desorption branches. Water mass was quantified at quasi-equilibrated RHw conditions, defined as when the mass rate of change was less than 0.0005% min−1 over the course of 10 min or after a maximum time of 1000 min per RHw step. For the aged soot samples in solutions, the liquid was evaporated in an oven at a constant temperature of 313 K. The residual soot material was dried in the DVS cell prior to water vapor sorption measurement for a period of 400 min, similar to the unaged samples, as described above. DVS measurements also allow to gravimetrically determine the accessibility of OH functional groups present on the soot samples through protium–deuterium exchange (see ESI Sect. S1†).73
3. Results and discussion
3.1 Unaged mCASTbl and mCASTbr
The AF as a function of RHw for the unaged soot samples at temperatures of 233 and 218 K is shown in Fig. 1. At T = 233 K, water supersaturation was required for both mCASTbl and mCASTbr to form ice. It is unlikely that heterogeneous freezing of unaged soot particles would occur at this or a higher temperature.75 At T = 218 K, the mCASTbr also required water saturation to nucleate ice. The AF of the unaged mCASTbl increased above the background level close to homogeneous freezing conditions, indicated by the grey shading in Fig. 1b. This suggests water uptake occured by the mCASTbl, followed by homogeneous freezing. Thermogravimetric analysis by Mahrt et al.33 however showed soluble material to be largely absent on this soot. Consequently, we interpret the observed macroscopic ice formation to be a deposition24 (adsorption freezing)7 or a PCF30–32,76–78 mechanism for this soot type. Furthermore, the tendency of the mCASTbl to have higher AF compared to the mCASTbr is consistent with previous findings of the ice nucleation ability of propane flame soot to decrease with increasing OC fraction.33,79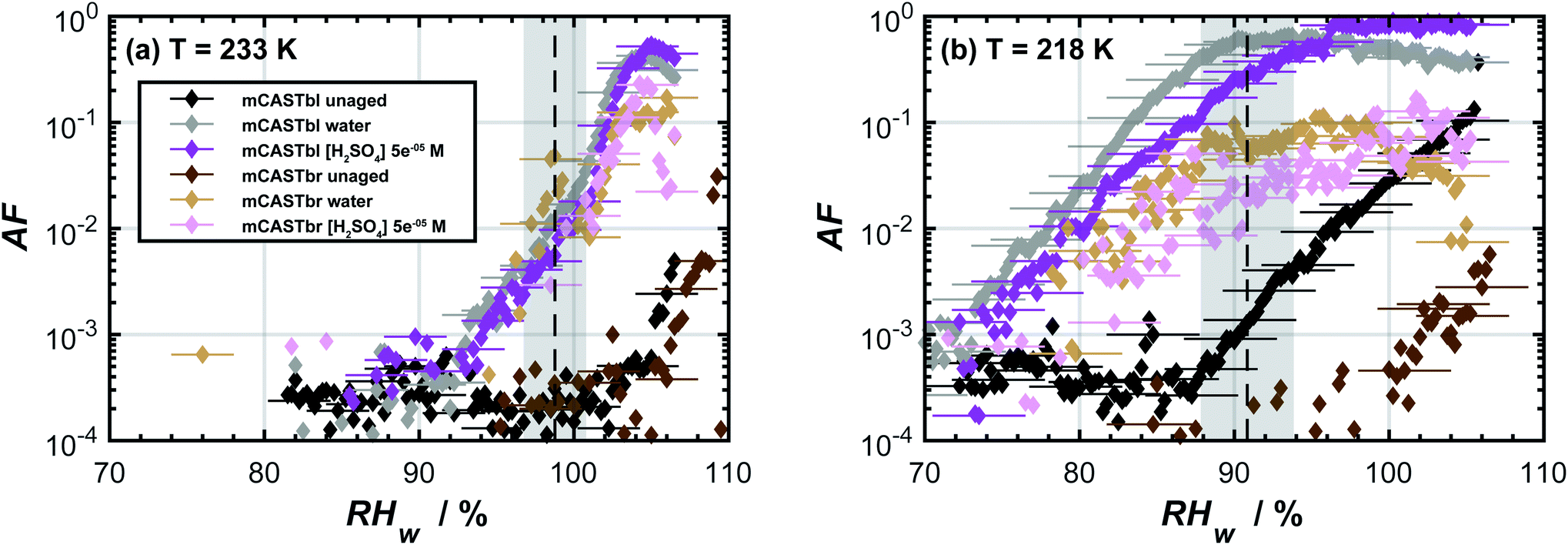 | ||
| Fig. 1 Ice nucleation activity of dm = 400 nm soot particles of mCASTbl and mCASTbr, showing the activated fraction (AF) as a function of relative humidity with respect to water (RHw) at temperatures of (a) 233 K and (b) 218 K. The dashed, black lines represent homogeneous freezing conditions according to Koop et al.74 Gray shaded regions indicate the calculated RHw variation across the aerosol lamina in the horizontal ice nucleation chamber, resulting from T uncertainties of 0.1 K and for an aerosol to sheath flow ratio of 1:10.33 Uncertainties in RHw are also indicated for every fifth data point. Data of unaged mCASTbl is taken from Mahrt et al.27 | ||
The difference in ice nucleation ability between mCASTbl and mCASTbr is also supported by the clear difference in the hydrophilicity of mCASTbl and mCASTbr, with the latter being significantly less hydrophilic, as observed in our DVS measurements (see Fig. 2). At RHw = 98%, the highest humidity of our sorption measurements, Δm = 10 ± 0.75% and 2 ± 0.75% for mCASTbl and mCASTbr, respectively. Throughout the whole RH range the difference in hydrophilicity is manifested by a separation of the water sorption isotherms of the mCASTbl and mCASTbr into two main sets of sorption curves, suggesting a different mechanism for the interaction with water vapor between these soot types. The inset in Fig. 2 shows the hydrophilicity at low RH more clearly. Previous studies reported water uptake at low RHw (<25%) to be mainly controlled by chemisorption.80 However, given the absence of a difference in the adsorption and desorption branch (hysteresis) at these RH conditions, we attribute the water uptake to reversible adsorption through van-der-Waals forces between the surface sites and water. This overall water uptake being largely reversible is consistent with previous studies on water uptake on soot.9,81 However, at RHw > 80% the isotherms of the mCASTbl samples clearly reveal a hysteresis. This sorption hysteresis is connected to capillary condensation of water into the cavities of the porous soot agglomerates, and results from the existence of thermodynamically metastable states of the condensate within the pores. While a perfectly reversible water uptake by adsorption on the soot particles would result in the water uptake and loss curves to overlap, the delay of the desorption branch can result from retention of the pore water in narrow confinements owing to the fact that the confinement has a lower saturation vapor pressure than the nominal bulk water due to the (inverse) Kelvin effect.31 In soot agglomerates with complex pore structure some pores might have access to the surrounding gas phase only through narrow necks (confinements), with the desorption vapor pressure depending on the size of these necks. Hence, the large pore body remains filled with water, until the narrow necks empty at lower relative humidities.29,31 The absence of a hysteresis for the unaged mCASTbr suggests the absence of pores that can fill with water. On the contrary, Mahrt et al.33 used transmission electron microscopy and revealed a general porous structure of aggregates from the same soot type, resulting from sintering of the primary spherules. To explain these observations, we interpret the absence of the hysteresis for mCASTbr here to result from water unable to penetrate into pores covered by hydrophobic moieties.
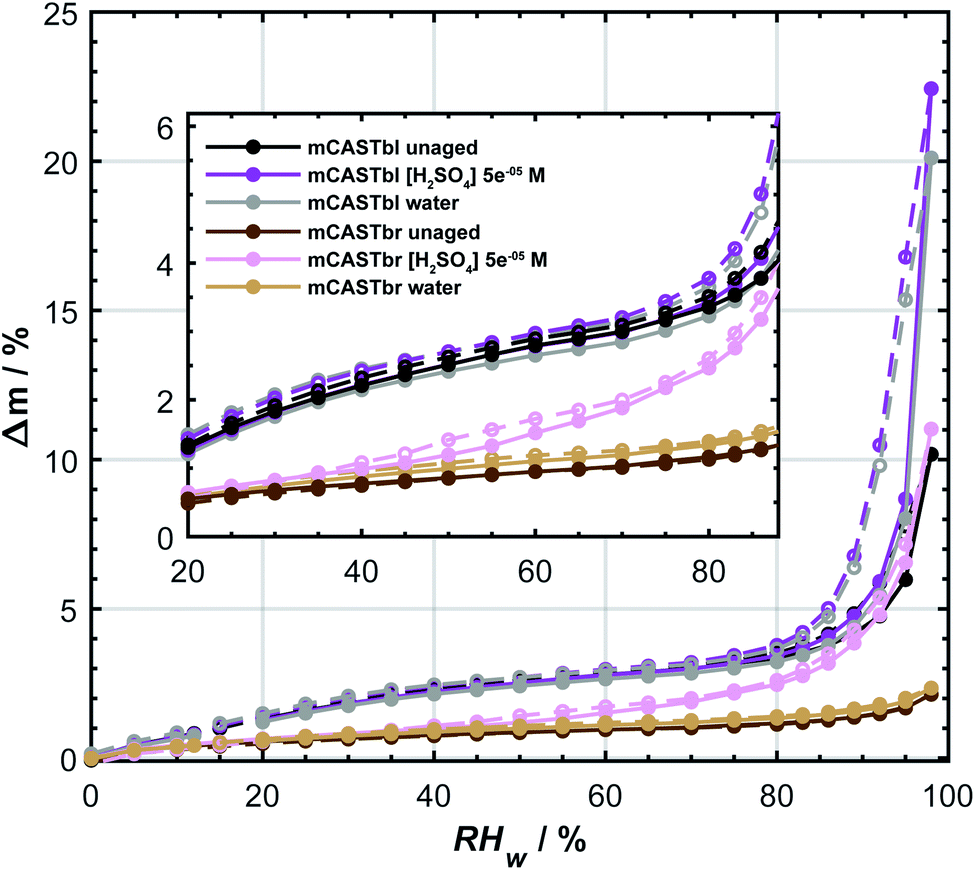 | ||
| Fig. 2 Water uptake (solid lines, filled symbols) and loss (dashed lines, open symbols) isotherms given as relative sample mass change, Δm, as a function of relative humidity with respect to water (RHw). Sorption isotherms were measured by dynamic vapor sorption at T = 298 K. All data points represent water uptake and loss at quasi-equilibrated RHw conditions and the difference between the adsorption and desorption branch at a given RHw level denotes the hysteresis. The curves are to guide the eye. Inset shows enlargement for the 20 ≤ RHw ≤ 88%. Data of unaged mCASTbl is taken from Mahrt et al.27 and uncertainty in Δm is ±0.75%. | ||
The NEXAFS spectra of both soot samples show strong absorption in the range between 284.9–285.5 eV (“graphitic peak”) and 292 eV (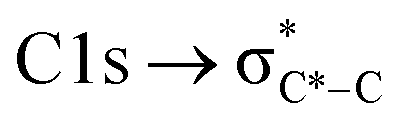 and “exciton peak”), which are identifying features for soot, denoting the
and “exciton peak”), which are identifying features for soot, denoting the 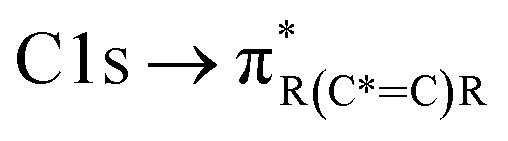 and
and 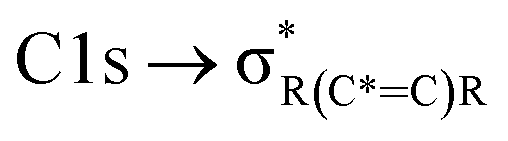 , respectively (see Fig. 3a and d and S3 in ESI†). These absorption peaks have been reported previously for other soot types from different sources.40 For instance, during the Carbonaceous Aerosols and Radiative Effects Study (CARES), an absorption peak at 285.4 eV was used as a tracer for particles dominated by EC or soot,82 suggesting that miniCAST particles can be a valid proxy for atmospheric soot. Despite this similarity in the overall spectral features, there are distinct differences between mCASTbl and mCASTbr, which might contribute to their different ice nucleation activity. For instance, it should be noticed that all C*
, respectively (see Fig. 3a and d and S3 in ESI†). These absorption peaks have been reported previously for other soot types from different sources.40 For instance, during the Carbonaceous Aerosols and Radiative Effects Study (CARES), an absorption peak at 285.4 eV was used as a tracer for particles dominated by EC or soot,82 suggesting that miniCAST particles can be a valid proxy for atmospheric soot. Despite this similarity in the overall spectral features, there are distinct differences between mCASTbl and mCASTbr, which might contribute to their different ice nucleation activity. For instance, it should be noticed that all C*![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peaks for mCASTbr are similarly around 285.0 eV while those of mCASTbl are at 285.4 eV (see Fig. 3a and d), revealing the chemical difference between both miniCAST samples. The larger contribution of the C*C bond (284.9–285.5 eV) in the case of mCASTbr compared to mCASTbl, confirms the less complete combustion (lower fraction of unsaturated carbons) of this soot type. The broadening (apparent shift) in peak absorption energy from 285.4 eV to 285.0 eV in the case of mCASTbr is evidence for the presence of aromatic functionalities. The intensity shoulder at the low energy side of the 285 eV peak, most clearly visible for mCASTbr, can be attributed to the resonance transition of the aromatic carbon atoms in benzoquinone
C peaks for mCASTbr are similarly around 285.0 eV while those of mCASTbl are at 285.4 eV (see Fig. 3a and d), revealing the chemical difference between both miniCAST samples. The larger contribution of the C*C bond (284.9–285.5 eV) in the case of mCASTbr compared to mCASTbl, confirms the less complete combustion (lower fraction of unsaturated carbons) of this soot type. The broadening (apparent shift) in peak absorption energy from 285.4 eV to 285.0 eV in the case of mCASTbr is evidence for the presence of aromatic functionalities. The intensity shoulder at the low energy side of the 285 eV peak, most clearly visible for mCASTbr, can be attributed to the resonance transition of the aromatic carbon atoms in benzoquinone 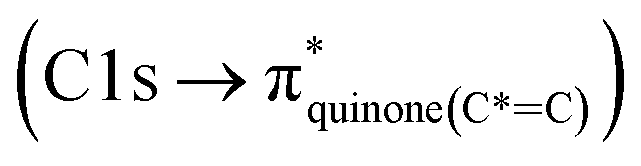 ,40 and has previously been reported for comparable miniCAST soots investigated by Ouf et al.83 and Zelenay et al.84 This high aromatic functionalities are in-line with the higher OC of the mCASTbr, which we interpret to cause the reduced ice nucleation ability of this sample compared to the mCASTbl. Absorption features associated with (resonant) electronic transitions in oxidized carbon or aliphatic carbon appear in the energy range 284–292 eV, i.e. in the valley between the graphitic and the
,40 and has previously been reported for comparable miniCAST soots investigated by Ouf et al.83 and Zelenay et al.84 This high aromatic functionalities are in-line with the higher OC of the mCASTbr, which we interpret to cause the reduced ice nucleation ability of this sample compared to the mCASTbl. Absorption features associated with (resonant) electronic transitions in oxidized carbon or aliphatic carbon appear in the energy range 284–292 eV, i.e. in the valley between the graphitic and the 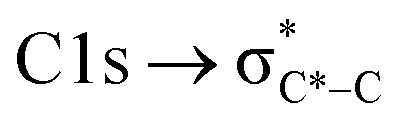 peak. For instance, in the range between 285.8–286.4 eV absorption can result from phenol functionalities
peak. For instance, in the range between 285.8–286.4 eV absorption can result from phenol functionalities 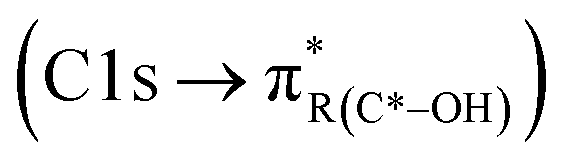 or ketone groups
or ketone groups 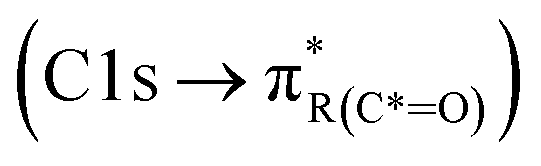 ,85 and carboxylic functionalities in atmospheric soot samples appear at 288.5 eV
,85 and carboxylic functionalities in atmospheric soot samples appear at 288.5 eV  .41
.41
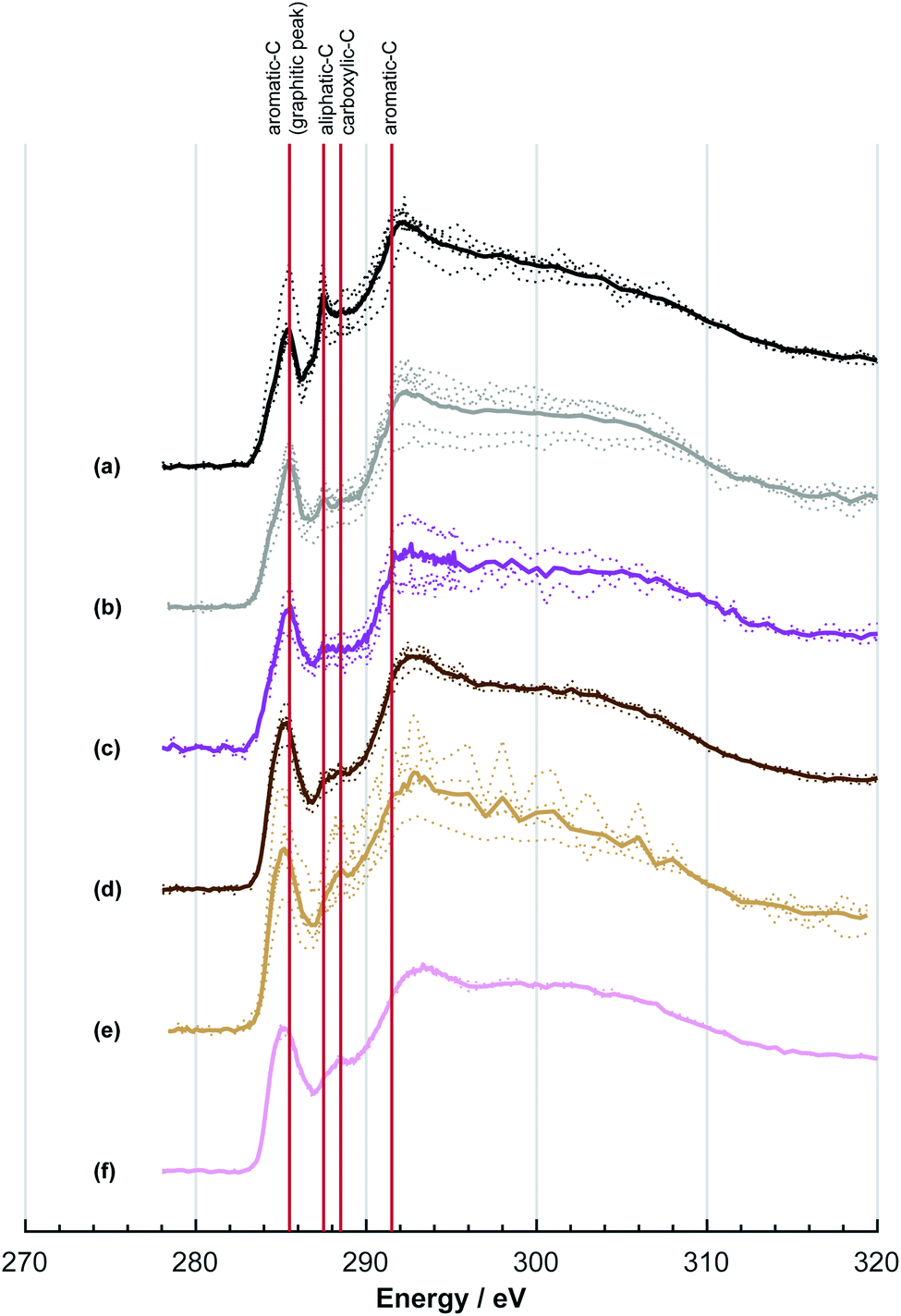 | ||
| Fig. 3 Near edge absorption fine structure (NEXAFS) spectra of (a) mCASTbl, (b) water aged mCASTbl, (c) acid aged mCASTbl, (d) mCASTbr, (e) water aged mCASTbr and (f) acid aged mCASTbr soot particles, measured at the carbon K-edge. All spectra have been corrected for pre-edge background absorption intensities and normalized to the total carbon content. Spectra are shifted vertically for clarity and better comparability. Solid colored lines denote averaged spectra and dotted lines spectra of individual particles of a given soot type. | ||
A distinct and sharp peak was observed at 287.5 eV for mCASTbl, but not for mCASTbr (see Fig. 3). Absorption at 287.5 eV can be associated with a variety of peaks, namely aliphatic carbon or hydroxyl, ketone and quinone functionalities taking into account our energy accuracy. These peaks overlap, rendering peak assignment difficult. However, oxygen NEXAFS spectra of the mCASTbl show a sharp peak at 538 eV, suggesting the presence of O–C functionalities as noted above, but lacking carbonyl or ketone structures (absorption around 531 eV, see Fig. S2a in ESI†). This in turn may be indicative of the 287.5 eV peaks in the carbon NEXAFS spectra to preliminarily result from OH functionalities, further supported by our protium–deuterium exchange measurements that revealed a higher amount of OH on the mCASTbl compared to the aged mCASTbl (see Table S1 in ESI;† see Sect. 3.2 for further analysis of the 287.5 eV peak). The availability of such OH functionalities may also help to explain the observed differences in ice nucleation ability between mCASTbl and mCASTbr.
3.2 Aged mCASTbl
The aged mCASTbl soot samples show a vastly different ice nucleation activity compared to the mCASTbl, with higher AF than for the unaged particles for the same experimental conditions. Just below homogeneous freezing at T = 233 K and RHw = 95%, the AF for aged mCASTbl is about one order of magnitude higher than for the unaged particles, irrespective of whether particles were aged in water or aqueous H2SO4. At T = 218 K, the AF for mCASTbl aged in water increased from about 10−4 to 10−1, when RHw increased from 75% to 88%. Hence, just before the onset of homogeneous ice nucleation (RHw ≈ 88%, grey shading), the AF of the water aged mCASTbl was about three orders of magnitude higher than for the unaged mCASTbl sample. The AF of mCASTbl aged in aqueous H2SO4 showed a similar trend T = 218 K and over the same RH range with a similar enhancement compared to those aged in water, even though the AF values reached at RHw = 88% are slightly lower.The enhanced ice nucleation could result from a change in physical or chemical particle properties, or a combination of both. Our DVS results show an increased amount of water uptake by the aged mCASTbl samples as compared to the unaged mCASTbl for RHw ≥ 92%. For the water aged mCASTbl, there is almost a twofold increase in water uptake. For the acid aged mCASTbl the amount of water being taken up at RHw = 98% is approximately a factor 2.2 higher than for the unaged mCASTbl, revealing an increased hydrophilicity of the aged samples. Yet, the DVS step at RHw = 98% for both the water and acid aged mCASTbl lasted for 1000 min, i.e. as long as the maximum time allowed per RHw level. Therefore, the amount of water taken up by these samples at RHw = 98% represent lower limits. More importantly, the water sorption isotherms of the aged mCASTbl samples both reveal a much stronger hysteresis compared to the unaged sample. The strengthening of this hysteresis indicates an increase in mesopore volume for the aged samples. An increase in mesopore volume is consistent with the observed enhanced ice nucleation ability of the aged samples, which we interpret as an enhanced ice formation due to PCF. The increase in pore water available to initiate ice formation via PCF can emanate from both physical and chemical changes of the aerosol and specifically pore properties upon aging.32 The aging in aqueous solutions likely caused a restructuring in the soot particle morphology, similar to the compaction of soot particles observed during cloud processing in other studies,27,86 that can contribute to an overall increase in the number of mesopores present and hence mesopore volume. At the same time, lowering the soot-water contact angle of the pores present could increase the mesopore volume accessible for water to condense at a lower RHw (compared to unaged soot) and hence PCF to occur even in the absence of any morphological change. The observed increase in pore water volume is likely a congruent combination of these effects.
Considering the NEXAFS spectra to assess chemical changes upon aging reveals the absence of an absorption peak at 287.5 eV in both the water and acid aged mCASTbl samples which coincide with the enhanced ice nucleation ability of these samples. This suggests that removal of the compound associated with the 287.5 eV peak, that we interpret to result from OH functionalities (see Sect. 3.1), and that is present on mCASTbl, may have allowed for the increased ice nucleation ability of the aged samples. In fact, a decrease in OH groups on the soot surface and a congruent increase in ice nucleation ability, is consistent with the result of Lupi and Molinero.87 They reported an increase in freezing temperature with a decrease in hydrophilicity in the form of an increase in the spacing between OH groups on an OH-decorated graphitic surface, using molecular dynamic simulations. A decrease of OH functionality upon aging is further supported by our protium–deuterium exchange measurements and also by the increased degree of graphitization for the aged samples (see Table S1 in ESI†).
A closer look at the results reveals that the ice nucleation in the aged mCASTbl samples proceed at higher RHw at T = 233 K than at T = 218 K. At T = 233 K and RHw = 95%, the ice saturation ratio is Si ≈ 1.4 and almost identical to 218 K and 85% RHw (Si ≈ 1.42). However, the latter had orders of magnitude greater values of AF. We believe this may be due to two effects. First, as T decreases holding RHi constant, the size of a critical ice nucleus decreases causing ice nucleation to be more probable.24,25 Following PCF, smaller pores can host the ice germ and allow for homogeneous ice nucleation within them, increasing the freezing probability.31 Second, pores that are filled with water at lower RHw values experience a stronger negative pressure (tension of the water meniscus) compared to pores that fill at RHw close to water saturation. The negative pressure increases the ice nucleation rates.88 The recent study by Marcolli88 reported that close to the homogeneous nucleation temperature, mesopores fill with water only very close to water saturation, causing the pore water to experience almost ambient pressure (no negative pressure). Consequently, the homogeneous nucleation rates are too low to cause freezing of the pore water on the timescale of the particle residence within HINC. Specifically, Marcolli88 showed that at T = 233 K pore water in a typical cylindrical mesopore of 3.3 nm in diameter takes about a day to freeze homogeneously. This likely explains the observed weak enhancement in ice nucleation ability of the aged mCASTbl samples at T = 233 K (see Fig. 1).
3.3 Aged mCASTbr
The effect of aging on the ice nucleation ability of mCASTbr is qualitatively similar to that of mCASTbl. For instance, at T = 218 K, the AF for mCASTbr aged in water increased from about 10−4 to 10−2, when RHw increased from 75% to 88%, resulting in an AF of two orders of magnitude higher just before the onset of homogeneous ice nucleation compared to the mCASTbr. The ice nucleation ability of the acid aged mCASTbr sample follows a similar trend. The mCASTbr particles nucleated ice only after bulk water saturation was reached whereas the aged samples nucleated ice at RHw significantly (≈10%) below water saturation, as shown in Fig. 1. This observation suggests that mCASTbr and the aged mCASTbr samples nucleate ice via different ice formation mechanisms.Despite the similarity in ice nucleation ability, the acid and water aged mCASTbr show vastly different hydrophilicity in the DVS experiments (see Fig. 2). This suggest that particle hydrophilicity alone is not a sufficient predictor for the ice nucleation activity, consistent with earlier findings,7,87 but rather indicates that relative sample hydrophilicity and other factors such as water uptake mechanism, ordering of the liquid water on the soot surface, spatial distribution of surface functional groups, and soot nano structure have to be considered in order to assess the ice nucleation potential and mechanism.87,89 Aging the mCASTbr in water results only in a marginal increase in water uptake capacity compared to the unaged sample. The absence of any hysteresis and the weak mass change for the water aged mCASTbr even at RHw = 98% is indicative of OC that is water-insoluble, blocking the pores on this soot type and preventing water uptake by capillary condensation. The presence of a weak hysteresis for the acid aged mCASTbr on the contrary suggests the presence of some mesopores, where water can be taken up, even though we note that the hysteresis for the acid aged mCASTbr is within our uncertainty of Δm (see Fig. 2). One possible explanation would be that the OC fraction gets largely dissolved/reacted in the acidic solutions (overall decrease in absorbance, see below), suggesting that the hydrophilic sites are associated with the EC skeleton of the soot particles. This would be consistent with Han et al.,59 who found no difference in hydrophilicity before and after heating (removing the OC fraction) of n-hexane soot. Given the higher hydrophilicity (and ice nucleation activity) of the mCASTbl compared to the mCASTbr, one might suppose that the hydrophilic sites of the miniCAST soots are indeed associated with the EC and not the OC fraction. Similarly, removal of the (hydrophobic) organic material, blocking the interaction of water molecules with the active sites and pores on the soot, could cause the enhanced water uptake of the acid aged mCASTbr compared to the unaged sample. This would be consistent with the findings of Persiantseva et al.,46 who report a lower water uptake capacity for kerosene soot in comparison to kerosene soot that was heated and out gassed, which was attributed to blocking of pores by organic material. Removal of some organic material is further supported by the increase in the degree of graphitization upon aging, indicating removal of non-graphitic material (see Table S1 in ESI†). However, a complete loss of all OC from the acid aged mCASTbr is unlikely, also for the scenario of a soot particle immersed in a haze or cloud droplet. While partitioning of fragmented, more volatile organics and reaction products from the condensed phase into the gas phase would contribute to loss of the OC, some dissolved organic material would remain on the soot aggregates upon evaporation of the liquid phase. A possible reason for the increased water uptake of the acid aged mCASTbr could be that the aged, oxidized organics cause a lowering in the soot water contact angle and/or that pores become accessible upon redistribution of the aged organic material, following the dissolution in the aqueous solution and evaporation of the aqueous and volatile components from the aerosol phase. Both processes would cause the pores on the aggregates to become more accessible for capillary condensation of water and in turn enhance the probability of ice formation via PCF. Nevertheless, given that the weak hysteresis is within our uncertainties in Δm, we conclude that the increased water uptake of the acid aged mCASTbr compared to the unaged sample results mainly from a decrease in contact angle. We can, however, not exclude that sulfuric acid residuals simply cause the initial water uptake, followed by capillary condensation and a dynamic increase in mesopore volume, i.e. water uptake through a positive feedback.
The NEXAFS results of the water and acid aged mCASTbr samples exhibit different spectral features (see Fig. 3e and f). The NEXAFS spectrum of the water aged mCASTbr indicates an overall increase in oxygenated functionalities compared to the mCASTbr. This includes phenolic functionalities (289.3–289.5 eV) and absorption in the region typical for carboxylic transitions (288.2–288.6 eV, see Fig. 3e). This is in-line with the slightly enhanced water uptake capacity of the water aged mCASTbr compared to the unaged sample (see above) and consistent with previous findings of Zelenay et al.,90 who reported an increased water uptake capacity for photochemically aged diesel soot particles and those derived from wood burning and attributed this to an increased amount of phenolic and carboxylic functionalities based on STXM/NEXAFS measurements. The trend towards overall higher absorbency of the water aged mCASTbr relative to the mCASTbr is also visible at the 1s → πR(C*C)R transition at 285.5 eV and the peak at 286.1 eV, that can also be associated with unsaturated carbon.91 With the exception of the increase in unsaturated carbons, we attribute the overall enhanced transitions in the energy range between 285.5–291 eV to the OC fraction associated with the mCASTbr to become more oxidized during water aging. The acid aged mCASTbr particles on the contrary show a general reduction in absorption compared to the mCASTbr, but overall do not indicate any bulk compositional change. This might be an indication that the stronger oxidation within the acidic environments largely removes the OC from the soot particles, as also suggested by our DVS results. Overall, the acid aged sample has a slightly higher O:C ratio compared to the mCASTbr sample (see Table S2 in ESI†). However, it is important to note, that it is likely that the oxygen in sulfate significantly contributes to the absorption in the oxygen spectra and thus discerning peak assignments using O–K edge NEXAFS for this sample are not discussed.
Overall, it remains inconclusive why the acid and water aged mCASTbr show different spectral features and water uptake abilities (see Fig. 2 and 3), while exhibiting similar ice nucleation properties (see Fig. 1). One way to reconcile the ice nucleation activity, chemical functionalities and water uptake capacities is to consider different ice nucleation mechanisms, as mentioned above, and to view the ice nucleation of water aged mCASTbr as a classical deposition nucleation process (the absence of PCF), where water vapor is directly deposited on the soot and forming the ice phase, whereas the acid aged mCASTbr can nucleate ice via PCF. In this regard, the enhancement of the ice nucleation ability of the water aged mCASTbr could be attributed to a lower contact angle compared to the mCASTbr. This interpretation would be consistent with the findings of Mahrt et al.,33 who find no PCF for the mCASTbr soot and in-line with the absence of a hysteresis in the DVS measurements of the water aged mCASTbr.
4. Atmospheric implications
The observed response of the ice nucleating ability of propane flame soot to aging in aqueous solutions suggests important modifications to the intrinsic ice nucleation activity (an enhancement) of combustion particles that should be considered when predicting the ice cloud-forming ability and associated radiative forcing.92 Aging in acidic aqueous environments can take place in fog and haze droplets forming on soot particles in polluted industrial environments.93,94 Such aqueous aging is atmospherically relevant,95 given that aerosol particles are involved in about three cloud cycles on average during their atmospheric lifetime,96 where they can be immersed into cloud droplets and undergo aging similar to that investigated here. For instance, Mahrt et al.27 reported a significant enhancement in soot ice nucleation ability after inclusion of soot particles in a single cloud cycle and identified the requirement of hydrometeor formation on the particles to largely contribute to this enhancement.The aging processes investigated here increase the range over which soot particles can act as INP to lower ice supersaturations at a given temperature. For instance, at T = 218 K the ice nucleation onset of the soot particles aged in aqueous solutions was found to be around RHw = 75–85%, i.e. significantly below homogeneous freezing conditions, suggesting that aged soot particles can impact ice cloud properties at upper tropospheric conditions.97 The consequence is that soot particles may become an important source of INPs after undergoing aging in liquid clouds. The ice nucleation activities of the aged soot observed here are comparable to those of mineral dust, which globally constitutes the largest mass fraction of aerosol particles98 and is generally regarded to be the predominant INP at these conditions.99 For instance, Welti et al.100 have reported a similar RHw = 78% as in our study to be required for 400 nm kaolinite particles to nucleate ice at T = 218 K. Apart from aging time and particle physicochemical properties, the hydrophilicity of soot can depend on environmental conditions other than RH. For instance, our water uptake measurements were performed at T = 298 K. However, the number of water molecules that could be adsorbed on soot particles was observed to increase with decreasing temperature.9,101 Hydrophilicity changes with temperature can have potential implications for ice cloud formation ability. That is, the reported water uptake capacity at 298 K in this work denotes a lower limit, given the lower temperatures within the upper troposphere, where cirrus cloud formation takes place.
5. Limitations
Soot particles are prone to atmospheric aging processes due to their relatively long atmospheric lifetime of several days (τ ≈ 5–7 days),102–104 but certainly the timescale of atmospheric aging can be much shorter than that.105,106 While soot particles in the upper troposphere can have lifetimes much longer than noted above, we point out the aging time scale investigated here is beyond the typical lifetime of atmospheric combustion particles, when considering the scenario of aqueous aging. Therefore, our results present an upper limit of the effect of soot aging in aqueous environments on the ice nucleation ability of these particles. Hence, while our results demonstrate the susceptibility of soot ice nucleation activity to the atmospheric aging processes studied here, whether the effect of aged soot particles on ice nucleation would be different if the ageing was shorter (e.g., 1 day, or 1 month) is beyond the scope of this work, but certainly a relevant question for future studies to address. In addition, our results are limited to the aging types investigated here. For instance, aging in aqueous particles of different pH or chemical composition, can change the particle chemistry. In addition, we recognize that realistic atmospheric aging is further complicated by other factors such as multiphase chemistry involving gas to particle partitioning and photochemical processes in the cloud water and/or aqueous aerosol phase, largely determining the aqueous chemistry of organic aqueous species, that can affect the aerosol aging107,108 and associated ice nucleation properties.109The largest challenge remains to further disentangle the contribution of changes from the physical properties versus those from chemical properties of the soot particles upon different aging processes, and the degree of their contribution to the enhanced ice nucleation ability. Certainly, one caveat of our analysis is that upon nebulizing soot particles from the bulk solutions, the particles can change their morphology. A compaction upon evaporation of the liquid has previously been reported,110–114 and can affect the pore volume and thus INP activity.
Finally, we note that soot particles emitted from real-world combustion sources, span a much wider range of complex physicochemical properties than the laboratory surrogates employed here. In particular, the exact chemical composition of the OC fraction associated with atmospheric aerosols from e.g. biomass burning, and comparability to the OC associated with laboratory generated soot particles constitute a relevant caveat in assessing the aging effect on the ice nucleation activity of such particles. Further laboratory studies in which the OC fraction (coating thickness) and OC type are varied systematically are needed to further our knowledge on how this affects the ice nucleation activity of these particles and how this in turn changes through atmospheric aging processes. Overall, we conclude that our results are relevant for the subset of soot particles of the size and type investigated here, and that similar aging processes affect atmospheric soot only under the premise that these behave like the particles studied here.
6. Conclusions
We have investigated the ice nucleation ability of soot particles at cirrus temperatures and how it changes upon particle aging. Two soot types, mCASTbl and mCASTbr, of low and high OC content respectively, were generated from a miniCAST diffusion flame propane burner. Atmospheric aging was mimicked through exposure of the soot particles to water and aqueous sulfuric acid. Acidic solutions were prepared to have a pH value of ∼4, typical of that frequently found in cloud or haze droplets in polluted environments. The ice nucleation measurements were performed on 400 nm size selected particles (mobility diameter) by means of RH-scans in a continuous flow diffusion chamber, at temperatures of 233 and 218 K. The range in RHw (RHi) was approximately 65–105% (100–180%). Ice nucleation activity of unaged and aged soots was investigated in relation to physicochemical particle properties. Particle chemical properties were measured by means of scanning transmission X-ray microscopy and near edge X-ray absorption fine structure (STXM/NEXAFS) measurements. In addition, particle hydrophilicity was determined through water sorption isotherms, using a dynamic vapor sorption (DVS) technique and OH accessibility measurements.We demonstrated that the ice nucleation of the two soot types is strongly linked to their physicochemical characteristics and found that alteration of such properties upon aging strongly impacts their ice cloud formation potential. Unaged particles were observed to form ice only at RH ≥ RHhom. In particular, the unaged soot type with larger OC content (mCASTbr) required water saturation conditions in order to nucleate ice. Aged soot particles showed enhanced ice nucleation abilities compared to unaged particles. More specifically the aged soot particles had significantly lower thresholds required for ice nucleation onset by up to 15% with respect to water (RHi ≈ 25%) and nucleated ice at conditions favorable for heterogeneous ice formation. These observations suggest that there is a need to consider atmospheric aging when assessing the ice nucleation ability of soot particles in global climate models,92,115 given that the ice nucleation of aged soot at the temperatures investigated here is comparable to that reported for mineral dust particles.100
Our water sorption measurements revealed that the mCASTbr sample is much less hydrophilic compared to the mCASTbl samples. The hysteresis observed at high RHw between the adsorption and desorption branches for the mCASTbl and the absence of such a hysteresis for the mCASTbr suggest different water sorption mechanisms. Namely, the presence and absence of water uptake through capillary condensation for mCASTbl and mCASTbr, respectively. Consistent with our ice nucleation measurements, the DVS revealed an increase in hydrophilicity upon aging irrespective of the soot and aging type. At the same time the aged mCASTbl samples both revealed enhanced hysteresis and we attribute the enhanced ice nucleation ability of the mCASTbl to result from an enhanced PCF mechanism. Interestingly, the acid aged mCASTbr also exhibited an increased water uptake capacity, associated with a weak hysteresis at RHw > 80%, which is within our measurement uncertainty. We attribute this to oxidation and/or removal of organic material from the pores (redistribution on the soot aggregate) and a general decrease in soot-water contact angle, allowing for formation of capillary condensates. Hence, aging of mCASTbr in acidic solutions not only enhanced the ice nucleation ability, but also changed the ice nucleation mechanism to a PCF mechanism. The mCASTbr aged in water, however, only revealed a marginal enhancement in water uptake capacity and did not exhibit any hysteresis (absence of mesopores accessible for water uptake). The observed enhanced ice nucleation ability of the water aged mCASTbr cannot be attributed to a PCF ice formation, but rather suggests a classical deposition nucleation mechanism.
Finally, our NEXAFS results revealed distinct chemical fingerprints of the two soot types and changes upon aging. Measurements at the O K-edge revealed little oxygen to be associated with all soot types. This was confirmed by protium–deuterium exchange measurements used to gravimetrically determine the accessibility of hydroxyl groups, which were found to be largely absent for both soot types and independent of aging. NEXAFS spectra taken on the C K-edge showed the propane flame soots to be characterized by high proportions of aromatic carbon (285.5 eV). In particular we found a marked peak at 287.5 eV observed for mCASTbl but not for the aged samples of the same soot type, suggesting a correlation between the ice nucleation activity and the feature of that peak. We attributed this to OH functionalities. The decrease in such OH functionalities is in line with the increased ice nucleation of the aged samples, and consistent with previous findings.87 Future studies should include soots from different combustion sources to further our understanding to what extent the impact of aqueous phase aging controls the ice nucleation activity of different soot types. In addition, it will be useful to more closely investigate the role of varying aging times. This could also help to elucidate to what extent different particle generation methods used in laboratory studies, in particular dry versus wet dispersion of particles, affect the comparability of the respective results, when assessing ice nucleation activity.
Author contributions
FM conducted the ice nucleation experiments, synthesized the figures and wrote the manuscript with contributions from PAA, ZAK and UL. PAA, JD, PCA, FM and ZAK performed STXM/NEXAFS measurements overseen and coordinated by PAA. PG performed DVS measurements and analyzed the data. MA conceived the idea to perform STXM/NEXAFS measurements, FM and ZAK designed the ice nucleation measurements. All authors discussed and interpreted data. PAA and ZAK oversaw the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from SLS grant application 20180678. Fabian Mahrt and Zamin A. Kanji acknowledge funding from ETH grant ETH-25-15-1 that supported this work. We acknowledge the Paul Scherrer Institut, Villigen, Switzerland for provision of synchrotron radiation beamtime at the PolLux beamline X07DA of the SLS and would like to thank Benjamin Watts for assistance. The PolLux end station was financed by the German Ministry für Bildung und Forschung (BMBF) through contracts 05K16WED and 05K19WE2. Peter A. Alpert acknowledges the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement (no. 701647). Claudia Marcolli is thanked for helpful discussions. We also thank Ingo Burgert for providing access to the DVS facility and funding the OH-accessibility measurements.Notes and references
- O. Gustafsson and V. Ramanathan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4243–4245 CrossRef CAS PubMed.
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Kaercher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, J. Geophys. Res.: Atmos., 2013, 118, 5380–5552 CAS.
- V. Ramanathan and G. Carmichael, Nat. Geosci., 2008, 1, 221–227 CrossRef CAS.
- A. Petzold, J. A. Ogren, M. Fiebig, P. Laj, S. M. Li, U. Baltensperger, T. Holzer-Popp, S. Kinne, G. Pappalardo, N. Sugimoto, C. Wehrli, A. Wiedensohler and X. Y. Zhang, Atmos. Chem. Phys., 2013, 13, 8365–8379 CrossRef CAS.
- M. O. Andreae and A. Gelencser, Atmos. Chem. Phys., 2006, 6, 3131–3148 CrossRef CAS.
- O. B. Popovicheva, E. D. Kireeva, M. A. Timofeev, N. K. Shonija and V. P. Mogil'nikov, Izvestiya Atmos. Ocean. Phys., 2010, 46, 339–346 CrossRef.
- K. A. Koehler, P. J. DeMott, S. M. Kreidenweis, O. B. Popovicheva, M. D. Petters, C. M. Carrico, E. D. Kireeva, T. D. Khokhlova and N. K. Shonija, Phys. Chem. Chem. Phys., 2009, 11, 7906–7920 RSC.
- L. Nichman, M. Wolf, P. Davidovits, T. B. Onasch, Y. Zhang, D. R. Worsnop, J. Bhandari, C. Mazzoleni and D. J. Cziczo, Atmos. Chem. Phys., 2019, 19, 12175–12194 CrossRef CAS.
- S. Seisel, A. Pashkova, Y. Lian and R. Zellner, Faraday Discuss., 2005, 130, 437–451 RSC.
- R. Zhang, A. F. Khalizov, J. Pagels, D. Zhang, H. Xue and P. H. McMurry, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10291–10296 CrossRef CAS PubMed.
- F. Friebel and A. A. Mensah, Langmuir, 2019, 35(45), 14437–14450 CrossRef CAS PubMed.
- M. Li, F. Bao, Y. Zhang, W. Song, C. Chen and J. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7717–7722 CrossRef CAS PubMed.
- G. Kulkarni, S. China, S. Liu, M. Nandasiri, N. Sharma, J. Wilson, A. C. Aiken, D. Chand, A. Laskin, C. Mazzoleni, M. Pekour, J. Shilling, V. Shutthanandan, A. Zelenyuk and R. A. Zaveri, Geophys. Res. Lett., 2016, 43, 3580–3588 CrossRef.
- J. Pagels, A. F. Khalizov, P. H. McMurry and R. Y. Zhang, Aerosol Sci. Technol., 2009, 43, 629–640 CrossRef CAS.
- K. S. Johnson, B. Zuberi, L. T. Molina, M. J. Molina, M. J. Iedema, J. P. Cowin, D. J. Gaspar, C. Wang and A. Laskin, Atmos. Chem. Phys., 2005, 5, 3033–3043 CrossRef CAS.
- M. Z. Jacobson, Nature, 2001, 409, 695–697 CrossRef CAS PubMed.
- C. D. Cappa, T. B. Onasch, P. Massoli, D. R. Worsnop, T. S. Bates, E. S. Cross, P. Davidovits, J. Hakala, K. L. Hayden, B. T. Jobson, K. R. Kolesar, D. A. Lack, B. M. Lerner, S. M. Li, D. Mellon, I. Nuaaman, J. S. Olfert, T. Petaja, P. K. Quinn, C. Song, R. Subramanian, E. J. Williams and R. A. Zaveri, Science, 2012, 337, 1078–1081 CrossRef CAS PubMed.
- Y. Yun, J. E. Penner and O. Popovicheva, Atmos. Chem. Phys., 2013, 13, 4339–4348 CrossRef.
- T. Tritscher, Z. Jurányi, M. Martin, R. Chirico, M. Gysel, M. F. Heringa, P. F. DeCarlo, B. Sierau, A. S. H. Prévôt, E. Weingartner and U. Baltensperger, Environ. Res. Lett., 2011, 6, 034026 CrossRef.
- A. F. Khalizov, R. Zhang, D. Zhang, H. Xue, J. Pagels and P. H. McMurry, J. Geophys. Res.: Atmos., 2009, 114, D05208 CrossRef.
- G. Lammel and T. Novakov, Atmos. Environ., 1995, 29, 813–823 CrossRef CAS.
- M. M. Dubinin, Carbon, 1980, 18, 355–364 CrossRef CAS.
- R. S. Vartapetyan and A. M. Voloshchuk, Russ. Chem. Rev., 1995, 64, 985 CrossRef.
- U. Lohmann, F. Lüönd and F. Mahrt, An Introduction to Clouds: From the Microscale to Climate, Cambridge University Press, Cambridge, 2016 Search PubMed.
- H. R. Pruppacher and D. J. Klett, Microphysics of Clouds and Precipitation, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2nd edn, 1997 Search PubMed.
- Y. Wei, Q. Zhang and J. E. Thompson, Atmos. Clim. Sci., 2017, 07, 11–22 CAS.
- F. Mahrt, K. Kilchhofer, C. Marcolli, P. Grönquist, R. O. David, M. Rösch, U. Lohmann and Z. A. Kanji, J. Geophys. Res.: Atmos., 2020, 125, e2019JD030922 Search PubMed.
- D. Ferry, J. Suzanne, S. Nitsche, O. B. Popovitcheva and N. K. Shonija, J. Geophys. Res.: Atmos., 2002, 107(D23), 4734 CrossRef.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- H. K. Christenson, CrystEngComm, 2013, 15, 2030–2039 RSC.
- C. Marcolli, Atmos. Chem. Phys., 2014, 14, 2071–2104 CrossRef.
- R. O. David, C. Marcolli, J. Fahrni, Y. Q. Qiu, Y. A. P. Sirkin, V. Molinero, F. Mahrt, D. Bruhwiler, U. Lohmann and Z. A. Kanji, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8184–8189 CrossRef CAS PubMed.
- F. Mahrt, C. Marcolli, R. O. David, P. Grönquist, E. J. B. Meier, U. Lohmann and Z. A. Kanji, Atmos. Chem. Phys., 2018, 18, 13363–13392 CrossRef CAS.
- S. D. Brooks, K. Suter and L. Olivarez, J. Phys. Chem. A, 2014, 118, 10036–10047 CrossRef CAS PubMed.
- L. Lacher, U. Lohmann, Y. Boose, A. Zipori, E. Herrmann, N. Bukowiecki, M. Steinbacher and Z. A. Kanji, Atmos. Chem. Phys., 2017, 17, 15199–15224 CrossRef CAS.
- E. Ganor, Z. Levin and D. Pardess, Atmos. Environ., Part A, 1993, 27, 1821–1832 CrossRef.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett Jr, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I. T. Ku, V. F. McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, Atmos. Chem. Phys. Discuss., 2019, 2019, 1–143 Search PubMed.
- A. Braun, F. E. Huggins, N. Shah, Y. Chen, S. Wirick, S. B. Mun, C. Jacobsen and G. P. Huffman, Carbon, 2005, 43, 117–124 CrossRef CAS.
- S. Di Stasio and A. Braun, Energy Fuels, 2006, 20, 187–194 CrossRef CAS.
- R. J. Hopkins, A. V. Tivanski, B. D. Marten and M. K. Gilles, J. Aerosol Sci., 2007, 38, 573–591 CrossRef CAS.
- R. C. Moffet, A. V. Tivanski and M. K. Gilles, in Scanning Transmission X-ray Microscopy: Applications in Atmospheric Aerosol Research, ed. R. Signorell and J. P. Reid, Taylor and Francis Books Inc., 2010, pp. 419–462 Search PubMed.
- D. A. Knopf, P. A. Alpert, B. Wang, R. E. O'Brien, S. T. Kelly, A. Laskin, M. K. Gilles and R. C. Moffet, J. Geophys. Res.: Atmos., 2014, 119, 10365–10381 Search PubMed.
- P. Parent, C. Laffon, I. Marhaba, D. Ferry, T. Z. Regier, I. K. Ortega, B. Chazallon, Y. Carpentier and C. Focsa, Carbon, 2016, 101, 86–100 CrossRef CAS.
- A. Liati, D. Schreiber, P. A. Alpert, Y. Liao, B. T. Brem, P. Corral Arroyo, J. Hu, H. R. Jonsdottir, M. Ammann and P. Dimopoulos Eggenschwiler, Environ. Pollut., 2019, 247, 658–667 CrossRef CAS PubMed.
- O. B. Popovitcheva, N. M. Persiantseva, M. E. Trukhin, G. B. Rulev, N. K. Shonija, Y. Y. Buriko, A. M. Starik, B. Demirdjian, D. Ferry and J. Suzanne, Phys. Chem. Chem. Phys., 2000, 2, 4421–4426 RSC.
- N. M. Persiantseva, O. B. Popovicheva and N. K. Shonija, J. Environ. Monit., 2004, 6, 939–945 RSC.
- O. B. Popovicheva, N. M. Persiantseva, E. E. Lukhovitskaya, N. K. Shonija, N. A. Zubareva, B. Demirdjian, D. Ferry and J. Suzanne, Geophys. Res. Lett., 2004, 31, L11104 CrossRef.
- O. Popovicheva, N. M. Persiantseva, N. K. Shonija, P. DeMott, K. Koehler, M. Petters, S. Kreidenweis, V. Tishkova, B. Demirdjian and J. Suzanne, Phys. Chem. Chem. Phys., 2008, 10, 2332–2344 RSC.
- Y. Liu, C. Liu, J. Ma, Q. Ma and H. He, Phys. Chem. Chem. Phys., 2010, 12, 10896–10903 RSC.
- R. H. Moore, L. D. Ziemba, D. Dutcher, A. J. Beyersdorf, K. Chan, S. Crumeyrolle, T. M. Raymond, K. L. Thornhill, E. L. Winstead and B. E. Anderson, Aerosol Sci. Technol., 2014, 48, 467–479 CrossRef CAS.
- A. Mamakos, I. Khalek, R. Giannelli and M. Spears, Aerosol Sci. Technol., 2013, 47, 927–936 CrossRef CAS.
- J. Yon, A. Bescond and F. X. Ouf, J. Aerosol Sci., 2015, 87, 28–37 CrossRef CAS.
- A. Bescond, J. Yon, F. X. Ouf, D. Ferry, D. Delhaye, D. Gaffie, A. Coppalle and C. Roze, Aerosol Sci. Technol., 2014, 48, 831–841 CrossRef CAS.
- M. N. Ess and K. Vasilatou, Aerosol Sci. Technol., 2018, 1–16 Search PubMed.
- I. Marhaba, D. Ferry, C. Laffon, T. Z. Regier, F.-X. Ouf and P. Parent, Combust. Flame, 2019, 204, 278–289 CrossRef CAS.
- T. Haller, C. Rentenberger, J. C. Meyer, L. Felgitsch, H. Grothe and R. Hitzenberger, Atmos. Meas. Tech., 2019, 12, 3503–3519 CrossRef CAS.
- A. R. Chughtai, N. J. Miller, D. M. Smith and J. R. Pitts, J. Atmos. Chem., 1999, 34, 259–279 CrossRef CAS.
- A. R. Chughtai, J. M. Kim and D. M. Smith, J. Atmos. Chem., 2002, 43, 21–43 CrossRef CAS.
- C. Han, Y. C. Liu, C. Liu, J. Z. Ma and H. He, J. Phys. Chem. A, 2012, 116, 4129–4136 CrossRef CAS PubMed.
- D. M. Murphy and T. Koop, Q. J. R. Metereol. Soc., 2005, 131, 1539–1565 CrossRef.
- J. Raabe, G. Tzvetkov, U. Flechsig, M. Boge, A. Jaggi, B. Sarafimov, M. G. C. Vernooij, T. Huthwelker, H. Ade, D. Kilcoyne, T. Tyliszczak, R. H. Fink and C. Quitmann, Rev. Sci. Instrum., 2008, 79, 113704 CrossRef CAS PubMed.
- J. Stöhr, NEXAFS spectroscopy, Springer, Berlin, 2003 Search PubMed.
- R. C. Moffet, T. Henn, A. Laskin and M. K. Gilles, Anal. Chem., 2010, 82, 7906–7914 CrossRef CAS PubMed.
- A. Braun, J. Environ. Monit., 2005, 7, 1059–1065 RSC.
- B. L. Henke, E. M. Gullikson and J. C. Davis, At. Data Nucl. Data Tables, 1993, 54, 181–342 CrossRef CAS.
- Y. Liu, T. P. Russell, M. G. Samant, J. Stöhr, H. R. Brown, A. Cossy-Favre and J. Diaz, Macromolecules, 1997, 30, 7768–7771 CrossRef CAS.
- S. G. Urquhart, H. Ade, M. Rafailovich, J. S. Sokolov and Y. Zhang, Chem. Phys. Lett., 2000, 322, 412–418 CrossRef CAS.
- O. Dhez, H. Ade and S. G. Urquhart, J. Electron Spectrosc. Relat. Phenom., 2003, 128, 85–96 CrossRef CAS.
- S. Takahama, S. Liu and L. M. Russell, J. Geophys. Res.: Atmos., 2010, 115, D01202 CrossRef.
- J. Bruley, P. Madakson and J. C. Liu, Nucl. Instrum. Methods Phys. Res., Sect. B, 1990, 45, 618–621 CrossRef.
- J. Diaz, O. R. Monteiro and Z. Hussain, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 094201 CrossRef.
- A. Liati, P. D. Eggenschwiler, D. Schreiber, V. Zelenay and M. Ammann, Combust. Flame, 2013, 160, 671–681 CrossRef CAS.
- P. Grönquist, M. Frey, T. Keplinger and I. Burgert, ACS Omega, 2019, 4, 12425–12431 CrossRef PubMed.
- T. Koop, B. P. Luo, A. Tsias and T. Peter, Nature, 2000, 406, 611–614 CrossRef CAS PubMed.
- B. Friedman, G. Kulkarni, J. Beranek, A. Zelenyuk, J. A. Thornton and D. J. Cziczo, J. Geophys. Res.: Atmos., 2011, 116, D17203 CrossRef.
- K. Higuchi and N. Fukuta, J. Atmos. Sci., 1966, 23, 187–190 CrossRef CAS.
- N. Fukuta, J. Atmos. Sci., 1966, 23, 741–750 CrossRef CAS.
- T. Koop, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 797–799 CrossRef CAS PubMed.
- I. Crawford, O. Moehler, M. Schnaiter, H. Saathoff, D. Liu, G. McMeeking, C. Linke, M. Flynn, K. N. Bower, P. J. Connolly, M. W. Gallagher and H. Coe, Atmos. Chem. Phys., 2011, 11, 9549–9561 CrossRef CAS.
- A. R. Chughtai, G. R. Williams, M. M. O. Atteya, N. J. Miller and D. M. Smith, Atmos. Environ., 1999, 33, 2679–2687 CrossRef CAS.
- C. A. Rogaski, D. M. Golden and L. R. Williams, Geophys. Res. Lett., 1997, 24, 381–384 CrossRef CAS.
- R. C. Moffet, R. E. O'Brien, P. A. Alpert, S. T. Kelly, D. Q. Pham, M. K. Gilles, D. A. Knopf and A. Laskin, Atmos. Chem. Phys., 2016, 16, 14515–14525 CrossRef CAS.
- F. X. Ouf, P. Parent, C. Laffon, I. Marhaba, D. Ferry, B. Marcillaud, E. Antonsson, S. Benkoula, X. J. Liu, C. Nicolas, E. Robert, M. Patanen, F. A. Barreda, O. Sublemontier, A. Coppalle, J. Yon, F. Miserque, T. Mostefaoui, T. Z. Regier, J. A. Mitchell and C. Miron, Sci. Rep., 2016, 6, 36495 CrossRef CAS PubMed.
- V. Zelenay, M. E. Monge, B. D'Anna, C. George, S. A. Styler, T. Huthwelker and M. Ammann, J. Geophys. Res.: Atmos., 2011, 116, D11301 CrossRef.
- G. D. Cody, H. Ade, S. Wirick, G. D. Mitchell and A. Davis, Org. Geochem., 1998, 28, 441–455 CrossRef CAS.
- J. Bhandari, S. China, K. K. Chandrakar, G. Kinney, W. Cantrell, R. A. Shaw, L. R. Mazzoleni, G. Girotto, N. Sharma, K. Gorkowski, S. Gilardoni, S. Decesari, M. C. Facchini, N. Zanca, G. Pavese, F. Esposito, M. K. Dubey, A. C. Aiken, R. K. Chakrabarty, H. Moosmüller, T. B. Onasch, R. A. Zaveri, B. V. Scarnato, P. Fialho and C. Mazzoleni, Sci. Rep., 2019, 9, 11824 CrossRef PubMed.
- L. Lupi and V. Molinero, J. Phys. Chem. A, 2014, 118, 7330–7337 CrossRef CAS PubMed.
- C. Marcolli, Atmos. Chem. Phys. Discuss., 2019, 2019, 1–26 Search PubMed.
- L. Lupi, A. Hudait and V. Molinero, J. Am. Chem. Soc., 2014, 136, 3156–3164 CrossRef CAS PubMed.
- V. Zelenay, R. Mooser, T. Tritscher, A. Krepelova, M. F. Heringa, R. Chirico, A. S. H. Prevot, E. Weingartner, U. Baltensperger, J. Dommen, B. Watts, J. Raabe, T. Huthwelker and M. Ammann, Atmos. Chem. Phys., 2011, 11, 11777–11791 CrossRef CAS.
- J. Lehmann, B. Q. Liang, D. Solomon, M. Lerotic, F. Luizao, J. Kinyangi, T. Schafer, S. Wirick and C. Jacobsen, Global Biogeochem. Cycles, 2005, 19, GB1013 Search PubMed.
- U. Lohmann, F. Friebel, Z. Kanji, F. Mahrt, A. A. Mensah and D. Neubauer, in preparation.
- Y. Wu, X. Wang, J. Tao, R. Huang, P. Tian, J. Cao, L. Zhang, K. F. Ho and R. Zhang, Atmos. Chem. Phys. Discuss., 2016, 2016, 1–25 Search PubMed.
- J. Hallett, J. G. Hudson and C. F. Rogers, Aerosol Sci. Technol., 1989, 10, 70–83 CrossRef CAS.
- W. M. Gong, C. Stroud and L. M. Zhang, Atmosphere, 2011, 2, 567–616 CrossRef CAS.
- H. R. Pruppacher and R. Jaenicke, Atmos. Res., 1995, 38, 283–295 CrossRef.
- P. J. DeMott, D. C. Rogers and S. M. Kreidenweis, J. Geophys. Res.: Atmos., 1997, 102, 19575–19584 CrossRef CAS.
- P. Ginoux, M. Chin, I. Tegen, J. M. Prospero, B. Holben, O. Dubovik and S.-J. Lin, J. Geophys. Res.: Atmos., 2001, 106, 20255–20273 CrossRef.
- P. J. DeMott, A. J. Prenni, X. Liu, S. M. Kreidenweis, M. D. Petters, C. H. Twohy, M. S. Richardson, T. Eidhammer and D. C. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11217–11222 CrossRef CAS PubMed.
- A. Welti, F. Lüönd, O. Stetzer and U. Lohmann, Atmos. Chem. Phys., 2009, 9, 6705–6715 CrossRef CAS.
- C. Alcala-Jornod, H. van den Bergh and M. J. Rossi, Phys. Chem. Chem. Phys., 2000, 2, 5584–5593 RSC.
- J. A. Ogren and R. J. Charlson, Tellus B, 1983, 35, 241–254 CrossRef.
- C. Textor, M. Schulz, S. Guibert, S. Kinne, Y. Balkanski, S. Bauer, T. Berntsen, T. Berglen, O. Boucher, M. Chin, F. Dentener, T. Diehl, R. Easter, H. Feichter, D. Fillmore, S. Ghan, P. Ginoux, S. Gong, J. E. Kristjansson, M. Krol, A. Lauer, J. F. Lamarque, X. Liu, V. Montanaro, G. Myhre, J. Penner, G. Pitari, S. Reddy, O. Seland, P. Stier, T. Takemura and X. Tie, Atmos. Chem. Phys., 2006, 6, 1777–1813 CrossRef CAS.
- M. S. Reddy and O. Boucher, Geophys. Res. Lett., 2007, 34, L11802 CrossRef.
- N. Riemer, H. Vogel and B. Vogel, Atmos. Chem. Phys., 2004, 4, 1885–1893 CrossRef CAS.
- A. C. Eriksson, C. Wittbom, P. Roldin, M. Sporre, E. Strm, P. Nilsson, J. Martinsson, J. Rissler, E. Z. Nordin, B. Svenningsson, J. Pagels and E. Swietlicki, Sci. Rep., 2017, 7, 12364 CrossRef CAS PubMed.
- V. F. McNeill, Environ. Sci. Technol., 2015, 49, 1237–1244 CrossRef CAS PubMed.
- S. W. Hunt, A. Laskin and S. A. Nizkorodov, Multiphase Environmental Chemistry in the Atmosphere, Oxford University Press, Oxford, New York, 2019 Search PubMed.
- N. Borduas-Dedekind, R. Ossola, R. O. David, L. S. Boynton, V. Weichlinger, Z. A. Kanji and K. McNeill, Atmos. Chem. Phys., 2019, 19, 12397–12412 CrossRef CAS.
- I. Colbeck, L. Appleby, E. J. Hardman and R. M. Harrison, J. Aerosol Sci., 1990, 21, 527–538 CrossRef.
- P. F. Huang, B. J. Turpin, M. J. Pipho, D. B. Kittelson and P. H. McMurry, J. Aerosol Sci., 1994, 25, 447–459 CrossRef CAS.
- A. C. Chamberlain, W. S. Clough, M. J. Heard, D. Newton, A. N. B. Stott and A. C. Wells, Proc. R. Soc., Ser. B, 1975, 192, 77–110 CAS.
- B. Miljevic, N. C. Surawski, T. Bostrom and Z. D. Ristovski, J. Aerosol Sci., 2012, 47, 48–57 CrossRef CAS.
- X. F. Ma, C. D. Zangmeister, J. Gigault, G. W. Mulholland and M. R. Zachariah, J. Aerosol Sci., 2013, 66, 209–219 CrossRef CAS.
- D. Koch, M. Schulz, S. Kinne, C. McNaughton, J. R. Spackman, Y. Balkanski, S. Bauer, T. Berntsen, T. C. Bond, O. Boucher, M. Chin, A. Clarke, N. De Luca, F. Dentener, T. Diehl, O. Dubovik, R. Easter, D. W. Fahey, J. Feichter, D. Fillmore, S. Freitag, S. Ghan, P. Ginoux, S. Gong, L. Horowitz, T. Iversen, A. Kirkev, Z. Klimont, Y. Kondo, M. Krol, X. Liu, R. Miller, V. Montanaro, N. Moteki, G. Myhre, J. E. Penner, J. Perlwitz, G. Pitari, S. Reddy, L. Sahu, H. Sakamoto, G. Schuster, J. P. Schwarz, Ø. Seland, P. Stier, N. Takegawa, T. Takemura, C. Textor, J. A. van Aardenne and Y. Zhao, Atmos. Chem. Phys., 2009, 9, 9001–9026 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00525k |
| ‡ Present address: Department of Chemistry, University of British Columbia, V6T1Z1 Vancouver, Canada. |
| § Present address: Department of Chemistry and Applied Biosciences, Laboratory of Physical Chemistry, ETH Zurich, 8093 Zurich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2020 |