
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple field-based biodegradation test shows pH to be an inadequately controlled parameter in laboratory biodegradation testing†
Matthew
Goss
 *,
Zhe
Li
and
Michael S.
McLachlan
*,
Zhe
Li
and
Michael S.
McLachlan
Department of Environmental Science (ACES), Stockholm University, Sweden. E-mail: mgoss@mit.edu
First published on 13th February 2020
Abstract
Biodegradation tests are essential for characterizing the behavior of organic micropollutants in the environment, but they are carried out almost exclusively in the laboratory. Test parameters such as temperature and test chemical concentration are often applied in ways that affect observed biodegradation, and laboratory testing requires sophisticated temperature-controlled facilities. We developed a field-based test based on OECD 309 which minimizes the need for laboratory resources such as temperature-controlled facilities by using bottles incubated in the natural water body. The test also utilized contaminant residues present in unspiked natural water to increase the relevance of the results to the local system. A test in a local river and a matching lab-based test were conducted in parallel. We quantified 26 of 40 targeted micropollutants and observed dissipation for 13. Significant differences in half-life (up to a factor of 3.5) between lab and field bottles were observed for 7 compounds, with 6 of 7 degrading more slowly in field bottles. For 4 of these, dissipation was positively correlated to the neutral fraction of the chemical. Differences in the neutral fraction arose due to a higher pH in the lab bottles induced by outgassing of CO2 from the oversaturated river water. We conclude that pH is an important parameter to control in biodegradation testing and that field-based tests may be more environmentally relevant.
Environmental significance statementOrganic micropollutants such as pharmaceuticals are widely released into the environment via wastewater treatment plants. Standard laboratory tests estimate biodegradation rates for these contaminants but biodegradation is rarely measured in the field. This manuscript compares a standard laboratory test with a novel field-based method, which attempts to measure biodegradation rates more directly relevant to a local environment. We observed that rising sample pH under typical laboratory conditions, due to outgassing of CO2 from oversaturated river water, was correlated with faster biodegradation of some compounds than was observed under constant pH in field incubations. This suggests standard laboratory tests may improperly estimate micropollutant half-lives in the environment when pH is not appropriately considered. |
Introduction
Biodegradation tests are essential for understanding the fate of organic contaminants in the aquatic environment. The information from biodegradation tests has two primary uses. The first of these is to support the assessment of persistence criteria that are defined in a number of chemical regulations.1 Here the purpose is to establish whether the chemical can be readily biodegraded and hence will not fulfill the persistence criteria. The second primary application of biodegradation test data is to support exposure assessment.2 Prospective exposure assessment is done with models which require quantitative biodegradation rate information to forecast the chemical levels in the environment.3Despite the goal of understanding contaminant behavior in the environment, biodegradation tests are carried out almost exclusively in a laboratory setting. A hierarchy of tests is available, ranging from simple screening tests such as the ready biodegradation test (OECD 301![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4) to more sophisticated tests that provide quantitative biodegradation rates such as OECD 3085 and 309.6 Aquatic biodegradation tests generally involve spiking a chemical into an aqueous system followed by some form of incubation. One feature of the higher tiered tests is that they use natural water and/or sediment in the laboratory incubation, providing a natural source of the microbial population in the test and thereby heightening the environmental relevance.
4) to more sophisticated tests that provide quantitative biodegradation rates such as OECD 3085 and 309.6 Aquatic biodegradation tests generally involve spiking a chemical into an aqueous system followed by some form of incubation. One feature of the higher tiered tests is that they use natural water and/or sediment in the laboratory incubation, providing a natural source of the microbial population in the test and thereby heightening the environmental relevance.
However, transferring the microbial population from the field to the lab has the potential to affect the microorganisms in ways that may influence biodegradation. Water temperature is one environmental variable that influences the composition of the microbial community and biodegradation of organic chemicals.7,8 OECD 309 recommends incubating biodegradation reactors at either the field temperature or 20 to 25 °C.6 These latter temperatures are well above typical natural water temperatures over much of the world. In these cases, incubation at laboratory room temperature would compromise the environmental relevance of the test. Hence, incubation at field temperature is frequently required.
The need to conserve the composition and activity of the microbial population during transfer from the field to the lab has practical consequences for conducting biodegradation tests. The test must be conducted in close proximity to the environment being studied to minimize the time for the transfer. In addition, the test requires some sophisticated laboratory facilities, in particular a regulatable constant-temperature environment in which the incubations at field temperature can be conducted. These facilities are not available in many areas of the world, and this is a significant obstacle to conducting biodegradation testing in these environments.
The spiking of test chemicals into the laboratory incubation is another factor that can compromise the environmental relevance of biodegradation tests. Test chemicals in OECD 308 and 309 are often spiked at concentrations substantially above those found in the natural environment. Differences in concentration have been shown to influence biodegradation rates.8–10 Furthermore, the act of spiking analytes itself can affect biodegradation as it removes the biological system from a steady state. If the chemical is a primary substrate for the degrading microorganisms, a lag phase will result during which the microbial population grows and adapts before substantial biodegradation occurs.11 Indeed, the OECD 309 test offers explicit instructions for treating the lag phase when evaluating test results.6 In comparing conventional OECD 309 tests to tests performed with unspiked water in which existing chemical contaminants were used as analytes, Li and McLachlan12 observed substantial differences in biodegradation rates and patterns, including the elimination of a lag phase and differing rate orders. In addition to illustrating that spiking can impact biodegradation test results, this work also showed that it is possible to overcome this problem in some cases by measuring biodegradation of contaminant residues already present in the environment being studied (“unspiked” water).
In this work we sought to develop a test method inspired by OECD 308/309 that would come as close as possible to measuring ongoing biodegradation in a natural body of water and that could be readily conducted around the world. We incubated bottles with unspiked water and a small amount of sediment by floating them in a river, using the local contaminant mixture, local microbial population, and local river temperature to simulate ongoing biodegradation. We compared this to a simultaneous laboratory experiment done using the same water, sediment, and approximate temperature. This field-based method eliminates the need for laboratory infrastructure to conduct the incubations.
Methods
Two-week biodegradation experiments were carried out for two parallel systems: a lab-based set-up with three replicates in a temperature-controlled room and a field-based set-up with three replicates incubated in a river. These experiments were based on the OECD 309 aerobic biodegradation test, with two significant modifications: the tested water was the source of experimental analytes (i.e., no spiking of test chemicals), as suggested by Li and McLachlan,12 and surface sediment was added to the tested water, as inspired by Shrestha et al.13 By adding sediment, the experiment more closely reflected a sediment-water system. The addition of sediment also increases the microbial population, making it more likely that measurable biodegradation would occur during the relatively short incubation period. Forty non-volatile organic contaminants selected in accordance with the OECD 309 guideline were targeted.Chemicals and reagents
Chemicals used as standards for analysis were purchased from Sigma-Aldrich (Steinheim, Germany) or Toronto Research Chemicals Inc. (North York, Canada). D- and 13C-labeled standards were purchased from Toronto Research Chemicals Inc. and CDN Isotopes (Pointe-Claire, Quebec, Canada). A solution containing all non-labeled standards and a solution containing all isotope-labeled standards were prepared at a concentration of 5 μg mL−1 in methanol and stored in the dark at −20 °C until use. LC/MS-grade acetonitrile and methanol were purchased from VWR (Stockholm, Sweden), and LC/MS-grade formic acid was obtained from Sigma-Aldrich. Sodium azide was purchased from Sigma-Aldrich. Milli-Q water was produced using a Milli-Q Integral Water Purification System (Merck Millipore, Stockholm, Sweden).Biodegradation experiments
All water samples were taken from the Fyris River in Uppsala, Sweden, approximately 1.1 km downstream of the Kungsängsverket wastewater treatment plant (59°49′56.1′′N 17°39′36.1′′E) on April 29, 2019. Uppsala is a small city with approximately 190000 inhabitants and one wastewater treatment plant, which releases ∼50000 m3 d−1 effluent. River flow at the start of the experiment was ∼460000 m3 d−1 (effluent diluted by a factor of 9). The sampling site was selected to be far enough downstream to ensure almost complete mixing of the effluent with the river water14 (see Text S1†) and where the water was deep enough (∼2 m) near the bank to allow for the planned set-up of river-incubated test chambers. Sediment was sampled 100 m upstream of the primary sampling site where the river bottom was more easily accessible (depth less than 0.3 m). Water samples were collected through grab-sampling 20 cm below the surface using 1 L HDPE bottles. Sediment was collected by taking the top 1 cm layer with a scoop. This was sieved wet to 3 mm and homogenized. Field-based incubation bottles were set-up immediately following sampling while water and sediment for lab-based bottles were transported in a cooled and insulated container and were set-up in the temperature-controlled room five hours after sampling.
All six experimental biodegradation reactors were prepared with 350 mL unspiked Fyris River water and 10 mL wet sediment (approximately 17 g wet/14 g dry solids; 40 g dry sediment per L) in 500 mL wide-mouth amber-glass bottles. Lab-based samples were incubated in the dark in a temperature-controlled room with the temperature held at 12.5 °C, based on the initial water temperature measured in the river using a handheld probe. These were stirred with magnetic stir bars at 100 rpm, and covered with cotton wool to prevent dust ingress. Field based samples were sealed with PTFE tape and HDPE lids, and then suspended in mesh bags attached to an anchor, roughly 0.5 m below the surface of the river. This set-up was chosen to minimize solar heating of the samples while maximizing movement of the bottles from waves to maintain oxygen exchange between the water phase and the headspace. Two sterile control incubations, one with sediment and one without sediment, were additionally set up in the temperature-controlled room with 250 mL Fyris River water and 10 mL wet sediment (56 g dry sediment per L). No sterile control was set up in the field due to the hazards of sodium azide. These were sterilized with sodium azide (final concentration 0.1%). The different concentration for the control bottles was selected to accommodate the smaller intermediate time-point sample size.
At six time points (day 0, 1, 3, 6, 10, and 14), a 20 mL water sample for vacuum-assisted evaporative concentration was removed from each field and lab incubation bottle using a sterile plastic syringe. For lab samples, these were frozen and stored immediately at −20 °C. For field samples, these were transported in a cooled and insulated container and placed in the freezer within three hours. The reduction in water volume resulted in a theoretical final sediment concentration in the incubation bottle of 61 g L−1. Due to the hazards of evaporating sodium azide to dryness, a direct injection analytical method was selected for the sterile control samples. Instead of 20 mL, three smaller 1.5 mL aliquots were removed from each bottle and frozen at each time point. This resulted in a theoretical final sediment concentration of 63 g L−1. Temperature, dissolved oxygen, conductivity, and pH were monitored at each sampling time point using handheld probes (Hach LDO101, CDC401, and PHC101).
Sample preparation and analysis
All experimental samples were concentrated using vacuum-assisted evaporation (Syncore, BÜCHI Labortechnik AG, Switzerland). The 20 mL samples were transferred to glass vials with 0.3 mL residual volume before they were spiked with 10 μL of an internal standard mixture (2.5 ng of each isotope-labeled standard in methanol). Concentration followed the method of Mechelke et al.,15 reducing samples to less than 0.5 mL under 20 mbar, 200 orbital revolutions per minute, and a slightly modified temperature of 60 °C over approximately 4 to 5 hours. The sides of the vials were washed down first twice with 0.75 mL methanol, and then with 1 mL Milli-Q water when the evaporation neared completion. Concentrates were then brought to 1 mL using Milli-Q water and filtered into LC vials using a 0.2 μm syringe filter (Thermo Fisher Scientific). Control samples were prepared for direct injection by spiking 1 mL aliquots with 10 μL of the internal standard mixture, before vortexing them and filtering them to 0.2 μm into LC vials. All prepared LC samples were stored frozen until analysis. Blank Milli-Q water samples were injected every 10 to 18 samples.Analysis was carried out as described in Li and McLachlan12 using an ultrahigh-performance liquid chromatography coupled to a Q-Exactive HF Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with electrospray ionization (ESI). All samples were separated on a reversed-phase Hypersil GOLD aQ C18 polar-end-capped column (2.1 mm × 100 mm; particle size of 1.9 μm; Thermo Fisher Scientific, San Jose, CA) with a water/acetonitrile gradient and were analyzed in both positive and negative mode. The injection volume for all samples was 100 μL.
Calculations
Quantification was carried out in Xcalibur 3.1 using a list of 40 polar micropollutants (Table S1†), 33 of which used matching isotope-labeled internal standards (Table S2†). A 10-point calibration curve was fitted with a weighted (1/x) least-squares regression. The calibration curve standards ranged from 5 ng L−1 to 5 μg L−1 in Milli-Q water for targeted analytes and included a fixed concentration of 2.5 μg L−1 of isotope-labeled compounds, enabling quantification of targeted analytes using an internal standard method. The calibration curve was run at the beginning of each sequence. When targeted analytes were detected near the limit of detection, the highest three calibration curve points were omitted to improve the linearity and reduce the residuals of the curve in the concentration range of interest.The limit of detection (LOD) and limit of quantification (LOQ) were calculated as described in Mechelke et al.15 with minor modifications (Table S2†). The LOD and LOQ in Milli-Q water were identified as the lowest standard concentration for which the signal-to-noise ratio was greater than 3:1 and 10:1 respectively, and that produced a chromatographic peak containing at least three data points. Due to the spacing of calibration curve points, LOD and LOQ were the same for many compounds. These levels were transformed to a matrix LOD (MLOD) and matrix LOQ (MLOQ) for each target compound by applying eqn (1). This uses a concentration-factor (CF) of 20 and the absolute yield, encompassing both pre-injection losses and matrix effects, determined by labeled internal standard peak areas. For the seven compounds without matching labeled internal standards, the nearest labeled standard in LC retention time was used for quantification and determination of MLOQ and MLOD. For direct injection samples, MLOQ and MLOD were calculated in the same way but with a CF of 1 (Table S3†).
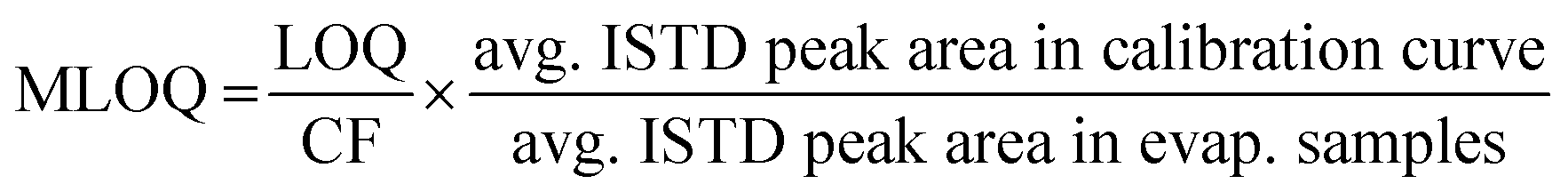 | (1) |
For compounds where dissipation of >25% was observed in either all field or all lab bottles, half-lives were determined assuming first order kinetics. Linear least squares regressions were performed independently for each incubation bottle using the natural logarithm of concentrations normalized to the starting concentration. All measurements below MLOQ were removed. Regressions were rejected when there were fewer than three data points above MLOQ or when R2 was below 0.7. Errors in individual half-lives were determined by propagating the standard error of the slope into days. Statistical differences between field and lab half-lives (two groups of n = 3) were determined using a T-test with a 95% confidence interval.
Results and discussion
Quality assurance
None of the targeted compounds were found in the blank samples, with the exception of diclofenac, which was found at a maximum of ∼20 ng L−1. This signal in the blank represented 4–11% of the peak area in experimental samples which had been concentrated by evaporation; we still considered this low enough to continue to use these data. Since this signal corresponded to an average of 50% of the peak area for abiotic controls which were directly injected without concentration, these data were discarded.While injections were not directly repeated, we assessed repeatability by calculating the relative standard deviation (RSD) between measured concentrations in parallel incubation bottles. The average RSD for each compound over the first three sampling time points ranged from 0.8% (metoprolol acid) to 23.6% (sitagliptin) with a median of 4.6%. This statistic encompasses both the between-bottle variability and the instrumental variability. Calibration curve fits were also consistent, ranging from R2 = 0.936 to 0.996 with a median of 0.989.
Due to the use of labeled internal standards, error due to the vacuum-assisted evaporation method was minimized. Average absolute yields for each targeted compound, combining both matrix effects and pre-injection loss, ranged from 3.6% to 86.3% with a median of 34.1% as calculated from the ratio of internal standard in evaporated samples to internal standard in calibration curve samples. For the 13 compounds of interest discussed below, absolute yields ranged from 19.4% to 86.3% with a median of 33.4% (Table S2†). Two compounds, methotrexate and hydrochlorothiazide, were incompatible with the evaporation method. Internal standard peak areas for hydrochlorothiazide were extremely low and inconsistent (average absolute yield of 2.0%), while evaporation enhanced baseline noise for methotrexate-D3 sufficiently such that no internal standard peak was visible. Mechelke et al.15 also reported a relatively low yield of 16.6% for hydrochlorothiazide in a surface water matrix.
We observed a slight rise in the water level in field sample A after the first day of the experiment, likely due to a leak. The jar was re-sealed and difference in water levels remained constant throughout the remainder of the experiment. We modeled the effect of a 10% leak (Fig. S1†) and determined it to have negligible effect on observed biodegradation rates. The good agreement between concentrations measured in parallel field bottles supports this conclusion.
The first sample from lab bottle B was lost during evaporation due to a broken flask. Regressions for this time series were calculated based on data normalized to the second time point.
Experimental conditions
While efforts were taken to make the experimental conditions in both the field and lab environments as similar as possible, differences between the set-ups arose. Fig. 1 shows dissolved oxygen, temperature, conductivity, and pH measurements for the field-based samples, the lab-based samples, and the Fyris River taken at each of the six sampling points (these data are provided in Table S4†).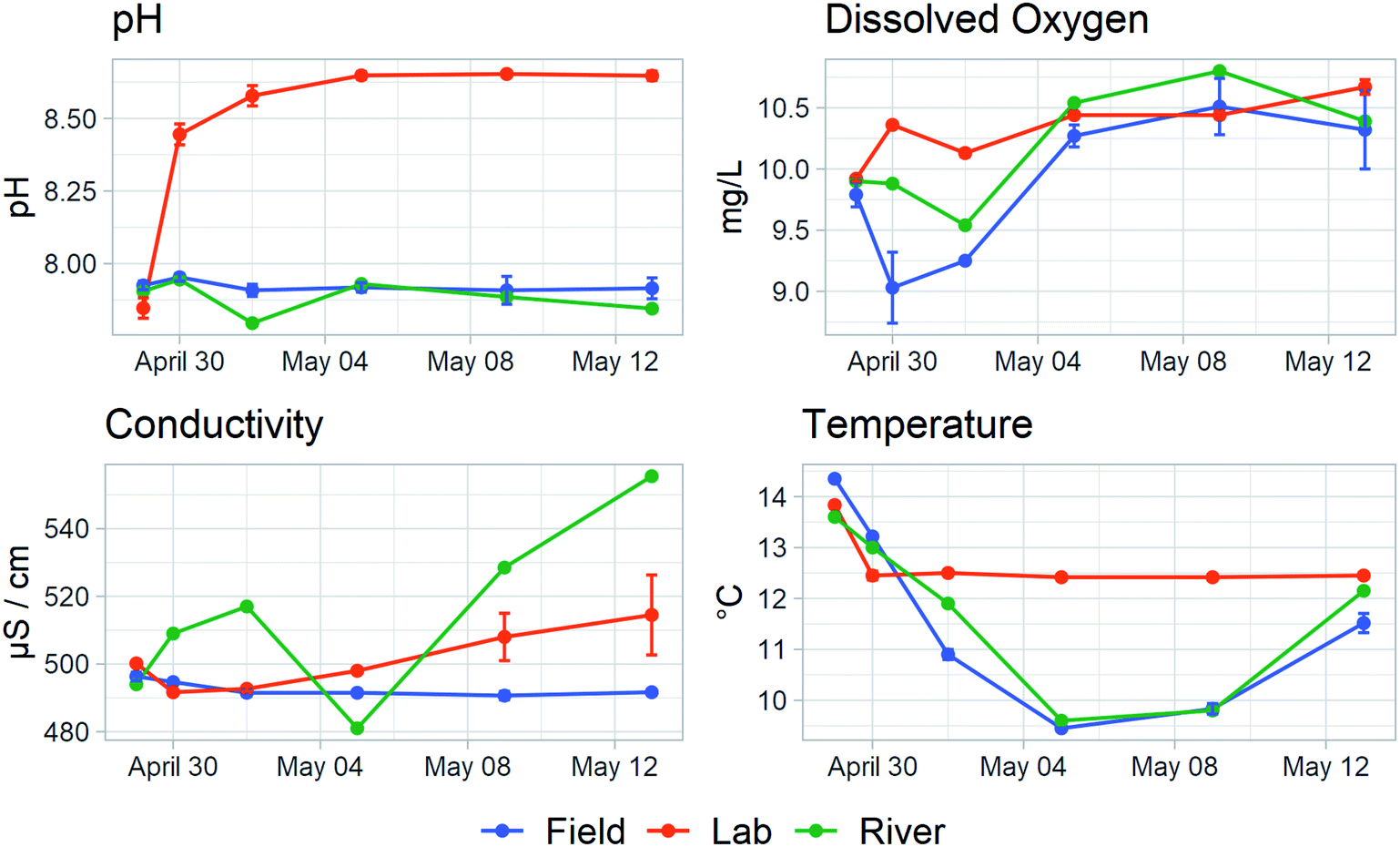 | ||
| Fig. 1 Dissolved oxygen concentration (mg L−1), temperature (°C), conductivity (μS cm−1), and pH measurements. The measurements for field- and lab-based samples are averaged and their standard deviations are plotted as error bars. Field bottle data are rendered in blue, lab bottle data in orange, and river data in green. | ||
The lab bottles were held at a time-weighted average temperature of 12.5 °C while the field bottles had a time-weighted average of 10.7 °C. The river and field bottle temperatures parallel each other, dropping slightly during the course of the experiment before rising again.
Conductivity varied slightly over the course of the experiment. While field-based bottles remained constant, the conductivity of lab-incubated bottles rose an average of 3% over the two-weeks. Variability in the conductivity of the river is likely due to changes in dilution of the upstream wastewater treatment plant effluent.
Dissolved oxygen concentration differed between the lab bottles and the field bottles. In the lab, it quickly increased to about 5% above the starting value while in the sealed field bottles it dipped about 7% below before climbing again. The dissolved oxygen concentration in the river lay in between, and neither the lab bottles nor the field bottles were clearly better at reproducing the concentrations in the river. Nevertheless, the differences between the lab bottles and the field bottles were small and are unlikely to have had a major influence on the microbial community.
The two systems differed substantially in pH. All systems started at pH 7.9 but in the lab bottles, pH quickly rose to and stayed at 8.6 by the third day while in the field bottles pH remained constant. This is most likely due to an oversaturation of CO2 in the river both at the time of sampling and over the duration of the incubation. Stirred bottles in the lab quickly allowed this excess to dissipate, resulting in a substantial rise in pH, while samples in the sealed bottles in the river remained oversaturated, matching the river.
In both systems, light wavelengths suitable for photosynthesis and photodegradation were excluded. All incubation bottles used amber glass, which effectively excludes light with wavelengths below 450 nm and laboratory bottles were additionally incubated in the dark.
Agitation also differed between the two test setups. We suspended the field test bottles in the water column to maximize agitation using water movement to keep dissolved oxygen levels raised. However, the Fyris River was slow moving and agitation in the field test bottle was most likely limited. The stirred lab setup had significantly more agitation, which would facilitate passive exchange of oxygen. However, the stir bars quickly cleared a path through the small amount of sediment and little to no suspended sediment was visible throughout the test despite the stirring.
Detection and observed dissipation of targeted compounds
We detected 30 of 38 targeted compounds, 26 of which were above MLOQ. The starting concentrations ranged from 2.1 ng L−1 (2-methyl-4-chlorophenoxyacetic acid [MCPA]) to 766.2 ng L−1 (metformin) (Fig. S2†). In directly injected abiotic control samples both with and without sediment, we detected 23 compounds, with 19 above MLOQ. Abiotic controls showed no dissipation for any detected compound except atenolol. This indicates that abiotic transformation and physical losses were minimal.Among quantified compounds, we observed first order degradation in both lab and field systems for 13 compounds (Fig. 2, S3 and Table S5†). One additional compound, irbesartan, was observed to dissipate in both lab (average half-life of 25 days) and field bottles but data points above the MLOQ in the field samples were insufficient to calculate a half-life. Within this set of 13 compounds, we observed statistically significant differences in half-lives between the field-incubated and the lab-incubated biodegradation chambers for 7 compounds. Six of these compounds dissipated more slowly in field-incubated bottles while metformin was found to degrade more quickly in the field. In the extreme case, metoprolol's half-life was measured to be on average 3.5 times greater in the field bottles than in the lab (22.0 and 6.3 days respectively). A one-day lag phase was observed for some bottles and some compounds, with concentration at the second sampling point rising slightly above the starting concentrations in some cases. This occurred most often in field bottles B and C. Of the 13 compounds of interest here, 11 were detected and 8 were above MLOQ in both abiotic controls. Measured half-lives for field bottles were more variable, with a median half-life RSD of 18% compared to 10% for lab bottles.
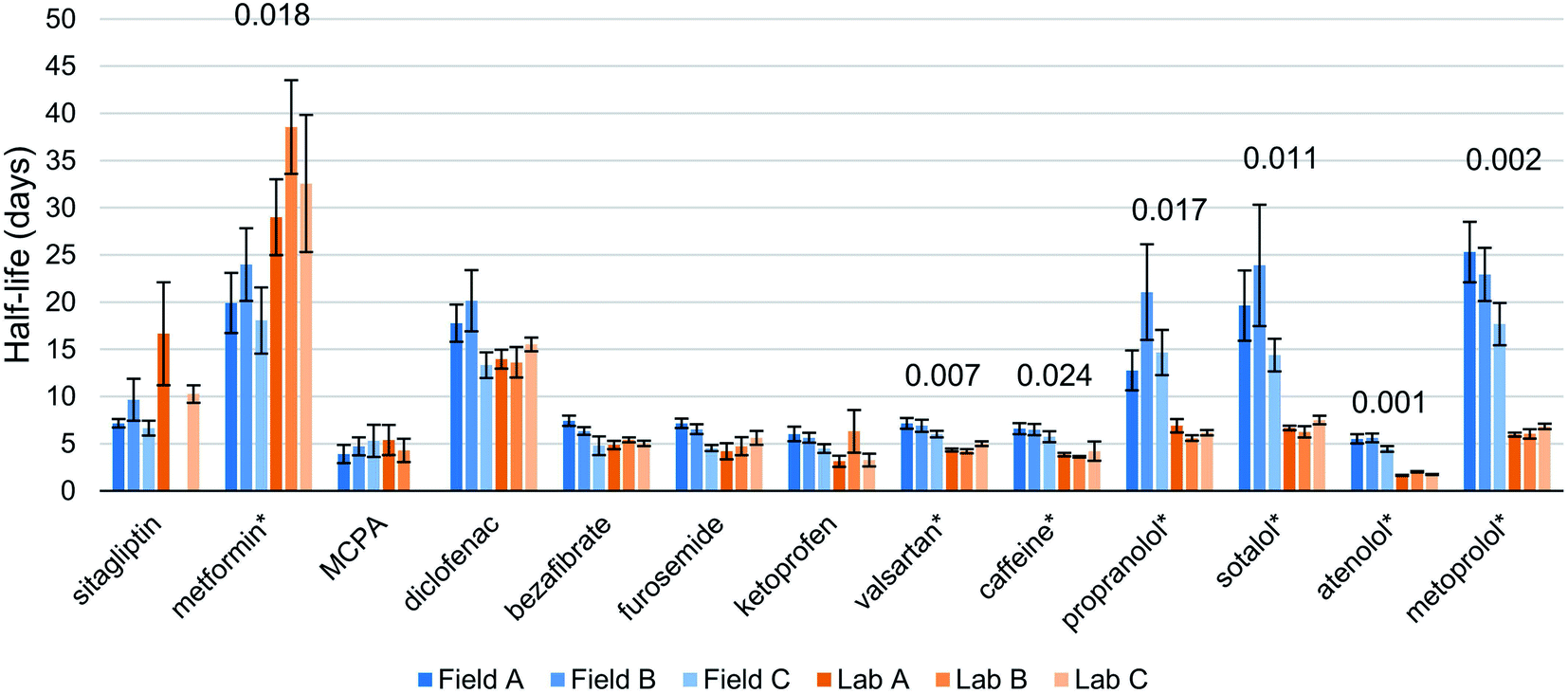 | ||
| Fig. 2 Half-lives of compounds that could be fitted with first-order degradation kinetics. Statistically significant differences between field and lab results are marked with an asterisk (*), and their respective p-values are labeled above. Error bars represent the standard error in the slope of the regression propagated into days. Missing half-lives for sitagliptin and MCPA are due to poor regressions (R2 < 0.7) (Table S6†). | ||
In lab-incubated bottles, furosemide concentrations were well-fitted by first-order kinetics until day 10 but then departed from this trend, with increasing dissipation rates after this time point (Fig. S3†). This deviation shows that the lab incubations were not able to sustain the biotransformation conditions in the river water for this compound beyond 10 days. Due to this, the half-lives for furosemide in the lab incubations were calculated from the data for the first 10 days only.
Unlike all other compounds detected in abiotic samples, atenolol was observed to dissipate in the abiotic control containing sediment. With a half-life of 3.1 days, atenolol dissipated in the abiotic bottle at a rate in between the field bottles and lab bottles, which were significantly different from each other. This dissipation of 74% over 6 days before it was below MLOQ is not explained by hydrolysis, since there was no dissipation in the abiotic control without sediment, or photolysis, since all lab-samples were kept in the dark. Sorption to the sediment also seems unlikely to be responsible for such a large dissipation since atenolol concentrations in unspiked water and sediment should be at least near equilibrium from the beginning of the experiment. It seems most probable then that the bottle was not completely sterile and that this dissipation was caused by biodegradation by a bacterial strain not fully eliminated by the 0.1% sodium azide concentration.
Analysis of differences between field and lab degradation patterns
The observed differences in dissipation between field and lab settings should logically be a result of differences in experimental conditions. As discussed above, differences in dissolved oxygen, temperature, and pH arose, and agitation was by design different between the two set-ups.Shrestha et al.13 identified stirring as a factor that can strongly affect biodegradation test results through grinding and exposing new sediment surfaces, causing the subsequent sorption of analytes to form non-extractable residues. During their experiments, they observed dramatic changes in sediment composition over 60 days (77.5% sand to 2.5% sand). In contrast, sediment in our stirred bottles did not appear qualitatively different by observation after two weeks of stirring. Furthermore, we observed no dissipation in our stirred abiotic/sediment bottle for all detectable compounds except atenolol. Given that sorption to any freshly exposed surfaces is a purely abiotic process and the mechanical stirring was identical in all lab bottles, this gives evidence that the sorption played little role in the observed dissipation of analytes.
Stirring could conceivably affect dissolved oxygen levels in the sediment layer, thereby affecting biodegradation rates. Shrestha et al.13 showed that dissolved oxygen in a sediment layer drops off dramatically within the first 2 mm under OECD 308 conditions. However, given the small quantity of sediment (10 mL per bottle, which corresponds to a ∼2 mm layer on the bottom of the bottle when fully settled, compared with 20 cm in Shrestha et al.13), such an effect would be expected to be weaker in our experiments. Dissolved oxygen in the water phase differed somewhat between systems but stayed well within an aerobic range (>85%). Field bottle A showed a less pronounced dip in dissolved oxygen at the beginning of the incubation than field bottles B and C (Table S4c†). However, this did not correspond to a consistent difference in chemical dissipation between the field bottles (Table S5†). This suggests that the dissipation rates were not strongly influenced by the differences in dissolved oxygen levels in water.
The temperature in the field bottles was on average 1.2 °C and at most 3 °C lower than in the lab. Correlations between temperature and biodegradation rate have been reported, but the influence of temperature is not readily predictable.1 Temperature can have a thermodynamic effect on the transformation reactions as described by the Arrhenius equation, whereby a higher temperature gives a faster reaction rate/shorter half-life. Temperature can also influence the composition and viability of the microbial community, and thereby, indirectly, the biotransformation rate. Although there has been no comprehensive assessment of the influence of temperature on biodegradation in aqueous systems, an extensive review of the influence of temperature on the degradation of plant protection products in soil has been conducted. It concluded that the temperature dependence can be described using the Arrhenius equation, and derived a median activation energy of 65.4 kJ mol−1.16 This corresponds to a 13% decrease in half-life for a 1.2 °C increase in temperature. In studies of the temperature dependence of biodegradation half-lives of a number of pharmaceuticals in municipal wastewater bioreactors, Meynet found trends consistent with the Arrhenius equation below 20 °C. According to the Arrhenius relationships reported, a 1.2 °C increase in temperature corresponded to a decrease in half-life of the order of 10%.17 We observed shorter half-lives in the warmer lab bottles for 6 of the test chemicals, but the differences were much greater (a factor of 1.5–3.5, Table S5†). This suggests that these differences in half-life were not primarily attributable to the differences in temperature.
The 0.7 unit difference in pH between the lab and field bottles may be particularly significant given that the micropollutants targeted in this study are ionizable and their dissociation is quite sensitive to pH changes in this range (Table 1). Neutral compounds pass more easily through cell membranes than ionic species and enzymatic biodegradation typically proceeds within the cell.11 A shift of 0.7 pH units therefore substantially affects the fraction of neutral micropollutants that are bioavailable. A pH-induced change in bioavailability aligns well with four of the five compounds where substantial differences are seen in the ionization states (Table 1). Atenolol, metoprolol, propranolol, and sotalol all showed significantly faster dissipation in the more basic conditions observed in the lab where the neutral fraction was larger. Gulde et al. investigated the effect of pH on the removal of cationic and neutral micropollutants in an activated sludge reactor and observed similar patterns, with significant increases in biodegradation rate between pH 7 and pH 8 for 11 of 15 compounds.18 While their analyte selection was largely different, atenolol and propranolol both showed rate increases similar to our results. Gulde et al. also observed that a simple model based only on the neutral fraction of micropollutants overestimated the observed biodegradation rate increases.18 Changes in pH will also change the bioavailability of other substrates, which in turn can lead to changes in the composition and viability of the microbial community. Therefore, pH can influence the biotransformation rate both by changing the bioavailable fraction of the chemical and by changing the composition of the microbial community. The latter is a potential explanation for differences in the dissipation rate for chemicals with a neutral fraction that was the same in the lab and field bottles, such as caffeine and valsartan. Given the magnitude of the difference in this parameter (0.7 pH units correspond to a factor of 5 difference in dissociation constant), we believe that this variable made the largest contribution to the differences in biodegradation rates between the field and laboratory bottles.
| Compound | Strongest acidic pKa | Strongest basic pKa | Neutral fraction (%) | |
|---|---|---|---|---|
| Field pH 7.9 | Lab pH 8.6 | |||
| a The pKa values and ionization were predicted using MarvinSketch 19.20 software from ChemAxon. These values are consistent with values given in http://https://www.drugbank.ca,19 some of which are experimentally derived. | ||||
| Metformin | — | 12.3 | 0.0 | 0.0 |
| Atenolol | 14.1 | 9.7 | 1.7 | 7.9 |
| Metoprolol | 14.1 | 9.7 | 1.7 | 7.9 |
| Propranolol | 14.1 | 9.7 | 1.7 | 7.9 |
| Sotalol | 10.1 | 9.4 | 2.9 | 12.9 |
| Sitagliptin | — | 8.8 | 11.5 | 39.5 |
| Caffeine | — | −1.2 | 100.0 | 100.0 |
| Valsartan | 4.4 | −0.6 | 0.0 | 0.0 |
| Furosemide | 4.3 | −1.5 | 0.02 | 0.0 |
| Diclofenac | 4.0 | — | 0.01 | 0.0 |
| Ketoprofen | 3.9 | — | 0.01 | 0.0 |
| Bezafibrate | 3.8 | −0.8 | 0.01 | 0.0 |
| MCPA | 3.4 | — | 0.0 | 0.0 |
Implications
For non-volatile compounds with Henry's law constants less than about 1 Pa m3 mol−1, the OECD 309 protocol recommends conducting the test under open and shaken or stirred conditions. However, for water samples taken from natural water bodies oversaturated with CO2, this procedure would have the effect of raising the pH above what is found in the environment. Since natural bodies of water oversaturated with CO2 are widespread,20–22 these lab tests may do a poor job of simulating pH conditions in the natural environment.While unspiked field-based biodegradation testing as we have carried out here is not applicable to regulatory testing where new chemicals must be evaluated, we suggest that it may be relevant to increasing our understanding of biodegradation as it actually takes place in the natural environment. Standardized laboratory tests often disregard many of the variables that can affect biodegradation rates including temperature, pH, contaminant concentrations, and composition of the local microbial community.8 A field-based test does not isolate the impact of these variables but instead incorporates them to generate what may be a better picture of ongoing biodegradation at a particular site and a particular time. Given the significant differences, as great as a factor of 3.5, shown here between closely matched laboratory and field tests, further exploring the application of field-based testing seems worthwhile to better understand the relevance and limitations of standard methods for describing contaminants' behavior in the real environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MG thanks the Swedish Fulbright Commission for financial support. Financial support was also provided by the Swedish Environmental Research Council Formas. We thank Uppsala Vatten & Avfall for providing wastewater effluent data.References
- R. Boethling, K. Fenner, P. Howard, G. Klečka, T. Madsen, J. R. Snape and M. J. Whelan, Environmental persistence of organic pollutants: guidance for development and review of POP risk profiles, Integr. Environ. Assess. Manage., 2009, 5(4), 539–556 CrossRef CAS PubMed.
- R. J. Larson and C. E. Cowan, Quantitative application of biodegradation data to environmental risk and exposure assessments, Environ. Toxicol. Chem., 1995, 14, 1433–1442 CrossRef CAS.
- J. P. A. Lijzen and M. G. J. Rikken, EUSES European Union system for the evaluation of substances, RIVM report no. 601900005/2004, Bilthoven, 2004, https://rivm.openrepository.com/handle/10029/259080 Search PubMed.
- OECD Guideline for the Testing of Chemicals 301, Organisation for Economic Co-operation and Development (OECD), Paris, 1992, pp. 1−62 Search PubMed.
- OECD Guideline for the Testing of Chemicals 308, Organisation for Economic Co-operation and Development (OECD), Paris, 2002, pp. 1−19 Search PubMed.
- OECD Guideline for the Testing of Chemicals 309, Organisation for Economic Co-operation and Development (OECD), Paris, 2004, pp. 1−21 Search PubMed.
- D. Ribicic, K. M. McFarlin, R. Netzer, O. G. Brakstad, A. Winkler, M. Throne-Holst and T. R. Størseth, Oil type and temperature dependent biodegradation dynamics – combining chemical and microbial community data through multivariate analysis, BMC Microbiol., 2018, 18, 83 CrossRef PubMed.
- A. Kowalczyk, T. J. Martin, O. R. Price, J. R. Snape, R. A. van Egmond, C. J. Finnegan, H. Schäfer, R. J. Davenport and G. D. Bending, Refinement of biodegradation tests methodologies and the proposed utility of new microbial ecology techniques, Ecotoxicol. Environ. Saf., 2015, 111, 9–22 CrossRef CAS PubMed.
- L. Toräng and N. Nyholm, Biodegradation rates in adapted surface water can be assessed following a preadaptation period with semi-continuous operation, Chemosphere, 2005, 61, 1–10 CrossRef PubMed.
- B. A. Kolvenbach, D. E. Helbling, H. P. E. Kohler and P. F. X. Corvini, Emerging chemicals and the evolution of biodegradation capacities and pathways in bacteria, Curr. Opin. Biotechnol., 2014, 27, 8–14 CrossRef CAS PubMed.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, John Wiley & Sons, Hoboken NJ, 2nd edn, 2003, pp. 739–750 Search PubMed.
- Z. Li and M. S. McLachlan, Biodegradation of chemicals in unspiked surface waters downstream of wastewater treatment plants, Environ. Sci. Technol., 2019, 53, 1884–1892 CrossRef CAS PubMed.
- P. Shrestha, T. Junker, K. Fenner, S. Hahn, M. Honti, R. Bakkour, C. Diaz and D. Hennecke, Simulation studies to explore biodegradation in water−sediment systems: from OECD 308 to OECD 309, Environ. Sci. Technol., 2016, 50, 6856–6864 CrossRef CAS PubMed.
- W. B. Mills, G. L. Bowie, T. M. Grieb, K. M. Johnson and R. C. Whittemore, Stream Sampling for Waste Load Allocation Applications, U.S. Environmental Protection Agency, Washington, 1986, pp. 1–63 Search PubMed.
- J. Mechelke, P. Longrée, H. Singer and J. Hollender, Vacuum-assisted evaporative concentration combined with LC-HRMS/MS for ultra-trace-level screening of organic micropollutants in environmental water samples, Anal. Bioanal. Chem., 2019, 4111(12), 2555–2567 CrossRef PubMed.
- D. B. Culleres, J. Boesten, C. Bolognesi, A. Boobis, A. Buchert, E. Capri, D. Coggon, A. Hardy, A. Hart, H. Koepp, M. Liess, R. Luttik, O. Meyer, S. Michaelidou-Canna, M. Montforts, A. Moretto, M. D. Mueller, B. Ossendorp, W. Steurbaut, M. Tasheva and C. Vleminckx, Opinion on a request from EFSA related to the default Q10 value used to describe the temperature effect on transformation rates of pesticides in soil, EFSA J., 2007, 622, 1–32 Search PubMed.
- P. Meynet, SETAC Europe 29th Annual Meeting, Helsinki, 2019 Search PubMed.
- R. Gulde, D. E. Helbling, A. Scheidegger and K. Fenner, pH-dependent biotransformation of ionizable organic micropollutants in activated sludge, Environ. Sci. Technol., 2014, 48, 13760–13768 CrossRef CAS PubMed.
- D. S. Wishart, C. Knox, A. C. Guo, S. Shrivastava, M. Hassanali, P. Stothard, Z. Chang and J. Woolsey, Drugbank: a comprehensive resource for in silico drug discovery and exploration, Nucleic Acids Res., 2006, 34, D668–D672 CrossRef CAS PubMed.
- E. G. Stets, D. Butman, C. P. McDonald, S. M. Stackpoole, M. D. DeGrandpre and R. G. Striegl, Carbonate buffering and metabolic controls on carbon dioxide in rivers, Global Biogeochem. Cycles, 2017, 31, 663–677 CrossRef CAS.
- D. Butman and P. A. Raymond, Significant efflux of carbon dioxide from streams and rivers in the United States, Nat. Geosci., 2011, 4, 839–842 CrossRef CAS.
- C. P. McDonald, E. G. Stets, R. G. Striegl and D. Butman, Inorganic carbon loading as a primary driver of dissolved carbon dioxide concentrations in the lakes and reservoirs of the contiguous United States, Global Biogeochem. Cycles, 2013, 27, 285–295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00491b |
| This journal is © The Royal Society of Chemistry 2020 |