
Thermal decomposition kinetics of glyphosate (GP) and its metabolite aminomethylphosphonic acid (AMPA)†
Milad
Narimani
and
Gabriel
da Silva
 *
*
Department of Chemical Engineering, University of Melbourne, Victoria 3010, Australia. E-mail: gdasilva@unimelb.edu.au; Fax: +61 3 8344 4153; Tel: +61 3 8344 6627
First published on 21st November 2019
Abstract
Glyphosate (GP) is a widely used herbicide worldwide, yet accumulation of GP and its main byproduct, aminomethylphosphonic acid (AMPA), in soil and water has raised concerns about its potential effects on human health. Thermal treatment, in which contaminants are vaporised and decomposed in the gas-phase, is one option for decontaminating material containing GP and AMPA, yet the thermal decomposition chemistry of these compounds remains poorly understood. Here, we have revealed the thermal decomposition mechanism of GP and AMPA in the gas phase by applying computational chemistry and reaction rate theory methods. The preferred decomposition channel for both substances involves the elimination of P(OH)3 to yield the imine N-methylene-glycine (from GP) or methanimine (from AMPA), with relatively low barrier heights (ca. 45 kcal mol−1). The half-life of GP and AMPA at 1000 K are predicted to be 0.1 and 4 ms respectively, and they should be readily destroyed via conventional incineration processes. The further decomposition of N-methylene-glycine is expected to also take place at similar temperatures, leading to N-methyl-methanimine + CO2, with a barrier height of ca. 48 kcal mol−1. The imine decomposition products of GP and AMPA are expected to react with water vapour to form simple amines and carbonyl compounds.
Environmental significanceRoundup is the world's most popular weed-killer, and the active component (glyphosate) as well as its metabolite (AMPA), are found throughout our soil, water, and wastes. With the recognition that Roundup is harmful to human health we need to understand how to treat contaminated materials and how it can be safely disposed of. Unfortunately, there is almost no information available on the thermal decomposition of Roundup, so that it's fate during incineration is presently unclear. We report the discovery of the mechanisms by which glyphosate and AMPA decompose in the gas phase, through several stages of reaction, obtained via detailed ab initio and reaction rate theory calculations. Importantly, we find that these compounds decompose to environmentally benign products at modest temperatures, guiding the way to design and optimize thermal treatment technologies. |
Introduction
Glyphosate (GP), N-(phosphonomethyl)glycine, is a non-selective, systematic, and post emergent herbicide capable of controlling more weed species than other herbicides,1–3 and gained widespread international use following its development in the 1970s. The introduction of glyphosate-resistant crops in 1996 further amplified its importance.3 The widespread use of GP has resulted in its accumulation in soil and water.4–7 The microbial degradation of GP in soil, natural waters and plants, or metabolization by select plant species, can lead to the formation of aminomethylphosphonic acid (AMPA); AMPA is more toxic and persistent than GP.8–12 In addition to GP, a number of other phosphonate compounds, including detergents, can degrade to AMPA.13,14 The structures of GP and AMPA are shown in Fig. 1. | ||
| Fig. 1 Structure of (a) glyphosate (GP) and (b) aminomethylphosphonic acid (AMPA). | ||
There are major concerns about residual GP and its major metabolite AMPA in aquatic and terrestrial environments. Numerous studies have demonstrated that GP has mutagenic and carcinogenic effects,15 genotoxic effects,16 and endocrinal effects17 on humans. As a result the World Health Organization (WHO) declared this herbicide to be carcinogenic to humans in 2015.18
Several treatment processes have been developed to remove pesticide contamination from soil and water. These remediation technologies can be classified into physical, biological, and chemical processes.19 Physical treatment processes, including adsorption20,21 and filtration,22,23 produce concentrated waste streams that require post-treatment processing in order to be converted to non-toxic products. Although bioremediation technologies can destroy the target contaminants, an obstacle to their applicability is long residence times.24 Chemical remediation processes are under development to remove GP from wastewater,25–27 however with chemical remediation there is a need for further treatment of degradation products.19,20
Thermal treatment methods including incineration and pyrolysis are well-established destruction strategies for solid waste management. The main advantages of thermochemical treatment processes are waste volume reduction with potentially complete destruction of toxic compound.28 Moreover, thermal treatment can be used to treat concentrated waste streams arising from physical separation processes. The initial stage in thermal treatment of soil and other solid material typically involves vaporization of the volatile components. GP is relatively volatile, with a boiling point of 230 °C, whereas AMPA is predicted to have a boiling point of 358 °C.35,36 Following vaporization, GP and AMPA are expected to undergo pyrolysis (thermal decomposition).
In order to develop and optimize thermal treatment processes for the destruction of GP and AMPA we require a fundamental understanding of their gas-phase decomposition reactions, particularly the products formed and reaction rate coefficients as a function of temperature. A wide variety of organophosphates are encountered in thermal systems, and insights into GP and AMPA decomposition will also aid in the development of broader chemical kinetic models for organophosphate combustion and pyrolysis.29–34
In this work, the detailed decomposition mechanism and kinetics of GP and AMPA are presented for the first time using computational chemistry and statistical reaction rate theory techniques. This study identifies the most probable products of the initial stages of the GP and AMPA decomposition mechanisms and their corresponding rate coefficients as a function of temperature.
Methods
Quantum chemistry calculations were conducted in Gaussian 16.37 Geometry optimizations, frequency calculations, and intrinsic reaction coordinate (IRC) calculations were carried out at the M06-2X/6-31G(2df,p) level of theory. Subsequently, the equilibrium structures were utilized in high-level composite G3X-K energy calculations.38 The structural coordinates, vibrational frequencies, and electronic energies of reactants and transition states were then used in the Multiwell-2016 program suit39,40 to calculate rate coefficients using canonical transition state theory. In these calculations the low frequency P–C internal rotational mode in GP and AMPA – which is lost in the key transition states – is treated as a free rotor, with the rigid-rotor and harmonic-oscillator approximations applied to the remaining degrees of freedom. Rate coefficients of all identified reaction channels were calculated in the temperature range of 300–2000 K for both GP and AMPA. Our calculated barrier heights, and therefore predicted activation energies, are expected to have 95% (2σ) uncertainties on the order of 1 kcal mol−1.38 Predicted pre-exponential factors are typically found to be accurate to within a factor of two. Thermochemical properties for key species and bond dissociation energies in GP are provided as ESI.†Results and discussion
GP decomposition
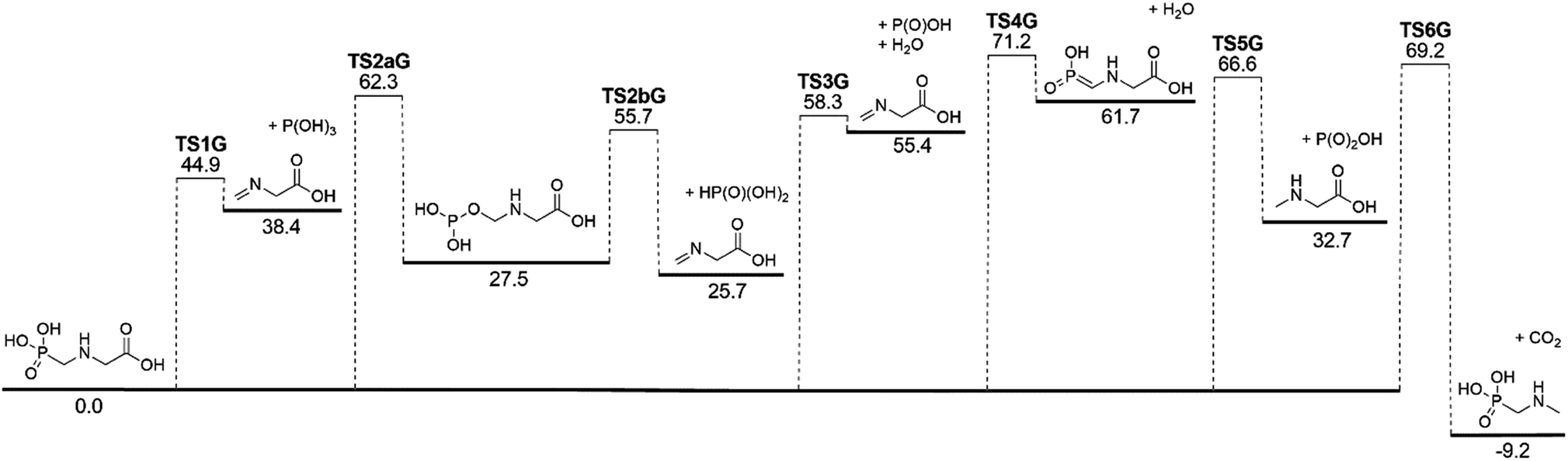
The lowest-energy reaction channels identified for the isomerization and decomposition of GP are demonstrated in Fig. 2. The process with the lowest barrier height corresponds to simultaneous C–P bond cleavage and proton transfer from the nitrogen to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen atom. This pathway requires 44.9 kcal mol−1 and leads to formation of P(OH)3 and N-methylene-glycine via TS1G (Fig. 3a). There is little available experimental information on the thermal decomposition of GP, although a calorimetric study identified that the first stage of decomposition proceeds with an activation energy of 48.1 kcal mol−1,41 in relative agreement with our calculations. Moreover, this step was found to be endothermic and to likely result in loss of a methylene group in GP, again consistent with our proposed mechanism.
O oxygen atom. This pathway requires 44.9 kcal mol−1 and leads to formation of P(OH)3 and N-methylene-glycine via TS1G (Fig. 3a). There is little available experimental information on the thermal decomposition of GP, although a calorimetric study identified that the first stage of decomposition proceeds with an activation energy of 48.1 kcal mol−1,41 in relative agreement with our calculations. Moreover, this step was found to be endothermic and to likely result in loss of a methylene group in GP, again consistent with our proposed mechanism.
 | ||
| Fig. 2 Potential energy diagram for the thermal decomposition of GP. Energies (0 K enthalpies, kcal mol−1) are calculated at the G3X-K level of theory. | ||
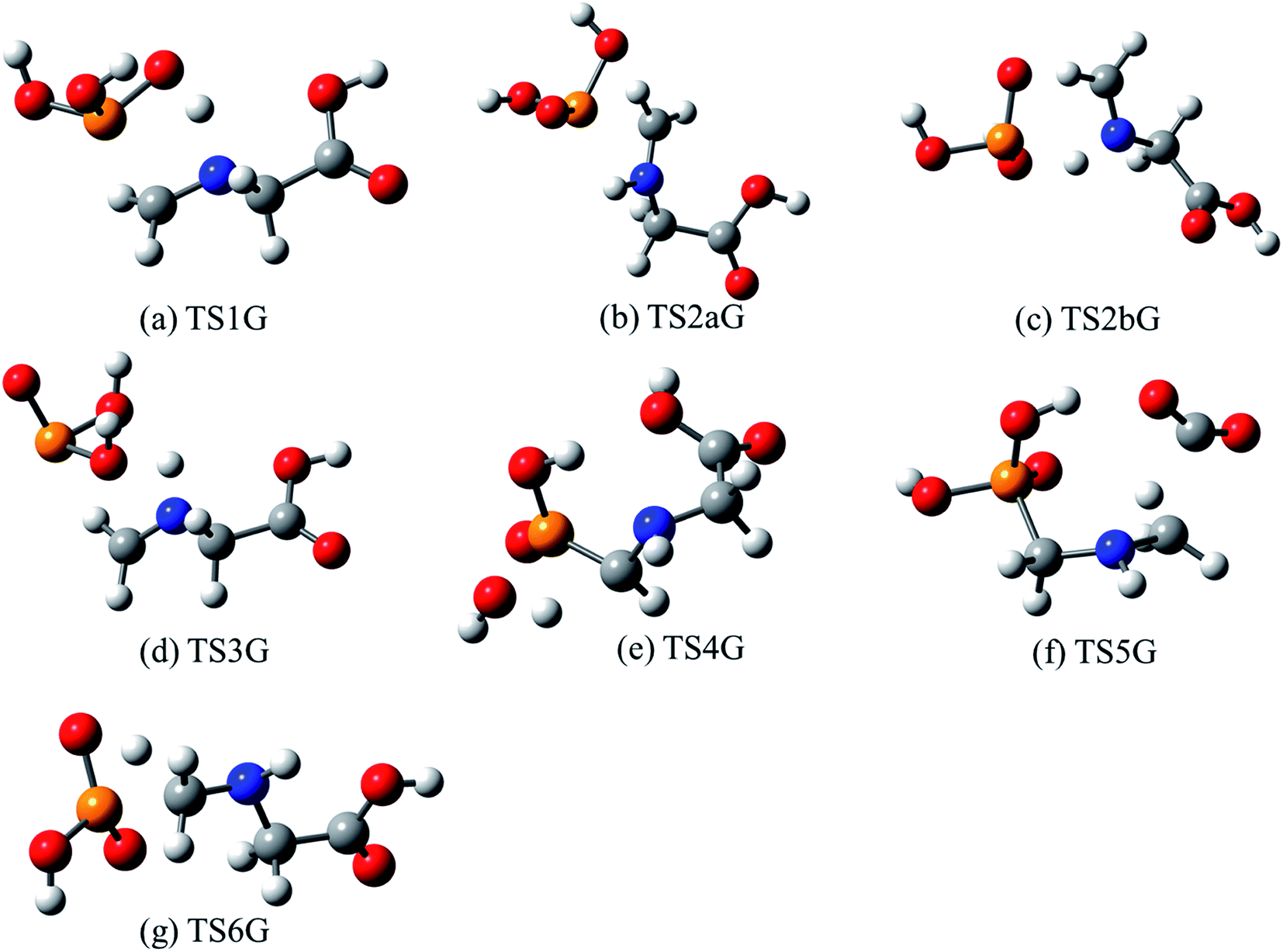 | ||
| Fig. 3 Optimized structures for transition states identified in the thermal decomposition of GP. Transition state numbering is identified in Fig. 2. | ||
Several higher-energy decomposition pathways are also identified in GP decomposition, and are included in Fig. 2. Pathway 2 (TS2aG, TS2bG) also leads to N-methylene-glycine formation, but with phosphoric acid (HP(O)(OH)2) as the co-product. Although HP(O)(OH)2 is more stable than P(OH)3, the higher barriers along pathway 2 mean that this pathway is unlikely to be kinetically favored. However, subsequent to the formation of P(OH)3 it is likely to rearrange to HP(O)(OH)2, where the barrier height is calculated to be 49.4 kcal mol−1 at the G3X-K level of theory. Pathway 3 demonstrates how water elimination can also lead to the production of N-methylene-glycine, along with P(O)OH, with a relatively large barrier height of 58.3 kcal mol−1. An alternative pathway for water elimination is through TS4G but with an even higher barrier energy of 71.2 kcal mol−1. Finally, pathways 5 and 6 lead to amine formation via elimination of the carboxylate (TS5G) or phosphate (TS6G) groups, respectively. Both reactions have similar barrier heights at ca. 70 kcal mol−1, and are unexpected to compete with pathway 1.
The illustrated potential energy surface in Fig. 2 was used to calculated reaction rate coefficients (k) for GP thermal decomposition. For the two-step process of pathway 2, the first step is assumed to be rate determining; i.e., the kinetic analysis treats the first step as leading directly to the dissociated products. Rate coefficients across the temperature range 300–2000 K are shown in Fig. 4. The dominant reaction channel at all temperatures is the formation of P(OH)3 and N-methylene-glycine via reaction channel 1. Rate coefficients for this process can be accurately expressed using the Arrhenius parameters A = 9.12 × 1013 s−1 and Ea = 46.3 kcal mol−1.
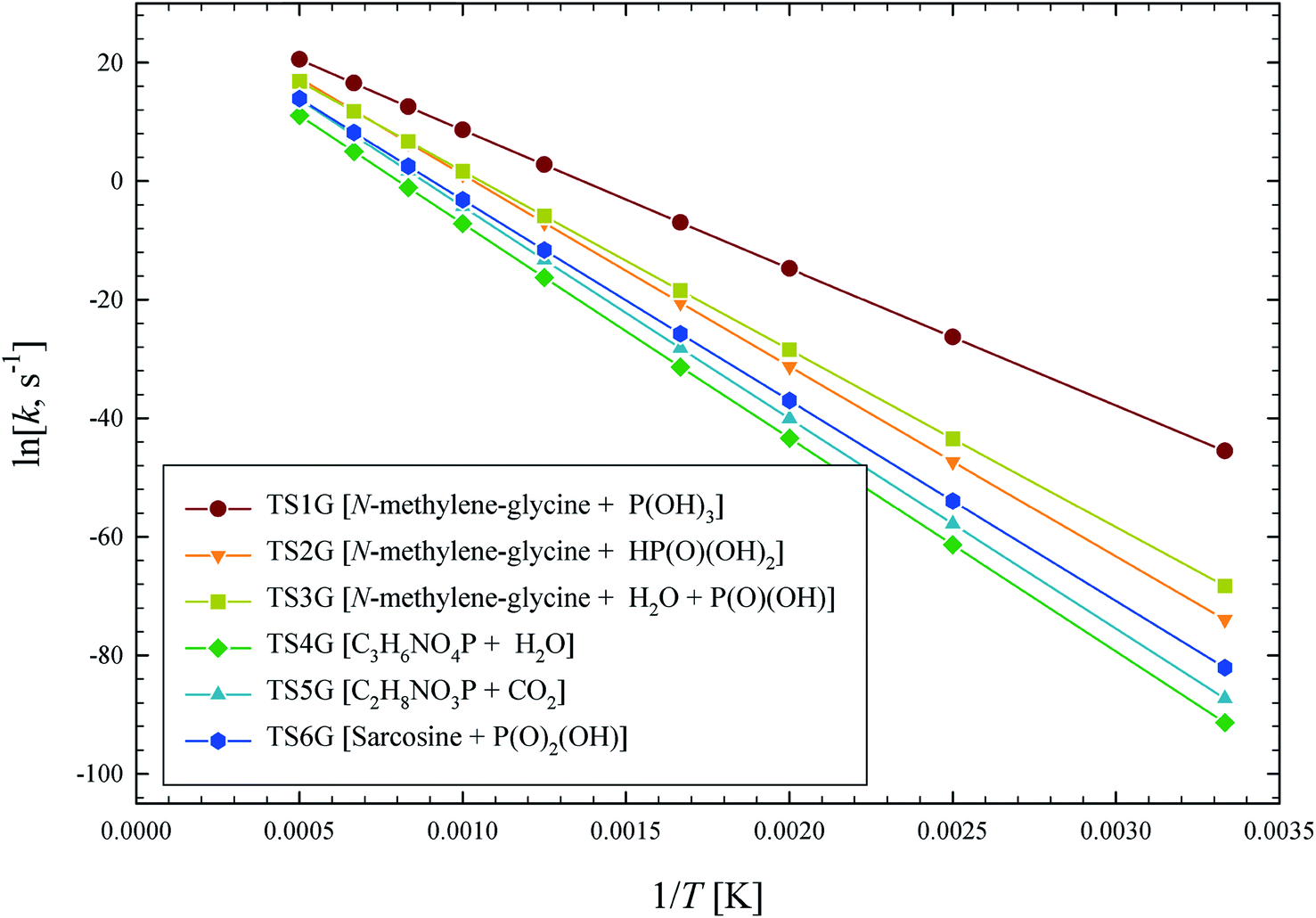 | ||
| Fig. 4 Arrhenius plot of calculated GP thermal decomposition reaction rate coefficients, k (s−1). Reaction pathways are identified in Fig. 2. | ||
The calculated rate coefficients illustrated in Fig. 4 were used to evaluate branching ratios in GP decomposition. The branching ratio (ki/kt) is defined as the fraction of each reaction channel's rate coefficient (ki) contributing to the overall rate coefficient (kt). The branching ratio (in percentage) for all decomposition channels of GP from 300 to 2000 K are shown in Fig. 5. This graph demonstrates that reaction channel 1 dominates across the examined temperature range, where the share of other channels at 1200 K (for example) is less than 1%, and this is likely to be the only process required to model the first stage of GP decomposition.
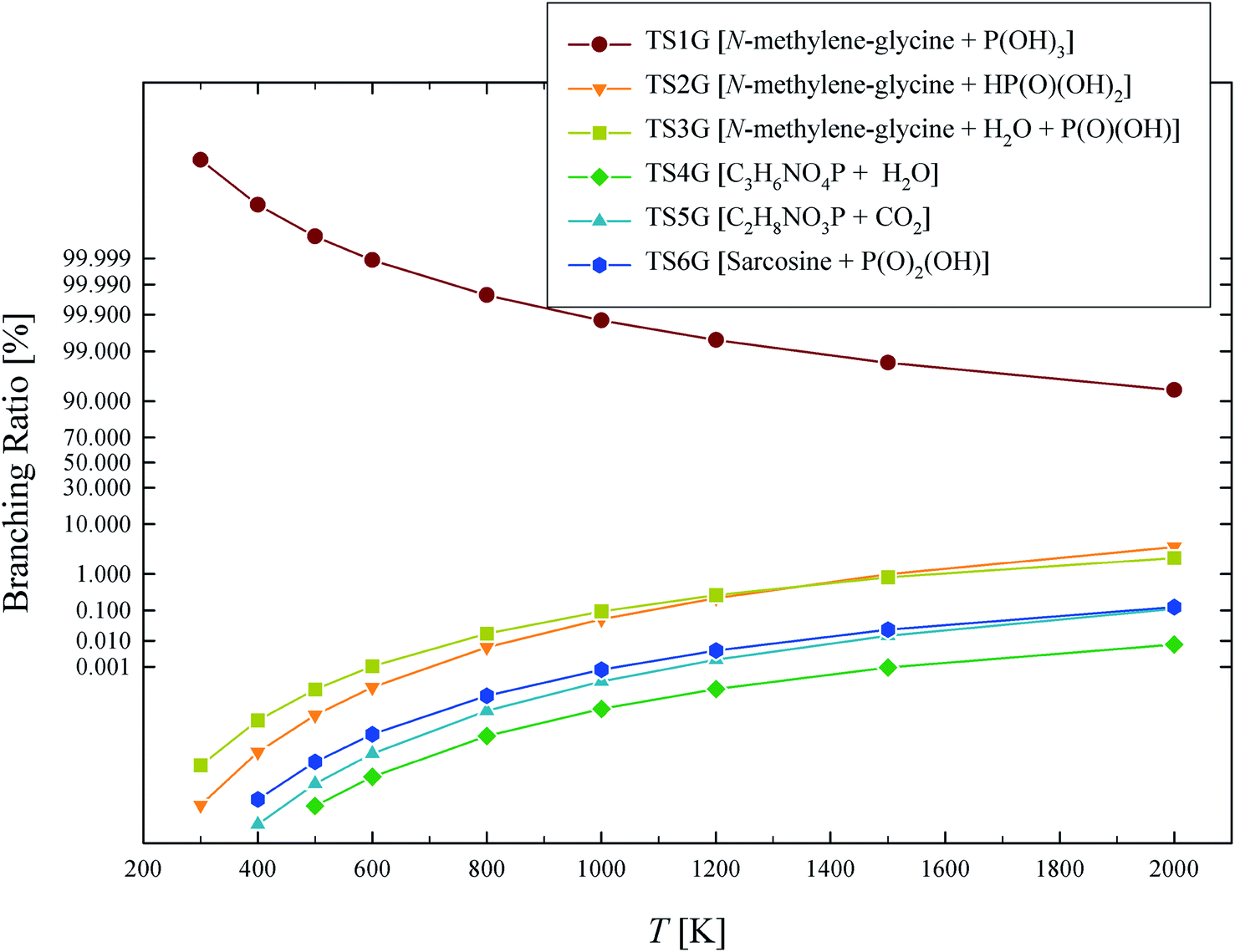 | ||
| Fig. 5 Branching ratio (%) for each of the reaction channels identified in GP decomposition across the temperature range of 300–2000 K. | ||
It is apparent that N-methylene-glycine will be the dominant decomposition product of GP, and we have therefore examined the further reaction chemistry of this important intermediate. A proposed decomposition mechanism for N-methylene-glycine involving decarboxylation is illustrated in Fig. 6a, with transition state structure shown in Fig. 7a. In this reaction, the carboylate moiety donates a proton to the unsaturated CH2N group, in concert with CO2 elimination, via TS1MG. This process leads to N-methyl methanimine and CO2 with barrier height of 48.2 kcal mol−1 and reaction exothermicity of 9.2 kcal mol−1. Given the similarity of barrier height to the first stage of GP decomposition, it is reasonable to expect that this would be relatively rapid at temperatures where GP forms N-methylene-glycine. This second step in the decomposition of GP is also qualitatively consistent with the experimental findings that the second stage of GP decomposition corresponds to an exothermic reaction with potential loss of the carbonyl (carboxylate) IR peak.41 So as to incorporate this reaction process into kinetic models, rate coefficients have been calculated between 300 and 2000 K, resulting in the Arrhenius expression 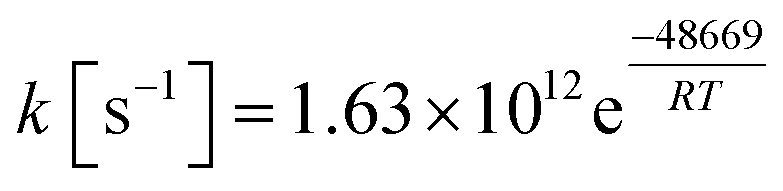 .
.
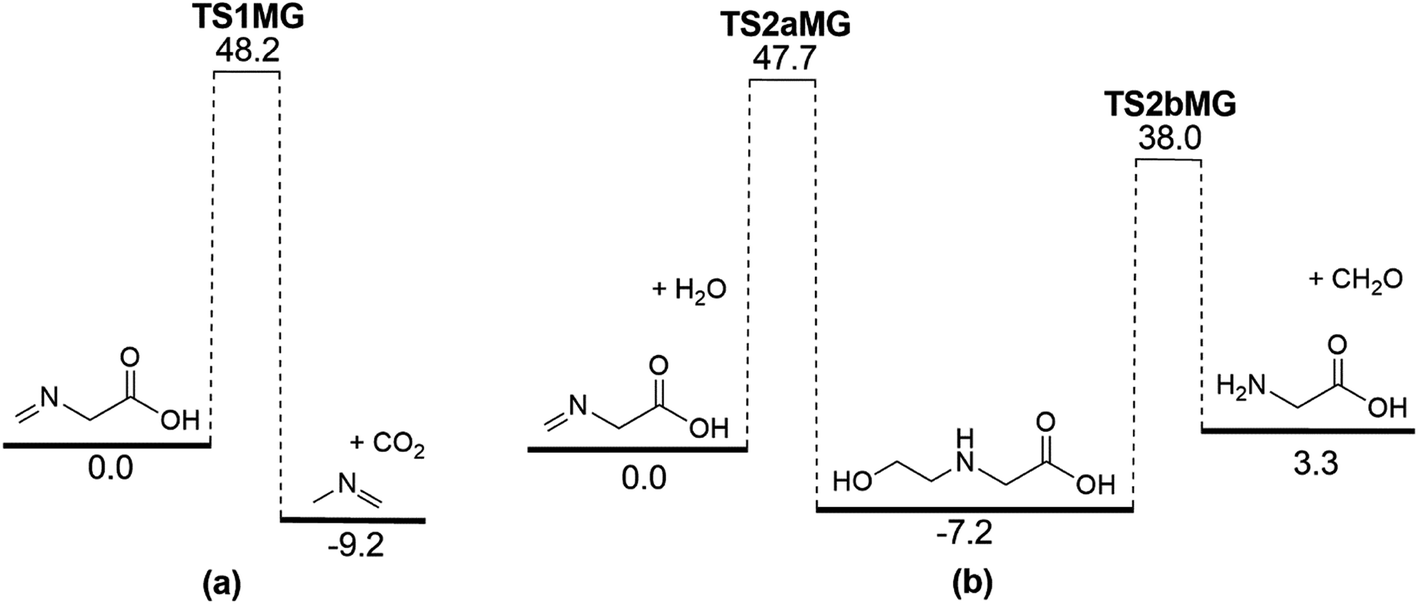 | ||
| Fig. 6 Potential energy diagram for (a) N-methylene-glycine thermal decomposition, and (b) N-methylene-glycine reaction with water vapour. Energies (0 K enthalpies, kcal mol−1) are calculated at the G3X-K level of theory. | ||
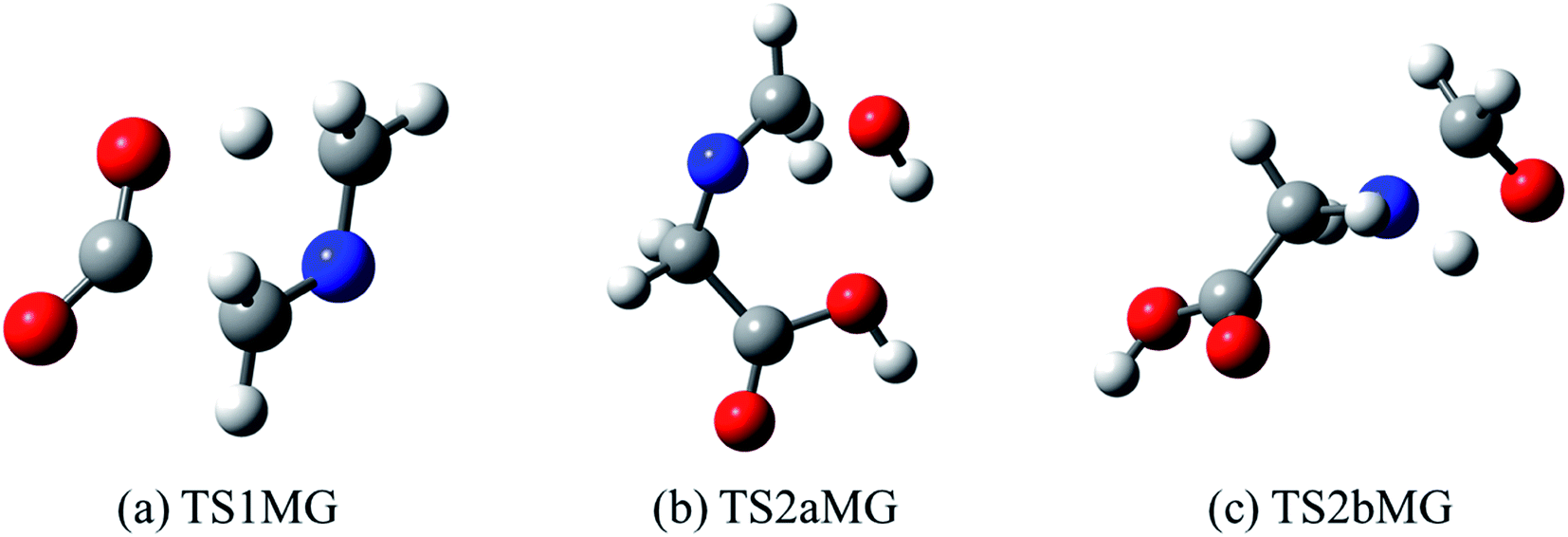 | ||
| Fig. 7 Optimized structures for transition states identified in (a) thermal decomposition, and (b and c) hydrolysis of N-methylene-glycine. | ||
Imines are known to undergo hydrolysis reactions, and we therefore also investigated the competing bimolecular reaction of N-methylene-glycine with water vapour. The potential energy surface for this reaction is shown in Fig. 6b (with transition states illustrated in Fig. 7b and c), where it is seen to involve a two-step reaction pathway. The first step of this process involves proton transfer from water to the nitrogen atom, along with C–OH bond formation (TS2aMG). This is the rate-determining step, with barrier of 47.7 kcal mol−1. Subsequent to this, proton transfer from the OH group to the nitrogen atom, along with N–C bond scission, transpires via TS2bMG with barrier of 38.0 kcal mol−1 relative to the reactants. Although this mechanism is comparable in energetics to that shown in Fig. 6a, the second order dependence on reagent concentrations means that it is unlikely to compete. However, in liquid water N-methylene-glycine may well follow this second decomposition pathway.
AMPA decomposition
AMPA is the major metabolite of GP, and these two compounds are likely to co-exist in contaminated materials. Decomposition reaction channels identified for AMPA are illustrated in Fig. 8, where it can be concluded that the decomposition mechanism is very similar to that of GP. The reaction channel with the lowest barrier (50.1 kcal mol−1) is again through P(OH)3 elimination (TS1A) (Fig. 9a), where the final products are methanimine (CH2NH) and P(OH)3. Of the higher-energy reactions depicted in Fig. 8, two again lead to the same imine, CH2NH, either with HP(O)(OH)2 (TS2aA and TS2bA) or HOPO + H2O (TS3A). The other pathway for water elimination, with energy barrier of 76.8 kcal mol−1, is via TS4A. As with GP, amine formation can occur via loss of the phosphate group, with barrier height of 62.5 kcal mol−1 (TS5A).
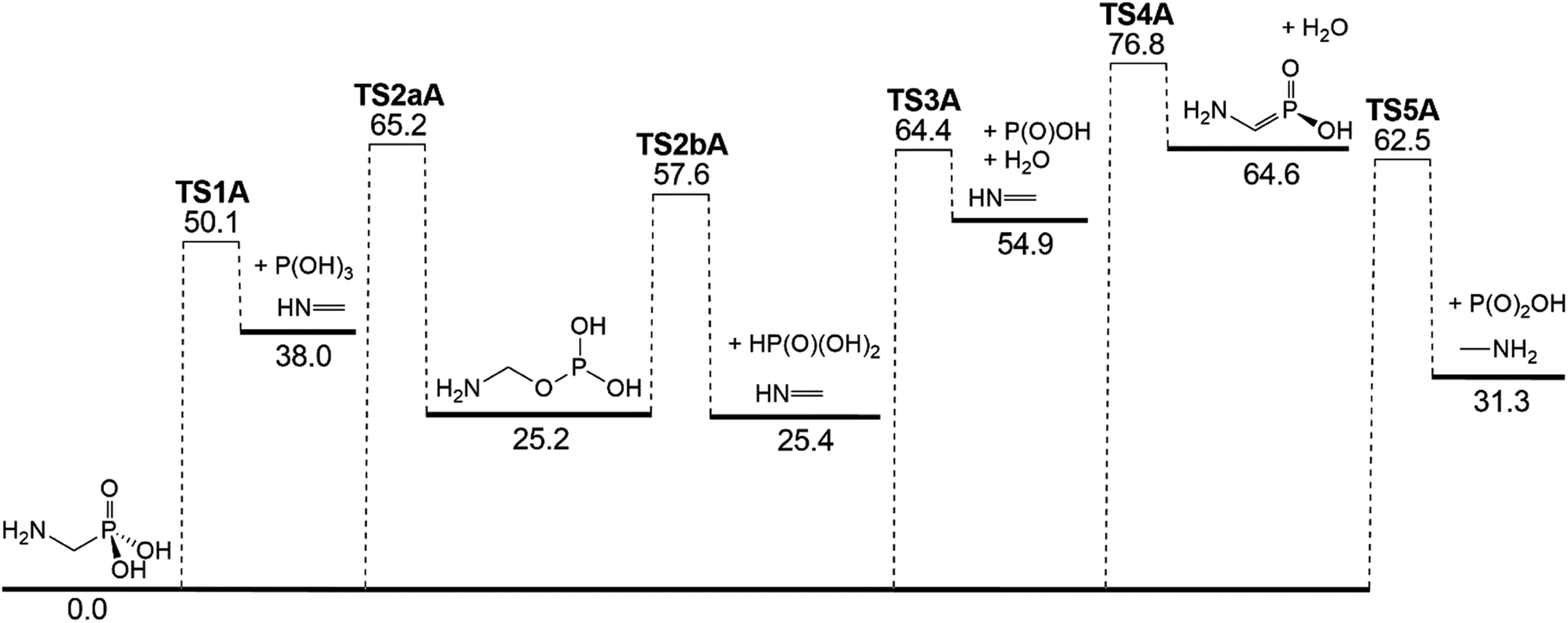 | ||
| Fig. 8 Potential energy diagram for the thermal decomposition of AMPA. Energies (0 K enthalpies, kcal mol−1) are calculated at the G3X-K level of theory. | ||
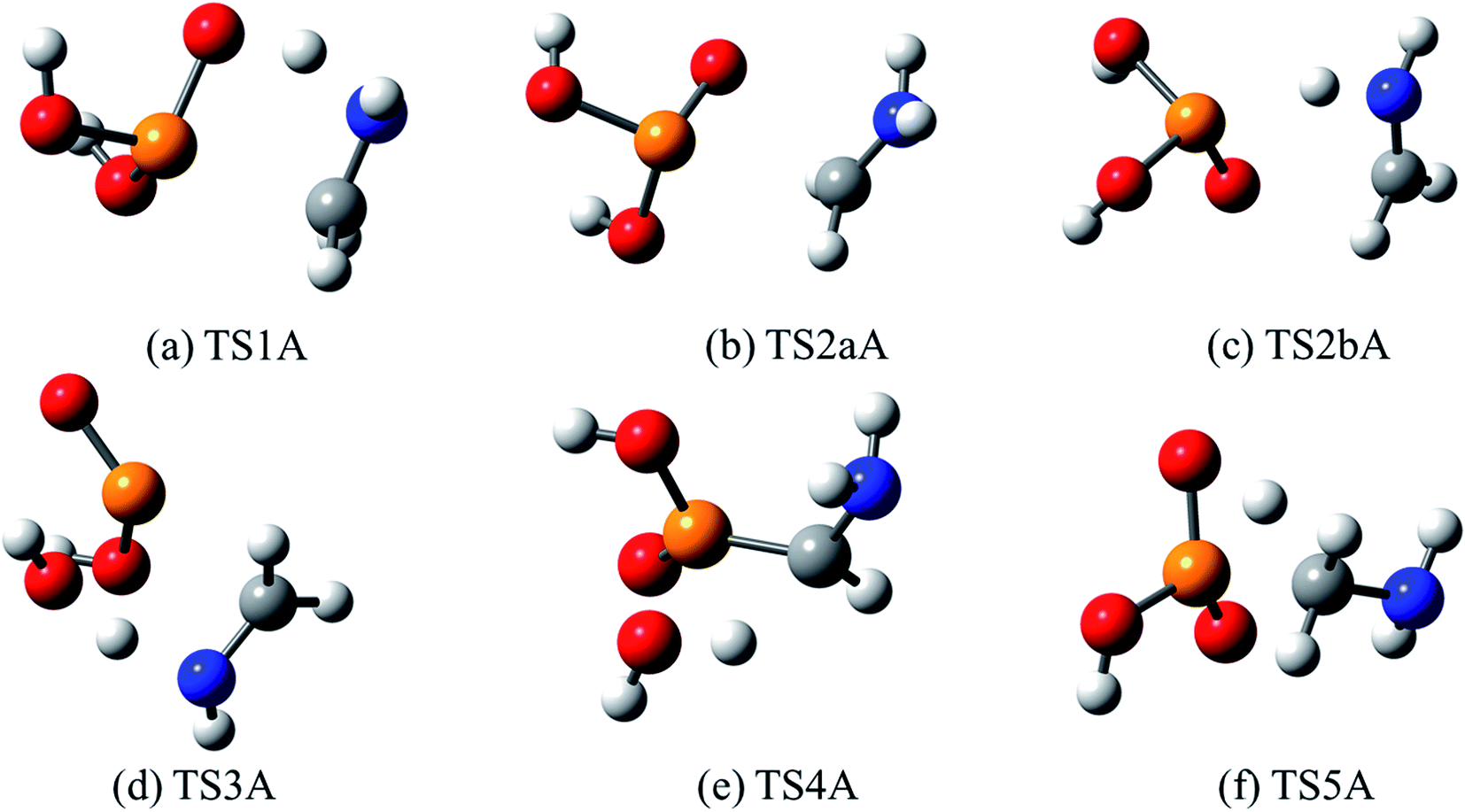 | ||
| Fig. 9 Optimized structures for transition states identified in the thermal decomposition of AMPA. Transition state numbering is identified in Fig. 8. | ||
The outlined potential energy surface for AMPA thermal decomposition was utilized to compute rate coefficients based on canonical transition state theory. The calculated rate coefficients for these five reaction channels over the temperature range 300–2000 K are shown in Fig. 10, and reaction channel 1 is kinetically dominant over all temperatures. Consequently, the compounds methanimine and phosphorous acid should be the main products of the first stage of AMPA decomposition. The Arrhenius parameters for this reaction over the examined temperature range are A = 2.32 × 1013 s−1 and Ea = 50.9 kcal mol−1. Branching ratios are shown as ESI,† and decomposition to methanimine and phosphorous acid is dominant across the entire temperature range. The reaction of methanimine with water vapour in the gas phase has also been studied, where the mechanism is similar to that shown in Fig. 6b. This process will lead to ammonia and formaldehyde, and rate coefficients between 300 and 2000 K are described by the expression 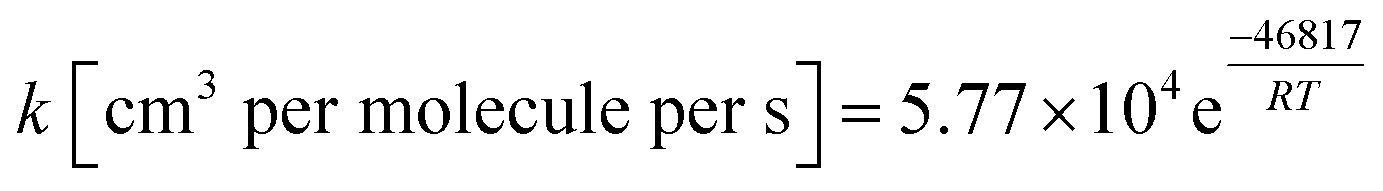 .
.
 | ||
| Fig. 10 Arrhenius plot of calculated AMPA thermal decomposition reaction rate coefficients, k (s−1). Reaction pathways are identified in Fig. 8. | ||
The half-life of dominant reaction channels for GP, N-methylene-glycine and AMPA decomposition are shown in Fig. 11. The half-life of GP is lower than that of its thermal decomposition intermediate, N-methylene-glycine, and metabolic product, AMPA. The half-life at 1000 K, which is a moderate temperature for conventional incineration processes, is below a millisecond for GP, and is several milliseconds for N-methylene-glycine and AMPA. These results reveal that the incineration or pyrolysis process can be applied for decomposition of GP aporised from waste material, with conversion to relatively benign products. Interestingly, AMPA has also been shown to have slower degradation kinetics than GP in catalytic destruction processes.42
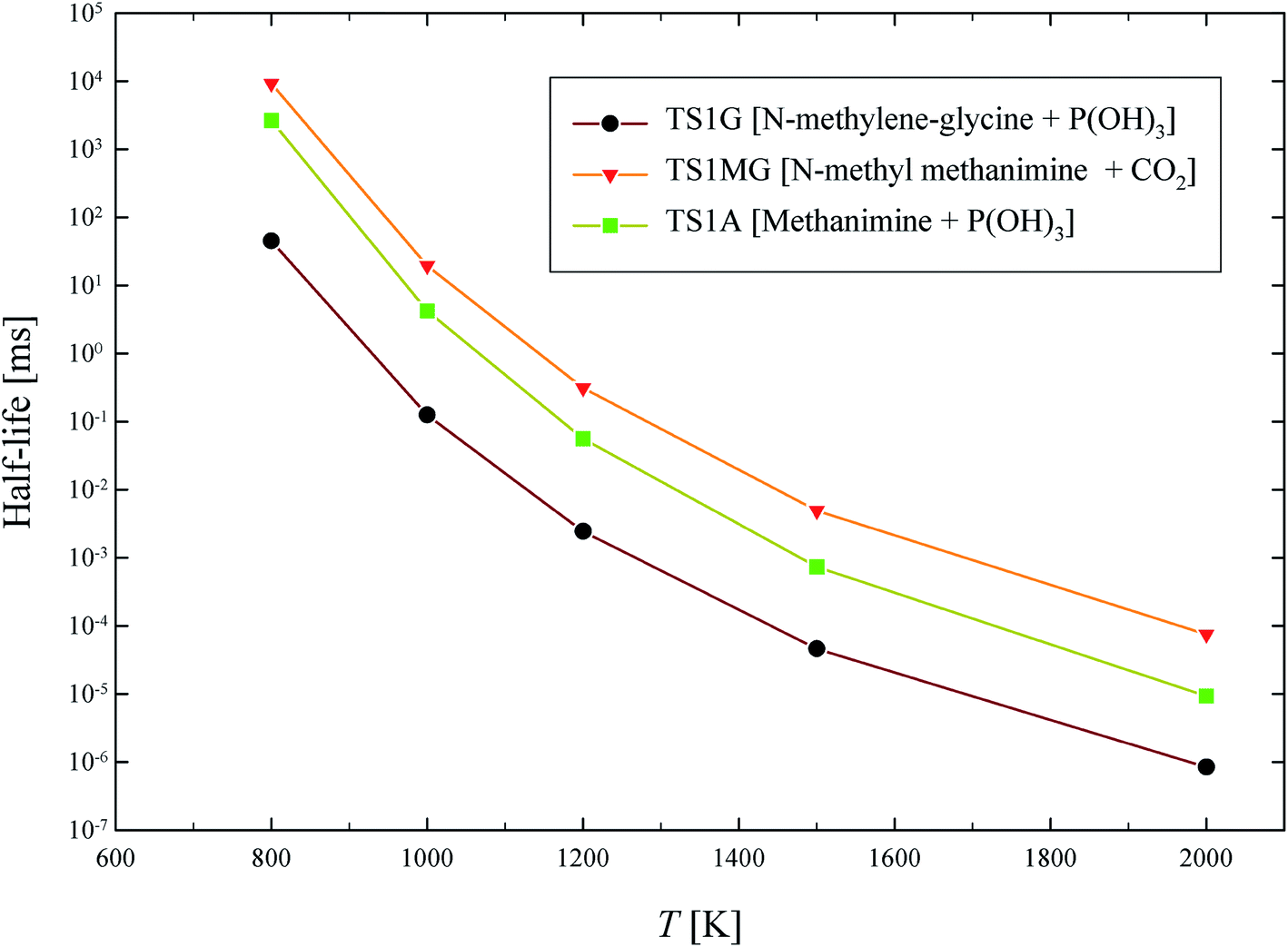 | ||
| Fig. 11 Calculated half-life (ms) for thermal decomposition of GP, N-methylene-glycine and AMPA through their dominant reaction channels. | ||
In summary, GP and its metabolite AMPA can be thermally decomposed rapidly in the gas phase through the chemical mechanism newly reported here, as summarized in Scheme 1. Moreover, the thermolysis mechanism of GP reveals that its direct decomposition will not proceed through its toxic metabolite AMPA, instead forming N-methylene-glycine and then N-methyl-methanimine. Decomposition of AMPA directly produces methanimine. The imine products are expected to undergo hydrolysis reactions to ultimately produce relatively benign amines and aldehydes. Importantly, none of these mechanisms proceed through free radical species, which may be environmentally persistent.43,44 These results will be helpful for developing incineration processes for the destruction of GP and AMPA, in addition to filling a gap in our fundamental understanding of their decomposition mechanisms.
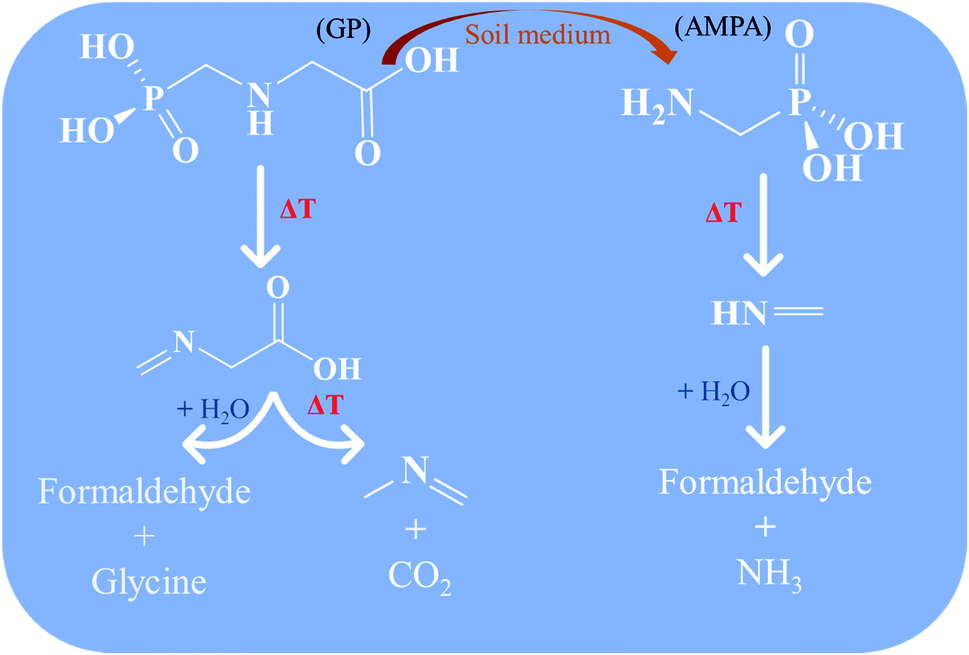 | ||
| Scheme 1 Proposed thermal decomposition mechanism for glyphosate (GP) and aminomethylphosphonic acid (AMPA). | ||
Conclusion
The thermochemical decomposition mechanism and kinetics of GP and AMPA have been examined by DFT and transition state theory calculations. Our findings reveal that the first stage of thermal decomposition of these organophosphorus compounds leads to elimination of phosphorous acid, with the co-product being N-methylene-glycine (for GP) or methanimine (for AMPA). Calculated rate coefficients from 300–2000 K confirm the dominant contribution of these pathways over other competing processes. The second stage of GP pyrolysis is shown to involve N-methylene-glycine decomposition to N-methyl-methanimine + CO2, whereas for AMPA the primary decomposition product methanimine is expected to react with water to yield formaldehyde and ammonia. These results can help to gain a better understanding of the fate of these organic phosphoric substances in thermal processes and to design and improve facilities efficiency for their destruction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through the Australian Research Council Future Fellowships scheme, FT130101304.References
- A. D. Baylis, Why glyphosate is a global herbicide: strengths, weaknesses and prospects, Pest Manage. Sci., 2000, 56, 299–308 CrossRef CAS.
- A. T. Woodburn, Glyphosate: production, pricing and use worldwide, Pest Manage. Sci., 2000, 56, 309–312 CrossRef CAS.
- S. O. Duke and S. B. Powles, Glyphosate: a once-in-a-century herbicide, Pest Manage. Sci., 2008, 64, 319–325 CrossRef CAS.
- D. R. Van Stempvoort, J. Spoelstra, N. D. Senger, S. J. Brown, R. Post and J. Struger, Glyphosate residues in rural groundwater, Nottawasaga River watershed, Ontario, Canada, Pest Manage. Sci., 2016, 72, 1862–1872 CrossRef CAS PubMed.
- H. Li, A. F. Wallace, M. Sun, P. Reardon and D. P. Jaisi, Degradation of glyphosate by Mn-oxide may bypass sarcosine and form glycine directly after C–N bond cleavage, Environ. Sci. Technol., 2018, 52, 1109–1117 CrossRef CAS.
- A. J. L. Catão and A. López-Castillo, On the degradation pathway of glyphosate and glycine, Environ. Sci.: Processes Impacts, 2018, 20, 1148–1157 RSC.
- A. Gosset, C. Durrieu, F. Orias, R. Bayard and Y. Perrodin, Identification and assessment of ecotoxicological hazards attributable to pollutants in urban wet weather discharges, Environ. Sci.: Processes Impacts, 2017, 19, 1150–1168 RSC.
- T. Poiger, I. J. Buerge, A. Bächli, M. D. Müller and M. E. Balmer, Occurrence of the herbicide glyphosate and its metabolite AMPA in surface waters in Switzerland determined with on-line solid phase extraction LC-MS/MS, Environ. Sci. Pollut. Res., 2017, 24, 1588–1596 CrossRef CAS PubMed.
- M. P. Gomes, E. Smedbol, A. Chalifour, L. Hénault-Ethier, M. Labrecque, L. Lepage, M. Lucotte and P. Juneau, Alteration of plant physiology by glyphosate and its by-product aminomethylphosphonic acid: an overview, J. Exp. Bot., 2014, 65, 4691–4703 CrossRef CAS PubMed.
- A. J. Al-Rajab and M. Schiavon, Degradation of 14C-glyphosate and aminomethylphosphonic acid (AMPA) in three agricultural soils, J. Environ. Sci., 2010, 22, 1374–1380 CrossRef CAS.
- B. Singh and K. Singh, Microbial degradation of herbicides, Crit. Rev. Microbiol., 2016, 42, 245–261 CrossRef CAS PubMed.
- D. P. Jaisi, H. Li, A. F. Wallace, P. Paudel, M. Sun, A. Balakrishna and R. N. Lerch, Mechanisms of bond cleavage during manganese oxide and UV degradation of glyphosate: results from phosphate oxygen isotopes and molecular simulations, J. Agric. Food Chem., 2016, 64, 8474–8482 CrossRef CAS PubMed.
- D. W. Kolpin, E. M. Thurman, E. A. Lee, M. T. Meyer, E. T. Furlong and S. T. Glassmeyer, Urban contributions of glyphosate and its degradate AMPA to streams in the United States, Sci. Total Environ., 2006, 354, 191–197 CrossRef CAS PubMed.
- C. Skark, N. Zullei-Seibert, U. Schöttler and C. Schlett, The occurrence of glyphosate in surface water, Int. J. Environ. Anal. Chem., 1998, 70, 93–104 CrossRef CAS.
- N. Lin and V. Garry, In vitro studies of cellular and molecular developmental toxicity of adjuvants, herbicides, and fungicides commonly used in Red River Valley, Minnesota, J. Toxicol. Environ. Health, Part A, 2000, 60, 423–439 CrossRef CAS.
- F. Mañas, L. Peralta, J. Raviolo, H. G. Ovando, A. Weyers, L. Ugnia, M. G. Cid, I. Larripa and N. Gorla, Genotoxicity of glyphosate assessed by the comet assay and cytogenetic tests, Environ. Toxicol. Pharmacol., 2009, 28, 37–41 CrossRef.
- C. Gasnier, C. Dumont, N. Benachour, E. Clair, M.-C. Chagnon and G.-E. Séralini, Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines, Toxicology, 2009, 262, 184–191 CrossRef CAS.
- S. H. Bai and S. M. Ogbourne, Glyphosate: environmental contamination, toxicity and potential risks to human health via food contamination, Environ. Sci. Pollut. Res., 2016, 23, 18988–19001 CrossRef CAS.
- M. Rodrigo, N. Oturan and M. A. Oturan, Electrochemically assisted remediation of pesticides in soils and water: a review, Chem. Rev., 2014, 114, 8720–8745 CrossRef CAS PubMed.
- Z. Ren, Y. Dong and Y. Liu, Enhanced glyphosate removal by montmorillonite in the presence of Fe(III), Ind. Eng. Chem. Res., 2014, 53, 14485–14492 CrossRef CAS.
- M. Mohsen Nourouzi, T. Chuah and T. S. Choong, Adsorption of glyphosate onto activated carbon derived from waste newspaper, Desalin. Water Treat., 2010, 24, 321–326 CrossRef.
- T. F. Speth, Glyphosate removal from drinking water, J. Environ. Sci., 1993, 119, 1139–1157 CAS.
- J. Song, X.-M. Li, A. Figoli, H. Huang, C. Pan, T. He and B. Jiang, Composite hollow fiber nanofiltration membranes for recovery of glyphosate from saline wastewater, Water Res., 2013, 47, 2065–2074 CrossRef CAS.
- M. D. Zhang, Y. F. Wei, K. Zhao, R. W. Mei and M. Huang, Presented in part at the Advanced Materials Research, 2011 Search PubMed.
- L. Cao, D. Ma, Z. Zhou, C. Xu, C. Cao, P. Zhao and Q. Huang, Efficient photocatalytic degradation of herbicide glyphosate in water by magnetically separable and recyclable BiOBr/Fe3O4 nanocomposites under visible light irradiation, Chem. Eng. J., 2019, 368, 212–222 CrossRef CAS.
- K. Barrett and M. McBride, Oxidative degradation of glyphosate and aminomethylphosphonate by manganese oxide, Environ. Sci. Technol., 2005, 39, 9223–9228 CrossRef CAS.
- A. Manassero, C. Passalia, A. C. Negro, A. E. Cassano and C. S. Zalazar, Glyphosate degradation in water employing the H2O2/UVC process, Water Res., 2010, 44, 3875–3882 CrossRef CAS.
- P. A. Vesilind and T. B. Ramsey, Effect of drying temperature on the fuel value of wastewater sludge, Waste Manage. Res., 1996, 14, 189–196 CrossRef.
- E. Zegers and E. Fisher, Gas-phase pyrolysis of diethyl methylphosphonate, Combust. Sci. Technol., 1996, 116, 69–89 CrossRef.
- E. Zegers and E. Fisher, Gas-phase pyrolysis of diisopropyl methylphosphonate, Combust. Flame, 1998, 115, 230–240 CrossRef CAS.
- P. Glaude, H. Curran, W. Pitz and C. Westbrook, Kinetic study of the combustion of organophosphorus compounds, Proc. Combust. Inst., 2000, 28, 1749–1756 CrossRef CAS.
- P. Glaude, C. Melius, W. Pitz and C. Westbrook, Detailed chemical kinetic reaction mechanisms for incineration of organophosphorus and fluoroorganophosphorus compounds, Proc. Combust. Inst., 2002, 29, 2469–2476 CrossRef CAS.
- J. H. Werner and T. A. Cool, Kinetic model for the decomposition of DMMP in a hydrogen/oxygen flame, Combust. Flame, 1999, 117, 78–98 CrossRef CAS.
- M. F. M. Nogueira and E. Fisher, Effects of dimethyl methylphosphonate on premixed methane flames, Combust. Flame, 2003, 132, 352–363 CrossRef CAS.
- R. P. Pohanish, Sittig's handbook of pesticides and agricultural chemicals, William Andrew, 2014 Search PubMed.
- Chemical Structure Substructure: (Aminomethyl)phosphonic acid, Scifinder, Version 2019, CAS Registry Number (RN) 1066-51-9 Search PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16 (Revision A), Gaussian Inc., Pittsburgh, PA, 2016 Search PubMed.
- G. da Silva, G3X-K theory: A composite theoretical method for thermochemical kinetics, Chem. Phys. Lett., 2013, 558, 109–113 CrossRef CAS.
- J. Barker, T. Nguyen, J. Stanton, C. Aieta, M. Ceotto, F. Gabas, T. Kumar, C. Li, L. Lohr and A. Maranzana, MultiWell-2016 Software Suite, JR Barker, University of Michigan, 2016 Search PubMed.
- J. R. Barker, Multiple-Well, multiple-path unimolecular reaction systems. I. MultiWell computer program suite, Int. J. Chem. Kinet., 2001, 33, 232–245 CrossRef CAS.
- F.-X. Chen, C.-R. Zhou and G.-P. Li, Study on thermal decomposition and the non-isothermal decomposition kinetics of glyphosate, J. Therm. Anal. Calorim., 2012, 109, 1457–1462 CrossRef CAS.
- H. Li, S. R. Joshi and D. P. Jaisi, Degradation and isotope source tracking of glyphosate and aminomethylphosphonic acid, J. Agric. Food Chem., 2016, 64, 529–538 CrossRef CAS PubMed.
- B. Dellinger, S. Lomnicki, L. Khachatryan, Z. Maskos, R. W. Hall, J. Adounkpe, C. McFerrin and H. Truong, Formation and stabilization of persistent free radicals, Proc. Combust. Inst., 2007, 31, 521–528 CrossRef PubMed.
- E. P. Vejerano, G. Rao, L. Khachatryan, S. A. Cormier and S. Lomnicki, Environmentally persistent free radicals: insights on a new class of pollutants, Environ. Sci. Technol., 2018, 52, 2468–2481 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated rate coefficients and branching ratios; potential energy diagram for methanimine hydrolysis; relative energies, moments of inertia, vibrational frequencies, and Cartesian coordinates of all species; thermochemical properties and bond dissociation energies in GP. See DOI: 10.1039/c9em00422j |
| This journal is © The Royal Society of Chemistry 2020 |