
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoted oxygen reduction kinetics on nitrogen-doped hierarchically porous carbon by engineering proton-feeding centers†
Guangbo
Chen‡
 a,
Tao
Wang‡
b,
Pan
Liu
c,
Zhongquan
Liao
d,
Haixia
Zhong
a,
Gang
Wang
a,
Panpan
Zhang
a,
Minghao
Yu
a,
Ehrenfried
Zschech
d,
Mingwei
Chen
e,
Jian
Zhang
*f and
Xinliang
Feng
*a
a,
Tao
Wang‡
b,
Pan
Liu
c,
Zhongquan
Liao
d,
Haixia
Zhong
a,
Gang
Wang
a,
Panpan
Zhang
a,
Minghao
Yu
a,
Ehrenfried
Zschech
d,
Mingwei
Chen
e,
Jian
Zhang
*f and
Xinliang
Feng
*a
aCenter for Advancing Electronics Dresden (Cfaed) and Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: xinliang.feng@tu-dresden.de
bSUNCAT Center for Interface Science and Catalysis, Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
cSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 200230 Shanghai, P. R. China
dFraunhofer Institute for Ceramic, Technologies and Systems (IKTS), 01109 Dresden, Germany
eDepartment of Materials Science and Engineering, Johns Hopkins University, Baltimore, MD 21218, USA
fDepartment of Applied Chemistry, School of Applied and Natural Sciences, Northwestern Polytechnical University, Xi'an, 710129, P. R. China. E-mail: zhangjian@nwpu.edu.cn
First published on 13th July 2020
Abstract
Electrocatalytic oxygen reduction reaction (ORR) is the vital process for next-generation electrochemical energy storage and conversion technologies, e.g., metal–air batteries and fuel cells. During the ORR, the O2* and O* intermediates principally combine with protons to form OOH* and OH* species, respectively, which are the proton-coupled electron transfer processes. Unfortunately, under alkaline conditions, the protons are essentially generated from the sluggish water dissociation process, which unavoidably limits the ORR kinetics. Herein, we design and synthesize a nitrogen-doped hierarchically porous carbon with homogeneously distributed ultrafine α-MoC nanoparticles (α-MoC/NHPC) as a model electrocatalyst. Theoretical investigations unveil that α-MoC on NHPC could efficiently reduce the energy barrier of the water dissociation process to generate protons, eventually promoting the proton-coupled ORR kinetics. In a 0.1 M KOH aqueous solution, α-MoC/NHPC exhibits excellent ORR performance with a high half-wave potential of 0.88 V (VS. reversible hydrogen electrode), which outperforms those for NHPC and commercial Pt/C. Moreover, as the air electrode in a zinc-air battery, α-MoC/NHPC presents a large peak power density of 200.3 mW cm−2 and long-term stability. Thereby, our approach to engineering proton-feeding centers paves a new avenue towards the understanding of ORR kinetics and the development of high-performance ORR electrocatalysts.
Broader contextTo enable large-scale commercialization of metal–air batteries, low-cost yet high-performance cathode catalysts for the sluggish oxygen reduction reaction (ORR) are urgently needed. During the ORR, the O2* and O* intermediates generally react with protons for the formation of OOH* and OH* species, respectively. Unfortunately, under alkaline conditions, the protons are principally generated from the sluggish water dissociation process, which unavoidably limits the ORR kinetics. In this work, by engineering the water dissociation centers on a model ultrafine α-MoC nanoparticles on nitrogen-doped hierarchically porous carbon electrocatalyst, we demonstrate the significant role of the water dissociation process in proton-feeding and enhancing ORR kinetics under alkaline environment. The viable design of the proton-feeding centers and the profound understanding of the ORR kinetics in this work will open up a new approach for exploring low cost and highly-active electrocatalysts for catalytic energy conversion reactions. |
The ever-increasing detrimental effects of traditional fuels on energy and environment concerns have stimulated extensive efforts for developing green and renewable energy technologies. Electrochemical oxygen reduction reaction (ORR) plays a significant role in next-generation sustainable energy storage and conversion systems, e.g., metal–air batteries and fuel cells.1–5 Up to now, platinum (Pt)-based catalysts still remain as the benchmark electrocatalysts for catalyzing the ORR.6,7 Unfortunately, the high cost, scarcity and poor durability of Pt seriously hinder its widespread utilization in practical energy conversion systems.8–10
To develop efficient and earth-abundant alternatives to the Pt as ORR electrocatalysts, the profound understanding on the ORR reaction mechanism is highly desirable.11 In alkaline solutions, the ORR principally proceeds according to the ever-proposed four-electron transfer pathways: (1) O2(g) + * → O2*; (2) O2* + H2O(l) + e− → OOH* + OH−; (3) OOH* + e− → O* + OH−; (4) O* + H2O(l) + e− → OH* + OH− and (5) OH* + e− → OH− + *, where * denotes the active site.12 In steps (2) and (4), the O2* and O* intermediates combine with protons to produce OOH* and OH*, respectively, which are the proton-coupled electron transfer (PCET) processes.13 Under alkaline conditions, protons intrinsically originate from the dissociation of water molecules (H2O → H* + OH*).14,15 The energy barrier of water dissociation in alkaline solutions undoubtedly determines the proton-feeding rates and overall ORR kinetics. Lately, nitrogen-doped hierarchically porous carbons (NHPCs) has emerged as appealing ORR catalysts alternative to the Pt owing to their low cost, earth abundance, and good electrochemical stability.16–18 Unfortunately, the water dissociation kinetic barrier on such NHPCs is extremely high,19,20 resulting in sluggish proton-feeding ability and low ORR activity in alkaline media (half-wave potential (E1/2): <0.85 V vs. reversible hydrogen electrode (RHE)).21–23 Recently, reported theoretical and experimental results disclose that α-MoC has superior water dissociation ability.24,25
In this work, by engineering the water dissociation centers on a model ultrafine α-MoC nanoparticles (NPs) on NHPC (α-MoC/NHPC) electrocatalyst, we demonstrate the significant role of water dissociation process in proton-feeding and enhancing ORR kinetics under alkaline environment. The α-MoC NPs with a diameter of ∼3 nm were in situ synthesized on NHPC by pyrolyzing a mixture of NaCl and phosphomolybdic acid/zeolitic imidazolate framework-8 (Mo12/ZIF-8) precursors. Density functional theory (DFT) calculations reveal that the α-MoC NPs in α-MoC/NHPC could effectively reduce the kinetic energy barrier of water dissociation and thereby provide plentiful protons for accelerating the PCET steps, eventually promoting the ORR kinetics in alkaline solutions (Scheme 1). As a result, α-MoC/NHPC electrocatalyst exhibits excellent ORR activity with an E1/2 of as high as 0.88 V in a 0.1 M KOH aqueous solution, which surpasses those values for NHPC (E1/2 = 0.84 V), Pt/C catalyst (E1/2 = 0.85 V) and previously reported metal-free electrocatalysts. Moreover, as the air electrode in an as-assembled Zn–air battery, α-MoC/NHPC demonstrates an extremely high peak power density of 200.3 mW cm−2, which is much higher than that for Pt/C (154.1 mW cm−2).
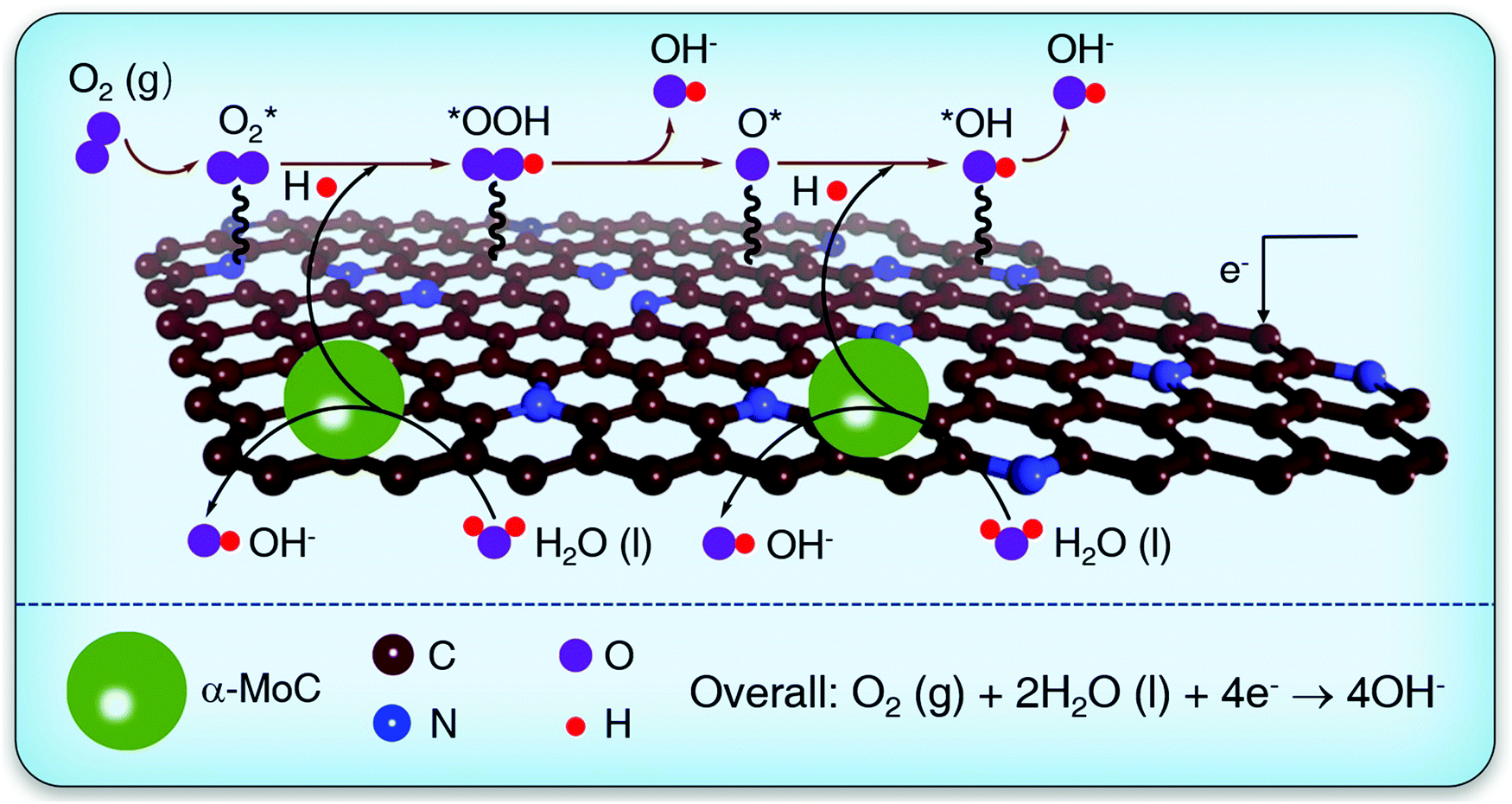 | ||
| Scheme 1 Schematic illustration for water dissociation step and the subsequent elementary processes for ORR in alkaline solutions over an α-MoC/NHPC electrocatalyst. | ||
As schematically illustrated in Fig. 1a, α-MoC/NHPC electrocatalyst was synthesized by using a NaCl-assisted pyrolysis of Mo12/ZIF-8 precursors. First, the Mo12/ZIF-8 was prepared via co-precipitation of Zn(NO3)2·6H2O (2.97 g), 2-methylimidazole (3.28 g) in a methanol solution (160 mL) containing phosphomolybdic acid (Mo12, 60 mg) at room temperature for 24 h (Fig. S1 and S2, ESI†). Second, the achieved Mo12/ZIF-8 (100 mg) was physically mixed with NaCl (35 mg) and then heated at 900 °C for 2 hours under nitrogen atmosphere. Here, NaCl served as templates and intercalating agents for the formation of hierarchically porous carbon nanostructures.26 After washing and drying, α-MoC/NHPC with an α-MoC loading amount of 5.0 wt% was obtained. For comparison, α-MoC/NHPCs with different loading amounts of α-MoC (α-MoC: 1.4, 2.4, 5.0, 6.8 and 8.1 wt%) while with a similar NP size (∼3 nm) were fabricated by changing the dosage of Mo12 (Fig. S3–S8, ESI†). Moreover, NHPC was prepared without the utilization of Mo12 (Fig. S9, ESI†).
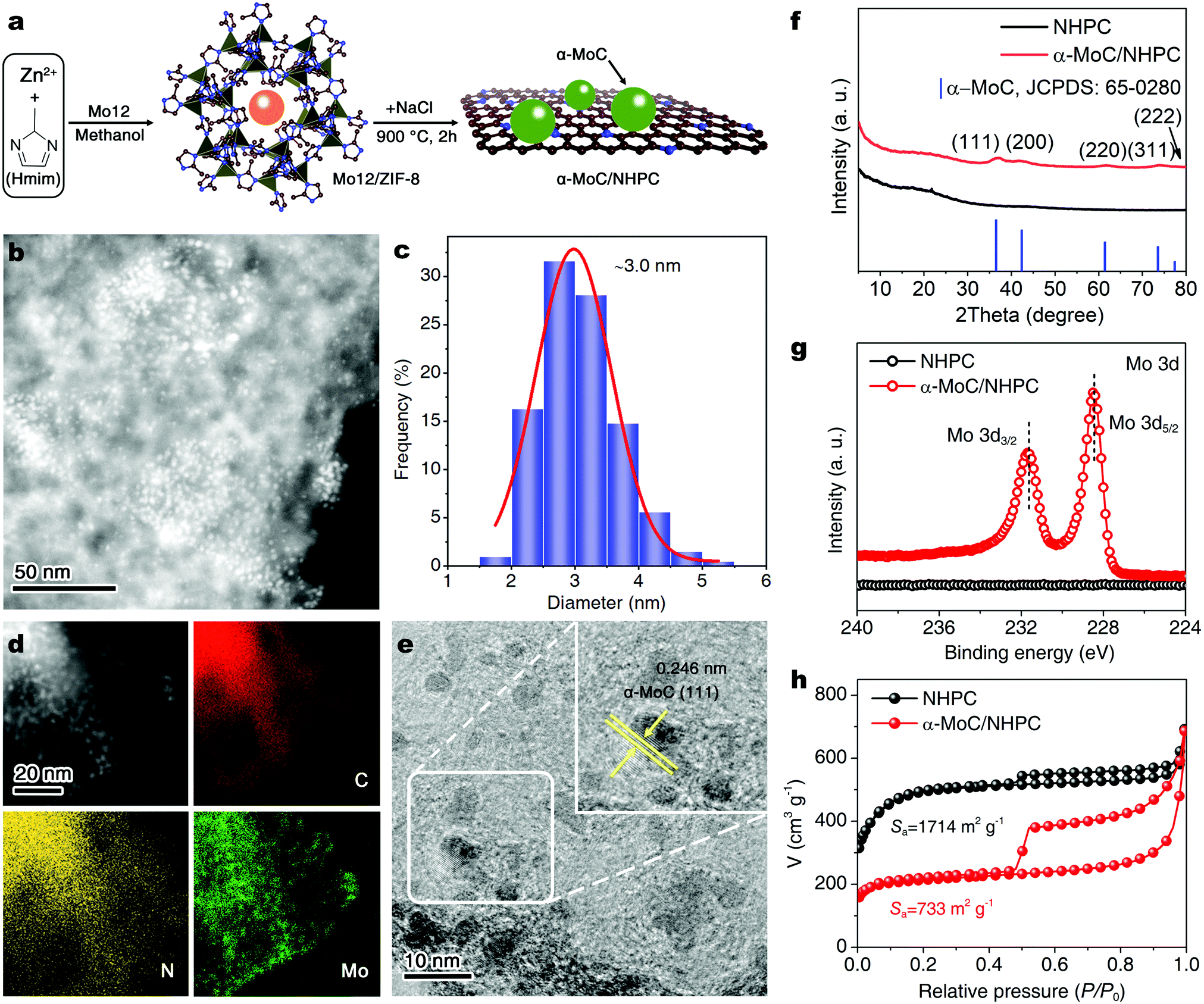 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of α-MoC/NHPC. (b) HAADF-STEM image of α-MoC/NHPC and (c) the corresponding α-MoC NPs size distribution. (d) Elemental mapping images showing the distribution of C (red), N (yellow), and Mo (green) elements. (e) HRTEM image of α-MoC/NHPC. (f) XRD patterns, (g) high-resolution Mo 3d XPS spectra, (h) N2 adsorption/desorption isotherms of NHPC and α-MoC/NHPC. | ||
The morphologies of resultant α-MoC/NHPC were firstly investigated using transmission electron microscopy (TEM). As shown in Fig. 1b and c, the α-MoC NPs with a mean diameter of 3.0 nm are uniformly decorated on the NHPC. The high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) and corresponding energy-dispersive X-ray spectroscopy (EDX) elemental mapping images reveal the distributions of C, N and Mo elements over the α-MoC/NHPC (Fig. 1d). The NPs show a homogeneous distribution of Mo element. High-resolution TEM (HRTEM) image displays clear lattice fringes with an interplanar spacing of 0.246 nm, corresponding to the (111) lattice plane of α-MoC (Fig. 1e and Fig. S10, ESI†). The crystalline structure of the achieved α-MoC/NHPC was further confirmed by X-ray diffraction (XRD) analysis. As disclosed in Fig. 1f, the diffraction peaks at 36.4°, 42.3°, 61.3°, 73.4° and 77.3° are indexed to the characteristic (111), (200), (220), (311) and (222) facets of the α-MoC (JCPDS No. 65-0280), respectively.24,25 The Raman spectrum of α-MoC/NHPC shows two peaks at 1350 and 1590 cm−1 (Fig. S11, ESI†), which are assigned to disordered sp3 carbon (D band) and graphitic sp2 carbon (G band) of NHPC, respectively.27
To gain insights into the surface chemical composition and elemental bonding configurations of the achieved α-MoC/NHPC and NHPC, X-ray photoelectron spectroscopy (XPS) analyses were conducted. As illustrated in Fig. S12 (ESI†), the survey spectrum of α-MoC/NHPC confirms the presence of C, N and Mo. High-resolution N 1s spectra of α-MoC/NHPC and NHPC can be well fitted into four characteristic peaks of pyridinic N (398.5 eV), pyrrolic N (400.3 eV), graphitic N (401.2 eV) and oxidized N (402.7 eV)28,29 (Fig. S13, ESI†). The pyridinic N content in α-MoC/NHPC is approximately 4.8 at%, which is lower than 5.5 at% for NHPC (Table S1, ESI†). In the high-resolution Mo 3d XPS spectrum of α-MoC/NHPC, the binding energies of Mo 3d5/2 and Mo 3d3/2 located at 228.4 eV and 231.6 eV are attributed to Mo2+ (Fig. 1g).24,25 The α-MoC content in α-MoC/NHPC was determined to be 5.0 wt% based on the inductively coupled plasma mass spectrometry (ICP-MS) analysis. The elemental analysis results reveal that the C and N contents in α-MoC/NHPC are 81.6 at% and 6.7 at%, respectively. (Table S1, ESI†). Next, N2 sorption analysis was carried out to assess the textural properties of the achieved NHPC and α-MoC/NHPC. The Barrett–Emmett–Teller (BET) surface area of α-MoC/NHPC was measured to be 733 m2 g−1, which was much smaller than 1714 m2 g−1 for NHPC (Fig. 1h). A detailed pore structure analysis reveals that α-MoC/NHPC shows a less microporous/mesoporous structure in comparison with that of NHPC, attributable to the partial blockage of pores by the α-MoC NPs (Fig. S14, ESI†). The specific micropore and mesopore surface areas of α-MoC/NHPC are 526 m2 g−1 and 143 m2 g−1, respectively.
The electrocatalytic ORR performances of α-MoC/NHPC were investigated by linear sweep voltammetry (LSV) in an O2-saturated 0.1 M KOH aqueous solution. By contrast, a commercial Pt/C (20% Pt, Fuelcellstore) and NHPC were also evaluated. All potentials were referenced to the RHE. As shown in Fig. 2a, the α-MoC/NHPC demonstrated excellent ORR activity with an onset potential of 0.98 V. The E1/2 of α-MoC/NHPC reached 0.88 V, which was higher than those for Pt/C (0.85 V), NHPC (0.84 V) and the previously reported ORR electrocatalysts, such as, nitrogen-doped three-dimensional graphene nanoribbon networks (N-GRW, E1/2 = 0.84 V),30 three-dimensional nitrogen and phosphorus co-doped mesoporous carbon (NPMC, E1/2 = 0.85),31 single-atom Co supported by porous nitrogen-doped carbon nanospheres (Co-ISAS/p-CN, E1/2 = 0.838 V),32 single atomic Cu in ultrathin nitrogenated carbon nanosheets (Cu–N–C, E1/2 = 0.85 V),33 atomically dispersed Fe atoms anchored on porous N- and S-codoped carbon framework (Fe SAs/NSC, E1/2 = 0.87 V).34 Meanwhile, the kinetic current density (Jk) of α-MoC/NHPC was up to 33.8 mA cm−2 at 0.80 V, which was considerably larger than those of NHPC (17.8 mA cm−2) and Pt/C (21.4 mA cm−2) (Fig. 2b and Table S2, ESI†). The corresponding Tafel slope of α-MoC/NHPC was determined to be ∼47.8 mV decade−1, which was much lower than 83.2 mV decade−1 for Pt/C and 56.1 mV decade−1 for NHPC, demonstrating an accelerated ORR kinetics on α-MoC/NHPC electrocatalyst (Fig. 2c). Moreover, the α-MoC/NHPC electrocatalyst also reveals superior selectivity toward OH− formation via a four-electron dominant transfer pathway and superb stability (Fig. S15–S19, ESI†).
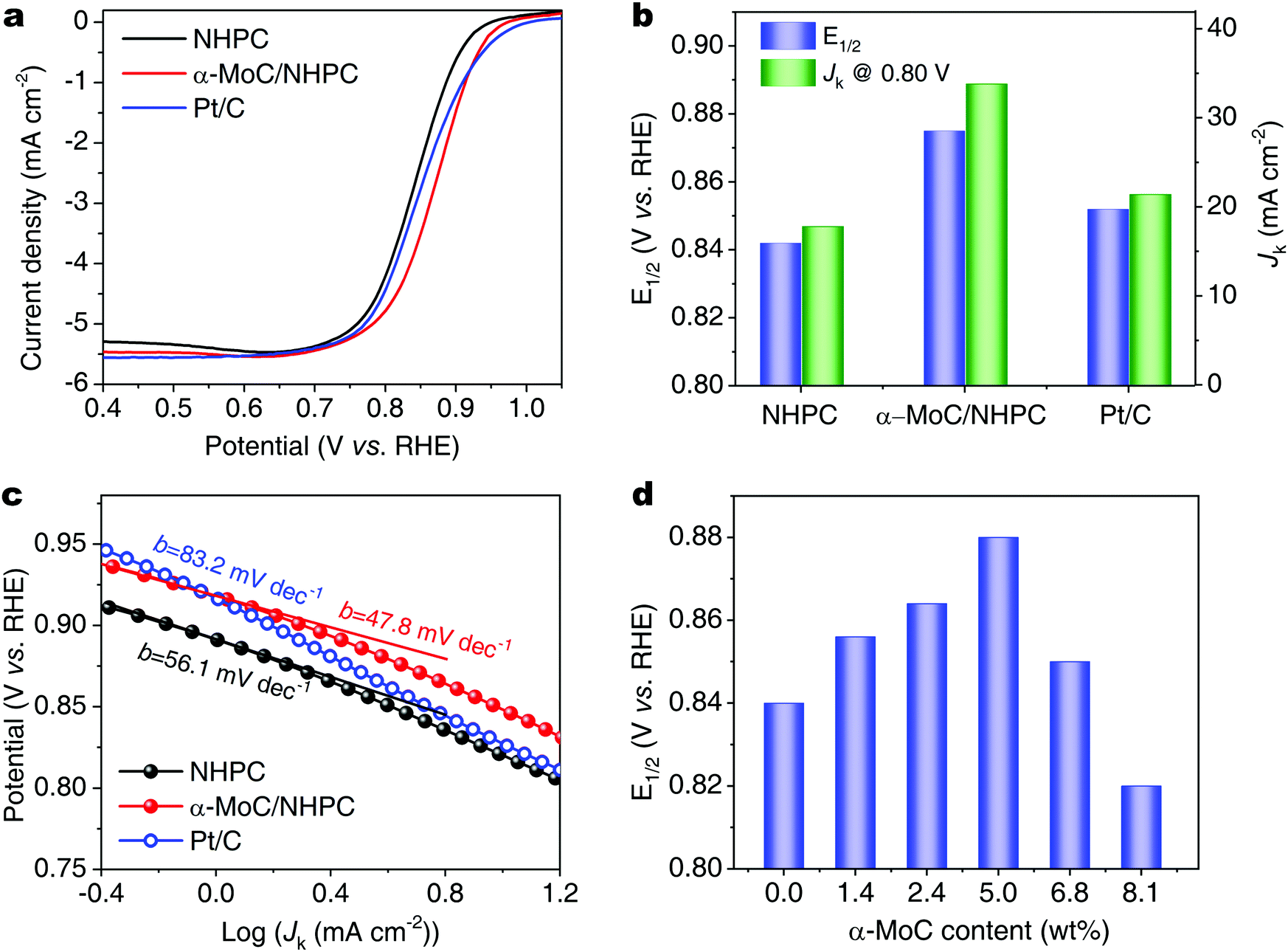 | ||
| Fig. 2 (a) ORR polarization curves, (b) Jk at 0.80 V and E1/2, and (c) corresponding Tafel slopes of NHPC, α-MoC/NHPC and Pt/C. (d) E1/2 of α-MoC/NHPCs with different α-MoC contents. | ||
In order to reveal the key role of α-MoC NPs during the alkaline ORR process, we investigated the ORR performance of a series of α-MoC/NHPCs with different α-MoC loading contents (1.4, 2.4, 5.0, 6.8 and 8.1 wt%) in a 0.1 M KOH aqueous solution. As shown in Fig. S20 (ESI†) and Fig. 2d, the E1/2 of α-MoC/NHPCs increased accompanied by larger α-MoC contents. When the weight content of α-MoC was 5.0 wt%, the α-MoC/NHPC showed the highest E1/2 (0.88 V). Then, the E1/2 of α-MoC/NHPC decreased when increasing the α-MoC content (6.8 wt%, 0.85 V), which was probably due to the blockage of the intrinsic ORR active site of NHPC by the high coverage of α-MoC NPs, as indicated by the reduced electrochemically active surface area (ECSA) and ORR kinetics (Fig. S21 and S22, ESI†).
To further elucidate the fundamental mechanism of the outstanding ORR activity of α-MoC/NHPC, the kinetic energy barrier of the water dissociation step and the subsequent ORR elementary processes were studied using DFT calculations based on the as-built electrocatalyst models including nitrogen doped carbon (NC), α-MoC(111) and α-MoC/NC (Fig. S23, ESI†). Fig. 3a–c present the water adsorption configurations on the as-constructed models. As illustrated in Fig. 3a, H2O can be only physically adsorbed on the NC and is hard to be dissociated, indicating an extremely sluggish water dissociation kinetics for the generation of protons.35 On the α-MoC(111), water is chemically adsorbed and can be dissociated into H* and OH* with an energy barrier of only 0.6 eV (Fig. 3b and Fig. S24, ESI†), suggesting an excellent water dissociation kinetics, which is consistent with the reported results.25 Surprisingly, water molecule demonstrates dissociative adsorption on the α-MoC/NC that can be spontaneously dissociated for the formation of H* and OH*,36 implying a superior property of water dissociation for proton feeding in comparison with those on NC and α-MoC (Fig. 3c and d). Fig. 3e displays the free energy diagram for the elementary steps of ORR at the overpotential of 0 V. For α-MoC, the formation of OOH* and O* species are all exothermic and downhill, evidencing thermodynamically favorable processes (Fig. S25, ESI†). However, the reduction of O* to OH* and the release of OH from the active sites on α-MoC are uphill processes with energy barriers of 1.45 eV and 2.27 eV, respectively, indicating that the pure α-MoC can be easily poisoned by O* and OH* intermediates that block the ORR kinetics. On NC and α-MoC/NC, the C atoms neighboring to N in NC are the real active site due to they demonstrate the most stable adsorption for reaction intermediates (OOH*, O* and OH*), according to the systematic DFT calculations (Fig. S26 and S27, ESI†). On NC, the energy barriers for O* to OOH* and O* to OH* are as high as 1.09 eV and 0.57 eV, respectively, which undoubtedly limit the overall ORR kinetics. Notably, on α-MoC/NC, the energy barriers for O* to OOH* and O* to OH* considerably decrease to 0.17 eV and 0.00 eV, respectively, indicating promoted PECT processes, as a result of an accelerated water dissociation kinetics for proton feeding.37
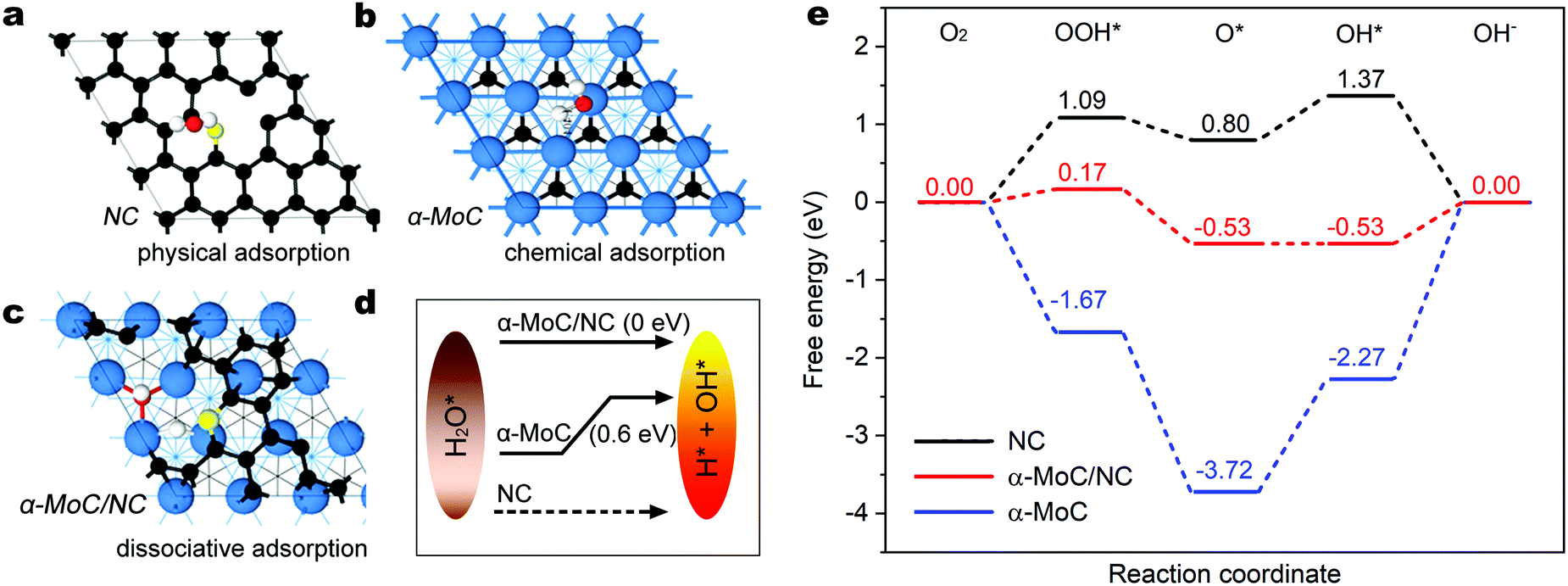 | ||
| Fig. 3 Adsorption configurations of water on (a) NC, (b) α-MoC(111) and (c) α-MoC/NC surfaces. (d) Schematic energy diagrams for water dissociation and (e) free energy diagrams for ORR on NC, α-MoC(111) and α-MoC/NC catalysts at the overpotential of 0 V. | ||
To investigate the practical applicability of the as-developed α-MoC/NHPC electrocatalyst in energy devices, a primary Zn–air battery was assembled utilizing α-MoC/NHPC as the oxygen electrocatalyst on air electrode in a 6.0 M KOH electrolyte containing 0.2 M Zn(OAc)2 (Fig. 4a).38 As depicted in Fig. S28 (ESI†), the open-circuit voltage of the α-MoC/NHPC was 1.44 V. The maximum power density of α-MoC/NHPC was up to 200.3 mW cm−2 (Fig. 4b), which substantially exceeded 154.1 mW cm−2 for Pt/C and those for the previously reported ORR electrocatalysts, e.g., N-GRW (65 mW cm−2),30 NMPC (55 mW cm−2),31 metal-free mesoporous N-doped carbons (192.7 mW cm−2),39 Mn/Fe-HIB-MOF (195 mW cm−2),40 MnO/Co in porous graphitic carbon (172 mW cm−2)41 and PdMo bimetallene (154.2 mW cm−2)42 (Table S3, ESI†). Meanwhile, the α-MoC/NHPC catalyst-based Zn–air battery delivered a specific capacity of 783.9 mA h gZn−1 at 10 mA cm−2, corresponding to a ∼95.6% utilization efficiency of the theoretical capacity (∼820 mA h gZn−1) (Fig. 4c). Furthermore, α-MoC/NHPC electrocatalyst could stably work in a mechanically rechargeable battery by only refueling the consumed zinc anode and electrolyte at the end of each discharge. No noticeable degradation was observed after 10 cycles over a period of 240 hours at a current density of 20 mA cm−2, highlighting the superior durability of α-MoC/NHPC electrocatalyst (Fig. 4d). Such two tandem Zn–air batteries steadily lighted up red light-emitting diodes (LEDs) in series more than ten days, promising their potential utilization for powering electronic devices (Fig. 4e).
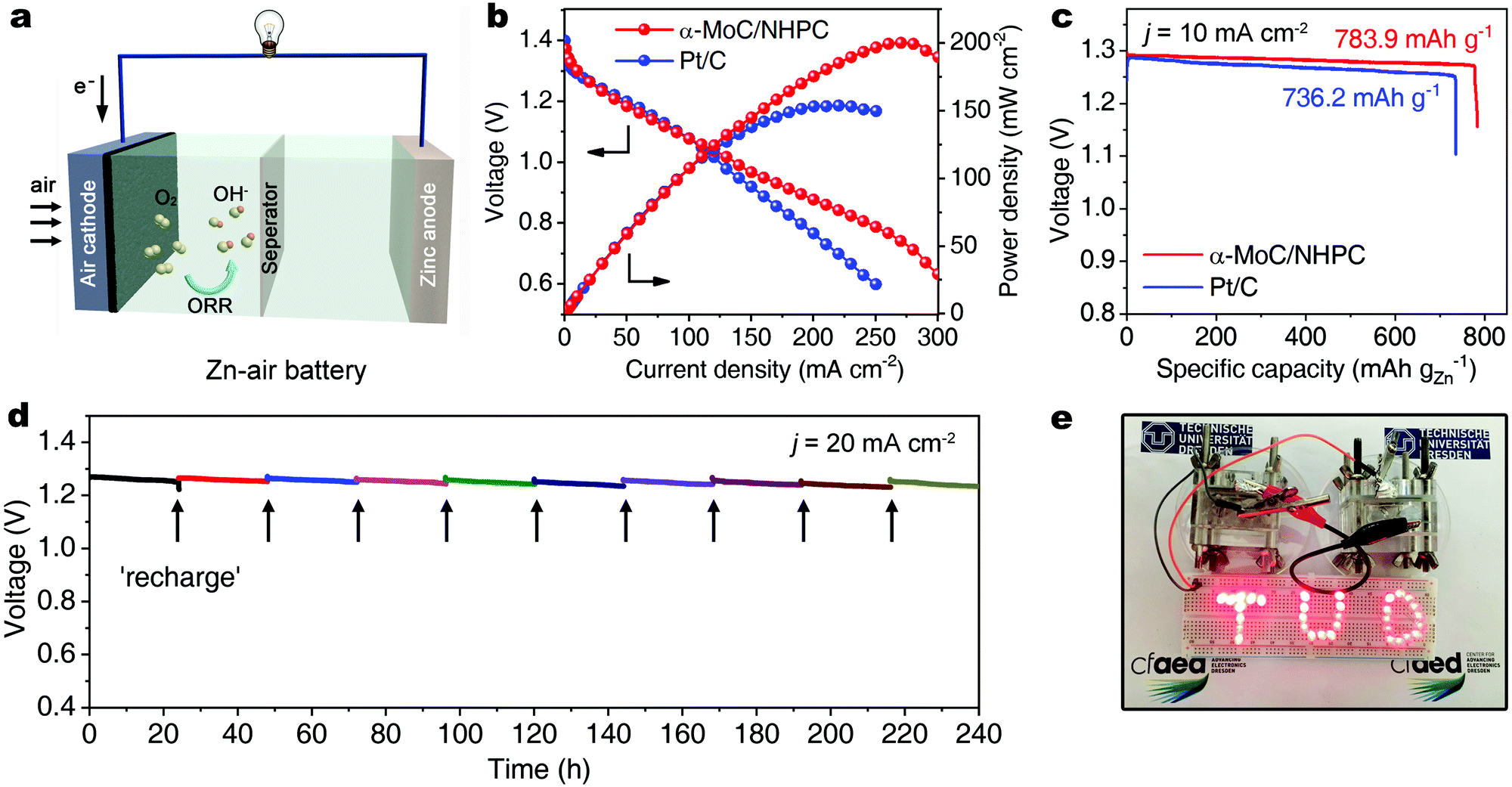 | ||
| Fig. 4 (a) Schematic illustration of an as-assembled Zn–air battery. (b) Discharge polarization curves and corresponding power density plots of the primary Zn–air batteries using α-MoC/NHPC and Pt/C as air electrode, respectively. (c) Long-time galvanostatic discharge curves of a Zn–air battery with α-MoC/NHPC as cathode catalyst until complete consumption of Zn anode. The specific capacity was normalized to the mass of consumed Zn. (d) Discharge plot of the Zn–air battery with α-MoC/NHPC as cathode catalyst by replenishing Zn anode and electrolyte. (e) Photograph of red LEDs in series powered by two tandem Zn–air batteries. | ||
Conclusions
In conclusion, we demonstrate that the water dissociation plays an essential role for proton-feeding and eventually accelerating the ORR kinetics in alkaline solutions by engineering the water dissociation centers on a model α-MoC/NHPC electrocatalyst. As a result, the as-constructed α-MoC/NHPC exhibits excellent ORR activity in alkaline solutions, which is superior to those of the noble metal Pt/C and the state-of-art noble metal-free electrocatalysts. Therefore, the viable design of the proton-feeding centers and the profound understanding of the ORR kinetics not only provide a promising alternative ORR catalyst to the Pt, but also open up a new window for exploring low cost and high-activity electrocatalysts for energy-conversion-related catalytic reactions including oxygen reduction, CO2 reduction, water splitting, and nitrogen reduction reaction, etc.Author contributions
G. C., J. Z. and X. F. conceived the idea, designed the experiments and wrote the paper. G. C. synthesized the materials. G. C. and H. Z. conducted the electrocatalytic tests and analyzed the data. T. W. did the DFT calculations. P. L., Z. L., E. Z. and M. C performed the TEM, HRTEM and HAADF-STEM measurements. P. Z. performed the SEM characterizations. G. W. and M. Y. rendered helpful discussions. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 819698 and GrapheneCore3 881603), Deutsche Forschungsgemeinschaft (MX-OSMOPED project) and Coordination Networks: Building Blocks for Functional Systems (SPP 1928, COORNET). We acknowledge the Center for Advancing Electronics Dresden (Cfaed), the Dresden Center for Nanoanalysis (DCN) at TU Dresden. G. Chen thanks China Scholarship Council for the financial support. Prof. J. Zhang acknowledges funding support from Shannxi National Science Foundation (No. 2020JQ-141) and the Fundamental Research Funds for the Central Universities (No. 310201911cx028).References
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed
.
- Y. Jiang, Y.-P. Deng, J. Fu, D. U. Lee, R. Liang, Z. P. Cano, Y. Liu, Z. Bai, S. Hwang, L. Yang, D. Su, W. Chu and Z. Chen, Adv. Energy Mater., 2018, 8, 1702900 CrossRef
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1907399 CrossRef CAS PubMed
- Y.-P. Deng, Y. Jiang, R. Liang, S.-J. Zhang, D. Luo, Y. Hu, X. Wang, J.-T. Li, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 1952 CrossRef CAS PubMed
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed
- M. Liu, L. Wang, K. Zhao, S. Shi, Q. Shao, L. Zhang, X. Sun, Y. Zhao and J. Zhang, Energy Environ. Sci., 2019, 12, 2890–2923 RSC
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed
- Y.-P. Deng, R. Liang, G. Jiang, Y. Jiang, A. Yu and Z. Chen, ACS Energy Lett., 2020, 5, 1665–1675 CrossRef CAS
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- Y. Zheng, Y. Jiao, A. Vasileff and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS
- J. Zhang, G. Chen, K. Müllen and X. Feng, Adv. Mater., 2018, 30, 1800528 CrossRef PubMed
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC
- C. Lu, D. Tranca, J. Zhang, F. N. Rodríguez Hernández, Y. Su, X. Zhuang, F. Zhang, G. Seifert and X. Feng, ACS Nano, 2017, 11, 3933–3942 CrossRef CAS PubMed
- W. Han, L. Chen, B. Ma, J. Wang, W. Song, X. Fan, Y. Li, F. Zhang and W. Peng, J. Mater. Chem. A, 2019, 7, 4734–4743 RSC
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 31, 1806403 CrossRef PubMed
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, 1805121 CrossRef PubMed
- S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu, Y. Ye, L. Lin, X. Wen, P. Liu, B. Chen, E. Crumlin, J. Guo, Z. Zuo, W. Li, J. Xie, L. Lu, C. J. Kiely, L. Gu, C. Shi, J. A. Rodriguez and D. Ma, Science, 2017, 357, 389–393 CrossRef CAS PubMed
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80 CrossRef CAS PubMed
- N. Fechler, T.-P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS PubMed
- L. Tao, M. Qiao, R. Jin, Y. Li, Z. Xiao, Y. Wang, N. Zhang, C. Xie, Q. He and D. Jiang, Angew. Chem., Int. Ed., 2019, 58, 1019–1024 CrossRef CAS PubMed
- H.-W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, 1501122 CrossRef PubMed
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed
- A. Han, W. Chen, S. Zhang, M. Zhang, Y. Han, J. Zhang, S. Ji, L. Zheng, Y. Wang, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1706508 CrossRef PubMed
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS
- P. J. Feibelman, Science, 2002, 295, 99–102 CrossRef CAS PubMed
- Q. Yang, Y. Jia, F. Wei, L. Zhuang, D. Yang, J. Liu, X. Wang, S. Lin, P. Yuan and X. Yao, Angew. Chem., Int. Ed., 2020, 59, 6122–6127 CrossRef CAS PubMed
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed
- X. Peng, L. Zhang, Z. Chen, L. Zhong, D. Zhao, X. Chi, X. Zhao, L. Li, X. Lu, K. Leng, C. Liu, W. Liu, W. Tang and K. P. Loh, Adv. Mater., 2019, 31, 1900341 CrossRef PubMed
- S. S. Shinde, C. H. Lee, J.-Y. Jung, N. K. Wagh, S.-H. Kim, D.-H. Kim, C. Lin, S. U. Lee and J.-H. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J.-Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee01613f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |