
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemporal trends of suspect- and target-per/polyfluoroalkyl substances (PFAS), extractable organic fluorine (EOF) and total fluorine (TF) in pooled serum from first-time mothers in Uppsala, Sweden, 1996–2017†
Luc T.
Miaz
 *a,
Merle M.
Plassmann
a,
Irina
Gyllenhammar
b,
Anders
Bignert
c,
Oskar
Sandblom
a,
Sanna
Lignell
b,
Anders
Glynn
d and
Jonathan P.
Benskin
*a
*a,
Merle M.
Plassmann
a,
Irina
Gyllenhammar
b,
Anders
Bignert
c,
Oskar
Sandblom
a,
Sanna
Lignell
b,
Anders
Glynn
d and
Jonathan P.
Benskin
*a
aDepartment of Environmental Science, Stockholm University, Stockholm, Sweden. E-mail: Luc@miaz.ch; Jon.Benskin@aces.su.se
bDepartment of Risk and Benefit Assessment, Swedish Food Agency, P.O. Box 622, SE-751 26 Uppsala, Sweden
cSwedish Museum of Natural History, Box 50007, 10405 Stockholm, Sweden
dDepartment of Biomedical Sciences and Veterinary Public Health, Swedish University of Agricultural Sciences (SLU), Box 7050, SE-750 07, Uppsala, Sweden
First published on 25th February 2020
Abstract
A combined method for quantitative analysis, along with suspect and non-target screening of per- and polyfluoroalkyl substances (PFAS) was developed using ultra-high pressure liquid chromatography-ultra-high resolution (Orbitrap) mass spectrometry. The method was applied together with measurements of total- and extractable organofluorine (TF and EOF, respectively), to pooled serum samples from 1996–2017 from first-time mothers living in the county of Uppsala, Sweden, some of which (i.e. 148 of 472 women sampled 1996–2012) were exposed to drinking water contaminated with perfluorohexane sulfonate (PFHxS) and other PFAS until mid-2012. Declining trends were observed for all target PFAS as well as TF, with homologue-dependent differences in year of onset of decline. Only 33% of samples displayed detectable EOF, and amongst these samples the percentage of EOF explained by target PFAS declined significantly (−3.5% per year) over the entire study period. This finding corroborates prior observations in Germany after the year 2000, and may reflect increasing exposure to novel PFAS which have not yet been identified. Suspect screening revealed the presence of perfluoro-4-ethylcyclohexanesulfonate (PFECHS), which displayed declining trends since the year 2000. Non-target time trend screening revealed 3 unidentified features with time trends matching PFHxS. These features require further investigation, but may represent contaminants which co-occurred with PFHxS in the contaminated drinking water.
Environmental significanceThe global occurrence of per- and polyfluoroalkyl substances (PFAS) is of concern for both human and wildlife health. Among the >4000 registered PFAS, only a few are commonly monitored, suggesting that exposure to these chemicals may be underestimated. The present work investigated temporal trends of suspect- and target PFAS, along with extractable organic fluorine (EOF) and total fluorine (TF) in pooled serum (1996–2017) from a cohort historically exposed to PFAS-contaminated drinking water. Declining temporal trends were observed for all target PFAS, demonstrating the positive impact of phase-outs and regulation, along with local drinking water clean-up initiatives. However, target PFAS also accounted for a smaller fraction of EOF in serum from more recent years, suggesting an increase in the relative contribution from some presently unidentified PFAS. Finally, non-target time trend screening revealed 3 unidentified features which are suspected to be contaminants which co-occurred in the contaminated drinking water supply. |
Introduction
Per- and polyfluoroalkyl substances (PFAS) are a family of synthetic compounds containing at least one fully fluorinated carbon atom in the alkyl chain.1 PFAS have been used since the 1950s in a wide variety of applications (e.g. firefighting-foams, food-contact materials, textiles, cosmetics, etc.) due to their stability along with surfactant and surface protection properties.2 In the early 2000s, however, concerns were raised about the toxicity, bioaccumulation, environmental occurrence and persistence of some perfluoroalkyl acids (PFAAs), in particular perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS).2 While production and use of PFOS and PFOA has been reduced, the over 4600 registered PFAS which exist on the global market3 has left questions regarding whether human and wildlife exposure to these chemicals is being underestimated. Moreover, recent organofluorine mass balance experiments have reported large quantities of unidentified extractable organofluorine (EOF) in human blood (15–67%)5 and wildlife (68–90%).5–7Non-target analysis or suspect-screening (collectively referred to herein as ‘NTA’) workflows offer the opportunity to elucidate novel PFAS which may account for unidentified organofluorine in samples. The application of NTA workflows within PFAS research have been reviewed recently.8 There are few examples of where NTA has been combined with organofluorine mass balance determination and only a single study9 which has applied NTA to elucidate novel PFAS in humans. In that work, a case–control design was used to identify novel features in the serum of firefighters from Australia. However, concentrations of PFOS and PFHxS ranged from 92–343 and 49–326 ng mL−1 in serum from firefighters, which is in some cases over 2 orders of magnitude higher than the general population. Identifying novel PFAS within the non-occupationally exposed population is considerably more challenging due to lower serum concentrations, which necessitates very low detection limits.
In the city of Uppsala, Sweden, residents in certain areas received drinking water (DW) contaminated with PFAS up until 2012, when the contamination was mitigated.10 The contamination was most likely due to release of PFAS from perfluorohexane sulfonate (PFHxS)-based aqueous film forming foam (AFFF) used for firefighting at the military training airport Uppsala-Ärna until at least 2003.10 PFHxS was the most prevalent PFAA in the contaminated well water (median 83 ng L−1), followed by PFOS (median 47 ng L−1), perfluorobutane sulfonate (PFBS; median 13 ng L−1), and perfluorohexanoate (PFHxA; median 10 ng L−1).11 Given the health-based guideline for drinking water issued by the Swedish Food Agency (<90 ng L−1 based on the sum of 11 PFAS), these concentrations are clearly of concern.12 Through the POPUP study (Persistent Organic Pollutants in Uppsala Primiparas), it was determined that 148 of 472 women (i.e. 31%) sampled 1996–2012 were exposed to PFAS-contaminated drinking water until mid-2012. Exposure of this population to DW contaminated with PFAS was linked to increased PFAS concentrations in the serum of both mothers and their children. However, given the number and diversity of PFAS, it remains unclear as to whether the full extent of exposure to PFAS has been elucidated.11,13,14
The present study builds upon prior targeted temporal trend analyses carried out on the POPUP cohort (1996–2016)15,16 by applying a combined target/suspect screening strategy together with fluorine mass balance determination to samples from the entire cohort (1996–2017). To the best of our knowledge, this is the first study to combine fluorine mass balance and suspect screening in human samples. These new methodologies provide a means of comprehensively assessing human exposure to PFAS and the effect of local drinking water clean-up initiatives. Moreover, this approach offers the possibility to identify novel contaminants not captured by traditional (targeted) methodologies.
Materials and methods
Target PFAS
Target PFAS included C4–C16 and C18 perfluoroalkyl carboxylic acids (PFCAs), C4–C11 perfluoroalkyl sulfonic acids (PFSAs), 32 perfluoroalkyl acid precursors/replacement PFAS (including (N-alkyl substituted) perfluorooctane sulfonamides/sulfonamidoalcohols/acetates, polyfluoroalkyl phosphate esters (PAPs), fluorotelomer sulfonates (FTSs), fluorotelomer acids (FTAs), perfluoroalkyl ethers) and 8 branched isomers. A full list, including abbreviations can be found in Table S1 (ESI†). In cases where isomers were quantified, branched isomers were denoted as ‘br-’ and linear isomers as ‘lin-’; in cases where multiple isomers were not reported, the measured target was assumed to be linear. All standards and reagents were purchased from Wellington Laboratories.Recruitment and sample preparation
In the POPUP study, first-time mothers (n = 622) from the general population living in Uppsala County were recruited between 1996 and 2017. All participants donated a blood sample 3 weeks after delivery. Blood sampling was done using 9 mL Vacutainer® or Vacuette® serum tubes and serum was stored at −20 °C, at the Swedish Food Agency. For details about recruitment and blood sampling see Glynn et al.17 and Lignell et al.18 The study was approved by the local ethics committee of Uppsala University, and the participating women gave informed consent prior to inclusion in the study (ethical approval number dnr 96114 and 2004:M-177).For each year of recruitment, 1–3 pooled serum samples were prepared, with serum from 6–25 individual mothers in each pool (Table 1). Three separate analyses were performed on each pooled serum sample: (1) targeted PFAS were measured in 0.5 mL serum using an acetonitrile (ACN) extraction followed by a newly developed Ultra high performance liquid chromatography-Orbitrap mass spectrometry (UHPLC-Orbitrap-MS) method; (2) extractable organofluorine (EOF) was determined in 0.5 mL portions of serum using the same ACN extraction as for targeted analysis (minus internal standards) followed by analysis by combustion ion chromatography (CIC); (3) total fluorine (TF) was determined in 150 μL portions of serum which were analysed directly by CIC. Details of these methods are provided below.
| Sampling year | N | No of pools | N in each pool | Age (yrs)b mean (range) |
|---|---|---|---|---|
| a Total number of serum samples from the specific sampling year. b Mean age of the women donating blood during the specific sampling year. | ||||
| 1996 | 19 | 3 | 6–7 | 30 (21–41) |
| 1997 | 62 | 3 | 20–21 | 28 (21–37) |
| 1998 | 74 | 3 | 24–25 | 29 (21–35) |
| 1999 | 17 | 3 | 5–6 | 27 (21–31) |
| 2000 | 20 | 2 | 10 | 30 (21–37) |
| 2001 | 9 | 1 | 9 | 29 (22–35) |
| 2002 | 31 | 3 | 10–11 | 30 (24–37) |
| 2004 | 32 | 3 | 10–11 | 29 (20–34) |
| 2006 | 30 | 3 | 10 | 30 (19–40) |
| 2007 | 29 | 3 | 9–10 | 30 (21–39) |
| 2008 | 30 | 3 | 10 | 29 (20–35) |
| 2009 | 30 | 3 | 10 | 29 (22–39) |
| 2010 | 30 | 3 | 10 | 30 (20–41) |
| 2011 | 29 | 3 | 9–10 | 30 (21–38) |
| 2012 | 30 | 3 | 10 | 29 (21–38) |
| 2013 | 30 | 3 | 10 | 29 (22–39) |
| 2014 | 30 | 3 | 10 | 30 (20–38) |
| 2015 | 30 | 3 | 10 | 30 (22–38) |
| 2016 | 30 | 3 | 10 | 30 (24–36) |
| 2017 | 30 | 3 | 10 | 29 (21–34) |
Sample extraction
Each serum sample (0.5 mL) was transferred to a polypropylene centrifuge tube and spiked with 50 μL of internal standard mix (see Table S1, ESI†). The extraction involved addition of 4 mL acetonitrile, mixing using a vortex-mixer (Vortex-Genie 2 G560E, Scientific Industries), placing the sample in an ultra-sonic bath (USC-TH, VWR) and finally centrifuging (Stathmos C312). The procedure was repeated and the two extracts were combined, transferred to a new vial, and concentrated down to 1 mL under a stream of nitrogen (40 °C; using a Turbovap LV from Biotage). Extracts were subsequently purified with ∼25 mg graphitized non-porous carbon (Supelclean™ ENVI-Carb™ SPE Bulk packing purchased from Sigma-Aldrich) and 50 μL glacial acetic acid. Thereafter each sample was vortex-mixed (same model) and centrifuged (Galaxy 14D from Stathmos), then 500 μL of supernatant were transferred to a new vial. Lastly, 50 μL of recovery standard mix (M8PFOS and M8PFOA) and 200 μL of 4 mM ammonium acetate in water were added prior to instrumental analysis.Instrumental analysis
The extracts were analyzed using a Dionex UltiMate 3000 Ultrahigh performance liquid chromatograph (Thermo Scientific) coupled to a Q Exactive HF hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific) (detailed parameters are listed on Table S2, ESI†). Separation of analytes was carried out using an injection volume of 5 μL into a BEH C18 column (2.1 × 50 mm, 1.7 μm particle size; waters) with a guard column BEH C18 (2.1 × 5 mm, 1.7 μm; Waters). In addition, an “isolator column” XBridge™ C18 (2.1 × 50 mm, 3.5 μm; Waters) was mounted before the injector. Mobile phase A consisted of 95% purified milli-Q water and 5% acetonitrile with 2 mM ammonium acetate while mobile phase B consisted of 95% acetonitrile with 2 mM ammonium acetate and 5% purified milli-Q water. A gradient elution with a flow rate of 0.4 mL min−1 was applied. The initial conditions were 90% of solvent (A) and 10% of solvent (B). The percentage of B was linearly increased to 80% from 1 min to 8 min then increased sharply to 100% at 8 min and held at 100% B until 11 min. Initial conditions at 90% A 10% B were regained quickly after 11 min and held until 13 min for column equilibration. All samples were run in negative ionization, full scan mode (200–1200 Da) at a resolution of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 full width at half maximum (fwhm) with data-dependant MS2 fragmentation using an inclusion list comprising 453 suspects from the NORMAN list plus 62 targeted compounds (50 of which authentic standards were available), 22 internal standards and 2 recovery standards (a full list is provided in Table S1, ESI†).
000 full width at half maximum (fwhm) with data-dependant MS2 fragmentation using an inclusion list comprising 453 suspects from the NORMAN list plus 62 targeted compounds (50 of which authentic standards were available), 22 internal standards and 2 recovery standards (a full list is provided in Table S1, ESI†).
Targeted data processing was carried out using TraceFinder version 4.1 (Thermo Scientific Version). A 9-point calibration curve (0.01–150 ng mL; linear, 1/x weighting) was used for quantification of target PFAS. The lowest concentration calibration standard for which a well-shaped peak (i.e. a bell curve with minimum peak/noise ratio over 3 and without substantial signal observable in the blanks) was used as the limit of quantification (LOQ). Solvent blanks were injected regularly throughout the run to verify that carry-over was negligible. Where a signal was observable in procedural blanks, the LOQ was defined as the mean concentration detected in the blanks plus three times the standard deviation of the blanks.
Targeted method validation, intercomparison, and ongoing QC
Initial validation of the newly developed UHPLC-Orbitrap method was carried out using replicate spike/recovery experiments (2.5 ng of individual PFAS spiked into 0.5 mL serum; n = 4) along with analysis of NIST standard reference materials (n = 2). Thereafter, a method intercomparison was conducted whereby duplicate spiked samples and n = 20 individual (unspiked) human serum samples were processed by each of 3 methods: method A involved an ACN extraction (described above) followed by analysis using the newly developed UHPLC-Orbitrap method. Method B utilized the same ACN extraction as in method A, with analysis by ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS; see ESI† for details). Finally, method C utilized a previously reported solid phase extraction (SPE) procedure combined with UPLC-MS/MS analysis, and was designed for an expanded suite of PFAS, including polyfluoroalkyl phosphate diesters15 (see ESI† for details). Following method validation, time trend samples were processed together with extraction blanks (one per batch, empty tubes processed in the same manner as samples) and quality control (QC) samples (spiked and unspiked human serum samples).Fluorine mass balance
For measurement of extractable organofluorine (EOF), sample processing was the same as for targeted PFAS analysis (i.e. using 0.5 mL of serum) but was performed without addition of internal standards. Following extraction, samples were analysed using a Thermo-Mitsubishi combustion ion chromatograph (CIC), details of which can be found in Schultes et al.19 A brief overview is provided here. For EOF measurements, sample extracts (∼200 μL) were placed in a ceramic sample boat containing glass wool. For TF measurements, 150 μL of serum was weighed directly into the sample boat. The samples were combusted at 1100 °C under a flow of oxygen (400 L min−1) and argon mixed with water vapor (200 L min−1) for ∼5 minutes. Combustion gases were collected in 10 mL water in an absorber unit (GA-210, Mitsubishi), after which an aliquot of the absorption solution (200 μL) was injected onto the IC (Dionex Integrion HPIC, Thermo Fisher Scientific) which was equipped with an anion exchange column (Dionex IonPac AS19 2 × 50 mm guard column and 2 × 250 mm analytical column, 7.5 μm particle size) operated at 30 °C. The mobile phase consisted of a single hydroxide solution which was maintained at 8 mM for 9 min, then increased linearly to 100 mM by 11 min, followed by a 3 min hold and then back to 8 mM for equilibration (total run time 20 min). The flow rate was set to 0.25 mL min−1 over the course of the run and measurement of fluorine was achieved by conductivity detection.Samples for EOF analysis were prepared together with blanks and spiked samples (250 ng PFOS; n = 4). EOF concentrations in samples were recovery-corrected by dividing each value by the average PFOS recovery measured for spiked samples by CIC (i.e. 73%; RSD = 6.1%). Additional spike/recovery experiments (n = 4; 1.1 μg sodium fluoride) confirmed that inorganic fluorine was completely removed during extraction (i.e. 0% recovery). For assessment of accuracy and precision of TF data, replicate standard reference material (BCR®-461, fluorine in clay; n = 5) was analysed (94% recovery; RSD = 4.6%). For comparison to EOF and TF data, individual PFAS concentrations are converted to fluorine equivalents using eqn (1).
| CF_PFAS = nF × MWF/MWPFAS × CPFAS | (1) |
The total known extractable fluorine concentration (∑CF_PFAS; ng F g−1) was obtained by summing the fluorine concentrations from all individual PFAS (except perfluoro-4-ethylcyclohexanesulfonate (PFECHS), which was identified later by suspect screening). Thereafter, the concentration of unidentified, extractable organic fluorine (CF_extr.unknown; ng F g−1) was determined by subtracting ∑CF_PFAS from the total extractable organic fluorine concentration (CF_EOF; ng F g−1), according to eqn (2).
| CF_EOF = ∑CF_PFAS + CF_extr.unknown | (2) |
The TF concentration (CF_TF; ng F g−1) is the sum of CF_EOF and the total non-extractable fluorine concentration (CF_non extr.; ng F g−1), as shown in eqn (3).
| CF_TF = CF_EOF + CF_non extr. | (3) |
Time trends of target PFAS, EOF, and TF
For determination of sum (∑) PFAS concentrations, values below LOQ were replaced with 0. For evaluating time trends of individual PFAS, concentrations below LOQ were substituted with LOQ/√2. Time trends for EOF were only evaluated using samples containing EOF above the detection limit since substituting with LOQ or LOQ/√2 could lead to artificial gaps between sum PFAS and EOF concentrations. While it has been pointed out that removing data below LOQ can lead to positive bias20 this was mitigated by focusing on the percentage of known EOF (rather than the absolute EOF), and the fact that samples with detectable EOF were not just present at early or late time points (i.e. they occurred throughout the study period). To test for significant changes in concentrations from 1997–2017, log-linear regression analyses were carried out. An approach adapted from Sturludóttir et al.21 was used for change-point (CP) analysis. Prior to CP analysis, the data were screened for outliers. Concentrations with a residual from a regression line covering the entire time period were excluded if the residual exceeded 3 times the interquartile range (IQR) of all the residuals. This is a conservative approach and only a few observations were excluded from the CP test. To detect the CP, the entire time-series was repeatedly divided into two sections with at least three years per section. Thereafter, log-linear regression lines were fitted to each section and the residual variance was recorded for each combination. The combination of regression lines that gained the smallest variance was compared with a log-linear regression line for the entire study period and the mean for the whole time period with F-tests. The degrees of freedom were reduced to compensate for the less constrained situation of two regression lines compared to a single regression line. Only a single change-point was determined for each substance because the time-series were generally too short for several change-points. The median concentration for the tested change-point year was included in both parts of the time series. This is a conservative approach which reduces the influence of abrupt changes from one year to the next but may also reduce the chance to detect significant trends on either side of the change-point. The two parts may not necessarily point in different directions (i.e. increasing-decreasing) and may not show significant slopes separately (only significant regressions lines were plotted) but they still show a significant decrease in residual variance, i.e. they explain significantly more of the variation in contaminant concentration than the mean or a regression line for the entire period. For time series without a significant CP, log-linear regression was carried out.Suspect screening
Suspect screening was carried out using Compound Discoverer (CD) 3.0 (Thermo Scientific). Software parameters are listed in Table S3 (ESI†). CD handles peak alignment, peak picking, blank subtraction and comparison with databases (suspect list of exact masses and mzCloud). For the peaks matching a molecular mass in the suspect list (within 5 ppm), the MS2 spectra were further inspected. The correspondence of the MS2 data from CD with the suspect compound was assessed in silico with both Sirius and MetFrag.22,23Non-target time trend screening
We previously utilized increasing time trends as a prioritization strategy to identify emerging bioaccumulative contaminants in human whole blood.4,24 In the present work, we postulated that unknown substances co-occurring with PFHxS in the contaminated drinking water supply may mimic the time trends of PFHxS. Therefore, we prioritized features with time trends that matched PFHxS (i.e. increasing until 2010 then decreasing) in our non-target data using Spearman rank correlation for all features against PFHxS.Results and discussion
Quality control and method validation
Blank concentrations were typically below LOQ for most targets and were not subtracted from samples. Method accuracy and precision (assessed via spiking of 2.5 ng of individual PFAS into 0.5 mL serum; n = 4) were excellent for most compounds, with mean percent recoveries typically ranging between 70 and 130%. The exceptions were for FOSAA, 6:2/8:2 diPAP, 10:2 diPAP, and 3:3, 5:3 and 7:3 FTAs, which all displayed lower average recoveries (21–63%) and PFBA, PFTriDA, PFTeDA PFHxDA, PFOcDA, which displayed higher recoveries (147–154%; Table S4, ESI†). The sub-optimal performance for these targets is likely due to the absence of exactly-matched, isotopically-labelled internal standards. Nevertheless, precision remained excellent for these substances, as seen in Table S4 (ESI†), and therefore they were included in the analysis of temporal trends. Measurements of NIST Standard Reference Material 1957 revealed good consistency with certified concentrations and previous studies15,25,26 (Table S5, ESI†). Furthermore, deviation from the values in Gebbink et al. were mostly under 13%, with the exception of PFDoDA (43%), PFTriDA (38%) and FOSA (88%), which is attributable to the very low concentrations of these substances in the NIST material. Overall these data indicate good performance of the method. Finally, average internal standard recoveries ranged between 59 and 92%, with the exception of M4PFBA (5.9 ± 1.8%), M3PFPeA (19 ± 4.2%) and M2PFHxA (35 ± 13%; Table S4, ESI†).Method intercomparison
A comparison of data generated using the newly developed UHPLC-Orbitrap method (i.e. method A) with two existing methodologies (methods B and C; both involving a triple quadrupole mass spectrometer), used in Glynn et al. 2015 (ref. 27) and Gyllenhammar et al. 2017 (ref. 10) revealed consistent percent recoveries for spiked duplicates for targets which were included in all 3 methods (Table S6, ESI†). The exceptions were fluorotelomer sulfonates, which displayed better overall recoveries and repeatability using the Orbitrap method, and PFBA, which was over-reported using previous methods B and C (albeit, over-reporting of PFBA was also noted during our initial Oribtrap method validation; see Table S4, ESI†). Notably, PFTriDA, PFTeDA, and FTSs displayed poor recoveries using method C, likely due to matrix-induced ionization effects, which have been previously noted with this method.28 Determination of monoPAPs was not possible using the Oribtrap method due to the necessity of an ion pairing agent in the mobile phase which was only used in method C. A comparison of n = 20 unspiked serum samples using all three methods demonstrated strong correlation coefficients between each method (r2 typically greater than 0.9 for substances detected by all three methods), and slopes and intercepts of linear regressions which were in most cases close to or not significantly different from ideal values (i.e. 1 and 0, respectively; Table S7†). Overall these data further demonstrate consistency among methods. In general, the biggest performance differences among methods were observed for low-abundance substances (e.g. PFHpA, br-PFOA, PFBS, etc.), which were more consistently detected by the Oribtrap method, due to lower LOQs (typically an order of magnitude lower; see Table S8, ESI†).Target PFAS profiles
Across all samples, concentrations were below LOQ for PFPeA, PFDoDA, PFTeDA, PFHxDA, PFOcDA, MeFOSA, 11Cl-PF3OUdS, NaDONA, 3:3 FTA, 5:3 FTA, 7:3 FTA, 4:2 FTS, 6:2/8:2 diPAP, 8:2 diPAP and 10:2 diPAP. PFDS, FOSA, EtFOSA, EtFOSE, 9-Cl-PF3ONS, and 6:2 diPAP were detected intermittently (i.e. ≤10% of all samples) and were therefore not investigated further. The remaining targets were detected in >30% of samples.Sum (∑) PFAS concentrations ranged from 8.1 (2017) to 32 ng g−1 (1999) and were dominated by PFOS, PFOA, and PFHxS over the entire study period (Fig. 1). Considerable changes were observed in the relative profile of these 3 PFAS over the study period. For example, from 1996–2002, profiles consisted primarily of PFOS, followed by PFOA and PFHxS. After 2002, a concomitant increase in PFHxS and decrease in PFOS led to approximately equal concentrations of these homologues by 2010. After 2010, PFHxS and PFOS concentrations remained approximately equivalent with the remaining ∼15–30% of the profile made up of PFOA and longer-chain PFCAs. Notably, the PFOS-precursors FOSAA, MeFOSAA, and EtFOSAA were clearly observable from 1996–2002; thereafter the occurrence of these substances declined rapidly and they were generally not observable after 2010 (Tables S9–S11, ESI†). A detailed discussion surrounding the factors controlling influencing the PFAS profile are presented in the following sections.
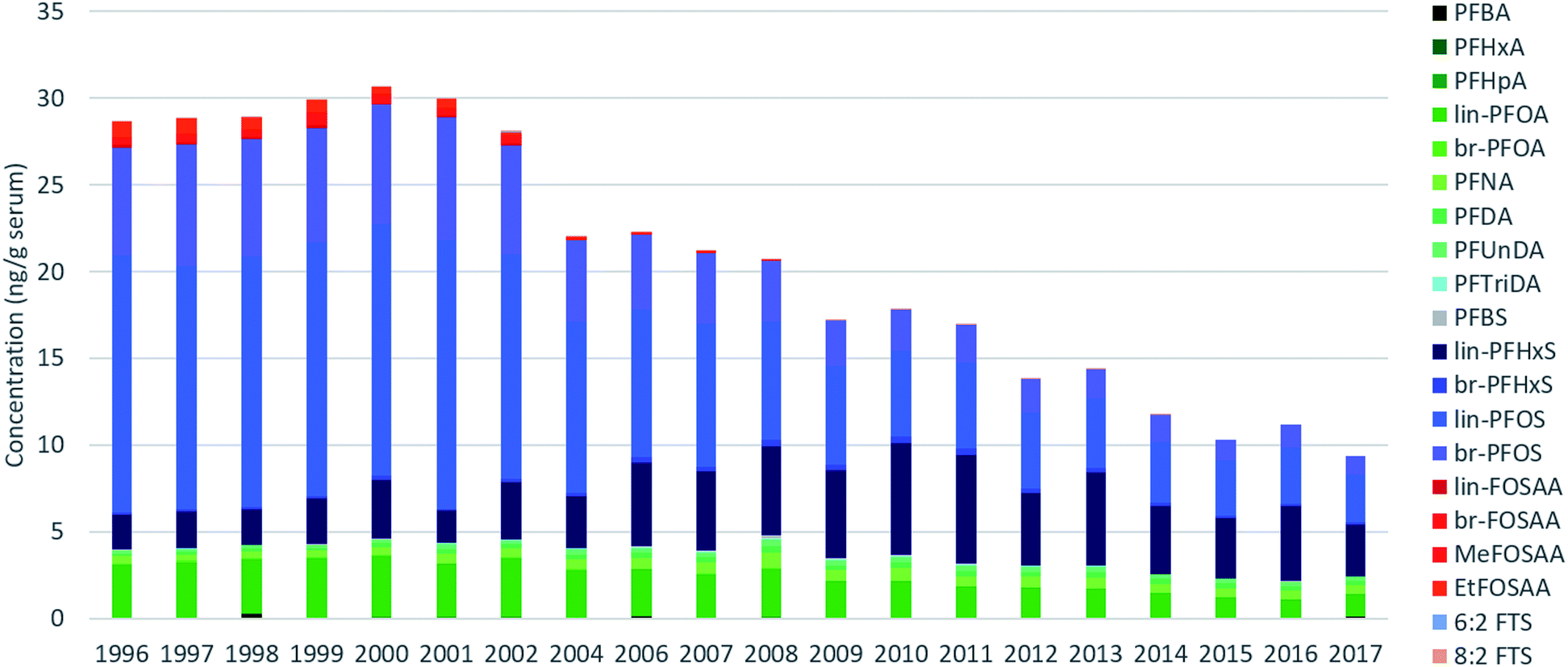 | ||
| Fig. 1 Average sum PFAS concentrations measured for each year in pooled human serum for PFAS detected in >30% of all samples. Concentrations below LOQs were replaced with 0. | ||
Target PFAS time trends
Among the PFSAs, statistically significant (i.e. p < 0.05) upwards-downwards trends were observed for PFBS, lin-PFHxS, br-PFHxS, lin-PFOS, and br-PFOS, with change-points occurring in 2007, 2011, 2010, 2001, and 2001, respectively (Fig. 2 and Table 2). The increasing PFBS and PFHxS trends at the beginning of the study period likely reflect increased PFAS contamination of the DW supply, originating from extensive use of aqueous film forming foams (AFFF) in certain areas of Uppsala.11 The PFAS contamination of the DW production wells was mitigated in 2012, but already in 2007 the DW distribution system of Uppsala was changed resulting in lower average PFBS and PFHxS concentrations in the areas that had previously received a high proportion of the contaminated drinking water.11 This change in DW distribution in Uppsala corresponds to the CP of PFBS (in 2007), br-PFHxS (in 2010) and lin-PFHxS (in 2011). The change-point of PFBS already in 2007 could be explained by the fact that PFBS is the most mobile and least biopersistent perfluoroalkyl sulfonic acid investigated here (serum elimination half-life of 25.8 days for PFBS vs. 1751 days for PFHxS and 2662 days for PFOS)29,30 and its concentration in human serum is therefore expected to respond more quickly to decreases in average exposure levels in 2007 than PFHxS (change-point 2010–11). The slightly earlier change-point for branched PFHxS isomers than of the linear isomer is likely due to the faster blood elimination kinetics of the branched isomers.11 The mitigation of PFAS contamination of the DW in 2012 explains the further decrease in PFBS and PFHxS towards the end of the study period (Fig. 2). Overall, the declining concentrations of PFHxS and PFBS highlights the effect of mitigation efforts on exposure to PFAS via drinking water in Uppsala. In contrast, the PFOS CP in 2001 likely reflects the phase-out of perfluorooctane sulfonyl fluoride-based substances in the early 2000s, which is also reflected in the rapid decline of PFOS precursors (FOSAA, MeFOSAA, and EtFOSAA) throughout the study period (average declines of 6.5–28% per year). This sharp drop in PFAA-precursor concentrations highlights the rapid response to phase-out initiatives in humans. Interestingly, the change-point for PFOS observed in this study is considerably later than that observed in human milk from Stockholm (1988), assessed between 1972 and 2016,31 most likely due to the additional PFOS exposure from DW in Uppsala.11| Target | N tot | Yrs | Year | Log-linear regression statistics | Change-point statistics | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Trend (%) | 95% C.I. | P(LR) | YRQ | LDT | CP | P(CP) | ||||
| a Detected by suspect screening. Not included in F mass balance calculations. | ||||||||||
| PFBA | 56 | 20 | 96–17 | −3.8 | (−9.6,2.4) | 0.3916 | 35 | 50 | 2000 | 0.0381 |
| PFHxA | 57 | 20 | 96–17 | −2 | (−3.6,−0.39) | 0.0498 | 15 | 10 | 2000 | 0.4985 |
| PFHpA | 57 | 20 | 96–17 | −5.2 | (−6.4,−3.9) | <0.0001 | 13 | 7.9 | 2004 | 0.0077 |
| lin-PFOA | 57 | 20 | 96–17 | −4.9 | (−5.6,−4.1) | <0.0001 | 9 | 4.4 | 2002 | 0.0002 |
| br-PFOA | 57 | 20 | 96–17 | −12 | (−13,−11) | <0.0001 | 15 | 9.8 | 2011 | 0.0537 |
| PFNA | 57 | 20 | 96–17 | 1.7 | (0.75,2.7) | 0.0193 | 10 | 5.4 | 2007 | <0.0001 |
| PFDA | 57 | 20 | 96–17 | 3 | (1.9,4.1) | 0.0009 | 11 | 6.3 | 2004 | 0.0001 |
| PFUnDA | 57 | 20 | 96–17 | 2.4 | (1.5,3.4) | 0.0005 | 11 | 5.5 | 2008 | 0.0011 |
| PFTriDA | 57 | 20 | 96–17 | 0.16 | (−2.0,2.3) | 1 | 18 | 13 | 2009 | <0.0001 |
| PFBS | 56 | 20 | 96–17 | −0.57 | (−2.3,1.2) | 0.6718 | 16 | 11 | 2007 | 0.0005 |
| lin-PFHxS | 57 | 20 | 96–17 | 3.6 | (2.2,5.1) | 0.0017 | 13 | 8.2 | 2011 | 0.0001 |
| br-PFHxS | 57 | 20 | 96–17 | 1.9 | (−0.08,3.9) | 0.2375 | 17 | 12 | 2010 | <0.0001 |
| lin-PFOS | 57 | 20 | 96–17 | −8.4 | (−9.0,−7.8) | <0.0001 | 9 | 3.9 | 2001 | <0.0001 |
| br-PFOS | 57 | 20 | 96–17 | −9 | (−9.8,−8.3) | <0.0001 | 10 | 4.8 | 2001 | <0.0001 |
| lin-FOSAA | 57 | 20 | 96–17 | −6.5 | (−7.5,−5.4) | <0.0001 | 12 | 6.4 | 2004 | <0.0001 |
| br-FOSAA | 57 | 20 | 96–17 | −27 | (−30,−23) | <0.0001 | 31 | 37 | 2007 | 0.0017 |
| EtFOSA | 57 | 20 | 96–17 | −16 | (−18,−15) | <0.0001 | 17 | 13 | 2009 | 0.0279 |
| MeFOSAA | 57 | 20 | 96–17 | −28 | (−30,−26) | <0.0001 | 22 | 19 | 2011 | 0.0001 |
| EtFOSAA | 57 | 20 | 96–17 | −8 | (−12,−4.2) | 0.0041 | 27 | 28 | 2004 | 0.1034 |
| 6:2 FTS | 57 | 20 | 96–17 | −9.6 | (−12,−6.9) | 0.0005 | 22 | 19 | 2004 | <0.0001 |
| 8:2 FTS | 56 | 20 | 96–17 | −3.8 | (−9.6,2.4) | 0.3916 | 35 | 50 | 2000 | 0.0381 |
| EOF | 19 | 12 | 96–16 | −0.86 | (−3.4,1.7) | 0.5261 | 19 | 17 | 2004 | 0.6944 |
| TF | 57 | 20 | 96–17 | −3.2 | (−4.5,−1.8) | 0.0033 | 13 | 8.3 | 2008 | 0.6517 |
| ∑PFAS (F-equivalents) | 57 | 20 | 96–17 | −5.4 | (−6.0,−4.8) | <0.0001 | 8 | 3.7 | 2001 | 0.0001 |
| % EOF explained by PFAS | 19 | 12 | 96–16 | −3.9 | (−6.7,−1.1) | 0.0252 | 21 | 19 | 2000 | 0.1585 |
| % TF explained by PFAS | 57 | 20 | 96–17 | −2.3 | (−3.8,−0.70) | 0.0409 | 15 | 9.7 | 2009 | 0.1148 |
| PFECHSa | 57 | 20 | 96–17 | −6.7 | (−7.2,−6.1) | <0.001 | 16 | 3.4 | 2000 | 0.011 |
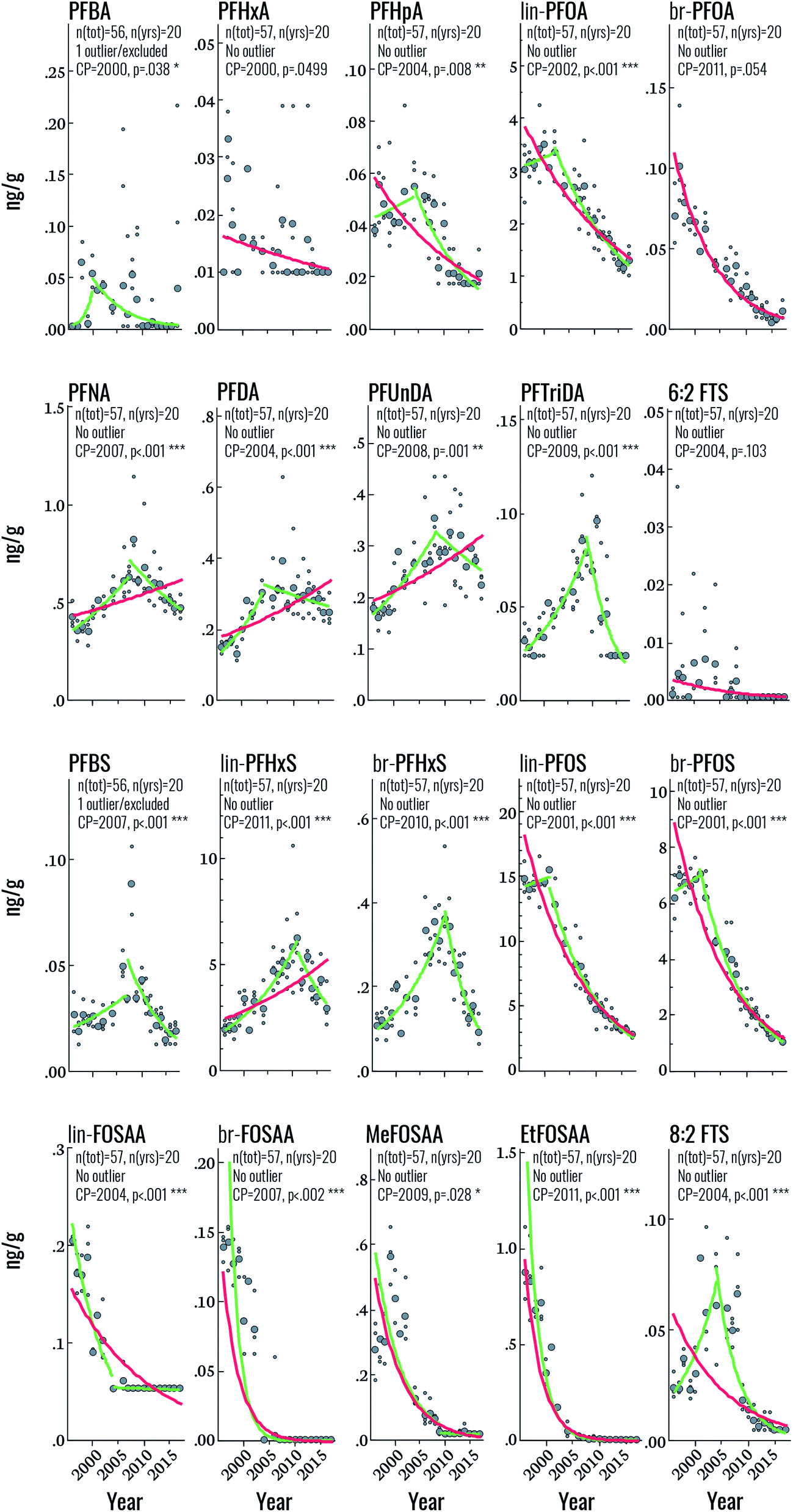 | ||
| Fig. 2 Temporal trends of individual PFAS (in units of ng g−1 blood serum). Large circles represent annual geometric means, small circles represent individual samples. Curves represent significant (p < 0.05) log-linear (red), and change-point (green) trends. | ||
Among the PFCAs for which temporal trend analysis was performed, all but PFHxA and br-PFOA (which both declined throughout the study period) displayed statistically significant upwards-downwards trends (Fig. 2). CPs occurred in the years 2000 (PFBA), 2002 (PFOA), 2004 (PFHpA, PFDA), 2007 (PFNA), 2008 (PFUnDA), and 2009 (PFTriDA) (Fig. 2 and Table 2). The declining trends in PFOA from 2002 and earlier are consistent with those of PFOS and likely also reflect the 2002 3M phase-out, when production of ECF PFOA (the source of branched PFOA isomers) was halted, and eventually replaced by telomerized (i.e. strictly linear) PFOA.32 The PFOA Stewardship Program, initiated in 2006, may have also contributed to the observed decline.33 The trends and CP for PFOA observed here also coincide with those observed for PFOA in human milk sampled from 1972–2016 in Stockholm (CP = 2000).31 The change point for C9, C11, C13 PFCAs (i.e. beginning 2007–2009) also indicates that exposure sources of these PFCAs have been restricted, but the delay relative to PFOA suggests that regulation of these substances occurred later than PFOA. Notably, Land et al. reviewed PFAS temporal trend data published up until 2015 and concluded that there was no evidence for significant declining trends in PFNA, PFDA, PFUnDA, PFDoDA, PFTrDA and PFTeDA in human sample at any global location.34 In the trend study on human milk from Stockholm, PFDA and PFTriDA did not display a change-point; instead these substances increased throughout the study period, while PFUnDA showed a CP in 1984, albeit with non-significant trends before and after this time. As mentioned before, differences in study design and sampling frequency may contribute to discrepancies between studies. The CP for PFNA in the present study (2007) is fairly consistent with that in human milk (2010).
Finally, both 6:2 and 8:2 FTS displayed CPs in 2004 (upwards–downwards), but only 8:2 FTS was statistically significant. Over the entire time period, statistically significant declines were observed for both substances, at a rate of 8 and 9.6% per year, respectively. Few data are available on FTS temporal trends but a recent report highlighted the widespread occurrence of FTSs in the Nordic environment, including sediment, groundwater, indoor dust and biota.35 FTSs generally occur at low concentrations with the exception of samples collected close to sites contaminated with AFFF. To the best of our knowledge, FTSs have not been reported previously in human serum from the Nordics; however, serum collected in 2009 from 50 individuals from the US contained 8:2 FTS at concentrations ranging from <0.005–0.231 ng mL−1 (detection in >95% of samples) and 6:2 FTS at concentrations ranging from <0.005–0.047 ng mL−1 (detection in >54% of samples).36 Sources of exposure to FTSs are thought to include commercial products containing FTSs (e.g. AFFF) or fluorotelomer mercaptoalkyl phosphate diester (e.g. food packaging).36
A comparison of time trends reported here to those of Glynn et al.,27 Gebbink et al.15 and Gyllenhammar et al.10 for the same cohort shows good overall agreement with a few exceptions (Fig. S1 and S2, ESI).† Among the carboxylic acids, PFOA, br-PFOA PFNA, PFDA and PFUnDA are well correlated (Pearson r > 0.78) with data from previous studies (Fig. S1†). Time series for PFBA and for PFHxA are poorly correlated due to low detection in the present study and relatively large standard deviations per year in previous studies. Correlation for PFHpA is also poor (r = −0.191), probably due to higher values detected for specific years (notably 2002, 2007 and 2009) in previous studies. PFDA, PFUnDA and PFDoDA show average correlation (r = 0.43–0.64). The trends are similar, but the deviations are high in both sets for most years. For PFDoDA, the present study found a similar trend but with lower concentrations. For PFTriDA, this study shows a steep increase around 2010 followed by a sharp decrease whereas values from previous studies suggest a steady increase throughout the period.
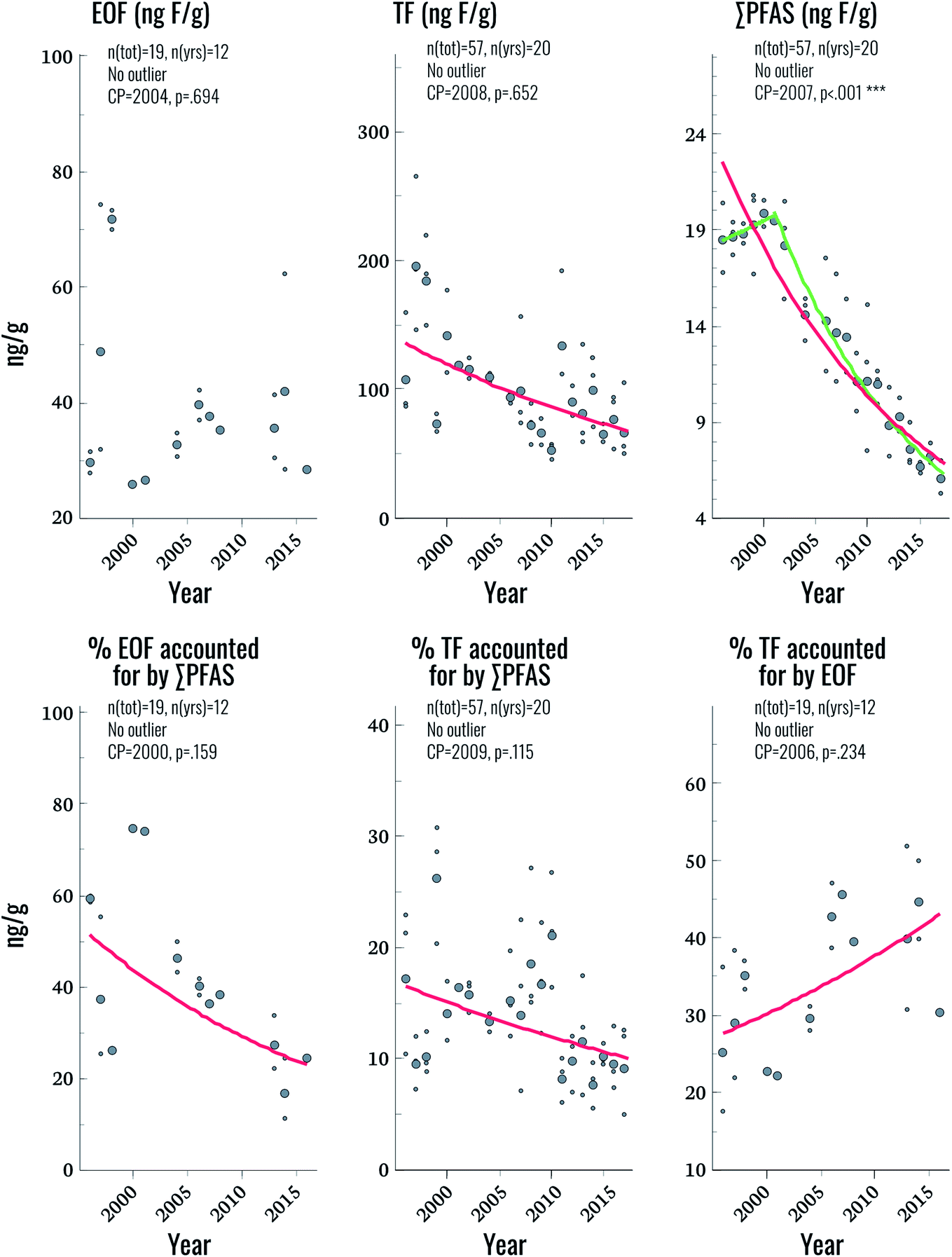 | ||
| Fig. 3 Temporal trends of EOF, TF, and ∑PFAS (in units of ng F g−1 blood serum) as well as % EOF accounted for by ∑PFAS, % of TF accounted for by ∑PFAS, and % of TF accounted for by EOF. Large circles represent annual geometric means, small circles represent individual samples. Curves represent significant (p < 0.05) log-linear (red), and change-point (green) trends. | ||
The correlation coefficients for sulfonamides (FOSAA, MeFOSAA and EtFOSAA) and for PFOS (branched and linear) measured in the present study vs. in previous studies were all over 0.91 and displayed similar time trends, indicating good accordance between the studies (Fig. S2†). The observed values are close across studies for the mentioned compounds except for FOSAA where the observations from this study are about a third of the values recorded previously. PFHxS (branched and linear) has a higher standard deviation per year in both studies. This results in lower correlation between the two study datasets (0.88 for PFHxS and 0.94 for br-PFHxS), even though time trends in Fig. S2† show close accordance. Correlations for PFDS and PFBS indicate poor agreement between studies (0.12 and −0.28). The former can be explained partly by limited observations prior to 2000 for this study with large coefficient of variation. The latter arises mainly from differences in the period 2005–2010. During that period, both present and previous studies recorded slightly higher values than during the rest of the studied period. However, the increase was more marked in the present study, leading to a discrepancy in the time frames, and thus, poorer correlation.
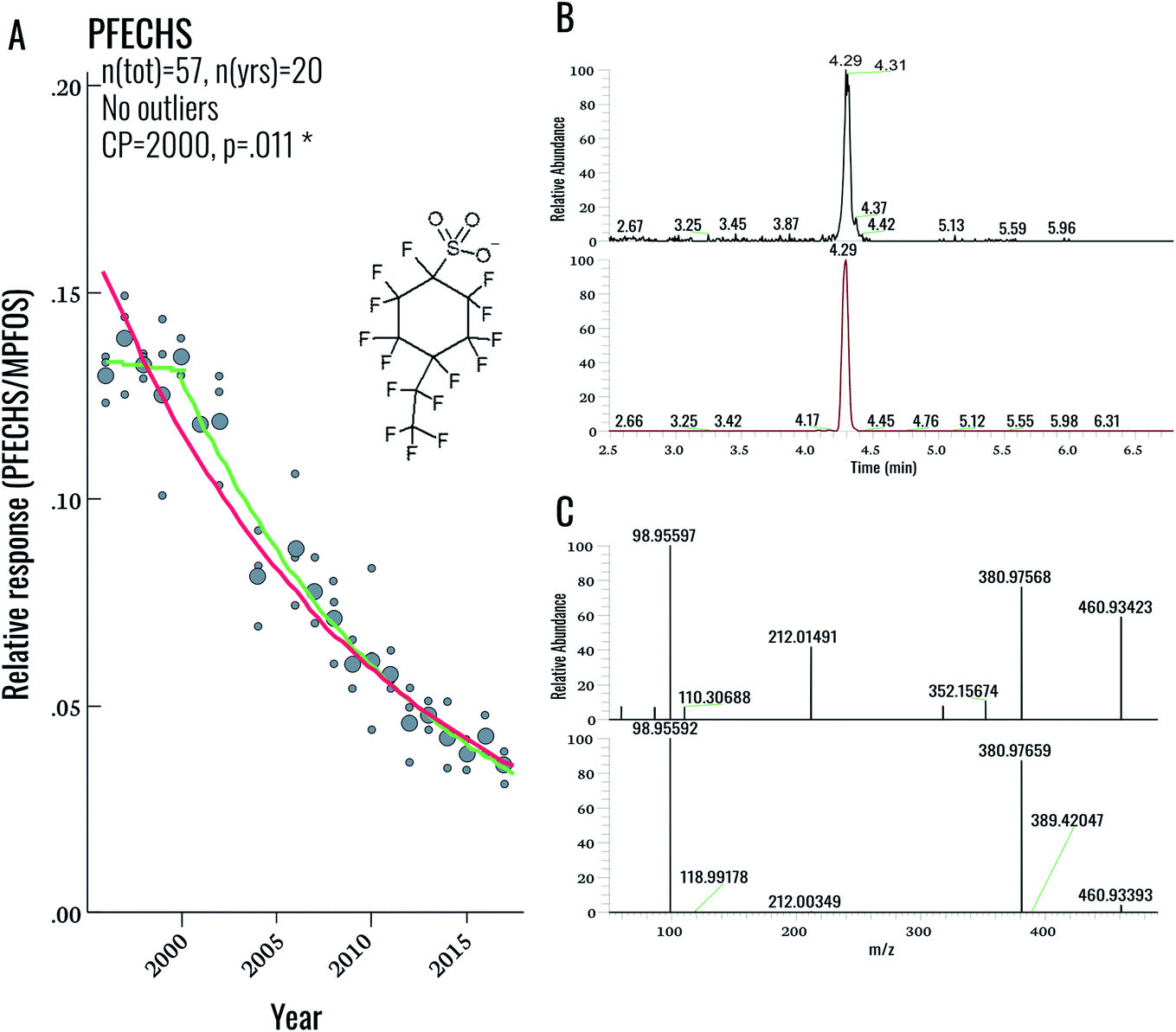 | ||
| Fig. 4 (A) Temporal trend and structure of PFECHS. Large circles represent annual geometric means, small circles represent individual samples. Curves represent significant (p < 0.05) log-linear (red), and change-point (green) trends. (B) Chromatogram of PFECHS in sample (top) vs. standard (bottom). (C) MS/MS spectrum of PFECHS in a real sample (top) versus standard (bottom). | ||
Finally, for 6:2 FTS, there were too few observations to compare between the studies. For 8:2 FTS the correlation is high for the comparable observations from 2007–2017. F-53B (9Cl-PF3ONS and 11Cl-PF3OUdS) datasets are both widely spread and show poor correlation.
Fluorine mass balance
EOF was above method detection limits (25 ng F g−1 serum) in only 33% of samples, hampering comparisons to TF and ∑CF_PFAS for the entire time period. Future work will focus on lowering detection limits to improve applicability to the general population. Nevertheless, some observations could be made by focusing on samples with detectable levels of EOF. Among these samples, the proportion of EOF explained by target PFAS ranged from 11–75%. Significant trends were not observed for EOF, despite the fact that significant declining trends in TF and ∑CF_PFAS were observed (3.2% and 5.4% per year, respectively; Fig. 3 and Table 2). The lack of a trend in EOF and the declining trends in ∑CF_PFAS resulted in a decline of 3.9% per year in the percentage of EOF explained by target PFAS over the study period (p = 0.025). This implies that samples collected in more recent years have a larger fraction of unknown EOF compared to older samples. The percentage of TF accounted for by EOF also increased significantly (2.2% per year) indicating that, collectively, known and unknown PFAS make up a larger percentage of TF in more recent years. Overall these findings corroborate observations in human plasma from Münster, Germany, which also showed an increasing proportion of unidentified organofluorine in samples collected after the year 2000.7 We are unaware of any PFAS not included in the present work which display increasing time trends over the last decade in human serum; therefore, the cause of this increase in unidentified organofluorine remains unclear at this time.Suspect screening
During our initial examination of the suspect screening data, a total of 5 exact masses were matched within 5 ppm mass error to suspects in our database (excluding targeted compounds). Subsequent inspection of MS2 data for these features ruled out all five tentative assignments. This result was not completely surprising, given the overall low PFAS concentrations in this population. Nonetheless a follow-up investigation of our suspect screening data using lower peak thresholds (carried out following the original submission of this manuscript) revealed the presence of an exact mass (460.93423) and MS/MS spectra corresponding to PFECHS. After obtaining a standard of PFECHS from Wellington Labs (Guelph, ON, Canada) and running it together with a serum sample, we confirmed the assignment (Fig. 4). PFECHS concentrations ranged from approximately 0.06–0.28 ng g−1, which would contribute to only a small fraction (i.e. an additional 0.03–0.21%) of our known EOF estimates (note that our original EOF calculations did not include PFECHS). However, since these concentrations are semi-quantitative they require confirmation using a method validated for PFECHS. Our data builds on the initial discovery of PFECHS in human serum from Sweden by Weiss et al.37 by demonstrating that human exposure to PFECHS occurred primarily during the 1990s-early 2000s and has been declining since the year 2000 (−6.7% per year from 1996–2017; p < 0.001; Table 2). PFECHS was also detected recently in 86% of Baltic sea water samples and was previously reported in fish and surface water from Canada,38,39 water, sediment, and fish from China,40,41 and water and snow from the High Arctic.42,43 According to De Silva et al. (2011), PFECHS was used as an erosion inhibitor in aircrafts and was phased out of production by the 3M Co. in 2002 along with PFOS and other perfluorooctyl-based substances.38 The phase-out of this product appears to align with our observed time trends in human serum from Sweden.Overall, this finding highlights the value in collecting NTA data that can be retrospectively mined. As PFAS databases are further developed, the present dataset can be continuously re-processed without re-analysing samples. The cost- and time-savings associated with retrospective mining (as opposed to sample re-analysis) represents a considerable advantage for NTA-based workflows.
Non-target time trend screening
Four targets (PFBS, PFNA, PFDA and PFTriDA) and three unidentified features (neutral masses 422.2307, 396.2066, and 436.3554) displayed time trends matching that of PFHxS (Spearman's rho > 0.5; Table 3). Unfortunately, MS2 data were not collected for the unidentified features, precluding structural elucidation. Nevertheless, it is notable that feature A2 (neutral mass 396.2066) displayed a chlorine isotope pattern. Previously Rotander et al. reported Cl-PFHxS in Australian firefighters who were exposed to AFFF, but the exact mass of Cl-PFHxS did not match that of feature A2.9 While we cannot confirm that any of the unidentified features were fluorinated, the consistency of their time trends to PFHxS is notable, and warrants further investigation.|
|
||||
|---|---|---|---|---|
| Trend matching PFHxS (Spearman's ρ > 0.5) | ||||
| ID | Exact mass (neutral) | ρ | RT (min) | Comment |
| A1 | 422.2307 | 0.63 | 3.84 | |
| A2 | 396.2066 | 0.58 | 7.16 | Cl isotope pattern |
| A3 | 436.3554 | 0.56 | 9.60 | |
| A4 | 463.9702 | 0.65 | 5.54 | PFNA |
| A5 | 299.9503 | 0.65 | 3.76 | PFBS |
| A6 | 663.9577 | 0.61 | 7.27 | PFTriDA |
| A7 | 513.9667 | 0.54 | 6.00 | PFDA |
Conclusions
Overall, the PFAS concentrations in our cohort were fairly low, despite receiving PFAS exposure additional to that of the general population. Moreover, declining trends were observed for all PFAS as well as EOF and TF, with homologue-dependent differences in year of onset of decline. While fluorine mass balance data indicate that the full extent of exposure to PFAS is not fully elucidated, further improvement in PFAS databases will help to identify this missing organofluorine. For now, we detected one novel PFAS (PFECHS) and several unknowns through suspect/non-target screening which should be included/prioritized in future studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish EPA (Environmental Protection Agency) is acknowledged for financial support. Appreciation is expressed to the participating women and to the midwives who assisted in recruitment, interviewing, and sampling. Ellen Edgren and Jane Karlsdotter are thanked for technical assistance.References
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins, Integr. Environ. Assess. Manage., 2011, 7(4), 513–541, DOI:10.1002/ieam.258.
- A. Ritscher, Z. Wang, M. Scheringer, J. M. Boucher, L. Ahrens, U. Berger, S. Bintein, S. K. Bopp, D. Borg, A. M. Buser, I. Cousins, J. DeWitt, T. Fletcher, C. Green, D. Herzke, C. Higgins, J. Huang, H. Hung, T. Knepper, C. S. Lau, E. Leinala, A. B. Lindstrom, J. Liu, M. Miller, K. Ohno, N. Perkola, Y. Shi, L. Småstuen Haug, X. Trier, S. Valsecchi, K. van der Jagt and L. Vierke, Zürich Statement on Future Actions on Per- and Polyfluoroalkyl Substances (PFASs), Environmental Health Perspectives, 2018, 126(8), 84502, DOI:10.1289/ehp4158.
- OECD (Organisation for Economic Co-operation and Development), Toward a New Comprehensive Global Database of Per- and Polyfluoroalkyl Substances (PFASs): Summary Report On Updating the OECD 2007 List of Per- and Polyfluoroalkyl Substances (PFASs), OECD, Paris, 2018 Search PubMed.
- M. M. Plassmann, S. Fischer and J. P. Benskin, Nontarget Time Trend Screening in Human Blood, Environ. Sci. Technol. Lett., 2018, 5(6), 335–340, DOI:10.1021/acs.estlett.8b00196.
- L. W. Y. Yeung, Y. Miyake, S. Taniyasu, Y. Wang, H. Yu, M. K. So, G. Jiang, Y. Wu, J. Li, J. P. Giesy, N. Yamashita and P. K. S. Lam, Perfluorinated Compounds and Total and Extractable Organic Fluorine in Human Blood Samples from China, Environ. Sci. Technol., 2008, 42(21), 8140–8145, DOI:10.1021/es800631n.
- Y. Miyake, N. Yamashita, M. K. So, P. Rostkowski, S. Taniyasu, P. K. S. Lam and K. Kannan, Trace analysis of total fluorine in human blood using combustion ion chromatography for fluorine: A mass balance approach for the determination of known and unknown organofluorine compounds, J. Chromatogr. A, 2007, 1154(1), 214–221, DOI:10.1016/j.chroma.2007.03.084.
- L. W. Y. Yeung and S. A. Mabury, Are humans exposed to increasing amounts of unidentified organofluorine?, Environ. Chem., 2016, 13(1), 102–110, DOI:10.1071/EN15041.
- Y. Liu, L. A. D'Agostino, G. Qu, G. Jiang and J. W. Martin, High-resolution mass spectrometry (HRMS) methods for nontarget discovery and characterization of poly- and per-fluoroalkyl substances (PFASs) in environmental and human samples, TrAC, Trends Anal. Chem., 2019, 121, 115420, DOI:10.1016/j.trac.2019.02.021.
- A. Rotander, A. Kärrman, L.-M. L. Toms, M. Kay, J. F. Mueller and M. J. Gómez Ramos, Novel Fluorinated Surfactants Tentatively Identified in Firefighters Using Liquid Chromatography Quadrupole Time-of-Flight Tandem Mass Spectrometry and a Case-Control Approach, Environ. Sci. Technol., 2015, 49(4), 2434–2442, DOI:10.1021/es503653n.
- I. Gyllenhammar, A. Glynn, J. Benskin, O. Sandblom, A. Bignert and S. Lignell, Temporal trends of poly- and perfluoroalkyl substances (PFASs) in pooled serum samples from first-time mothers in Uppsala 1997–2016, Livsmedelsverket, Uppsala, 2017 Search PubMed.
- I. Gyllenhammar, U. Berger, M. Sundström, P. McCleaf, K. Eurén, S. Eriksson, S. Ahlgren, S. Lignell, M. Aune, N. Kotova and A. Glynn, Influence of contaminated drinking water on perfluoroalkyl acid levels in human serum – A case study from Uppsala, Sweden, Environ. Res., 2015, 140, 673–683, DOI:10.1016/j.envres.2015.05.019.
- Livsmedelsverket, PFAS, in drinking water and fish - risk management, 2018 Search PubMed.
- I. Gyllenhammar, J. P. Benskin, O. Sandblom, U. Berger, L. Ahrens, S. Lignell, K. Wiberg and A. Glynn, Perfluoroalkyl Acids (PFAAs) in Children's Serum and Contribution from PFAA-Contaminated Drinking Water, Environ. Sci. Technol., 2019, 53(19), 11447–11457, DOI:10.1021/acs.est.9b01746.
- I. Gyllenhammar, J. P. Benskin, O. Sandblom, U. Berger, L. Ahrens, S. Lignell, K. Wiberg and A. Glynn, Perfluoroalkyl Acids (PFAAs) in Serum from 2–4-Month-Old Infants: Influence of Maternal Serum Concentration, Gestational Age, Breast-Feeding, and Contaminated Drinking Water, Environ. Sci. Technol., 2018, 52(12), 7101–7110, DOI:10.1021/acs.est.8b00770.
- W. A. Gebbink, A. Glynn and U. Berger, Temporal changes (1997–2012) of perfluoroalkyl acids and selected precursors (including isomers) in Swedish human serum, Environ. Pollut., 2015, 199, 166–173, DOI:10.1016/j.envpol.2015.01.024.
- A. Glynn, U. Berger, A. Bignert, S. Ullah, M. Aune, S. Lignell and P. O. Darnerud, Perfluorinated Alkyl Acids in Blood Serum from Primiparous Women in Sweden: Serial Sampling during Pregnancy and Nursing, And Temporal Trends 1996–2010, Environ. Sci. Technol., 2012, 46(16), 9071–9079, DOI:10.1021/es301168c.
- A. Glynn, M. Aune, P. O. Darnerud, S. Cnattingius, R. Bjerselius, W. Becker and S. Lignell, Determinants of serum concentrations of organochlorine compounds in Swedish pregnant women: a cross-sectional study, Environ. Health, 2007, 6(1), 2, DOI:10.1186/1476-069X-6-2.
- S. Lignell, M. Aune, P. O. Darnerud, S. Cnattingius and A. Glynn, Persistent organochlorine and organobromine compounds in mother's milk from Sweden 1996–2006: compound-specific temporal trends, Environ. Res., 2009, 109(6), 760–767, DOI:10.1016/j.envres.2009.04.011.
- L. Schultes, R. Vestergren, K. Volkova, E. Westberg, T. Jacobson and J. P. Benskin, Per- and polyfluoroalkyl substances and fluorine mass balance in cosmetic products from the Swedish market: implications for environmental emissions and human exposure, Environ. Sci.: Processes Impacts, 2018, 20(12), 1680–1690, 10.1039/C8EM00368H.
- R. A. Hites, Correcting for Censored Environmental Measurements, Environ. Sci. Technol., 2019, 53(19), 11059–11060, DOI:10.1021/acs.est.9b05042.
- E. Sturludóttir, H. Gunnlaugsdottir, O. K. Nielsen and G. Stefansson, Detection of a changepoint, a mean-shift accompanied with a trend change, in short time-series with autocorrelation, Communications in Statistics – Simulation and Computation, 2017, 46(7), 5808–5818, DOI:10.1080/03610918.2014.1002849.
- K. Dührkop and S. Böcker, Fragmentation Trees Reloaded BT - Research in Computational Molecular Biology, ed. T. M. Przytycka, Springer International Publishing, Cham, 2015, pp. 65–79 Search PubMed.
- C. Ruttkies, E. L. Schymanski, S. Wolf, J. Hollender and S. Neumann, MetFrag relaunched: incorporating strategies beyond in silico fragmentation, J. Cheminf., 2016, 8(1), 3, DOI:10.1186/s13321-016-0115-9.
- M. M. Plassmann, E. Tengstrand, K. M. \AAberg and J. P. Benskin, Non-target time trend screening: a data reduction strategy for detecting emerging contaminants in biological samples, Anal. Bioanal. Chem., 2016, 408(16), 4203–4208, DOI:10.1007/s00216-016-9563-3.
- L. W. Y. Yeung, S. J. Robinson, J. Koschorreck and S. A. Mabury, Part I. A Temporal Study of PFCAs and Their Precursors in Human Plasma from Two German Cities 1982–2009, Environ. Sci. Technol., 2013, 47(8), 3865–3874, DOI:10.1021/es303716k.
- L. W. Y. Yeung, S. J. Robinson, J. Koschorreck and S. A. Mabury, Part II. A Temporal Study of PFOS and Its Precursors in Human Plasma from Two German Cities in 1982–2009, Environ. Sci. Technol., 2013, 47(8), 3875–3882, DOI:10.1021/es4004153.
- A. Glynn, J. P. Benskin, I. Gyllenhammar, M. Aune, T. Cantillana and O. Sandblom, Temporal trends of perfluoroalkyl substances (PFAS) in individual serum samples from first-time mothers in Uppsala 1996-2014, Livsmedelsverket, Uppsala, 2015 Search PubMed.
- S. Ullah, T. Alsberg and U. Berger, Simultaneous determination of perfluoroalkyl phosphonates, carboxylates, and sulfonates in drinking water, J. Chromatogr. A, 2011, 1218(37), 6388–6395, DOI:10.1016/j.chroma.2011.07.005.
- G. W. Olsen, S.-C. Chang, P. E. Noker, G. S. Gorman, D. J. Ehresman, P. H. Lieder and J. L. Butenhoff, A comparison of the pharmacokinetics of perfluorobutanesulfonate (PFBS) in rats, monkeys, and humans, Toxicology, 2009, 256(1), 65–74, DOI:10.1016/j.tox.2008.11.008.
- G. W. Olsen, J. M. Burris, D. J. Ehresman, J. W. Froehlich, A. M. Seacat, J. L. Butenhoff and L. R. Zobel, Half-Life of Serum Elimination of Perfluorooctanesulfonate,Perfluorohexanesulfonate, and Perfluorooctanoate in Retired Fluorochemical Production Workers, Environmental Health Perspectives, 2007, 115(9), 1298–1305, DOI:10.1289/ehp.10009.
- E. Nyberg, R. Awad, A. Bignert, C. Ek, G. Sallsten and J. P. Benskin, Inter-individual, inter-city, and temporal trends of per- and polyfluoroalkyl substances in human milk from Swedish mothers between 1972 and 2016, Environ. Sci.: Processes Impacts, 2018, 20(8), 1136–1147, 10.1039/C8EM00174J.
- J. P. Benskin, A. O. De Silva and J. W. Martin, Isomer Profiling of Perfluorinated Substances as a Tool for Source Tracking: A Review of Early Findings and Future Applications, in Reviews of Environmental Contamination and Toxicology Volume 208: Perfluorinated alkylated substances, ed. P. De Voogt, Springer, New York, New York, NY, 2010, pp. 111–160 Search PubMed.
- United States Environmental Protection Agency (EPA), Fact Sheet: 2010/2015 PFOA Stewardship Program, 2016 Search PubMed.
- M. Land, C. A. de Wit, A. Bignert, I. T. Cousins, D. Herzke, J. H. Johansson and J. W. Martin, What is the effect of phasing out long-chain per- and polyfluoroalkyl substances on the concentrations of perfluoroalkyl acids and their precursors in the environment? A systematic review, Environmental Evidence, 2018, 7(1), 4, DOI:10.1186/s13750-017-0114-y.
- S. Posner, S. Roos, P. Brunn Poulsen, H. Ólína Jörundsdottir, H. Gunnlaugsdóttir, D. Xenia Trier, A. Astrup Jensen, A. A. Katsogiannis, D. Herzke, E. Cecilie Bonefeld-Jörgensen, C. Jönsson, G. Alsing Pedersen, M. Ghisari and S. Jensen, Per- and polyfluorinated substances in the Nordic Countries: Use, occurence and toxicology, Nordic Council of Ministers, Copenhagen, 2013 Search PubMed.
- H. Lee and S. A. Mabury, A Pilot Survey of Legacy and Current Commercial Fluorinated Chemicals in Human Sera from United States Donors in 2009, Environ. Sci. Technol., 2011, 45(19), 8067–8074, DOI:10.1021/es200167q.
- J. Weiss, M. Öst, H. Håkansson, M. Herlin, L. Rylander, M. Plassmann and J. P. Benskin, Target-, and suspect screening of per- and polyfluoroalkyl substances (PFASs) in serum from an elderly population from the Swedish East coast with high fish intake, in preparation.
- A. O. De Silva, C. Spencer, B. F. Scott, S. Backus and D. C. G. Muir, Detection of a Cyclic Perfluorinated Acid, Perfluoroethylcyclohexane Sulfonate, in the Great Lakes of North America, Environ. Sci. Technol., 2011, 45(19), 8060–8066, DOI:10.1021/es200135c.
- S. R. de Solla, A. O. De Silva and R. J. Letcher, Highly elevated levels of perfluorooctane sulfonate and other perfluorinated acids found in biota and surface water downstream of an international airport, Hamilton, Ontario, Canada, Environ. Int., 2012, 39(1), 19–26, DOI:10.1016/j.envint.2011.09.011.
- Y. Wang, R. Vestergren, Y. Shi, D. Cao, L. Xu, Y. Cai, X. Zhao and F. Wu, Identification, Tissue Distribution, and Bioaccumulation Potential of Cyclic Perfluorinated Sulfonic Acids Isomers in an Airport Impacted Ecosystem, Environ. Sci. Technol., 2016, 50(20), 10923–10932, DOI:10.1021/acs.est.6b01980.
- Y. Liu, Y. Zhang, J. Li, N. Wu, W. Li and Z. Niu, Distribution, partitioning behavior and positive matrix factorization-based source analysis of legacy and emerging polyfluorinated alkyl substances in the dissolved phase, surface sediment and suspended particulate matter around coastal areas of Bohai Bay, China, Environ. Pollut., 2019, 246, 34–44, DOI:10.1016/j.envpol.2018.11.113.
- J. J. MacInnis, K. French, D. C. G. Muir, C. Spencer, A. Criscitiello, A. O. De Silva and C. J. Young, Emerging investigator series: a 14-year depositional ice record of perfluoroalkyl substances in the High Arctic, Environ. Sci.: Processes Impacts, 2017, 19(1), 22–30, 10.1039/C6EM00593D.
- G. L. Lescord, K. A. Kidd, A. O. De Silva, M. Williamson, C. Spencer, X. Wang and D. C. G. Muir, Perfluorinated and Polyfluorinated Compounds in Lake Food Webs from the Canadian High Arctic, Environ. Sci. Technol., 2015, 49(5), 2694–2702, DOI:10.1021/es5048649.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00502a |
| This journal is © The Royal Society of Chemistry 2020 |