
Lithium-activated SnS–graphene alternating nanolayers enable dendrite-free cycling of thin sodium metal anodes in carbonate electrolyte†
Wei
Liu
 *ab,
Zidong
Chen
a,
Zheng
Zhang
a,
Pingxian
Jiang
a,
Yungui
Chen
*ab,
Eunsu
Paek
c,
Yixian
Wang
d and
David
Mitlin
*d
*ab,
Zidong
Chen
a,
Zheng
Zhang
a,
Pingxian
Jiang
a,
Yungui
Chen
*ab,
Eunsu
Paek
c,
Yixian
Wang
d and
David
Mitlin
*d
aInstitute of New-Energy and Low-Carbon Technology (INELT), Sichuan University, Chengdu, Sichuan 610065, China. E-mail: weiliu@scu.edu.cn; chenyungui@scu.edu.cn
bEngineering Research Center of Alternative Energy Materials & Devices, Ministry of Education, Sichuan University, Chengdu, Sichuan 610065, China
cChemical & Biomolecular Engineering, Clarkson University, Potsdam, New York 13699, USA
dMaterials Science and Engineering Program & Texas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: David.Mitlin@austin.utexas.edu
First published on 13th November 2020
Abstract
Sodium metal battery (SMB, NMB) anodes can become dendritic due to an electrochemically unstable native Na-based solid electrolyte interphase (SEI). Herein Li-ion activated tin sulfide graphene nanocomposite membrane (A-SnS–G) is employed as an artificial SEI layer, allowing cyclability of record-thin 100 μm Na metal foils. The thin Na metal is prepared by a self-designed metallurgical rolling protocol. A-SnS–G is initially placed onto the polypropylene (PP) separator but becomes in situ transferred onto the Na metal surface. Symmetric metal cells protected by A-SnS–G achieve low-overpotential extended high-rate cycling in a standard carbonate electrolyte (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC = 1:1, 5% FEC). Accumulated capacity of 1000 mA h cm−2 is obtained after 500 cycles at 4 mA cm−2, with accumulated capacity-to-foil capacity (A/F) ratio of 90.9. This is among the most favorable cycle life, accumulated capacity, and anode utilization combinations reported. Protection by non-activated SnS–G membrane yields significantly worse cycling, albeit still superior to the baseline unprotected sodium. Post-mortem and dedicated light optical analysis indicate that metal swelling, dendrite growth and dead metal formation is extensive for the unprotected sample, but is suppressed with A-SnS–G. Per XPS, post-100 cycles near-surface structure of A-SnS–G is rich in metallic Sn alloys and inorganic carbonate salts. Even after 300 cycles, Li-based SEI components ROCO2-Li, Li2CO3 and LiF are detected with A-SnS–G. As a proof of principle, an SMB with a high mass loading (6 mg cm−2) NVP cathode and a A-SnS–G protected anode delivered extended cyclability, achieving 74 mA h g−1 after 400 cycles at 0.4C.
:DEC = 1:1, 5% FEC). Accumulated capacity of 1000 mA h cm−2 is obtained after 500 cycles at 4 mA cm−2, with accumulated capacity-to-foil capacity (A/F) ratio of 90.9. This is among the most favorable cycle life, accumulated capacity, and anode utilization combinations reported. Protection by non-activated SnS–G membrane yields significantly worse cycling, albeit still superior to the baseline unprotected sodium. Post-mortem and dedicated light optical analysis indicate that metal swelling, dendrite growth and dead metal formation is extensive for the unprotected sample, but is suppressed with A-SnS–G. Per XPS, post-100 cycles near-surface structure of A-SnS–G is rich in metallic Sn alloys and inorganic carbonate salts. Even after 300 cycles, Li-based SEI components ROCO2-Li, Li2CO3 and LiF are detected with A-SnS–G. As a proof of principle, an SMB with a high mass loading (6 mg cm−2) NVP cathode and a A-SnS–G protected anode delivered extended cyclability, achieving 74 mA h g−1 after 400 cycles at 0.4C.
Broader contextRoom temperature sodium metal batteries (SMBs) are an exciting emerging area of electrochemical energy storage research, offering approximately 50% higher energy than traditional sodium ion batteries (SIBs). A major problem for SMBs is the cycling instability of the Na metal anode's solid electrolyte interphase (SEI), leading to dendrites that cause electrical shorting and cell failure. The motivation for this study was the hypothesis that a Li-based SEI will tremendously improve the Na metal anode's stability. A reinforcing nanocomposite membrane of alternating SnS and graphene nanolayers was firstly lithium ion activated and then in situ transferred onto the Na metal surface during cycling. Lithium activation created a unique robust microstructure that allowed rapid Na ion flux during cycling, with an inherently more stable Li-based SEI that replaced the conventional Na-based SEI. The SnS and the graphene performed in a complementary manner: the Li-activated Sn alloy and S phases “breathe” allowing for facile Na ion diffusion, while the graphene effectively stays fixed, acting as a rigid membrane scaffolding. A synergistic performance improvement was achieved, with state-of-the-art stable cycling and fast charging of symmetrical Na–Na cells and of full SMBs adopting 100 micron thin Na metal. |
Introduction
Room temperature sodium metal batteries (SMBs) (e.g., Na–S, Na–Se and Na–ceramic cathode) are an exciting emerging area of electrochemical energy storage research.1–4 While sodium ion batteries (SIBs) have received intensive scientific attention over the last decade, room temperature SMBs have gained attention primarily in the last several years.5,6 Sodium metal provides a low electrochemical voltage (−2.7 V vs. SHE) and a high theoretical specific capacity (1165 mA h g−1), allowing for relatively high energy when paired with a ceramic or a sulfur-based cathode. Sodium metal anodes are essential for SMBs and are also necessary for laboratory sodium ion battery (SIB, NIB) half-cells. A major impediment towards using sodium metal in rechargeable SMBs is the intrinsic sodium metal–electrolyte interfacial instability during cycling, ultimately being manifested as growth of dendrites of various morphologies. Authors reported that during cycling of Na metal the ultimate failure was caused by an unacceptable increase in the overpotential, rather than an overpotential that went to zero.7 This indicates severe SEI growth leading to impedance rise fails the cell before dendrite-induced electrical shorting occurs. In fact, Na metal cells normally show poor CE during cycling, leading to a generalization that severe SEI formation is an intrinsic feature of the system.7 This is especially true with commercial-type carbonate-based, rather than ether-based electrolytes, presumably due to a less stable SEI in the former.3,4Significant gains have been achieved in promoting stable dendrite – free cycling of Li metal anodes.8–12 By contrast, cycling stability with Na metal anodes remains more limited due to its greater reactivity in battery electrolytes. Both Li and Na metals occupy a higher energy level compared with the lowest unoccupied molecular orbital (LUMOs) of organic electrolytes. Since the number of electron shells in Na is larger than in Li, there is a lower constraining force on the outermost electrons. This causes Na to be a more reductive agent than Li in comparable electrolytes. As compared to Li metal, Na metal anodes in ether and ester solvents possess accelerated SEI growth, worse coulombic efficiency (CE), larger overpotentials for plating and stripping, and more severe growth of dendrites.13–15 Numerous metal anode protection strategies have been employed for Li, enabling dendrite-free cycling with a range of electrolytes.16–19 Multiple approaches have been employed to stabilize Na metal anodes as well. For example, graphene,15,20 Al2O3/alucone,6,21,22 NaBr,23 NaxSy,24 Na3PS425 and composite films26 have been used as effective protection layers. High surface area sodiophilic current collectors have been employed to reduce the local current density.27–30 A number of electrolyte additives, like fluoroethylene carbonate (FEC),31,32 ionic liquid,7 bis(2,2,2-trifluoroethyl)ether (BTFE)33 and potassium salts34 have reduced Na dendrite growth. However many electrolyte functional additives are known to be progressively consumed during prolonged cycling.35,36
The morphology of Na dendrites is reported as dense forests of needles or moss, rather than as more isolated.7,15,32,37 This is likely due to defect catalyzed base growth at currents low enough where ion diffusional limitations do not yet dominate,38 rather than the Sand's Time concentration polarization scenario. With glyme-based solvents, the SEI structure of Na is stabilized, due to the relatively higher fraction of the stable and adherent Na2O and NaF, versus a more carbonate – rich SEI.39 However, a carbonate-based electrolyte is essential for enabling high voltage (>3 V vs. Li/Li+) cathodes due to its high oxidation voltage and is used in commercial lithium ion batteries.40 The major issue is the intrinsic reactivity of Na metal in ethylene carbonate (EC), propylene carbonate (PC), diethyl carbonate (DEC), and dimethyl carbonate (DMC) solvents.41 To date, achieving stable Na metal plating–stripping with carbonate electrolytes remains challenging. Studies that directly compare Li and Na metal cycling stability in similar electrolyte combinations report that the Li system is consistently more stable.42,43 At open-circuit voltage (OCV), the Li metal interface exhibited minimal changes in impedance with time. At comparable conditions, the Na metal interface showed a monotonic increase in impedance and changes in its surface morphology.43 Authors reported effective suppression of Na dendrites by adding Li-salts into the electrolyte, taking advantage of the electrostatic shield effect of Li+.44 In a different study, authors employed Li–Na alloy anode and an electrolyte additive to realize aprotic bimetal Li–Na alloy–O2 battery with improved cycling stability.45
From such observations regarding Li vs. Na reactivity, one can hypothesize that a Li-based SEI will tremendously improve the stability of sodium metal anodes. In this study, achieving stable sodium metal plating/stripping is based on this hypothesis, combined with a reinforcing nanocomposite membrane of alternating SnS and graphene nanolayers. A synergistic effect is sought, with the effectiveness of the membrane in preventing dendrites being enhanced by electrochemical Li activation. Lithium activation creates a unique robust microstructure that allows for rapid Na ion flux during cycling, with an inherently more stable Li-based SEI that replaces the conventional Na-based SEI. The SnS–graphene membrane is initially placed onto the separator but becomes in situ transferred onto Na anode during electrochemical cycling. During plating/stripping, the SnS and the graphene perform in a complementary manner: The activated Sn alloy and S phases “breathe” allowing for facile ion diffusion, while the graphene effectively stays fixed, acting as a rigid membrane scaffolding. Stable cycling is achieved in a standard carbonate electrolyte with record thin (100 μm) Na metal foils, both as Na–Na symmetric cells and as full battery cells with high cathode mass loading. Such an architecture and its performance are unprecedented for SMB applications.
Experimental section
Thin Na anode fabrication
Thin Na metal foils are prepared by a self-designed metallurgical rolling protocol. A section of Na metal was firstly compressed onto a standard Cu foil current collector and placed into a PE seal bag, this step being performed inside an Ar-filled glovebox. The PE sealed Na–Cu was then taken out of the glovebox and subjected to metallurgical rolling. After rolling the Na metal to 100 μm thickness, the sealed package was transferred back into glovebox. After removing the PE seals, the Na–Cu laminate foils were punched into 12 mm circular discs to be used for electrochemical testing. Cell assembly was performed inside the same glove box.Membrane synthesis
Bulk herzenbergite was used as the precursor for the SnS nanosheets. A liquid exfoliation method was adopted for nanosheets synthesis.46,47 For the process, 0.3 g of herzenbergite was firstly ground for 30 min into finer particles. The fine SnS powder was added into an isopropanol (IPA) aqueous solution (30 vol% IPA) in a concentration of 10 mg ml−1. Hexadecyl trimethyl ammonium bromide (CTAB) was used as surfactant in the solution at 0.37 wt%. The suspension was shaken and subject to bath sonication for 6 hours. After sonication, the suspension was centrifuged at 2000 rpm for 10 min, which removed the non-exfoliated large particles. The supernatant liquid was repeatedly filtrated-washed with ethanol through a pre-weighted filter membrane. After being dried in an oven at 80 °C for overnight, the filter cake was re-dispersed into 30% IPA aqueous solution for future use.Graphene layers (G) were produced from expanded graphite, which was in turn fabricated from flaky graphite (1200 mesh, Aladdin Chemicals). The below reaction scheme summarizes the transformation of graphite flakes to expanded graphite. The individual layers of the graphite were positively charged by S2O82− oxidant, allowing for intercalation of H2SO4 molecules between the (001) layers and the associated expansion of their spacing.48,49
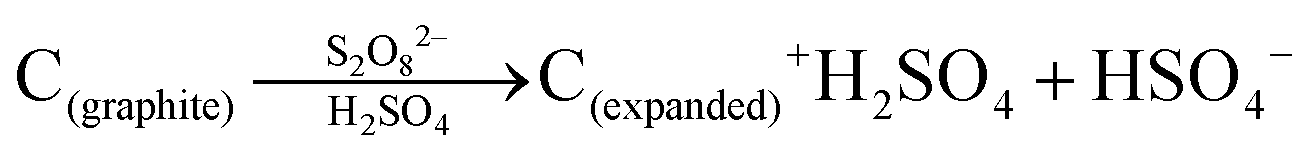
H2SO4 and oleum were firstly mixed together in a volume ratio of 1:1 by magnetic stirring in cold water bath. Two grams of (NH4)2S2O8 were then dissolved into 16 mL of this acid solution. Then 0.1 g of graphite powder was step wise added into the beaker, which was then sealed and transferred to a 60 °C water bath that was magnetically stirred. The expansion of graphene requires several hours of holding at temperature. Once the foam-like expanded mixture displayed no further obvious volume changes, the product was repeatedly rinsed with deionized water and centrifuged. The obtained material was re-dispersed into an IPA aqueous solution (30 vol%). To yield the end-product pristine (largely defect-free) graphene dispersion, this suspension was subject to 4 hours of mild bath sonication. Poly(dimethyldiallyl ammonium chloride) (PDDA), was then employed to modify the graphene layers to have a positive surface charge. The PDDA–graphene dispersion was prepared by dispersing 50 mg graphene powder into 20 ml 2 wt% PDDA aqueous solution through 5 min of bath sonication. Then the suspension was centrifuged (2000 rpm) for 20 min. The remaining sediment was re-dispersed into aqueous solution and reused.
To synthesize the tin sulfide–graphene nanocomposite (SnS–G), the PDDA–graphene dispersion was drop-wise added into the SnS dispersion, with a weight ratio of SnS:G = 1.8:1. The suspension was then filtered through a commercial polypropylene PP membrane (Celgard 2400) to form the final coating layer. The mass loading of the SnS–G composite on the PP was ∼0.76 mg cm−2. Lithium activation of SnS–G to create A-SnS–G was done in a coin cell using a Li metal counter electrode, with the membrane placed on the PP separator. The blank side of PP was placed towards the Li metal, with the SnS–G coating layer in direct contact with bottom shell. A solution of 1 M LiPF6 dissolved in a 1:1:1 (volume ratio) mixture of ethylene carbonate (EC), ethyl methyl carbonate (EMC) and diethyl carbonate (DEC) with 5 vol% fluoroethylene carbonate (FEC) was used as the activation electrolyte. The single lithiation–delithiation cycle was done at a current density of 30 μA cm−2 and a voltage range of 0.01–2.5 V vs. Li/Li+. The same electrochemical profile was employed to fabricate the A-Na baseline, which had a Li-based SEI but without the SnS–G on its surface. After activation, the A-SnS–G or A–Na were disassembled from the button cell and rinsed with dimethyl carbonate (DMC) and then dried in the glove box. The A-SnS–G or control SnS–G (not activated) membranes on the PP separator were then placed facing the Na metal anode. This allowed for cycling-induced in situ transfer of the membranes onto the metal surface, creating in effect an artificial SEI.
Analytical characterization
Scanning Electron Microscopy (SEM) analysis was performed using a JSM-7500F Field Emission SEM operated at 15 kV, equipped with an energy dispersive X-ray spectrum (EDXS, Oxford X-Max). TEM analysis was performed using a Tecnai G2-F20 operated at 200 kV. X-ray photoelectron spectroscopy (XPS) was performed employing a Thermo-Fisher EScalab 250Xi XPS system, with Al Kα radiation. An Ar+ beam with beam energy of 1 kV and beam current of 0.5 μA was employed for depth profiling. Peak fitting was done using mixed Gaussian/Lorentzian peak shapes after subtraction of Shirley background. X-ray diffraction was carried out in DX2700 (Dandong Haoyuan with Cu Kα radiation). The data was analyzed and processed using Jade 6 software package. Thickness and morphology were further analyzed using atomic force microscopy (AFM). AFM was performed in tapping mode, using a Bruker Multimode 8.Transmission electron microscopy (TEM) samples were prepared by pipetting several drops of the SnS:G dispersion onto a lacey carbon mesh grid. AFM specimens were fabricating by dispersing a drop of graphene ink onto freshly cleaved mica substrate. For post-mortem XPS analysis, the cycled electrodes were disassembled in a glove box and thoroughly rinsed with dimethyl carbonate (DMC). This washed away the remnant electrolyte and soluble SEI components. The cleaned and dried electrodes were then put into seal bag, held in screw-bottle in the glovebox, and then transferred to the XPS load-lock. For SEM analysis, the cleaned and dried electrodes were transferred by a dedicated SEM sample transfer tool. Specimens employed for light optical analysis were cycled 100 times in conventional symmetric button cells prior to being disassembled inside the glove box. Inside the glove box, the working electrodes were then cut into strips and assembled into a quartz-cuvette symmetric cells employed for light optical analysis.
Electroanalytical characterization
The electrochemical performance of electrodes was measured in 2025-type coin cells. The cells were assembled in an argon-filled glovebox with oxygen and water content lower than 0.1 ppm. A standard research electrolyte was employed: 1 M NaClO4 dissolved in 1:1 (volume ratio) mixture EC and DEC, with 5 vol% FEC as an additive. The cathode powder Na3V2(PO4)3 (NVP) was supplied by Canrd group, Shenzheng, China. The cathode electrodes were prepared by mixing NVP powder with polyvinylidene difluoride (PVDF) and CB in N-methyl pyrrolidone (NMP) with a weight ratio of 8:1:1. This slurry was coated onto a standard Al foil using the doctor blade technique. Electrochemical tests were carried out using a LAND-CT2001A battery tester. Electrochemical impedance spectroscopy (EIS) tests were carried out using AUTOLAB M204 (Metrohm, Switzerland).
Results and discussion
A self-designed rolling process is employed to fabricate 100 μm thin Na foil anodes, shown in Fig. S1 (ESI†). It is recognized that employing thin metal anodes, rather than the standard laboratory-grade thick foils, is critical for commercial applications of metal batteries.10 While advances have been made in rolling Li to sub-100 μm thickness, there are no prior reports where rolling was employed to achieve thin Na foils. Sodium metal foils employed in previous studies are in the 300–1000 micrometer range, or unspecified. Per Table S1 in ESI† a 100 μm Na foil is unprecedented.Scheme 1 summarizes the synthesis process of the Li ion activated SnS–graphene nanolayered composite membrane, termed A-SnS–G. The SnS–G membrane is based on alternating stacking of positively charged graphene and negatively charged SnS nanosheets. The membrane was synthesized from two building blocks, well-ordered graphene and SnS nanosheets. An expansion-exfoliation technique was employed to firstly fabricate expanded graphite, which was the precursor for the graphene in SnS–G. The expansion of graphite greatly weakens the van der Waals interaction between graphene layers, leading to the preliminary exfoliation of the graphite structure.48,49 The graphene used for SnS–G was obtained by liquid phase sonication exfoliation of this expanded graphite. The resultant few layered graphene possessed a low level of structural defects, nanopores, oxygen groups, or other heteroatoms. This is opposite to the signature features of traditional Hummer's-based reduced graphene oxide (r-GO). Herzenbergite, an abundant mineral with a 2D structure, was employed as the precursor to SnS. A surfactant-assisted liquid exfoliation method was adopted to produce the SnS nanosheets. After fabrication, both the SnS nanosheets and the graphene are negatively charged, as indicated by the respective zeta-potentials of −59.9 and −48.9 mV, per Fig. S2 (ESI†). Without functionalization the SnS and graphene repel each other and would segregate rather than order. A cationic polymer, poly(dimethyldiallyl ammonium chloride) (PDDA), was therefore employed to modify the graphene layers so as to have a positively charged surface.50 The +117 mV zeta-potential of PDDA modified graphene is also shown in Fig. S2 (ESI†). The positively charged graphene–negatively charged SnS nanosheets are energetically favored to alternate, creating a nanolayered composite in the process.
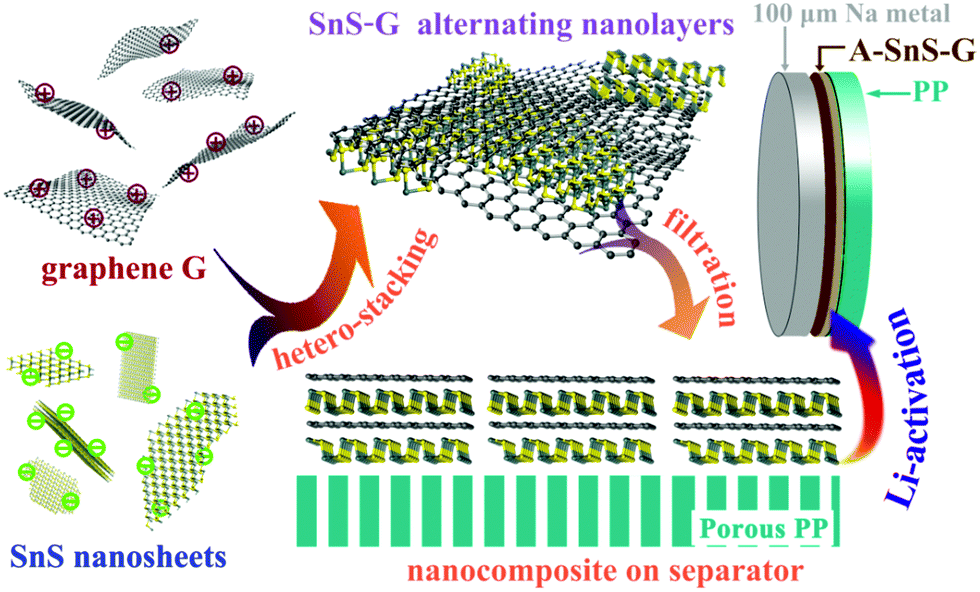 | ||
| Scheme 1 Summary of the synthesis and application of the Li-activated SnS–graphene nanolayer composite membranes (A-SnS–G) to stabilize the 100 μm Na foils during electrochemical cycling. Rolling process to obtain the record thin Na foils is shown in Fig. S1 (ESI†). | ||
Each material has differing and complementary electrochemical activity with ions. The two-dimensional SnS is a mixed conversion – alloying compound with both Li and Na.51,52 Upon lithiation/sodiation the SnS forms a Na2S (Li2S) compound and alloys to either the terminal Na15Sn4 (Li15Sn4) or to a sub-stoichiometric intermetallic. Upon delithiation/desodiation, the two elements don't recombine, but rather form a nanoscale dispersion of amorphous Sn and S, the scale being re-refined at every cycle.51 The graphene complements the ion-active Sn and S by serving as a strengthening phase within the nanocomposite membrane. Due to the chosen synthesis route, the graphene employed has a low level of structural and oxygen defects, as compared to reduced graphene oxide (rGO), etc. Namely the graphene, termed “G”, possesses a Raman D/G band ratio of 0.38, and an O content of ∼5 at%. These numbers are substantially lower than the typical values for rGO, which are ∼1 and ∼8 at%, respectably.53 Structural and chemical defects (oxygen groups, dangling bonds, etc.) in carbons are known to catalyze excessive decomposition of carbonate solvent molecules.54,55 This is something to be avoided if the long-term stability of the SEI is sought. Capacity of pristine graphene for Na is minimal since there is no Na ion intercalation and few defects for Na ion adsorption.56 During cycling with Na, graphene should undergo minimal volume changes and will retain its structure.
Fig. S3(a) and (b) (ESI†) show a bright field TEM image and the associated [001] zone axis SAED of the as-synthesized graphene. Fig. S3(c) and (d) (ESI†) show an AFM image and height map of the as-synthesized graphene deposited on mica substrate. The corresponding AFM height profile measures the graphene layers to be 4–6 nm thick. By tapping-mode AFM the measured thickness of a true monolayer of graphene is 0.8–1.5 nm.57 The measured 4–6 nm thickness therefore corresponds to the graphene in SnS–G being about 5 monolayers. Fig. S4 (ESI†) shows the XPS C 1s spectra and the Raman spectra of these graphene layers in the as-synthesized condition. The XPS C 1s spectra shows a prominent C–C peak overlapped with a relatively low intensity C–O peak, indicating minimal oxygen within the structure. The Raman spectra shows a predominant G peak and an IG/ID intensity ratio of 2.63. The spectra also shows a relatively intense 2D peak, its presence also being indicative of high degree of graphitic ordering. Fig. S5(a) (ESI†) shows a bright field TEM image of as-synthesized SnS nanosheets, highlighting their lateral dimensions which span from sub-50 nm to around 500 nm. Fig. S5(b) (ESI†) shows a HRTEM image in the [001] zone axis and the associated FFT, highlighting that the sheets are initially single crystal. The AFM images shown in Fig. S5(c) and (d) (ESI†) give an individual SnS sheet thickness of 17 nm.
A membrane based on SnS–G was deposited onto the polypropylene (PP) separator by vacuum filtration. This membrane was Li-activated by a single lithiation–delithiation cycle, creating A-Sn–G membrane. A range of 0.01–2.5 V vs. Li/Li+ ensured that a stable Li-based SEI was formed but that no Li metal was plated. This galvanostatic cycle is shown in Fig. S6 (ESI†), indicating that the irreversible capacity of the layer is 0.16 mA h cm−2. This irreversible capacity is primarily attributed to the formation of a Li-based SEI. More details of the Li-activation process are provided in the Experimental section.
Fig. 1(a) shows the standard porous structure of the PP separator, employed as a baseline. Fig. 1(b) shows the PP separator with the SnS–G nanolayered composite which coats the separator in a macroscopically conformal manner. The coating consisting of packed arrays of nanosheets is expected to be porous and well-wetted by the electrolyte. Cross section SEM characterization shown in Fig. S7 (ESI†) reveals that the membrane is approximately 5 μm in thickness. As can be seen from Fig. 1(c), the A-SnS–G is still macroscopically conformal, but now with the Li-based SEI layer acting as the binder to hold the sheets together. Fig. 1(d) shows the SEM images and the EDXS C, S and Sn elemental maps of A-SnS–G. After Li activation, the layer thickness remains near 5 microns. Post activation, the three elements remain well-dispersed (within the resolution of the SEM EDXS detector). This is expected since the immobile graphene sheets should anchor the lithiating and then the delithiating phases, preventing their agglomeration.
 | ||
| Fig. 1 (a)–(c) Top-down SEM images and photographs (inset) of baseline unmodified PP separator, SnS–G on PP, and A-SnS–G on PP, respectively. (d) Cross-section SEM images and EDXS elemental maps (same scale) of A-SnS–G, highlighting the morphology of the 5 μm thick membrane, and the C, S and Sn elemental distribution within it. | ||
Fig. 2(a) highlights transmission electron microscopy (TEM) analysis for SnS–G. The panel shows a bright field micrograph highlighting the overall nanolayered composite morphology. The selected area electron diffraction (SAED) pattern inset identifies the equilibrium (wurtzite) structure of the SnS sheets. The SAED pattern displays orthorhombic SnS(142) (040) and (021) reflections, as well as (100) for graphene. That the two patterns are well aligned confirms the stacking of the two phases. Fig. 2(b) shows the high resolution TEM (HRTEM) image and the associated Fast Fourier Transforms (FFTs) of SnS–G, further highlighting the well-ordered crystalline structure of SnS and of graphene. In this case, both the SnS and the graphene are aligned along their [001] zone axis. The TEM HADDF image and EDXS elemental maps of SnS–G are shown in Fig. S8 (ESI†). These further verify the initial layer-by-layer arrangement of the composite per the map of Sn near the arrowed region. During extended electrochemical cycling the nanolayer periodicity is likely to be disrupted, although an ultrafine nanocomposite structure should largely remain due to the graphene scaffolding.
 | ||
| Fig. 2 (a) TEM images with associated SAED patterns for as-synthesized SnS–G. (b) HRTEM image and FFTs of SnS–G, highlighting the well-ordered crystalline structure of SnS and G. (c) HRTEM image and FFTs of A-SnS–G, demonstrating the amorphization of SnS due to activation. Fig. S9 (ESI†) shows additional TEM analysis for A-SnS–G, depicting the intact structure of the graphene. (d) XRD analysis of A-SnS–G and SnS–G, demonstrating the amorphization of SnS due to activation, but the retained structure of G. (e) XPS C 1s spectra of A-SnS–G and SnS–G. (f) and (g) XPS F 1s and Li 1s spectra of A-SnS–G. The overview XPS spectra are shown in Fig. S10 (ESI†). | ||
The Li-activated structure of SnS in A-SnS–G is fully amorphized, per the HRTEM and FFT analysis in Fig. 2(c). Additional TEM analysis of A-SnS–G is shown in Fig. S9 (ESI†). The intact structure of the graphene is evidenced by the bright field images showing the characteristic sheet-like morphology, and the associated [001] zone axis SAED pattern displaying hexagonal symmetry. Amorphization of Sn and S agrees with reports for transition metal dichalcogenides that don’t re-crystallize after delithiation.51,58 The activation induced amorphization of SnS is further confirmed by XRD analysis, shown in Fig. 2(d). The XPS C 1s spectra of A-SnS–G and SnS–G are shown in Fig. 2(e). The XPS F 1s and Li 1s spectra of A-SnS–G are shown in Fig. 2(f) and (g). The associated overview XPS spectra for A-SnS–G and SnS–G are shown in Fig. S10 (ESI†). The activation process creates a Li-based SEI surrounding and interspersed with a nanocomposite of graphene with Sn and S species. Per the XPS spectra, the SEI contains substantial levels of the usual electrolyte decomposition products, Li2CO3, Li2O, ROCO2Li as well as LiF. The LiF is unambiguously identified by F 1s and Li 1s XPS spectra at 684.8 eV and 55.9 eV, respectively. LiF is known to stabilize both Li metal and Li ion anodes. It forms a structurally resilient layer within the SEI that is adjacent to the active material.59–61 As will be demonstrated, the Li-based SEI remains during ongoing Na metal plating/stripping.
Significant differences were observed in the electrical conductivity of as-synthesized SnS–G versus A-SnS–G, as characterized by 4-probe conductivity tests. These results are shown in Table S2 (ESI†). The specimen SnS–G is moderately conductive, measuring 0.22 S cm−1. By contrast, the A-SnS–G membrane is insulating, measuring ∼0.001 S cm−1. After activation, the membrane is amorphized with its internal surfaces becoming covered by the numerous Li-based SEI phases which are all electrically insulating. This ensures the A-SnS–G will not act as a support for the plating Na, i.e. that it cannot sustain an appreciable electrical current necessary for Na ion reduction. Likely the SnS–G@Cu also becomes insulating with extended cycling due to the Na-based SEI that is formed.
When these membranes are placed in contact with Na metal anodes, the architectures are termed “A-SnS–G@Na” and “SnS–G@Na”. Fig. 3 shows the electrochemical behavior of symmetric cells, A-SnS–G@Na||A-SnS–G@Na and SnS–G@Na||SnS–G@Na. Two baselines are also tested. The first baseline is unprotected thin Na foils, termed Na||Na. The second baseline is the unprotected Na foil but that was Li activated through the same process, termed A–Na||A–Na. The galvanostatic data for the Li – activation process of the Na foil is shown in Fig. S11(a) (ESI†). Fig. 3(a) and (b) display the voltage-time profiles of these four symmetric cell architectures, tested at 1 mA cm−2 and 4 mA cm−2. The plating time was fixed at 0.5 hour per cycle. The associated average plating–stripping overpotential results at 4 mA cm−2 are shown in Fig. 3(c). In symmetric cells, the average overpotential is defined as the average of the absolute values of highest anodic and cathodic overpotential (1 V being the limit).
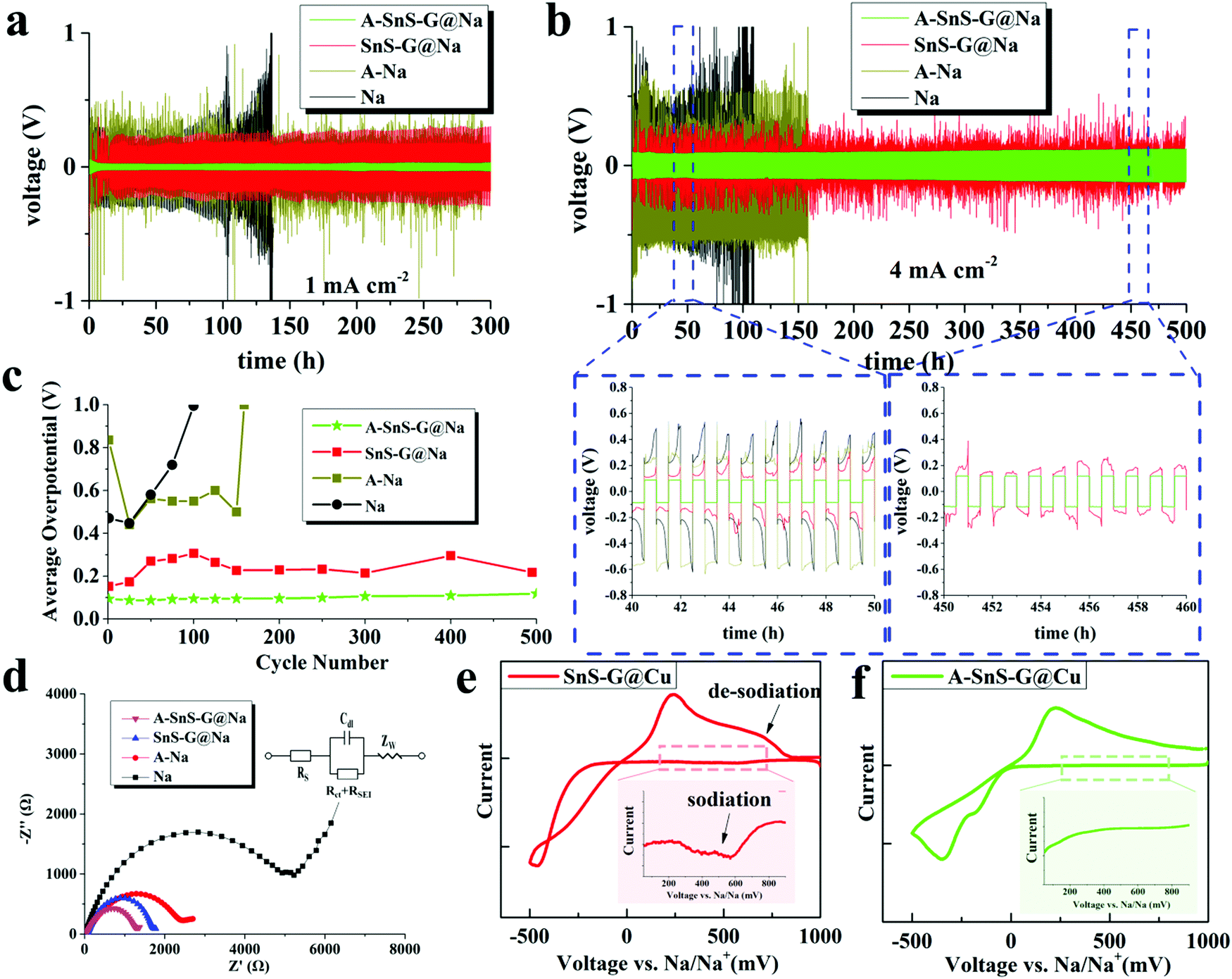 | ||
| Fig. 3 (a) and (b) Electrochemical performance of symmetric cells based on A-SnS–G@Na||A-SnS–G@Na, Sn-G@Na||Sn-G@Na, and baselines A–Na||A–Na and Na||Na, at 1 mA cm−2 and 4 mA cm−2, respectively. (c) Average overpotentials of the symmetric cells, tested at 4 mA cm−2. (d) Nyquist plots of the post-cycled cells, with equivalent circuit shown as inset. (e and f) Cyclic voltammetry curves of A-SnS–G and SnS–G membranes tested directly on the Cu collector without underlying Na metal, obtained at 0.2 mV s−1. The insets spotlight the cathodic current prior to Na plating, which is associated with the sodiation reaction of SnS–G. The CVs of Li-activated Cu and bare Cu are provided in Fig. S15 (ESI†). | ||
By far the most stable cycling and the lowest voltage overpotential is observed with A-SnS–G@Na||A-SnS–G@Na. This is evident from the cycling curves, as well as from the plot of the average overpotentials which stay nearly flat for the 500 cycles. The average overpotential never goes above 0.35 V at either 1 mA cm−2 (shown in Fig. S12, ESI†) or 4 mA cm−2 (Fig. 3c). The A-SnS–G@Na||A-SnS–G@Na cell survives the 300 cycles at 1 mA cm−2 and the 500 cycles at 4 mA cm−2. The SnS–G@Na||SnS–G@Na cell also shows relatively stable cycling behavior at both currents, but with markedly higher overpotentials. At 1 mA cm−2 the baseline Na||Na cell exhibits an unstable voltage from the onset, displaying a severe overpotential increase. The rapid drop in the overpotential at cycle ∼105 may be interpreted at a “soft” electrical shorting event, where there is mixed electronic and ionic conduction between the two metal anodes.10 With both A-SnS–G@Na||A-SnS–G@Na and SnS–G@Na||SnS–G@Na the overpotentials do not appreciably increase when the current density is raised from 1 to 4 mA cm−2. For instance, the SnS–G@Na||SnS–G@Na cell displayed overpotentials in the range of 0.22–0.30 V at 1 mA cm−2, and in the range of 0.25–0.31 V at 4 mA cm−2. The overpotential behavior in metal electrodeposition is complex, having the initial overpotential spike being associated with nucleation events followed by an overpotential plateau that is associated with secondary nucleation and grain growth.62,63 Moreover, at high currents and/or large SEI thicknesses, there is an additional overpotential associated with concentration polarization of ions in electrolyte and/or in the SEI.64,65 The difference between 1 mA cm−2 and 4 mA cm−2 may not be substantive enough in terms of concentration polarization or nucleation barriers. In addition, Joule heating at 4 mA cm−2 may enhance solid-state diffusion and lead to stress relaxation,66,67 both effects reducing the measured overpotentials.
During cycling the A-SnS–G and SnS–G membranes are in situ transferred onto the Na metal and become incorporated into the anode's surface. This is evidenced in Fig. S13 (ESI†), which presents light optical images of both the membrane and of the corresponding Na metal in as-fabricated state, after cycle 1, and after cycle 100. The current density employed for these tests was 1 mA cm−2. Fig. S13(a) (ESI†) shows the conformal A-SnS–G layer placed on the PP separator prior to cycling. Fig. S13(b) (ESI†) shows the post-1 cycle (plated, stripped) separator, now with a section of the A-SnS–G layer missing from its surface. Fig. S13(c) (ESI†) shows the post-100 cycle separator, with more of the A-SnS–G layer missing. Fig. S13(d)–(f) (ESI†) show the Na metal anode corresponding to conditions in Fig. S13(a)–(c) (ESI†), indicating progressive transfer of A-SnS–G onto its surface. The surface of the Na metal becomes covered with a brownish-black coating, which coincides well with the section of the membrane missing from the post-cycled PP separator. Transfer is driven by the formation of additional SEI that binds the Na metal surface to the A-SnS–G layer.
The role of A-SnS–G could be understood by comparing the cycling performance of A-SnS–G@Na versus A–Na. The large voltage fluctuations with A–Na are eliminated by the incorporation of A-SnS–G surface film. The Li-based SEI layer in A–Na is likely prone to fracture and disintegrate during Na metal cycling due to the volume changes. A-SnS–G provides a robust scaffold for the Li-SEI species, making the Na–electrolyte interface resilient to the repeated expansion–contraction. The A-SnS–G@Na||A-SnS–G@Na specimen remains stable when the plating time at 1 mA cm−2 is extended from 0.5 to 2 hours. Fig. S14 (ESI†) shows the galvanostatic voltage profiles of symmetric cells, tested at 1 mA cm−2 with 2 hours of plating time per cycle. By contrast, with Na||Na, A–Na||A–Na and SnS–G@Na||SnS–G@Na cells, a continuous overpotential increase to the voltage limit is observed. The limit was reached first in the Na||Na at 56 hours (cycle 14), followed by A–Na||A–Na at 104 hours (cycle 26), and then SnS–G@Na||SnS–G@Na at 172 hours (cycle 43). For A-SnS–G@Na||A-SnS–G@Na there is minimal increase in the overpotentials even at 200 hours (cycle 50).
Fig. 3(d) shows the electrochemical impedance spectroscopy (EIS) Nyquist plots of all four the post-cycled cells, after 50 cycles at 1 mA cm−2. The A–Na||A–Na specimen has a lower combination of charge transfer resistance and SEI resistance than the Na||Na specimen, being at 2.58 kΩ vs. 4.59 kΩ. This may be directly related to a Li-based rather than a Na-based SEI and will correlate with a lower overpotential. For A-SnS–G@Na||A-SnS–G@Na versus SnS–G@Na||SnS–G@Na the EIS findings are likewise consistent. The A-SnS–G@Na specimen is at 1.37 kΩ while the SnS–G@Na is at 1.89 kΩ. Comparing the overall electrochemical performance improvement, it may be concluded that a Li-based SEI is preferred over a Na-based SEI. In a carbonate-based electrolyte, a Li-based SEI has been shown to be less soluble than its Na-based analogue,68 indirectly supporting the current findings.
Fig. 3(e) and (f) show cyclic voltammetry (CV) plating/stripping analysis of the A-SnS–G and SnS–G membranes, placed directly on bare Cu collectors without underlying Na metal. Also analyzed as a baseline for SEI formation is a Li-activated Cu collector (Fig. S11(b), ESI†), and untreated bare Cu. Those results are shown in Fig. S15 (ESI†). The CV tests were carried out at −0.5 to 1 V vs. Na/Na+, with a scanning rate of 0.2 mV s−1. The main peaks corresponding to the plating/stripping of Na metal with A-SnS–G@Cu and SnS–G@Cu were at −346/217 mV and −463/236 mV vs. Na/Na+, respectively. A discernable series of cathodic peaks in the range of 200–750 mV is present for SnS–G@Cu. This is highlighted in the inset of Fig. 3(e), and is ascribed to the sodiation reaction of SnS–G. Per the inset in Fig. 3(f), A-SnS–G@Cu does not display this series of cathodic peaks, indicating that the Li-based Sn and S phases are relatively stable.
Achieving stable cycling with a limited amount of Na metal is vital for achieving the oft quoted gravimetric and volumetric energy values for SMBs (and LMBs). Hence it is important to determine how much Na was cycled with respect to the available capacity of the Na foil. The total accumulated capacity “A” is the plated capacity per cycle times the total number of cycles. The foil capacity “F” is the total weight of Na foil times 1165 mA h g−1. An indicator that can be then employed to measure the utilization of the Na metal anode is the “A/F ratio”, i.e. the accumulated capacity/foil capacity. As may be observed from Table S1 (ESI†), the A/F ratio of A-SnS–G@Na reached 90.9. This is an exceptionally high value in respect to the typically reported values which are below 10.5,20,21,26–29 The difference is due to a combination of extended stability (numerator) and low foil thickness (denominator).
The role of the membranes in preventing dendrites can be further understood by characterizing the post-cycled metal surfaces. Fig. 4(a)–(c) show top-down SEM analysis of post-100 cycles (1 mA cm−2) A-SnS–G@Na and SnS–G@Na specimens, as well as the unprotected Na baseline. Per Fig. 4(a), the surface of the post-cycled A-SnS–G@Na specimens is smooth and dendrite-free. This agrees with the cycling and the EIS results and highlights the efficacy of a Li-based SEI combined with a SnS–G nanocomposite. According to Fig. 4(b) while SnS–G does improve the overall surface smoothness, some dendrites do protrude in isolated regions. Per the cycling results, the dendrites present on the surface of the SnS–G@Na were not severe enough to lead to shorting, or to a catastrophic impedance rise. According to Fig. 4(c), a dense forest-like dendritic morphology is present on the surface of baseline Na. The dendrites appear rounded due to the remnant SEI that covers the metal and is not removed by washing the electrode. To further unravel the structure of the SEI during cycling, XPS depth profiling was performed on the post-100 cycles A-SnS–G@Na and SnS–G@Na. Each etch level corresponded to a sputtering time of 210 s. Since the etch speed of Ar+ is about 4 nm min−1, each etch step corresponds to approximately 12–15 nm. As shown in Fig. 4(d) and (e), for A-SnS–G–Na there are discernable peaks at 489.5 eV and 484.3 eV, corresponding to the Sn 3d3/2 and Sn 3d5/2 of metallic Sn and Sn alloys. Conversely, the SnS–G–Na only exhibits peaks of Sn2+ at 494.1 eV (Sn 3d3/2) and 485.2 eV (Sn 3d5/2). This indicates that the valence state of Sn is mostly not altered and that the conversion reaction is not run to completion.
 | ||
| Fig. 4 Top-down SEM images of post-cycled (100 cycles at 1 mA cm−2) surfaces of (a) A-SnS–G@Na, (b) SnS–G@Na and (c) baseline Na. (d) and (e) XPS sputter depth profiling analysis of post-cycled A-SnS–G@Na and SnS–G@Na, showing Sn 3d spectra as a function of etch time. (f) and (g) Ex situ light optical microscope observation of A-SnS–G@Na||A-SnS–G@Na and Na||Na cells. Scale bar: 600 μm. | ||
To further understand the role of A-SnS–G protection, optical plating–stripping experiments were performed. A-SnS–G@Na||A-SnS–G@Na and Na||Na baseline were tested in quartz-cuvette symmetric cell, the sample preparation process being described in the Experimental section. In the quartz-cuvette cell with interelectrode distance of 1.5 mm, which is much wider than the typical 20 micrometers in a button cell. According to Sand's time space charge theory, the enlarged inter-electrode distance will exacerbate dendrite growth allowing for an accelerated testing comparison of the two samples. Per Fig. 4(f), the A-SnS–G@Na||A-SnS–G@Na cell displays planar deposition morphologies, with no evidence of dendrites at cycles 1, 3 and 10 (top to bottom images). As shown in Fig. 4(g), the Na||Na cell shows an unstable interface even at cycle 1, which gets worse at cycle 3 and even worse at cycle 10. There is also evidence of significant levels of “Dead Metal”, i.e. electrically isolated Na metal particles fully or partially imbedded in the SEI.
Fig. 5(a) and (b) show SEM cross-sectional images of post-300 cycles A-SnS–G@Na electrode. Fig. 5(d) and (e) show SEM cross-sectional images post 131 cycles (cycled until failure) unprotected Na electrode. The difference in the cycling-induced expansion of the A-SnS–G protected versus unprotected metal can now by clearly visualized. After 300 cycles, the protected metal foil expands from the initial 100 μm to the final 131 μm, without appreciable cracking or decrepitation. When the unprotected electrode fails at cycle 131, it has already swelled from the 100 μm initial thickness to 181 μm. Moreover, the electrode is heavily cracked throughout, with porosity in the bulk structure. The SEM EDXS analysis performed on the post-cycled A-SnS–G@Na is shown in Fig. 5(c). Shown are the EDXS spectra at spot 1 which is near the electrode surface and spot 2 which is in the bulk Na metal. The EDXS analysis confirms that the cycled Na metal remains underneath the A-SnS–G membrane, with the Sn, S species being at the top surface.
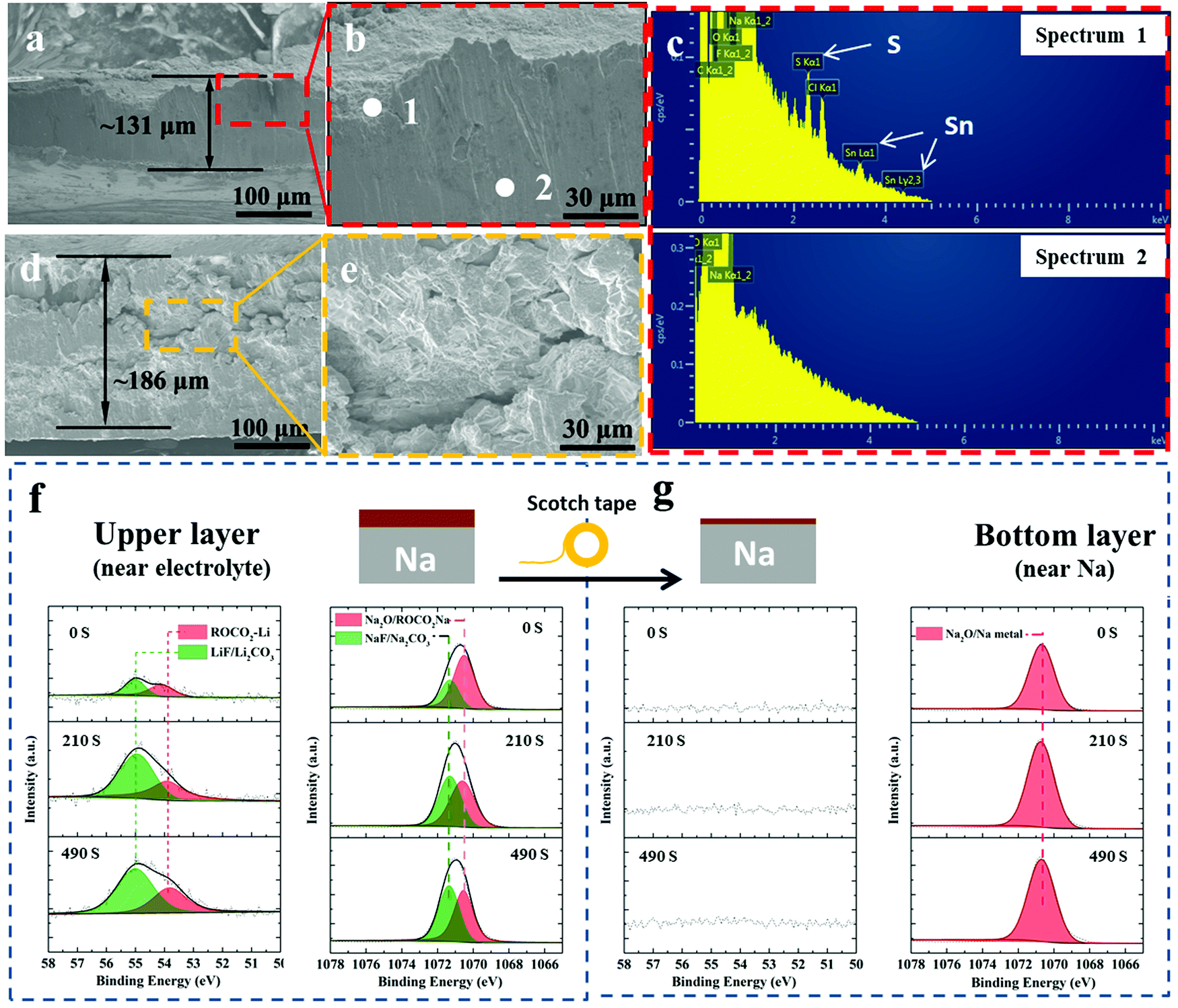 | ||
| Fig. 5 (a and b) SEM cross-sectional images of post-300 cycles A-SnS–G@Na highlighting its final thickness. (c) EDXS spectra of cycled A-SnS–G@Na, with spot 1 being near the electrode surface and spot 2 being in the bulk Na metal. (d and e) SEM cross-sectional images of post 131 cycles (cycled until failure) unprotected Na electrode highlighting the expansion and cracking. (f) XPS depth profiles of the post-cycled A-SnS–G@Na, showing the Li 1s and Na 1s spectra. (g) Same XPS analysis but with most of the A-SnS–G layer removed using the scotch-tape method, so as to expose the near-Na surface that is underneath. | ||
Additional XPS ion-sputter depth analysis was performed on the post 300-cycles A-SnS–G@Na. As shown in Fig. 5(f) the Li 1s spectra demonstrated Li-based SEI components at all the sputter depths (sputter etch times 0, 210, 490 s). Given the ∼4 nm min−1 etch speed, a 490 s etch time corresponds to ∼35 nm of analysis. In both as-fabricated A-SnS–G membrane and the cycled A-SnS–G@Na, the Li-based SEI components are ROCO2-Li, Li2CO3 and LiF. Per Fig. 2(g) for the as fabricated A-SnS–G, within the Li 1s spectra it is possible to differentiate the Li2CO3 peak at 55.4 eV from the LiF peak at 55.9 eV. According to Fig. 5(f), for post-cycled A-SnS–G@Na these two peaks become inseparable within the relatively less intense Li 1s signal. The Na 1s spectra demonstrates the existence of Na2O, ROCO2-Na, Na2CO3 and NaF. These Na-based SEI components were formed during repeated Na metal cycling and are interspersed with the Li-based SEI components that were formed during Li activation. In order to probe deeper into the SEI, a classical scotch tape method was employed to remove the top-most section of A-SnS–G. This is illustrated in the schematic inset at the top of Fig. 5(f) and (g). Per Fig. 5(g), the XPS depth profiling on the scotch tape exfoliated A-SnS–G@Na specimen shows intense Na metal and Na2O signal. The Li-related XPS signals are absent. This indicates that the Li-based SEI remains near the electrolyte interface while the Na metal plates underneath, which is expected since the membrane is ionically conductive but electrically insulating.
Scheme 2 summarizes our approach for protecting the Na metal during extended plating/stripping. The A-SnS–G membrane combines improved mechanical properties with improved Na ion transport to stabilize the Na metal–electrolyte interface. One role of the Li activation process in facilitating Na ion transport is to transform crystalline SnS nanosheets into amorphous sulfides and tin based species (Li2S, Sn), which are inherently good ion conductors and interfacial stabilizers.69–71 Another aspect of Li activation is that it generates a robust and highly ionic conductive Li-based SEI. The presence of LiF in A-SnS–G@Na is an important factor. Both LiF and NaF are beneficial for mechanical integrity of the SEI. However, per prior reports, it is the LiF that gives more dramatic improvement.31,60 In addition, ion-diffusion may be faster in Li-based SEI components (Li2O, LiF, Li2CO3, etc.) than in the Na analogues (Na2O, NaF, Na2CO3, etc.). Ionic conductivity in LiF (∼10−7 S cm−1) is five orders of magnitude higher than in NaF (∼10−13 S cm−1).72 Recent density functional theory (DFT) calculations have shown that Na ion can readily migrate through Li-based SEI components.73 Adopting a direct hopping mechanism, the activation barrier for Na ion migration through LiF was calculated as 0.38 eV. This value is actually lower than the 0.73 eV calculated for Li ion migration through LiF.74 Sodium ions were calculated to migrate readily through Li2CO3 by knock-off and by direct hopping mechanisms, with 0.19 eV and 0.9 eV diffusion barriers respectively.73 Overall it may be concluded that Na ion conduction through Li-based inorganic SEI phases is kinetically favorable. This will be useful in facilitating and homogenizing the solid-state Na ion flux during repeated plating and stripping, leading to uniform deposition/dissolution of the metal front.
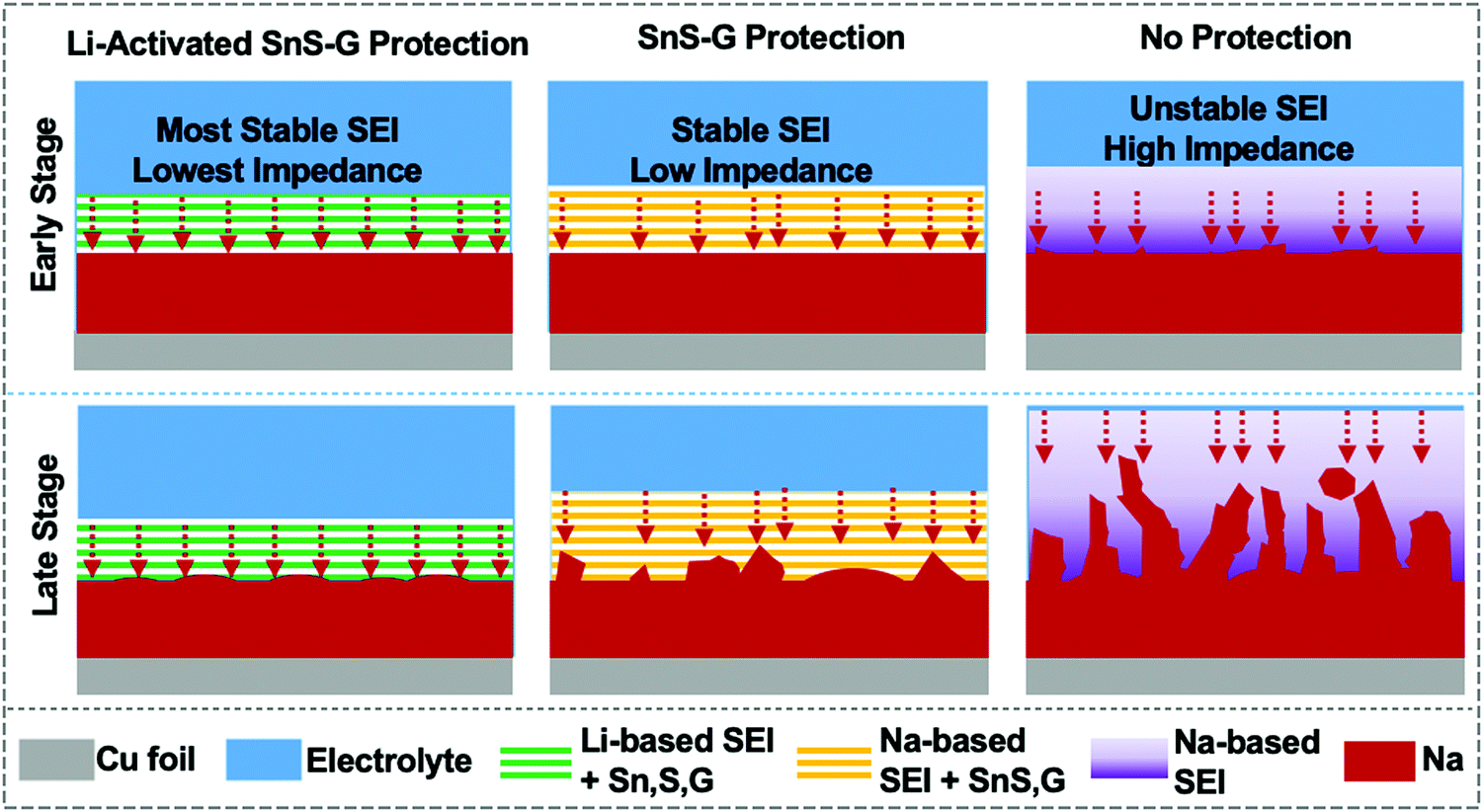 | ||
| Scheme 2 Schematic illustration of early stage cycling and late stage cycling behavior of A-SnS–G@Na, SnS–G@Na and unprotected Na. Key aspects captured are the differences in the SEI stability with cycling and the resultant planar versus dendritic morphology of the metal front. | ||
The activated membrane protection strategy is verified in a full SMB cell configuration by pairing the A-SnS–G@Na, SnS–G@Na and baseline bare Na foils with a ceramic cathode Na3V2(PO4)3 (NVP). The NVP cathode is attractive for SMBs because of its high working voltage of 3.4 V vs. Na/Na+ and its good cyclability in carbonate electrolytes.41Fig. 6(a) shows the cycling performance of the three cells, each with a cathode mass loading of 2 mg cm−2. According to the figure, Na||NVP cell is the least stable, displaying rapid capacity decay in the initial 20 cycles. The SnS–G@Na||NVP cell is intermediate, showing continuous capacity decay from cycle 1 to cycle 350. The poor CE in both Na||NVP and SnS–G@Na||NVP is indicative of parasitic side reactions on the Na metal anode during charging (plating) and discharging (stripping). The A-SnS–G@Na||NVP cell is by far the most stable, achieving an average CE of 99.69% between cycle 1 and cycle 600. This extended stability agrees with the symmetric cell data shown earlier. In fact, the A-SnS–G@Na||NVP cell stability is limited by the cathode, rather than by the anode. The observed capacity/voltage fade at cycle 375 is fully recovered by interrupting the test and replacing the cathode.
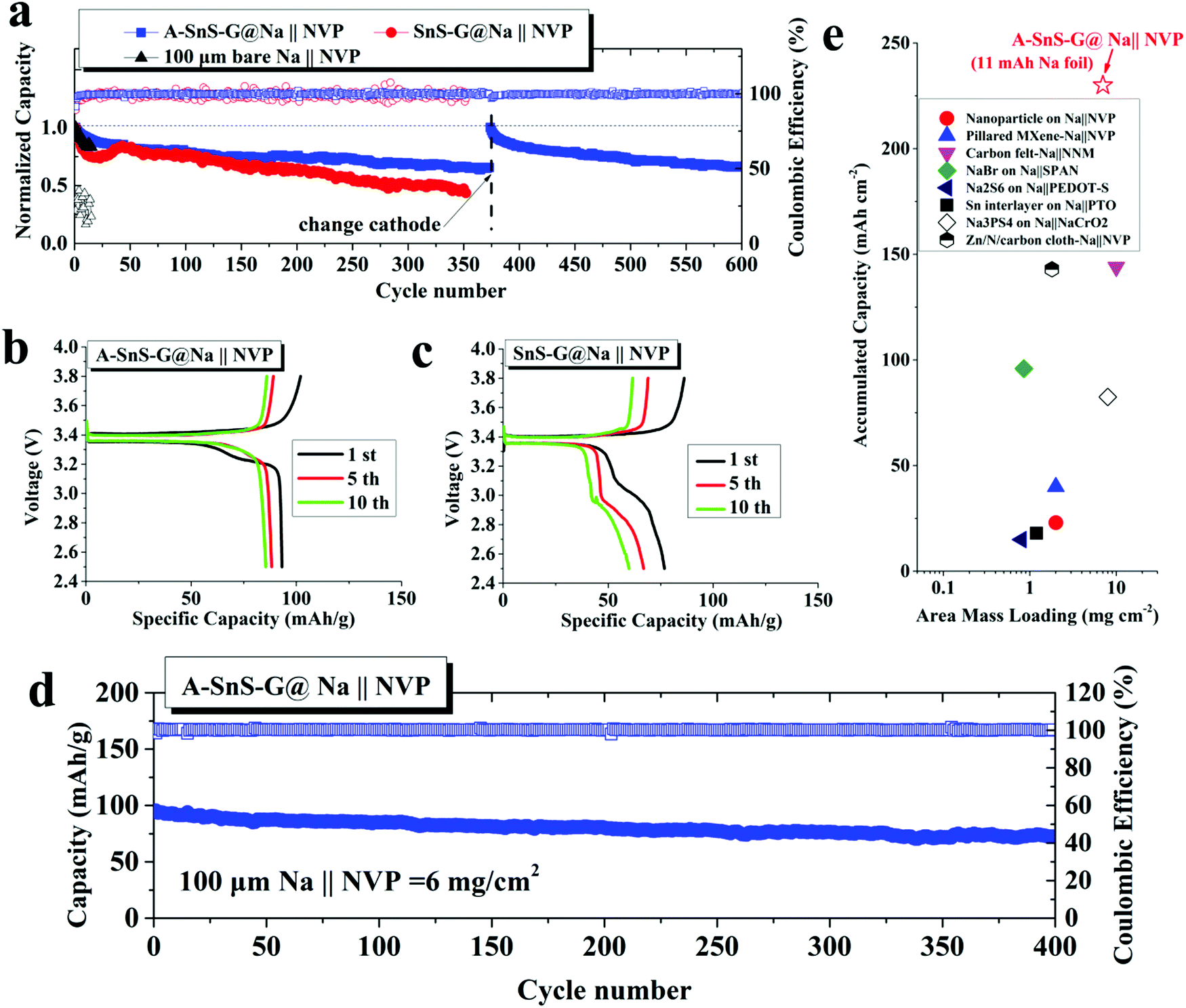 | ||
| Fig. 6 (a) Galvanostatic cycling performance for full sodium metal battery (SMB) cells, with thin A-SnS–G@Na, SnS–G@Na or Na anodes are paired to 2 mg cm−2 mass loading NVP cathodes. The comparison is at 0.2C, with C being the theoretical capacity of NVP, 110 mA h g−1. (b and c) Charge–discharge profiles of the full cells based on A-SnS–G@Na and SnS–G@Na anodes, respectively. (d) Fast charge (0.4C) cycling of high mass loading (6 mg cm−2) NVP paired with A-SnS–G@Na. (e) Comparison over achieved accumulated cycle capacity of current full cell with state-of-the-art previous advances, note high accumulated cycle capacity (225 mA h cm−2) with respect to the 100 μm thin Na metal (11 mA h cm−2) is unprecedented. | ||
Fig. 6(b) and (c) show the early cycle charge–discharge voltage profiles for A-SnS–G@Na||NVP and SnS–G@Na||NVP at 2 mg cm−2 respectively. The A-SnS–G@Na||NVP cell demonstrates a distinctly flat discharge plateau at ∼3.36 V throughout cycling. Per Fig. 6(c), the SnS–G@Na||NVP cell shows evidence of unstable behavior. Such unstable full cell voltage profiles have been reported to arise from the Na metal anode side due to excessive SEI formation and “Dead Metal”.35 Similar double plateau behavior was reported for Li metal full cells and was attributed to similar origins.75 The stable cycling behavior of A-SnS–G@Na||NVP is also evidenced by a lack of such secondary plateau.
It is known that thin Na foils may possess intrinsic problems that are often masked by conventional laboratory conditions. Therefore, cycling thin Na foils is even more challenging than foils of conventional thickness. Likely the extra difficulty is related to the higher percentage of stripped Na in the thin foils with respect to the overall Na metal inventory, i.e. the depth of discharge. This agrees with a discussion published regarding analysis of thin versus thick Li foils for commercial application.10 To further evaluate the performance of A-SnS–G@Na anode at deeper discharge, a high mass loading (6 mg cm−2) NVP cathode was adopted in the SMB. For these high NVP mass loading cells, a formation cycle of 0.1C for 10 cycles was employed prior to extended cycling at 0.4C for 400 cycles. These results are shown in Fig. 6(d). With this deep discharge configuration, a reversible capacity of 73.8 mA h g−1 with stable CE approaching 100% is retained after 400 cycles. This corresponds to an accumulated cycle capacity of 225 mA h cm−2.
Fig. 6(e) plots the accumulated capacity versus area mass loading for A-SnS–G@Na||NVP versus advanced high performance SMBs from literature.24,25,28,29,76–79 It may be observed that the combination of high mass loading and extended cyclability gives the current full cell system favorable characteristics relative to prior art. We note that most proof-of-principal SMB full cell configurations adopt mass loading levels of ∼1 mg cm−2 at the cathode side and a much thicker metal foil anode. This leads to cycling the cells at capacities insignificant relative to the overall Na metal reservoir, i.e. at a very shallow plating/stripping conditions. For the A-SnS–G@Na||NVP cell, an A/F ratio of 20.5 is achieved. Based on NVP cathode alone (anode-free configuration), the specific energy of the cell is 307 W h kg−1. A specific energy of 118 W h kg−1 is achieved based on the summed weight of the NVP cathode and the 100 μm Na metal foil. Adopting more advanced metallurgical rolling techniques, a further reduction of the Na metal foil thickness down to 50 μm should be possible. This would roughly double the specific energy of the cell to >200 W h kg−1.
Conclusions
Stable plating/stripping performance of Na metal anodes is especially difficult to achieve in standard carbonate-based electrolytes due to the instability of the resultant SEI layer. The current approach to stabilize Na metal anodes synergizes a nanolayered SnS–graphene membrane with a stable surface structure and improved impedance of a Li-based SEI, achieved by electrochemical Li-activation. In parallel, a novel inert atmosphere rolling process is employed to fabricate 100 μm Na metal anodes, which represent the thinnest Na foils reported but add a further challenge to stability due to the increased plating/stripping depth per cycle. It is demonstrated that these thin Na metal cells once protected by the Li-activated membrane will achieve stable cycling in a carbonate electrolyte, with negligible dendrite growth, and with only minor levels of cycling-induced swelling. The Li-activated SnS–graphene membrane is in situ transferred onto the Na anode during electrochemical cycling, forming a stable artificial SEI. XPS and SEM analyses are employed to unravel the details of the complex post-cycled membrane structure, including the decomposition of SnS into various alloys and compounds. Metal anodes utilizing SnS–G membranes that were not Li-activated show inferior electrochemical performance. Full SMB cells adopting Li-activated SnS–graphene protected anodes coupled to NVP cathodes deliver extended cycling life along with high anode utilization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. L. (research conception and manuscript preparation) is supported by National Natural Science Foundation of China (51702223), International Collaborative Project of Chengdu (2019-GH02-00031-HZ), China Postdoctoral Science Foundation (2019T120839). D. M. and E. P. (research co-conception and guidance, manuscript preparation) and Y. W. (manuscript co-preparation) are supported by the National Science Foundation, Division of Materials Research, Award Number 1938833. The authors thank Shanlin Wang, Hui Wang and Yingming Zhu in Analytical & Testing Center and INELT of SCU for their assistance in electron microscopy.References
- Y. Zhao, K. R. Adair and X. Sun, Energy Environ. Sci., 2018, 11, 2673–2695 RSC.
- Y. X. Wang, B. W. Zhang, W. H. Lai, Y. F. Xu, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- X. Zheng, C. Bommier, W. Luo, L. Jiang, Y. Hao and Y. Huang, Energy Storage Mater., 2019, 16, 6–23 CrossRef.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS.
- B. Sun, P. Li, J. Zhang, D. Wang, P. Munroe, C. Wang, P. H. L. Notten and G. Wang, Adv. Mater., 2018, 30, 1801334 CrossRef.
- Y. Zhao, L. V. Goncharova, A. Lushington, Q. Sun, H. Yadegari, B. Wang, W. Xiao, R. Li and X. Sun, Adv. Mater., 2017, 29, 1606663 CrossRef.
- S. Y. Wei, S. Choudhury, J. Xu, P. Nath, Z. Y. Tu and L. A. Archer, Adv. Mater., 2017, 29, 1605512 CrossRef.
- B. Wu, S. Wang, J. Lochala, D. Desrochers, B. Liu, W. Zhang, J. Yang and J. Xiao, Energy Environ. Sci., 2018, 11, 1803–1810 RSC.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- W. Liu, Y. Xia, W. Wang, Y. Wang, J. Jin, Y. Chen, E. Paek and D. Mitlin, Adv. Energy Mater., 2019, 9, 1802918 CrossRef.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS.
- H. J. Yoon, N. R. Kim, H. J. Jin and Y. S. Yun, Adv. Energy Mater., 2018, 8, 1701261 CrossRef.
- W. Liu, P. Li, W. Wang, D. Zhu, Y. Chen, S. Pen, E. Paek and D. Mitlin, ACS Nano, 2018, 12, 12255–12268 CrossRef CAS.
- T. Liu, X.-L. Feng, X. Jin, M.-Z. Shao, Y.-T. Su, Y. Zhang and X.-B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 18240–18245 CrossRef CAS.
- J. Wang, G. Huang, J.-M. Yan, J.-L. Ma, T. Liu, M.-M. Shi, Y. Yu, M.-M. Zhang, J.-L. Tang and X.-B. Zhang, Natl. Sci. Rev., 2020 DOI:10.1093/nsr/nwaa150.
- Y. Yu and X.-B. Zhang, Matter, 2019, 1, 881–892 CrossRef.
- Y. Yu, Y.-B. Yin, J.-L. Ma, Z.-W. Chang, T. Sun, Y.-H. Zhu, X.-Y. Yang, T. Liu and X.-B. Zhang, Energy Storage Mater., 2019, 18, 382–388 CrossRef.
- H. Wang, C. Wang, E. Matios and W. Li, Nano Lett., 2017, 17, 6808–6815 CrossRef CAS.
- W. Luo, C.-F. Lin, O. Zhao, M. Noked, Y. Zhang, G. W. Rubloff and L. Hu, Adv. Energy Mater., 2017, 7, 1601526 CrossRef.
- Y. Zhao, L. V. Goncharova, Q. Zhang, P. Kaghazchi, Q. Sun, A. Lushington, B. Wang, R. Li and X. Sun, Nano Lett., 2017, 17, 5653–5659 CrossRef CAS.
- S. Choudhury, S. Y. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Y. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 CrossRef.
- H. Wang, C. Wang, E. Matios and W. Li, Angew. Chem., Int. Ed., 2018, 57, 7734–7737 CrossRef CAS.
- Y. Zhao, J. Liang, Q. Sun, L. V. Goncharova, J. Wang, C. Wang, K. R. Adair, X. Li, F. Zhao, Y. Sun, R. Li and X. Sun, J. Mater. Chem. A, 2019, 7, 4119–4125 RSC.
- D. Lei, Y.-B. He, H. Huang, Y. Yuan, G. Zhong, Q. Zhao, X. Hao, D. Zhang, C. Lai, S. Zhang, J. Ma, Y. Wei, Q. Yu, W. Lv, Y. Yu, B. Li, Q.-H. Yang, Y. Yang, J. Lu and F. Kang, Nat. Commun., 2019, 10, 4244 CrossRef.
- W. Luo, Y. Zhang, S. Xu, J. Dai, E. Hitz, Y. Li, C. Yang, C. Chen, B. Liu and L. Hu, Nano Lett., 2017, 17, 3792–3797 CrossRef CAS.
- S. S. Chi, X. G. Qi, Y. S. Hu and L. Z. Fan, Adv. Energy Mater., 2018, 8, 1702764 CrossRef.
- T. Yang, T. Qian, Y. Sun, J. Zhong, F. Rosei and C. Yan, Nano Lett., 2019, 19, 7827–7835 CrossRef CAS.
- L. Zhang, S. Peng, Y. Ding, X. Guo, Y. Qian, H. Celio, G. He and G. Yu, Energy Environ. Sci., 2019, 12, 1989–1998 RSC.
- M. Han, C. Zhu, T. Ma, Z. Pan, Z. Tao and J. Chen, Chem. Commun., 2018, 54, 2381–2384 RSC.
- R. Rodriguez, K. E. Loeffler, S. S. Nathan, J. K. Sheavly, A. Dolocan, A. Heller and C. B. Mullins, ACS Energy Lett., 2017, 2, 2051–2057 CrossRef CAS.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 9069–9072 CrossRef CAS.
- R. Dugas, A. Ponrouch, G. Gachot, R. David, M. R. Palacin and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A2333–A2339 CrossRef CAS.
- S. A. Webb, L. Baggetto, C. A. Bridges and G. M. Veith, J. Power Sources, 2014, 248, 1105–1117 CrossRef CAS.
- D. Ruiz-Martínez, A. Kovacs and R. Gómez, Energy Environ. Sci., 2017, 10, 1936–1941 RSC.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- Z. W. Seh, J. Sun, Y. M. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS.
- H. Y. Che, S. L. Chen, Y. Y. Xie, H. Wang, K. Amine, X. Z. Liao and Z. F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- Y.-S. Hong, N. Li, H. Chen, P. Wang, W.-L. Song and D. Fang, Energy Storage Mater., 2018, 118–126 CrossRef.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS.
- Q. Zhang, Y. Lu, L. Miao, Q. Zhao, K. Xia, J. Liang, S.-L. Chou and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 14796–14800 CrossRef CAS.
- J.-L. Ma, F.-L. Meng, Y. Yu, D.-P. Liu, J.-M. Yan, Y. Zhang, X.-B. Zhang and Q. Jiang, Nat. Chem., 2019, 11, 64–70 CrossRef CAS.
- J. R. Brent, D. J. Lewis, T. Lorenz, E. A. Lewis, N. Savjani, S. J. Haigh, G. Seifert, B. Derby and P. O’Brien, J. Am. Chem. Soc., 2015, 137, 12689–12696 CrossRef CAS.
- J. N. Coleman, M. Lotya, A. O’Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS.
- W. Liu, H. Li, J. Jin, Y. Wang, Z. Zhang, Z. Chen, Q. Wang, Y. Chen, E. Paek and D. Mitlin, Angew. Chem., Int. Ed., 2019, 58, 16590–16600 CrossRef CAS.
- A. M. Dimiev, G. Ceriotti, A. Metzger, N. D. Kim and J. M. Tour, ACS Nano, 2016, 10, 274–279 CrossRef CAS.
- P. Xiong, R. Ma, N. Sakai and T. Sasaki, ACS Nano, 2018, 12, 1768–1777 CrossRef CAS.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS.
- C. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713–2731 RSC.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- C. Bommier, D. Mitlin and X. Ji, Prog. Mater. Sci., 2018, 97, 170–203 CrossRef CAS.
- C. Backes, T. M. Higgins, A. Kelly, C. Boland, A. Harvey, D. Hanlon and J. N. Coleman, Chem. Mater., 2017, 29, 243–255 CrossRef CAS.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- E. Markevich, G. Salitra, F. Chesneau, M. Schmidt and D. Aurbach, ACS Energy Lett., 2017, 2, 1321–1326 CrossRef CAS.
- Z. Xue-Qiang, C. Xin-Bing, C. Xiang, Y. Chong and Z. Qiang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Lett., 2017, 2, 1337–1345 CrossRef CAS.
- W. Liu, P. Liu and D. Mitlin, Chem. Soc. Rev., 2020, 49, 7284–7300 RSC.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS.
- A. Mistry, C. Fear, R. Carter, C. T. Love and P. P. Mukherjee, ACS Energy Lett., 2019, 4, 156–162 CrossRef CAS.
- W. Liu, P. Liu and D. Mitlin, Adv. Energy Mater., 2020, 10, 2002297 CrossRef CAS.
- L. Li, S. Basu, Y. Wang, Z. Chen, P. Hundekar, B. Wang, J. Shi, Y. Shi, S. Narayanan and N. Koratkar, Science, 2018, 359, 1513–1516 CrossRef CAS.
- P. Hundekar, S. Basu, X. Fan, L. Li, A. Yoshimura, T. Gupta, V. Sarbada, A. Lakhnot, R. Jain, S. Narayanan, Y. Shi, C. Wang and N. Koratkar, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5588–5594 CrossRef CAS.
- R. Mogensen, D. Brandell and R. Younesi, ACS Energy Lett., 2016, 1, 1173–1178 CrossRef CAS.
- X. Zheng, H. Fu, C. Hu, H. Xu, Y. Huang, J. Wen, H. Sun, W. Luo and Y. Huang, J. Phys. Chem. Lett., 2019, 10, 707–714 CrossRef CAS.
- H. Chen, A. Pei, D. Lin, J. Xie, A. Yang, J. Xu, K. Lin, J. Wang, H. Wang, F. Shi, D. Boyle and Y. Cui, Adv. Energy Mater., 2019, 9, 1900858 CrossRef.
- S. Xia, X. Zhang, C. Liang, Y. Yu and W. Liu, Energy Storage Mater., 2020, 24, 329–335 CrossRef.
- H. Yildirim, A. Kinaci, M. K. Y. Chan and J. P. Greeley, ACS Appl. Mater. Interfaces, 2015, 7, 18985–18996 CrossRef CAS.
- F. A. Soto, A. Marzouk, F. El-Mellouhi and P. B. Balbuena, Chem. Mater., 2018, 30, 3315–3322 CrossRef CAS.
- Y. C. Chen, C. Y. Ouyang, L. J. Song and Z. L. Sun, J. Phys. Chem. C, 2011, 115, 7044–7049 CrossRef CAS.
- K.-H. Chen, K. N. Wood, E. Kazyak, W. S. LePage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, J. Mater. Chem. A, 2017, 5, 11671–11681 RSC.
- S. Tang, Y.-Y. Zhang, X.-G. Zhang, J.-T. Li, X.-Y. Wang, J.-W. Yan, D.-Y. Wu, M.-S. Zheng, Q.-F. Dong and B.-W. Mao, Adv. Mater., 2019, 31, 1807495 CrossRef.
- J. Luo, C. Wang, H. Wang, X. Hu, E. Matios, X. Lu, W. Zhang, X. Tao and W. Li, Adv. Funct. Mater., 2019, 29, 1805946 CrossRef.
- S. Choudhury, S. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 CrossRef.
- X. Chi, F. Hao, J. Zhang, X. Wu, Y. Zhang, S. Gheytani, Z. Wen and Y. Yao, Nano Energy, 2019, 62, 718–724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02423f |
| This journal is © The Royal Society of Chemistry 2021 |