
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective CO production from aqueous CO2 using a Cu96In4 catalyst and its integration into a bias-free solar perovskite–BiVO4 tandem device†
Motiar
Rahaman
 a,
Virgil
Andrei
a,
Chanon
Pornrungroj
a,
Demelza
Wright
ab,
Jeremy J.
Baumberg
b and
Erwin
Reisner
*a
a,
Virgil
Andrei
a,
Chanon
Pornrungroj
a,
Demelza
Wright
ab,
Jeremy J.
Baumberg
b and
Erwin
Reisner
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk; Web: http://www-reisner.ch.cam.ac.uk Tel: +44-1223336323
bNanoPhotonics Centre, Cavendish Laboratory, Department of Physics, University of Cambridge, J J Thomson Avenue, Cambridge CB3 0HE, UK
First published on 17th August 2020
Abstract
Sunlight-driven production of fuels is attracting attention for the generation of storable renewable energy, but the design of selective catalysts for CO2 utilization and the assembly of unassisted devices for selective and efficient CO2-to-fuel conversion remains challenging. In this study, we report a bimetallic Cu96In4 alloy with a dendritic foam morphology for the reduction of aqueous CO2 to CO at an onset potential of −0.3 V vs. the reversible hydrogen electrode (RHE) and with >70% selectivity. Operando Raman spectroscopy reveals weaker *CO adsorption on the Cu96In4 alloy surface compared to bare Cu and supports the immediate release of CO(g) as the product from the electrocatalyst surface. The Cu96In4 catalyst is subsequently employed in an overall bias-free tandem device for CO2 conversion using water as an electron donor. The buried photovoltaic-biased photoelectrochemical cell relies on state-of-the-art triple-cation mixed halide perovskite and BiVO4 photoabsorbers that can also be assembled in an artificial leaf configuration. The device reaches a solar-to-CO energy conversion efficiency of 0.19% with a selectivity of 75% for CO after 10 h simulated sunlight irradiation using bias-free conditions. The buried perovskite|Cu96In4 cathode shows robust, unaltered PEC activity at different solar intensities, which also allows it to function under low and diffuse sunlight. This study highlights the potential of alloying to improve catalytic performance and strategies to integrate such catalysts into solar-driven PEC devices.
Broader contextStorage of solar energy into chemical energy vectors provides a pathway to a sustainable, circular economy by diminishing the global energy reliance on fossil fuels. Sunlight is the most abundant and exploitable energy source and can power the direct conversion of the greenhouse gas CO2 and H2O into renewable fuels. However, the practical usage of this technology still faces several challenges such as developing efficient catalysts and light harvesters and their combination to produce solar fuels in a selective, scalable, and cost-effective manner. Here, we present a novel approach of interfacing an inexpensive and selective Cu alloy catalyst into a state-of-the-art lead halide perovskite‖BiVO4 tandem device for sunlight-driven CO2-to-CO conversion without the need of any other energy input. Solar fuel generators that synthesize CO are of interest as this product can be converted into liquid hydrocarbon fuels by the Fischer–Tropsch process. The light-driven device shows robust performance towards CO production over a wide range of light intensities, which indicates its compatibility for operation under varying daylight conditions. This prototype presents a proof-of-concept for the integration of selective transition metal catalysts into solar fuel devices and motivates the development of solar reactors for multicarbon products from CO2 in the future. |
Introduction
The conversion of CO2 into value-added products is of contemporary interest for the development of a carbon-neutral energy cycle, but the underlying catalytic reaction is thermodynamically challenging and kinetically complex. Sunlight is an abundant and readily available energy source that can be directly used for the generation of useful molecules from CO2, thereby storing energy in the chemical bonds of the product.1,2 The bio-inspired process of converting CO2 and H2O into energy carriers with sunlight is known as artificial photosynthesis.3–5 Among all the possible CO2 reduction products, CO has particular importance since it can be combined with H2 to form alcohols and long chain hydrocarbons by established industrial processes.6,7 A substantial energy input is required to obtain CO electrochemically, but light-driven CO formation presents an alternative and sustainable way for its production from sunlight.Lead halide perovskites are emerging light absorbers for solar fuel production and can either be indirectly coupled as external photovoltaic cells to an electrolyzer or integrated as encapsulated component in a photoelectrochemical (PEC) device.8–17 The combination of a buried perovskite-biased cathode with a water oxidizing BiVO4 photoanode in a tandem configuration (perovskite‖BiVO4) can perform water splitting under bias-free conditions.18–20 Recently, we have reported a molecular catalyst-based perovskite‖BiVO4 tandem system for unassisted CO2 reduction to syngas using water as an electron donor.21 However, the PEC tandem device showed a limited bias-free solar-to-CO conversion efficiency of 0.02%, with a relatively low CO selectivity (CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio of ∼3:10). Therefore, an improved catalyst and a redesign of the device architecture are required to increase the CO2 reduction activity and selectivity of the PEC tandems.
:H2 ratio of ∼3:10). Therefore, an improved catalyst and a redesign of the device architecture are required to increase the CO2 reduction activity and selectivity of the PEC tandems.
In this study, a bimetallic alloy is employed as an efficient catalyst on an improved perovskite‖BiVO4 tandem to achieve an increased efficiency and selectivity for solar CO2 conversion under bias-free conditions. We report the design of a Cu96In4 alloy with a unique dendritic foam morphology, which confers high selectivity toward CO production at low overpotentials. To improve the onset bias of the PEC tandem device, we also incorporate an additional hole-transporting layer into the perovskite architecture. The newly designed Cu–In alloy catalyst was employed on electrodes, perovskite-biased cathodes, and perovskite‖BiVO4 PEC tandems for bias-free selective CO formation coupled to O2 evolution. Fig. 1a shows the schematic diagram of a standalone tandem device, revealing the perovskite device architecture, metal alloy catalyst morphology, BiVO4 photoanode and their assembly.
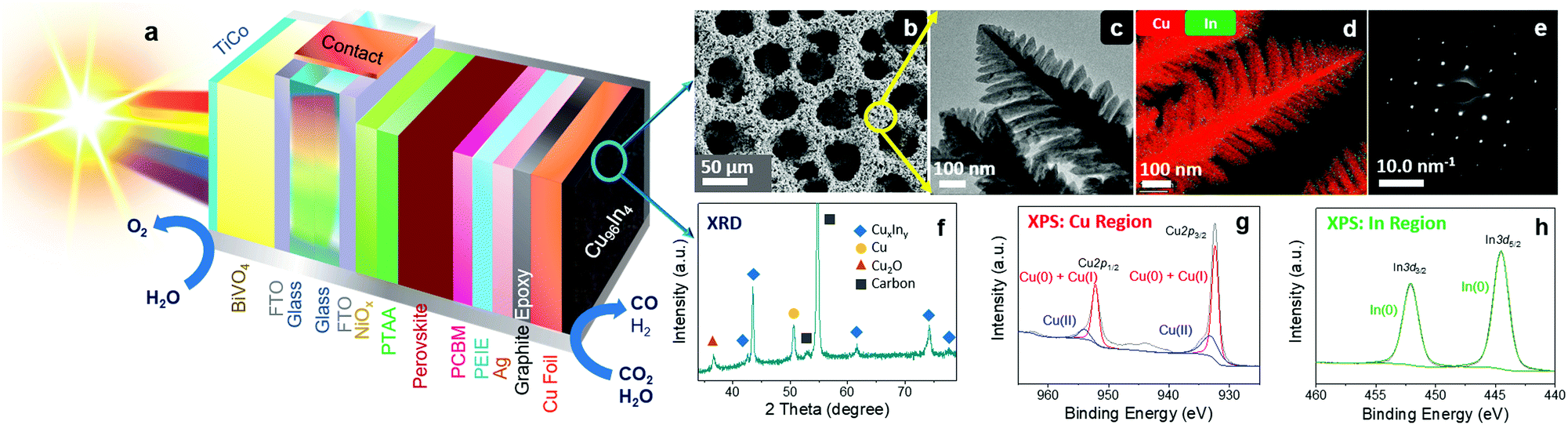 | ||
| Fig. 1 Schematic model illustrating a BiVO4‖perovskite|Cu96In4 tandem device assembled in a leaf configuration (a). The CuIn catalyst is characterized by low-resolution SEM (b), high-resolution TEM (c), STEM mapping (d), SAED pattern analysis (e), powder XRD analysis (f), and XPS analysis in the Cu (g) and In (h) regions. The Cu 2p peaks (g) are deconvoluted into Cu(0) + Cu(I) peaks (red) and a Cu(II) peak (blue) as indicated in the figure. Cu(I) and Cu(II) components are assigned to surface oxide formation due to aerial oxidation. | ||
Results and discussion
Preparation and characterization of the catalyst
Transition metal alloys, especially Cu-based bimetallic systems are well-known as potentially active and inexpensive catalysts for selective electrochemical CO2 conversion.22–25 Here, we have used a template assisted electrodeposition method to prepare a bimetallic copper–indium foam catalyst. Galvanostatic electrodeposition was carried out by applying −3 A cm−2 using a deposition bath containing Cu2+ and In3+ precursors in aqueous H2SO4 (1.5 M), where the co-produced H2 bubbles acted as template for the porous architecture (see Fig. S1 and Methods for details, ESI†).26,27Scanning electron microscopy (SEM) shows a porous structure of the catalyst with an average surface pore size of 35 ± 4.5 μm (Fig. 1b), whereas high resolution transmission electron microscopy (HR-TEM) reveals that the pore side walls consist of dendritic fern-like structures (Fig. 1c). The Cu:In metallic ratio of the alloy was found to be ∼96:4 by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis and energy-dispersive X-ray spectroscopy (EDX) mapping (in both TEM and SEM mode; Fig. S2, ESI†). As In is a scarce element that is commonly used by the electronics industry, we calculated the amount of In present in our catalyst. Gravimetric analyses show that 1 cm2 active area contains around 1.7 mg of catalyst which corresponds to only ∼70 μg of metallic In, considering the bulk composition of Cu and In as 96:4 according to the ICP-OES and EDX analyses. Scanning transmission electron microscopy (STEM) mapping shows a uniform distribution of Cu and In on the branches of the dendrite nanostructures (Fig. 1d). The selected area electron diffraction (SAED) pattern in Fig. 1e indicates a face centered cubic (fcc) lattice system, where a particular preferential crystal plane orientation in the bimetallic polycrystalline material is shown. Powder X-ray diffraction (XRD) analysis of the as-prepared material indicates the presence of a major CuIn alloy phase, as well as some residual Cu and Cu2O (from aerial oxidation) phases in the catalyst (Fig. 1f; JCPDS: 42-1476 and 35-1150 for CuIn alloy, 85-1326 for Cu, and 75-1531 for Cu2O). No surface contamination (traces of other metals) was identified by X-ray photoelectron spectroscopy (XPS) survey analysis (Fig. S3, ESI†). The Cu 2p1/2, Cu 2p3/2 peaks from the Cu region and In 3d3/2, In 3d5/2 peaks from In region are shown in Fig. 1g and h, respectively, which indicates the presence of both metals on the surface of the material. The XPS study also indicates the presence of CuxO components on the surface due to aerial oxidation. However, these residual oxide phases are unstable under cathodic conditions as they are being reduced back to the metallic state when applying a negative potential (see below). We termed the material as a Cu96In4 alloy to indicate the actual bulk composition of Cu and In and also to denote the major active catalytic phase, which is a mixed alloy.
Electrochemical analysis of the catalyst
The Cu96In4 alloy was electrochemically characterized by linear sweep voltammetry (LSV) and controlled potential electrolysis (CPE), using a three electrode configuration with a Pt mesh counter and Ag/AgCl(Satd.NaCl) reference electrode. Fig. 2a shows LSV curves in N2 saturated (green, pH 8.3) and CO2 saturated (red, pH 7.2) 0.5 M aqueous KHCO3 electrolyte solution. The onset potential (at approximately −0.3 V vs. RHE) in CO2 saturated electrolyte solution shows a positive shift compared to that in N2 saturated solution, which indicates that the catalytic activity of the material starts at a low overpotential.
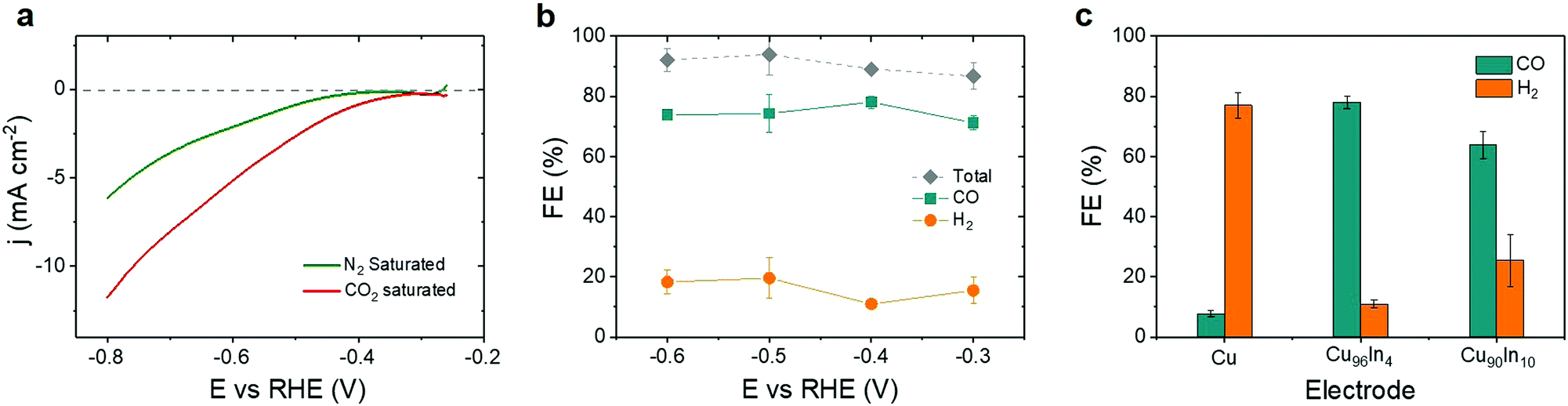 | ||
| Fig. 2 Electrochemistry of the Cu96In4 alloy catalyst: (a) LSV in N2 and CO2 saturated 0.5 M KHCO3 electrolyte at a scan rate of 10 mV s−1, (b) CPE showing the potential dependent faradaic efficiency of CO and H2 on the Cu96In4 catalyst, (c) composition dependent electrochemical performance of pure Cu and different CuxIny alloys at −0.4 V vs. RHE. CPE in (b) and (c) was carried out in CO2 saturated aqueous 0.5 M KHCO3 medium (pH 7.2) for 4 h at room temperature. | ||
CPE under CO2 at applied potentials ranging from −0.3 V to −0.6 V vs. RHE shows CO as the dominant product (Fig. 2b). The faradaic efficiency for CO (FECO) was roughly constant over the applied potential window with a maximum of (78.1 ± 2.1)% at −0.4 V vs. RHE. The remaining charge was consumed for the generation of H2 as a secondary product from proton reduction with an overall combined FE for CO and H2 (FECO+H2) of close to 100% (Table S1, ESI†). Cu is the only metal that can form multicarbon products from CO2, whereas In is known as formate producing metal.28 Selective CO formation from CO2 without any multicarbon products at more negative potentials (−0.6 V vs. RHE) supports the assignment of the active catalyst as a mixed alloy phase. The total amount of gaseous products gradually increased with more negative applied potentials (Fig. S4 and Table S2, ESI†). To study the effect of catalyst composition on the selectivity, CPE was carried out at −0.4 V vs. RHE with pristine Cu, Cu96In4, and Cu90In10 foam (Fig. 2c). Pristine Cu produced H2 with a FE (FEH2) of (77.0 ± 4.2)%, whereas Cu90In10 shows a FECO of (63.8 ± 4.6)% (see Tables S3 and S4 for FEs and amounts of products, Fig. S5 for SEM and EDX analysis of Cu90In10, ESI†). Hence, an optimal catalytic behavior for selective CO production can be achieved by incorporating 4% In into Cu.
The conversion of CO2 to CO in aqueous medium depends on the interaction of the reaction intermediates with the active sites of the catalyst material. It is typically considered to consist of the following elementary steps:29–31
| CO2(g) + H+(aq) + e− + * → *COOH | (1) |
| *COOH + H+(aq) + e− → *CO + H2O | (2) |
| *CO → CO(g) + * | (3) |
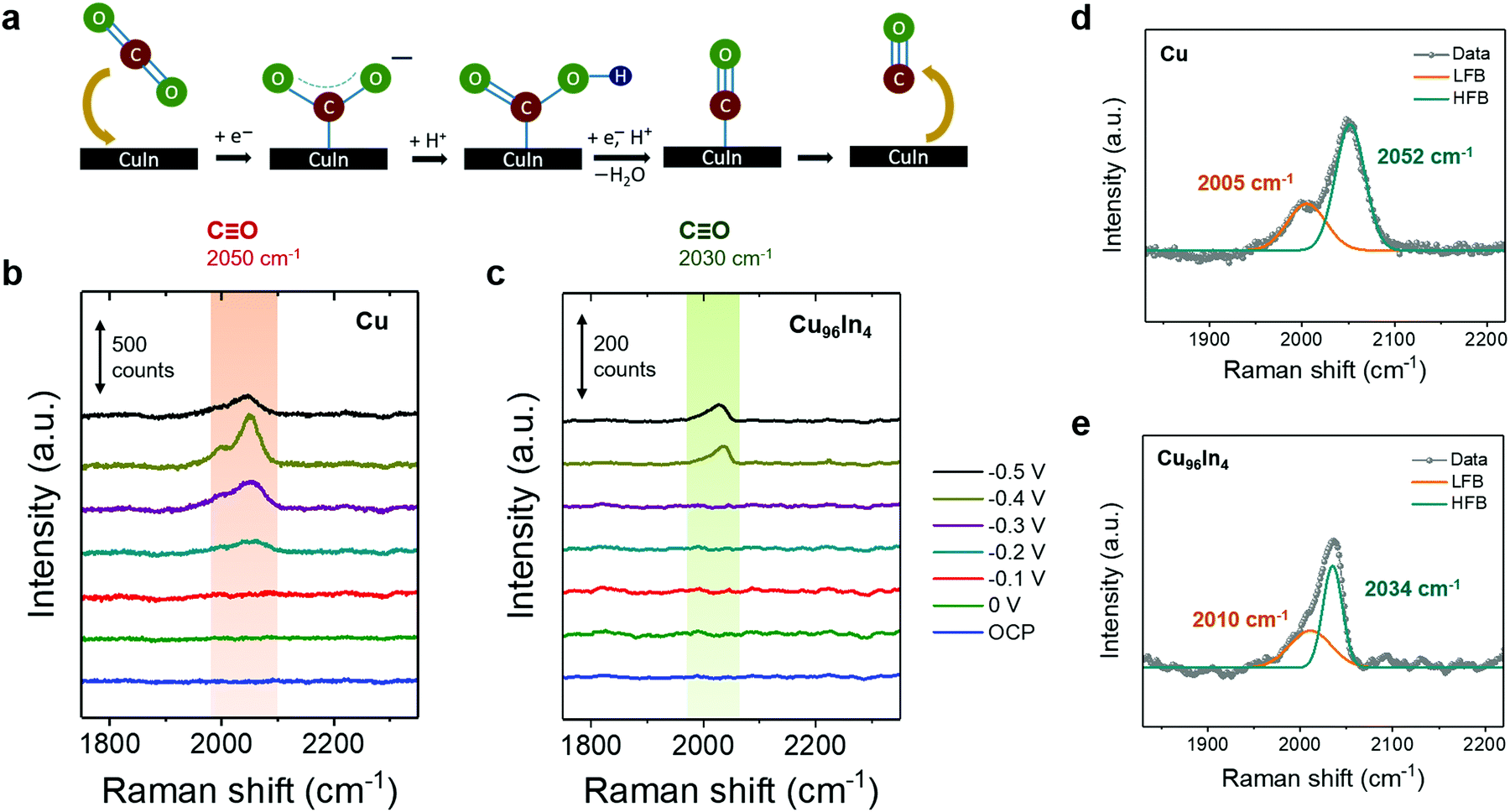 | ||
| Fig. 3 (a) Schematic representation of proposed CO forming mechanism on CuIn surface. Operando Raman spectroscopy studies on (b) porous Cu foam and (c) porous Cu96In4 foam surfaces in CO2 saturated 0.5 M KHCO3 (pH 7.2) at room temperature using a 633 nm laser. The applied potentials shown here are with respect to the RHE scale. The ν(CO) peak at −0.4 V vs. RHE was deconvoluted into a lower frequency band (LFB) and a higher frequency band (HFB) for both pristine Cu (d) and Cu96In4 alloy surface (e). | ||
Operando Raman spectroscopy
To identify intermediate species and characterize the exposed alloy phase on the catalyst surface, we performed operando Raman spectroscopy during stepped-potential chronoamperometry. Fig. 3b and c show operando Raman spectra of Cu and Cu96In4, respectively, in CO2 saturated 0.5 M aqueous KHCO3 showing the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching mode, ν(CO). The full range spectra are presented in Fig. S6 (ESI†), where reduction of the CuxO layer under cathodic potentials as well as CO32−/HCO3− adsorption peaks are shown. For the pure Cu sample, a broad band appeared when the potential was stepped to −0.2 V vs. RHE at around 2050 cm−1, which can be deconvoluted into two overlapping bands at 2005 and 2052 cm−1 (Fig. 3b and d). The lower frequency band (LFB, 2005 cm−1) has been previously assigned as atop bound CO on terraces, based on Cu(100) surface studies.33,34 The higher frequency band (HFB, 2052 cm−1) has been more confidently assigned as undercoordinated defect site-bound ν(CO), which is the major key component of CO adsorption on a 3D hierarchical surface.34–36 In contrast, an asymmetric band around 2030 cm−1 emerges when the potential is stepped to −0.4 V vs. RHE for the Cu96In4 sample (Fig. 3c). This band may be deconvoluted into two peaks at 2010 cm−1 and 2034 cm−1 (Fig. 3e).
O stretching mode, ν(CO). The full range spectra are presented in Fig. S6 (ESI†), where reduction of the CuxO layer under cathodic potentials as well as CO32−/HCO3− adsorption peaks are shown. For the pure Cu sample, a broad band appeared when the potential was stepped to −0.2 V vs. RHE at around 2050 cm−1, which can be deconvoluted into two overlapping bands at 2005 and 2052 cm−1 (Fig. 3b and d). The lower frequency band (LFB, 2005 cm−1) has been previously assigned as atop bound CO on terraces, based on Cu(100) surface studies.33,34 The higher frequency band (HFB, 2052 cm−1) has been more confidently assigned as undercoordinated defect site-bound ν(CO), which is the major key component of CO adsorption on a 3D hierarchical surface.34–36 In contrast, an asymmetric band around 2030 cm−1 emerges when the potential is stepped to −0.4 V vs. RHE for the Cu96In4 sample (Fig. 3c). This band may be deconvoluted into two peaks at 2010 cm−1 and 2034 cm−1 (Fig. 3e).
While this experiment does not allow for direct comparison of Raman intensities between the two different samples due to fundamental differences in site density and intrinsic interaction with the laser, we can compare the ν(CO) mode frequencies. It has been previously reported that, as the atop site bound CO on terraces results in a lower ν(CO) than a defect site bound CO, a stronger CO adsorption on Cu defect sites leads to a counter-intuitive blue-shift of ν(CO).34,36–38 A blue-shift of ν(CO) with increasing Cu–CO interaction has also been predicted by theory.39 We observe that the pure Cu defect-site ν(CO) at 2052 cm−1 is red-shifted to 2034 cm−1 in the Cu96In4 sample, suggesting a weaker CO adsorption on the alloy surface.
Despite significant CO formation at −0.3 V vs. RHE, ν(CO) is not observed until −0.4 V vs. RHE on the Cu96In4 sample, and persists at −0.5 V vs. RHE. This indicates that a large local CO concentration is required for the surface-bound species to accumulate, further supporting a weaker CO adsorption on this surface compared to pure Cu. Although Raman spectroscopy is used and particularly useful in this study to observe the nature of CO adsorption to the whole catalyst surface, it cannot be employed to provide unequivocal support for a particular catalytic site.40 Nevertheless, our electrochemical studies support an alloy as catalytic site.
Cu is a unique material that can form multicarbon products from CO2 because it stabilizes the surface bound *CO sufficiently to allow time for dimerization.41 This contrasts with Cu96In4 as we observe excellent CO selectivity over the entire range of applied potentials (−0.3 to −0.6 V vs. RHE, Fig. 2b) without any multicarbon product formation. We conclude that Cu96In4 can be highly selective towards CO due to beneficially weaker CO adsorption on the surface. The Cu96In4 bimetallic alloy with a dendritic foam morphology shows electroreduction of CO2 with 70% FECO even at −0.3 V vs. RHE, which is a key advance over previously reported Cu–In materials.25,32,42
PEC with perovskite|CuxIny cathodes
The CuxIny catalysts were next interfaced with cesium formamidinium methylammonium (CsFAMA) triple cation mixed halide perovskite inverse structure devices in an integrated configuration, which can be named a buried PV-biased cathode according to Nielander et al.43 The perovskite architecture was modified with a doped poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA) hole transport layer, which improved the open-circuit voltage (Voc) compared to previously reported devices (see the experimental procedure, photovoltaic histograms and J–V examples in Fig. S7 and S8, ESI†).18 The PV-biased cathodes were encapsulated using a conductive graphite epoxy paste, which provides increased stability in aqueous media and enables a direct catalyst adhesion to the underlying perovskite without additional binders.44Fig. 4a shows cyclic voltammetry (CV) scans of a buried perovskite|Cu96In4 cathode in CO2 saturated 0.5 M aqueous KHCO3 electrolyte solution. The photocurrent density reached −15 mA cm−2 at 0 V vs. RHE during irradiation (AM1.5G, 100 mW cm−2), without any notable dark current. A potential of +0.55 V vs. RHE was selected for further CPE studies, as this is the potential at which the photocurrents of the buried perovskite-biased cathode and BiVO4 photoanode overlap in the tandem assembly (Fig. S9, ESI†), and this potential is therefore comparable with the bias-free conditions of the tandem device (see below).
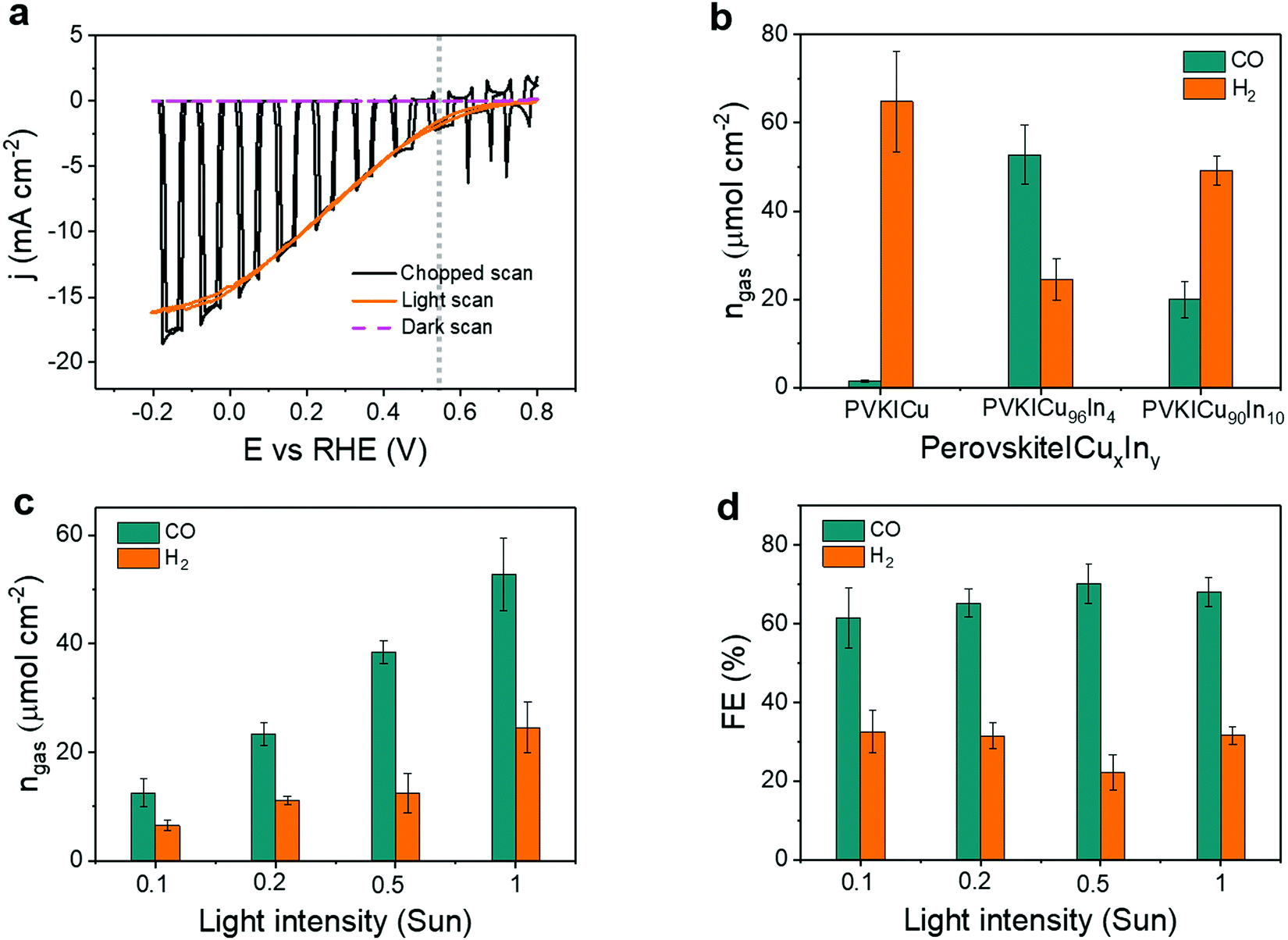 | ||
| Fig. 4 Photoelectrochemistry with perovskite|CuxIny cathodes: (a) CV scan with perovskite|Cu96In4 cathode under chopped, continuous and no artificial sunlight illumination (1 sun, AM1.5G, 100 mW cm−2, scan rate: 10 mV s−1). (b) Activity of buried PV-biased cathodes containing catalysts with different composition under chopped 1 sun illumination (50 min on, 10 min off). (c) Amounts of products formed, and (d) their faradaic efficiencies on perovskite|Cu96In4 cathodes under different light intensities (chopped; 50 min on, 10 min off). All experiments were performed at room temperature and CO2 saturated aqueous 0.5 M KHCO3 (pH 7.2) was used as reaction medium. The applied bias in (b), (c) and (d) was +0.55 V vs. RHE and duration of all the PEC experiments was 4 h. | ||
To study the effect of the catalyst composition on the PEC response, we have performed CPE using different perovskite|CuxIny cathodes with varying In percentage in the catalyst. Fig. 4b shows the amounts of CO and H2 from different buried PV-biased cathodes under 1 sun illumination. The corresponding FEs are shown in Fig. S10, ESI† (see also Tables S5 and S6, ESI†). A perovskite|CuxIny cathode containing a pure Cu catalyst (Cu foam, Cu100In0) shows selective H2 production with only minor CO formation, whereas perovskite|CuxIny cathodes with a Cu96In4 catalyst displayed a good CO selectivity over H2 with a CO:H2 ratio of 2.2. However, when a catalyst with higher In percentage (Cu90In10) was employed on the buried perovskite|CuxIny cathode, the CO:H2 ratio dropped to 0.4, showing a similar trend to the electrolysis data above (Fig. 2c). From PEC analysis we conclude that Cu96In4 is the optimum catalyst composition for a high CO selectivity of the buried perovskite|CuxIny cathodes, thus Cu96In4 was used in the tandem devices for bias-free CO production.
Next, the influence of light intensity on the performance of perovskite|Cu96In4 cathodes was studied. CPE and CV scans showed that the photocurrent density increased almost linearly with the light intensity (Fig. S11–S13, ESI†). Hence, the amount of gaseous products also increased with the light intensity (Fig. 4c). No major changes in FECO were observed for light intensities between 1 sun and 0.1 sun (Fig. 4d). This behavior is consistent with the small change in selectivity observed with different applied potentials at the electrodes (Fig. 2b), as the light intensity affects the perovskite photovoltage, and hence the applied potential at the catalyst interface.21 The best CO:H2 ratio of 3.2 ± 0.6 was obtained under 0.5 sun, corresponding to a (70.1 ± 5.0)% FECO, where the amount of CO was 38.45 ± 2.12 μmol cm−2 (Tables S7 and S8, ESI†). The results demonstrate a robust PEC activity of the perovskite|Cu96In4 cathodes toward CO2 conversion under different light irradiation, which makes them suitable for operation under low, diffuse sunlight.
BiVO4‖perovskite|Cu96In4 tandem device
To achieve bias-free solar CO2-to-CO conversion using water as an electron donor, a tandem device was assembled in a compact, back-to-back ‘artificial leaf’ configuration by incorporating a BiVO4 photoanode (containing an immobilized TiCo water oxidation catalyst) onto the integrated perovskite|Cu96In4 cathode.18,21 This configuration can be described as a buried PV-biased PEC cell as proposed by Nielander et al.,43 standing out from wired perovskite PV-electrode assemblies as both perovskite and BiVO4 photoelectodes are immersed under water during operation, which simplifies the overall device design. Thus, this integrated tandem system is technically different from a classical PV + electrolyser system, where catalysts are immersed in the electrolyte solution as anode and cathode and the required energies are supplied externally by a solar cell to perform CO2 electrolysis.45Optical images of a tandem device are shown in Fig. S14 (ESI†). CV of the individual buried perovskite-biased cathode and BiVO4 photoanode shows an overlap at +0.55 V vs. RHE with a photocurrent density of 208 μA cm−2, while the photoanode exhibits an early onset potential at 0.3 V vs. RHE (Fig. S9, ESI†). Since the perovskite solar cells achieve a Voc of 1.07 ± 0.02 V (Fig. S7, ESI†), the tandem system would apply an effective potential around −0.5 V vs. RHE onto the Cu96In4 alloy catalyst, which is in the optimal potential range (−0.4 V to −0.6 V vs. RHE) for selective CO production (Fig. 2b). CV of the tandem device reveals an early onset bias of −0.4 V and a photocurrent density of around 212 μA cm−2 under no applied bias (Fig. 5a), which matches with the current density expected from the cathodic and anodic photocurrent overlap.
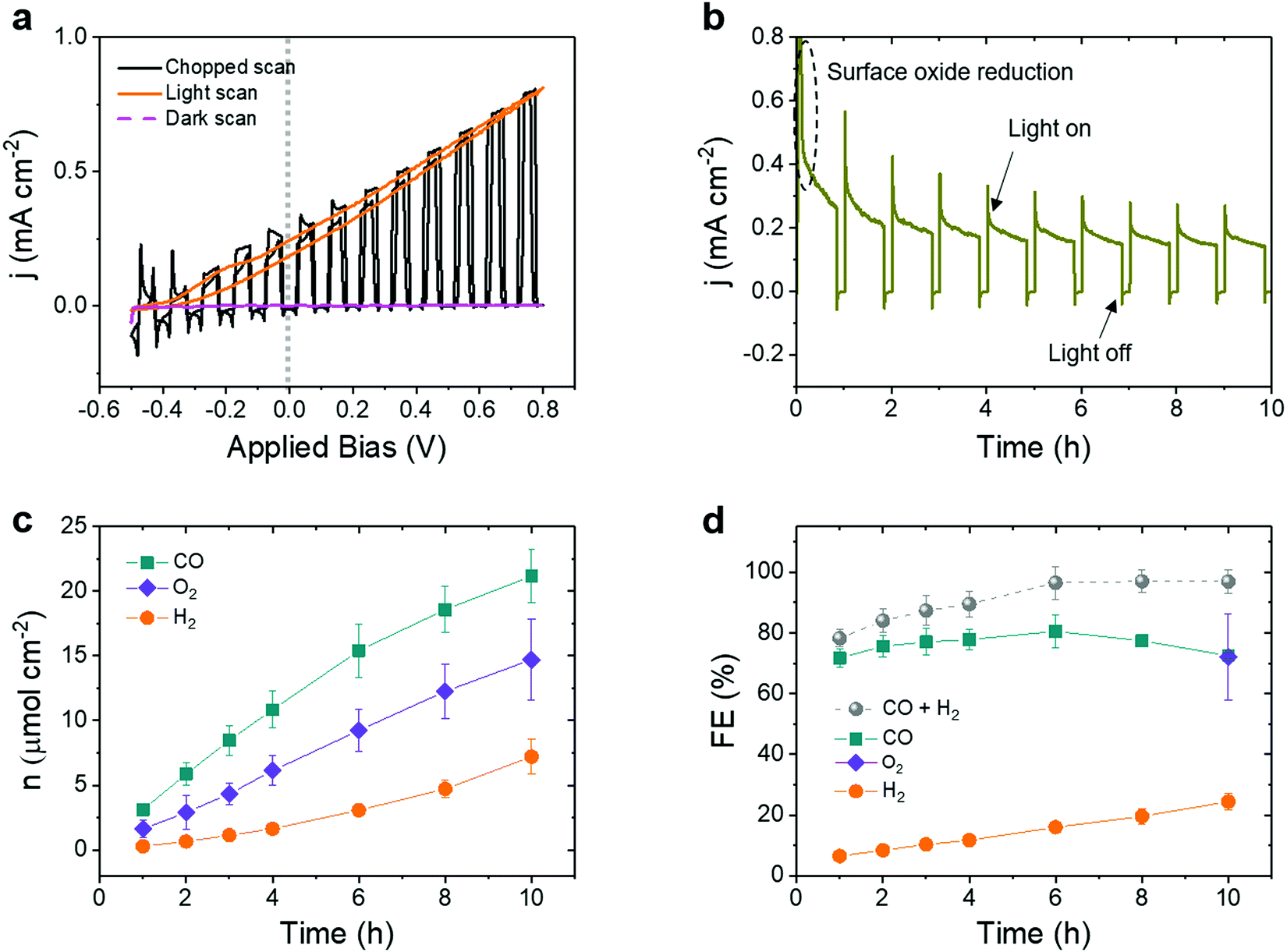 | ||
| Fig. 5 Bias-free solar CO2 conversion on a tandem BiVO4‖perovskite|Cu96In4 assembly. (a) CV scan under chopped, continuous and no artificial sunlight illumination (1 sun, AM1.5G, 100 mW cm−2, scan rate: 10 mV s−1), (b) representative photocurrent for 10 h Bias-free operation under chopped 1 sun irradiation (50 min on, 10 min off), (c) amounts of products formed and (d) faradaic efficiency over the 10 h experiment. Bias-free (Uapp = 0 V) experiments were carried out at room temperature in a one compartment cell using CO2 saturated 0.5 M KHCO3 electrolyte (pH 7.2). | ||
A stable photocurrent was maintained over 10 h experiment under no applied bias (Uapp = 0 V, Fig. 5b), reaching an average steady-state photocurrent density of 195 ± 20 μA cm−2. A higher initial photocurrent accounts for the reduction of a surface oxide layer, which forms on the Cu96In4 catalyst under air (Fig. S3 and S6, ESI†).
The amounts of gaseous products increased gradually during the course of the experiment, accumulating to 21.17 ± 2.04 μmol cm−2 CO, 7.21 ± 1.34 μmol cm−2 H2 and 14.71 ± 3.15 μmol cm−2 O2 after 10 h operation under zero applied bias (Fig. 5c and Table S9, ESI†). After the experiment, the electrolyte solution was analyzed by 1H NMR spectroscopy and ion exchange chromatography (IC) for the presence of any liquid products. Only a negligible trace of formate (0.9 ± 0.2 ppm) was detected by IC (see Fig. S15, ESI† for representative chromatograms). A FECO of (72.5 ± 1.1)% and FEH2 of (24.4 ± 2.7)% was obtained at the perovskite|Cu96In4 cathode and a faradaic efficiency for O2 (FEO2) of (72.1 ± 14.3)% at the BiVO4 photoanode (Fig. 5d). FECO was roughly constant during the course of the experiment, whereas FEH2 showed a slight increase over time (Table S10, ESI†). The tandems showed an excellent CO:H2 ratio of 3:1 after 10 h, which is an order of magnitude increase of the CO selectivity compared to a molecular catalyst-based tandem under bias-free conditions.21 Due to the improved perovskite device architecture and lower catalyst overpotential, the tandem device also showed a noteworthy increase in the solar-to-fuel conversion, reaching 0.19% solar-to-CO (ten times higher) and 0.06% solar-to-H2 efficiencies.13,46
Some minor chemical and morphological transformations of the Cu96In4 catalyst were also observed during the 10 h bias-free PEC experiment. The as-prepared catalyst is a uniform alloy, where Cu and In are well distributed on the dendrite surface. During the experiment, the catalyst undergoes an in situ phase transformation, where the Cu and In phases partially separate from each other. XRD analysis reveals that some of the alloy phases even disappeared after the experiment (Fig. S16, ESI†). Post-experimental STEM mapping and XPS analysis confirmed that the In phase migrates towards the periphery as well as into the bulk of the dendrites (Fig. S17 and S3, ESI†). This phenomenon leads to an exposure of Cu phase on the catalyst surface, which may explain the slight increase of FEH2 over time as pristine Cu phase is more active for H2 evolution under these conditions (see Fig. 2c and 4b). However, the catalyst's bulk composition was unchanged as post-experiment ICP-OES analysis showed similar ratio of Cu and In (∼96:4) as the freshly prepared material. The overall porous dendritic morphology of the catalyst was intact after the experiment, but some structural agglomeration was observed in the nano-branches due to constant gas evolution (Fig. S18, ESI†).
Conclusions
A porous dendritic Cu96In4 foam is reported and shown to be electrocatalytically active towards CO production from aqueous CO2 at low overpotentials. The weak adsorption of CO on the bimetallic alloy surface makes it a selective CO forming material, as supported by operando Raman spectroscopy. The excellent performance of the Cu-alloy electrocatalyst can be exploited in a perovskite‖BiVO4 device for bias-free CO2 reduction to produce CO with high selectivity using sunlight as energy input and water as the electron donor. A relatively high photocurrent density and early onset bias was obtained for the tandem device due to the lower CO2 reduction overpotential of the catalyst and an improved perovskite device architecture providing a higher photovoltage. A CO selectivity of ∼75% and solar-to-CO conversion efficiency of 0.19% were obtained after 10 h of photoelectrocatalysis under zero applied bias, which are an order of magnitude higher compared to a previous report.21 This study demonstrates that transition metal catalysts in combination with PEC systems allow for bias-free solar fuel production from CO2 using sunlight as the sole energy source. The proof-of-concept can be further extended by interfacing different Cu-based catalysts onto the tandem to form various mono and multicarbon products under bias-free conditions.47–51Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a SNSF EPM Fellowship Grant (P2BEP2_184483 code to M. R.), the OMV group (to C. P., V. A., E. R.), the Cambridge Trust (Cambridge Thai Foundation Award to C. P. and Vice-Chancellor's Award to V. A.), a Trinity-Henry Barlow Scholarship (to C. P.) as well as the Winton Programme (to V. A.) and the EPSRC (EP/L027151, EP/S003053, EP/S025308, NanoDTC, EP/L015978/1 and EP/S022953 to D. W., J. B., E. R.). We acknowledge Prof. Dominic S. Wright (University of Cambridge) for providing the water oxidation catalyst precursor (TiCo). We also thank Dr Qian Wang and Dr Tengfei Li (University of Cambridge) for helpful feedback on the manuscript.References
- P. K. Nayak, S. Mahesh, H. J. Snaith and D. Cahen, Nat. Rev. Mater., 2019, 4, 269–285 CrossRef CAS.
- J. Warnan, J. Willkomm, Y. Farré, Y. Pellegrin, M. Boujtita, F. Odobel and E. Reisner, Chem. Sci., 2019, 10, 2758–2766 RSC.
- A. Listorti, J. Durrant and J. Barber, Nat. Mater., 2009, 8, 929–930 CrossRef CAS.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS.
- J. J. Concepcion, R. L. House, J. M. Papanikolas and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560–15564 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- M. E. Dry, Appl. Catal., A, 1996, 138, 319–344 CrossRef CAS.
- H. Zhang, Z. Yang, W. Yu, H. Wang, W. Ma, X. Zong and C. Li, Adv. Energy Mater., 2018, 8, 1800795 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- M. Crespo-Quesada, L. M. Pazos-Outón, J. Warnan, M. F. Kuehnel, R. H. Friend and E. Reisner, Nat. Commun., 2016, 7, 12555 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS.
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 7326 CrossRef CAS.
- J. Chen, J. Yin, X. Zheng, H. Ait Ahsaine, Y. Zhou, C. Dong, O. F. Mohammed, K. Takanabe and O. M. Bakr, ACS Energy Lett., 2019, 4, 1279–1286 CrossRef CAS.
- Q. Wang, L. Tao, X. Jiang, M. Wang and Y. Shen, Appl. Surf. Sci., 2019, 465, 607–613 CrossRef CAS.
- Y.-S. Chen, J. S. Manser and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 974–981 CrossRef CAS.
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS.
- V. Andrei, R. L. Z. Hoye, M. Crespo-Quesada, M. Bajada, S. Ahmad, M. De Volder, R. Friend and E. Reisner, Adv. Energy Mater., 2018, 8, 1801403 CrossRef.
- S. Zhang, L. Shen, T. Ye, K. Kong, H. Ye, H. Ding, Y. Hu and J. Hua, Energy Fuels, 2020, 34, 5016–5023 CrossRef CAS.
- E. Edwardes Moore, V. Andrei, S. Zacarias, I. A. C. Pereira and E. Reisner, ACS Energy Lett., 2020, 5, 232–237 CrossRef CAS.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS.
- Y. Wang, D. Wang, C. J. Dares, S. L. Marquard, M. V. Sheridan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 278–283 CrossRef CAS.
- X. Zheng, Y. Ji, J. Tang, J. Wang, B. Liu, H.-G. Steinrück, K. Lim, Y. Li, M. F. Toney, K. Chan and Y. Cui, Nat. Catal., 2019, 2, 55–61 CrossRef CAS.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS.
- H.-C. Shin and M. Liu, Chem. Mater., 2004, 16, 5460–5464 CrossRef CAS.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS.
- W. Luo, W. Xie, R. Mutschler, E. Oveisi, G. L. De Gregorio, R. Buonsanti and A. Züttel, ACS Catal., 2018, 8, 6571–6581 CrossRef CAS.
- Y. Hori, O. Koga, Y. Watanabe and T. Matsuo, Electrochim. Acta, 1998, 44, 1389–1395 CrossRef CAS.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- Z. Pan, K. Wang, K. Ye, Y. Wang, H.-Y. Su, B. Hu, J. Xiao, T. Yu, Y. Wang and S. Song, ACS Catal., 2020, 10, 3871–3880 CrossRef CAS.
- W. Akemann and A. Otto, J. Raman Spectrosc., 1991, 22, 797–803 CrossRef CAS.
- B. Eren, Z. Liu, D. Stacchiola, G. A. Somorjai and M. Salmeron, J. Phys. Chem. C, 2016, 120, 8227–8231 CrossRef CAS.
- S. Vollmer, G. Witte and C. Wöll, Catal. Lett., 2001, 77, 97–101 CrossRef CAS.
- P. S. Bagus and G. Pacchioni, Electrochim. Acta, 1991, 36, 1669–1675 CrossRef CAS.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2016, 6, 6265–6274 CrossRef.
- A. C. Nielander, M. R. Shaner, K. M. Papadantonakis, S. A. Francis and N. S. Lewis, Energy Environ. Sci., 2015, 8, 16–25 RSC.
- V. Andrei, K. Bethke and K. Rademann, Phys. Chem. Chem. Phys., 2016, 18, 10700–10707 RSC.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- H. Dotan, N. Mathews, T. Hisatomi, M. Grätzel and A. Rothschild, J. Phys. Chem. Lett., 2014, 5, 3330–3334 CrossRef CAS.
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee01279c. Raw data related to this publication are available at the University of Cambridge data repository: https://doi.org/10.17863/CAM.56420. |
| This journal is © The Royal Society of Chemistry 2020 |