
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High energy density anodes using hybrid Li intercalation and plating mechanisms on natural graphite†
Yeonguk
Son
a,
Taeyong
Lee
b,
Bo
Wen
ac,
Jiyoung
Ma
 b,
Changshin
Jo
ad,
Yoon-Gyo
Cho
e,
Adam
Boies
a,
Jaephil
Cho
*b and
Michael
De Volder
*a
b,
Changshin
Jo
ad,
Yoon-Gyo
Cho
e,
Adam
Boies
a,
Jaephil
Cho
*b and
Michael
De Volder
*a
aDepartment of Engineering, University of Cambridge, 17 Charles Babbage Road, CB3 0FS Cambridge, UK. E-mail: mfld2@cam.ac.uk
bDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: jpcho@unist.ac.kr
cCambridge Graphene Centre, University of Cambridge, 9 JJ Thomson Avenue, CB3 0FA Cambridge, UK
dSchool of Chemical Engineering and Materials Science, Chung-Ang University, Seoul 06974, Republic of Korea
eDepartment of Nanoengineering, University of California, San Diego, CA 92093-0448, USA
First published on 17th September 2020
Abstract
Lithium plating on conventional graphite anodes in lithium-ion batteries is typically considered an undesirable side reaction, a safety hazard or a degradation mechanism. However, lithium plating and stripping allow for efficient energy storage, and therefore various new porous anode designs with tailored surface coatings and electrolyte systems have been proposed to achieve reversible Li plating and stripping. Unfortunately, these material designs often rely on highly porous plating scaffolds with an overall lower volumetric energy and power density than conventional graphite anodes. Herein, a novel anode design is presented which leverages the good volumetric performance of industrial graphite anodes and further enhances their capacity by allowing for a reversible Li plating on their surface. The latter is achieve by conformally coating them with a nanoscale lithiophilic Si coating. As a result, excellent volumetric energy densities of 656 mA h cm−3 and gravimetric capacities of 551 mA h g−1 are achieved, which are a clear improvement compared to the commercial graphite anode (app. 570 mA h cm−3 and 360 mA h g−1 respectively). Moreover, by carefully balancing the thickness of the Si layer and the plating capacity, a capacity retention close to 100% is achieved after 200 cycles in half cells. Overall, this approach leverages the advances in industrial graphite anode manufacturing while at the same time embracing the additional capacity offered by reversible plating and stripping of Li metal, resulting in full cells energy densities of 474 W h kg−1 and 912 W h L−1, which is a step forward compared to previous Li metal and graphite anodes.
Broader contextThe demand for higher energy density lithium ion batteries (LIBs) is growing steadily as these batteries are powering ever more energy hungry applications. To increase the energy density of LIB anodes, silicon and lithium metal are considered as a replacement for graphite anodes that are used commercially. However these new anodes suffer from degradation processes associated with their volume expansion and side reactions. Moreover, lithium metal based anodes also suffer from safety issues due to dendritic lithium growth. In addition, it is difficult to exceed the areal and volumetric capacities of commercial graphite anodes, which have undergone years of industrial optimisation. Herein, we leverage the advances made in industrial graphite LIB anode manufacturing and coat these with a thin silicon layer that allows for stable lithium metal plating and stripping inside the pores that are present in graphite anodes. As a result, our anodes store energy through a combination of intercalation in graphite, alloying with Si and Li plating within the same electrode volume where standard commercial graphite anodes only use intercalation. As a result, we achieve full cell energy densities of 474 W h kg−1 and 912 W h L−1, which is a step forward compared to previous work using either graphite or Li-metal anodes. |
Introduction
Lithium (Li) metal is being considered as a next-generation anode material for lithium-ion batteries (LIBs) because of its high theoretical capacity (3860 mA h g−1 or 2061 mA h cm−3) and low electrochemical potential (−3.04 V versus standard hydrogen electrodes).1–3 However, these anodes rely on repeated Li metal plating and stripping, which results in large volume changes with excessive solid electrolyte interphase (SEI) and dead Li formation, as well as Li dendrite growth which can short-circuit the battery and can cause fires.1,4–6 Various approaches have been reported to address these degradation and safety issues, for example: (i) new types of electrolyte effectively suppressing inhomogeneous Li plating and non-flammable electrolytes to address safety issues such as polymer, ceramic and innovate liquid electrolytes.7–13 (ii) Alternatively, Li metal anodes have been coated with MoS2, Si, graphitic-C3N4, and artificial SEI to stabilize side reactions by preventing Li metal from directly contacting the electrolyte.14–17 However, this approach relies on having some Li metal pre-loaded in the anodes when the battery is assembled, which makes their handling and manufacturing challenging. In addition, this often requires a delithiated cathode,18–20 which is the opposite of current practice. (iii) Finally, the above solutions still suffer from changes in electrode volume while cycling and therefore numerous porous host materials have been suggested to improve the Li plating process.21–23 These scaffolds typically provide a high surface area to reduce the average Li plating and provide ample voids to accommodate volume expansion without changing the overall electrode volume (Fig. 1a). Additionally, lithiophilic materials are often coated on the surface of the plating scaffolds to further smooth the plating process and improve the cycle life.24–26 Nevertheless, the performance of these electrodes can be misleading as the volume and weight of the porous hosts are typically ignored in capacities calculation and the overall gravimetric, areal, and volumetric capacities are not reported.27,28 In practice, however, the high volume and weight of these porous hosts often negate the benefits of Li metal's high specific capacity, resulting in anodes with an overall volumetric and gravimetric performance lower than that of commercial graphite anodes.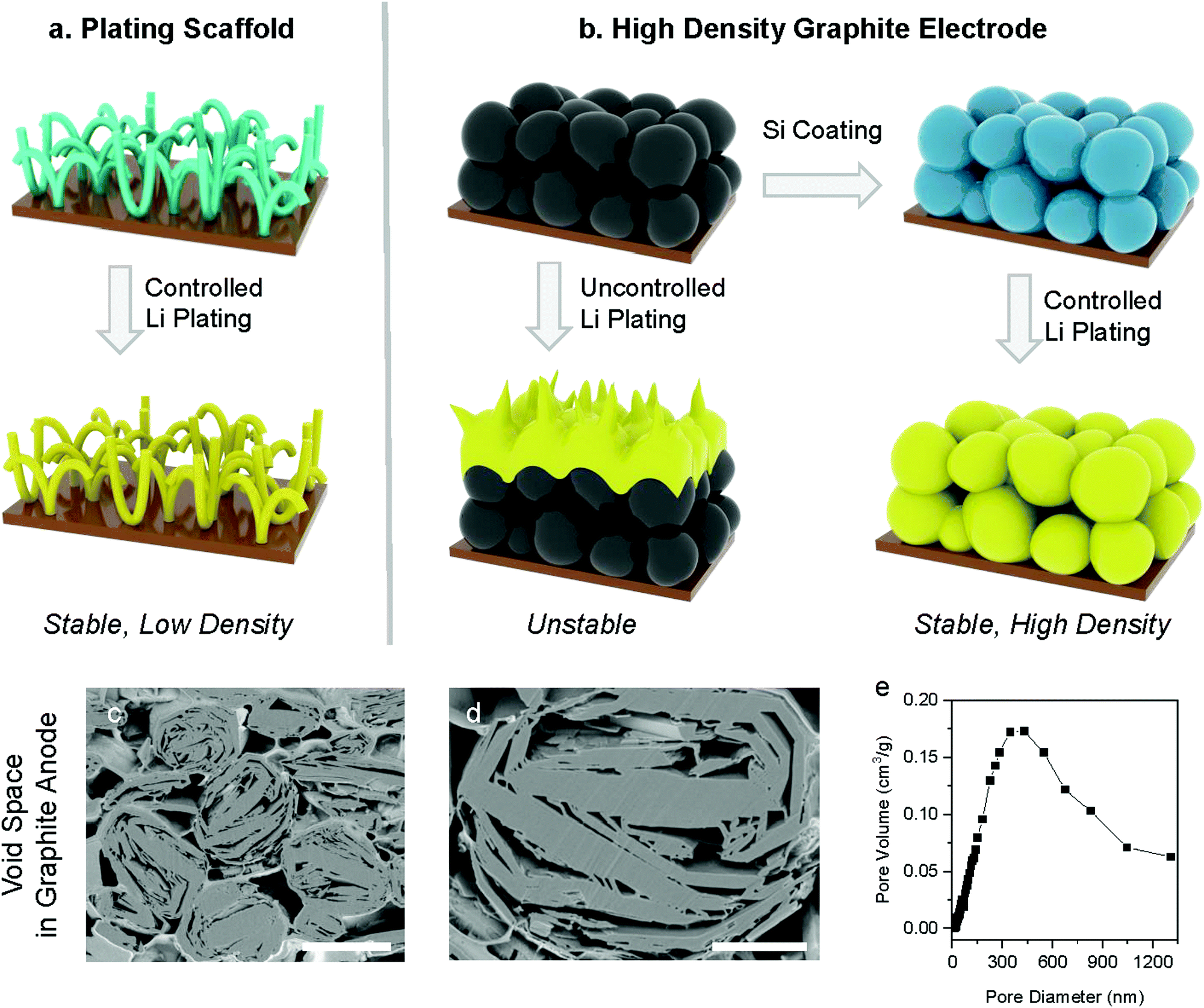 | ||
| Fig. 1 Schematic view of typical Li plating scaffolds, graphite anodes, and the proposed anode concept combining intercalation and plating mechanisms. Schematic view of Li plating (a) on carbon nanotube scaffold design and (b) on conventional graphite and proposed concept anode. Cross-sectional image of (c) pristine graphite electrode and (d) pristine graphite particle. (e) the pore volume of pristine graphite depending on pore diameter measured by a mercury porosimeter. Scale bar = 15 μm for (c) and 5 μm for (d). | ||
Although there have been reports overcoming intrinsic drawbacks of each of the above approaches in Li anode separately, there have been no reports showing all critical parameters determining performance of LIBs including gravimetric, areal, and volumetric capacities as well as the initial coulombic efficiency (ICE), full-cell configuration (e.g. N/P ratio), and the specific energy and energy density. This is an important issue because if any of the above metrics are subpar, the proposed electrode will be challenging to implement commercially. For example, enhanced gravimetric capacity is thought to decrease the cell weight, but this is not necessarily the case if the areal capacity is decreased as this requires more current collector, separator, etc. Even if the gravimetric and areal capacities are improved but the volumetric capacity decreases due to excessive pore sizes, the electrodes could still be too voluminous to be attractive. Likewise, if a new anode formulation improves the gravimetric, areal, and volumetric capacities, but at the cost of a low ICE, the overall gravimetric and volumetric energy densities of the battery could still be reduced as it would require higher amounts of cathode materials to compensate for the low ICE.
Commercial natural graphite anodes have been extensively optimised over the past decades and achieve well-balanced performance metrics (360 mA h g−1, 3.3 mA h cm−2, 570 mA h cm−3, ICE of 92%), which overall outperform Li-metal anodes taking into account the considerations discussed above as well as the electrode manufacturability.4,29 In this paper, we take a new approach to leverage the good performance of commercial graphite anodes and further increase their gravimetric capacity by allowing for a controlled amount of Li plating on the graphite surface. In classic graphite anodes, Li plating is an unwanted phenomenon taking place during fast charging or long-term cycling, which leads to degradation or failure of the battery.30,31 Hence several papers report coatings to prevent lithium plating on graphite such as amorphous Al2O3, TiO2 and polyvinylidene difluoride (PVDF).32–34 A recent paper reported an anode combining Li plating/stripping and graphite intercalation/deintercalation using a combination of dual salt and mechanical pressure to stabilise the electrode operation.35 Here we propose instead to coat graphite with a lithiophilic layer in conjunction with an organo-gel electrolyte to promote reversible Li plating (Fig. 1b). A thin conformal Si layer (∼15 nm) was selected as the lithiophilic coating because it allows for some additional energy storage through Si alloying, and the layer is sufficiently thin to allow for efficient Li transport.14,26,36 By judiciously balancing the amount of energy stored by lithium intercalation in graphite, alloying with Si and Li plating, we demonstrate stable LIB anodes with gravimetric capacities of 551 mA h g−1, areal capacities of 3.35 mA h cm−2, and volumetric capacities of 656 mA h cm−3 in half cell and specific energy of 474 W h kg−1 and energy density of 912 W h L−1 in full cell (N/P ratio of 1.05) based on the weight and volume of anode and cathode materials. Finally, this new anode concept is compatible with established industrial expertise and capital equipment investments for commercial graphite anode manufacturing.
Results and discussion
Commercial natural graphite for LIB anodes typically undergoes a number of chemical and mechanical refinement steps, including spheroidization and grinding steps.37 For instance, the graphite used in this work is treated by impact milling to create spherical particles with low surface area and high electrode density.38 The resulting graphite has a porous structure as shown in cross-section SEM (Fig. 1c and d), which offers an opportunity for Li plating. Mercury porosimetry and cross-section SEM of our electrodes show that the dominant pore size in the graphite particles is 300–500 nm, and in addition, there are larger inter-particle pores (Fig. 1e). The BET surface area of the graphite used in this work is 4.45 m2 g−1. Even when these spherical natural graphite electrodes are calendered to a density of 1.35 g cm−3, their porosity is still 36% (see the Supplementary Note 1, ESI† for calculation details), which shows that graphite anodes have substantial voids that will be used in this paper for Li plating. Theoretically, if all the voids in our graphite anodes would be filled Li metal plating, then the gravimetric capacity would reach 963 mA h g−1 which is over 2.5 times higher than that of pure natural graphite (360 mA h g−1), assuming 7.5 mg cm−2 of mass loading (see the Supplementary Note 1, ESI† for calculation details).In order to access some of this additional capacity offered by Li plating on graphite, we need to suppress the undesirable side effects caused by Li plating such as excessive SEI formation and Li-dendrites causing short circuits.31,39 These challenges are due to the localised nature of the plating process, typically from defect sites. As discussed above, we coated our graphite particles conformally with Si to ensure a uniform plating process, which is achieved via Chemical Vapour Deposition (CVD, see details in ESI†). The best results were obtained by coating approximately 5 wt% of Si, which results in a 15 nm thick amorphous Si layer (see Fig. 2d). We observed that thinner Si coatings (approximately 2 wt%) result in poor battery lifetime, probably due to partially uncoated graphite surface that comes in direct contact with Li. On the other hand, thicker Si coatings (approximately 7 wt%) fails due to excessive Si swelling during lithiation.40,41 In what follows, we denote pristine graphite as ‘G’ and silicon-coated graphite as ‘Si/G’. The morphologies of G and Si/G were very similar as shown in Fig. 2 and Fig. S1 (ESI†), and we found as a result that Si/G can also be processed via the same procedures developed for G, which is important for industrial applications. We tested different amounts of Li plating by setting the total capacity to 600–800 mA h g−1 in half-cells and in full cells we control the N/P ratio to 1.05 (using capacity results in half cell test). Finally, we stabilised the plating process using an organogel electrolyte for which we added 2 wt% of cyanoethyl polyvinyl alcohol (PVA-CN) to a standard liquid electrolyte, which allows for in situ polymerization by heating our coin cells to 60 °C after assembly (see ESI† for details).
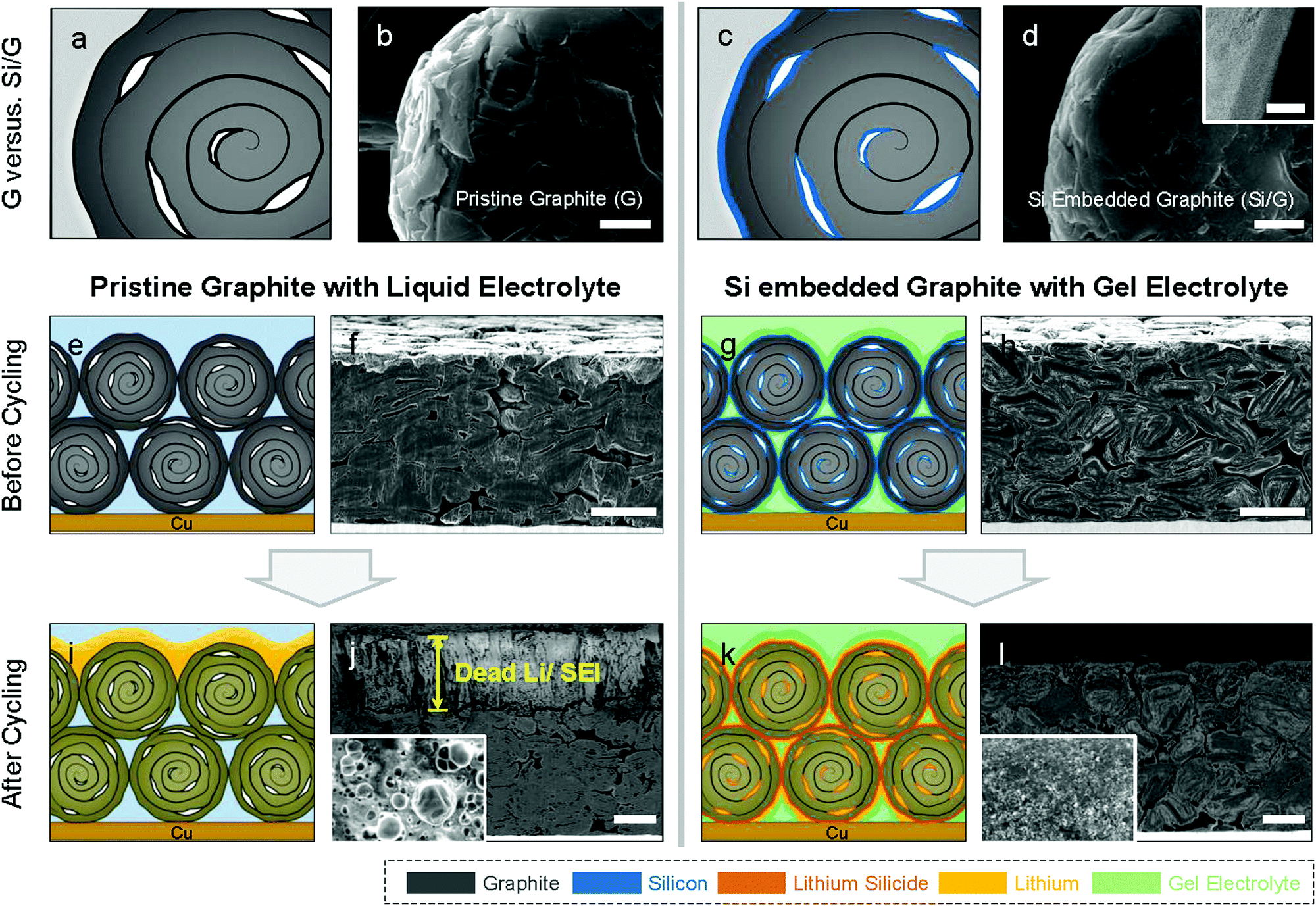 | ||
| Fig. 2 Conceptual views and correspondence SEM images of graphite (G) and Si embedded graphite (Si/G) before/after cycling. (a) and (b): G, (c) and (d): Si/G (inlet: cross-sectional TEM image of Si/G showing Si nanolayer on graphite), (e) and (f): G electrode before cycling, (g) and (h): Si/G electrode before cycling, (i) and (j): Li/G@liquid electrode after 20 cycling, and (k) and (l): Li/Si/G@gel electrode after 50 cycling. Scale bar = 2 μm for (b) and (d), 20 μm for (f), (h), (j), and (l). | ||
In what follows, we denote Li plating on G electrode as ‘Li/G’ and lithium plating on Si/G electrode as ‘Li/Si/G’, in addition we tag ‘@liquid’ or ‘@gel’ to cells testified with liquid or gel electrolyte respectively. Fig. 2e–l compare a standard G electrode with our proposed Si/G architecture combining intercalation, alloying, and plating. Fig. 2j and l, show cross-section SEM images of G and Si/G electrodes which we cycled to a fixed capacity of 600 mA h g−1 (0.05C for formation followed by cycling at 0.5C or 300 mA g−1). After 20 cycles, the top of the Li/G@liquid electrode is covered with over 30 μm of by-products including dead Li and SEI, which brought about severe cell degradation (Fig. 2j). In top-view, the Li/G@liquid electrodes shows characteristic rounded dead lithium morphologies (Fig. 2j, inset).42 After 50 cycles, the Li/G@liquid electrodes have become too brittle to transfer onto a SEM holder. Post-mortem cross-section SEM imaging suggested that Li plating on the conventional G anodes occurred predominantly on top of the electrode, which is in accordance with theoretical predictions.43,44 On the other hand, Li/Si/G@gel electrodes do not show any sign of excessive ‘dead Li’ or SEI formation even after 50 cycles, which is a first indication that our proposed strategy seems effective (Fig. 2l). For reference, cross-sectional images of Li/G@gel and Li/Si/G@liquid electrode after formation and after cycling are provided in Fig. S2 (ESI†).
Fig. 3a and b show typical galvanostatic voltage profiles of standard electrodes (Li/G@liquid) and the electrode formulation suggested in this work (Li/Si/G@gel) when a capacity of 600 mA h g−1 is imposed on the electrode (for reference, Li/G@gel voltage profiles are provided in Fig. S3, ESI†). For all electrochemical tests, the electrode composition has a ratio of active material to super-P to CMC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SBR of 90:4:3:3. The mass loading of the anodes is 6–8 mg cm−2 to achieve industrial level areal capacities of over 3.3 mA h cm−2 (for high energy LIBs). In addition, calendering is used to increase the electrode density to 1.3–1.4 g cm−3. The lithiation was cut off when a capacity of ∼600 mA h g−1 is reached, and the delithiation was cut off at 1.5 V versus Li+/Li (for comparison, voltage profiles using standard CC (constant current) with cut off voltages at 0.005 V and CV (constant voltage) with cut off C-rate at 0.02C during lithiation process are shown in Fig. S4, ESI†). The Li/G@liquid, Li/G@gel, and Li/Si/G@gel electrodes show reversible capacities and ICE of 520.9 mA h g−1 (86.82%), 524.4 mA h g−1 (87.56%), and 551.0 mA h g−1 (91.84%) respectively (Fig. S3, ESI†). The profiles of Fig. 3a and b clearly show three different energy storage mechanisms upon delithiation (i) Li stripping (<0.1 V vs. Li+/Li), (ii) graphite deintercalation (0.1–0.3 V), and (iii) Si dealloying (>0.3 V).45 When a standard Li/G@liquid electrode is charged to 600 mA h g−1, the delithiation capacity (Fig. 3a) comprises roughly 230 mA h g−1 of Li stripping and 290 mA h g−1 of graphite deintercalation. Under the same conditions, Li/Si/G@gel electrodes (Fig. 3b) offer roughly 65 mA h g−1 Li stripping, 320 mA h g−1 of graphite deintercalation, and 165 mA h g−1 of Si dealloying. A striking difference between these curves is the limited capacity from graphite deintercalation in the Li/G@liquid electrode, which suggests it degrades due to Li plating.31 After formation, the top of the Li/G@liquid electrodes is entirely covered with by-products such as SEI and dead lithium (Fig. S5a, ESI†). On the other hands, Li/Si/G@gel electrode maintained a clear electrode surface (Fig. S5b, ESI†). This is also reflected in linear sweep voltammetry (LSV) (Fig. 3c and Fig. S6, ESI†) which show clear distinct redox peaks in Li/Si/G@gel electrodes for the three energy storage mechanisms Li stripping, deintercalation, and dealloying, whereas the graphite intercalation peak is spread over a much larger voltage window in the Li/G@liquid electrodes.
:SBR of 90:4:3:3. The mass loading of the anodes is 6–8 mg cm−2 to achieve industrial level areal capacities of over 3.3 mA h cm−2 (for high energy LIBs). In addition, calendering is used to increase the electrode density to 1.3–1.4 g cm−3. The lithiation was cut off when a capacity of ∼600 mA h g−1 is reached, and the delithiation was cut off at 1.5 V versus Li+/Li (for comparison, voltage profiles using standard CC (constant current) with cut off voltages at 0.005 V and CV (constant voltage) with cut off C-rate at 0.02C during lithiation process are shown in Fig. S4, ESI†). The Li/G@liquid, Li/G@gel, and Li/Si/G@gel electrodes show reversible capacities and ICE of 520.9 mA h g−1 (86.82%), 524.4 mA h g−1 (87.56%), and 551.0 mA h g−1 (91.84%) respectively (Fig. S3, ESI†). The profiles of Fig. 3a and b clearly show three different energy storage mechanisms upon delithiation (i) Li stripping (<0.1 V vs. Li+/Li), (ii) graphite deintercalation (0.1–0.3 V), and (iii) Si dealloying (>0.3 V).45 When a standard Li/G@liquid electrode is charged to 600 mA h g−1, the delithiation capacity (Fig. 3a) comprises roughly 230 mA h g−1 of Li stripping and 290 mA h g−1 of graphite deintercalation. Under the same conditions, Li/Si/G@gel electrodes (Fig. 3b) offer roughly 65 mA h g−1 Li stripping, 320 mA h g−1 of graphite deintercalation, and 165 mA h g−1 of Si dealloying. A striking difference between these curves is the limited capacity from graphite deintercalation in the Li/G@liquid electrode, which suggests it degrades due to Li plating.31 After formation, the top of the Li/G@liquid electrodes is entirely covered with by-products such as SEI and dead lithium (Fig. S5a, ESI†). On the other hands, Li/Si/G@gel electrode maintained a clear electrode surface (Fig. S5b, ESI†). This is also reflected in linear sweep voltammetry (LSV) (Fig. 3c and Fig. S6, ESI†) which show clear distinct redox peaks in Li/Si/G@gel electrodes for the three energy storage mechanisms Li stripping, deintercalation, and dealloying, whereas the graphite intercalation peak is spread over a much larger voltage window in the Li/G@liquid electrodes.
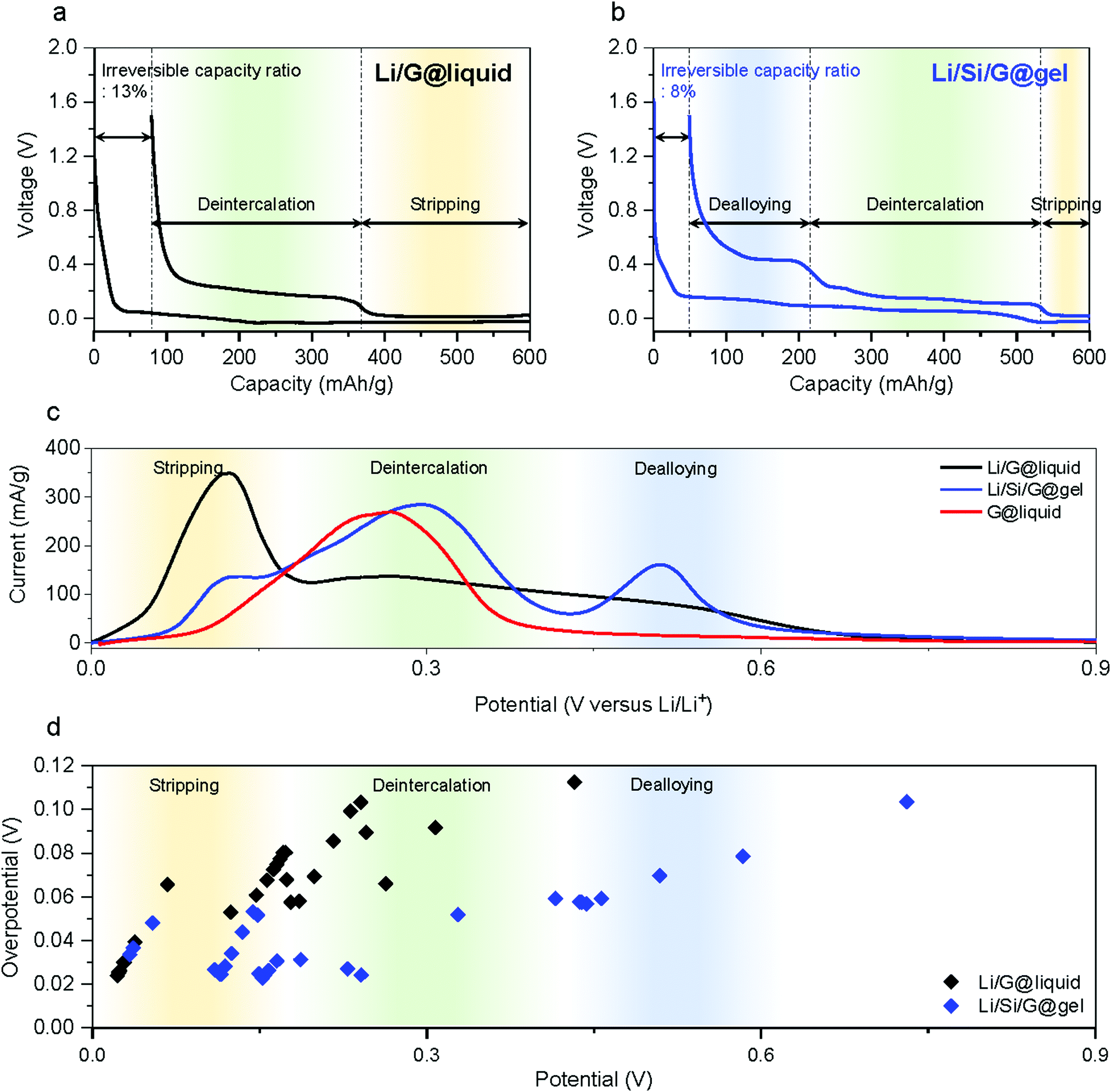 | ||
| Fig. 3 Electrochemical properties of proposed Si/G anodes. Delithiation voltage profiles of (a) Li/G@liquid and (b) Li/Si/G@gel. (c) LSV results of Li/G@liquid, Li/Si/G@gel, and G@liquid. (d) Overpotentials of Li/G@liquid and Li/Si/G@gel electrodes derived from GITT measurements. | ||
To better understanding the above potential shifts and peak broadening, we use Galvanostatic Intermittent Titration Technique (GITT) analysis (see the Supplementary Note 2 for the detailed analysis method and raw data is provided in Fig. S7, ESI†). Fig. S8 (ESI†) shows the measured overpotentials and the IR drops for Li stripping (<0.08 V), Li deintercalation (0.1–0.4 V), and Li dealloying (>0.4 V) based on the formation results. The overpotential and IR drops of Li/G@liquid electrode are 75 and 20 mV, respectively, in the intercalation voltage range. Those of Li/G@gel are 85 and 25 mV. Those of Li/Si/G@gel are 32 and 11 mV. This confirms that the plating process severely affects the operation of graphite anodes. Further, we observed that the gel electrolyte slightly increases the overpotential compared to the liquid electrolyte, but it stabilises interfacial side reactions as will be demonstrated by long-term galvanostatic cycling (see further).46,47
In most studies on Li metal anodes, the change in plating and stripping overpotential is reported as the cell is charged and discharged to a fixed capacity. However, because our anodes are relying on a combination of different energy storage mechanisms, we also galvanostatically cycling batteries to the following capacities: (i) lithiation capacity of Si/G with further Li plating/stripping capacity of 100 mA h g−1 denoted ‘Li100/Si/G’ (total capacity ∼600 mA h g−1); (ii) lithiation capacity of G (without Si coating) with a further Li plating/stripping capacity of 100 mA h g−1 denoted ‘Li100/G’ (total capacity ∼460 mA h g−1, same amount of plating as Si/G electrodes); (iii) lithiation capacity of G (without Si coating) with a further Li plating/stripping of 240 mA h g−1 denoted ‘Li240/G’ (total capacity ∼ 600 mA h g−1, same capacity as Si/G electrodes)., All tests are performed at 300 mA g−1 for over 150 hours. As shown in Fig. 4a, the Li stripping potential of Li/G electrodes soars up after tens of hours while that of Li/Si/G remains unchanged for 150 hours, reconfirming the advantages of Li/Si/G for Li plating compared to Li/G electrodes with either the same total gravimetric capacity or the same amount of Li plating and stripping. In addition, we also conducted galvanostatic charge–discharge experiments where the lithiation is cut off when the desired capacity is reached (600 mA h g−1 or 800 mA h g−1) while the delithiation is cut off when a voltage of 1.5 V against Li is reached (Fig. 4b and 5a). As expected from the previous experiments, the electrode using pure graphite with a classic electrolyte (Li/G@liquid) failed within the first 20 cycles (∼80% capacity fade),which can be extended to about 75 cycles using a gel electrolyte (Li/G@gel). On the other hand, our suggested Li/Si/G@gel with a capacity of 551 mA h g−1 shows outstanding capacity retention up to 200 cycles (retention: 100% and CE over 99% after 37th cycle). These trends are also clearly visible in cycling CE in Fig. 5b. For reference, cycling results of Li/Si/G@liquid electrode are provided in Fig. S9 (ESI†). These experiments show that the proposed gel electrolyte improves the coulombic efficiency and cycle life.
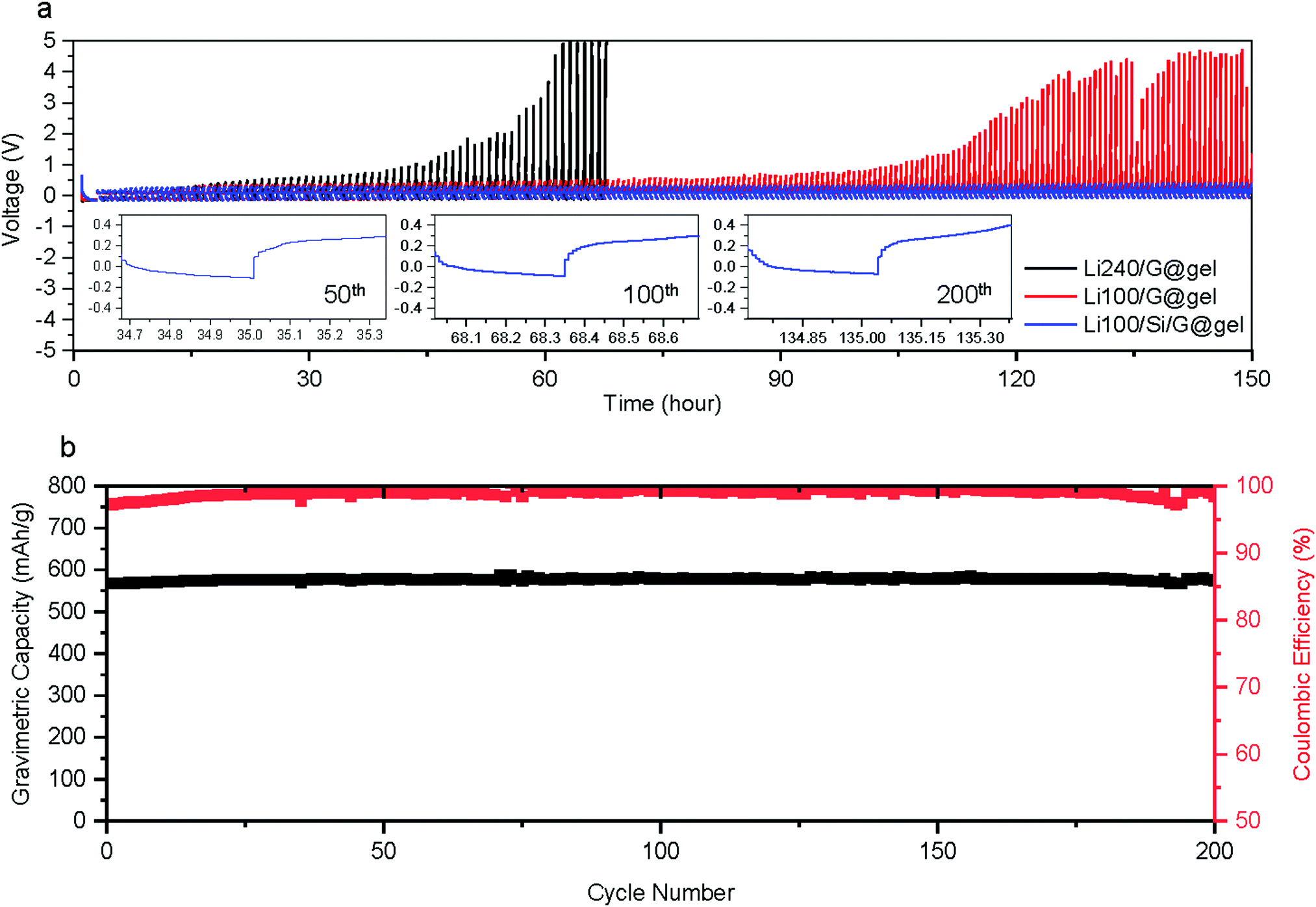 | ||
| Fig. 4 Cycling properties of the proposed anode. (a) Lithiation/delithiation capacity cut-off cycling performance. (b) Cycling performance of Li/Si/G@gel electrode samples at a 0.5C rate with CE (black: gravimetric capacity, red: CE). | ||
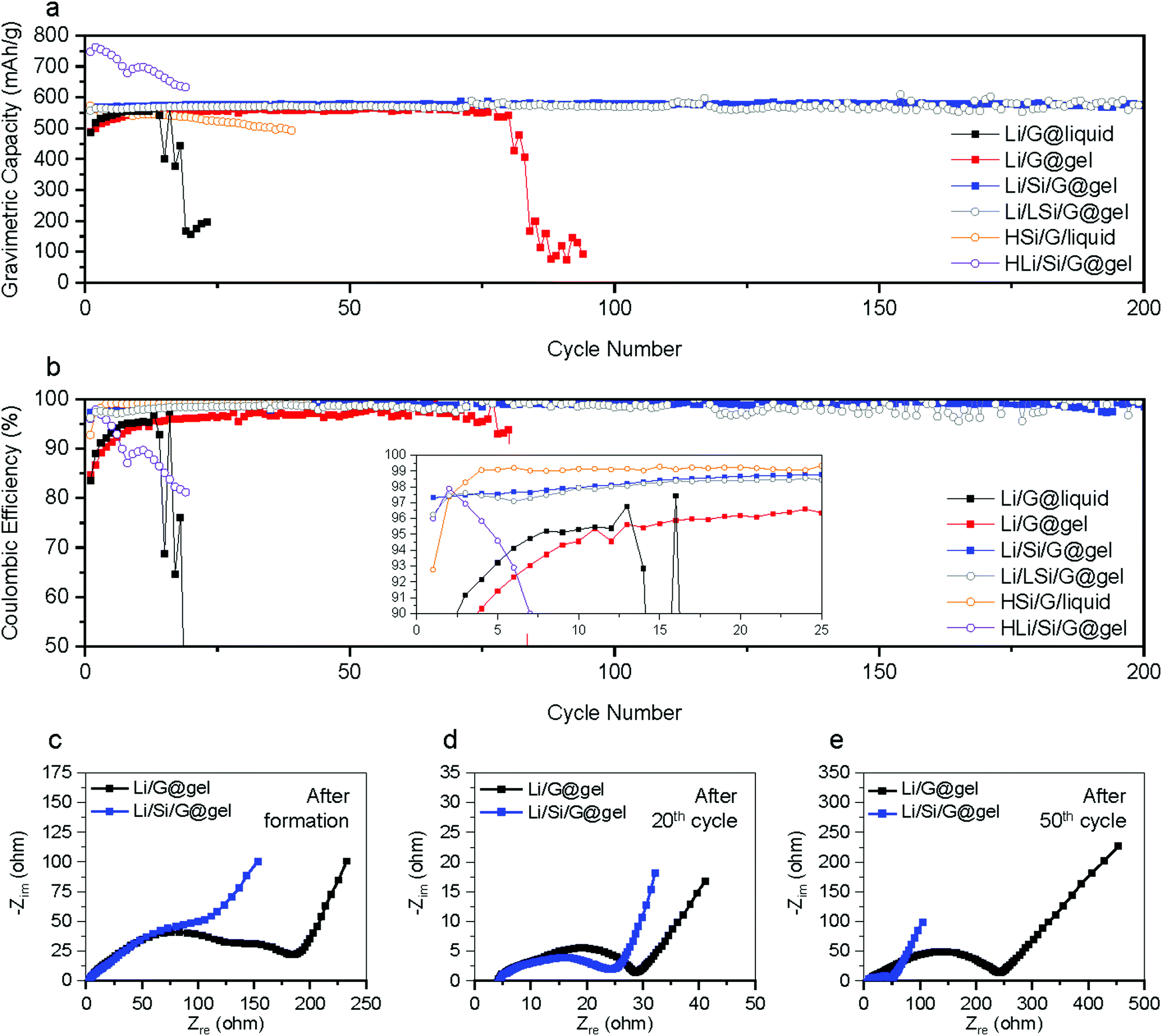 | ||
| Fig. 5 Comparison of cycling properties with various anodes. (a) Cycling performance of various electrodes samples at a 0.5C rate. (b) Cycling CE of various electrodes at 0.5C rate (inlet: magnified CE). EIS spectra of Li/G@liquid and Li/Si/G@gel after (c) formation, (d) 20th, and (e) 50th cycle. | ||
We also tested electrodes where about half as much of Si is coated (roughly 2 wt% of Si) referred to as Li/LSi/G@gel in Fig. 4b. This sample, however, shows fluctuations in capacity and poor CE. Conversely, samples with a thicker coating were tested to achieve approximately our targeted gravimetric capacity without the need for Li plating referred to as HSi/G@liquid (Fig. 5a). However, within the first 20 cycles, these started losing capacity, possibly due to excessive mechanical stress in these thick Si coatings. We also tried to use these anodes in conjunction with Li plating to achieve 800 mA h g−1, but these immediately lost their capacity referred to as HLi/Si/G@gel (Fig. 5a). Note that all the first formation cycles of Li/LSi/G@gel, HSi/G@liquid, and HLi/Si/G@gel are provided in Fig. S10 (ESI†).
To further analyse the operation and degradation of our electrodes, electrochemical impedance spectroscopy (EIS) was recorded after the formation, 20th, and 50th cycle for Li/G@gel and Li/Si/G@gel electrodes as shown in Fig. 5c–e. It is challenging to convert the EIS spectra into an accurate equivalent circuit because three different redox mechanisms are taking place in our electrodes. Hence, we simply report a series resistance (Rs) and a combined SEI and charge transfer resistance (RSEI+CT) (see the Supplementary Note, ESI† for the detailed method). The Rs of Li/G@gel and Li/Si/G@gel after formation are 3.4 and 3.2 Ω, respectively, and the RSEI+CT of Li/G@gel and Li/Si/G@gel after formation are 186 and 105 Ω, respectively. After 20 cycles, the Rs of Li/G@gel and Li/Si/G@gel increase to 4.2 and 4.5 Ω respectively, and the RSEI+CT decreases to 28 and 24 Ω respectively. These decreases in resistance are typically attributed to activation and stabilization of the cycling process. After 50 cycles, clear signs of degradation set in with the Rs and RSEI+CT of Li/G@gel electrode increasing to 20.6 and 242 Ω respectively, while Li/Si/G@gel electrodes retain 8.3 Ω and 51 Ω respectively. The EIS might indicate a lower amount of SEI formation in Li/Si/G@gel electrodes, which is in agreement with observations made in cross-sectional SEM images shown in Fig. 2 and Fig. S5 (ESI†).
Finally, we tested full cells with commercial LiCoO2 cathodes (Fig. S11, ESI†). After the formation cycle, the cells were cycled at a rate of 0.5C (Fig. S11, ESI†), showing energy retentions of Li/G@liquid and Li/Si/G@gel at 50th cycle of 62.5 and 74.5%, respectively. Note that these results were achieved with N/P ratios of 1.05 which is remarkable for Li metal anodes, where typically much higher N/P ratios over 1.5 are utilized, wasting significant anode capacity.10,23 These high N/P ratios mean that the anode capacity is 50% larger than what the cathode can deliver, and whilst these over-dimensioned anodes help stabilising the battery, they drastically reduce the energy density of the overall cell which we avoid here. Table S1 (ESI†) summarizes the electrochemical performances of our cells compared to recent leading Li-metal anode papers.10,11,23 We have not found any paper listing reporting all the metrics listed in our table, which reflects the fact that several technical problems remain to be solved for Li plating batteries. Note that specific energy and energy density can vary depending on whether the whole weight and volume of miscellaneous components are considered. Further, specific energy and power density of batteries are different depending on the test conditions, cell size and type, which can vary from one research institution to another. The N/P ratio for charging is 1.048 and discharging N/P ratio is 1.053 (see the Supplementary Note 3, ESI† for detailed full cell design). As a result, the full cell of Li/G@liquid and Li/Si/G@gel shows gravimetric capacities and ICE of 118.8 mA h g−1 (85.9%) and 126.5 (87.8%) respectively based on both weight of cathode and anode materials (Fig. S11, ESI†). In the case of Li/Si/G@gel, the specific energy is 474 W h kg−1 and energy density 912 W h L−1, as shown in Table S1 (ESI†). Compared to recent Li based anode publications, our works showed remarkably high ICE in half cell test, and in addition, energy densities and specific energy considerably higher than commercial graphite anodes was achieved.
Conclusions
Industrial development of graphite anodes has resulted in electrodes with a very impressive performance combining high specific capacity, mass loading, and electrode density (360 mA h g−1, 3.3 mA h cm−2, and 570 mA h cm−3). Many reports improve one of these metrics at the cost of others, which often make these batteries unsuitable for commercial applications. In this paper, we leverage the industrial research and development work that went in the reliable manufacture of high areal capacity and density graphite anodes, and show these can be hybridised with emerging academic developments in Li metal anodes to develop high performance anodes that combine the best properties of both technologies. This is achieved by conformally coating a Si nanolayer on graphite that results in a homogeneous plating process. The electrodes achieved specific capacity, mass loading, and electrode density of 551 mA h g−1, 3.35 mA h cm−2 and 656 mA h cm−3 respectively while maintaining high ICEs of 91.8% and stable capacity retention over 200 cycles in half cell. We demonstrate that these can operate in full cells with an N/P ration of 1.05, and specific energies of 474 W h kg−1 and 912 W h L−1, which is a step forward in Li metal anode performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by IUK (UKRI: 104174), the EPSRC Graphene CDT (EP/L016087/1) and an ERC Consolidator Grant (MIGHTY-866005). Also, finical support from the 2020 Research Funds (1.200029.1) of Ulsan National Institute of Science and Technology (UNIST) is greatly acknowledged.Notes and references
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 Search PubMed.
- C. C. Fang, J. X. Li, M. H. Zhang, Y. H. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Y. C. Yang, B. Y. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. F. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 Search PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J. G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 Search PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 Search PubMed.
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 16114 Search PubMed.
- C. Yang, K. Fu, Y. Zhang, E. Hitz and L. Hu, Adv. Mater., 2017, 29, 1701169 Search PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2018, 3, 22–29 Search PubMed.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L. Q. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 Search PubMed.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 Search PubMed.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 Search PubMed.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J. G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 Search PubMed.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J. G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 Search PubMed.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 Search PubMed.
- W. Tang, X. Yin, S. Kang, Z. Chen, B. Tian, S. L. Teo, X. Wang, X. Chi, K. P. Loh, H. W. Lee and G. W. Zheng, Adv. Mater., 2018, 30, 1801745 Search PubMed.
- M. S. Kim, J. H. Ryu, Deepika, Y. R. Lim, I. W. Nah, K. R. Lee, L. A. Archer and W. Il Cho, Nat. Energy, 2018, 3, 889–898 Search PubMed.
- Z. Lu, Q. Liang, B. Wang, Y. Tao, Y. Zhao, W. Lv, D. Liu, C. Zhang, Z. Weng, J. Liang, H. Li and Q. H. Yang, Adv. Energy Mater., 2019, 9, 1803186 Search PubMed.
- E. Cha, M. D. Patel, J. Park, J. Hwang, V. Prasad, K. Cho and W. Choi, Nat. Nanotechnol., 2018, 13, 337–343 Search PubMed.
- F. Wu and G. Yushin, Energy Environ. Sci., 2017, 10, 435–459 Search PubMed.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 Search PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 Search PubMed.
- G. Li, Z. Liu, Q. Huang, Y. Gao, M. Regula, D. Wang, L. Q. Chen and D. Wang, Nat. Energy, 2018, 3, 1076–1083 Search PubMed.
- D. Lin, Y. Liu, Z. Liang, H. W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 Search PubMed.
- C. Niu, H. Pan, W. Xu, J. Xiao, J. G. Zhang, L. Luo, C. Wang, D. Mei, J. Meng, X. Wang, Z. Liu, L. Mai and J. Liu, Nat. Nanotechnol., 2019, 14, 594–601 Search PubMed.
- P. Shi, T. Li, R. Zhang, X. Shen, X. B. Cheng, R. Xu, J. Q. Huang, X. R. Chen, H. Liu and Q. Zhang, Adv. Mater., 2019, 31, 1807131 Search PubMed.
- C. B. Jin, O. W. Sheng, J. M. Luo, H. D. Yuan, C. Fang, W. K. Zhang, H. Huang, Y. P. Gan, Y. Xia, C. Liang, J. Zhang and X. Y. Tao, Nano Energy, 2017, 37, 177–186 Search PubMed.
- Z. Liang, D. Lin, J. Zhao, Z. Lu, Y. Liu, C. Liu, Y. Lu, H. Wang, K. Yan, X. Tao and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2862–2867 Search PubMed.
- Y. F. Li, Y. H. Shi, S. G. Wang, J. H. Liu, J. Lin, Y. Xia, X. L. Wu, C. Y. Fan, J. P. Zhang, H. M. Xie, H. Z. Sun and Z. M. Su, Adv. Energy Mater., 2019, 9, 1803690 Search PubMed.
- M. Wang, Z. Peng, W. Luo, F. Ren, Z. Li, Q. Zhang, H. He, C. Ouyang and D. Wang, Adv. Energy Mater., 2019, 9, 1802912 Search PubMed.
- S. Chae, S. H. Choi, N. Kim, J. Sung and J. Cho, Angew. Chem., Int. Ed., 2020, 59, 110–135 Search PubMed.
- T. Waldmann, B. I. Hogg and M. Wohlfahrt-Mehrens, J. Power Sources, 2018, 384, 107–124 Search PubMed.
- C. Uhlmann, J. Illig, M. Ender, R. Schuster and E. Ivers-Tiffée, J. Power Sources, 2015, 279, 428–438 Search PubMed.
- J. Luo, C. E. Wu, L. Y. Su, S. S. Huang, C. C. Fang, Y. S. Wu, J. Chou and N. L. Wu, J. Power Sources, 2018, 406, 63–69 Search PubMed.
- D. S. Kim, D. J. Chung, J. Bae, G. Jeong and H. Kim, Electrochim. Acta, 2017, 258, 336–342 Search PubMed.
- D. S. Kim, Y. E. Kim and H. Kim, J. Power Sources, 2019, 422, 18–24 Search PubMed.
- C. Martin, M. Genovese, A. J. Louli, R. Weber and J. R. Dahn, Joule, 2020, 4, 1296–1310 Search PubMed.
- K. Yan, Z. Lu, H. W. Lee, F. Xiong, P. C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 Search PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 Search PubMed.
- W. Guoping, Z. Bolan, Y. Min, X. Xiaoluo, Q. Meizheng and Y. Zuolong, Solid State Ionics, 2005, 176, 905–909 Search PubMed.
- Q. Liu, C. Du, B. Shen, P. Zuo, X. Cheng, Y. Ma, G. Yin and Y. Gao, RSC Adv., 2016, 6, 88683–88700 Search PubMed.
- H. Kim, M. Seo, M. H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 Search PubMed.
- M. Ko, S. Chae, J. Ma, N. Kim, H. W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 16113 Search PubMed.
- F. F. Shi, A. Pei, A. Vailionis, J. Xie, B. F. Liu, J. Zhao, Y. J. Gong and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12138–12143 Search PubMed.
- A. Kushima, K. P. So, C. Su, P. Bai, N. Kuriyama, T. Maebashi, Y. Fujiwara, M. Z. Bazant and J. Li, Nano Energy, 2017, 32, 271–279 Search PubMed.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 Search PubMed.
- J. K. Lee, C. Oh, N. Kim, J. Y. Hwang and Y. K. Sun, J. Mater. Chem. A, 2016, 4, 5366–5384 Search PubMed.
- Y. S. Kim, Y. G. Cho, D. Odkhuu, N. Park and H. K. Song, Sci. Rep., 2013, 3, 1917 Search PubMed.
- Y. G. Cho, C. Hwang, D. S. Cheong, Y. S. Kim and H. K. Song, Adv. Mater., 2019, 31, 1804909 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02230f |
| This journal is © The Royal Society of Chemistry 2020 |