
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A global atmospheric chemistry model for the fate and transport of PFCAs and their precursors†
Colin P.
Thackray‡
 *a,
Noelle E.
Selin§
a and
Cora J.
Young
b
*a,
Noelle E.
Selin§
a and
Cora J.
Young
b
aDepartment of Earth, Atmospheric, and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, USA. E-mail: thackray@seas.harvard.edu
bDepartment of Chemistry, York University, Toronto, Canada
First published on 24th December 2019
Abstract
Perfluorocarboxylic acids (PFCAs) are environmental contaminants that are highly persistent, and many are bio-accumulative and have been detected along with their atmospheric precursors far from emission sources. The overall importance of precursor emissions as an indirect source of PFCAs to the environment is uncertain. Previous studies have estimated the atmospheric source of PFCAs using models and degradation pathways of differing complexities, leading to quantitatively different results. We present results from simulations of atmospheric PFCA formation and fate using the chemical transport model GEOS-Chem. We simulate the most up-to-date chemistry available to our knowledge for the degradation of the precursors fluorotelomer alcohol (FTOH), fluorotelomer olefin (FTO), and fluorotelomer iodide (FTI), as well as the deposition and transport of the precursors, intermediates and end-products of the formation chemistry. We calculate yields of C3–C13 PFCAs formed from 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 12:2 fluorotelomer precursors and their deposition to the surface. We find that the ratio of long-chain to short-chain PFCAs increases strongly with distance from source regions. We compare our model results to remote deposition measurements and mid-latitude rainwater measurements. The model captures the observed relationship between rainwater abundance and PFCA chain length, as well as the average deposition rates at mid-latitude and Arctic sites, but underestimates the deposition of PFDoA, PFDA, and TFA at mid-latitudes and PFNA at the Devon Ice Cap. We provide estimates of cumulative PFCA deposition globally. We find that given the most recent emission inventory, the atmospheric source of PFCAs is 6–185 tonnes per year globally and 0.1–2.1 tonnes per year to the Arctic.
:2 to 12:2 fluorotelomer precursors and their deposition to the surface. We find that the ratio of long-chain to short-chain PFCAs increases strongly with distance from source regions. We compare our model results to remote deposition measurements and mid-latitude rainwater measurements. The model captures the observed relationship between rainwater abundance and PFCA chain length, as well as the average deposition rates at mid-latitude and Arctic sites, but underestimates the deposition of PFDoA, PFDA, and TFA at mid-latitudes and PFNA at the Devon Ice Cap. We provide estimates of cumulative PFCA deposition globally. We find that given the most recent emission inventory, the atmospheric source of PFCAs is 6–185 tonnes per year globally and 0.1–2.1 tonnes per year to the Arctic.
Environmental significancePerfluorocarboxylic acids (PFCAs) are persistent environmental contaminants that bio-accumulate, and, along with their atmospheric precursor species, have been detected far from emission sources. Measurements of the deposition of PFCAs, especially over more than a year at a single location, are rare and spatially sparse. In this work, we present a global atmospheric model for simulating the atmospheric transport and deposition of PFCAs as well as their formation through the detailed chemistry of their atmospheric precursors. We use our model to estimate deposition of PFCAs globally through time, complementing the existing measurement record, and find that atmospherically generated PFCAs are the majority of remotely deposited PFCAs. We also show that the model can be used in combination with measurements to infer the importance of experimentally highly uncertain processes in PFCA formation chemistry. |
1. Introduction
Perfluorocarboxylic acids (PFCAs) are persistent environmental contaminants that bio-accumulate,2–4 and, along with precursor species, have been detected far from emission sources.5–10 Atmospheric PFCA precursors such as fluorotelomer alcohols (FTOHs) undergo photochemical reactions11 which form PFCAs after a multiple-step reaction mechanism.12 The magnitude of this indirect source of environmental PFCAs is uncertain. The atmospheric formation of PFCAs depends on the reaction of intermediate species13,14 with photochemically important species that are common in the atmosphere, such as HOx and NOx. In different atmospheric environments, these species vary over orders of magnitude, affecting the ability of the atmosphere to produce PFCAs.15,16 This means that the connection between chemistry, transport, and deposition of PFCAs, their precursors, and intermediates in their formation is important to the quantity of PFCAs formed in the atmosphere as well as deposited to remote locations such as the Arctic. Atmospheric chemical transport models are therefore an important tool for quantifying the formation and fate of PFCAs in the atmosphere.Long-chain PFCAs (lcPFCAs, perfluorinated chain length ≥7) are particularly important environmentally because the detrimental effects of PFCAs increase with chain length.2–4 Reducing lcPFCA emissions has been the intent of policy actions due to their health effects,17 resulting in decreasing direct emissions globally. In contrast, emissions of PFCA atmospheric precursors are rising,1 leading to an increasing importance of indirect environmental sources of PFCAs. Emissions may also be shifting toward shorter chain compounds (including precursor compounds), and short-chain PFCAs are still an environmental and health concern. Previous studies have modeled lcPFCA formation from the degradation of FTOHs in the atmosphere.15,16,18 However, other emitted atmospheric precursors exist in the form of other fluorotelomer compounds.1,12,19–21 These different precursors and emissions from the most recent comprehensive inventory1 have not been included in previous chemical transport models.
Wallington et al.18 simulated the atmospheric degradation of 8:2 FTOH using the IMPACT atmospheric chemistry model on a 4° × 5° global grid. They found that the yield of PFOA (C8) from its FTOH precursors ranges from 1% to 10% depending on time of year and location, but did not report PFNA (C9) or shorter-chain PFCA yields and deposition from the same precursors. Yarwood et al.,15 using a high-resolution (72 km × 72 km) atmospheric chemistry model with North American coverage, estimated yields of PFOA from FTOH degradation at approximately 6% on average due to precursor degradation and yields of PFNA of much less than 1%, but did not consider shorter chain PFCAs. Using a global-scale multispecies mass-balance model and simplified chemistry, Schenker et al.22 found that precursor atmospheric transport followed by degradation could contribute to observed Arctic PFCAs.
Thackray and Selin16 found using a detailed box model of updated chemistry that the capacity of the atmosphere to form PFOA and PFNA from fluorotelomer precursors in the absence of non-chemical removal varies over orders of magnitude based on the location and season due to strong dependence on the photochemical environment, and that in regions far from NOx pollution sources, the yields of PFOA and PFNA can be larger than 30%.
In this work, we present results from simulations of atmospheric PFCA formation and fate using the global chemical transport model GEOS-Chem. GEOS-Chem is a community-developed atmospheric chemistry model, which has transport governed by re-analysis meteorology and a spatial resolution conducive to site-specific comparisons. We simulate the most up-to-date chemistry available to our knowledge for the degradation of the emitted PFCA precursors 12:2, 10:2, 8:2, 6:2 and 4:2 FTOH, fluorotelomer olefin (FTO) and fluorotelomer iodide (FTI), as well as deposition and transport of the precursors, intermediates and end-products of the degradation chemistry with global coverage.
2. Methods
We use the GEOS-Chem model23 to simulate the transport and fate of fluorotelomer precursors, from their chemical degradation to the deposition of their reaction products, including PFCAs.2.1. PFCA formation chemistry model
We add PFCAs to the GEOS-Chem 12.2 chemistry simulation.24 GEOS-Chem calculates the concentrations of HOx, NOx, RO2, etc. on a global Eulerian grid using a detailed set of chemical reactions, detailed emission inventories, prescribed meteorology, and wet25 and dry26 deposition schemes. We run the model at 4° × 5° horizontal resolution and 46 vertical levels and use assimilated GEOS-FP meteorology. We use a precursor degradation mechanism16 which builds on the work of previous modeling efforts15,18 with additional reactions from more recent literature.12 The chemical reactions included are listed in Table S2.† The mechanism defines the degradation of X:2 FTOH, X:2 FTO, and X:2 FTI (where X = 12, 10, 8, 6, and 4), which are the most commonly emitted fluorotelomer PFCA precursors,1,27 to form FT-aldehyde and further intermediate and stable end products including PFOA and other PFCAs. While the degradation mechanism presented by Thackray et al.16 addressed only 8:2 precursors, we assume the rates of reaction in the chemical mechanism are chain-length independent, due to the reactions involving exclusively the functional group.11,12
FT-aldehyde can be oxidized by OH or photolyzed to form peroxy and acylperoxy radicals. These radicals react with NO, NO2, RO2, and HO2 to form intermediates which can again react with OH and ultraviolet light (at wavelengths <350 nm), with further radical reactions leading to stable PFCAs or intermediates with reduced chain length.
Due to the solubility of PFCAs and some of the intermediates in their formation, wet deposition and scavenging are important non-chemical removal processes that we account for in our simulations. For wet removal, we use a scheme analogous to that for existing soluble GEOS-Chem species such as H2O2. Wet removal can occur as rainout and washout from both large-scale and convective precipitation, as well as scavenging in updrafts.25 Fluorinated species in the model partition to the aqueous phase according to their Henry's Law coefficients, assuming equilibrium. We calculate Henry's Law coefficients using the Henrywin application in the EPISUITE software package, which uses a group additive method for estimating the physicochemical properties of species for which experimental determinations are unavailable. For some highly fluorinated species, including FTOHs, this method can introduce errors in the magnitude of the Henry's Law coefficient.28 While different methods of estimation give Henry's Law coefficients differing by orders of magnitude, the estimates agree that precipitation will rapidly remove PFCAs present in the atmosphere, meaning that the fate of PFCAs in the model is much more strongly dependent on the presence of precipitation than the value of Henry's Law coefficients.
For emissions to air of FTOH, FTO, FTI, PFOA, and PFNA, we use the inventory of Wang et al.1 This inventory gives total fluorotelomer production estimates for the period 1962 to the present, as well as the fraction of total production accounted for by each homolog, which we use to calculate annual releases of fluorotelomer precursors. We assume that the proportion of released precursors is 48% FTOH, 48% FTO, and 4% FTI.1,27 The inventory gives constant average annual emissions for groups of years, and we interpolate these average values to result in smooth changes between years while conserving the inventory's total cumulative emissions. We assume that the global spatial distribution of emissions due to use and disposal of fluorotelomer products is the same as that of the anthropogenic emissions of NOx over land. This results in a pattern of emissions similar to those of Stemmler and Lammel29 who weighted their emissions by GDP.
Simulations are conducted for emission years 1960–2015, with meteorological fields corresponding to years 2012–2017, which are repeated in a cycle throughout the simulated period. We simulate minimum and maximum emission scenarios1 for both precursor and direct emissions, with and without the hydration reaction of perfluoroaldehyde, due to the reaction's high rate constant uncertainty and importance to odd-carbon-number PFCA formation.12 This reaction can take place either in the gas phase or heterogeneously, and only the upper limit of the reaction rate has been determined, for the gas phase.12 To calculate bounds on this reaction's importance, we perform simulations both with and without this gas-phase reaction at its upper-limit rate (see Table S1† for full list of model scenarios). Sea spray-generated particles have been shown to be a pathway of PFCA to the atmosphere from the ocean.30 We do not model this process here, but it could act as an additional source to the atmosphere as well as a pathway of re-suspension of previously deposited atmospheric PFCA.
2.2. Comparison to observations
We compare our model output PFCA depositions to observations at measurement sites both in the Arctic and at mid-latitudes. PFCA annual deposition fluxes were measured for the years 1977–2015 at the Devon Ice Cap in the Arctic Archipelago by analyzing snow samples.31 We use these particular Devon Ice Cap measurements because they represent a consistent record of deposition over more than three decades. PFCA wet deposition fluxes were also reported for mid-latitudes, and we directly compare our deposition fluxes to the reported fluxes.9,32–49 We directly compare our deposition fluxes for each of these sites to the fluxes measured. In these comparisons, we include a “representativeness error”50 in the measurements which estimates the effect of comparing a point measurement to a model grid-box using the local spatial variability in the model output.At lower latitudes, rainwater concentration measurements and wet deposition fluxes are compared to modeled wet deposition fluxes at the corresponding locations and times. The complete list of rainwater observations is shown in Table S2.†
3. Results
3.1. Measurement comparison at remote sites
Fig. 1(a) shows the chain-length-dependence of rainwater abundance of PFCAs in modeled and observed rainwater. Rainwater measurements span the years 1998–2010 and include multiple locations in North America, Asia, and Europe. Both model and observations show PFOA as the dominant PFCA in rainwater and PFTrA (C13) as the least abundant. Both also show an increase in abundance with decreasing chain lengths from C13–C9, and C7–C5 are tightly grouped. The consistency in the ordering between modeled wet deposition and observed rainwater concentration demonstrates that the deposition rate is driven by abundance in rainwater, and not by the co-location of particular PFCA homologs and higher precipitation rates. The rainwater observations show greater variability than the model. Because of the temporal as well as spatial separation of available measurements, a time-resolved emission inventory and atmospheric response is important, and previous modeling studies have not been able to conduct this type of comparison.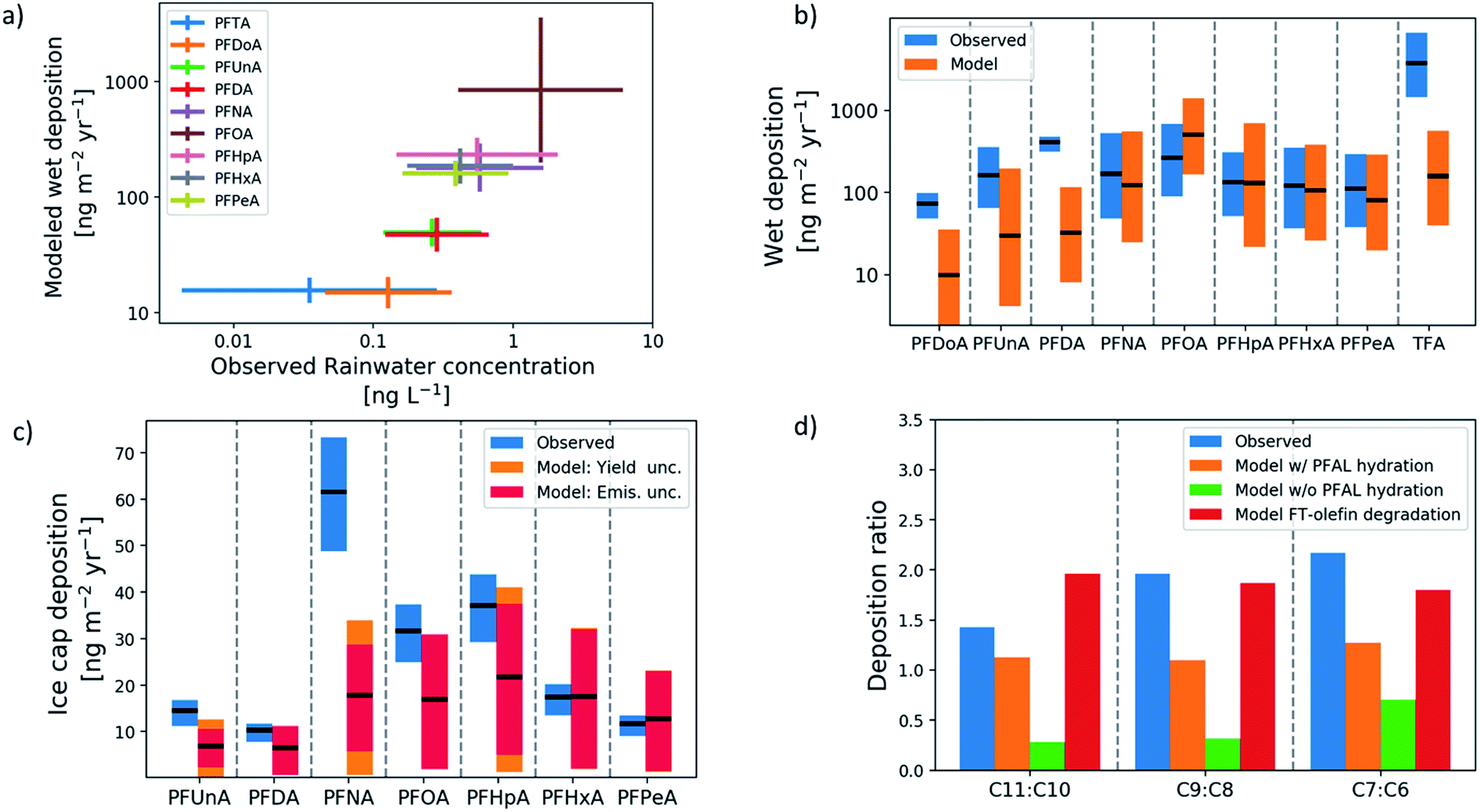 | ||
| Fig. 1 Measurement-model comparison. (a) PFCA abundance in rainwater across sites globally. (b) Mid-latitude wet deposition of C3–C12 PFCA across measurement locations. Observed geometric means and standard deviations across sites in blue, and modeled geometric means and uncertainty in geometric means in orange. (c) Average deposition of C5–C11 PFCAs to the Devon Ice Cap over the years 1977–2015. Observed mean deposition rates and their uncertainty are in blue. Modeled deposition rates and their uncertainty range are in red/orange, with color shading representing the fraction of the total uncertainty range contributed by uncertainty in yields (orange) and uncertainty in emissions magnitude (red). (d) Odd-even homolog deposition ratios over the Devon Ice Cap record. Observed ratios (blue), with modeled ratios using BASE (orange), NO_HYDR (green) and FTO_ONLY (red) scenarios. | ||
Fig. 1(b) shows the model-measurement comparison of wet deposition fluxes at mid-latitude locations. The extent of the bars for the measurements represents the mean and standard deviation between locations for each homolog, and the model bars represent the uncertainty in the mean over the same locations. All but three of the homologs, PFDoA (C12), PFDA (C10), and TFA (C2), show agreement within uncertainty between modeled and observed means. The model underestimates these three homologs, with TFA underestimated by an order of magnitude. The underestimate of TFA is likely due to the influence of additional TFA sources, including atmospheric oxidation of hydrochlorofluorocarbons and hydrofluorocarbons.12
Fig. 1(c) shows the model-measurement comparison for deposition to the Devon Ice Cap in the Canadian Arctic Archipelago. Each bar shows the average annual deposition flux for each PFCA homolog for the time period 1980–2015. The extent of the bars for the measurements represents the representativeness errors for the model-measurement comparison, which estimate variability within a grid box using spatial variability between grid boxes.50,51 The figure shows that the model agrees within uncertainty with the observed deposition of PFHxA (C6) and PFPeA (C5). The uncertainty bounds in the observations and model overlap for PFUnA (C11), PFDA, PFOA, and PFHpA (C7), but the model underestimates PFNA. Because of uncertainty in the perfluorinated aldehyde (PFAL) hydration reaction12 and the relatively direct pathway from the FTO precursor to odd-carbon PFCAs through this reaction, uncertainty in the rate of hydration makes uncertainty in odd-carbon deposition larger than the even-chain counterparts. In all three odd-even pairs (PFUnA-PFDA, PFNA-PFOA, and PFHpA-PFHxA), the observed deposition is on average 70% higher for the odd homolog (Fig. 1(d)). The model does not reproduce this and predicts more even-homolog deposition unless the perfluoroaldehyde hydration reaction is included. Including the hydration reaction increases the odd-even ratio above parity, but the observed ratios most resemble the modeled degradation of FT-olefins (including PFAL hydration) in particular. Full time-series comparisons for each homolog are shown in Fig. S1.† All modeled homologs show an underestimate in the 1990s. For all PFCAs except PFNA, the model reproduces the observed deposition in the 2000s. For PFNA, the model underestimates deposition for the whole time series. Direct emission of PFNA was also included and represents an additional source of uncertainty.
3.2. Deposition of PFCAs
Fig. 2(a) shows the modeled spatial distribution of 2013–2015 annual PFOA deposition due to precursor degradation and direct emission globally. Total deposition is considered below, with dry deposition making up 35% of deposition globally (see Fig. S3†). Deposition rates vary across orders of magnitude globally, due to both variations in PFOA formation and rates of precipitation, as wet deposition is the primary means for atmospheric PFCA removal. The highest deposition rates occur over source regions and the oceans downwind of strong emission sources such as eastern North America and eastern Asia. Far from sources, the highest deposition occurs near the equator, especially in the region of the Intertropical Convergence Zone (ITCZ). This is due to the high rates of precipitation in this region and the photochemical environment of the tropical oceans being the best for long-chain PFCA formation.16 Desert areas show less deposition, highlighting the importance of rain and wet removal for PFCAs. Fig. 2(b) shows the same information for the sum of all PFCAs with fluorinated chains longer than PFOA. Across the globe, these PFCAs show a very similar spatial pattern to PFOA, with the highest deposition rates over the equatorial oceans and downwind of densely populated regions. The deposition of all shorter-chain PFCAs (Fig. 2(c)) is highest near sources and where NOx can most quickly shorten the fluorinated chain length, but decreases more rapidly than the longer chains as the distance from sources increases. Farther from emissions sources, the ratio of short-chain to long-chain PFCA (Fig. 2(d)) deposition decreases, highlighting the relationship between long-chain PFCA yield and formation time discussed in previous work.16 The distance from sources also has a direct effect on the ratio because short-chain PFCAs are removed more efficiently by wet deposition and therefore undergo less efficient transport in the atmosphere. Farther from sources and the presence of NOx, long-chain compounds become an increasingly large fraction of deposition, with the highest ratios over the equatorial and southern oceans.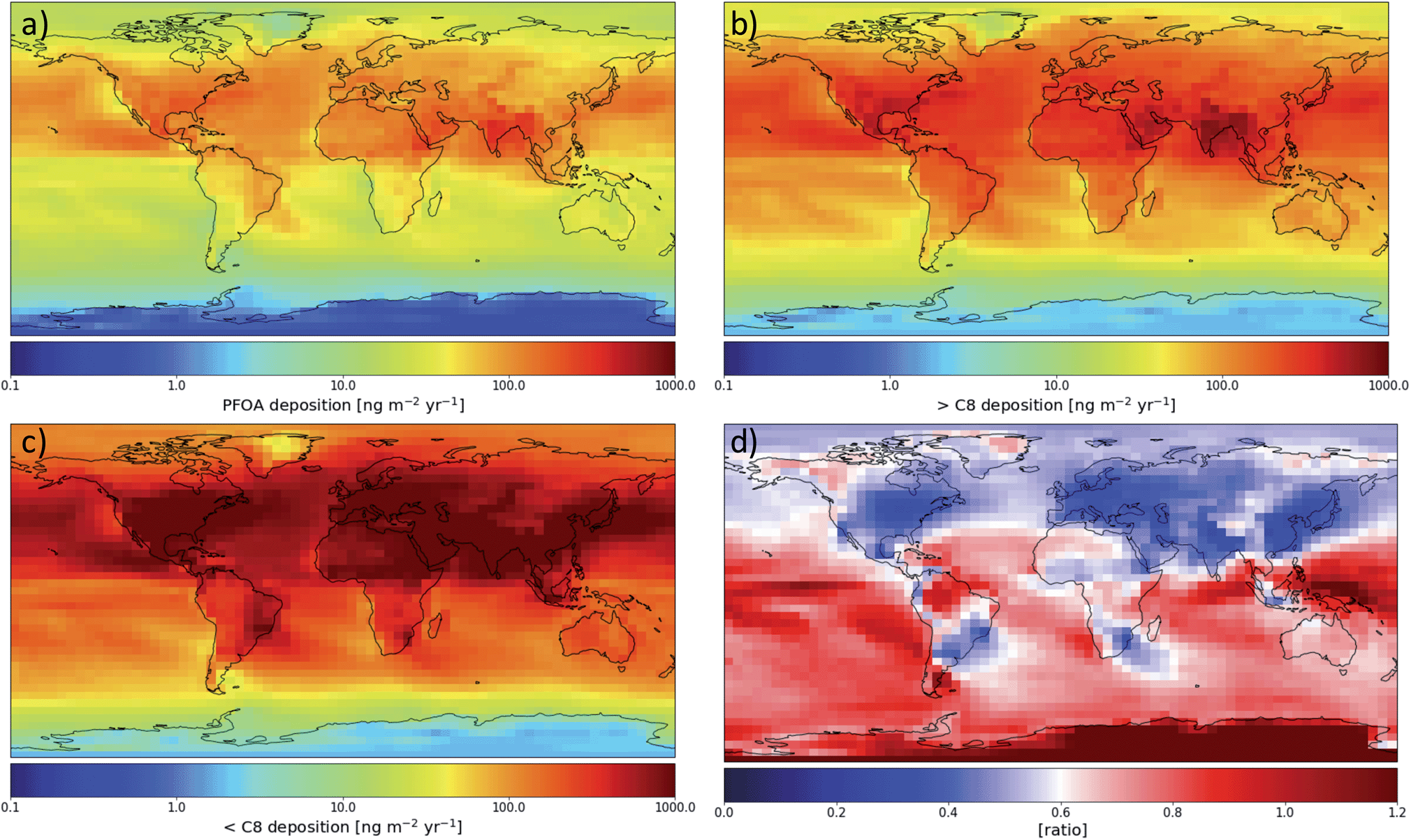 | ||
| Fig. 2 Spatial distribution of modeled 2013–2015 annual average (a) PFOA deposition (ng per m2 per year), (b) deposition of all longer-chain PFCAs (ng per m2 per year), (c) deposition of all shorter-chain PFCAs, and (d) precursor-produced long-chain (>C7) to short-chain deposition ratio. | ||
Over North America, we see a spatial pattern of PFOA consistent with the pattern simulated by Yarwood et al.,15 with the highest values over the eastern U.S. and decreasing deposition westward and northward. PFNA deposition (Fig. S2†) follows a very similar pattern to that of PFOA; previous work15 did not simulate the degradation of FTO to form PFNA.
Table 1 shows the fraction of total fluorinated precursor emissions that culminate in the deposition of each modeled PFCA for each fluorotelomer chain length group (with ranges bounded by the NO_HYDR and FTO_ONLY scenarios). This value is between 1 and 7% globally for PFOA, 1–16% for PFNA and longer-chain PFCAs, and 6–35% for the sum of PFHpA and shorter-chain PFCAs, depending on the precursor chain length and uncertainty in yields.
| 12:2 |
10:2 |
8:2 |
6:2 |
|
|---|---|---|---|---|
| PFTA (C13) | 0.01–0.16 | 0.00 | 0.00 | 0.00 |
| PFDoA (C12) | 0.05–0.07 | 0.00 | 0.00 | 0.00 |
| PFUnA (C11) | 0.04–0.05 | 0.01–0.15 | 0.00 | 0.00 |
| PFDA (C10) | 0.03–0.04 | 0.05–0.07 | 0.00 | 0.00 |
| PFNA (C9) | 0.02–0.03 | 0.04–0.05 | 0.01–0.15 | 0.00 |
| PFOA (C8) | 0.01–0.02 | 0.03–0.04 | 0.05–0.07 | 0.00 |
| PFHpA (C7) | ≤0.01 | 0.02–0.03 | 0.04–0.05 | 0.01–0.15 |
| PFHxA (C6) | ≤0.01 | ≤0.01 | 0.03–0.04 | 0.03–0.05 |
| PFPeA (C5) | ≤0.01 | ≤0.01 | 0.01–0.02 | 0.02–0.03 |
| <C5 PFCA | 0.03–0.05 | 0.02–0.05 | 0.04–0.10 | 0.04–0.12 |
Table 2 shows Arctic (>66 N) and mid-latitude total PFCA deposition due to direct emissions and the degradation of precursors over the period 1960–2015, bounded by NO_HYDR_LOW and FTO_HIGH scenarios. Globally, one of the largest cumulative depositions is that of PFOA (122–1632 t), due to its direct emission and the large fraction of fluorotelomer precursors emitted as 8:2 homologs, but in total, longer- and shorter-chain PFCAs are deposited in larger quantities globally than PFOA (74–4129 t and 124–4390 t, respectively). PFOA deposits to Northern Hemisphere mid-latitudes more than any other PFCA, but in the Southern Hemisphere, mid-latitude PFOA deposition (7–165 t) is less than that of PFNA (6–485 t) and PFHpA (8–272 t). Farther from direct emission sources, fluorotelomer degradation becomes the most import source of PFCA, and the C7 homolog has the added source from the 6:2 fluorotelomer precursors which cannot form C8 and larger PFCAs.
| PFCA | Arctic | N.H. Mid-latitude | S.H. Mid-latitude |
|---|---|---|---|
| PFTA (C13) | 0.0–3.9 | 1.0–261.4 | 0.3–87.1 |
| PFDoA (C12) | 0.1–2.2 | 5.3–118.1 | 1.5–33.9 |
| PFUnA (C11) | 0.1–11.5 | 6.8–736.9 | 2.2–236.2 |
| PFDA (C10) | 0.3–6.9 | 17.1–380.5 | 4.9–108.9 |
| PFNA (C9) | 0.3–24.0 | 18.5–1575.7 | 5.7–484.8 |
| (Direct) | 0.0–0.2 | 8.6–55.6 | 0.0–0.0 |
| PFOA (C8) | 0.5–11.4 | 40.4–906.7 | 7.4–165.0 |
| (Direct) | 0.2–1.6 | 73.6–546.9 | 0.0–0.0 |
| PFHpA (C7) | 0.5–16.5 | 27.9–991.7 | 7.7–272.0 |
| PFHxA (C6) | 0.4–10.5 | 21.8–607.5 | 5.7–159.8 |
| PFPA (C5) | 0.2–5.7 | 9.0–325.6 | 2.2–80.9 |
| PFBUA (C4) | 0.3–10.6 | 15.4–605.0 | 3.8–149.7 |
| PFPRA (C3) | 0.2–7.3 | 10.8–422.8 | 2.5–97.9 |
| TFA (C2) | 0.2–8.9 | 12.8–501.2 | 2.8–111.3 |
4. Discussion
Our model simulations capture within uncertainty the measured values at the Devon Ice Cap for deposition of all PFCAs except PFNA, which is underestimated. This homolog was one of the leading directly emitted PFCAs, and our underestimate compared to observations could point to an underestimate of direct emissions. The model captures the production and deposition of even-carbon PFCAs but underestimates the relative rate of odd-carbon PFCA deposition, especially if the perfluorinated aldehyde (PFAL) hydration reaction is excluded. Odd-even homolog deposition ratios in the observations are better represented in the model with PFAL hydration included. Along with gas-phase hydration, heterogeneous hydration of PFAL, which is not resolved in the model, can contribute to formation. The high odd-even deposition ratio in the observations indicates the importance of PFAL hydration in the atmosphere and could be the result of a large fraction of observed deposited PFCAs having FT-olefin origin. With better experimental determinations of the gas-phase and heterogeneous hydration of PFAL, the atmospheric source of odd-carbon PFCAs could be better quantified.At mid-latitude sites, the relationship between modeled PFCA deposition and chain length follows the observed relationship between rainwater concentration and chain length. The rainwater concentration of a specific PFCA depends on two factors: (1) the relative atmospheric abundance of the PFCA and (2) its affinity for liquid water. Longer-chain PFCAs partition to water less and therefore will be less prevalent in rainwater at the same atmospheric abundance. At the same time, the PFCAs larger than PFNA also have lower emissions of their precursors and negligible direct emissions. PFOA is an outlier from this chain-length ordering in both model and observations because of direct emission having a large impact on its abundance, especially for Northern-Hemisphere mid-latitude measurements.
Our model predicts that high deposition rates of long-chain PFCAs occur over the equatorial oceans. Wallington et al.'s18 model predicted that the highest PFOA fractions would occur over the equatorial oceans and the Southern Ocean. We find a similar pattern which depends on the PFCA chain length. Over the equatorial oceans and the Southern Ocean, we calculate substantially higher fractions of C8 PFCA and longer than other regions of the globe. This echoes previous work16 that showed that these environments are conducive to long-chain PFCA formation.
We simulate deposition of both long- and short-chain PFCAs. Short-chain PFCAs have not been included in previous studies. Longer-chain PFCAs, however, are more bioaccumulative, and are therefore important to quantify. We show that both longer and shorter chain PFCAs are, on aggregate, globally deposited in larger quantities than PFOA and therefore should not be ignored when considering impacts of precursor emissions. As shorter chain PFCAs are less often measured than their longer-chain counterparts, our model fills a gap in the knowledge of the distribution of PFCAs.
We consider here only the degradation of 4:2 through 12:2 fluorotelomer precursors as well as direct PFOA and PFNA emissions, accounting for most of the proposed sources of atmospheric PFCAs. PFCAs can also be formed through the degradation of other precursor molecules (e.g. perfluoroalkanesulfonamides12). Sensitivity tests show that the distribution of emissions between different precursor compounds can affect yields of PFCA and thereby deposition. Fluorotelomer species other than alcohols, olefins and iodides are also emitted, but how large or small a fraction of the fluorotelomer total they comprise is uncertain. While we neglect them in the current study, the different chemistry of fluorotelomer acrylates and methacrylates, for instance, could yield different quantities of PFCA per unit emission than the precursors studied here due to their initial degradation reactions. Future consideration of these species could be important if they are found to be emitted in significant quantities. Degradation of fluorotelomer polymers in waste is a source of FTOH which we do not consider, but that has recently been shown to be relevant for environmental PFCA contamination.52,53 Non-polymeric compounds make up about half of the fluorotelomer production for C6, but 15% or less for other chain lengths.1 Some of these non-polymeric species may break down, after use and release, into simpler molecules such as FTI, FTO, and FTOH,1 but we neglect this as a source of atmospheric precursors in this study. The observations that we compare to our model represent the actual contribution of all these unquantified and quantified emissions, as well as the atmospheric processes both included in the model and omitted. Our comparison to observations shows a wide model uncertainty range that reflects this fact, and further work to build more model details would decrease uncertainty and increase comparability with observations. We do not include direct emissions of PFUnA and PFTA, which could contribute to our modeled wet deposition of PFUnA being underestimated compared to observations.
The chemistry of atmospheric PFCA formation and subsequent deposition is highly uncertain. The leading source of uncertainty in deposition across all PFCA homologs is the overall magnitude of precursor emissions. On top of this, the particular speciation of precursor emissions impacts the efficiency of PFCA formation and is also highly uncertain. Uncertainty in the rate at which perfluorinated aldehyde hydration occurs contributes significantly to the uncertainty in the atmospheric production of odd-carbon-number PFCAs and is unimportant for even-carbon homologs. Uncertainty due to unresolved processes such as oceanic aerosol generation, further precursor species, unquantified emission sources, and the spatial distribution of emissions could also potentially play an important role.
We estimate the source of PFOA to the Arctic to be 1–13 tonnes (0.02–0.24 tonnes per year average) over the simulated time period. Previous modeling work18 estimated the atmospheric indirect source of PFOA (0.4 tonnes per year) to the Arctic using a chemical transport model for a representative year with hypothetical emissions. While the chemistry simulated by that model was the most detailed of its time, it did not include the complete set of reactions used by our model. The hypothetical emission scenario used in that study for 8:2 FTOH is above more recent emissions estimates,1 but we include contributions from other chain lengths of precursors not included in that study. That work did not report the source of other PFCAs to the Arctic, which we calculate to be less than that of PFOA for each homolog longer than PFOA, but the sum of longer-chain PFCAs is larger than that of PFOA. Wania54 estimated with a zonally averaged transport model 150 kg per year deposited to the Arctic because of atmospherically generated PFCA. This falls within the uncertainty bounds of our modeled annual PFCA deposition to the Arctic.
Given the diversity of PFCA formation across the Northern Hemisphere and the spatial differences we see across the Arctic, as well as the deposition of atmospherically formed PFCA to the Southern Ocean, having global coverage in our model is an asset to quantifying both Arctic and global sources of PFCA. While detailed chemistry including most of our model's reactions was included in a regional-scale modeling study15 which focused on North American impacts of atmospherically formed PFCAs, the effect on the rest of the Northern Hemisphere was estimated as a single average quantity.
Globally, approximately 25% of fluorotelomer releases to air are deposited as PFCAs and 4% are deposited as PFOA specifically. At lower latitudes over land, this number is dominated by short-chain PFCAs. Long-chain PFCA deposition is on par with short-chain deposition over the equatorial oceans and the Southern Ocean. This comparatively high fraction of deposition as long-chain PFCAs agrees with yield calculations in previous work,16 which showed that conditions over the oceans far from NOx sources are conducive to large yields of PFNA and PFOA. While fluxes of PFCAs are lower at the southern latitudes, the longer chain PFCAs of more consequence make up a much higher fraction. This higher long-chain fraction at southern latitudes means that while total PFCA deposition rates are lower, there will be more bioaccumulation potential per PFCA molecule deposited.
Our work accentuates the importance of measurements of PFCAs at remote locations. Over the time period of the Devon Ice Cap measurements, more than half of the modeled PFOA and essentially all of the other PFCAs deposited in the Canadian Arctic come from precursor degradation rather than direct emissions, meaning that this remote area is key to our understanding of atmospherically generated PFCAs.
As a source to the Arctic, our work shows that while atmospheric production and deposition of long-chain PFCAs is potentially larger than previous estimates,54 it is likely that the atmosphere is secondary to ocean transport of directly emitted PFCAs. Previous estimates of ocean transport of directly emitted PFCAs to the Arctic (11–20 tonnes per year)54 are larger than our upper bound estimate of total annual Arctic deposition of PFCAs from the atmosphere (2.1 tonnes per year), though atmospheric transport could be larger for specific homologs.
We assume a spatial distribution of emissions which is correlated with NOx emissions. Given the relationship of reduced yields of longer chain PFCAs under higher NOx conditions,12,18 this assumption will lead to lower modeled atmospheric production of long-chain PFCAs than an emissions distribution which is less correlated with NOx emissions. This assumption, which leads to similar distributions as previous estimates29 is reasonable, but while much of the product use emissions will occur where the highest populations are, manufacturing sites of PFCA precursors are not located in the largest urban areas. This may mean that we underestimate the atmospheric yield of PFCA per emission.
While we use the most up-to-date chemistry available, the reactions studied are focused on fast photochemical reactions that are most prevalent during day-time conditions. Possible night-time chemistry of PFCA precursors, which has not been studied, is likely to be less important than the included chemistry under most conditions, but for polar winters, any reactions could create important differences for the production of PFCAs. We also do not resolve partitioning of PFCAs and their precursors to aerosol particles. Particle-bound PFCAs could experience transport differently than their gas-phase counterparts. Association with particulate matter becomes important for long-chain PFCAs and increases with chain length.55 For this particle association to become important, the PFCA must be able to dissociate into the ionic form, meaning that partitioning would be to aqueous aerosol in particular.55 We did not consider the acid and conjugate base speciation and partitioning of PFCAs in atmospheric aerosol.
5. Conclusions
Given the declining importance of direct emissions of long-chain PFCAs, the signal of atmospherically produced PFCA in the future should be most easily detectable in precipitation in the tropics. Sites that are both remote and tropical such as Pacific islands see high annual deposition rates which could be observed through measurements of PFCAs in precipitation. High precipitation rates combined with relatively long times since emission would allow detection across the full suite of PFCA chain lengths. We also see in our model results varying relative abundances of different PFCA homologs in deposition in different regions. This means that measuring many chain lengths of PFCAs in a single location would help quantitatively validate and improve our knowledge of precursor chemistry. In particular, the ratio of long- to short-chain PFCA predicted by our model is strongly dependent on location, varying on average between hemispheres by close to an order of magnitude. Observations of this ratio at a variety of remote locations could be used to constrain our understanding of PFCA formation.The simulations of atmospheric PFCA formation and fate that we introduce in the GEOS-Chem model can be used in future work to investigate further elements of atmospheric PFCA formation, such as yet-unstudied gas-phase and heterogeneous reactions, future emission scenarios, and uncertainty in the spatial distribution of emissions.
Our model predicts a strong gradient in the long-chain to short-chain ratio moving from more-polluted to less-polluted air masses to be the signature of atmospheric formation of PFCAs via the degradation of precursors. The modeled deposition of PFCAs due to the degradation of precursors is highest close to sources, and near the equator and over temperate oceans, marking a continuing supply of PFCAs to the ocean even if their direct emissions cease entirely.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the US National Science Foundation Arctic Natural Sciences Program (1203526), a fellowship from the National Science and Engineering Research Council of Canada (to C.P.T.) and by the National Institute of Environmental Health Sciences Superfund Basic Research Program, National Institute of Health, 3P42ES027707-03S1.Notes and references
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbuhler, Environ. Int., 2014, 70, 62–75 CrossRef CAS PubMed.
- J. W. Martin, S. A. Mabury, K. R. Solomon and D. C. G. Muir, Environ. Toxicol. Chem., 2003, 22, 189–195 CrossRef CAS PubMed.
- J. W. Martin, S. A. Mabury, K. R. Solomon and D. C. G. Muir, Environ. Toxicol. Chem., 2003, 22, 196–204 CrossRef CAS PubMed.
- J. M. Conder, R. A. Hoke, W. de Wolf, M. H. Russell and R. C. Buck, Environ. Sci. Technol., 2008, 42, 995–1003 CrossRef CAS PubMed.
- C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2007, 41, 3455–3461 CrossRef CAS PubMed.
- M. Shoeib, T. Harner and P. Vlahos, Environ. Sci. Technol., 2006, 40, 7577–7583 CrossRef CAS PubMed.
- N. L. Stock, V. I. Furdui, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2007, 41, 3529–3536 CrossRef CAS PubMed.
- Z. Xie, Z. Wang, W. Mi, A. Moller, H. Wolschke and R. Ebinghaus, Sci. Rep., 2015, 5, 8912 CrossRef CAS PubMed.
- B. F. Scott, C. Spencer, S. A. Mabury and D. C. G. Muir, Environ. Sci. Technol., 2006, 40, 7167–7174 CrossRef CAS PubMed.
- M. Houde, J. W. Martin, R. J. Letcher, K. R. Solomon and D. C. G. Muir, Environ. Sci. Technol., 2006, 40, 3463–3473 CrossRef CAS PubMed.
- D. A. Ellis, J. W. Martin, S. A. Mabury, M. D. Hurley, M. P. S. Andersen and T. J. Wallington, Environ. Sci. Technol., 2003, 37, 3816–3820 CrossRef CAS PubMed.
- C. J. Young and S. A. Mabury, Rev. Environ. Contam. Toxicol., 2010, 208, 1–109 CrossRef CAS PubMed.
- R. L. Waterland and K. D. Dobbs, J. Phys. Chem. A, 2007, 111, 2555–2562 CrossRef CAS PubMed.
- M. S. Chiappero, F. E. Malanca, G. A. Arguello, S. T. Wooldridge, M. D. Hurley, J. C. Ball, T. J. Wallington, R. L. Waterland and R. C. Buck, J. Phys. Chem. A, 2006, 110, 11944–11953 CrossRef CAS PubMed.
- G. Yarwood, S. Kemball-Cook, M. Keinath, R. L. Waterland, S. H. Korzeniowski, R. C. Buck, M. H. Russell and S. T. Washburn, Environ. Sci. Technol., 2007, 41, 5756–5762 CrossRef CAS PubMed.
- C. P. Thackray and N. E. Selin, Atmos. Chem. Phys., 2017, 17, 4585–4597 CrossRef CAS.
- L. Vierke, C. Staude, A. Biegel-Engler, W. Drost and C. Schulte, Environ. Sci. Eur., 2012, 24, 1–11 CrossRef.
- T. J. Wallington, M. D. Hurley, J. Xia, D. J. Wuebbles, S. Sillman, A. Ito, J. E. Penner, D. A. Ellis, J. Martin, S. A. Mabury, O. J. Nielsen and M. P. S. Andersen, Environ. Sci. Technol., 2006, 40, 924–930 CrossRef CAS PubMed.
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbuhler, Environ. Int., 2014, 69, 166–176 CrossRef CAS PubMed.
- C. J. Young, M. D. Hurley, T. J. Wallington and S. A. Mabury, J. Phys. Chem. A, 2008, 112, 13542–13548 CrossRef CAS PubMed.
- C. M. Butt, C. J. Young, S. A. Mabury, M. D. Hurley and T. J. Wallington, J. Phys. Chem. A, 2009, 113, 3155–3161 CrossRef CAS PubMed.
- U. Schenker, M. Scheringer, M. Macleod, J. W. Martin, I. T. Cousins and K. Hungerbuhler, Environ. Sci. Technol., 2008, 42, 3710–3716 CrossRef CAS PubMed.
- I. Bey, D. J. Jacob, R. M. Yantosca, J. A. Logan, B. Field, A. M. Fiore, Q. Li, H. Liu, L. J. Mickley and M. Schultz, J. Geophys. Res.: Atmos., 2001, 106, 23073–23096 CrossRef CAS.
- GEOS-Chem wiki, wiki.seas.harvard.edu/geos-chem.
- H. Liu, D. J. Jacob, I. Bey and R. M. Yantosca, J. Geophys. Res.: Atmos., 2001, 106, 12109–12128 CrossRef CAS.
- Y. Wang, D. J. Jacob and J. A. Logan, J. Geophys. Res.: Atmos., 1998, 103, 10713–10726 CrossRef CAS.
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Environ. Sci. Technol., 2006, 40, 32–44 CrossRef CAS PubMed.
- H. P. Arp, C. Niederer and K.-U. Goss, Environ. Sci. Technol., 2006, 40, 7298–7304 CrossRef CAS PubMed.
- I. Stemmler and G. Lammel, Atmos. Chem. Phys., 2010, 10, 9965–9980 CrossRef CAS.
- J. Johansson, M. Salter, J. A. Navarro, C. Leck, E. Nilsson and I. Cousins, Environ. Sci.: Processes Impacts, 2019, 21, 635–649 RSC.
- H. M. Pickard, A. S. Criscitiello, C. Spencer, M. J. Sharp, D. C. G. Muir, A. O. D. Silva and C. J. Young, Atmos. Chem. Phys., 2018, 18, 5045–5058 CrossRef CAS.
- K. Kwok, S. Taniyasu, L. Yeung, M. Murphy, P. Lam, Y. Horii, K. Kannan, G. Petrick, R. Sinha and N. Yamashita, Environ. Sci. Technol., 2010, 44, 7043–7049 CrossRef CAS PubMed.
- T. Kirchgeorg, A. Dreyer, J. Gabrieli, N. Kehrwald, M. Sigl, M. Schwikowski, C. Boutron, A. Gambaro, C. Barbante and R. Ebinghaus, Environ. Pollut., 2013, 178, 367–374 CrossRef CAS PubMed.
- C. Barton, M. Kaiser and M. Russell, J. Environ. Monit., 2007, 9, 839–846 RSC.
- S.-K. Kim and K. Kannan, Environ. Sci. Technol., 2007, 41, 8328–8334 CrossRef CAS PubMed.
- R. Loos, J. Wollgast, T. Huber and G. Hanke, Anal. Bioanal. Chem., 2007, 387, 1469–1478 CrossRef CAS PubMed.
- S. Taniyasu, K. Kannan, L. Yeung, K. Kwok, P. Lam and N. Yamashita, Anal. Chim. Acta, 2008, 619, 221–230 CrossRef CAS PubMed.
- W. Liu, G. Dong, Y. Jin, K. Sasaki, N. Saito, I. Sato, S. Tsuda and S. Nakayama, Chin. Sci. Bull., 2009, 54, 2440–2445 CrossRef CAS.
- W. Liu, Y. Jin, X. Quan, K. Sasaki, N. Saito, S. Nakayama, I. Sato and S. Tsuda, Environ. Int., 2009, 35, 737–742 CrossRef CAS PubMed.
- M. Mahmoud, A. Karrman, S. Oono, K. Harada and A. Koizumi, Chemosphere, 2009, 74, 467–472 CrossRef CAS PubMed.
- M. Murakami, H. Shinohara and H. Takada, Chemosphere, 2009, 74, 487–493 CrossRef CAS PubMed.
- A. Dreyer, V. Matthias, I. Weinberg and R. Ebinghaus, Environ. Pollut., 2010, 158, 1221–1227 CrossRef CAS PubMed.
- B. Scott, A. D. Silva, C. Spencer, E. Lopez, S. Backus and D. Muir, J. Great Lakes Res., 2010, 36, 277–284 CrossRef CAS.
- X. Zhang, H. Niu, Y. Pan, Y. Shi and Y. Cai, Anal. Chem., 2010, 82, 2363–2371 CrossRef CAS PubMed.
- T. Meyer, A. D. Silva, C. Spencer and F. Wania, Environ. Sci. Technol., 2011, 45, 8113–8119 CrossRef CAS PubMed.
- C. Muller, N. Spiess, A. Gerecke, M. Scheringer and K. Hungerbuhler, Environ. Sci. Technol., 2011, 45, 9901–9909 CrossRef CAS PubMed.
- V. Nguyen, M. Reinhard and G.-H. Karina, Chemosphere, 2011, 82, 1277–1285 CrossRef CAS PubMed.
- H. Nishikoori, M. Murakami, H. Sakai, K. Oguma, H. Takada and S. Takizawa, Chemosphere, 2011, 84, 1125–1132 CrossRef CAS PubMed.
- X. Zhao, Y. Cai, F. Wu, Y. Pan, H. Liao and B. Xu, Microchem. J., 2011, 2011, 98 Search PubMed.
- C. P. Thackray, C. L. Friedman, Y. Zhang and N. E. Selin, Environ. Sci. Technol., 2015, 49, 9185–9193 CrossRef CAS PubMed.
- Y.-H. Chen and R. G. Prinn, J. Geophys. Res.: Atmos., 2006, 111, D10307 Search PubMed.
- L. Li, J. Liu, J. Hu and F. Wania, Environ. Sci. Technol., 2017, 51, 4461–4470 CrossRef CAS PubMed.
- J. Washington, K. Rankin, E. Libelo, D. Lynch and M. Cyterski, Sci. Total Environ., 2019, 651, 2444–2449 CrossRef CAS PubMed.
- F. Wania, Environ. Sci. Technol., 2007, 41, 4529–4535 CrossRef CAS PubMed.
- L. Ahrens, T. Harner, M. Shoeib, D. A. Lane and J. G. Murphy, Environ. Sci. Technol., 2012, 46, 7199–7206 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00326f |
| ‡ Harvard John A. Paulson School of Engineering & Applied Sciences, Harvard University, Cambridge, USA |
| § Institute for Data, Systems, and Society, Massachusetts Institute of Technology, Cambridge, USA |
| This journal is © The Royal Society of Chemistry 2020 |