
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePower-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: a review
Vincent
Dieterich
 *a,
Alexander
Buttler
a,
Andreas
Hanel
a,
Hartmut
Spliethoff
ab and
Sebastian
Fendt
*a
*a,
Alexander
Buttler
a,
Andreas
Hanel
a,
Hartmut
Spliethoff
ab and
Sebastian
Fendt
*a
aTechnical University of Munich, Department of Mechanical Engineering, Chair of Energy Systems, Boltzmannstr. 15, 85748 Garching, Germany. E-mail: Vincent.Dieterich@tum.de; Sebastian.Fendt@tum.de
bBavarian Centre for Applied Energy Research, Walther-Meißner-Str. 6, 85748 Garching, Germany
First published on 13th August 2020
Abstract
The conversion of H2 and CO2 to liquid fuels via Power-to-Liquid (PtL) processes is gaining attention. With their higher energy densities compared to gases, the use of synthetic liquid fuels is particularly interesting in hard-to-abate sectors for which decarbonisation is difficult. However, PtL poses new challenges for the synthesis: away from syngas-based, continuously run, large-scale plants towards more flexible, small-scale concepts with direct CO2-utilisation. This review provides an overview of state of the art synthesis technologies as well as current developments and pilot plants for the most prominent PtL routes for methanol, DME and Fischer–Tropsch-fuels. It should serve as a benchmark for future concepts, guide researchers in their process development and allow a technological evaluation of alternative reactor designs. In the case of power-to-methanol and power-to-FT-fuels, several pilot plants have been realised and the first commercial scale plants are planned or already in operation. In comparison power-to-DME is much less investigated and in an earlier stage of development. For methanol the direct CO2 hydrogenation offers advantages through less by-product formation and lower heat development. However, increased water formation and lower equilibrium conversion necessitate new catalysts and reactor designs. While DME synthesis offers benefits with regards to energy efficiency, operational experience from laboratory tests and pilot plants is still missing. Furthermore, four major process routes for power-to-DME are possible, requiring additional research to determine the optimal concept. In the case of Fischer–Tropsch synthesis, catalysts for direct CO2 utilisation are still in an early stage. Consequently, today's Fischer–Tropsch-based PtL requires a shift to syngas, benefiting from advances in co-electrolysis and reverse water-gas shift reactor design.
 Vincent Dieterich | Vincent Dieterich studied Chemical Engineering at the Technical University of Munich (TUM). During his studies he worked as a visiting student at the Massachusetts Institute of Technology. He currently works as a Doctoral candidate at the Chair of Energy Systems at the TUM under Prof. Hartmut Spliethoff, where he joined the research-group of Dr-Ing. Sebastian Fendt. His research interest is in the field of electricity-based fuels, focusing on the simulation, integration and techno-economic analysis of Power-to-X systems. |
 Alexander Buttler | Alexander Buttler studied Mechanical Engineering at the Technical University of Munich (TUM). After finishing his studies, he started his work as a Doctoral candidate at the Chair of Energy Systems at the TUM under Prof. Hartmut Spliethoff, where he finished his dissertation in 2018. Alexander Buttler authored and co-authored a number of publications in various renowned scientific journals focusing on polygeneration, electrolysis and renewable fuel production. He currently works in an industrial position at MTU Friedrichshafen GmbH continuing his work on energy systems optimisation and modeling of power-to-fuel plants. |
 Andreas Hanel | Andreas Hanel studied Physics at the LMU Munich and Mechanical Engineering at the Technical University of Munich (TUM). He currently works as a Doctoral candidate at the Chair of Energy Systems at the TUM under Prof. Hartmut Spliethoff, where he joined the research-group of Dr-Ing. Sebastian Fendt. His research interest is in the field of polygeneration plants, focusing on the optimized interaction of possible synthesis routes and power generation. |
 Hartmut Spliethoff | Hartmut Spliethoff studied mechanical engineering at the universities of Kaiserslautern and Stuttgart. In 1991 he was appointed head of the Boiler Technology department at the IVD, University of Stuttgart. In 1999 he finished his postdoctoral thesis and in 2000 he was appointed as full time professor at the Chair of Energy Conversion of the Technical University of Delft. Since 2004 he is full time professor at the Chair of Energy Systems (TUM). Prof. Spliethoff has over 30 years of research experience and has co-authored more than 200 scientific articles. Furthermore he has been coordinator of, both national and international multidisciplinary centres and consortia. |
 Sebastian Fendt | Sebastian Fendt studied Chemical Engineering at Technical University of Munich (TUM) and University of California, Berkeley. After finishing his studies, he started his work at the Chair of Energy Systems (TUM) first as Doctoral candidate and since 2015, he heads a research group with the focus on energetic utilization of biomass, renewable fuels and Power-to-X systems as postdoc. Besides acting as project manager and coordinator of various research projects, he acts as coordinator for the TUM.PtX Network and holds lectures and seminars. Sebastian Fendt authored and co-authored a number of publications in various renowned scientific journals. |
Broader contextIn their effort to achieve the goals set by the Paris Climate agreement, governments worldwide have facilitated the expansion of renewable energy sources and increased research funding for technologies such as solar and wind power, energy storage or carbon capture. At the same time, particularly the mobility sector is growing strongly, which results in increased greenhouse gas emissions and for many other sectors (e.g. chemical industry) a path towards emission reduction is not in sight. Power-to-Liquid (PtL) processes are seen as a promising solution to tackle these challenges. Using renewable electricity to produce liquid fuels with high energy densities, PtL can offer carbon neutral fuels for the mobility sector and new production routes for the chemical industry. Furthermore, PtL can contribute to grid stability and act as a long-term energy storage solution. In this review the most prominent PtL routes are considered. The review focuses on the synthesis step of each product and compares the state of the art with current research trends in the PtL context. In addition, an overview of PtL pilot plants is given and economic aspects are discussed. The goal is to identify research gaps and promote development steps towards commercialisation of PtL processes. |
1. Introduction
Reducing greenhouse gas emissions has become a central target of worldwide energy policies. The Paris Climate agreement in 2015 set the aim of limiting global warming to below 2 °C compared to pre-industrial levels.1 An increase of renewable energies, especially wind and solar power, represents an important component to realise this ambitious target. Their fluctuating feed-in results in an increased flexibility requirement of the energy system to balance supply and demand. Frontrunners like Denmark already face excess power production with wind generation supplying 130% of the load within a 24-hour window, giving an outlook for future renewable energy systems.2 Large-scale short- and long-term energy storages will play a crucial role besides other flexibility options like demand side management to ensure electricity supply. Power-to-Gas (PtG) is often discussed as a potential long-term storage option by linking the power grid with the gas grid.3 Several reviews concerning commercial methanation technologies,4 technological and economic reviews of PtG-routes,5 comparison of different renewable SNG pathways6 and pilot plants7–9 have been published. In addition entire books on the topic are available.10–12 However, the conversion of electrolytic H2 and CO2 into liquid fuels (Power-to-Liquid, PtL) gains rising attention as a storage and flexibility option as well as carbon capture and utilisation (CCU) technology.Fig. 1 shows that compared to Li-ion batteries and PtG products (hydrogen or methane), PtL products have the advantage of higher energy densities both in terms of weight and volume, making PtL processes especially interesting for hard-to-abate sectors e.g. aviation, shipping or heavy goods traffic. Moreover many PtL products represent a value added product, which otherwise has to be produced from natural gas (or coal). Consequently, PtL leads to a coupling of the electricity sector with the mobility sector and chemical industry. It enables excess renewable power from the electricity sector to be taken up and substitute fossil fuels in the mobility sector and chemical industry, resulting in a reduction of greenhouse gas emissions. The basic process chain of PtL as discussed within this review is shown in Fig. 2. Main process steps are: water electrolysis, carbon capture, synthesis and product upgrade.
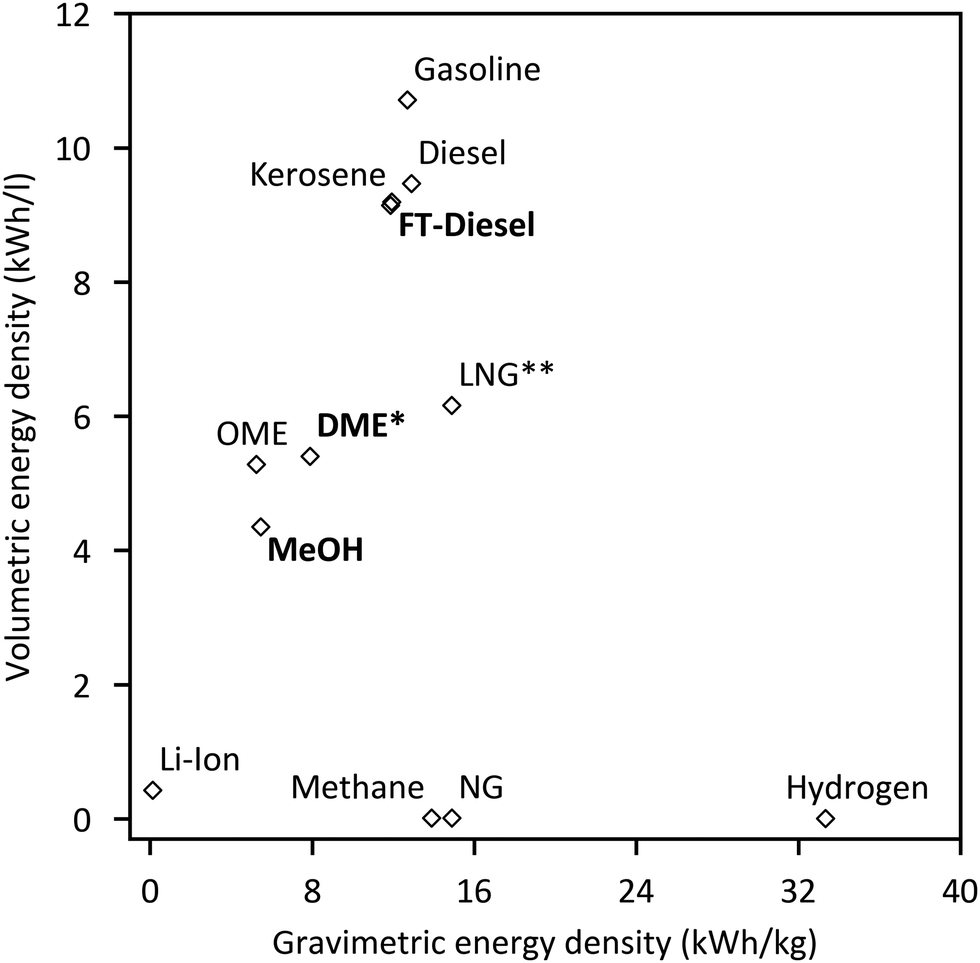 | ||
| Fig. 1 Gravimetric and volumetric energy density of different fuels and Li-Ion batteries for comparison (*at 10 bar; **at −160 °C).13–15 | ||
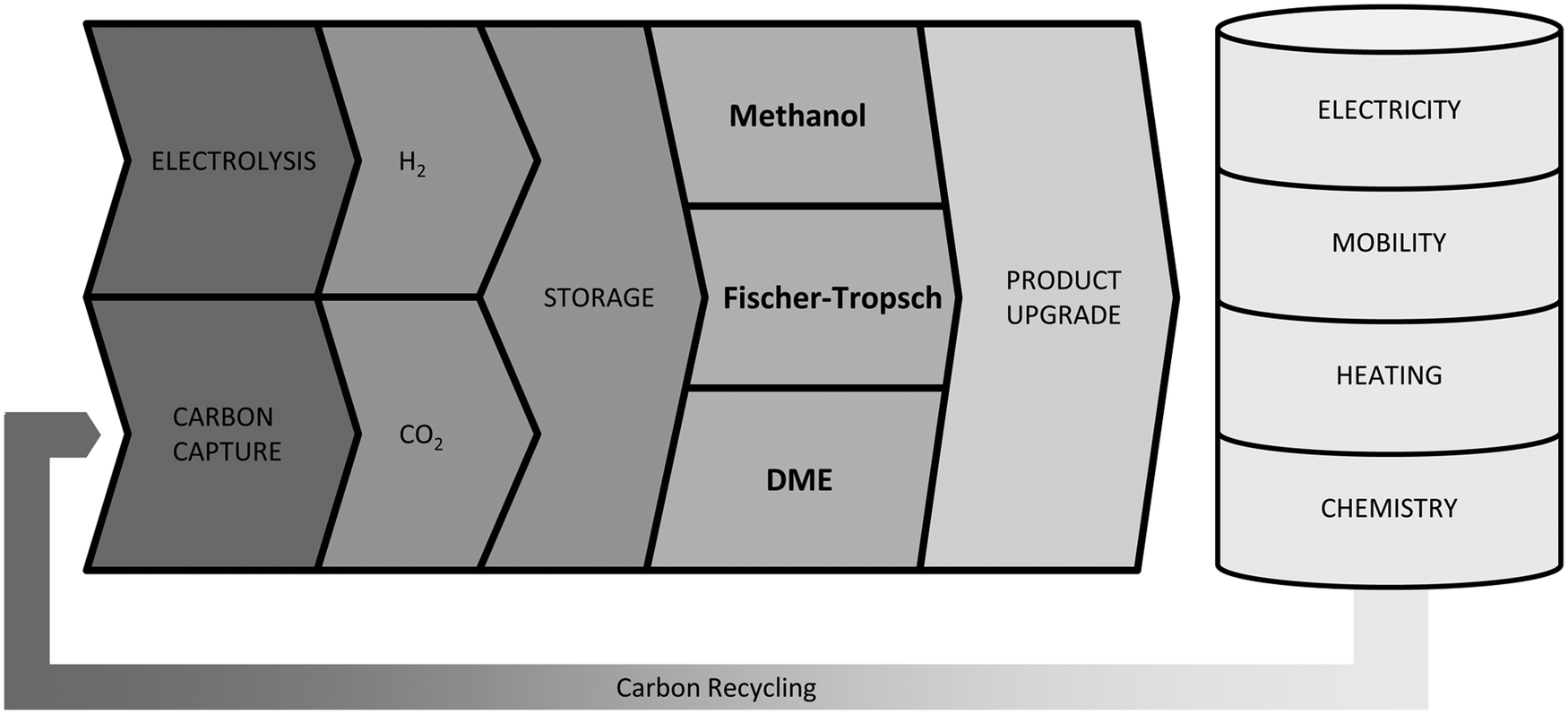 | ||
| Fig. 2 Power-to-X pathways to transform electricity to chemicals via electrolysis and synthesis. The power-to-X principle connects different energy sectors via carbon capture from industrial plants or chemical/natural (photosynthesis) CO2 scrubbing from the air, the carbon cycle can be closed so that no additional CO2 is emitted. This paper regards paths to transform electricity to liquid fuels for the mobility sector. | ||
Until now the commercial application of PtL processes is limited due to the remaining technological challenges regarding major process steps e.g. higher electrolysis efficiencies or improvements in carbon capture methods. In addition experience regarding the interaction between the process steps is still scarce. While Section 1.1.1 will give a short introduction about water electrolysis, and Section 1.1.2 will summarise current carbon capture technologies, the article will focus on the synthesis and product upgrade within the PtL context. Three of the most prominent examples of PtL products will be discussed: methanol, dimethyl ether (DME) and Fischer–Tropsch-fuels (FT-fuels). For each, synthesis from CO and H2 (syngas)† is an established technology with decades of operational experience.
However, using PtL as CCU technology raises the question of a direct synthesis from CO2. There are several elaborate reviews and books on fuel production routes from CO2 in general,16–20 catalysts for the hydrogenation of CO2 to methanol,21–23 DME or FT-fuels, as well as conventional and innovative processes for the production of methanol,24–29 DME27,30–34 or FT-fuels35–37 from natural gas, coal or biomass derived syngas. More recently reviews that evaluate the state of PtL with regard to the life cycle,38 advances in catalysis and reaction routes,39 as well as the area of application40,41 have been published. However, a comprehensive technology review summarizing the status of synthesis technologies and evaluating new developments and research needs to address challenges within the context of PtL is missing.
Therefore, the review will provide an extensive overview of the synthesis for each product considering thermodynamics, catalysts, commercial converter designs and processes as well as the product upgrade. The goal is to provide boundary conditions for future concepts with regard to performances, catalyst life times and reactor and process designs. It should guide further developments and enable benchmarking. In addition, current research addressing the main challenges of the synthesis within the PtL process, i.e. CO2 utilisation and flexible operation, will be discussed. An overview of existing PtL pilot plants is given and operation experiences are summarised. Moreover economic aspects and techno-economic analysis for medium-term commercialisation are discussed.
1.1. Power-to-liquid main process steps
| AEL | PEMEL | SOEC | |
|---|---|---|---|
| a Operating at thermoneutral voltage. b Including auxiliaries and heat supply (SOEC). c High uncertainty due to pre-commercial status of SOEC. | |||
| Operation parameters | |||
| Cell temperature (°C) | 60–90 | 50–80 | 700–900 |
| Typical pressure (bar) | 10–30 | 20–50 | 1–15 |
| Current density (A cm−2) | 0.25–0.45 | 1.0–2.0 | 0.3–1.0 |
| Flexibility | |||
| Load flexibility (% of nominal load) | 20–100 | 0–100 | −100/+100 |
| Cold start-up time | 1–2 h | 5–10 min | Hours |
| Warm start-up | 1–5 min | <10 s | 15 min |
| Efficiency | |||
| Nominal stack efficiency (LHV) | 63–71% | 60–68% | 100%a |
| Nominal systemb efficiency (LHV) | 51–60% | 46–60% | 76–81% |
| Available capacity | |||
| Max. nominal power per stack (MW) | 6 | 2 | <0.01 |
| H2 production per stack (N m3 h−1) | 1400 | 400 | <10 |
| Durability | |||
| Life time (kh) | 55–120 | 60–100 | (8–20) |
| Efficiency degradation (% per a) | 0.25–1.5 | 0.5–2.5 | 3–50 |
| Economic parameters | |||
| Investment cost (€ per kW) | 800–1500 | 1400–2100 | (>2000)c |
| Maintenance costs (% of investment costs per year) | 2–3 | 3–5 | |
As can be seen, AEL and PEMEL are available on MW-scale and several flexible operated pilot plants have been realised in the last years. The hydrogen produced reaches very high purity of more than 99% (above 99.999% possible). Moreover pressurised electrolysers up to an operational pressure of 60–80 bar are commercially available reducing the need for compression.
SOEC on the other hand offers thermodynamic advantages due to operation at high temperatures theoretically enabling electric efficiencies above 100% based on LHV. However, industrial production is only in an early stage and pressurised operation is still in the research phase. In addition, SOEC can be used for syngas production via high temperature co-electrolysis of H2O and CO2. In this case hydrogen and carbon monoxide are formed. This variant is especially interesting for PtL processes using syngas and the application of co-electrolysis has already been discussed for PtSNG,43 PtMeOH,44 and PtFT-fuels.45 However, degradation of the cell remains a challenge for commercialisation, making a better understanding of the reaction and degradation mechanism necessary as well as further material developments. An overview of the technology, material and current developments is given by Zheng et al.46
| Methods | Description |
|---|---|
| Capture from point sources | |
| Absorption technologies | CO2 is removed from flue gases by absorption in liquid solvents. Typically aqueous amine solutions are used and water-soluble salts are formed in the process. Afterwards CO2 can be recovered in a desorption column. |
| Adsorption technologies | Solid adsorbents are used for the CO2 capture. Typical adsorbents are carbons, alumina, silica and zeolites but also new adsorbents are investigated e.g. polymers. Cyclic processes are implemented for the loading and regeneration of the adsorbents in particular pressure vacuum swing adsorption and temperature swing adsorption. Furthermore, so-called carbonate looping processes, which are based on chemisorption can be used. The most prominent process is calcium looping (using CaO as sorbent). CO2 is captured forming calcium carbonate. The sorbent is afterwards regenerated via calcination releasing the CO2. |
| Direct air capture | |
| Wet air capture | CO2 from ambient air is absorbed in a liquid solution within packed-columns, convection towers or spray-tower contractor systems. An example is the soda/lime process based on a sodium hydroxide solution. |
| Dry air capture | Typically solid organoamine based adsorbents are used for the CO2 capture. Desorption occurs at elevated temperatures in an inert gas stream, however only a diluted CO2 stream is generated. Newer developments focus on improving regeneration conditions. |
In general CO2 for the synthesis can either be captured from point sources with a high CO2 partial pressure, e.g. power plant or industry exhaust gases, or can be obtained directly from the air via direct air capture (DAC) technologies. DAC requires much higher energy inputs and the processing of greater gas volumes.49 Consequently, costs are expected to be up to ten times higher,50 however, due to learning effects and process improvement cost competitiveness could be reached by 2040.51
It is difficult to estimate the costs of CO2 from different carbon sources due to the variety of capture technologies, location-specific circumstances and variable energy and utility costs. For a first indication Table 3 summarises recent cost estimates for CO2 from different sources.
| Carbon source | CO2 cost in € per tCO2 |
|---|---|
| a In €2015, exchange rate €/$ of 1.11. b Approx. costs for the seperation of CO2 from biogas, currently an unused waste product. c Estimation for 8000 full load hours in 2020 and 10% learning curve effects. | |
| Coal gasification power plant | 28–40 |
| Coal-fired power plant | 31–49 |
| Gas-fired power planta | 47–90 |
| Refineries & NG processinga | 18–71 |
| Steel mill | 70–73 |
| Cement production | 58–87 |
| Biogas plantb | 0–90 |
| Direct air capturec | 222–268 |
A simplified flowsheet of a conventional synthesis loop applied in most cases is depicted in Fig. 3. The fresh feed gas is mixed with the recycle gas stream. This feed stream is optionally preheated before entering the synthesis reactor partially converting the syngas. The reacted gas is then cooled and liquids are separated by condensation. This crude product has to be further purified to meet the given specifications. The unreacted syngas is recycled with a small part being purged to avoid an enrichment of inert gases.
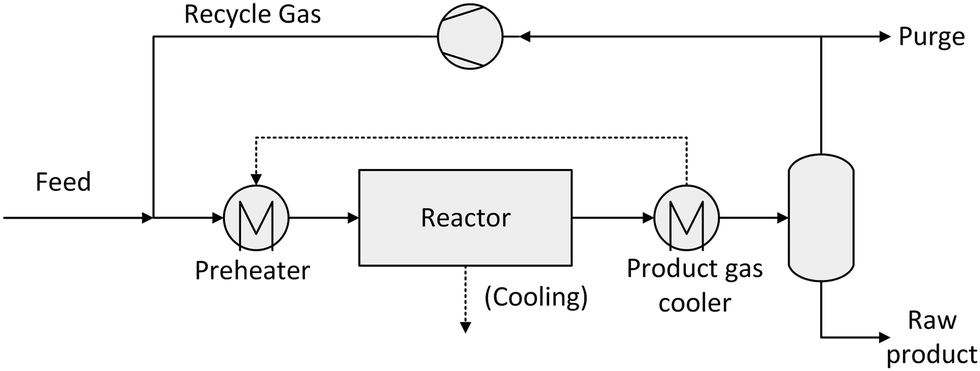 | ||
| Fig. 3 Conventional synthesis loop in methanol, DME and FT-processes. | ||
The optimal feed gas composition in terms of maximum conversion is usually defined by the stoichiometric number (SN) determined by the synthesis reactions. The stoichiometric number is given by the ratio of the reactants hydrogen, carbon monoxide and carbon dioxide (in molar fraction):
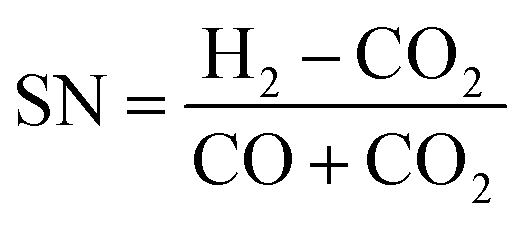 | (1) |
In general, similar reactor designs (and combinations of them) are applied for methanol, DME and Fischer–Tropsch synthesis. Table 4 gives an overview of typically used reactor designs. The various commercial reactor designs and the particular characteristics of methanol, DME and FT-fuels synthesis routes are discussed in the following sections in detail.
| Fixed bed reactor (FB) | Fluidized bed reactor | Liquid-phase reactor | ||||
|---|---|---|---|---|---|---|
| Adiabatic FB | Quench | SRCa | GCCb | |||
| a SRC = steam raising converter. b GCC = gas-cooled converter. | ||||||
| Operation mode | Adiabatic | Polytropic | Isothermal | Polytropic | Isothermal | Isothermal |
| State of catalyst | Catalyst bed | Catalyst bed divided into sections | Catalyst filled into shell or tube side | Catalyst filled into shell or tube side | Catalyst in fluidized bed | Catalyst suspended in liquid hydrocarbons |
| Cooling concept | Series of reactors with intercooling | Cold feed gas is injected between sections | Cooled by evaporation of water on opposing side of the catalyst | Cooled by preheating cold gas passing through the catalyst bed in tubes | Submerged coil heat exchanger | Liquid hydrocarbons as heat transfer medium; heat removed in internal heat exchanger |
| Advantages | Simple scale up, no mechanical stress of catalyst, defined residence time | No mechanical stress of catalyst, improved temperature control | Very efficient heat recovery, good heat transfer and temperature control | High heat transfer coefficients, uniform temperature distribution | Excellent heat transfer performance, isothermal reaction profile | |
| Disadvantages | Lower heat transfer, danger of hotspots, several pressure vessels and piping | Large catalyst volume | Large number of tubes, expensive | Limited heat transfer | Non-uniform residence time (bubble formation), attrition of catalyst, erosion of internals, difficult scale up | High mechanical stress of catalyst |
One alternative is the electrochemical conversion of CO2 to value added products. CO2 can be directly reduced to different products including lower alkanes, formic acid or methanol.60 However, low solubility of CO2 and CO species in aqueous electrolytes currently limits the formation of long-chain hydrocarbons.61 Thus, of the products under consideration in this review, only methanol has been discussed for direct electrochemical production. Alternatively, a two-stage process is possible where CO2 is converted to CO in a first electrolyser, which then reacts to other products in a second electrolyser under alkaline conditions.61 Spurgeon et al.45 compared the direct and two-stage electrochemical production of ethanol as well as the direct production of formic acid and a PtL process with an electrochemical reduction to CO with subsequent thermochemical FT-synthesis. Their results indicate that the PtFT-fuels process offers cost advantages over electrochemical conversion and that a two-stage electrochemical synthesis has slight cost benefits over a direct conversion. Current advances and remaining challenges for the direct electrochemical conversion as well as economic aspects have been reviewed by Birdja et al.60 and Durst et al.62 A review of the two-stage process is provided by Jouny et al.61
In comparison to electrochemical conversion, photocatalytic CO2 conversion mimics natural photosynthesis by using solar energy to directly convert CO2 without producing electricity first. The photocatalytic synthesis occurs in three main steps, the solar-light absorption, charge separation and migration followed by the catalytic CO2 reduction.63 Similar to electrochemical CO2 conversion, the main products are CO, methane and methanol.64 Long-chain hydrocarbons comparable to FT-fuels or DME have not been produced yet. The main advantage of photocatalytic CO2-conversion is the independence from an external electricity source, allowing a higher flexibility in the choice of the location.65 However, solar-to-fuel efficiency remains low and costs of the photocatalytic devices high (mainly due to noble metal-based catalysts).66,67 A review on photocatalytic devices has been given by Jiang et al.66 Wang et al. discuss the recent progress for different photocatalytic CO2 conversion pathways64 and reviews on recent catalyst developments have been published.63,67
In addition to the above-mentioned pathways of fuel synthesis there are biological processes which can be used for the production of biofuels. Hereby the use of bio membrane-reactors and biocatalysts (enzymes) can be differentiated.68 For example, biological conversion of methane to methanol,69,70 the synthesis of petroleum-like hydrocarbons71 or biofuels from microalgae72 could be achieved. Currently, there are several challenges that need to be addressed, such as dissolving CO2 in or poisoning of the used solution.70 In a recent review by Bhatia et al., several biological ways of CO2 capture and conversion as well as their associated challenges are discussed.73
1.2. Considered power-to-liquid fuels and comparison with conventional fuels
The properties of methanol, DME and FT-fuel (diesel fraction) are compared with conventional fuels in Table 5. Methanol can be used in both diesel and Otto motors. It is miscible with gasoline and has a higher octane number, which increases the efficiency of combustion. While the local emissions are lower in methanol combustion, the energy density is only about 50% of gasoline and methanol is disadvantageous in terms of corrosion. Furthermore, methanol has no lubricating effect on the motor. Methanol-gasoline blends with high methanol percentage (M85) as well as pure methanol (M100) have been successfully used in Otto motors with little modification for example in Brazil and Sweden. Methanol can also be used in fuel cells (direct or with reformer) with a very good efficiency.| Properties | Gasoline | Methanol | Diesel | FT-fuel (diesel) | LPG | DME |
|---|---|---|---|---|---|---|
| Aggregate | Liquid | Liquid | Liquid | Liquid | Gaseous (liquid under pressure, 5–10 bar) | Gaseous (liquid under pressure, 5 bar) |
| Chem. formula | C5–C12 | CH3OH | C10–C23 | C10–C23 | C3–C4 | CH3OCH3 |
| Miscibility | In gasoline and diesel | In diesel | In LPG; in diesel | |||
| Degradable | No | Yes | No | Yes | Yes | |
| Pollution | Oxygen content reduces local emissions | High soot and NOX emissions | Less hydrocarbon, CO and particle emissions | NOX emissions −80%, KW emissions −50% compared to gasoline | No C–C binding → almost no particle emissions | |
| Density (g l−1) | 715–780 | 791 | 815–855 | 770–860 | 540 (at 10 bar) | 668 |
| Boiling point at 1 atm (°C) | 25–215 | 64.7 | 170–380 | 150–320 | −42 to −0.5 | −24.9 |
| Vapour pressure at 20 °C (bar) | 0.45–0.9 | 0.37 | 0.01–0.1 | 0.01–0.1 | 2.1–8.3 | 5.3 |
| LHV (MJ l−1) | 31.2–32.2 | 15.4–15.6 | 35.3–36 | 33.1–34.3 | 24.84 | 18.2–19.3 |
| Octane number74 | 90–95 | 110–112 | — | — | 105–115 | — |
| Cetane number | — | 5 (low) | 45–53 | 70–80 | — | 55–60 |
| Ref. | 75 and 76 | 75 and 76 | 75 and 76 | 74–76 | 75 and 76 | 75 and 76 |
Dimethyl ether is the simplest ether in nature. At ambient conditions, it is gaseous, just like LPG, but under moderate pressure (5 bar) it is in a liquid state. As it has no carbon bindings and contains no sulphur, soot and sulphur emissions are avoided. DME can be used in diesel engines, but modifications have to be made to the infrastructure to keep the fuel in a liquid state.77 The energy density is lower, but the cetane number is higher than for conventional diesel, hence DME is seen as a promising option for heavy-load traffic. Further, DME might have advantages in terms of maximum energy efficiency compared to MeOH and FT-fuels.78
FT-based diesel has very similar properties compared to conventional diesel, except that it contains no sulphur and no aromatics, which decreases emissions drastically. Hydrocarbon, CO, NOx and particle emissions are only 40–80% of those in conventional diesel combustion.35 In the production process, the product can be adapted to the motor, which can increase the combustion efficiency. Due to the very similar properties, infrastructure and motors of conventional diesel can be used without adaption. FT-synthesis can be realised with biogenic feedstock or, to increase the output of conventional fuel production, it can be applied to stranded gas. Its products can be blended with low-quality conventional diesel to increase the yield of refineries.74 The similarity with conventional diesel also causes some drawbacks. The product of FT-synthesis has to be fractionated to gain valuable products. Also, some products have to be treated further, e.g. in hydrocrackers. However, this also means that other products like diesel, gasoline and waxes can be produced on the same route.
2. Methanol synthesis
2.1. State of the art methanol synthesis
| CO2 + 3H2 ⇌ CH3OH + H2O ΔH = −49.4 kJ mol−1 | (2) |
While CO2 hydrogenation incorporates the formation of water, CO is converted to CO2via the reverse water gas shift reaction (RWGS) by consumption of water:
| CO2 + H2 ⇌ CO + H2O ΔH = 41.2 kJ mol−1 | (3) |
The CO conversion can be expressed as:
| CO + 2H2 ⇌ CH3OH ΔH = −90.6 kJ mol−1 | (4) |
Both synthesis reactions are exothermal and involve a decrease of volume. Thus, methanol formation is favoured by low temperatures and elevated pressures. CO hydrogenation is significantly more exothermic than CO2 hydrogenation resulting in a higher cooling demand. The maximum conversion is determined by the chemical equilibrium shown in Fig. 4 for a CO- and CO2-based feed gas. As seen, the equilibrium limited methanol yield of CO2 (18–58% at 200–250 °C, 50–100 bar) is substantially lower than that of CO (55–89%).
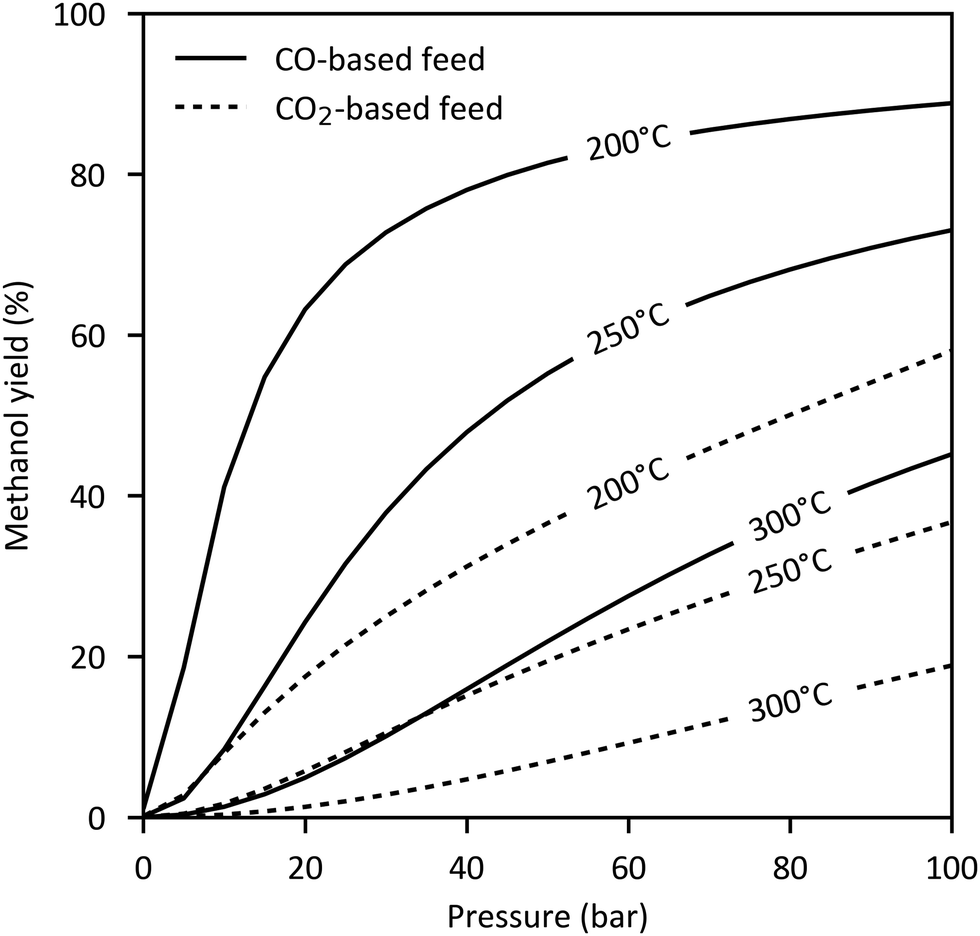 | ||
| Fig. 4 Equilibrium conversion of CO- or CO2-based feed gas to methanol. | ||
Based on equilibrium considerations, the highest methanol yield results from an educt gas composition with a stoichiometric number of 2 in the absence of CO2, while for CO2-based feed gas a H2/CO2 ratio of 3 to 1 is optimal. In practice, a slight excess of hydrogen given by an adjusted stoichiometric number of 2.02–2.1 is used, as this has been found to improve the space time yield of the catalyst and avoids by-product formation by a deficiency in hydrogen.55,59,94,95 Additionally, experience with commercial copper-based catalysts reveal a maximum conversion at low CO2 contents between 2–5%.55,81,85,88,96 For higher CO2 concentrations the methanol synthesis is inhibited by water formed in the reverse water gas shift reaction.26,92
An overview of the composition, operating conditions and performance of some catalysts is listed in Table 6. The reported space time yields (STY) for CO syngases are in the order of 0.7–2.3 kg of methanol per litre of catalyst per hour (40–100 bar GHSV around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) which is reported from commercial plants as well.97–99 CO2-based feed gases usually show lower STY in the order of 0.4–0.8 kg lcat−1 h−1.
000 h−1) which is reported from commercial plants as well.97–99 CO2-based feed gases usually show lower STY in the order of 0.4–0.8 kg lcat−1 h−1.
| Licensor | Compositiona CuO/ZnO/Al2O3 (wt%) | T (°C) | p (bar) | GHSVb (×103 h−1) | STY (kg lcat−1 h−1) | Byprod.c (wt%) | Ref. |
|---|---|---|---|---|---|---|---|
|
a Partially calculated based on given atomic ratio.
b Gas hourly space velocity as ratio of feed gas volume flow under standard conditions to catalyst volume.
c Except water.
d H2:CO > 2:1 (H2 65–90%, CO 5–25%, CO2 4–14%).
e Applicable temperature range 200–310 °C, pressure range 20–150 bar.
f Activity at start of run, decreased to 1.28 kg l−1 h−1 after 1000 h.
g Activity at start of run, decreased to 2.20 kg l−1 h−1 after 1000 h.
|
|||||||
| CO-based feed gasd | |||||||
| Süd Chemie | 65–68/21–23/10–12 | 250 | 50 | 10 | 1.03–1.10 | 0.3–0.4 | 104 |
| BASF | 40/52/9 | 230 | 50 | 10 | 1.43 | <0.6 | 106 |
| ICI | 62/32/6 | 240 | 51 | 9.6 | 0.43 | <0.2 | 107 |
| Shell | 65/29/0/6 (di) | 250 | 53 | 11.5 | 0.69 | Trace | 108 |
| 300 | 53 | 10.9 | 1.01 | <1.3 | |||
| Casale | 30/50/3/16 (Cr2O3) | 250 | 100 | 12.5 | 1 | n.a. | 109 |
| MGC | 34/26/3/36 (Zr) | 264 | 100 | 10 | 2.22 | n.a. | 103 |
| Topsoe (MK-121) | >55/21–55/8–10 | 200–310 | 39–122 | n.a. | n.a. | n.a. | 100 |
| Topsoe (MK-101) | ×/×/× | 221e | 67e | n.a. | 1.02 | n.a. | 105 |
| ICI | 40/41/0/19 (Cr2O3) | 260 | 41 | 6–7 | 0.26 | <0.1 | 110 |
| 260 | 41 | 10 | 0.38 | <0.1 | |||
| 260 | 81 | 10 | 0.77 | <0.1 | |||
| Metallgesellschaft | 60/30/0/10 (Cr2O3) | 250 | 41 | 9.8 | 1.42f | n.a. | 111 |
| 250 | 101 | 9.8 | 2.28g | n.a. | |||
| NIRE/RITE | ×/×/×/ZrO2/SiO2 | 250 | 50 | 10 | 1.7 | n.a. | 112 |
The main suppliers of conventional methanol catalysts are Johnson Matthey (Katalco 51-series), Clariant (MegaMax 700/800/NJ-1), Haldor Topsøe (MK 121 and MK 151-Fence) and Mitsubishi Gas Chemicals (M5-5, M6).24 The catalysts are usually offered in a tablet shaped (e.g. 6 × 4 mm100,101 or 5.1 × 5.3 mm102) and have a bulk density in the range of 1000–1300 kg m−3.98,102–106
Modern copper catalysts achieve a high selectivity (apart from water) of over 99%24,113 (up to 99.9% reported by Haldor Topsøe)114 for standard syngas. Generally the low content of impurities is remarkable as the formation of typical by-products like higher alcohols (predominantly ethanol), esters (mainly methyl formate), ether (dimethyl ether), ketones (mainly acetone) and hydrocarbons are thermodynamically favoured over methanol (except for formaldehyde and formic acid).26 By-product formation is promoted by catalyst impurities (residual amounts of alkalis), high pressures, high temperatures (requiring adequate control of the converter temperature), higher CO/H2 and CO/CO2 ratios, as well as lower space velocities (higher residence time).115,116
In industrial applications the common catalyst life time is 4–6 years95,117,118 with up to 8 years being reported.102 Life time is limited by catalyst deactivation caused by poisoning and thermal sintering. Major poisons for copper catalysts are sulphur compounds (blocking of active sites) and chlorides (acceleration of sintering). The tolerance against sulphur is higher than chlorides, as sulphur is scavenged by the ZnO component of the common Cu/ZnO catalysts.119 Other poisons reported in industrial applications are, for example, arsenic or carbonyls (formed at high CO concentrations at inappropriate types of steel)120 resulting in a decrease of selectivity due to promotion of Fischer–Tropsch side reactions.121 Typical gas purity requirements are summarised in Table 7. These impurities are commonly removed by prior gas cleaning. Additionally, guard beds (e.g. ZnO targeting sulphur) are often installed to protect the catalyst.113,120,122 As a result, in normal operation of methanol synthesis plants deactivation is dominated by thermal sintering.113 A growth of copper crystallite size is reported at temperatures higher than 227 °C and above 300 °C a sintering of ZnO is also observed.119
Typical curves of deactivation of commercial methanol catalysts are illustrated in Fig. 5. After an initial strong decline in catalyst activity during the first 1000 h (−14 to −21%/1000 h), the deactivation slows down, resulting in a mean deactivation rate of about 2%/1000 h over 3 years (based on presented data). Operation with CO2-based feed gas indicates comparable stability to syngas, with a decrease in catalyst activity of 8% in the first 400 h,124,125 but there is still a lack of long-term studies of CO2-based pilot plants.126 Intensive research on catalysts results in enhanced activity and stability. Haldor Topsøe reports a 20 percentage-point increase in activity at the end of life in each case from the MK-101 to the MK-121 to their latest methanol catalyst MK-151 Fence.127 To maintain a constant production of methanol in practice, the operating temperature (or the pressure) is gradually increased during time on stream.105,121,128
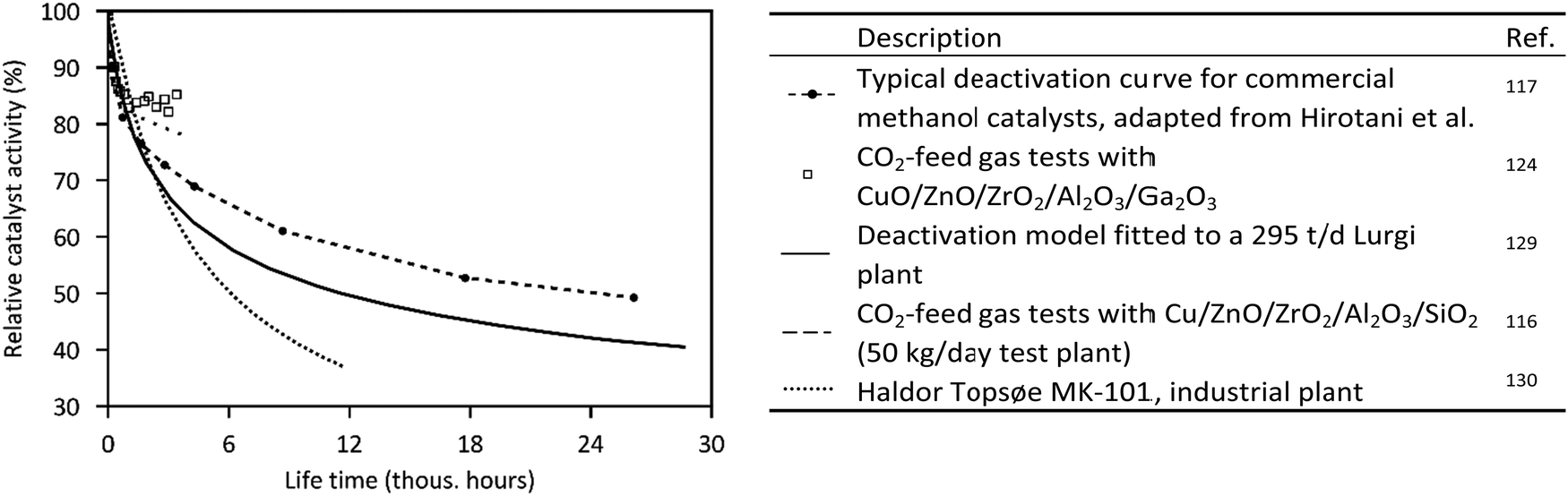 | ||
| Fig. 5 Deactivation curves of commercial methanol catalysts.116,117,124,129,130 | ||
000 t per d have been developed. Compared to a 2000 t per d plant the specific costs can be reduced by 30% for a 5000 t per d plant and by 40% for a 10000 t per d plant.131 Hereby the maximum capacity of the methanol converter is limited energetically by the pressure drop as well as by manufacturing and transportation constraints of the pressure vessel. Methanol production is mainly based on syngas from reforming natural gas. In the last few years methanol production via coal gasification has been growing too, driven by China.
The main challenge in reactor design is to economically remove the heat of reaction avoiding by-product formation and achieving a high conversion rate by low outlet temperatures and a good energy efficiency by internal heat recovery.26
A summary of the main commercial reactor types (Johnson Matthey Davy Technologies is missing due to scarce information)132 is given in Table 7. Respective simplified reactor schemes are illustrated in Fig. 6 and detailed temperature profiles are shown in Fig. 7.
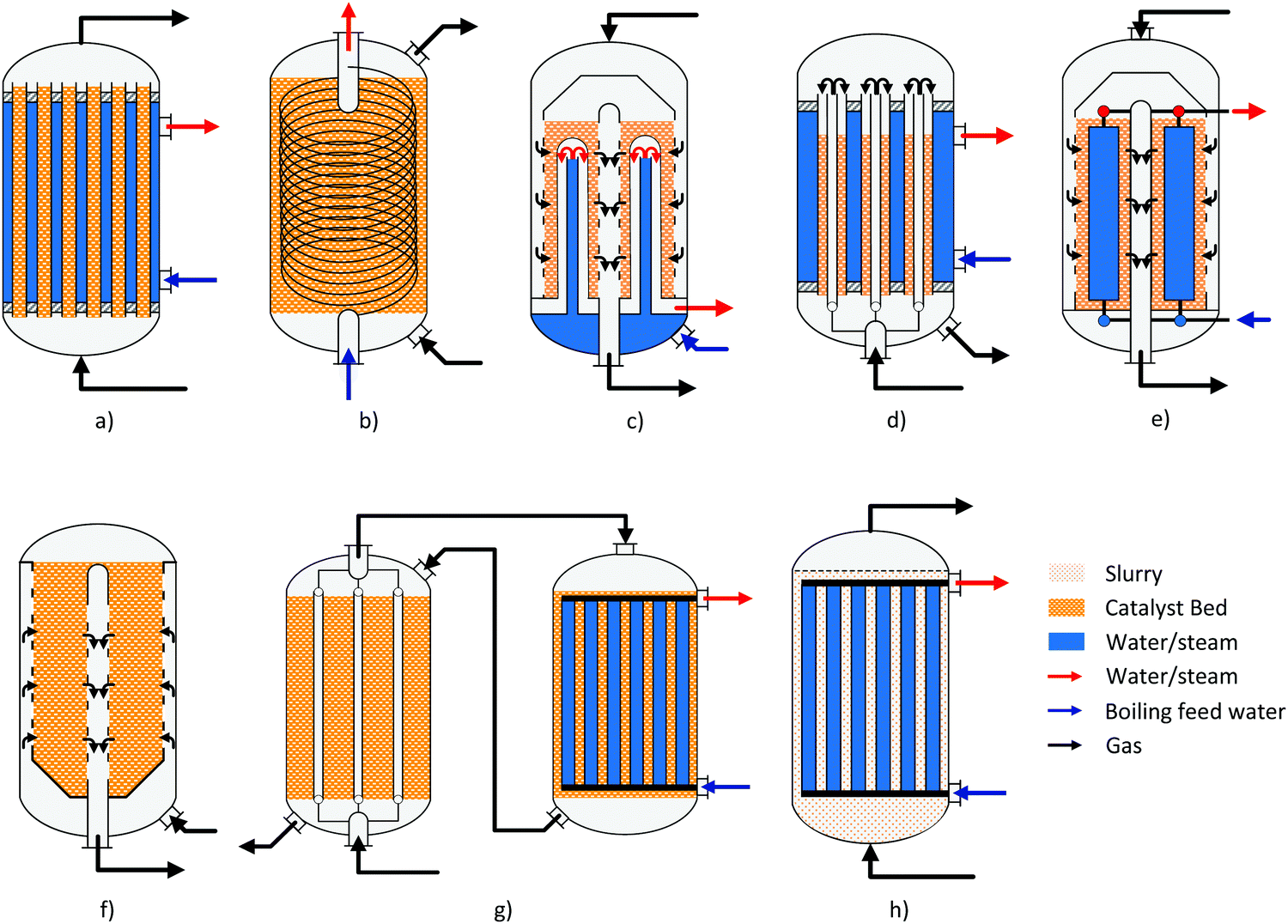 | ||
| Fig. 6 Simplified reactor layout of (a) Lurgi tubular reactor, (b) Linde Variobar, (c) Toyo MRF, (d) Mitsubishi Superconverter, (e) Methanol Casale IMC, (f) Haldor Topsøe adiabatic reactor, (g) Lurgi MegaMethanol and (h) Air Products LPMEOH, adapted from Buttler 2018.138 | ||
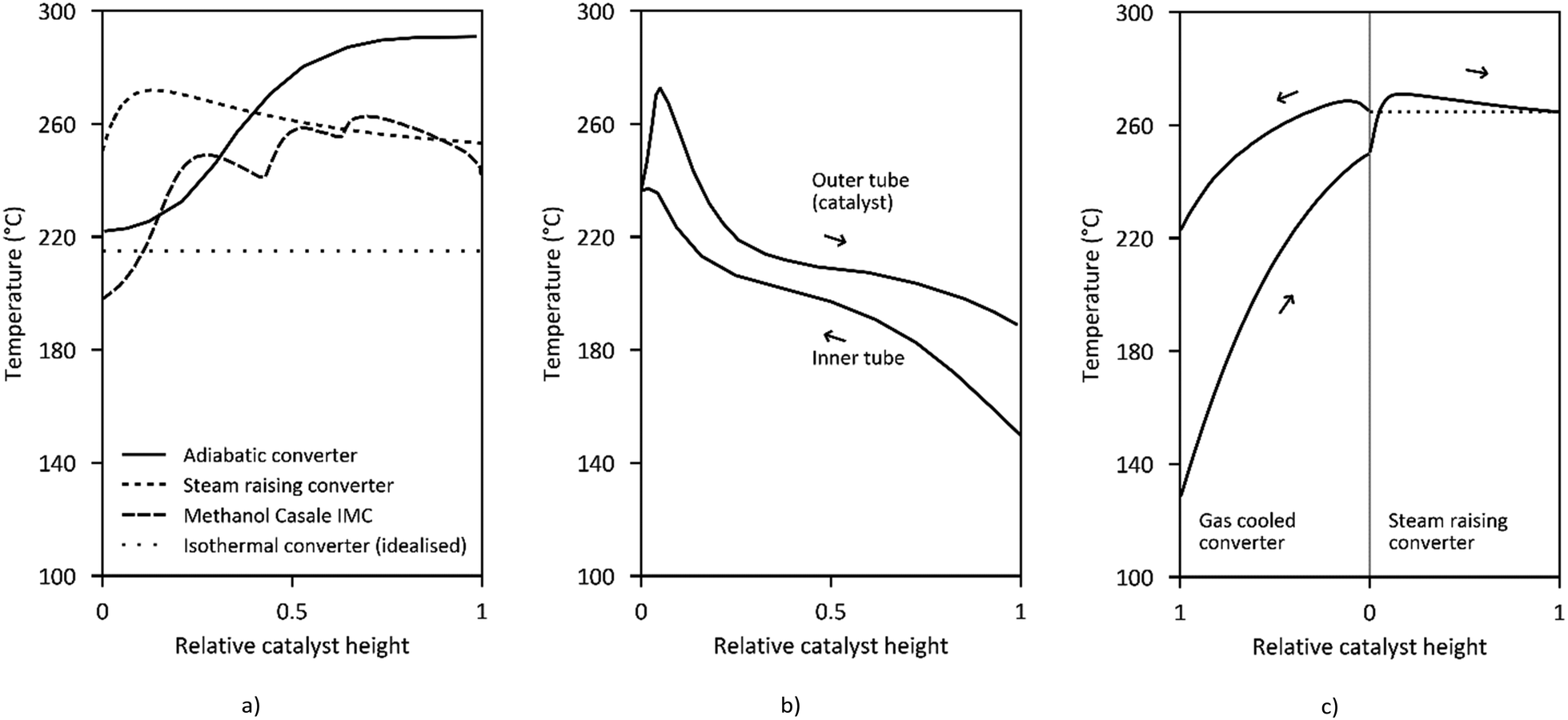 | ||
| Fig. 7 Temperature profiles along the reactor length of (a) adiabatic converter,105 steam raising converter,126 Methanol Casale IMC,142 and isothermal converter (idealized), (b) Mitsubishi Superconverter,143 (c) Lurgi MegaMethanol.99 | ||
Current reactor designs are dominated by quasi-isothermal steam-raising fixed bed reactors (SRC) which replaced the former design of quench reactors due to the higher catalyst volume utilisation, improved heat recovery and temperature control. SRC were introduced for methanol synthesis for the first time by Lurgi (now part of Air Liquide) at the beginning of the 1970s.133 The Lurgi design is based on a tubular reactor (Fig. 6a). The feed gas flows in an axial direction through the tubes filled with catalyst cooled by the surrounding boiling water on the shell-side. About 80% of the reaction heat is converted to medium pressure steam.99 Similar designs are currently offered by Johnsson Matthey Davy Technologies132 and Haldor Topsøe.114 Haldor Topsøe optionally proposes an adiabatic catalyst bed installed upstream in a separate vessel or on top of the upper tube sheet resulting in an optimised temperature profile.134 The maximum single-train capacity of SRCs is limited to 1500–2200 t per d (the range mainly depends on the syngas activity).114,117,135 Further quasi-isothermal fixed bed reactor concepts are licensed by Linde, Toyo, Methanol Casale and Mitsubishi Heavy Industries as discussed in the following.
The Linde Variobar process is based on their coil-wound heat exchanger with catalyst loaded on the shell-side and boiling water circulating through the tubes (Fig. 6b).136 Linde claims that this design features the highest catalyst per reactor volume ratio of all isothermal reactors and high heat transfer coefficients due to the cross flow reducing the heat transfer area.137
Toyo Engineering Corporation (TEC) developed a Multi-stage Radial Flow (MRF) reactor jointly with Mitsui Toatso Chemicals (MTC).139 It is characterised by the radial flow of syngas across the catalyst bed loaded in the shell-side, which is indirectly cooled by bayonet boiler tubes (Fig. 6c). Thermal stress is avoided by using duplex tubes with the coolant first flowing up the inner tube before being evaporated in the outer tube. The arrangement of the tubes results in concentric cooling zones. This allows the temperature profile to be adjusted close to the path of the maximum reaction rate curve.117 The radial flow arrangement results in a very low pressure drop across the reactor (<0.5 bar).117,139 This has the advantage of a simple scale-up by vertical extension of the reactor with single-train capacities of 5000 t per d.117
Mitsubishi Gas Chemical and Mitsubishi Heavy Industries have jointly developed the Mitsubishi Methanol Process based on the so-called Superconverter (Fig. 6d). This complex reactor design combines the features of a tube gas cooling reactor and a steam-raising tubular reactor by using double-pipe tubes in a boiling water vessel (or even triple pipes).140 The feed gas first flows upwards through the inner tube before passing downwards through the outer tube which is filled with catalyst.141 Preheating the feed gas in the inner tube results in a specific temperature profile (Fig. 7b) along the catalyst bed with a high temperature (maximum 250–260 °C) near the inlet that gradually declines towards the outlet (240–250 °C) closely following the maximum reaction rate line.140
The Methanol Casale (pseudo) Isothermal Methanol Converter (IMC) is cooled by feed gas, boiling water or a combination of both inside hollow plates which are immersed in the catalyst bed (Fig. 6e). Independent temperature control of different parts of the plates is possible by adjusting the cooling fluid flow at different heights.144 Thereby the quasi-isothermal temperature profile (Fig. 7a.3) can be modified to fit the maximum reaction rate curve.128,145 For capacities up to 2000 t per d axial flow of the process gas is preferred.146 For larger capacities up to 7000–10000 t per d in a single converter, axial-radial flow configuration is used due to the lower pressure drop.146–148
Another concept which is especially suited for large capacities of 10000 t per d in a single line are multi-stage adiabatic fixed bed reactors with intercooling as offered by Haldor Topsøe (Fig. 6f) and Kellogg (now part of KBR).26 Very large capacities based on axial flow steam-raising converters are either realised by several reactors in parallel or by dual stage concepts like the MegaMethanol concept of Lurgi (Fig. 6g), with a capacity of 5000 t per d.94 The syngas is partially converted to methanol in a steam raising reactor before it is further converted in a tubular reactor cooled in counter-current (or co-current) flow with the cold feed gas for the first reactor.149 This has the advantage that only a small feed gas preheater (up to about 130 °C) is required which results in a decreasing temperature along the reaction path maintaining the equilibrium driving force for methanol production (Fig. 7c).
Innovative reactor concepts. Beside conventional fixed bed reactor design, alternative concepts have been investigated which are very well discussed by Hansen and Nielsen26 and Riaz et al.28 Examples are membrane reactors or sorption-enhanced reactor concepts150,151 to overcome the equilibrium conversion by removal of products or fluidized bed reactors (e.g. 10 t per d demo plant152), trickle bed reactors153 and slurry reactors,121 which eliminate the diffusion limitations and offer a good heat transfer. To date, none of the alternative concepts have any industrial relevance. Membrane reactors modules are still in the fundamental research stage and suffer from low conversion (experimentally below 9%)154–162 as well as durability and cost issues, while fluidized bed reactors have problems with attrition of the catalyst.26
The only one of these alternative concepts that has been tested on an industrial scale is the Liquid Phase Methanol (LPMeOH) process developed by Air Products and Chemicals (1997–2002, Eastman Chemical's Coal Gasification Complex, 235 t per d, documented in ref. 121 and 163–165). It is based on a slurry bubble column reactor with fine catalyst particles suspended in an inert mineral oil acting as an efficient heat transfer medium (Fig. 6h). The heat removal via an internal heat exchanger results in an almost isothermal operation of the reactor. The superior temperature control compared to conventional fixed bed converters enables the use of CO-rich syngases (in excess of 50% tested) without damaging the catalyst by excessive temperature peaks. However, the LPMeOH process has a poor reactor volume utilisation due to the limited catalyst loading (slurry concentration of 25–50 wt%).166
Haldor Topsøe demonstrated a novel methanol fixed bed reactor synthesis as part of the small-scale BioDME pilot plant in Pitea commissioned in 2010.167,168 Once-through operation is achieved by a second radial converter after the conventional steam raising converter. The second converter is operated under methanol condensing conditions.169 This results in a once-through methanol yield higher than 95% (130 bar) as the equilibrium constraint can be overcome.170 The first experiments of this technology were already published in 1991.171
Operating conditions and performance of conventional converters. The operating conditions are mainly determined by the applied catalysts while the temperature profile along the reaction path and the heat recovery is determined by the reactor design (a good overview of characteristic conversion profiles of different converter types is given by Hirotani et al.).117 Reported exemplary operating parameters and performance of converter designs are summarised in Table 8.
| Lurgi | Variobar | MRF-Za | Super-converter | IMCb | Haldor Topsøe | Mega Methanol | LPMeOHc | |
|---|---|---|---|---|---|---|---|---|
| a Multi-stage indirect cooling and radial flow. b Isothermal methanol converter. c Liquid phase methanol. d Simplified reactor schemes are presented in Fig. 6. e Haldor Topsøe also offers steam raising converters. f Catalyst powder suspended in mineral oil. g <2000 t per d axial flow, >2000 t per d axial–radial configuration.146 h Outlet/Peak catalyst temperature, temperature profiles see Fig. 7. i Due to catalyst aging. The demonstration unit was tested from 214–259 °C.121 j <1000 t per d, p = 50–80 bar, >1000 t per d, p = 70–150 bar.137 k Revamping projects with operating pressures of 46–100 bar128,148 realised. l Defined as ratio of recycle gas to fresh make-up gas based on volume flow. m Reactor only. n Pressure loss of the overall synthesis loop is 3.5–4 bar.55 o Pressure loss of the overall synthesis loop is 3 bar.139 p 2000 t per d. q 3000 t per d. r Per pass conversion = 1 − (COout + CO2,out)/(COin + CO2,in) based on given molar flow rates (partially own calculations based on given inlet and outlet compositions). s Methanol concentration at reactor outlet. t Depending on the operating pressure (65–79 bar).120 u >5000 t per d by two SRC in parallel and condensation of methanol before one GCC.24 v Data from 2013.142 In total 23 new projects and 22 revamping projects since 1993.175 w First MegaMethanol plant with 5000 t per d commissioned in 2004. | ||||||||
| Licensor | Lurgi | Linde | Toyo (TEC) | MGC and MHI | Methanol Casale | Haldor Topsøe | Lurgi | Air products and chemicals |
| Reactor typed | SRC | SRC | SRC | GCC/SRC | SRC | Adiabatice | GCC and SRC | Slurry |
| Catalyst location | Tube-side | Shell-side | Shell-side | Double-pipes | Shell-side | Fixed bed | Shell-side | Shell-sidef |
| Heat exchanger | Tubular | Tubular (spiral) | Bayonet tubes | Tubular | Plate | Intercooler | Tubular | Tubular |
| Flow | Axial | Radial | Radial | Axial | Axial/radialg | Radial | Axial | Axial |
| Stages | 1 | 1 | 1 | 1 | 1 | 2–4 | 2 | 1 |
| T (°C) | 255/270 | n.a. | 240/280 | 190/270 | 225/280 | 290/n.a. | 220/270 | 215i |
| p (bar) | 50–100 | 50–150j | 80–100 | 55–100 | 65–80k | 50–100 | 75 | 30–50 |
| Recycle ratiol | 3–4 | n.a. | n.a. | 2–3 | 3 | 3–5 | 2–2.7 | 1–5 |
| Pressure lossm (bar) | 3n (295 t per d) | n.a. | 0.3–0.5o (300–2500 t per d) | 2.4–7.5 (10 t per d) | 1.1 (axialp), 0.3 (radialq) | n.a. | n.a. | 3–4.8 (235 t per d) |
| Per pass conversionr (%) | 36 | n.a. | 60 | 55–67 | n.a. | n.a. | >80 | 20–50 |
| Y MeOH (mol%) | 6–7 | n.a. | 10 | 10–15 | 10.1–13.3t | 7 | 11 | 8–12 |
| p Steam (bar) | 29–43 | 40 | n.a. | 19–45 | 25–32 | n.a. | 50–60 | 16–25 |
| Max. capacity (t per d) | 1500–2200 | 4000 | 5000 | n.a. | 7000–10000 |
10000 |
5000–10000u |
Low |
| Industrial references | >55 plants licensed | 8 plants | 6 projects, 315–3000 t per d | 9 plants, 520–5000 t per d | 9 plants, 1350–3000 t per dv | >40 plants | >10 plants since 2001w | 235 t per d demo-plant |
| Ref. | 55, 98, 99, 114, 117, 126, 131, 135 and 177 | 24, 29, 118, 137 and 182 | 117, 139 and 181 | 97, 140, 143 and 178–180 | 95, 120, 128 and 146–148 | 55, 105, 122, 127 and 134 | 99, 173, 176 and 177 | 121 |
High operating temperatures improve the reaction kinetics (at the risk of by-product formation) while low temperatures at the reactor outlet are favoured by the equilibrium, maximising the overall conversion. Detailed temperature profiles of exemplary converters are given in Fig. 7. As seen, the Superconverter design as well as the MegaMethanol process achieve relatively low temperatures at the outlet and only a small preheater is necessary due to the internal preheating of the feed gas. The heat transfer coefficient and the peak catalyst temperature determine the steam pressure of the cooled reactors, which is typically below 300 °C. Resulting steam pressures are in the range of 16–60 bar (Tsat ∼ 200–275 °C). Steam raising converters produce about 0.7–1.4 t of medium pressure steam per tonne of methanol.55,121,143,148
In order to achieve an almost complete carbon conversion rate (typically 93–98%, dependent on purge gas losses)114,148,172 the rate of recycled unconverted gas to fresh feed gas must be between 3 and 5 (Table 8). The recycle gas ratio can be reduced in case of the two-stage MegaMethanol process to 2–2.7.24,173 By condensation of methanol after the first reactor, the driving force of the methanol reaction is increased, allowing for a reduced size of the gas cooled reactor and a lower recycle gas ratio of about 1.7.174 The recycle gas ratio affects the size of the equipment as well as the power requirement of the recycle compressor. Beside the reactor design (and catalyst activity), the recycle gas ratio is also dependent on the reactivity of the gas. Reported conversion per pass lie in the range of 20–50% for the LPMeOH-Process,121 below 40% for conventional SRC (own calculation based on98), 55–67% for advanced SRC like Toyo MRF and Mitsubishi Superconverter (own calculations based on ref. 97 and 117) and above 80% for the two-stage MegaMethanol process.94 The resulting methanol concentrations at the reactor outlet are in the range of 6–14 mol%.
Due to the energy penalty of compressing the recycle gas, the pressure loss across the reactor (and the preheater and product cooler) is important. Typical pressure losses are below 0.5 bar for radial flow converters (overall pressure loss of 3 bar for a 2500 t per d synthesis loop139) or between 2.4 and 7.5 bar for axial flow converters (Table 8).
The amount of purge gas is dependent on the inert gas fraction (N2 and CH4) in the feed gas. In the LPMeOH experiments about 2–6% of the recycle gas were purged (corresponding to 1–5% of the reactor inlet gas or 5–20% of the feed gas) keeping the fraction of N2 and CH4 below 5% in the reactor inlet gas.121
Selected gas hourly space velocities (GHSV), the ratio of feed gas volume flow under standard conditions to catalyst volume, are in the range of 6000–12000 h−1 for single-stage steam raising reactors.55,97,98,173 For two-stage concepts, increased GHSV of 14000–24000 h−1 are reported.173 The LPMeOH demonstration plant was operated at lower space velocities in the range of 2000–5000 h−1 (own calculations based on ref. 121).
These operation experience of CO-syngas converters represent the basis for CO2 conversion pilot plants as discussed in the next section.
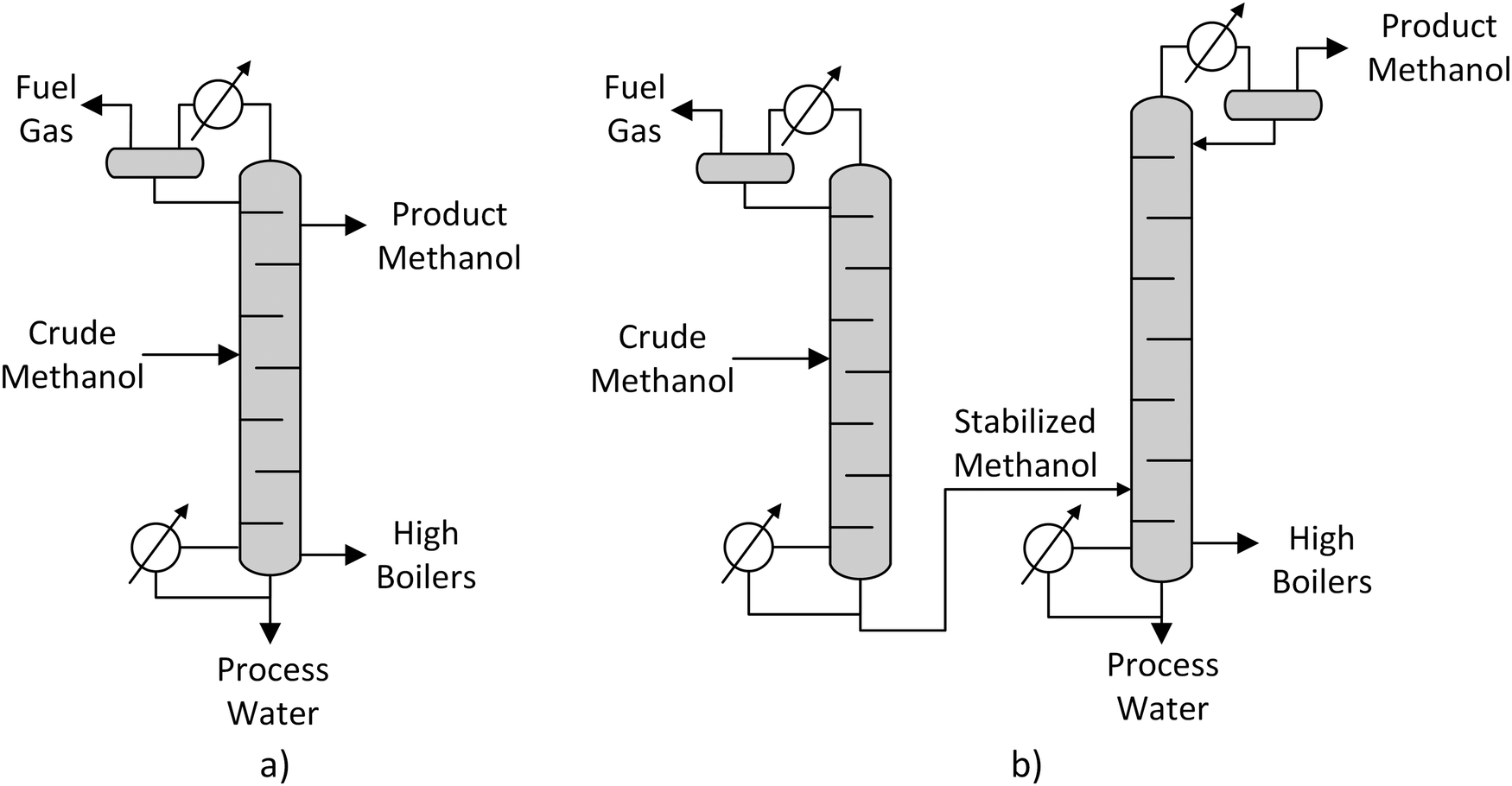 | ||
| Fig. 8 Single column (a) and two-column (b) methanol distillation, own illustration based on Supp.55 | ||
2.2. Current research on power-to-methanol
| Licensor | Composition CuO/ZnO/Al2O3 (wt%) | T (°C) | p (bar) | GHSVb (×103 h−1) | STY (kg lcat−1 h−1) | Byprod.c (wt%) | Ref. |
|---|---|---|---|---|---|---|---|
|
a H2/CO2 of 3:1(H2 = 75%, CO2 = 25%) with 22% CO2 and 3% CO, H2/(CO + CO2) = 2.4 and CO2/(CO + CO2) = 2.4 after RWGS reactor (H2 = 70.6%, CO = 17.6%, CO2 = 11.8%).
b Activity at start of run, decreased to 0.63 kg l−1 h−1 after 1000 h.
c Activity at start of run, decreased to 0.70 kg l−1 h−1 after 1000 h.
d In volume percentage.
e Calculated based on given catalyst volume and reactor inlet flow.
|
|||||||
| Süd Chemie | n.a. | 250 | 80 | 10.5 | 0.6 | 0.04 | 126 |
| NIRE/RITEa | x/x/x/ZrO2/Ga2O3 | 250 | 50 | 18 | 0.75b | 0.04 | 124 |
| NIRE/RITEa | x/x/x/ZrO2/SiO2 | 250 | 30–70 | 10 | 0.4–0.8 | 0.05 | 112 |
| NIRE/RITEa | 45/27/5/23(ZrO2)/0.6 (SiO2) | 250 | 50 | 10 | 0.76c | n.a. | 186 |
| JM Katalco 51-8 | ×/×/× | 240 | 69–97 | 3.3–8.3 | 0.06–0.24 | <0.05d | 183 |
| Süd Chemie (C79-05-GL) | ×/×/× | 260 | 80 | 8.1 | 0.66 | n.a. | 189 |
| Commerciala | 60/30/10 | 250 | 30 | 7.9e | 0.6 | n.a. | 190 |
Typical compositions of crude methanol produced from syngas and CO2-based feed gas are compared in Table 10. The CO2 conversion in reaction (2) is accompanied by formation of water resulting in significantly higher water contents of 30–40% of the crude methanol compared to 10–15% for syngas.126,191 However, the formation of other by-products (see Tables 6 and 9) is lower for CO2-based feed gas with contents below 0.05 wt% in crude methanol compared to 0.1–0.6 wt% for CO-syngas.
| CO-Syngas | CO2-based feed gas (H2:CO2 = 3) |
||||||
|---|---|---|---|---|---|---|---|
| a Mainly methyl formate. | |||||||
| Temperature (°C) | 250 | 250 | 255 | 260 | 230 | 250 | 270 |
| Main components (wt%) | |||||||
| Methanol | 84.5 | 63.7 | 63.7 | 63.7 | 63.3 | 63.0 | 63.4 |
| Water | 15.4 | 36.2 | 36.3 | 36.3 | 36.7 | 36.9 | 35.6 |
| Impurities (wt ppm) | |||||||
| n-Paraffins | 78 | 0 | 0 | 0 | |||
| Higher alcohols | 626 | 89 | 105 | 148 | 28 | 45 | 92 |
| Estersa | 582 | 145 | 140 | 129 | 450 | 290 | 270 |
| Ketones | 24 | 0 | 0 | 0 | |||
| Dimethylether | 61 | 14 | 18 | 24 | |||
| Total impurities (wt ppm) | 1371 | 248 | 263 | 301 | 478 | 335 | 362 |
| Methanol selectivity, except water (%) | 99.84 | 99.96 | 99.96 | 99.95 | 99.92 | 99.95 | 99.94 |
The higher selectivity of CO2 conversion compared to CO conversion can be partly explained by lower peak temperatures in the reactor at the same jacket temperature as CO2 hydrogenation is less exothermic. However, even at comparable peak temperatures by-product formation from syngas is about three times higher than for CO2-based feed gas. Based on this, Pontzen et al. concluded that CO2 conversion can be run at higher temperatures by less intense cooling in order to increase productivity.126 Further, Göhna and König from Lurgi concluded that the energy demand of distillation can be reduced by about 20%.191
An overview of main operating and performance parameters of CO2 to methanol pilot plants is given in Table 11. Operating parameters are in the range of conventional syngas processes (T ∼ 250 °C, p = 50–100 bar, GHSV ∼ 10000 h−1). However, the reported carbon conversion rate is lower in several pilots ranging from 40–97% (with recycle). Most of the pilot plants are on a laboratory or small demonstration scale (<30 t per a). The only commercial plant has been commissioned by Carbon Recycling International in 2012 with a capacity of 4000 t per a.193
One of the first industrial-scale oriented investigations of the conversion of CO2 to methanol was presented in the beginning of the 1990s by Lurgi AG which developed a two-stage process (Fig. 9).191,194 The make-up gas is pre-converted (10–30% of carbon oxides) in an adiabatic fixed bed reactor in a once-through operation before entering a synthesis loop incorporating a steam raising quasi-isothermal reactor. In the adiabatic reactor the temperature is only slightly increased by maximum 35 °C as the exothermic methanol formation and the endothermic water gas shift reaction proceed simultaneously. Water and methanol formed in the adiabatic reactor are separated, promoting the methanol formation in the second reactor. The overall carbon conversion to methanol is 58–61% (based on data of ref. 194). Results of a single-stage CO2 to methanol pilot plant presented by Lurgi (now part of Air Liquide) in 2011126 showed a total CO2 conversion of 94–97% (per pass conversion of 30–45%).
 | ||
| Fig. 9 Simplified scheme and process parameters of the Lurgi two-stage process for methanol synthesis from CO2 (own illustration based on König and Göhna194). | ||
Specht et al. reported the successful conversion of atmospheric CO2 (sequestrated by caustic scrubber and electro dialysis) and hydrogen to methanol in a bench-scale test plant in 1996.195 Per pass conversion of 23%189 is stated for the methanol converter and a total conversion of 98% is estimated by recycling unreacted syngas.123
The CAMERE process (carbon dioxide hydrogenation to form methanol via a reverse water–gas shift reaction) is another two-stage concept that has been developed at the Korean Institute of Science and Technology (KIST). In contrast to the Lurgi process, the first reactor focuses on the endothermic reverse water–gas shift reaction only. It is electrically heated and operated at higher temperatures of 600–700 °C with the goal of 60% conversion of CO2 to CO.190,196 Due to the increased CO content and removal of by-product water prior to the methanol synthesis loop, the per pass methanol yield is increased and recycle gas ratio is reduced. In minipilot experiments a methanol yield of 53% (own calculations based on ref. 190) was achieved. Although a significant increase in methanol production is claimed, the STY of 0.6 kg lcat−1 h−1190 (excluding the catalyst volume of the RWGS reactor) is in line with the results from direct hydrogenation pilot plants.126,189,197,198 A 75 kg per d pilot plant has been constructed in collaboration with POSCO (Korean Pohang Iron and Steel Company) and KEPRI (Korea Electric Power Institute) in combination with a pilot plant for CO2 separation from a power plant. The pilot plant reached a methanol yield of 70% at a CO2 conversion in the RWGS reactor of 35%.196
| Institute/company, country, year | Capacity (kg d−1) | Reactor volume (l) | T (°C) | p (bar) | GHSV (×103 h−1) | Carbon efficiencya (%) | Recycle (ratiob) | Descriptionc | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Carbon efficiency = CH3OHProduct/(CO + CO2)MUG. b Recycle gas ratio = Recycle (mol s−1)/MUG (mol s−1). c All plants based on fixed bed reactor technologies. d First CO2 results by Göhna and König191 in 1994. e Peak temperature in catalyst bed of 260–264 °C. f Per pass conversion of 35–45%. g Calculated based on catalyst volume and STY. h Catalyst volume. i d = 38.4 mm, l = 4 m; given catalyst volume = 0.5–3 l. j Per pass conversion of 13.6–16.7%. k Purge ratio = 0.5–1% of reactor inlet flow. l Designed for 100 kg d−1. m Calculated based on cat. volume and inlet flow. n Calculation based on given material balance of ref. 190 who states a carbon efficiency of 89% not taking into account purge gas losses. Per pass conversion of 30%. o Due to prior RWGS, Purge ratio = 40% of MUG or 22% of reactor inlet flow. p 100 t per a. q Including recycling of the combusted purge gas. r Purge = 27–37% of MUG, 7–10% of reactor inlet flow. s Based on given dimensions (l = 30.5 cm, d = 2.5 cm). t Based on flow (1.5–30 NLPM) and reactor volume. u Plant opening with 1300 t per year, expanded in 2014 to 4000 t per year. | |||||||||
| Lurgi AG Germany, 1994d/2010 | n.a. | n.a. | 250e | 80 | 10.5 | 94.0–96.5f | Yes (4.5) | Heated water jacket | 126 |
| NITE, RITE Japan, 1996/1998 | 0.9g | 0.05h | 250 (200–275) | 50 | 18 | n.a. | Yes | Reactor immersed in a sand bath | 124 and 185 |
| 50 | 4.6i | 250 (230–270) | 50 (30–70) | 10 (5–20) | n.a.j | Yesk | Oil cooled reactor | 112, 116, 184 and 192 | |
| Centre for Solar Energy and Hydrogen Research (ZSW) Germany, 1996 | 6.1g | 0.4h | 260 | 80 | 8.1 | 23 | No | Electrical heated jacket | 189 and 199 |
| Korea Institute of Science and Technology (KIST) Korea, 1999/2004 | 75l | 8.1 | 250–300n | 51/61 | n.a. | 66.9–70.5 | Yes | Prior RWGS reactor (electrical heated) and four SRC in parallel | 196 |
| n.a. | n.a. | 250 | 30 | 7.9m | 53n | Yes (1o) | Minipilot-plant | 190 | |
| Mitsui Chemicals Inc. Japan, 2009 | 274p | n.a. | 250 | 50 | 10 | 72–88q | Yes (2.6–3.2r) | Pilot plant | 197, 198 and 200 |
| Northern Arizona University (NAU) USA, 2009/2014 | <0.5g | 0.05–0.08h | 240 | 69–97 | 3.3–8.3 | 2.6–14.3 | No | Electrical heated jacket | 183 |
| 1.6 | 0.15s | 240(/260) | 90 | 1.0 (0.6–12.0)t | 40 | Yes | Mobile test rig, electrolysis, electrical heated reactor jacket | 201–203 | |
| Silicon Fire AG Switzerland, 2010 | 40 | n.a. | 265 | 80 | n.a. | n.a. | n.a. | Pilot plant | 204 |
| Carbon Recycling International (CRI) Island, 2012 | 12000u |
n.a. | 250 | 100 | n.a. | n.a. | Yes | Commercial plant converting CO2 from geothermal flue gas | 205 and 206 |
| CRI, MHI Germany, 2019 | 1000 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | Integrated in a coal power plant | 207 and 208 |
A research group of RITE (Research Institute of Innovative Technology for the Earth) and NITE (National Institute for Resources and Environment) conducted short and long-term experiments of in-house catalysts with a 50 kg d−1 oil-cooled test plant.112,116,184,192 Based on the Toyo MRF-Z converter, a double-train 8000 t per d methanol synthesis plant was designed showing that the size of the reactor is almost the same as for natural gas based plants.116
Mitsui Chemicals, an industrial partner of the CO2 utilisation project led by RITE, continued with the catalyst development and constructed a 100 t per a pilot plant in 2009 based on the established fixed bed synthesis loop scheme.209,210 It converts by-product hydrogen and CO2 separated from the exhaust gas both originating from naphtha cracking for an adjacent ethylene oxide unit.200 For future applications hydrogen production from photo-catalysis211 and biomass212 are investigated. Low-cost supply of renewable hydrogen is currently identified to be the major hurdle for commercialisation.212
Northern Arizona University has developed a laboratory-scale mobile methanol synthesis test rig for the conversion of decentralised CO2-sources to methanol. It includes a 30 bar pressurised electrolyser (1.4 kW) for on-site production of hydrogen. The overall efficiency from electricity to methanol (LHV) of first experiments is very poor with only 16.5%. The methanol yield is stated to be 40% although a commercial catalyst has been used.202
Silicon Fire AG (Switzerland) has developed a modular container concept, which they claim is ready for series production. The idea is that the container, which includes an electrolyser and methanol synthesis unit, can be placed close to renewable energy sources e.g. wind parks, or close to CO2-sources. More details about the design have not been reported.203,204
Carbon Recycling International (CRI) commissioned a carbon dioxide to methanol plant with a capacity of 1300 t methanol per year in 2012 which was expanded to 4000 t per a in 2014.193 The plant is based on state of the art technology using Cu/ZnO-catalysts operated at 250 °C and 100 atm.206 The commercial plant is located in Svartsengi, Iceland and recycles 5600 t per a of CO2 released by a nearby geothermal power plant. The H2S containing geothermal flue gas has to be desulphurised first.213 Hydrogen is produced by an alkaline electrolysis unit with a production capacity of 1200 N m3 h−1 (6 MWel).193
In 2015 CRI and MHPSE (Mitsubishi Hitachi Power Systems Europe) announced a strategic partnership to offer industrial solutions for carbon capture and power-to-fuel production.207 Within the EU Horizon 2020 project MefCO2, a demonstration plant was set up and opened in 2019 (1 MWel, 500 t per a CO2, 400 t per a MeOH).207,208,214 CO2 is sequestrated from a coal power plant in Niederaussem (Germany) and hydrogen is produced by electrolysis (supplied by Hydrogenics).
Blue Fuel Energy (Canada)215 plans to integrate hydrogen and oxygen from electrolysis in a large-scale methanol-to-gasoline plant (2.5 million litres of gasoline per day) based on natural gas reforming. Blue Fuel Energy and Siemens entered a memorandum of understatement for the supply of a 20 MW PEM electrolysis system in 2014. A final investment decision was announced for the end of 2016 but no further information have been reported until now.
One main motivation for power-to-methanol (PtMeOH) represents the CO2 abatement by CO2 recycling. Based on the stoichiometry of methanol formation from CO2 (2), 1.37 tCO2/tMeOH are utilized. Assuming an overall carbon conversion of 96%, this results in a CO2 demand of 1.43 tCO2/tMeOH. The CO2 emissions of the conventional production process via natural gas reforming or coal gasification correspond to 0.52 and 2.83 tCO2/tMeOH respectively.216 According to Rivera-Tinoco et al. the weighted average emissions of conventional synthesis plants in Europe is 0.77 tCO2/tMeOH.217 A lifecycle analysis for Blue Fuel Energy indicates an 84.3% reduction of emissions of PtMeOH compared to gasoline and 81% compared to natural gas based methanol.218 The overall emissions were determined to be 14.3 gCO2eq MJ−1 (0.28 tCO2/tMeOH) for PtMeOH using wind energy (including CO2 separation) and combustion of the methanol as a fuel. The methanol produced in the commercial plant of CRI received certification for a 90% reduction of CO2 emissions compared to fossil fuels according to the EU Renewable Energy Directive.193 The specific emissions were determined to be 8.5 gCO2eq MJ−1 (0.17 tCO2/tMeOH) based on electricity from the Icelandic grid and including transportation by ship from Iceland to Rotterdam.
Steam raising converters are claimed to be very flexible by Lurgi AG with a minimal part-load of 10–15% of the design capacity and a load change rate from zero to full load within a few minutes.133 Fast load changes are possible due to the large thermal capacity of the surrounding water ensuring a constant behaviour of the reactor. Easy start-up is realised by steam injection initiating the natural water circulation of the cooling cycle and resulting in a uniform heating of the reactor. Dynamic simulation of the two-stage process for methanol synthesis from CO2 (Fig. 9) by Air Liquide within the project VIteSSe2 implied a transient production rate step between 20–100% within 6–7 minutes.219
The LPMeOH converter was demonstrated to be suited for load-following operation with variable feed flows (5% of design flow per minute) and compositions as well as on/off operation.164
For CO2 operation, Göhna and König observed an activation phase after start-up and concluded that there is a reversible change in the catalyst caused by the reagents or the products.191 It takes about 60 h (at 95 bar) to 80 h (at 80 bar) starting from about 90% of the full activity until steady state is reached. This was also reported by Pontzen et al. using a catalyst from Südchemie.126 The slow increase of conversion after restart has to be investigated in more detail and has to be taken into account for flexible operation.
Taking the thermal energy penalty of CO2 separation into account, the efficiency from PtMeOH with low temperature electrolysis is in the range of 38% from atmospheric CO2225 and 46–48% from flue gas derived CO2.15,218,222,225 Mignard et al.228 report higher efficiencies of 52–58% due to a high electrolysis efficiency of 72% and temporally part-load operation (with 81% at 20% load). The assumed or simulated performance of the electrolysis has a major impact on overall efficiency as electrolysis represents by far the major consumer (97% of electric energy demand).222 High temperature electrolysis promises higher efficiencies. Rivera-Tinoco et al.217 showed an improvement of overall efficiency of 9.5 percentage-points for SOEC (54.8%) compared to PEM electrolysis. However, the efficiency of the PEM-electrolysis based process not taking CO2 separation into account is low with 45.3% compared to 49–52% determined by others24,216,222 and 61% claimed by CRI and Mitsubishi Hitachi Power Systems.193,235 Integrated concepts offer an efficiency potential due to the integration of waste heat streams (e.g. for methanol distillation), utilisation of by-product oxygen from electrolysis (e.g. in a blast furnace234 or in a gasifier225,231) or avoidance of CO2 separation in the case of biomass or coal gasification.225
The major economic assumptions and determined production costs of some studies are compared in Table 12. The given specific CAPEX of PtMeOH plants excluding CO2 separation unit is in the range of 330–410 € per kgMeOH production per d (except for the SOEC case217). Specific investment costs for methanol synthesis and purification from literature (including NG and coal studies) are compared in Fig. 10. One major assumption regarding the methanol production price represents the capacity utilisation. Based on a capacity factor of more than 85% (>7450 h per a) in most cases, a specific price in the range of 300–900 € per tMeOH is determined. It is mainly affected by the electricity price. The market price of methanol was highly variable in the past ranging from 150 € per t to 525 € per t in the last years (Fig. 11), with prices of around 260 € per t at the beginning of 2020.236 Regional benchmark methanol production costs were identified by Boulamanti and Moya237 to vary between 51 € per tMeOH in Saudi Arabia and 408 € per tMeOH in Europe in 2013 mainly dependent on the regional feedstock costs.
| Description | Capacity (tMEOH d−1) | CAPEXa (103 € per tMeOH per d) | Capacity factor | Electricity price (€ per MW per h) | CO2 cost (€ per t) | Methanol price (€ per t) | Ref. |
|---|---|---|---|---|---|---|---|
| LTEL, low temperature electrolysis, BM Biomass gasification. Unit conversion: 19.9 MJ kgMeOH−1, 0.792 kg lMeOH−1.a Based on given total investment costs and capacity, (a1) including CO2 separation; (a2) CO2 provided externally; (a3) CO2 and H2 provided externally.b Average spot price of used electricity price duration curve.c CO2 supply included in CAPEX and OPEX.d Based on given prices in $ per barrel with 1.3 €/$ in 2011.e Supplied from hydropower plant.f Estimated based on given efficiency.g Including a 50% cost reduction of the electrolyser (2940 € per kW for SOEC, 300 € per kW for PEM).h 1956 DM per €. | |||||||
| LTEL + BM | 900 | 210–290a1 | 91 | 40 | 15 | 252–316 | 231 |
| LTEL + CO2 | 900 | 380a2 | 91 | 40 | 15 | 555 | |
| SOEC + BM | 1053 | 325a1 | 91 | 61/82b | —c | 325–375d | 232 |
| LTEL + CO2 (flue gas) | 2400 | 393a2 | 88 | n.a.e | 35 | 294 | 238 |
| LTEL + CO2 (flue gas) | 178 | 410a2 | 91 | 25 | —c | 515 | 228 |
| CCU-plant | 1300 | 181a3 | 91 | 95.1 | 0 | 724 | 229 |
| SOEC (20 MW) + CO2 | 50f | 2350a2 | 91 | 20–50 | 3–10 | 700g–5460 | 217 |
| PEM (24 MW) + CO2 | 50f | 390a2 | 92 | 20–50 | 3–10 | 404g–890 | |
| LTEL + CO2 | 140 | 330a2 | 68 | 93 | 50 | 980 | 78 |
| LTEL + CO2 (flue gas) | n.a. | n.a. | 95 | 26h | n.a. | 525h | 225, 189 and 239 |
| LTEL + CO2 (atm.) | 170 | 355a2,h | 95 | 26h | —c | 715h | 225, 189 and 239 |
| LTEL + BM | 53 | 410a1,h | 95 | 26h | —c | 385h | 225, 189 and 239 |
| LTEL + CO2 | 1485 | n.a. | 92 | 10–50 | n.a. | 400–820 | 24 |
| LTEL + CO2 | 1486 | n.a. | 46 | 10–50 | n.a. | 700–1100 | |
| LTEL (140 MW) + CO2 | n.a. | n.a. | 85 | 50 | —c | 913 | 226 |
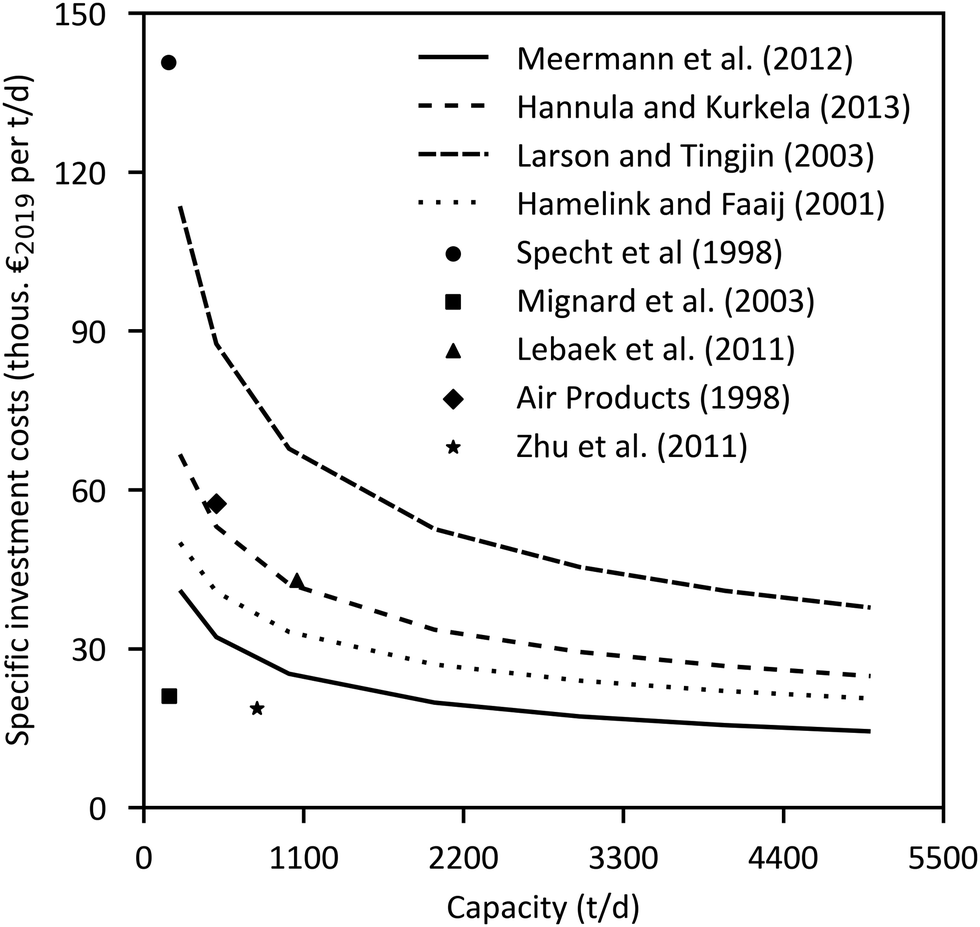 | ||
| Fig. 10 Specific investment cost curves of methanol synthesis loop including purification normalized to the year 2019.228,232,239–245 | ||
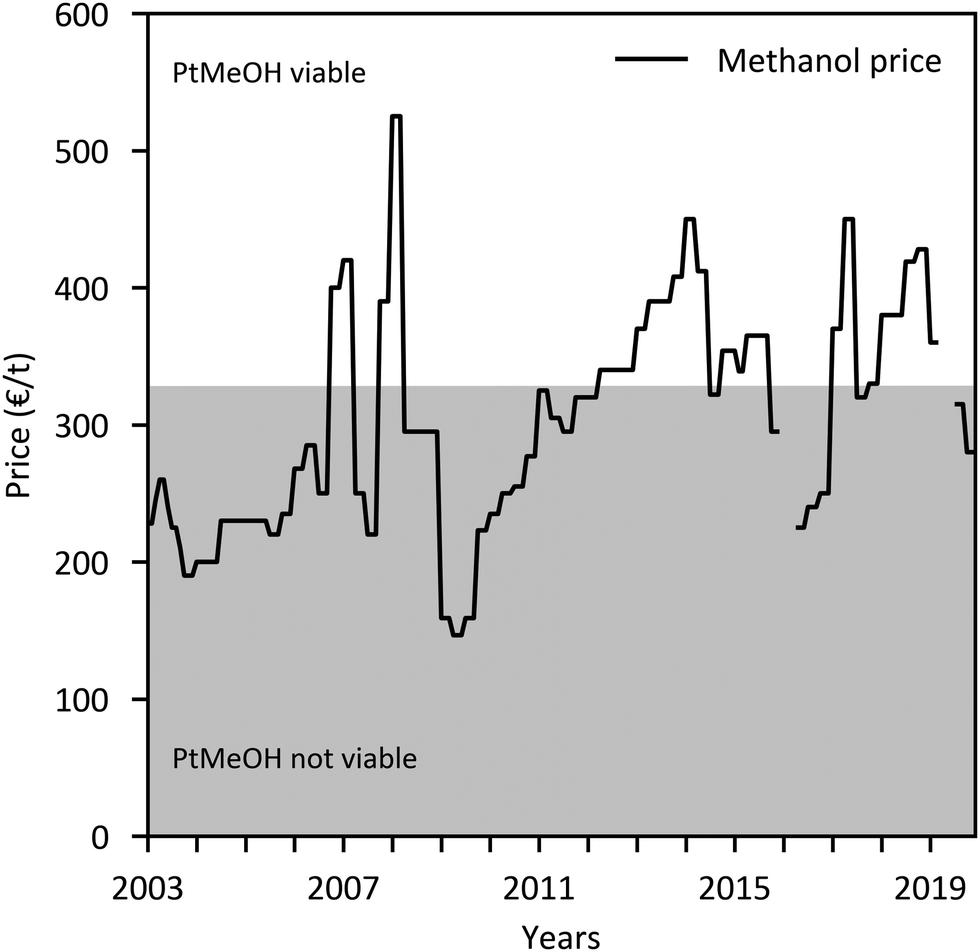 | ||
| Fig. 11 Historical European methanol prices236 with price area where PtMeOH might be economically viable (based on the featured techno economic analyses). | ||
2.3. Discussion
Methanol production from CO2 and hydrogen was already conducted at the beginning of industrial production in the 1920's. More intensive research began in the 1990's. However, information from pilot plants is still scarce. Despite of some laboratory and pilot plant tests, long-term pilot plant studies on catalyst deactivation and on the optimal operating conditions are still missing. Further, new catalysts need to be developed to increase the methanol yield from CO2 hydrogenation and to reduce problems caused by increased water production. In addition, efforts to develop reactor concepts that improve the equilibrium yield of the process should be intensified. Due to less by-product formation, cost and energy savings for the product upgrade need to be considered as well.Currently, commercial converter designs are largely influenced by the necessity for efficient heat removal. The lower heat released by the methanol synthesis from CO2 can enable more simple reactor designs and therefore also reduce the complexity of the process design in general.
For state of the art methanol processes a trend towards larger-scale plants to benefit from economy of scale effects exists. Though, using PtMeOH as CCU technology or an operation close to renewable energy sources will require profitable small-scale plants. Consequently, additional efforts are necessary to adapt state of the art reactor and process designs for small-scale applications and develop new concepts better suited for a decentralized and flexible operation.
The identification of new business cases with concepts integrated in other processes and low cost supply of hydrogen play a crucial role in the commercialisation of CO2 utilisation-based methanol plants. In addition, the feasibility of dynamic operation has to be investigated, to make use of electricity price variations and to act as an energy storage in renewable energy systems.
3. DME synthesis
3.1. State of the art DME synthesis
| 2CH3OH ⇌ CH3OCH3 + H2O ΔH = −23.4 kJ mol−1 | (5) |
However, DME can also be produced directly from syngas. In this case, methanol synthesis reaction (4), water gas shift reaction (3) and methanol dehydration reaction (5) take place simultaneously and form a synergistic system.88,247–249 On the one hand, the dehydration or consumption of methanol relieves the equilibrium constraints of methanol synthesis. On the other hand, water formed in reaction (5) is consumed to some extent by the water gas shift reaction and hydrogen is produced which increases the rate of methanol synthesis. Additionally, the consumption of water protects the catalyst from degradation due to water accumulation.250 The enhanced syngas conversion compared to the methanol synthesis reaction is shown in Fig. 12 for the two overall reaction routes from syngas:
| 3CO + 3H2 ⇌ CH3OCH3 + CO2 ΔH = −246.0 kJ mol−1 | (6) |
| 2CO + 4H2 ⇌ CH3OCH3 + H2O ΔH = −205.0 kJ mol−1 | (7) |
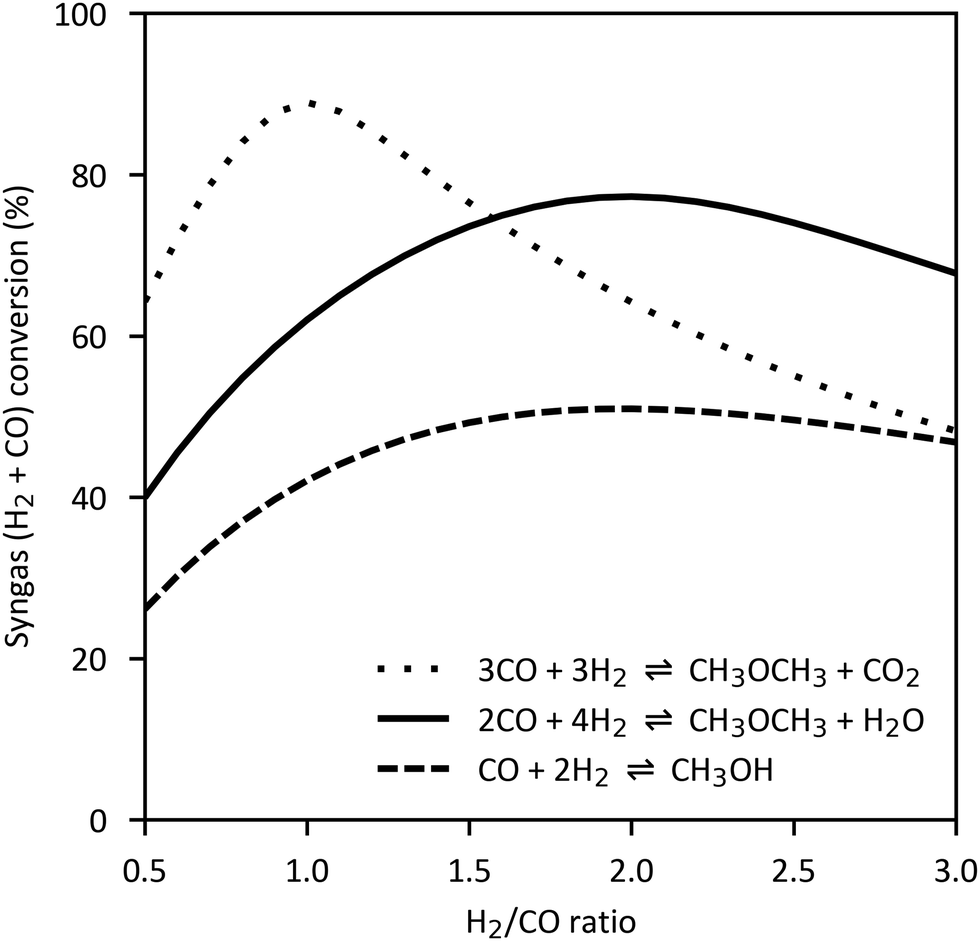 | ||
| Fig. 12 Syngas conversion for methanol synthesis and DME synthesis reactions as function of H2/CO ratio (260 °C, 50 bar, own illustration of ref. 30 based on Aspen Plus Simulation). | ||
The direct process of Haldor Topsøe follows reaction (7) while the processes of JFE, Air Products and KOGAS follow reaction (6) resulting in a H2O or CO2-rich product respectively. This poses different requirements for the product upgrade discussed later. Based on the reaction stoichiometry, the H2/CO ratio should be adjusted to 1.0 for reaction (6) and to 2.0 for reaction (7). As the reactions (6) and (7) are exothermic and reduce the number of moles, direct DME synthesis is favoured by low temperature and high pressure. Operation conditions of direct DME synthesis plants have a temperature range of 200–300 °C and a pressure range of 30–70 bar.
In case of CO2-based DME synthesis, the optimal stoichiometry for direct hydrogenation is given by a H2/CO2 ratio of 3 to 1 following on from the stoichiometry of the combination of methanol synthesis reaction (2) and DME synthesis reaction (5). Equilibrium limited conversion of CO2 and selectivity of DME decreases at a higher temperature and a lower pressure while CO selectivity increases due to the RWGS reaction as shown in Fig. 13.
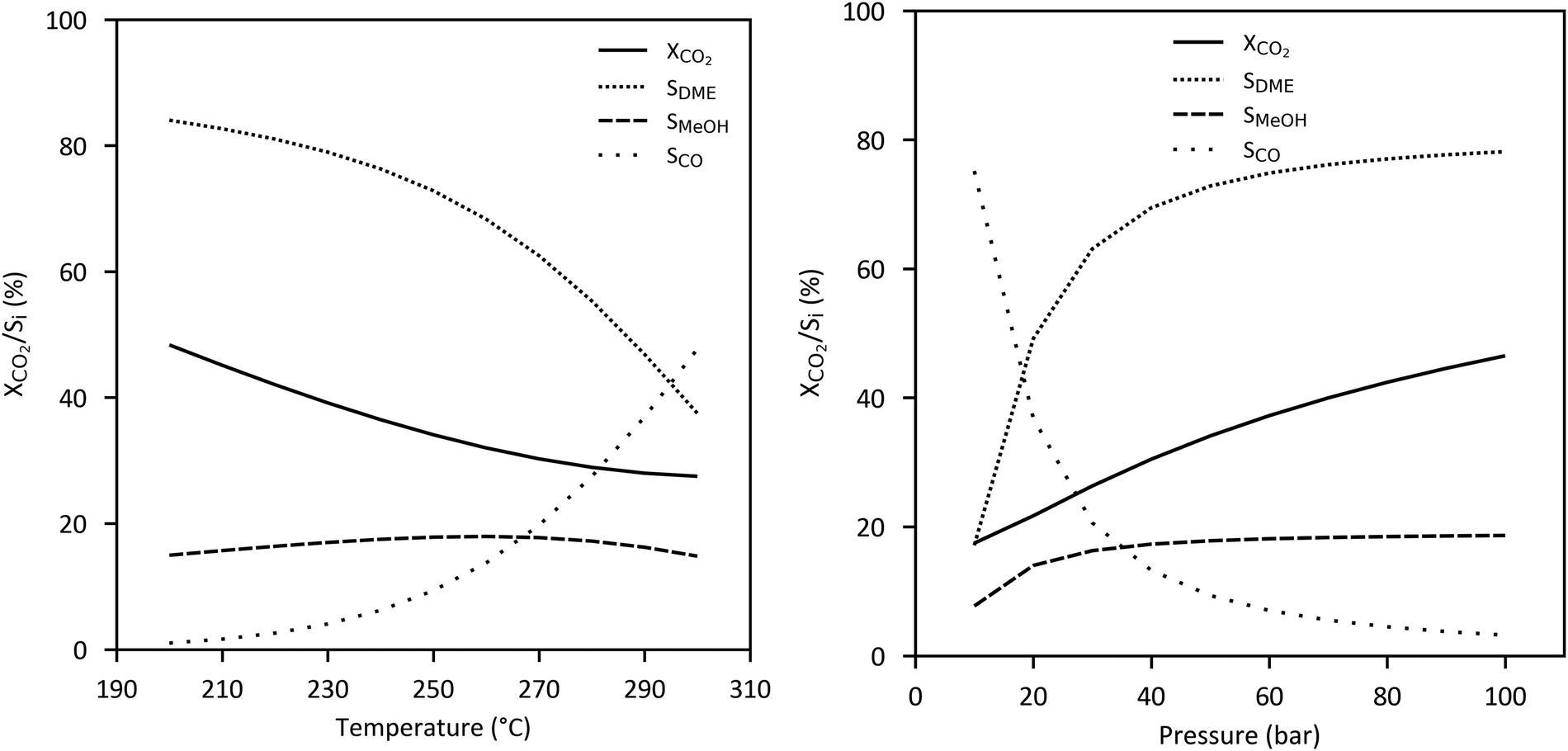 | ||
| Fig. 13 Equilibrium conversion of CO2 (= [CO2,in − CO2,out]/CO2,in) and selectivity of DME, methanol and CO (Si = i/[CO2,in − CO2,out], with i = 2 × DME/MeOH/CO, based on amount of C-atoms) dependent on (a) temperature and (b) pressure (H2:CO2 = 3:1, based on Aspen Plus Simulation). | ||
The direct conversion of syngas to DME is realised by using a hybrid or supported bifunctional catalyst. Hybrid catalysts are prepared by physical mixing of powder or granules of conventional methanol synthesis (e.g. CuO/ZnO/Al2O3) and dehydration catalysts (e.g. γ-alumina).30,32 Supported bifunctional catalysts commonly comprise Cu/ZnO as a metal side and dehydration catalyst as a support.252 The methanol synthesis reaction (2) and the WGS reaction (3) are catalysed by the methanol synthesis catalyst. The dehydration reaction (5) is catalysed by the acidic component. As a result, the product ratio of methanol and DME can be adjusted by the ratio of dehydration and methanol synthesis catalyst, which represents an important design parameter also with regard to the consumption and production of water.88 Catalyst mixtures applied for the production of DME as the main product contain 33–80 wt% DME catalyst (mainly ranging from 33–50 wt%).88,251,253–256
According to MGC, the expected life time of the dehydration catalyst in a commercial two-step process is 4–6 years or more, comparable to the methanol synthesis catalyst.30 ENN reports a life time of their dehydration catalyst of more than 24 months.257 Haldor Topsøe operated a 50 kg per d direct DME synthesis bench plant with a hybrid catalyst charge for more than 14000 h.130,249 The results indicate higher stability compared to their commercial methanol synthesis catalyst MK-101, which was in operation in a large methanol plant for more than 5 years. For the JFE direct DME synthesis process it is expected that the catalyst life is at least one year for a commercial scale plant, maintaining DME production by gradually increasing the operation temperature.258
Deactivation of dehydration and hybrid catalysts is caused by sintering active copper sites, coke deposition, poisoning by contaminants in the syngas or adsorption of water (also formed from CO2) and thus blockage of acidic sites.259 The requirements of the syngas purity to avoid deactivation by contaminants is specified in Table 13. While Wang et al. reported that the deactivation of hybrid catalysts is caused mainly by the deactivation of the Cu-based methanol catalyst,260 results from Air Products indicate an almost similar deactivation rate of methanol catalyst and gamma alumina (γ-Al2O3).261 γ-Al2O3 and Cu-based methanol synthesis catalysts show a higher degradation in slurry reactors than fixed bed reactors which represent the main reactor types of direct DME synthesis.32 According to Fujimoto et al.30 the rate of water transfer from alumina to the methanol catalyst is much lower in the slurry phase reaction than in a fixed bed system. However, the problem of increased deactivation could be solved by adding an active component, which promotes the shift reaction and thus efficiently removes water as hydrogen from the alumina surface. Moreover, in the beginning of the direct DME synthesis development of Air Products (LPDME), rapid degradation of γ-Al2O3 and methanol synthesis catalyst in a physical mixture was observed due to cross metal contamination.262 A comparison of degradation over time of a catalyst from Haldor Topsøe and JFW for direct synthesis is shown in Fig. 14. As seen, in the first 1000 h, deactivation is in the range of 10–25% of the initial value.
| Component | Purity requirement |
|---|---|
| NH3, HCN | <1.0 ppm (vol) |
| H2S, COS, CS2, Fe carbonyl, Ni carbonyl, Cl, F | <0.1 ppm (vol) |
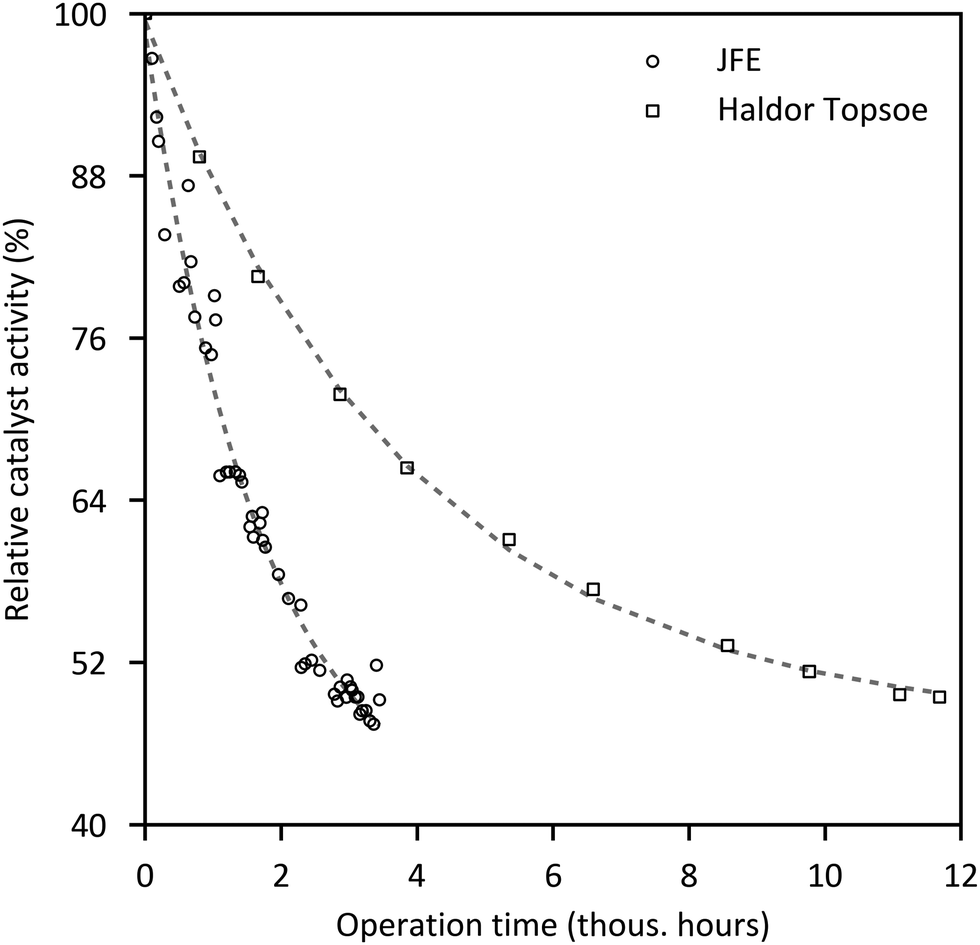
The two-step process benefits from the simple extension of the well-established methanol process available on a very large scale associated with low additional investment costs. Moreover the two-step process offers advantages in product upgrade discussed later and the possibility to change the product ratio of methanol and DME according to the market demand.30,263 Lurgi claims that the investment costs of the two-step process including product upgrades are 10–20% lower on a 3000–5000 t per d scale than the direct synthesis process.264
Most methanol technology licensors like Haldor Topsøe, Lurgi, Toyo Engineering Corporation (TEC), Mitsubishi Gas Chemical or Johnson Matthey Technology also offer two step DME processes up to the scale of their methanol technology. For example, TEC states that a single train JumboDME plant can be realised based on their JumboMethanol scheme with a DME capacity of 3500 t per d and a radial reactor similar to the MRF-Z converter with a capacity up to 7000 t per d is feasible.
Beside the established companies there are some emerging Chinese companies like China Energy,265 Tianyi266 or ENN.267 A typical flowsheet of a two-step DME synthesis is shown in Fig. 15. Stabilised methanol (after the first distillation column), further upgraded methanol (Grade A or AA) or even raw methanol directly after the methanol converter268,269 are used as feedstock. The methanol is vaporized and fed to the dehydration reactor where it is partly converted to DME, water and very small amounts of light ends (CH4, CO, CO2, H2). Methanol consumption is about 1.4 tMethanol/tDME, which is close to the theoretical minimum of 1.39 tMethanol/tDME. Unconverted methanol is separated from the DME and recycled to the DME reactor. Per pass conversion is in the range of 70–85% at typical operating conditions of 220–400 °C and a low pressure of 1–30 bar (see Table 14). Apart from China Energy all technologies are based on methanol dehydration in a fixed bed reactor.
 | ||
| Fig. 15 Typical flowsheet of the DME section of a two-step synthesis process. | ||
| Company | MGC | Lurgi (air liquide) | Toyo (TEC) | Haldor Topsøe | ENN |
|---|---|---|---|---|---|
| a <50 wt ppm carbonyl compounds, about 20% water. b Trademark DK-500 and DME-99 Eco. c Two-stage DME conversion process. Per pass conversion is 70–80% in the first stage and 85–92% in the second stage. | |||||
| Methanol | Grade AA | Stabilised methanola | Stabilized methanol | Crude, fuel grade, grade A | >50% MeOH |
| Reactor design | Adiabatic fixed bed | (Adiabatic or cooled) fixed bed | Adiabatic fixed bed (radial) | Adiabatic fixed bed | (Adiabatic or cooled) fixed bed |
| p (bar) | 10–25 | 1–30 | 10–20 | 10–15 | 6–12 |
| T (°C) | 250–400 | 250–360 | 220–250 (inlet), 300–350 (outlet) | 290–400 | 260–360 (1st stage), 180–240 (2nd stage). |
| Per pass conversion | 70–80% | 70–85% | 70–85% | 80% | >80%c |
| Selectivity | 98.60% | n.a. | 99.9% | n.a. | >99% |
| Catalyst | γ-Alumina | n.a. | γ-Alumina | Activated aluminab | Alumina-silica |
| Plants | Niigata (80 ktpy), Trinidad and Tobaga (20 ktpy) | — | 4 plants in China (total 470 ktpy) | BioDME Pitea (5 tpdy), Iran (800 ktpy) | Zhangjjagang (200 ktpy), Bengbu (20 ktpy) |
| Ref. | 30, 179 and 270 | 126 and 271–273 | 263 and 274–277 | 167–169, 249, 278 and 279 | 257, 267, 280 and 281 |
Direct DME synthesis processes have been developed and demonstrated on a pilot plant scale by JFE, KOGAS, Haldor Topsøe and Air Products and Chemicals Inc. These processes are compared in Table 15 and will be discussed in the following in more detail.
| Haldor Topsøe | KOGAS | JFE | Air products | |
|---|---|---|---|---|
| a Product C-mol ratio: 2 × DME/(2 × DME + MeOH). b Product H-mol ratio: 2 × H2O/(2 × H2O + 6 × DME + 4 × MeOH). c Calculation based on patent WO96/23755.301 d 65–82% dependent on CO2 in feed gas.31 e About 6 gcat h mol−1 for 100 t per d and 3000 t per d concept,31 4 (3–8 gcat h mol−1 for 5 t per d plant).250 f Based on patent US 2006/52647295 when the methanol catalyst substitutes the WGS catalyst. g Commercial catalyst/t BASF S3-86. h Own calculations based on Air Products and Chemicals Inc.261,288 | ||||
| H2/CO | 2 | 0.8–1.5 | 1 | 0.5–0.7 |
| Reactor design | Adiabatic fixed bed | Tubular SRC | Slurry | Slurry |
| p (bar) | 37–42 | 30–60 | 50 (30–70) | 52 |
| T (°C) | 240–330 | 260 (200–300) | 260 (240–280) | 250 |
| GHSV | n.a. | 2000–10000 h−1 |
4000 sl per kgcat per h (3000–8000)e | 6000 sl per kgcat per h (5700/9000) |
| Catalyst MeOH/DME | Cu-based/aluminosilicate | CuO/ZnO/Al2O3 AlPO4/γ-alumina | CuO/ZnO/Al2O3 γ-alumina | CuO/ZnO/Al2O3g γ-alumina |
| Catalyst ratio (MeOH:DME) |
n.a. | 0.8–4:1 |
2:1 (0.75–20:1)f |
95:5 (80:20) |
| Product (DME purity %) | MeOH/DME mixture | 99.6 | >99.5% (99.9) | MeOH/DME mixture |
| Recycle ratio | 4–5c | n.a. | 1.6–1.8 | n.a. |
| CO conversion (per pass) | 40–70c | 67d | 50–60 | 19–24 (21–31)h |
| Overall CO conversion (%) | 94–97c | n.a. | 94–96 | n.a. |
| H2O selectivitya (%) | 18–28c | n.a. | 1.3 | <1h |
| DME selectivityb (%) | 74–76c | 60–70 | 91 | 61–67 (40–80)h |
| Plants | 1993: 50 kg per d Bench | 2003: 50 kg per d Bench 2008: 10 t per d Demo | 1995: 50 kg per d Bench 1997: 5 t per d Pilot 2003: 100 t per d Demo | 1991: 4–6 t per d Pilot 1999: 8–9 t per d Demo |
| Ref. | 130, 171 and 301 | 251, 255, 256, 283, 284 and 300 | 30, 250, 254, 258 and 295–299 | 261, 288 and 294 |
Haldor Topsøe have tested their self-developed dual function catalyst (e.g. aluminosilicate catalyst)282 in a 50 kg per d cooled reactor pilot plant as presented in 1995 by Hansen et al. showing a comparable durability to their methanol catalyst MK-101.249 Laboratory experiments at 37 bar and 220–330 °C were reported by Hansen and Joensen in 1991.171 These experiments indicated a very low amount of by-products even with hot spot temperatures above 300 °C. The Haldor Topsøe process was based on reaction (7) resulting in a product with a high water content. For large-scale applications, three adiabatic reactors with interstage cooling are proposed by Dybkjaer and Hansen.130 This enables single train capacities of up to 10000 t per d methanol equivalent. However, Haldor Topsøe's commercial projects are all based on their two-step process presented before.
KOGAS (Korea Gas Corporation) began the development of DME synthesis from natural gas based on a shell and tube steam raising converter design in 2000.283 The catalyst is filled in tubes surrounded by boiling water on the shell-side. In addition, KOGAS examined a mixture of methanol and dehydration catalyst pellets and developed a proprietary hybrid catalyst. A higher activity and DME selectivity of the hybrid catalyst was shown in laboratory experiments as well as reactor simulations.251,256 This proprietary hybrid catalyst consists of a mixture of fine powdered methanol catalyst (CuO/ZnO/Al2O3 with added promoters Mg, Zr, Ga, Ca) and dehydration catalyst (γ-alumina with aluminum phosphate).251,255 In 2003, a 50 kg per d pilot plant was set-up and a 10 t per d demonstration plant was brought into operation in combination with a natural gas tri-reformer in 2008. Based on flowsheet modelling284 and numerical simulations of the reactor,253 a conceptual design of 3000 t per d DME process was completed by 2009.283,285 However, problems with degradation during the demo plant tests were reported due to hot spots.30 As a result, for the 3000 t per d concept the CO2 content was planned to be 25–30%, resulting in a lower conversion and a reduced increase in temperature.
The Japanese Corporation JFE (formally NKK) developed a direct DME synthesis process based on a slurry reactor in various collaborations.250,258 It started with the development of a new catalyst in 1989.250 Afterwards, a 50 kg per d bench unit was brought into operation in 1995, followed by a 5 t per d pilot plant in 1997286 and a 100 t per d demonstration plant (including autothermal reformation of natural gas, process scheme see Fig. 16) which was operated from 2003 to 2006 with a total production of 19520 t of DME.30,258 JFE selected the slurry reactor design due to its excellent heat transfer capabilities, resulting in almost isothermal temperature profiles within the reactor.250,258 The once-through CO conversion is in the range of 50–60% (at standard conditions of 260 °C, 50 bar and about 6 gCat h−1 mol−1).30,31 The overall CO conversion reaches up to 96% at a recycling ratio of about 1.8.250 The cold gas efficiency of the DME section in the 100 t per d demonstrations tests was 81.7%.258 A 3000 t per d reactor concept was presented with an inner diameter of 7 m and a height of 50 m (slurry 46 m), which is a scale-up of the 10 t per d reactor which had an inner diameter of 2.3 m and a height of 22 m (slurry 15 m).258,287
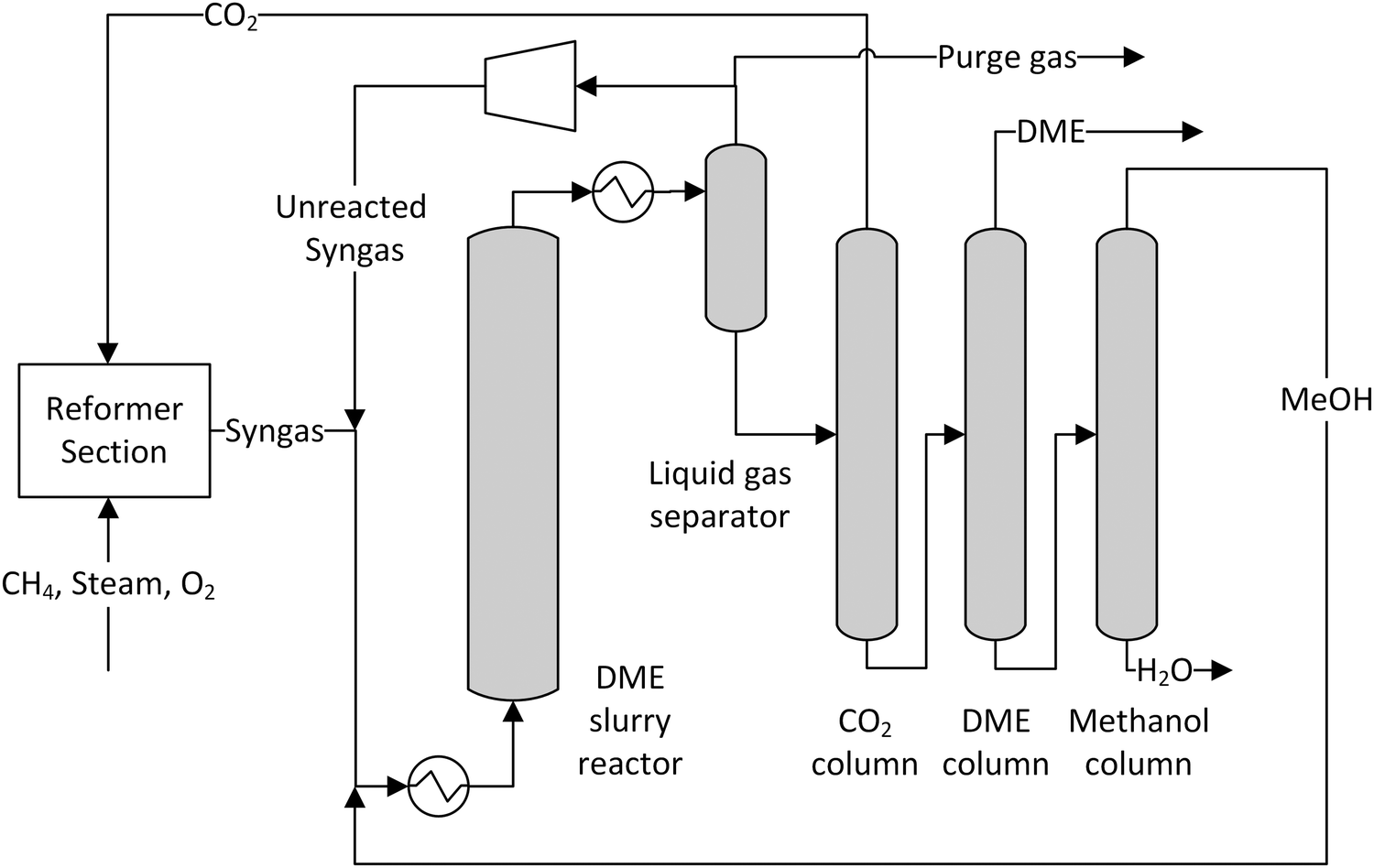 | ||
| Fig. 16 Schematic process flow of the JFE 100 t per d demonstration plant (own illustration based on Yagi et al.258). | ||
Air Products and Chemicals investigated the co-production of DME within the LPMEOH-Project. The proof of concept was demonstrated in 1991 at a production capacity of 4–6 t per d of methanol and DME.288 A physical mixture of γ-Al2O3 (6.6–19.3 wt%) and a commercial methanol synthesis catalyst (BASF S3-86) were used. The CO conversion was identified to be in the range of 21–31%. The DME selectivity is determined to be 40–80% dependent on the amount of γ-Al2O3 and the space velocity. In the following, efforts were spent in the investigation of reaction kinetics and improvement of stability of the catalyst system.88,247,248,289 In 1999, the Liquid Phase Dimethyl Ether (LPDME) process was demonstrated in the 10 t per d (4–5 t per d DME) Alternative Fuels Development Unit (AFDU) of the U.S Department of Energy (DOE).261 A methanol to dehydration catalyst ratio of 95 to 5 by weight was found to be the optimum for catalyst stability. Selectivity was in the range of 61–67%. The deactivation rate for both catalysts was identified to be 0.7% per d. In 2019 BASF, Linde and Lutianhua announced a partnership to build a pilot plant for an energy-efficient one-step DME process.290 Currently no further details have been reported. A comparison of once-through CO conversion for JFE and LPDME plant concepts is given in Fig. 17.
 | ||
| Fig. 17 Dependency of once-through CO conversion on catalyst loading ratio W/F (catalyst weight/reactor inlet gas flow rate) of JFE plants (260 °C, 50 bar)258 and LPDME 10 t per d demonstration (own calculation based on ref. 261). | ||
Although direct DME synthesis is ready for commercialisation and the cold gas efficiency of DME synthesis can be increased by up to 10 percentage-points compared to the two-step process,30,291 no commercial project has been realised until now. However, due to the high potential, direct DME synthesis still attracts research and commercial interest. For example, Linde AG and BASF SE recently investigated direct DME synthesis technology and catalysts within the project DMEEXCO2 (2012–2015).291–293
Direct DME synthesis features a more complex product upgrade compared to the two-step process due to the reactor effluent mixture of DME, methanol, CO2, water and unconverted syngas. The removal of CO2 especially from the DME product stream is very difficult. Several ways have been proposed to solve this problem. Examples are the separation of CO2 by membranes293 or to avoid the formation of large amounts of CO2 by operation in a H2-rich regime (e.g. H2/CO > 5).301
Because of the low boiling point of DME, a cryogenic separation of liquid products and unconverted syngas is typically applied in the recycle loop.271 To condense most of the DME along with the methanol and water, the syngas is cooled down to temperatures of about −40 °C in the JFE and KOGAS processes.284,296 CO2 is dissolved in the product DME. The unconverted syngas is recycled to the DME synthesis reactor. The liquids are depressurised and directed to the CO2 column followed by the DME column and the methanol recovery column.258,284,296 In the KOGAS process, CO2 is separated from the liquid stream in the CO2 column operated at a pressure of 35 bar and DME is recovered in the DME column at a pressure of 18 bar.284
In a US patent of Air Products,304 the reactor effluent is fed to a high-pressure flash column. The vapour mixture comprises DME, CO2 and unconverted syngas. A mixture of DME and methanol is used in a scrubber to remove DME and CO2 from the unconverted syngas, which is recycled to the DME reactor.
Another concept is disclosed in a US patent of Haldor Topsøe305 removing the CO2 in a potassium carbonate scrubber from the reactor effluent. In a subsequent distillation column, methanol and water are discharged in the bottoms and DME is obtained by condensation of the top product. The remaining syngas depleted in CO2 is recycled to the DME reactor.
3.2. Current research on power-to-DME
The extent of synergy of the direct synthesis declines with rising CO2 content due to the high formation of water via the RWGS reaction inhibiting methanol production and the dehydration reaction.30,88,248 Experiments with varying CO2 content show a significant decrease of CO conversion.30,259 Direct hydrogenation of CO2 to DME has been investigated mainly by using a physical mixture of Cu-based methanol catalysts and zeolites in fixed bed reactors306–320 as zeolites are not sensitive to water. Hybrid catalysts containing γ-alumina were investigated to a lesser extent.88,313,321,322 An overview of CO2 conversion and selectivity of several investigated catalysts is shown in Fig. 18. Per pass CO2 conversion of maximum 35% (at 210 °C, 50 bar) is achieved. The selectivity of DME lies mainly between 40 and 70%. Naik et al.313 tested two hybrid catalysts (Cu-based catalyst with γ-alumina or HZSM-5) in a fixed bed and slurry reactor. The γ-alumina catalyst shows an inferior performance regarding activity, selectivity and durability compared to the zeolite catalyst. Moreover, CO2 conversion, DME selectivity and catalyst stability is higher for the fixed bed reactor than for the slurry reactor. This can be explained by water accumulation on the catalyst surface due to the additional mass transfer resistance through the hydrocarbon oil in the slurry reactor. More detail can be found in Alvarez et al., which provide an overview of hybrid catalysts investigated in the last 10 years.323
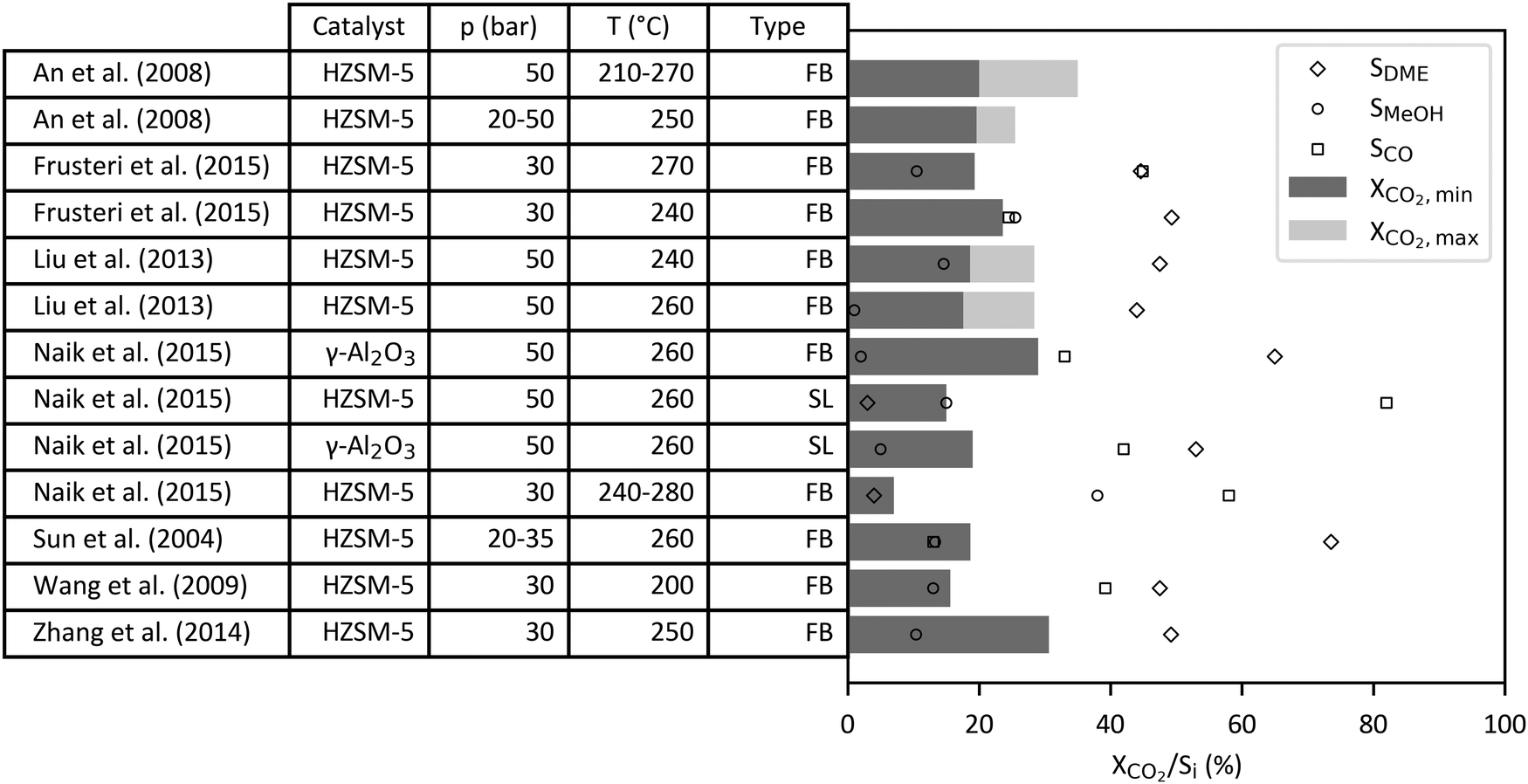 | ||
| Fig. 18 Overview of CO2 conversion and selectivity of DME, methanol and CO (Si = i/[CO2,in − CO2,out], with i = 2 × DME/MeOH/CO, based on amount of C-atoms) of direct hydrogenation experiments of CO2 to DME.306,310,313,314,317–319 | ||
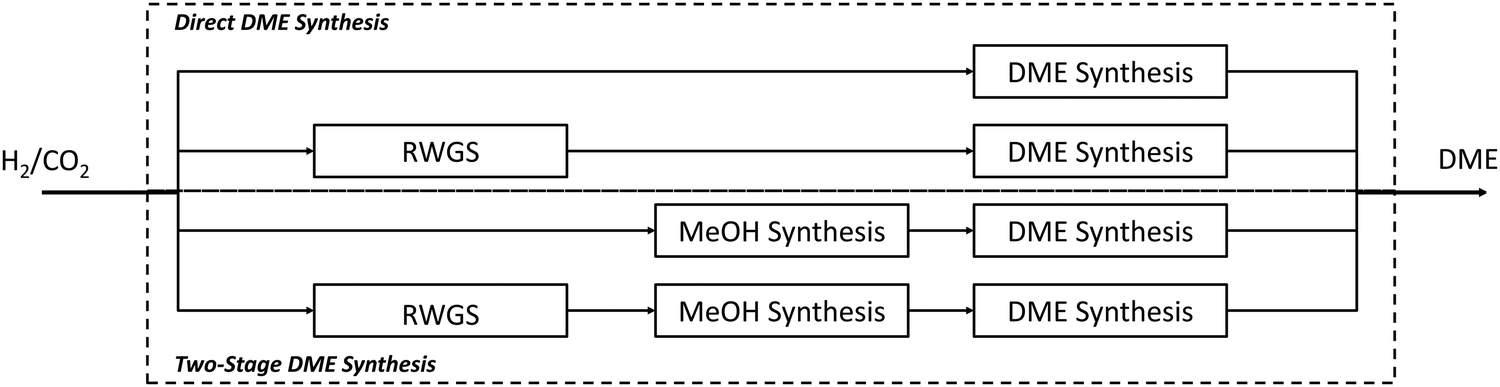 | ||
| Fig. 19 Possible process pathways from CO2 to DME. | ||
Ateka et al.324 investigated alternative promoters for bifunctional catalysts (Cu-based with Zr or Mn promoters on SAPO-18 zeolite). Zr and Mn metallic functions of the catalyst showed a similar behaviour for the methanol synthesis step, however provided higher yields and selectivity for the DME step. In addition, the catalysts showed a lower deactivation for H2/CO2-based feed gas compared to conventional syngas. Catizzone et al. give a good summary of different bifunctional catalysts that have been investigated recently for direct CO2 hydrogenation.325
A feasibility study of a 500 t per d DME production from CO2 in Iceland has been commissioned by the Icelandic Ministry of Industry, Mitsubishi Heavy Industries (MHI) and others as part of the long-term vision of a zero percent hydrocarbon fuel emissions society in Iceland.326 The CO2 should be captured from the exhaust gas of the ELKEM ferrosilicon plant by MHI's CO2 recovery process. Hydrogen is supplied by water electrolysis. DME is produced via MHI's two-step process from CO2 and H2 comprising the superconverter and methanol dehydration technology. The authors conclude that this project is considered to be feasible with dedicated support from the government.
At RWE's Innovation Centre at Niederaussem power station in Germany, a Power-to-X-to-Power research facility was established as part of the multi-partner ALIGN-CCUS project in November 2019. The synthesis of DME investigated at the new pilot plant is based on captured CO2 from the lignite-fired power plant and electrolysis-based hydrogen. The aimed production rate is up to 50 kg of DME per d in a one-step process.327 The synthetic energy carrier will then be used and evaluated both for re-electricity generation and use as fuel in vehicles that are difficult to electrify.328,329
Matzen and Demirel330 conducted a life-cycle assessment of the production of renewable methanol and DME (via the two-step process) based on Aspen Plus simulations. The use of fermentation-based CO2 and H2 from wind-powered electrolysis results in a reduction of greenhouse gas emissions by 86% for methanol and 82% for DME compared to conventional petroleum.
Vibhatavata et al. discussed the production of DME from CO2 emissions of the cement industry and hydrogen produced by nuclear-powered water electrolysis in France. The overall efficiency of the CO2-to-DME process was 53%, comprising CO2 capture (MEA wash) from flue gas of the cement industry, reverse water gas shift reactor, electrolysis, methanol and DME synthesis. A similar efficiency in the range of 52–55% from electricity to DME was estimated for a reference case based on electrolysis, RWGS and direct DME synthesis (excluding CO2 capture effort) by Ohno et al.31,331 For the RWGS autothermal operation without catalyst at a temperature of 1000 °C by adding O2 from the electrolysis unit is proposed.
Sun et al.332 determined the feasible operating conditions of a pressurised SOEC integrated in a DME synthesis process. The thermodynamic analysis indicates that a high operating temperature of the SOEC is necessary (e.g. >900 °C at 80% reactant utilisation and 50 bar) to avoid carbon formation due to the required high pressure and high CO2/H2 ratio (≥1) of the DME process.
An et al.306 showed that the recycling of unconverted syngas in direct synthesis of DME from CO2 not only increases overall conversion but also increases space-time yield of DME. This is because the reactor effluent contains CO as a result of the fast water gas shift reaction and the recycling of CO increases methanol and DME yield.
Peng et al. investigated the influence of changes of the H2/CO2 ratio on DME production for a single step DME synthesis with recycle.333 They conclude that the DME productivity is very sensitive to changes in the H2/CO2 ratio due to an amplifying effect of the recycle.
Gonzáles et al. investigated the flexible operation of fixed-bed reactors for CO2 hydrogenation reactions. DME is identified as a promising route to be operated under flexible conditions due to its high and robust product selectivity. In addition, the limited sensitivity of the DME synthesis towards temperature variations and the high catalyst stability for DME synthesis are seen as beneficial.334
Farsi and Jahanmiri modelled and simulated the behaviour of a fixed bed MeOH to DME reactor (second stage of the two-stage DME synthesis). They applied different disturbances and investigated the open loop behaviour of the reactor. Their results show that a 2 bar step change in pressure does not significantly affect the product composition. Feed composition and temperature, however, have a large influence on the product composition.
Hadipour and Sohrabi developed a dynamic model of a direct DME reactor based on experimental data. They conducted experiments at 230–300 °C and 9 bar. The experimental data shows that for all temperatures it takes around 300 min from start up to reach a steady state.335
While first cost estimates of PtDME differ in their approach and investigated process chain, they agree on DME costs above 1000 € per t, therefore significantly higher than DME from biomass or coal. Michailos et al. examine the two-stage synthesis of DME using CO2 captured from a cement plant and hydrogen from a PEM electrolyser.343 The simulated plant, designed for a throughput of 740 t of DME per d, can synthesise DME at costs five times the price of conventional diesel (based on the LHV). A Monte Carlo simulation-based sensitivity analysis indicates that the 95% confidence interval for the DME minimal sales price ranges from 1828–2322 € per t.343
Schemme et al. compared different H2-based synthetic fuels, indicating DME and MeOH as the fuels with the lowest manufacturing cost. They consider direct DME synthesis from CO2. Compared to the LHV-based diesel equivalent (DE), they estimate DME manufacturing costs to be 1.49 € per lDE or 1490 € per t, which is in good agreement with Tremel et al.,78 who also considers the direct synthesis of DME and estimates costs of 1390 € per t. The sensitivity analysis shows that after hydrogen and CO2 costs, the fixed capital investment has the biggest effect on DME costs.344 In China with its big DME market, prices in 2020 varied between 329 € per t und 276 € per t, leading to a big spread in PtDME production costs.345,346
3.3. Discussion
Currently, DME is almost exclusively produced from syngas in a two-stage process with methanol as an intermediate. Consequently, equivalent PtDME processes can benefit from the long-term operational experience. However, an additional RWGS-reactor is necessary, increasing capital expenditures. An alternative two-stage design might include the direct conversion of CO2-based feed gas to methanol in the first reactor, thus benefitting from experiences from power-to-methanol pilot plants. Both variants offer the advantage that the product ratio of methanol-to-DME can be adjusted based on current market prices. However, this is accompanied by a more complex process design and disadvantages in terms of chemistry.The one-step process can offer a synergetic effect between the ongoing reactions, however, an increasing CO2 content in the feed gas reduces these benefits due to higher formation of water. Hence, a shift of CO2 to CO before the DME reactor might be necessary. Therefore, it is not certain that process complexity decreases with the one-step-process, especially if the more challenging product upgrade is considered. While catalyst development for the direct conversion to DME has already attracted research interest, additional long-term catalyst tests are necessary for a better understanding of catalyst stability and deactivation behaviour. In particular, bifunctional catalysts, which have already shown their high potential regarding selectivity and stability, should be further investigated.
In order to evaluate the different production pathways for PtDME, extensive techno-economic analyses are needed. Different demands determined by the use-case as well as plant location require not only the consideration of classical process factors (such as process pathway, energy consumption or operational expenditures), but in addition the process surroundings (e.g. CO2-source) and market situation (e.g. optimal product ratio). Furthermore, a lack of long-term experiments, pilot plant operation and flexible operations are evident to enable a full assessment of the different pathways.
4. Fischer–Tropsch synthesis
4.1. State of the art Fischer–Tropsch-synthesis
Historically, FT-synthesis was developed in the early 20th century as an alternative fuel production route from coal gasification. After the work on the hydrogenation of CO published by Sabatier in 1902,347 Fischer and Tropsch worked on the catalytic hydrogenation of CO to various products and published their findings in 1923.348,349 Soon after its discovery it was recognised that temperature control of the exothermal reaction would become far easier in a liquid phase synthesis. In 1930 the first pilot plant ran in liquid phase modus.350 The first commercial plant based on a nickel catalyst was constructed in 1933 at Ruhrchemie. In the 1940s, a total capacity of 0.6 million t per a was installed in Germany.36 All these plants were shut down after WWII due to economic reasons.36 In the second half of the 20th century, large-scale processes were applied: a multi-tubular fixed bed reactor with gas recycle was developed by Lurgi and Ruhrchemie. Compared to precursor reactors, this one uses increased temperatures and pressures, a stable reaction rate profile over the length and a better heat removal due to higher gas velocities. This process is called Arge process and was the basis for the Sasolburg plant.
Generally, syngas with a H2/CO ratio of 2–2.2 is processed on a Fe- or Co-catalyst at temperatures of at least 160–200 °C at ambient pressure. Higher pressures and temperatures are advantageous. Reactions (8)–(10) take place during FT-synthesis with n as the resulting carbon chain length (typically ranging between 10 and 20). Reaction (8) is the desired one for the production of alkanes. Water, which is always a by-product, can be converted via WGS reaction with CO to CO2 and H2 by using Fe-catalysts.123
| nCO + (2n + 1)H2 ⇌ CnH2n+2 + nH2O ΔH = n(−146.0) kJ mol−1 | (8) |
| nCO + (2n)H2 ⇌ CnH2n + nH2O | (9) |
| nCO + (2n)H2 ⇌ CnH2n+1OH + (n − 1)H2O | (10) |
The FT-synthesis takes place at temperatures between 150–300 °C. Higher temperatures lead to undesired short chains and methane, the catalyst may be damaged and carbon deposition can occur. Lower temperatures are limited by reaction velocities and the conversion rate. The pressure can be increased to favour long-chain products and to increase the conversion rate. If the pressure is too high, coke formation can lead to catalyst deactivation and expensive high-pressure equipment is needed. The educt gas composition should have a H2/CO ratio around 2 for Co-catalysts, Fe-catalysts tolerate higher CO concentrations. Usually, the carbon source on which the FT-process is based on is CO from gasification of coal or other solid feedstock or natural gas. If natural gas is used as a feedstock, steam reforming has to take place to transform the methane molecule to CO and H2. By using the WGS reaction even CO2 can be used as a carbon source for the synthesis. Regardless of the carbon source, the desired H2/CO ratio is set via the WGS reaction to the ideal level of two. If no pure H2 and CO gas mixture is used as feedstock, a separation of sulphur, which is a catalyst poison, must be ensured.
As mentioned above, the products of the FT-process are carbon chains of different length. The chain length distribution of the alkanes is described by the Anderson–Schulz–Flory distribution (11), which is shown in Fig. 20 for different chain length regions.351,352
| Wn = n(1 − α)2αn−1 | (11) |
 | ||
| Fig. 20 Anderson–Schulz–Flory distribution: weight fraction of different carbon number products as a function of the chain growth α. α is among other things influenced by the catalyst, the reaction conditions, the temperature and the syngas stoichiometry. Adapted from ref. 351–353. | ||
Table 16 shows the chain growth probability that is necessary to obtain a certain product range (number of C atoms). Depending on the α value, the minimum temperature to avoid condensation is indicated. This can be seen as the minimum operation temperature for fluidized bed reactors. Temperatures above 350 °C cannot be achieved in fluidized bed reactors due to carbon formation, which limits its application to medium range carbon chain length. Low-Temperature-Fischer–Tropsch (LT-FT) reactors contain liquid hydrocarbons and can hence be operated below condensation temperature. The main products in these reactors are waxes, whereas High-Temperature-Fischer–Tropsch (HT-FT) produces mainly alkenes and gasoline.36
| Product number of C atoms | Chain growth probability α | Minimum temperature in °C |
|---|---|---|
| 2–5 | 51% | 109 |
| 5–11 | 76% | 329 |
| 5–18 | 82% | 392 |
| 12–18 | 87% | 468 |
Liquid products (in the range of 10–23 carbon atoms per chain) can be used to substitute gasoline or diesel and have advantages compared to conventional fuels, due to their low aromatics content and the absence of sulphur. This results in lower emission values regarding particles, SO2 and aromatics.
As Fig. 21 shows, depending on the chain growth probability, only a small product fraction can directly be used as a diesel fuel. Other by-products must be upgraded first, which mainly refers to hydro-cracking of waxes.
 | ||
| Fig. 21 Fischer–Tropsch product distribution as a function of the chain growth probability, calculated on a mass fraction basis. The number of carbon atoms per chain is shown. | ||
Co-based catalysts are mostly used for natural gas as feedstock and have a high activity.35 Natural gas has a high H2/CO ratio and no WGS reaction is needed to achieve a suitable syngas composition. Ryttera et al. give an overview of different parameters on the selectivity of Co-based catalysts for higher carbons.356 At lower H2/CO ratios, less hydrogen is adsorbed on the catalyst surface increasing the chain growth probability. The same is true for lower temperatures, however the CO conversion decreases.356 At higher water partial pressures methane formation is inhibited improving selectivity as well.357,358 The influence of CO conversion (controlled by the GHSV) depends on the reactor type. For slurry reactors selectivity for higher carbons improves until a conversion of around 75% when CO2 is formed by WGS, which is catalysed due to oxidation of the catalyst.356 The oxidation is facilitated within the slurry reactor through backmixing. Therefore the same effect does not occur in fixed bed or micro-channel reactors.356 CO2 methanation is not possible with Co-catalysts, as Co does not catalyse the WGS reaction and blends of the methanation catalyst with a WGS catalyst are not efficient in CO2 conversion.123 For this reason CO2 is either adsorbed or hydrogenated and therefore only acts as a dilutor.
Compared to Co-catalysts, Fe also catalyses the WGS reaction and is therefore better suited for coal and biomass feedstocks359 as well as for CO2 as a feedstock.123 In this application, higher temperatures above 300 °C are needed due to the higher CO equilibrium concentration. Alkali metals are used as promoters, especially potassium. However their effect highly depends on the interaction with the support material.360 The best conversion rate was achieved with alkalised Fe on γ-alumina.361 H2O on Fe catalysts promotes product inhibition, which leads to a restricted conversion and complicated gas recycling.359
Compared to the Fe catalysts, the Co-based catalysts have a higher productivity at higher conversion levels and an equal productivity at intermediate levels.362 As can be seen in Fig. 22, iron catalysts are favourable at increased pressure and space velocity.
 | ||
| Fig. 22 Productivity comparison between iron (240 °C) and cobalt (220 °C) based catalysts. Adapted from Steynberg and Dry.35 | ||
In the first years after discovery (1925–1950), Co-catalysts were used exclusively. Afterwards, Fe-catalysts were developed. The still higher activity and stability of Co-catalysts lead to a revival, e.g. in the Sasol slurry-phase distillate process.123
In terms of selectivity, two different paths can be realised. In the Sasol-Synthol process, low-molecular-weight products are preferred at high temperatures and all products are in gaseous state at reaction conditions (HTFT). It is operated above 330 °C with Fe-catalysts.363 The second reaction path takes place at lower temperatures with the highest catalytic activities and produces high-molecular-weight products, which are liquid at reaction conditions (LTFT). Both Fe- and Co-based catalysts are used for this route, which need to have big pore sizes to prevent liquid product plugging.363 As the product waxes can be hydrocracked to diesel fuels, this process is very flexible. The diesel fuel selectivity of the latter process reaches 80%.364 One example for a LTFT process is Shell's GtL plant in Malaysia.365
The catalysts can be manufactured by impregnation and precipitation. For the former, the metal oxide is impregnated with a metal salt solution and calcinated afterwards.366 The catalyst activity is influenced by the pore size distribution and the conditions of calcination, but also by promoters e.g. alkali metals for Fe-catalysts or potassium and copper for Co-catalysts.
For Co-catalysts, the main causes of catalyst deactivation are poisoning, re-oxidation of Co-active-sites, formation of surface carbon species (e.g. coke formation at elevated pressures), carbidisation, surface reconstruction, sintering of Co-crystallites, metal-support solid state reactions and attrition.367 Two regimes of deactivation can be distinguished: In the initial deactivation regime, during the first days to weeks, reversible deactivation takes place. In the subsequent long-term deactivation regime, irreversible deactivation decreases the syngas conversion rate more slowly but continuously.367 For Co-catalysts, alkali metals are a catalyst poison. While they increase the chain growth probability, activity is decreased consequently an optimum concentration balancing these effects is necessary.367 Sulphur is a catalyst poison, especially for Co-catalysts, but also for Fe. Sulphur atoms cause a geometric blockage of the active sites.367 Therefore, a limit of 1–2 mg m−3 or 0.02 mg m−3 in modern applications has to be kept.36,347 The impact of sulphur poisoning depends strongly on the sulphur species.367
Fe-catalyst share the same causes of catalyst deactivation, however the oxidation of the active iron phases by water and CO2 play a more prominent role.368 Furthermore, deposition of inactive carbonaceous compounds, a loss of surface area by sintering and catalyst poisoning by sulphur are seen as the main deactivation mechanism for Fe-based catalyst.369
Converter designs. An overview over four currently used converter designs is given in Table 17. Assigned to the HTFT processes, the Sasol's Synthol (SAS) process has been developed from the Argen process (low temperatures, medium pressures, fixed bed) since the 1950s. It involves higher temperatures (300–600 °C) and medium pressures in a circulating fluidized bed and produces light olefins. The updated version is the Advanced Synthol process.370
| Fixed bed (trickle bed) | Slurry phase | Fluidized bed | Circulating fluidized bed (CFB) | |
|---|---|---|---|---|
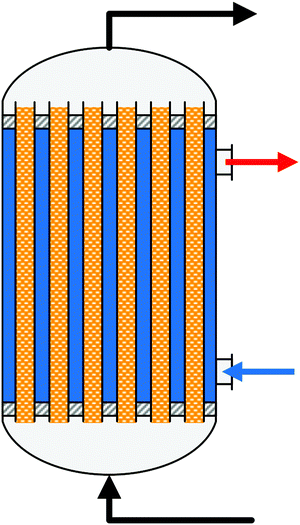
|
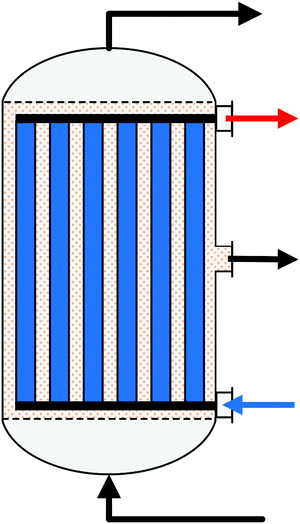
|
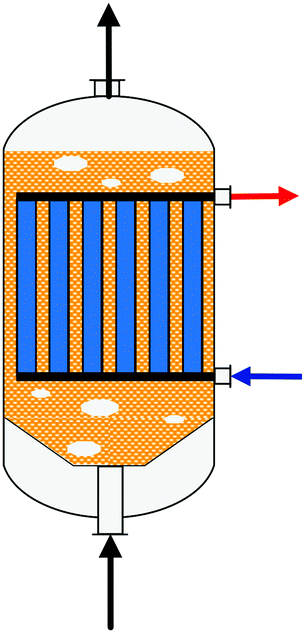
|
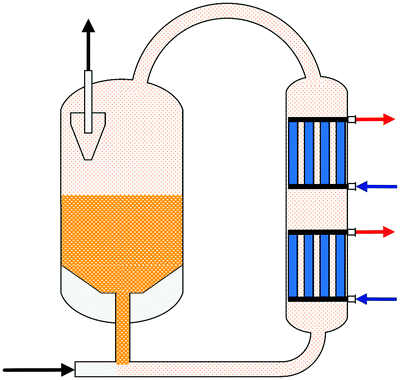
|
|
| Description | Tubes filled with catalyst, boiling water on the shell side. The products are mostly liquid, therefore the reactor is processed as a trickle bed reactor. | Waxy products are used as heat carrier and suspension fluid for fine catalyst particles. The syngas bubbles through the liquid and cooling coils remove the heat. | Low-chain molecules are produced at high temperatures (>300 °C) | Low-chain molecules are produced at high temperatures (>300 °C) |
| Temperature limit | <260 °C, higher temperatures lead to catalyst swelling and hence tube blocking | <260 °C, higher temperatures lead to hydrocracking of the waxes | >300 °C, to avoid agglomerating of catalysts by heavy waxes | >300 °C, to avoid agglomerating of catalysts by heavy waxes |
| Temperature control | Small tube diameters (5 mm for Fe-catalysts, narrower for Co due to activity) and high gas velocities increase the heat flux, medium temperature has to be lower to avoid hotspot temperatures above limit; big temperature gradients in radial and axial direction. | Well mixed reaction phase leads to isothermal conditions and excellent temperature control, therefore average temperature can be higher than in fixed bed. | High degrees of turbulence exhibit very high rates of heat exchange; bed is virtually isothermal | High degrees of turbulence exhibit very high rates of heat exchange; bed is virtually isothermal |
| Reactor cooling coils in suspension fluid generating steam | ||||
| Process conditions | 220–240 °C | 220–240 °C | 320–350 °C | 320–350 °C |
| 20–25 bar | 20–25 bar (SASOL SPD) | Higher pressure up to 40 bar possible because reaction section is much wider | 25 bar | |
| Pressure drop | High in large-scale reactors | Lower than fixed bed | Much lower than in CFB, due to lower velocity | |
| 4 bar | 1 bar | |||
| Selectivity | Similar product distribution to crude oil (high carbon number products) | Similar product distribution to crude oil (high carbon number products) | Mainly low carbon number products (80% < C10) | Mainly low carbon number products (80% < C10) |
| No condensation on outer side of particles (agglomeration) | No condensation on outer side of particles (agglomeration) | |||
| Conversion | ∼60% per pass, recycling is used | ∼60% per pass; carbon efficiency 75% | >85% (higher than CFB due to more catalyst participation in reaction, which increases the conversion) | ∼85% |
| GHSV | 3–4 times higher velocities than in FFB | |||
| Space charge | Low | Higher than fixed bed | ||
| Construction, operation | High construction costs and high weight for large reactors. | 25% more cost-efficient compared to fixed bed (for Fe-catalysts and same capacity); biggest challenge is separation of wax and catalyst; viscosity limits catalyst loading; Thermal efficiency 60% | Construction cost 40% lower than CFB, support structure 95% cheaper; much smaller | Narrower sections are ceramic lined because of abrasive catalyst, regular maintenance is essential |
| Easy separation of liquid products | Less maintenance than CFB, therefor longer on-line times and higher production rates | |||
| Catalyst | Fe or Co-based | Fe or Co-based | Fe-based | Fe-based |
| Catalyst loading and consumption | Only catalyst in first centimetres is affected by catalyst poisons | Catalyst loading 4 times lower than in fixed bed as particle size is 1–2 orders of magnitude smaller than in fixed bed while pore diffusion is rate determining | Carbon deposition is less significant, therefore a lower rate of catalyst removal than in CFB is necessary | Carbon deposition results in lower catalyst loading which leads to declining conversion |
| Small catalyst loading increases sensibility against catalyst poisons like H2S | ||||
| Catalyst removal/exchange | Offline | Online; longer reactor runs, but less advantage with Co-catalysts | Online; due to less carbon deposition, less replacement is necessary than for CFB | Online |
| Upscaling | Easy by adding tubes, limited to 3000–4500 barrel per d | Easy, up to 20000 barrels per d |
||
| Example process | Shell Middle Distillate Synthesis (SMDS) | SASOL SPD | SASOL SAS | Synthol |
| Ref. | 123 and 371 | 35, 123, 307, 371 and 372 | 35, 36, 371 and 373 | 35, 36 and 371 |
In Sasol's slurry-phase distillate (SSPD) process, hot syngas bubbles through a slurry of catalyst particles and liquid products at low temperatures (200–250 °C) and pressures. Whereas in the beginning Fe-based catalysts were used, Co-based catalysts are in favour today.370 The second listed LTFT process is the Shell middle-distillate synthesis (SMDS) process. In this two-step process, paraffinic waxes are synthesised and subsequently hydrocracked to gain a middle-distillate product. In the heavy paraffin synthesis stage, a tubular fixed bed reactor and a catalyst with high selectivity towards heavier products is used followed by a trickle flow hydrocracking reactor.370
Processes. Starting in the 1950s, Sasol built the first commercial, large-scale FT plant for the production of synthetic fuels (Sasol I) in Sasolburg (South Africa). The knowledge gained from operating the first generation of HTFT Circulating Fluidized Bed (CFB) reactors was used to build two more plants (Sasol II and Sasol III) in Secunda in the 1980s, which were based on the second generation of CFB reactors. While Sasol II and III were converted at the end of the 20th century to the Sasol Advanced Synthol reactor type (stationary fluidised bed instead of circulating) which are used until today, Sasol I was completely converted to the production of waxes (LTFT). For this purpose, the CFB reactors were dismantled and a reactor based on the Sasol slurry phase distillate (SPD) process was added to the ARGE Type reactors which had already been operated since the 1950s. The three plants are still in operation today. Whereas in the beginning all synthesis plants used gas from coal gasification, Sasol I is now operated entirely with natural gas. In addition to Sasol, Shell built a comparable high capacity FT plant in the 1990s based on their SMDS fixed bed reactors in Bintulu (Malaysia). Both the SMDS (Shell) and the SPD (Sasol) processes were used by Qatar Petroleum in 2011 and 2007 respectively to install two large-scale GtL plants in Ras Laffan. Sasol itself is currently moving away from coal and natural gas-based synthesis processes towards the production of chemicals.374 Another HTFT plant focusing on the use of low grade coal is the Shenhua Ningxia CtL plant (China), which was commissioned in 2016. The plant, built with the participation of China Fuels, has a capacity of 100
000 bpd and produces diesel, naphtha and alcohols.375 An overview of the process parameters of the HTFT and LTFT plants mentioned is given in Tables 18 and 19 respectively.
| Sasol | Sasol | Mossgas (PetroSA) | ||
|---|---|---|---|---|
| Location | Sasolburg | Secunda | Mossel Bay | |
| Plant Name | Sasol I | Sasol II & III (CtL) | ||
| Product | Gasoline, diesel, C2–C4 gases | Gasoline, distillates, kerosene, alcohols and LPG | ||
| Temperature | 340 °C | 340 °C | 340 °C | |
| Pressure | 20 bar | 20–40 bar | 20–40 bar | |
| Catalyst | Fe-based | Fe-based | Fe-based | |
| Product capacity | 160000 bpd |
1 million t per a | ||
| Feed | Coal | Coal | NG | |
| Reactor type | CFB 1st generation | CFB 2nd generation | SAS | CFB (Sasol Synthol) |
| Status | Shut down | Replaced | Running | Running |
| Time of operation | 1950s–1990s | 1980s–1990s | Since 1990s | Since 1992 |
| No. of reactors | 3 | 16 | 10 (formerly 8) | |
| Ref. | 36 and 376–378 | 376, 377 and 379 | 36, 376, 377 and 379 | 35, 36, 376 and 380 |
| Sasol | Shell | Qatar Petroleum, Shell | Qatar Petroleum, Sasol | |||
|---|---|---|---|---|---|---|
| Location | Sasolburg | Bintulu | Ras Laffan | Ras Laffan | ||
| Plant name | Sasol I | Pearl GTL | Oryx GTL | |||
| Product | Waxes, alkanes | Waxes | Diesel, naphtha | Diesel, naphtha | ||
| Reactor type | ARGE (FB) multitubular | FB (high pressure) | Sasol SPD (slurry) | SMDS fixed bed multitubular | SMDS fixed bed multitubular | Sasol SPD (slurry) |
| Temperature | 230 °C | 220–240 °C | 200–230 °C | 230 °C | 230 °C | |
| Pressure | 27 bar | 45 bar | 20–30 bar | 30 bar | 30 bar | 20–30 bar |
| Status | Running | Running | Running | Running | Running | Running |
| Catalyst | Fe-based | Co-based | Co-based | Fe-based | ||
| Product capacity | 2500 bpd (500 bpd each) | 2500 bpd | 12500 bpd (8000 bpd each) |
140000 bpd |
34000 bpd |
|
| Feed | Coal/NG since 2004 | NG | NG | NG | ||
| Time of operation | Since 1950s | 1987 | Since 1993 | Since 1993 | Since 2011 | Since 2007 |
| No. of reactors | 5 | 4 | 24 | 2 | ||
| Sources | 35 | 381 | 372, 382 and 383 | 376 and 384 | 385 and 386 | 387 and 388 |
High molecular weight products (wax fraction) are either processed to speciality wax products by hydrogenation, hydro-isomerisation or controlled oxidation, or converted to naphtha, diesel and fuel jet in a hydro cracker389–391 Diesel fuel can be gained not only from fractionating, but also from wax hydrocracking and olefin oligomerisation.
Lower molecular weight fractions are hydrogenated and fractionated. The naphtha can be upgraded in a conventional refining step to produce gasoline.35
Several distillation and absorption columns separate the resulting products. These processes are well established from petrochemical industry and their influence on the process performance is rather small.392
Ethen and propene can be used for polymer production and offer good market prices. Propane and butane can be sold as LPG. The naphtha fraction can be used to produce light olefins.
In a first step, light hydrocarbons and gases are removed to ease atmospheric storage. Then oxygenated compounds are removed in a liquid–liquid extraction, for example. In the next step, olefins can be removed by fractionating and extractive distillation. For the remaining liquid products and waxes, the conventional petroleum refinery process can be utilised35,363 even though there are big differences in terms of feed composition, focus of refining and heat management.393
4.2. Current research on power-to-FT-fuels
For the hydrogenation of CO2 to alkanes, kinetic equations have been developed.398,399 As CO2 is a thermodynamically stable molecule, higher synthesis temperatures are required to improve conversion, which lead to a lower chain growth probability and result in more low-carbon number products.400 An increase in pressure and a decrease of the gas velocity lead to more saturated products. Furthermore, lower conversion rates as well as higher inert gas and CO2 content are increasing the chain length.401,402
Regarding conventional FT catalysts, Fe catalysts are more promising for CO2-based feed gases than Co-catalysts because they also catalyse the WGS reaction,398,400,403–405 whereas the feed gas has to be shifted first for a Co-catalyst. If CO2 is fed directly to a Co-catalyst, product selectivity is strongly shifted to methane, even at typical FT temperatures. Fe-based catalysts can be used for CO2 hydrogenation in all three reactor types (fixed bed, fluidized bed, slurry reactor). If the process is aiming for saturated, high boiling products, slurry reactors are advantageous over fixed bed reactor. Fluidized bed reactors are better suited for unsaturated low boiling components. Both slurry and fluidized bed reactors increase the heat transfer from the highly exothermic reaction and are therefore advantageous for the CO2 conversion and product selectivity.406 More recently promising Fe-based multifunctional catalysts have been reported, which allow a highly selective conversion of CO2-based feed gas to gasoline-range hydrocarbons,407,408 however, tests on a pilot or industrial scale have not been conducted yet.
Due to the limited experiences with CO2-based feed gases for FT synthesis and the early stage of FT catalyst development for direct CO2 conversion, realised and planned Power-to-FT-fuels (PtFT-fuels) plants exclusively rely on a shift from CO2 to CO via RWGS-reactor or co-electrolysis. An overview of several projects is given in Table 20.
| Sunfire – fuel 1 plant | Soletair | Integrated PtL test facility | Nordic Blue Crude | The Hague Airport Demo-Plant | |
|---|---|---|---|---|---|
| a Planned capacity. | |||||
| Location | Dresden (Germany) | Lappeenranta (Finland) | Karlsruhe (Germany) | Heroya Industripark (Norway) | Rotterdam (Netherlands) |
| Year | 2014 | 2017 | 2019 | 2022 | Announced 2019 |
| Project partner | Sunfire, EIFER, Fraunhofer, GETEC,HGM, FZ Jülich, Kerafol, Lufthansa, Univ. Bayreuth, Univ. Stuttgart | VTT, LUT | KIT, Climeworks, Ineratec, Sunfire | Nordic Blue Crude AS, Sunfire, Climeworks, EDL Anlagenbau | Rotterdam The Hague Airport, Climeworks, SkyNRG, EDL, Schiphol, Sunfire, Ineratec, Urban Crossovers |
| Operating time | >1500 h | 300 h | — | — | |
| Fuel production | 159 l per d | 10 l per d | 8000 t per aa | 1000 l per da | |
| Process | |||||
| Carbon source | DAC, 3.8 kg CO2 per d | DAC | 28000 t per a |
DAC | |
| Electrolysis | SOEC, 10 kW | PEM electrolysis, 5 kW, 50 bar | Co-electrolysis (SOEC), 10 kW | Co-electrolysis, 20 MW | Co-electrolysis (SOEC) |
| Syngas production | RWGS | RWGS-reactor, 850 °C, 1–5 bar | Co-electrolysis (SOEC) | Co-electrolysis | |
| Reactor | Microstructured heat exchanger reactor cooled by high pressure boiling water | Microstructured reactor | Microstructured reactor | ||
| Ref. | 409–411 and 417 | 412, 418 and 419 | 413–415 | 415 and 416 | 420 |
Sunfire GmbH together with several partners developed a first demonstration plant called ‘Fuel 1’, which was opened in 2014 in Dresden (Germany). The plant was operated for more than 1500 h and can produce 1 barrel (159 l) of FT product per day. A high-temperature SOEC is used for hydrogen production. CO2 is converted in a RWGS reactor. The energy efficiency is around 60% and a CO2 mitigation potential of 85% has been determined compared to conventional fuels.409–411
VTT and LUT operated a first demonstrator plant for 300 h in 2017 within the Soletair project. CO2 was obtained by DAC and H2 produced by PEM electrolysis. The CO2 was converted in a RWGS reactor before entering the FT-reactor. A Co-based catalyst was applied. The microstructured heat exchanger reactor was operated between 200–250 °C and 20–30 bar. Due to a mismatch of production capacities, additional CO and H2 needed to be supplied from gas bundles, also explaining the low carbon efficiency of 38%.412
In 2019 a so-called integrated PtL test facility has been opened as part of the ‘Kopernikus P2X’ initiative funded by the German Federal Ministry of Education and Research. Within a container DAC, co-electrolysis, microstructured FT-reactor and a hydrocracking unit are combined to produce liquid fuels. While the current configuration is only able to supply 10 l per d, a larger pilot plant for the second project phase and a pre-commercial plant design in the third project phase is planned.413–415
A first industrial sized PtL-plant based on FT-synthesis is scheduled to open by 2022 in the Heroya industrial park (Norway). The plant will feature a 20 MW co-electrolysis for syngas production and will produce 8000 t per year of FT-crude, which can be further processed in existing refineries. The power for the electrolysis will be supplied by hydropower plants.415,416
Another project announced in 2019 is a demo-plant at the Hague airport in Rotterdam. The goal is to produce 1000 l of jet fuel per d. CO2 will be directly captured from the air and co-electrolysis will be used to produce syngas. A concrete opening date has not been reported yet.
Feimer et al. published a series of articles about forced cycling in the 1980s. The first article investigates the response of the reaction rate for different products to feed concentration step changes.421 It observes that a steady state is reached not less than 20 minutes after changing the hydrogen content in the feed from 1 to 0.65 in the reference case. Whereas in forced cycling this is an aspired effect, it means unsteady product concentrations in regular FT-reactors.
For moderate changes of the H2/CO ratio (1.1–2), the consumption and formation rates determined under stationary conditions can be used for periodic changes in the H2/CO ratio, as can the resulting product distribution.422 Nevertheless, it must be noted that the synthesis of long-chain hydrocarbons follows the changes in process conditions much more slowly than the synthesis of CH4.423 However, considering the entire process of FT synthesis itself, clear dependencies on changes in process conditions become apparent with regard to process control (e.g. educt separation and recirculation) as well as catalyst activity and product distribution.422
A common method to gain a required flexibility in the synthesis is the integration into an overall process. For example, a BtL and PtL based process can achieve flexibility by providing a storage system between the educt supply and the synthesis reactor.422 Instead of storing excess electricity in H2 storage facilities after electrolysis, current work indicates that the direct utilisation by adapting the H2/CO ratio can be used while increasing the productivity and simultaneously keeping the product distribution range almost unchanged.424
As already mentioned, the product distribution strongly depends on the contact time between the catalyst and educt gas. Therefore increasing GHSV result in a decrease of the CO conversion as well as the C5+.425
Variable H2 supply costs (electricity costs) and plant capacities given, a change from pure BtL over combined processes to pure PtL can be shown for falling electricity costs and smaller plant capacities as well as decreasing production costs for increasing plant scales.336,432 The supply of hydrogen in pure PtL processes accounts for the vast majority of the production costs, which also displays the strong coupling to the electricity as shown above.336,344
In general, the economic feasibility of conventional as well as pure electricity based FT-plants is strongly connected to the market price of diesel and therefore the crude oil price. The crude oil price represents (as shown in Fig. 23) at least a share of 30–50% of the final diesel pricing. FT can be an economic alternative if some requirements are fulfilled. First of all the carbon source must be large enough and cheap, for example stranded or associated natural gas, which is mostly flared now, or to be economically friendly based on carbon capture or biomass gasification. For the history of the FT process, the development of crude oil prices was crucial. In times of an increasing oil price, like in the 1940s and during the oil crisis of the 1970s, FT plants were commissioned.36 At least a product market, e.g. fuel-driven cars, must be accessible. If currently expected PtL prices are taken as a basis, it can be seen that even if FT-based diesel based solely on electricity were to be fully released from taxes, it would not be able to compete in Germany. In order to make FT-diesel marketable, an additional charge on fossil fuels would therefore have to be raised in addition to the tax release for sustainable fuels.
 | ||
| Fig. 23 Development of crude oil and diesel prices in Germany over time, own illustration based on ref. 434 and 435. | ||
In addition to pure techno-economic approaches, Tremel et al. used a ranking system, which includes acceptance as a third group of indicators, to compare different H2 based fuels. The ranking system contains about 20 different weighted indicators, including available infrastructure, number of product upgrading stages or market prices. Compared to SNG, ammonia, DME and methanol, FT-fuels achieved the second highest ranking in the final evaluation.78 This shows in particular that, mainly in a phase-out period in which vehicles still powered by internal combustion engines are used, FT-fuels can make a significant contribution to reducing the demand for fossil fuels. In conclusion, FT-diesel would be economically usable if the price lies below the crude oil price.
Neglecting H2-production coupled costs the synthesis capital costs can be indicated as one of the next most costly factors, whereas the CO2 capture or water costs can be neglected.336
4.3. Discussion
Fischer–Tropsch synthesis is a well-known process that has been developed for almost a century. Big plants with coal and natural gas as a feedstock exist and can be economic, depending on the framework conditions. Smaller plants with biomass-based feedstock have been built in the last decade. For PtFT-fuels plant concepts an opposing trend compared to state of the art FT-processes can be observed. While conventional FT plants have increased in scale to achieve profitability by economy-of-scale effects, many realised PtFT-fuels pilot plants are modular, small-scale, all-in-one container concepts. Their goal is to become profitable by allowing a more dezentrailzed, flexible operation and an easy scale up through modularity.There is still need for research on FT synthesis based on CO2. On the one hand, direct FT-fuel synthesis from CO2-based feed gas is still at a very early stage, requiring further catalyst developments and first lab scale plant tests before becoming an option for future PtL plants. On the other hand, several PtFT-fuels demo plants that include a shift from CO2 to CO have been operated successfully and further larger-scale plants have been announced. For the near term future this will remain the dominant process design for FT-based PtL plants. Consequently, FT-based PtL can benefit substantially from developments in RWGS reactor design and co-electrolysis technologies.
FT-synthesis has not been operated in a flexible mode at a larger scale and research on flexibility and dynamic operation is limited. As a result first PtFT-fuels pilot plants have usually been operated continuously and large hydrogen as well as CO2 storages seem to be currently unavoidable for large-scale PtFT-fuels plants. Further research is necessary to determine the influence of pressure and temperature changes, variations in the syngas quality and part-load performance to determine an operation window in which a satisfying amount of liquid fuels can still be produced. In addition, plant and reactor concepts that allow a dynamic switch between part load and full load operation should be investigated to be able to benefit from low electricity prices without high storage costs. The current state of research does not yet permit cost-optimised or competitive operation of PtFT-fuels plants. Further techno-economic analyses must be undertaken in order to identify possible markets or routes to market entrance and to guide the research accordingly.
5. Remarks on future power-to-liquid applications
Identifying the optimal PtL process route and selecting the most promising product is still challenging and general recommendations remain difficult because the final decision depends on numerous factors including technological, political and economical aspects. Nonetheless some general aspects that should be considered can be derived.Regarding water electrolysis technologies, currently AEL offers the highest technology readiness. In the short-term therefore it can be seen as the obvious choice for relatively low-cost, reliable electrolysis technology for PtL, in particular for large-scale plants. As improvements in the technology and cost reductions have been rapid for PEMEL and with its higher flexibility, it seems especially suited for PtL processes providing grid stability through flexible operation. In the long-term SOEC is very promising for PtL due to the ability to use the process heat generated by the exothermal synthesis reactions thereby achieving higher overall process efficiencies. This will be particularly beneficial in cases where the PtL plant is not integrated into existing industrial processes that can use the excess heat and by-products. Furthermore, the possible application of SOEC for co-electrolysis to produce syngas is favorable for PtFT-fuels and the direct PtDME synthesis route. However, the technology readiness still lacks behind and pressurized operation remains difficult. Therefore, it is a confirmation of the advantages SOEC has to offer for PtL that some of the large-scale projects that have been announced are planning to use co-electrolysis in a MW-scale in only a few years (see Section 4.2.1).
The provision of hydrogen via water electrolysis is the major cost factor for all PtL products. Therefore reducing the hydrogen cost is a significant challenge for the commercialisation of PtL processes. However, depending on the PtL product, different cost reductions are necessary to achieve cost competitiveness. Based on the evaluations by Schemme et al.344 cost shares of the H2 supply for the discussed PtL process routes can be estimated. In the case of methanol synthesis, the H2 supply covers a share of approximately 83%. Assuming that the continuous development of electrolysis and falling electricity prices are the greatest potential for cost reduction, a cost decrease of 25% for the H2 supply may already be sufficient to make PtMeOH economically competitive. Similar to the PtMeOH process, the PtDME process with a H2 supply share of 81% would need a substantial reduction of the H2 supply costs. In contrast, PtFT-fuels would not be viable as a Diesel substitute even with free hydrogen supply due to the low crude oil prices. This, however, may not be the case if FT-waxes are considered as main synthesis product. The change from fuel-focused Coal- or Gas-to-Liquids to the production of chemical products was also announced in 2017 by Sasol's joint chief as a new corporate strategy.374 Especially in combination with existing FT plants, modifying reactors towards wax production and a change to CO2/H2-based feedgas could lead to an economically viable introduction of PtFT-waxes processes.
Currently and in the medium-term CO2 point sources that offer a high partial pressure of CO2 in their flue gases remain the most cost-efficient carbon source for PtL (see Section 1.1.2). Due to the high capital investment necessary to build a PtL plant, however, security of the CO2-supply must be considered. This can be critical for PtL as CCU technology integrated into existing lignite, coal or gas plants that might be shut down in government efforts to achieve climate targets. As a consequence, industrial CO2-sources e.g. from the cement or steel industry as well as CO2 from biogas plants are specifically interesting for PtL. In the long-term, with sufficient cost reductions DAC can provide a certain geographic independence. This might be particularly important for small-scale PtL plants designed to provide grid flexibility as well as large-scale plants build close to large solar or wind power plants away from industrial CO2-sources.
Regarding the purity of the CO2-source, all considered syntheses require very low concentrations of sulphur and chloride compounds making gas cleaning necessary for most carbon sources. PtMeOH offers the advantage that CO2-based feedgases can be used directly and most catalysts are able to tolerate CO as well as CO2 allowing for fluctuations in the feed. The same is true for two-stage PtDME processes. In contrast PtFT-fuels currently requires pure H2/CO syngas with low CO2 content as feed.
Regarding the scale of PtL plants, opposing trends can be observed. Many PtMeOH and PtFT-fuels research projects are focusing on small-scale, modular container concepts. First commercial projects e.g. George Olah plant or Nordic Blue Crude, however, rely on large-scale processes. This illustrates that the scale of future PtL plants is still up for discussion and will probably depend on the surrounding factors of the project as well as the purpose of the plant. Commercial PtL processes trying to provide a carbon-neutral substitute to fossil fuel based products might require a large scale to become cost competitive and producing the required volumes. PtL as a sector coupling technology providing flexibility to the power grid, however, can benefit from small-scale plants that can be implemented close to wind or solar power plants and have the flexibility to use excess electricity. Under this premise, large-scale PtL concepts should focus on reducing costs, enhancing reliability and maximising operation hours, while small-scale, modular concepts also need to consider flexibility.
The location of the project will significantly influence which product is more desirable. FT-fuels might be particularly interesting for locations were the infrastructure for oil processing is already in place. Hence, for many sun-rich oil producing countries, PtFT-fuels can be considered as a promising technology for the transition away from fossil fuels. Large-scale solar power plants can provide affordable, carbon-neutral electricity and during the transition flue gases from refineries and natural gas processing can be used as CO2-sources. In the long-term these CO2-sources can be substituted by DAC to achieve carbon neutrality and allow the production independent from other industrial CO2-sources. As a consequence PtFT-fuels might integrate especially well into the current economic model of these countries. PtMeOH and PtDME however could become more interesting close to their target market. Methanol already is an important platform chemical, which makes an integration into existing industrial parks particularly interesting, saving transportation costs and allowing an optimal by-product and heat integration. The same could be true for DME which has been popular in China were it has a large market as a LPG substitute in cosmetics, lighters or as transportation fuel (see Section 3.2.3).
6. Conclusion
PtL is a promising technical solution for the defossilisation of hard-to-abate sectors. It enables sector coupling by carbon capture and utilisation and can be used to produce valuable products for the chemical industry. With regard to the synthesis step, major challenges are the direct utilisation of CO2, flexible and dynamic operation as well as techno-economic guidance to improve design choices. To which extent these challenges have been addressed depends on the PtL product.Direct synthesis of methanol from CO2-based feed gas has already been tested in PtMeOH pilot plants and even on a commercial scale. For DME direct and two-stage synthesis from CO2 have been investigated, however, pilot plants are still missing and the optimal process route for PtDME remains to be identified. Finally, several PtFT-fuels demo plants based on FT-synthesis have been implemented but all include a shift from CO2 to CO. Direct utilisation of CO2 in the FT-synthesis is at an early stage and additional catalyst development as well as lab-scale testing is necessary.
As state-of-the-art methanol, DME and FT synthesis are typically operated at full-load and part-load operation has not been a focus historically, research on flexibility and dynamic synthesis behaviour is generally limited. The economic viability of PtL processes, however, will depend to a large extent on a more dynamic and flexible operation that enables the optimal use of excess power and low electricity prices. Consequently, additional research concerning the catalyst behaviour under flexible conditions, the influence of reactor and process design choices on the dynamics and the determination of the dynamic plant behaviour should be given more attention. In addition, the scope of future analyses should be extended to the entire system and thus the individual subsystems should be examined from a system integration perspective. Among others, this includes the use of by-products, heat integration and synergy effects.
With regards to techno-economic analyses, several studies have been conducted for each PtL product. Independent of the product, several studies have identified the supply of hydrogen and thus both electrolysis and electricity costs as the main cost factors. Likewise, the CO2-capture is the second major part of the manufacturing costs. Nonetheless detailed cost estimates of the processes are important to support future design choices. This is often made difficult through the extreme variation in boundary conditions. Future studies should therefore strive for more consistent boundary conditions and indicate the influence of changes in their economic assumptions.
Existing pilot plants and future concepts indicate that the development of synthesis concepts for PtL is turning away from the conventional approach in the chemical industry: striving for ever larger-scale plants operated centrally in industry parks. Instead more modular, small-scale concepts dominate, that allow a decentralised operation close to renewable energy sources and CO2-sources. This trend is particularly pronounced for FT-based PtL concepts, where several container-based concepts have been tested and smaller, microchannel reactors replace the large-scale, commercial fixed bed or slurry reactors. Nevertheless, large-scale projects are still being planned, which leads to a split in the development trend. On the one hand, small concepts in container size, which are designed for decentralized application, can be assigned to the electricity sector. The ideas are usually based on a highly flexible operation to use low electricity prices and to act as operating reserve. The large-scale plants, on the other hand, can be allocated to the chemical or mobility sector and aim for a substitution of current energy carriers and basic chemicals. Within these trends, further investigations into possible business cases must be carried out.
Overall, the commercialisation of PtL will require further advancements of the product synthesis together with improvements in electrolysis and carbon capture.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was carried out in the framework of the projects E2Fuels (project no.: 03EIV011G) and HotVeGas III (project no.: 0327731) sponsored by the Federal Ministry for Economic Affairs and Energy (Germany). The financial support is gratefully acknowledged. Furthermore this study was created within the TUM.PtX Network. The authors would like to thank Miriam Sepke-Vogt for her contributions to the DME and FT sections of the paper. Thanks also go to Liu Wei for his help with Chinese patents and Alexander Fröse for his help with literature research.References
- United Nations Framework Convention on Climate Change, Paris Agreement, 2015, https://unfccc.int/files/meetings/paris_nov_2015/application/pdf/paris_agreement_english_.pdf, (accessed March 2019).
- Energinet, Wind turbines cover Denmark's power demand for 24 hours, https://en.energinet.dk/About-our-news/News/2019/09/23/Wind-turbines-cover-power-demand-for-24-hours, (accessed October 2019).
- H. Blanco and A. Faaij, A review at the role of storage in energy systems with a focus on Power to Gas and long-term storage, Renewable Sustainable Energy Rev., 2018, 81, 1049–1086 CrossRef.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Production of synthetic natural gas (SNG) from coal and dry biomass – A technology review from 1950 to 2009, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Power-to-Gas, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- S. Fendt, A. Buttler, M. Gaderer and H. Spliethoff, Comparison of synthetic natural gas production pathways for the storage of renewable energy, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 327–350 CAS.
- G. Gahleitner, Hydrogen from renewable electricity, Int. J. Hydrogen Energy, 2013, 38, 2039–2061 CrossRef CAS.
- M. Bailera, P. Lisbona, L. M. Romeo and S. Espatolero, Power to Gas projects review, Renewable Sustainable Energy Rev., 2017, 69, 292–312 CrossRef CAS.
- M. Thema, F. Bauer and M. Sterner, Power-to-Gas: Electrolysis and methanation status review, Renewable Sustainable Energy Rev., 2019, 112, 775–787 CrossRef CAS.
- M. Lehner, R. Tichler, H. Steinmüller and M. Koppe, Power-to-Gas: Technology and Business Models, Springer International Publishing, Cham, 2014 Search PubMed.
- M. Zapf, Stromspeicher und Power-to-Gas im deutschen Energiesystem, Rahmenbedingungen, Bedarf und Einsatzmöglichkeiten, Springer Vieweg, Wiesbaden, 2017 Search PubMed.
- M. Boudellal, Power-to-Gas. Renewable hydrogen economy for the energy transition, De Gruyter, Berlin, Boston, 2018 Search PubMed.
- A. Tremel, Electricity-based fuels, Springer, Cham, Switzerland, 2018 Search PubMed.
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, Lithium ion, lithium metal, and alternative rechargeable battery technologies: the odyssey for high energy density, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef CAS.
- M. Held, Y. Tönges, D. Pélerin, M. Härtl, G. Wachtmeister and J. Burger, On the energetic efficiency of producing polyoxymethylene dimethyl ethers from CO 2 using electrical energy, Energy Environ. Sci., 2019, 12, 1019–1034 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes, Energy Environ. Sci., 2013, 6, 3112 RSC.
- G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, Hydrogenation of CO2 to value-added products—A review and potential future developments, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS.
- I. Ganesh, Conversion of carbon dioxide into methanol – a potential liquid fuel, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Recent advances in catalytic hydrogenation of carbon dioxide, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- K. A. Ali, A. Z. Abdullah and A. R. Mohamed, Recent development in catalytic technologies for methanol synthesis from renewable sources, Renewable Sustainable Energy Rev., 2015, 44, 508–518 CrossRef CAS.
- S. G. Jadhav, P. D. Vaidya, B. M. Bhanage and J. B. Joshi, Catalytic carbon dioxide hydrogenation to methanol, Chem. Eng. Res. Des., 2014, 92, 2557–2567 CrossRef CAS.
- X.-M. Liu, G. Q. Lu, Z.-F. Yan and J. Beltramini, Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO 2, Ind. Eng. Chem. Res., 2003, 42, 6518–6530 CrossRef CAS.
- M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H.-J. Wernicke, Methanol. The basic chemical and energy feedstock of the future: Asinger's vision today, Springer, Heidelberg, 2014 Search PubMed.
- Methanol production and use, ed. W.-H. Cheng and H. H. Kung, M. Dekker, New York, 1994, vol. 57 Search PubMed.
- J. B. Hansen and P. E. Hojlund Nielsen, Methanol Synthesis, Handbook of Heterogeneous Catalysis, 2008, vol. 6, 13–2949 Search PubMed.
- D. Seddon, Advances in Clean Hydrocarbon Fuel Processing, Elsevier, 2011, pp. 363–386 Search PubMed.
- A. Riaz, G. Zahedi and J. J. Klemeš, A review of cleaner production methods for the manufacture of methanol, J. Cleaner Prod., 2013, 57, 19–37 CrossRef CAS.
- P. J. A. Tijm, F. J. Qaller and D. M. Brown, Methanol technology developments for the new millennium, Appl. Catal., A, 2001, 221, 275–282 CrossRef CAS.
- DME handbook, ed. K. Fujimoto, Y. Ohno, S. Goto, S. Kajitani, M. Konno, T. Shikada and S. Suzuki, Japan DME Forum, Tokyo, 2007 Search PubMed.
- DME handbook supplement, ed. K. Fujimoto, Y. Ohno, S. Kajitani, Y. Mikita, M. Oguma, H. Yagi and T. Yanagawa, Japan DME Forum, Tokyo, 2011 Search PubMed.
- Z. Azizi, M. Rezaeimanesh, T. Tohidian and M. R. Rahimpour, Dimethyl ether, Chem. Eng. Process., 2014, 82, 150–172 CrossRef CAS.
- G. Bozga, I. T. Apan and R. E. Bozga, Dimethyl Ether Synthesis Catalysts, Processes and Reactors, Recent Pat. Catal., 2013, 2, 68–81 CrossRef CAS.
- T. H. Fleisch, A. Basu and R. A. Sills, Introduction and advancement of a new clean global fuel, J. Nat. Gas Sci. Eng., 2012, 9, 94–107 CrossRef CAS.
- Fischer-Tropsch Technology, ed. A. Steynberg and M. Dry, Elsevier, Amsterdam, 2006, vol. 152 Search PubMed.
- M. E. Dry, The Fischer–Tropsch process, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- A. de Klerk, Fischer–Tropsch refining, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Hoboken, 2011 Search PubMed.
- J. C. Koj, C. Wulf and P. Zapp, Environmental impacts of power-to-X systems - A review of technological and methodological choices in Life Cycle Assessments, Renewable Sustainable Energy Rev., 2019, 112, 865–879 CrossRef.
- B. R. de Vasconcelos and J. M. Lavoie, Recent advances in power-to-X technology for the production of fuels and chemicals, Front. Chem., 2019, 7, 392 CrossRef PubMed.
- S. Hänggi, P. Elbert, T. Bütler, U. Cabalzar, S. Teske, C. Bach and C. Onder, A review of synthetic fuels for passenger vehicles, Energy Rep., 2019, 5, 555–569 CrossRef.
- P. Schmidt, V. Batteiger, A. Roth, W. Weindorf and T. Raksha, Power-to-Liquids as Renewable Fuel Option for Aviation: A Review, Chem. Ing. Tech., 2018, 90, 127–140 CrossRef CAS.
- A. Buttler and H. Spliethoff, Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- L. Wang, M. Pérez-Fortes, H. Madi, S. Diethelm, J. van herle and F. Maréchal, Optimal design of solid-oxide electrolyzer based power-to-methane systems: A comprehensive comparison between steam electrolysis and co-electrolysis, Appl. Energy, 2018, 211, 1060–1079 CrossRef CAS.
- R. Andika, A. B. D. Nandiyanto, Z. A. Putra, M. R. Bilad, Y. Kim, C. M. Yun and M. Lee, Co-electrolysis for power-to-methanol applications, Renewable Sustainable Energy Rev., 2018, 95, 227–241 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, A comparative technoeconomic analysis of pathways for commercial electrochemical CO 2 reduction to liquid products, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, A review of high temperature co-electrolysis of H2O and CO2 to produce sustainable fuels using solid oxide electrolysis cells (SOECs): advanced materials and technology, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Carbon capture and storage (CCS): the way forward, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, An overview of CO2 capture technologies, Energy Environ. Sci., 2010, 3, 1645 RSC.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Carbon capture and storage update, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- S. Brandani, Carbon Dioxide Capture from Air: A Simple Analysis, Energy Environ., 2012, 23, 319–328 CrossRef.
- M. Fasihi, O. Efimova and C. Breyer, Techno-economic assessment of CO2 direct air capture plants, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- S. Budinis, S. Krevor, N. M. Dowell, N. Brandon and A. Hawkes, An assessment of CCS costs, barriers and potential, Energy Strateg. Rev., 2018, 22, 61–81 CrossRef.
- T. Trost, S. Horn, M. Jentsch and M. Sterner, Erneuerbares Methan: Analyse der CO2-Potenziale für Power-to-Gas Anlagen in Deutschland, Z. Energiewirtsch., 2012, 36, 173–190 CrossRef.
- A.-M. Cormos, S. Dragan, L. Petrescu, V. Sandu and C.-C. Cormos, Techno-Economic and Environmental Evaluations of Decarbonized Fossil-Intensive Industrial Processes by Reactive Absorption & Adsorption CO2 Capture Systems, Energies, 2020, 13, 1268 CrossRef.
- E. Supp, How to produce methanol from coal, Springer-Verlag, Berlin, 1990 Search PubMed.
- P. J. A. Tijm, F. J. Qaller and D. M. Brown, Methanol technology developments for the new millennium, Appl. Catal., A, 2001, 221, 275–282 CrossRef CAS.
- J. Werther, in Ullmann's Encyclopedia of Industrial Chemistry, ed. B. Elvers, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2020, pp. 320–366 Search PubMed.
- K.-D. Henkel, in Ullmann's Encyclopedia of Industrial Chemistry, ed. B. Elvers, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2020, pp. 293–327 Search PubMed.
- J. Ott, V. Gronemann, F. Pontzen, E. Fiedler, G. Grossmann, D. B. Kersebohm, G. Weiss and C. Witte, in Ullmann's Encyclopedia of Industrial Chemistry, ed. B. Elvers, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2020 Search PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. Jouny, G. S. Hutchings and F. Jiao, Carbon monoxide electroreduction as an emerging platform for carbon utilization, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- J. Durst, A. Rudnev, A. Dutta, Y. Fu, J. Herranz, V. Kaliginedi, A. Kuzume, A. A. Permyakova, Y. Paratcha, P. Broekmann and T. J. Schmidt, Electrochemical CO2 Reduction - A Critical View on Fundamentals, Materials and Applications, Chimia, 2015, 69, 769–776 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Cocatalysts in Semiconductor-based Photocatalytic CO2 Reduction: Achievements, Challenges, and Opportunities, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- C. Wang, Z. Sun, Y. Zheng and Y. H. Hu, Recent progress in visible light photocatalytic conversion of carbon dioxide, J. Mater. Chem. A, 2019, 7, 865–887 RSC.
- O. S. Bushuyev, P. de Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, What Should We Make with CO2 and How Can We Make It?, Joule, 2018, 2, 825–832 CrossRef CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Photoelectrochemical devices for solar water splitting - materials and challenges, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Low-dimensional catalysts for hydrogen evolution and CO2 reduction, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- Z. Zakaria and S. K. Kamarudin, Direct conversion technologies of methane to methanol: An overview, Renewable Sustainable Energy Rev., 2016, 65, 250–261 CrossRef CAS.
- H. J. Kim, J. Huh, Y. W. Kwon, D. Park, Y. Yu, Y. E. Jang, B.-R. Lee, E. Jo, E. J. Lee, Y. Heo, W. Lee and J. Lee, Biological conversion of methane to methanol through genetic reassembly of native catalytic domains, Nat. Catal., 2019, 2, 342–353 CrossRef CAS.
- C. E. Bjorck, P. D. Dobson and J. Pandhal, Biotechnological conversion of methane to methanol: evaluation of progress and potential, AIMS Bioeng., 2018, 5, 1–38 CAS.
- T. P. Howard, S. Middelhaufe, K. Moore, C. Edner, D. M. Kolak, G. N. Taylor, D. A. Parker, R. Lee, N. Smirnoff, S. J. Aves and J. Love, Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7636–7641 CrossRef CAS PubMed.
- L. Brennan and P. Owende, Biofuels from microalgae—A review of technologies for production, processing, and extractions of biofuels and co-products, Renewable Sustainable Energy Rev., 2010, 14, 557–577 CrossRef CAS.
- S. K. Bhatia, R. K. Bhatia, J.-M. Jeon, G. Kumar and Y.-H. Yang, Carbon dioxide capture and bioenergy production using biological system – A review, Renewable Sustainable Energy Rev., 2019, 110, 143–158 CrossRef CAS.
- P. K. Swain, L. M. Das and S. N. Naik, Biomass to liquid, Renewable Sustainable Energy Rev., 2011, 15, 4917–4933 CrossRef CAS.
- I. Martinez, Fuel Properties, http://webserver.dmt.upm.es/~isidoro/bk3/c15/Fuel%20properties.pdf, (accessed March 2017).
- R. P. Verbeek and A. van Doorn, Global assessment of Dimethyl-ether as an automotive fuel, http://repository.tudelft.nl/view/tno/uuid%3A3ca1fa57-4226-4442-b005-86f092e7e5b4/, (accessed 23 March 2017).
- Introduction to Chemicals from Biomass, ed. J. H. Clark and F. E. I. Deswarte, John Wiley & Sons, Ltd, Chichester, West Sussex, 2015 Search PubMed.
- A. Tremel, P. Wasserscheid, M. Baldauf and T. Hammer, Techno-economic analysis for the synthesis of liquid and gaseous fuels based on hydrogen production via electrolysis, Int. J. Hydrogen Energy, 2015, 40, 11457–11464 CrossRef CAS.
- I. Wender, Reactions of Synthesis Gas, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- M. L. Kastens, J. F. Dudley and J. Troeltzsch, Synthetic Methanol Production, Ind. Eng. Chem., 1948, 40, 2230–2240 CrossRef CAS.
- K. Klier, V. Chatikavanij, R. G. Herman and G. W. Simmons, Catalytic Synthesis of Methanol from CO/H2, J. Catal., 1982, 74, 343–360 CrossRef CAS.
- G. H. Graaf, J. G. M. Winkelman, E. J. Stamhuis and A. A. C. M. Beenackers, Kinetics of the Three Phase Methanol Synthesis, Chem. Eng. Sci., 1988, 43, 2161–2168 CrossRef CAS.
- J. S. Lee, K. H. Lee, S. Y. Lee and Y. G. Kim, A Comparative Study of Methanol Synthesis from CO2/H2 and CO/H2 over a Cu/ZnO/AI20 3 Catalyst, J. Catal., 1993, 144, 414–424 CrossRef CAS.
- G. Liu, D. Willcox, M. Garland and H. H. Kung, The Rate of Methanol Production on a Copper-Zinc Oxide Catalyst:The Dependence on the Feed Composition, J. Catal., 1984, 90, 139–146 CrossRef CAS.
- M. Sahibzada, I. S. Metcalfe and D. Chadwick, Methanol Synthesis from CO/CO2/H2 over Cu/ZnO/Al2O3 at Differential and Finite Conversions, J. Catal., 1998, 174, 111–118 CrossRef CAS.
- J. Skrzypek, M. Lachowska and H. Moroz, Kinetics of Methanol Synthesis over Commercial Copper/Zinc Oxide/Alumina Catalysts, Chem. Eng. Sci., 1991, 46, 2809–2813 CrossRef CAS.
- K. M. Vanden Bussche and G. F. Froment, A Steady-State Kinetic Model for Methanol Synthesis and the Water Gas Shift Reaction on a Commercial Cu/ZnO/Al2O3 Catalyst, J. Catal., 1996, 161, 1–10 CrossRef CAS.
- K. L. Ng, D. Chadwick and B. A. Toseland, Kinetics and modelling of dimethyl ether synthesis from synthesis gas, Chem. Eng. Sci., 1999, 54, 3587–3592 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, D. G. Parker, M. S. Spencer and D. A. Whan, Mechanism of methanol synthesis from CO2/CO/H2 mixtures over copper/zinc oxide/alumina catalysts: use of14C-labelled reactants, Appl. Catal., 1987, 30, 333–338 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Review, Appl. Catal., 1988, 36, 1–65 CrossRef CAS.
- A. Y. Rozovskii, Modern problems in the synthesis of methanol, Russ. Chem. Rev., 1989, 58, 68–93 CAS.
- M. Saito, T. Fujitani, M. Takeuchi and T. Watanabe, Development of copper/zinc oxide-basedmulticomponent catalysts for methanol synthesisfrom carbon dioxide and hydrogen, Appl. Catal., A, 1996, 138, 311–318 CrossRef CAS.
- A. Y. Rozovskii, Novye dannye o mehanizme kataliticheskih reakcij s uchastiem okislov ugleroda, Kinet. Katal., 1980, 21, 97–107 CAS.
- J. Haid and U. Koss, Lurgi's Mega-Methanol technology opens the door for a new era in down-stream applications, Stud. Surf. Sci. Catal., 2001, 399–404 CrossRef CAS.
- E. Filippi and M. Badano, Methanol Converter and Synloop Designs for Gasification Plants, Orlando, Florida, USA, 2007.
- C. J. Schack, M. A. McNeil and R. G. Rinker, Methanol Synthesis from Hydrogen, Carbon Monoxide, and Carbon Dioxide over a CuO/ZnO/Al2O3 Catalyst, Appl. Catal., A, 1989, 50, 247–263 CrossRef CAS.
- H. Makihara, K. Niwa, H. Nagai, K. Morita, H. Horizoe, K. Kobayashi and C. Kuwada, Characteristics of a New Methanol Synthesis Reactor, Energy Prog., 1987, 7, 51–58 CAS.
- L. Chen, Q. Jiang, Z. Song and D. Posarac, Optimization of Methanol Yield from a Lurgi Reactor, Chem. Eng. Technol., 2011, 34, 817–822 CrossRef CAS.
- T. Wurzel, Lurgi MegaMethanol Technology – Delivierung the building blocks for future fuel and monomer demand, Dresden, Germany, 2006 Search PubMed.
- Haldor Topsøe A/S, MK-121, https://www.topsoe.com/products/catalysts/mk-121, (accessed April 2020).
- Clariant International Ltd, Catalysts for Syngas, http://www.catalysts.clariant.com, (accessed 24 May 2016).
- Johnson Matthey Catalysts, Katalco, http://www.jmcatalysts.cn/en/pdf/Methanol-top-level.pdf, (accessed 18 May 2016).
- F. J. Broecker, K.-H. Gruendler, L. Marosi, M. Schwarzmann, B. Triebskorn and G. Zirker, US Pat., 4666945, 1987 Search PubMed.
- M. Schneider, K. Kochloefl and J. Ledbeck, EP Pat., 0125689, 1987 Search PubMed.
- I. Dybkjær, Topsoe Methanol Technology, CEER, Chem. Econ. Eng. Rev., 1981, 13, 17–25 Search PubMed.
- F. J. Broecker, K.-H. Gruendler, L. Marosi and M. Schwarzmann, US Pat., 4436833, 1984 Search PubMed.
- T. Gallagher and J. M. Kidd, Br. Pat., 1159035, 1969 Search PubMed.
- E. F. Magoon and L. Henry, DE Pat., 2154074, 1972 Search PubMed.
- A. Passariello, DE Pat., 3238845, 1983 Search PubMed.
- P. Davies and F. F. Snowdon, US Pat., 3326956, 1963 Search PubMed.
- G. Baron, G. Bechtholdt, K. Bratzler, H. Liebgott and E. Erhard, DE Pat., 1300917, 1967 Search PubMed.
- M. Saito, M. Takeuchi, T. Fujitani, J. Toyir, S. Luo, J. Wu, H. Mabuse, K. Ushikoshi, K. Mori and T. Watanabe, Advances in joint research between NIRE and RITE for developing a novel technology for methanol synthesis from CO2 and H2, Appl. Organomet. Chem., 2000, 14, 763–772 CrossRef CAS.
- M. V. Twigg and M. S. Spencer, Deactivation of supported copper metal catalysts for hydrogenation reactions, Appl. Catal., A, 2001, 212, 161–174 CrossRef CAS.
- P. J. Dahl, T. S. Christensen, S. Winter-Madsen and S. M. King, Proven autothermal reforming technology modern large for modern large scale methanol plants, Paris, 2014 Search PubMed.
- I. Dybkjær and J. B. Hansen, New Developments in Methanol Technology, CEER, Chem. Econ. Eng. Rev., 1985, 17, 13–17 Search PubMed.
- K. Ushikoshi, K. Mori, T. Kubota, T. Watanabe and M. Saito, Methanol synthesis from CO2 and H2 in a bench-scale test plant, Appl. Organomet. Chem., 2000, 14, 819–825 CrossRef CAS.
- K. Hirotani, H. Nakamura and K. Shoji, Optimum catalytic reactor design for methanol synthesis with TEC MRF-Z reactor, Catal. Surv. Jpn., 1998, 2, 99–106 CrossRef CAS.
- P. Grimm, Six years successful operation of Linde isothermal reactor, Rep. Sci. Technol., 1991, 49, 57–59 Search PubMed.
- H. H. Kung, Deactivation of Methanol Synthesis Catalysts – A Review, Catal. Today, 1992, 11, 443–453 CrossRef CAS.
- F. Baratto, H. Rong and H. Anxin, Casale Coal-Based Methanol Synloops, Barcelona, Spain, 2010 Search PubMed.
- E. C. Heydorn, B. W. Diamond and R. D. Lilly, Commercial-Scale Demonstration of the Liquid Phase Methanol (LPMEOH) Process. Final Report (Volume 2: Project Performance and Economics), U.S. Department of Energy, 2003.
- C. S. Nielsen, Topsøe Methanol Technology for Coal Based Plants, http://www.topsoe.com/sites/default/files/topsoe_methanol_coal_based_plants.ashx_pdf, (accessed 4 May 2016).
- A. Bandi and M. Specht, in Renewable Energy, ed. K. Heinloth, Springer, Berlin, Heidelberg, 2006, pp. 414–482 Search PubMed.
- M. Saito, M. Takeuchi, T. Watanabe, J. Toyir, S. Luo and J. Wu, Methanol Synthesis from CO2 and H2 over a Cu/ZnO-Based Multicomponent Catalyst, Energy Convers. Manage., 1997, 38, 403–408 CrossRef.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- F. Pontzen, W. Liebner, V. Gronemann, M. Rothaemel and B. Ahlers, CO2-based methanol and DME – Efficient technologies for industrial scale production, Catal. Today, 2011, 171, 242–250 CrossRef CAS.
- J. B. Hansen, Methanol Production Technology: Todays and future Renewable Solutions, Lund, 2015 Search PubMed.
- P. L. Golosnichenko, S. M. Kononov, E. Filippi and N. Ringer, Revamping of the Nevinnomyssk Azotmethanol Synthesis Converter. The First Application of the Casale ‘IMC’ Design, Phoenix, USA, 2003.
- M. R. Rahimpour, B. Moghtaderi, A. Jahanmiri and N. Rezaie, Operability of an Industrial Methanol Synthesis Reactor with Mixtures of Fresh and Partially Deactivated Catalyst, Chem. Eng. Technol., 2005, 28(2), 226–234 CrossRef CAS.
- I. Dybkjær and J. B. Hansen, Large-scale Production of Alternative Synthetic Fuels from Natural Gas, Stud. Surf. Sci. Catal., 1997, 107, 99–116 CrossRef.
- U. Dengel, Global E&C Solution, Buenos Aires, Argentina, 2013 Search PubMed.
- Johnson Matthey Davy Technologies, Company Website, http://davyprotech.com/, (accessed 7 June 2016).
- E. Supp, Improved Methanol Process, Hydrocarbon Process., 1981, 71–75 CAS.
- K. Aasberg-Petersen, C. S. Nielsen, I. Dybkjær and J. Perregaard, Large Scale Methanol Production from Natural Gas, http://www.topsoe.com/file/large-scale-methanol-production-natural-gas, (accessed 4 May 2016).
- Johnson Matthey Process Technologies, Delivering world class methanol plant performance, http://www.jmprotech.com/, (accessed 7 June 2016).
- H. Schneider, H. Schmid, A. Watson and W. Witter, DE Pat., 2504343, 1975 Search PubMed.
- A. G. Linde, The Linde-Variobar® Process for the Production of Methanol with the New Isothermal Reactor, CEER, Chem. Econ. Eng. Rev., 1983, 15, 14–16 Search PubMed.
- A. Buttler, Technoökonomische Bewertung von Polygenerationskraftwerken und Power-to-X-Speichern in einem nachhaltigen Energiesystem, Verlag Dr Hut, München, 2018 Search PubMed.
- K. Ohsaki, K. Shoji, O. Okuda, Y. Kobayashi and H. Koshimizu, A Large-Scale Methanol Plant Based on ICI Process, CEER, Chem. Econ. Eng. Rev., 1985, 17, 31–38 CAS.
- K. Kobayashi, H. Osora, H. Nagai and H. Ohira, US Pat., 7189379, 2001 Search PubMed.
- T. Shinkawa, D. Shozen, K. Kobayashi, H. Makihara, K. Niwa, K. Morita, M. Kuwa and M. Katsutoshi, US Pat., 4938930, 1987 Search PubMed.
- I. Muscionico, P. Moreo and P. Talarico, 3000 MTD Methanol Synloop in Coal-Based Plant, Singapore, 2013 Search PubMed.
- I. Takase and K. Niwa, Mitsubishi (MGC/MHI) Methanol Process, CEER, Chem. Econ. Eng. Rev., 1985, 17, 24–30 CAS.
- F. Baratto, H. Rong and H. Anxin, Casale Coal-Based Methanol Synloops, Barcelona, Spain, 2010 Search PubMed.
- E. Filippi, Challenges in Designing Synthesis Converters for very large Methanol Production Capacity, Tehran, Iran, 2003 Search PubMed.
- I. Muscionico and P. Moreo, Jiu Tai Energy. First Methanol Casale Axial-Radial IMC Converter in Coal-Based Methanol Plant, 2013.
- F. Zardi, Latest Casale technologies for grass-root fertilizer and methanol plants, Tehran, Iran, 2008 Search PubMed.
- E. Filippi, Methanol Casale's Latest Converters: New Milestones for the Methanol Industry, Prague, Czech Republic, 2006 Search PubMed.
- H. Kopetsch, P. M. Hackel, V. Gronemann and R. Morgenroth, US Pat., 8629191, 2009 Search PubMed.
- A. Arora, S. S. Iyer, I. Bajaj and M. M. F. Hasan, Optimal Methanol Production via Sorption-Enhanced Reaction Process, Ind. Eng. Chem. Res., 2018, 57, 14143–14161 CrossRef CAS.
- J. Terreni, M. Trottmann, T. Franken, A. Heel and A. Borgschulte, Sorption-Enhanced Methanol Synthesis, Energy Technol., 2019, 7, 1801093 CrossRef.
- H. Matsumoto, H. Watanabe, H. Nagai, K. Morita and H. Makihara, Advanced Technology for Large Scale Methanol Plant, Tech. Rev. – Mitsubishi Heavy Ind., 1997, 34, 58–62 Search PubMed.
- G. Pass, C. Holzhauser, A. Akgerman and R. G. Anthony, Methanol synthesis in a trickle-bed reactor, AIChE J., 1990, 36, 1054–1060 CrossRef CAS.
- R. P. W. J. Struis and S. Stucki, Verification of the membrane reactor concept for the methanol synthesis, Appl. Catal., A, 2001, 216, 117–129 CrossRef CAS.
- R. P. W. J. Struis, M. Quintilii and S. Stucki, Feasibility of Li-Nafion hollow fiber membranes inmethanol synthesis: mechanical and thermal stabilityat elevated temperature and pressure, J. Membr. Sci., 2000, 177, 215–223 CrossRef CAS.
- R. P. W. J. Struis, S. Stucki and M. Wiedorn, A membrane reactor for methanol synthesis, J. Membr. Sci., 1996, 113, 93–100 CrossRef CAS.
- B. Sea and K.-H. Lee, Methanol synthesis from carbon dioxide and hydrogen using a ceramic memebrane reactor, React. Kinet. Catal. Lett., 2003, 80, 33–38 CrossRef CAS.
- G. Chen, Methanol synthesis from CO2 using a silicone rubber/ceramic composite membrane reactor, Sep. Purif. Technol., 2004, 34, 227–237 CrossRef CAS.
- G. Barbieri, G. Marigliano, G. Golemme and E. Drioli, Simulation of CO2 hydrogenation with CH3OH removal in a zeolite membrane reactor, Chem. Eng. J., 2002, 85, 53–59 CrossRef CAS.
- F. Galluci and A. Basile, A theoretical analysis of methanol synthesis from CO2 and H2 in a ceramic membrane reactor, Int. J. Hydrogen Energy, 2007, 32, 5050–5058 CrossRef.
- F. Galluci, L. Paturzo and A. Basile, An experimental study of CO2 hydrogenation into methanol involving a zeolite membrane reactor, Chem. Eng. Process., 2004, 43, 1029–1036 CrossRef.
- M. R. Rahimpour and S. Ghader, Theoretical Investigation of a Pd-membrane Reactor for Methanol Synthesis, Chem. Eng. Technol., 2003, 26, 902–907 CrossRef CAS.
- W. R. Brown, D. P. Drown and F. S. Frenduto, Commercial-Scale Demonstration of the Liquid Phase Methanol (LPMEOH) Process. Final Report, Volume 1: Public Design, U.S. Department of Energy, 2000 Search PubMed.
- National Energy Technology Laboratory, Commercial-Scale Demonstration of the Liquid PhaseMethanol (LPMEOH™) Process. A DOE Assessment, U.S. Department of Energy, 2003 Search PubMed.
- U.S. Department of Energy and Air Products Liquid Phase Conversion Company, Commercial-Scale Demonstration of the Liquid Phase Methanol (LPMEOHTM) Process, U.S. Department of Energy, 1999 Search PubMed.
- D. W. Studer and E. S. Schaub, US Pat., 5284878, 1993 Search PubMed.
- E. L. Sorensen, Production of DME from Synthesis Gas, http://www.solandersciencepark.se/, (accessed 6 October 2016).
- E. L. Sorensen, Valorisation of Synthesis Gas from Biomass - The Pitea DME pilot, http://tu-freiberg.de/fakult4/iec/evt/valorisation-of-synthesis-gas-from-biomass-gasification-the-pitea-biodme-pilot-plant, (accessed 6 October 2016).
- I. Landälv, R. Gebart, B. Marke, F. Granberg, E. Furusjö, P. Löwnertz, O. G. W. Öhrman, E. L. Sørensen and P. Salomonsson, Two years experience of the BioDME project-A complete wood to wheel concept, Environ. Prog. Sustainable Energy, 2014, 33, 744–750 CrossRef.
- I. Landälv, Two Years Experience of the BioDME Project- A complete Wood to Wheel Concept, Chicago, USA, 2013 Search PubMed.
- J. B. Hansen and F. Joensen, High Conversion of Synthesis Gas into Oxygenates, Stud. Surf. Sci. Catal., 1991, 61, 457–467 CrossRef CAS.
- J. R. LeBlanc, R. V. Schneider and R. B. Strait, in Methanol production and use, ed. W.-H. Cheng and H. H. Kung, M. Dekker, New York, 1994, pp. 51–132 Search PubMed.
- P. König and H. Göhna, US Pat., 5827901, 1997 Search PubMed.
- Nitrogen + Syngas, Methanol plants keep getting bigger, BCInsights, 2012.
- S. A. Casale, Company website, http://www.casale.ch/, (accessed 2 May 2016).
- Lurgi GmbH, Lurgi MegaMethanol, http://www.zeogas.com/files/83939793.pdf, (accessed 3 May 2016).
- Air Liquide Global E&C Solutions, Lurgi Methanol. Proven technology flexible in feedstock, (accessed 3 May 2016).
- Brunei Methanol Company Sendirian Berhard, Company Website, http://brunei-methanol.com/, (accessed 29 April 2016).
- Mitsubishi Corporation, Company Website, http://www.mitsubishicorp.com/jp/en/, (accessed 29 April 2016).
- Mitsubishi Gas Chemical, Company Website, http://www.mgc-a.com/, (accessed 29 April 2016).
- Toyo Engineering Corporation, Company Website, http://www.toyo-eng.com/jp/en/, (accessed 29 April 2016).
- Linde AG, Company Website, http://www.linde-engineering.com/en/, (accessed 4 May 2016).
- B. Doss, C. Ramos and S. Atkins, Optimization of Methanol Synthesis from Carbon Dioxide and Hydrogen, Energy Fuels, 2009, 23, 4647–4650 CrossRef CAS.
- J. Toyir, R. Miloua, N. E. Elkadri, M. Nawdali, H. Toufik, F. Miloua and M. Saito, Sustainable process for the production of methanol from CO2 and H2 using Cu/ZnO-based multicomponent catalyst, Phys. Procedia, 2009, 2, 1075–1079 CrossRef CAS.
- M. Saito and K. Murata, Development of high performance Cu/ZnO-based catalysts for methanol synthesis and the water-gas shift reaction, Catal. Surv. Asia, 2004, 8, 285–294 CrossRef CAS.
- M. Takeuchi, H. Mabuse, T. Watanabe, M. Umeno, T. Matsuda, K. Mori, K. Ushikoshi, J. Toyir, S. Luo, J. Wu and M. Saito, US Pat., 6048820, 2000 Search PubMed.
- S. Dang, H. Yang, P. Gao, H. Wang, X. Li, W. Wei and Y. Sun, A review of research progress on heterogeneous catalysts for methanol synthesis from carbon dioxide hydrogenation, Catal. Today, 2019, 330, 61–75 CrossRef CAS.
- R. Guil-López, N. Mota, J. Llorente, E. Millán, B. Pawelec, J. L. G. Fierro and R. M. Navarro, Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis, Materials, 2019, 12, 3902 CrossRef PubMed.
- M. Specht, A. Bandi, M. Elser and F. Staiss, Comparison CO2 sources for the synthesis of renewable methanol, Stud. Surf. Sci. Catal., 1998, 114, 363–366 CrossRef.
- O.-S. Joo, K.-D. Jung, I. Moon, A. Y. Rozovskii, G. I. Lin, S.-H. Han and S.-J. Uhm, Carbon Dioxide Hydrogenation To Form Methanol via a Reverse-Water-Gas-Shift Reaction (the CAMERE Process), Ind. Eng. Chem. Res., 1999, 38, 1808–1812 CrossRef CAS.
- H. Goehna and P. König, Producing methanol from CO2, CHEMTECH, 1994, 36–39 CAS.
- K. Ushikoshi, K. Mori, T. Watanabe, M. Takeuchi and M. Saito, A 50 kg/day class test plant for methanol synthesis from CO2 and H2, Stud. Surf. Sci. Catal., 1998, 114, 357–362 CrossRef.
- B. Stefansson, Methanol fuel from power and CO2 emissions Opportunities and Challenges, Brussels, 2015 Search PubMed.
- P. König and H. Göhna, US Pat., 5631302, 1995 Search PubMed.
- M. Specht, Power-to-Gas - Speicherung erneuerbarer Energie im Ergasnetz, Emmerthal, 2012 Search PubMed.
- O.-S. Joo, K.-D. Jung and Y. Jung, Camere process for methanol synthesis from CO2 hydrogenation, Stud. Surf. Sci. Catal., 2004, 153, 67–72 CrossRef CAS.
- T. Matsushita, T. Haganuma and D. Fujita, WO Pat., 136345, 2011 Search PubMed.
- T. Matsushita, T. Haganuma and D. Fujita, US Pat., 0237618, 2013 Search PubMed.
- M. Specht and A. Bandi, Der “Methanol-Kreislauf” – nachhaltige Bereitstellung flüssiger Kraftstoffe, Forschungsverbund Sonnenenergie “Themen 98/99”, 1998, 59–65.
- Y. Nawa, Greenhouse Gas to Chemical Resources. Mitsui Chemical's Challenge for Sustainable Growth, Tianjin, China, 2011 Search PubMed.
- E. R. Morgan and T. Acker, Practical Experience with a mobile methanol synthesis device, Proceedings of the ASME 2014 8th International Conference on Energy Sustainability & 12thFuel Cell Science, Engineering and Technology Conference, 2014.
- E. R. Morgan and T. L. Acker, Practical Experience With a Mobile Methanol Synthesis Device, J. Sol. Eng., 2015, 137, 064506 CrossRef.
- P. Grauer and R. Meyer-Pittroff, EP Pat., 2647596, 2013 Search PubMed.
- R. Meyer-Pittroff, Chemische Energiespeicherung mittels Methanol, BWK, 2012, 64, 36–39 Search PubMed.
- O. F. Sigurbjörnsson, Sustainable Fuels and Chemicals by Carbon Recycling, 2015 Search PubMed.
- J. Gale, IEAGHG Information Paper (2016-IP4): Developments in Renewable Methanol Production, http://documents.ieaghg.org/index.php/s/9q8FudyD9HOVrax, (accessed 5 July 2019).
- Mitsubishi Hitachi Power Systems Europe, Carbon Recycling Interantional, Power-to-Fuel Technologies, http://www.eu.mhps.com/media/files/broschueren_final/Power-to-Fuel-2015-10-19.pdf, (accessed September 2018).
- MefCO – Project Website, http://www.mefco2.eu/project_progress.php#VIDEOS, (accessed 9 January 2020).
- Mitsui Chemicals Inc., CSR Report 2009, http://www.mitsuichem.com/csr/report/index.htm, (accessed 28 June 2016).
- Y. Goto, N. Takahashi, M. Yoshinaga and M. Murakami, US Pat., 9314774, 2016 Search PubMed.
- Mitsui Chemicals Inc., CSR Report 2010, http://www.mitsuichem.com/csr/report/index.htm, (accessed 28 June 2016).
- Mitsui Chemicals Inc., CSR Report 2015, http://www.mitsuichem.com/csr/report/index.htm, (accessed 28 June 2016).
- O. F. Sigurbjörnsson, Recycling of Geothermal Carbon and Sulfur Emissions for Chemical Production, 2013 Search PubMed.
- European Commission Community Research and Development Information Service, Projects & Results Service: Synthesis of methanol from captured carbon dioxide using surplus electricity, http://cordis.europa.eu/project/rcn/193453_en.html, (accessed 5 July 2016).
- Blue Fuel Energy, Company Website, http://bluefuelenergy.com/, (accessed 28 June 2016).
- IEA and International Energy Agency, Energy and GHG Reductions in the Chemical Industry via Catalytic Processes: Annexes, http://www.iea.org/, (accessed 1 September 2016).
- R. Rivera-Tinoco, M. Farran, C. Bouallou, F. Auprêtre, S. Valentin, P. Millet and J. R. Ngameni, Investigation of power-to-methanol processes coupling electrolytic hydrogen production and catalytic CO2 reduction, Int. J. Hydrogen Energy, 2016, 41, 4546–4559 CrossRef CAS.
- (S&T)2 Consultants Inc., GHG Emissions of Blue Fuel Methanol Production Process, http://www.methanolfuels.org/wp-content/uploads/2013/05/Blue-Fuel-RM-Carbon-Intensity-S+T2-Consultants.pdf, (accessed 29 August 2016).
- S. Valentin, Energy Storage Methanol, http://www.isel-logistique.fr/wp-content/uploads/2014/09/RIH_2013_VALENTIN_Sol%C3%A8ne.pdf, (accessed 13 October 2016).
- B. Anicic, P. Trop and D. Goricanec, Comparison between two methods of methanol production from carbon dioxide, Energy, 2014, 77, 279–289 CrossRef CAS.
- A. A. Kiss, J. J. Pragt, H. J. Vos, G. Bargeman and M. T. de Groot, Novel efficient process for methanol synthesis by CO2 hydrogenation, Chem. Eng. J., 2016, 284, 260–269 CrossRef CAS.
- É. S. Van-Dal and C. Bouallou, Design and simulation of a methanol production plant from CO2 hydrogenation, J. Cleaner Prod., 2013, 57, 38–45 CrossRef.
- H. Al-Kalbani, J. Xuan, S. García and H. Wang, Comparative energetic assessment of methanol production from CO2, Appl. Energy, 2016, 165, 1–13 CrossRef CAS.
- J. B. Hansen, N. Christiansen and J. U. Nielsen, Production of Sustainable Fuels by Means of Solid Oxide Electrolysis, ECS Trans., 2011, 35, 2941–2948 CrossRef CAS.
- M. Specht, A. Bandi, F. Baumgart, C. N. Murray and J. Gretz, in Greenhouse Gas Control Technologies, ed. B. Eliasson, P. W. F. Riemer and A. Wokaun, Pergamon, Amsterdam, 1999 Search PubMed.
- K. Atsonios, K. D. Panopoulos and E. Kakaras, Investigation of technical and economic aspects for methanol production through CO2 hydrogenation, Int. J. Hydrogen Energy, 2016, 41, 2202–2214 CrossRef CAS.
- D. Mignard and C. G. Pritchard, On the use of electrolytic hydrogen from variable renewable energies for the enhanced conversion of biomass to fuels, Chem. Eng. Res. Des., 2008, 86, 473–487 CrossRef CAS.
- D. Mignard, M. Sahibzada, J. M. Duthie and H. W. Whittington, Methanol synthesis from flue-gas CO2 and renewable electricity: a feasibility study, Int. J. Hydrogen Energy, 2003, 28, 455–464 CrossRef CAS.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Methanol synthesis using captured CO2 as raw material, Appl. Energy, 2016, 161, 718–732 CrossRef.
- M. Asif, X. Gao, H. Lv, X. Xi and P. Dong, Catalytic hydrogenation of CO2 from 600 MW supercritical coal power plant to produce methanol: A techno-economic analysis, Int. J. Hydrogen Energy, 2018, 43, 2726–2741 CrossRef CAS.
- L. R. Clausen, N. Houbak and B. Elmegaard, Technoeconomic analysis of a methanol plant based on gasification of biomass and electrolysis of water, Energy, 2010, 35, 2338–2347 CrossRef CAS.
- J. Lebæk, H. O. Hansen, A. Mortensgaard, J. B. Hansen, A. S. Petersen, I. Loncarevic and C. Torbensen, GreenSynFuels Report. Final Project Report, Danish Technological Institute, 2011.
- Y. Sakamoto and W. Zhou, Energy analysis of a CO2 recycling system, Int. J. Energy Res., 2000, 24, 549–559 CrossRef CAS.
- G. Harp, K. C. Tran, O. F. Sigurbjörnsson, C. Bergins, T. Buddenberg, I. Drach and E.-I. Koytsoumpa, Proceedings of the 2nd European Steel Technology and Application Days: Duesseldorf, Germany, TEMA Technologie Marketing AG, Aachen, 2015, pp. 15–19.
- C. Bergins, K. C. Tran, E.-I. Koytsoumpa, E. Kakaras, T. Buddenberg and O. F. Sigurbjörnsson, Power to Methanol Solutions for Flexible and Sustainable Operations in Power and Process Industries, Amsterdam, 2015 Search PubMed.
- Methanex Corp., Methanex Monthly Average Regional Posted Contract Price History, https://www.methanex.com/our-business/pricing, (accessed 10 March 2020).
- A. Boulamanti and J. A. Moya, Production costs of the chemical industry in the EU and other countries: Ammonia, methanol and light olefins, Renewable Sustainable Energy Rev., 2016, 68(Part 2), 1205–1212 Search PubMed.
- L. Barbato, G. Iaquaniello and A. Mangiapane, CO2: A Valuable Source of Carbon, ed. M. D. Falco, G. Iaquaniello and G. Centi, Springer London, London, 2013, pp. 67–79 Search PubMed.
- M. Specht, F. Staiss, A. Bandi and T. Weimer, Comparison of the renewable transportation fuels, liquid hydrogen and methanol, with gasoline – energetic and economic aspects, Int. J. Hydrogen Energy, 1998, 23, 387–396 CrossRef CAS.
- J. C. Meerman, A. Ramírez, W. C. Turkenburg and A. P. C. Faaij, Performance of simulated flexible integrated gasification polygeneration facilities, Part B, Renewable Sustainable Energy Rev., 2012, 16, 6083–6102 CrossRef.
- I. Hannula and E. Kurkela, Liquid transportation fuels via large-scale fluidised-bed gasification of lignocellulosic biomass, VTT Technical Research Centre of Finland, Espoo, 2013 Search PubMed.
- C. N. Hamelinck and A. Faaij, Future prospects for production of methanol and hydrogen from biomass. System analysis of advanced conversion concepts by ASPEN-plus flowsheet modelling, Utrecht University, Copernicus Institute, Science Technology Society, Utrecht, 2001 Search PubMed.
- E. D. Larson and R. Tingjin, Synthetic fuel production by indirect coal liquefaction, Energy Sustainable Dev., 2003, 7, 79–102 CrossRef.
- Y. Zhu, S. A. Tjokro Rahardjo, C. Valkenburgm, L. J. Snowden-Swan, S. B. Jones and M. A. Machinal, Techno-economic Analysis for the Thermochemical Conversion of Biomass to Liquid Fuels, U.S. Department of Energy, 2011 Search PubMed.
- Air Products and Chemicals, Inc., Economic analysis: LPMeOH process as an add-on to integrated gasification combined cycle (IGCC) for coproduction, US Department of Energy, 1998 Search PubMed.
- M. Müller and U. Hübsch, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- X. D. Peng, B. A. Toseland, A. W. Wang and G. E. Parris, Progress in Development of LPDME Process: Kinetics and Catalysts. Coal Liquefaction & Solid Fuels Contractors Review Conference, Pittsburgh, 1997, (accessed 20 November 2016).
- X. D. Peng, B. A. Toseland and P. J. A. Tijm, Kinetic understanding of the chemical synergy under LPDME conditions—once-through applications, Chem. Eng. Sci., 1999, 54, 2787–2792 CrossRef CAS.
- J. B. Hansen, B. Voss, F. Joensen and I. D. Siguroardóttir, SAE Technical Paper, SAE International, 1995.
- T. Ogawa, N. Inoue, T. Shikada and Y. Ohno, Direct Dimethyl Ether Synthesis, J. Nat. Gas Chem., 2003, 12, 219 Search PubMed.
- Y. Baek, W. Cho, Y. B. Yan, Y. G. Mo, K. H. Lee and E. M. Jang, US Pat., 8450234, 2013 Search PubMed.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, Catalysis Chemistry of Dimethyl Ether Synthesis, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
- D. Song, W. Cho, G. Lee, D. K. Park and E. S. Yoon, Numerical Analysis of a Pilot-Scale Fixed-Bed Reactor for Dimethyl Ether (DME) Synthesis, Ind. Eng. Chem. Res., 2008, 47, 4553–4559 CrossRef CAS.
- T. Shikada, Y. Ohno, T. Ogawa, M. Mizuguchi, M. Ono and K. Fujimoto, US Pat., 6147125, 1997 Search PubMed.
- Y. Baek, W. Cho, B. H. Cho, J. C. Suh, D. H. Kim, H. Kim, J. H. Lee and W. S. Ju, US Pat., 0287405, 2006 Search PubMed.
- D. Song, W. Cho, D. K. Park and E. S. Yoon, Comparison of the performance of a fixed bed reactor in the two cases, mixture of catalyst pellets and a hybrid catalyst, for Dimethyl Ether synthesis, J. Ind. Eng. Chem., 2007, 13, 815–826 CAS.
- C. Junshi and Y. Xingan, DME Production & Standardization in China, Niigata, Japan, 2011 Search PubMed.
- H. Yagi, Y. Ohno, N. Inoue, K. Okuyama and S. Aoki, Slurry Phase Reactor Technology for DME Direct Synthesis, Int. J. Chem. React. Eng., 2010, 8, A109 Search PubMed.
- M. Stiefel, R. Ahmad, U. Arnold and M. Döring, Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas, Fuel Process. Technol., 2011, 92, 1466–1474 CrossRef CAS.
- D. Wang, Y. Han, Y. Tan and N. Tsubaki, Effect of H2O on Cu-based catalyst in one-step slurry phase dimethyl ether synthesis, Fuel Process. Technol., 2009, 90, 446–451 CrossRef CAS.
- Air Products and Chemicals, Inc., Liquid Phase Dimethyl Ether Demonstration in the Laporte Alternative Fuels Development Unit, 2001 Search PubMed.
- X. D. Peng, B. A. Toseland and R. P. Underwood, Catalyst Deactivation, Proceedings of the 7th International Symposium, Elsevier, 1997, pp. 175–182 Search PubMed.
- T. Mii and M. Uchida, Fuel DME Plant in East Asia, Dhahran, Saudi Arabia, 2005 Search PubMed.
- H. Koempel, W. Libner and M. Rothaemel, Progress report on MTP with focus on DME, 2014.
- China Energy Limited, Company Website, http://www.chinaenergy.com.sg/about.html, (accessed 10 October 2016).
- China Haohua Chemical Group Co., Ltd, Company Website, http://www.chinahaohua.com, (accessed 10 October 2016).
- ENN Energy Holdings, Company Website, http://www.enn.cn/, (accessed 14 November 2016).
- P. J. Dahl and H. O. Stahl, US Pat., 9187393, 2015 Search PubMed.
- J. Haugaard and B. Voss, US Pat., 6191175, 2001 Search PubMed.
- A. Ishiwada, DME Promotion Project in Japan. As A Future Alternative Clean Energy, 2011 Search PubMed.
- M. K. Strätling, New Methanol Technologies Offer Alternatives to Dirty Fuels, World Energy, 2002, 5, 66–71 Search PubMed.
- B. Ahlers, M. Gil de Tober and E. Seidel, US Pat., 0038745, 2015 Search PubMed.
- P. Mitschke, E. Seidel, T. Renner and M. Rothaemel, US Pat., 0220804, 2012 Search PubMed.
- T. Mii, DME Projects in East Asia, Tehran, Iran, 2004.
- T. Mii and M. Uchida, DME Synthesis Technology Ready for Market, Bilbao, Spain, 2005.
- Toyo Engineering Corporation, Company Website, Dimethyl Ether, http://www.toyo-eng.com/jp/en/products/energy/dme/, (accessed 13 October 2016).
- S. Kazuo and T. Satoshi, US Pat., 0034255, 2004 Search PubMed.
- J. Perregaard, DME Production Technology Overview, Stockholm, Sweden, 2010 Search PubMed.
- H. Topsoe, DME-99 ECO - Topsoe synthesis catalyst, http://www.topsoe.com/products/dme-99-ecotm, (accessed 14 November 2016).
- C. Junshi, S. Lijie, L. Xuefei and Z. Jianxiang, WO Pat., 130214, 2010 Search PubMed.
- L. Jinlai and Y. Xingan, DME in China and ENN DME Technology, Stockholm, Sweden, 2010 Search PubMed.
- J. Topp-Jorgensen, US Pat., 4536485, 1985 Search PubMed.
- S. H. Lee, W. Cho, T. Song and Y. J. Ra, Scale up study of direct DME synthesistechnology, Proceedings of 24th World Gas Conference, 2009.
- I. H. Kim, S. Kim, W. Cho and E. S. Yoon, in 20th European Symposium on Computer Aided Process Engineering – ESCAPE20, ed. S. Pierucci and G. B. Ferraris, Elsevier, Amsterdam, 1st edn, 2010, pp. 799–804 Search PubMed.
- W. Cho, Introduction of KOGAS'sActivities on DME, Stockholm, Sweden, 2010 Search PubMed.
- Y. Ohno, T. Shikada, T. Ogawa, M. Ono and M. Mizuguchi, in Preprints of Papers Presented at the 213th ACS National Meeting, ed. G. P. Huffman, ACS, Washington, DC, 1997, pp. 705–709 Search PubMed.
- Y. Ohno, New Clean Fuel DME, Ho Chi Minh City, Vietnam, 2008 Search PubMed.
- Air Products and Chemicals, Inc., Development of Alternative Fuels from Coal Derived Syngas. Topical Report, US Department of Energy, 1993.
- X. D. Peng, G. E. Parris, B. A. Toseland and P. J. Battavio, US Pat., 5753716, 1998 Search PubMed.
- BASF, Linde and Lutianhua, BASF and Lutianhua plan to pilot a new production process that significantly reduces CO2 emissions, https://www.basf.com/at/de/media/news-releases/2019/06/p-19-249.html, (accessed November 2019).
- E. Schwab, N. Schödel, A. Peschel, J. Fendt, H. Klein, F. Schüth, W. Schmidt, D. Wendt, M. Marzi, T. Schulzke, H. Kaiser, T. Mäurer, S. Schunk, S. Altwasser and S. Schuster, Integrierte Dimethylethersynthese aus Methan und CO2 (DMEEXCO2), Königswinter, Germany, 2014 Search PubMed.
- E. Schwab, S. Schunk, S. Schuster, A. Peschel, H. Schmaderer, N. Schödel, J. Fendt and H. Klein, Direct Synthesis of Dimethlyether from Syngas: Catalysts as Enablers of Energy-efficient Process Design, Dresden, Germany, 2015 Search PubMed.
- N. Schödel, EP Pat., 2809639, 2013 Search PubMed.
- J. J. Lewnard, T. H. Hsiung, J. F. White and B. L. Bhatt, US Pat., 5218003, 1993 Search PubMed.
- T. Shikada, Y. Ohno, T. Ogawa, M. Mizuguchi, M. Ono and K. Fujimoto, US Pat., 0052647, 2005 Search PubMed.
- Y. Ohno, H. Yagi, N. Inoue, K. Okuyama and S. Aoki, Slurry Phase DME Direct Synthesis Technology-100 tons/day Demonstration Plant Operation and Scale up Study, Natural Gas Conversion VIII, 2007, 403–408.
- T. Ogawa, N. Inoue, T. Shikada, O. Inokoshi and Y. Ohno, Direct Dimethyl Ether (DME) synthesis from natural gas, Stud. Surf. Sci. Catal., 2004, 147, 379–384 CrossRef CAS.
- Y. Ohno, M. Yoshida, T. Shikada, O. Inokoshi, T. Ogawa and N. Inoue, New Direct Synthesis Technology for DME (Dimethyl Ether) and Its Application Technology, JFE Technical Report, 2006.
- Y. Ohno, N. Takahashi, T. Shikada and Y. Ando, An Ultra Celan Fuel: Dimethyl Ether (DME), Proceedings of the 18th World Energy Council, Buenos Aires, 2001.
- W. Cho, KOGAS DME Activities for Commercialization, Niigata, Japan, 2011 Search PubMed.
- B. Voss, F. Joensen and J. B. Hansen, WO Pat., 96/23755, 1996 Search PubMed.
- P. J. Dahl and J. Ostergaard, US Pat., 0239813, 2015 Search PubMed.
- B. Ahlers, G. Birke, H. Kömpl, H. Bach, M. Rothaemel, W. Liebner, W. Boll and V. Gronemann, US Pat., 0295043, 2011 Search PubMed.
- X. D. Peng, B. W. Diamond, T. C. R. Tsao and B. L. Bhatt, US Pat., 6458856, 2002 Search PubMed.
- J. Madsen, US Pat., 7652176, 2010 Search PubMed.
- X. An, Y.-Z. Zuo, Q. Zhang, D.-Z. Wang and J.-F. Wang, Dimethyl Ether Synthesis from CO2 Hydrogenation on a CuO–ZnO–Al2O3–ZrO2/HZSM-5 Bifunctional Catalyst, Ind. Eng. Chem. Res., 2008, 47, 6547–6554 CrossRef CAS.
- G. Bonura, M. Cordaro, C. Cannilla, A. Mezzapica, L. Spadaro, F. Arena and F. Frusteri, Catalytic behaviour of a bifunctional system for the one step synthesis of DME by CO2 hydrogenation, Catal. Today, 2014, 228, 51–57 CrossRef CAS.
- G. Bonura, M. Cordaro, L. Spadaro, C. Cannilla, F. Arena and F. Frusteri, Hybrid Cu–ZnO–ZrO2/H-ZSM5 system for the direct synthesis of DME by CO2 hydrogenation, Appl. Catal., B, 2013, 140-141, 16–24 CrossRef CAS.
- J. Ereña, R. Garoña, J. M. Arandes, A. T. Aguayo and J. Bilbao, Effect of operating conditions on the synthesis of dimethyl ether over a CuO-ZnO-Al2O3/NaHZSM-5 bifunctional catalyst, Catal. Today, 2005, 107-108, 467–473 CrossRef.
- F. Frusteri, G. Bonura, C. Cannilla, G. Drago Ferrante, A. Aloise, E. Catizzone, M. Migliori and G. Giordano, Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation, Appl. Catal., B, 2015, 176–177, 522–531 CrossRef CAS.
- F. Frusteri, M. Cordaro, C. Cannilla and G. Bonura, Multifunctionality of Cu–ZnO–ZrO2/H-ZSM5 catalysts for the one-step CO2-to-DME hydrogenation reaction, Appl. Catal., B, 2015, 162, 57–65 CrossRef CAS.
- W. Gao, H. Wang, Y. Wang, W. Guo and M. Jia, Dimethyl ether synthesis from CO2 hydrogenation on La-modified CuO-ZnO-Al2O3/HZSM-5 bifunctional catalysts, J. Rare Earths, 2013, 31, 470–476 CrossRef CAS.
- S. P. Naik, T. Ryu, V. Bui, J. D. Miller, N. B. Drinnan and W. Zmierczak, Synthesis of DME from CO2/H2 gas mixture, Chem. Eng. J., 2011, 167, 362–368 CrossRef CAS.
- K. Sun, W. Lu, M. Wang and X. Xu, Low-temperature synthesis of DME from CO2/H2 over Pd-modified CuO–ZnO–Al2O3–ZrO2/HZSM-5 catalysts, Catal. Commun., 2004, 5, 367–370 CrossRef CAS.
- S. Wang, D. Mao, X. Guo, G. Wu and G. Lu, Dimethyl ether synthesis via CO2 hydrogenation over CuO–TiO2–ZrO2/HZSM-5 bifunctional catalysts, Catal. Commun., 2009, 10, 1367–1370 CrossRef CAS.
- T. Witoon, T. Permsirivanich, N. Kanjanasoontorn, C. Akkaraphataworn, A. Seubsai, K. Faungnawakij, C. Warakulwit, M. Chareonpanich and J. Limtrakul, Direct synthesis of dimethyl ether from CO 2 hydrogenation over Cu–ZnO–ZrO2/SO42−–ZrO 2 hybrid catalysts, Catal. Sci. Technol., 2015, 5, 2347–2357 RSC.
- Y. Zhang, D. Li, S. Zhang, K. Wang and J. Wu, CO2 hydrogenation to dimethyl ether over CuO–ZnO–Al2O3/HZSM-5 prepared by combustion route, RSC Adv., 2014, 4, 16391 RSC.
- R.-W. Liu, Z.-Z. Qin, H.-B. Ji and T.-M. Su, Synthesis of Dimethyl Ether from CO 2 and H 2 Using a Cu–Fe–Zr/HZSM-5 Catalyst System, Ind. Eng. Chem. Res., 2013, 52, 16648–16655 CrossRef CAS.
- G.-X. Qi, J.-H. Fei, X.-M. Zheng and Z.-Y. Hou, DME synthesis from carbon dioxide and hydrogen over Cu-Mo/HZSM-5, Catal. Lett., 2001, 72(1–2), 121–124 CrossRef CAS.
- K. W. Jun and K. W. Lee, US Pat., 6248795, 2001 Search PubMed.
- K. Takeishi and Y. Wagatsuma, in Recent advances in energy and environmental management, ed. V. Mladenov, T. Tashev, H. Wang, I. Kralov, S. Stankevich, P. Yildiz and J. Burley, WSEAS Press, Rhodes Island, Greece, 2013, pp. 134–136 Search PubMed.
- M. Hirano, T. Imai, T. Yasutake and K. Kuroda, Dimethyl Ether Synthesis from Carbon Dioxide by Catalytix Hydrogenation (PArt 2) Hybrid Catalyst Consisting of Methanol Synthesis and Methanol Dehydration Catalysts, J. Jpn. Pet. Inst., 2004, 47, 11–18 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- A. Ateka, I. Sierra, J. Ereña, J. Bilbao and A. T. Aguayo, Performance of CuO–ZnO–ZrO 2 and CuO–ZnO–MnO as metallic functions and SAPO-18 as acid function of the catalyst for the synthesis of DME co-feeding CO2, Fuel Process. Technol., 2016, 152, 34–45 CrossRef CAS.
- E. Catizzone, G. Bonura, M. Migliori, F. Frusteri and G. Giordano, CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives, Molecules, 2018, 23(1), 31 CrossRef PubMed.
- Ministry of Industry, Orkustofnun, The National Energy Authority, The Innovation Center Iceland, Mitsubishi Heavy Industries, Mitsubishi Corporation, Hekla, NordicBlueEnergys, A Feasibility Study Report For A DME Project in Iceland (Summary), 2010, http://www.nea.is/media/eldsneyti/A-DME-feasibility-study-in-Iceland-summary-report.pdf, (accessed 2 December 2016).
- P. Moser, G. Wiechers, S. Schmidt, K. Stahl, M. Majid, S. Bosser, A. Heberle, H. Kakihira, M. Maruyama, R. Peters, S. Weiske, P. Zapp, S. Troy, B. Lehrheuer, M. Neumann, S. Schaub, J. Vente, J.-P. Pieterse, J. Boon and E. Goetheer, Demonstrating the CCU-chain and sector coupling as part of ALIGN-CCUS. Dimethyl ether from CO2 as chemical energy storage, fuel and feedstock for industries, Melbourne, 2018 Search PubMed.
- ALIGN-CCUS, Making fuels from CO2, http://www.alignccus.eu/news/making-fuels-co2-rwe-unveils-new-synthesis-pilot-plant-germany, (accessed April 2020).
- ALIGN-CCUS, Work Package 4 Description - CO2 Re-Use, https://www.alignccus.eu/about-project/work-package-4-co2-re-use-0, (accessed April 2020).
- M. Matzen and Y. Demirel, Methanol and dimethyl ether from renewable hydrogen and carbon dioxide, J. Cleaner Prod., 2016, 139, 1068–1077 CrossRef CAS.
- Y. Ohno, DME as a carrier of Renewable Energy, 2013 Search PubMed.
- X. Sun, M. Chen, S. H. Jensen, S. D. Ebbesen, C. Graves and M. Mogensen, Thermodynamic analysis of synthetic hydrocarbon fuel production in pressurized solid oxide electrolysis cells, Int. J. Hydrogen Energy, 2012, 37, 17101–17110 CrossRef CAS.
- X. D. Peng, A. W. Wang, B. A. Toseland and P. J. A. Tijm, Single-Step Syngas-to-Dimethyl Ether Processes for Optimal Productivity, Minimal Emissions, and Natural Gas-Derived Syngas, Ind. Eng. Chem. Res., 1999, 38, 4381–4388 CrossRef CAS.
- M. I. González, H. Eilers and G. Schaub, Flexible Operation of Fixed-Bed Reactors for a Catalytic Fuel Synthesis—CO2 Hydrogenation as Example Reaction, Energy Technol., 2016, 4, 90–103 CrossRef.
- A. Hadipour and M. Sohrabi, Kinetic Parameters and Dynamic Modeling of a Reactor for Direct Conversion of Synthesis Gas to Dimethyl Ether, J. Ind. Eng. Chem., 2007, 13, 558–565 CAS.
- S. Brynolf, M. Taljegard, M. Grahn and J. Hansson, Electrofuels for the transport sector: A review of production costs, Renewable Sustainable Energy Rev., 2018, 81, 1887–1905 CrossRef.
- L. R. Clausen, B. Elmegaard and N. Houbak, Technoeconomic analysis of a low CO2 emission dimethyl ether (DME) plant based on gasification of torrefied biomass, Energy, 2010, 35, 4831–4842 CrossRef CAS.
- G. H. Huisman, G. L. M. A. van Rens, H. de Lathouder and R. L. Cornelissen, Cost estimation of biomass-to-fuel plants producing methanol, dimethylether or hydrogen, Biomass Bioenergy, 2011, 35, S155–S166 CrossRef.
- L. Tock, M. Gassner and F. Maréchal, Thermochemical production of liquid fuels from biomass: Thermo-economic modeling, process design and process integration analysis, Biomass Bioenergy, 2010, 34, 1838–1854 CrossRef CAS.
- D. Cocco, A. Pettinau and G. Cau, Energy and economic assessment of IGCC power plants integrated with DME synthesis processes, Proc. Inst. Mech. Eng., Part A, 2006, 220, 95–102 CrossRef CAS.
- A. Farooqui, F. Di Tomaso, A. Bose, D. Ferrero, J. Llorca and M. Santarelli, Techno-economic and exergy analysis of polygeneration plant for power and DME production with the integration of chemical looping CO2/H2O splitting, Energy Convers. Manage., 2019, 186, 200–219 CrossRef CAS.
- C. Mevawala, Y. Jiang and D. Bhattacharyya, Techno-economic optimization of shale gas to dimethyl ether production processes via direct and indirect synthesis routes, Appl. Energy, 2019, 238, 119–134 CrossRef CAS.
- S. Michailos, S. McCord, V. Sick, G. Stokes and P. Styring, Dimethyl ether synthesis via captured CO2 hydrogenation within the power to liquids concept: A techno-economic assessment, Energy Convers. Manage., 2019, 184, 262–276 CrossRef CAS.
- S. Schemme, J. L. Breuer, M. Köller, S. Meschede, F. Walman, R. C. Samsun, R. Peters and D. Stolten, H2-based synthetic fuels: A techno-economic comparison of alcohol, ether and hydrocarbon production, Int. J. Hydrogen Energy, 2020, 45, 5395–5414 CrossRef CAS.
- Market Price Dimethyl-Ether, https://www.ceicdata.com/en/china/china-petroleum--chemical-industry-association-petrochemical-price-organic-chemical-material/cn-market-price-monthly-avg-organic-chemical-material-dimethyl-ether-990-or-above, (accessed 18 June 2020).
- China DME Spot Price, http://www.sunsirs.com/, (accessed 18 June 2020).
- P. Sabatier and J. B. Senderens, Hydrogenation of CO Over Nickel to Produce Methane, J. Soc. Chem. Ind., 1902, 21, 504–506 Search PubMed.
- A. A. Adesina, R. R. Hudgins and P. L. Silveston, Fischer-Tropsch synthesis under periodic operation, Catal. Today, 1995, 25, 127–144 CrossRef CAS.
- F. Fischer and H. Tropsch, Über die Reduktion des Kohlenoxyds zu Methan am Eisenkontakt unter Druck, Brennst.-Chem., 1923, 4, 193–197 CAS.
- Studies in Surface Science and Catalysis: Heterogeneous Catalysis of Mixed OxidesPerovskite and Heteropoly Catalysts, ed. M. Misono, Elsevier, 2013 Search PubMed.
- P. J. Flory, Molecular size distribution in linear condensation polymers, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS.
- G. V. Schulz, Über die Beziehung zwischen Reaktionsgeschwindigkeit und Zusammensetzung des Reaktionsproduktes bei Makropolymerisationsvorgängen, Z. Phys. Chem. B, 1935, 30, 379–398 Search PubMed.
- R. A. Friedel and R. B. Anderson, Composition of synthetic liquid fuels. I. Product distribution and analysis of C5-C8 paraffin isomers from cobalt catalyst, J. Am. Chem. Soc., 1950, 72, 2307 CrossRef CAS.
- L. Caldwell, Selectivity in the Fischer-Tropsch Synthesis using heterogeneous catalysts, ChemSA, 1981, 7, 88–92 CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Advances in the Development of Novel Cobalt Fischer—Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels, Chem. Rev., 2007, 107(5), 1692–1744 CrossRef CAS PubMed.
- E. Ryttera, N. E. Tsakoumisa and A. Holmena, On the selectivity to higher hydrocarbons in Co-based Fischer–Tropsch synthesis, Catal. Today, 2016, 261, 3–16 CrossRef.
- H. Schulz, M. Claeys and S. Harms, Effect of water partial pressure on steady state Fischer-Tropsch activity and selectivity of a promoted cobalt catalyst, Stud. Surf. Sci. Catal., 1997, 107, 193–200 CrossRef CAS.
- H. Schulz, E. vein Steen and M. Claeys, Selectivity and mechanism of Fischer-Tropsch synthesis with iron and cobalt catalysts, Stud. Surf. Sci. Catal., 1994, 81, 455–460 CrossRef CAS.
- H. Schulz, Short history and present trends of Fischer–Tropsch synthesis, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS.
- F. Jiang, M. Zhang, B. Liu, Y. Xu and X. Liu, Insights into the influence of support and potassium or sulfur promoter on iron-based Fischer–Tropsch synthesis: understanding the control of catalytic activity, selectivity to lower olefins, and catalyst deactivation, Catal. Sci. Technol., 2017, 7, 1245–1265 RSC.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S.-S. Nam, K.-W. Jun, M.-J. Choi, G. Kishan and K.-W. Lee, Comparative study of Fischer-Tropsch synthesis with H2/CO and H2/CO2 syngas using Fe- and Co-based catalysts, Appl. Catal., A, 1999, 186, 201–213 CrossRef CAS.
- S. S. Ail and S. Dasappa, Biomass to liquid transportation fuel via Fischer Tropsch synthesis – Technology review and current scenario, Renewable Sustainable Energy Rev., 2016, 58, 267–286 CrossRef CAS.
- O. O. James, A. M. Mesubi, T. C. Ako and S. Maity, Increasing carbon utilization in Fischer–Tropsch synthesis using H2-deficient or CO2-rich syngas feeds, Fuel Process. Technol., 2010, 91, 136–144 CrossRef CAS.
- B. Eisenberg, R. A. Fiato, H. Mauldin, G. R. Say and S. L. Soled, Exxon's Advanced Gas-To-Liquids Technology, Stud. Surf. Sci. Catal., 1998, 119, 943–948 CrossRef CAS.
- W. Al-Shalchi, Gas to Liquid Technology, Bagdad, 2006 Search PubMed.
- S. Storsater, B. Totdal, J. Walmsley, B. Tanem and A. Holmen, Characterization of alumina-, silica-, and titania-supported cobalt Fischer–Tropsch catalysts, J. Catal., 2005, 236, 139–152 CrossRef.
- N. E. Tsakoumis, M. Rønning, Ø. Borg, E. Rytter and A. Holmen, Deactivation of cobalt based Fischer–Tropsch catalysts, Catal. Today, 2010, 154, 162–182 CrossRef CAS.
- D. B. Bukura, B. Todica and N. Elbashira, Role of water-gas-shift reaction in Fischer–Tropsch synthesis on iron catalysts: A review, Catal. Today, 2016, 275, 66–75 CrossRef.
- E. D. Smit and B. M. Weckhuysen, The renaissance of iron-based Fischer–Tropsch synthesis: on the multifaceted catalyst deactivation behaviour, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- Handbook of alternative fuel technologies, ed. S. Lee, J. G. Speight and S. K. Loyalka, CRC Press, Boca Raton, 2nd edn, 2014 Search PubMed.
- A. de Klerk, Gas-To-Liquids Conversion, Houston, Texas, 2012 Search PubMed.
- B. Jager and R. Espinoza, Advances in low temperature Fischer-Tropsch synthesis, Catal. Today, 1995, 23, 17–28 CrossRef CAS.
- A. P. Steynberg, R. L. Espinoza, B. Jager and A. C. Vosloo, High temperature Fischer–Tropsch synthesis in commercial practice, Appl. Catal., A, 1999, 186, 41–54 CrossRef CAS.
- Will Beacham, Sasol's switch to specialties, https://www.icis.com/explore/resources/news/2019/02/28/10325360/sasol-s-switch-to-specialties, (accessed 2 March 2020).
- U.S. DOE Gasification Plant Databases.
- P. L. Spath and D. C. Dayton, Preliminary Screening - Technical and Economic Assessment of Synthesis Gas to Fuels and Chemicals with Emphasis on the Potential for Biomass-Derived Syngas, National Renewable Energy Laboratory, 2003 Search PubMed.
- D. Vallentin, Coal-to-Liquids (CtL) – Driving forces and barriers - synergies and conflicts from an energy and climate policy perspective including country studies on the United States, China and Germany, ibidem-Verlag, Stuttgart, 2009 Search PubMed.
- Sasol, Sasolburg Operations, https://www.sasol.com/about-sasol/regional-operating-hubs/southern-africa-operations/sasolburg-operations/operations, (accessed April 2020).
- Sasol, Sasol Facts 12/13, http://www.sasolnorthamerica.com/Images/Interior/news/3-12695%20sasol_facts.pdf, (accessed 25 February 2020).
- M. E. Dry, Practical and theoretical aspects of the catalytic Fischer-Tropsch process, Appl. Catal., A, 1996, 138, 319–344 CrossRef CAS.
- M. E. Dry, The Fischer-Tropsch process – commercial aspects, Catal. Today, 1990, 6, 183–206 CrossRef CAS.
- R. L. Espinoza, A. P. Steynberg, B. Jager and A. C. Vosloo, Low temperature Fischer–Tropsch synthesis from a Sasol perspective, Appl. Catal., A, 1999, 186, 13–26 CrossRef CAS.
- D. J. Moodley, PhD thesis, Technische Universiteit Eindhoven, 2008.
- Shell GTL Fuel Geschichte und Herstellung, https://www.shell.de/geschaefts-und-privatkunden/shell-kraftstoffe-fuer-geschaeftskunden/shell-gas-to-liquids-fuel/shell-gtl-fuel-history-and-process.html, (accessed 26 March 2020).
- Shell Qatar, Pearl Gas-to-Liquids (GTL), https://www.shell.com.qa/en_qa/about-us/projects-and-sites/pearl-gtl.html, (accessed March 2020).
- B. S. Rahardjo, The Assessment of Syngas Utilization by Fischer Tropsch Synthesis in the Slurry Reactor using Co/SiO2 Catalyst, Int. J. Eng. Appl. Sci., 2013, 4, 20–39 Search PubMed.
- J. Furness, Sasol GTL reactors shipped to Qatar, http://www.engineeringnews.co.za/article/sasol-gtl-reactors-shipped-to-qatar-2005-03-30/rep_id:4136, (accessed 19 February 2020).
- P. J. A. Tijm, J. M. Marriott, H. Hasenack, M. M. G. Senden and T. van Herwijnen, The Markets for Shell Middle Distillate Synthesis Products, Vancouver, Canada, 1995.
- C. Zhang, K.-W. Jun, K.-S. Ha, Y.-J. Lee and S. C. Kang, Efficient utilization of greenhouse gases in a gas-to-liquids process combined with CO2/steam-mixed reforming and Fe-based Fischer-Tropsch synthesis, Environ. Sci. Technol., 2014, 48, 8251–8257 CrossRef CAS PubMed.
- B. Bao, M. M. El-Halwagi and N. O. Elbashir, Simulation, integration, and economic analysis of gas-to-liquid processes, Fuel Process. Technol., 2010, 91, 703–713 CrossRef CAS.
- C. Knottenbelt, Mossgas “gas-to-liquid” diesel fuels—an environmentally friendly option, Catal. Today, 2002, 71, 437–445 CrossRef CAS.
- K. S. Ha, J. W. Bae, K. J. Woo and K. W. Jun, Efficient utilization of greenhouse gas in a gas-to-liquids process combined with carbon dioxide reforming of methane, Environ. Sci. Technol., 2010, 44, 1412–1417 CrossRef CAS PubMed.
- A. de Klerk, 2nd Sub-Saharan Africa Catalyst Symposium, 2001.
- EBTP-SABS, Biomass to Liquids (BtL), 2016, http://www.biofuelstp.eu/btl.html.
- Fulcrum BioEnergy, Company Website – Sierra Biofuels Plant, 2017, http://fulcrum-bioenergy.com/facilities/, (accessed 12 April 2017).
- T. Jungbluth, BioTfuel. Gas geben mit Abfall im Tank, 2015, https://engineered.thyssenkrupp.com/biotfuel-gas-geben-mit-abfall-im-tank/, (accessed 12 April 2017).
- R. Rauch, A. Kiennemann and A. Sauciuc, in The Role of Catalysis for the Sustainable Production of Bio-fuels and Bio-chemicals, ed. K. S. Triantafyllidis, A. A. Lappas and M. Stöcker, Elsevier Science, Amsterdam, 2013, pp. 397–443 Search PubMed.
- T. Riedel, G. Schaub, K.-W. Jun and K.-W. Lee, Kinetics of CO 2 Hydrogenation on a K-Promoted Fe Catalyst, Ind. Eng. Chem. Res., 2001, 40, 1355–1363 CrossRef CAS.
- M. Rohde, D. Unruh, P. Pias, K.-W. Lee and G. Schaub, Fischer-Tropsch synthesis with CO2-containing syngas from biomass – Kinetic analysis of fixed bed reactor model experiments, Stud. Surf. Sci. Catal., 2004, 153, 97–102 CrossRef CAS.
- Carbon dioxide utilization for global sustainability, ed. S.-E. Park, J.-S. Chang and K.-W. Lee, Elsevier Science, Amsterdam, 2004, vol. 153 Search PubMed.
- J.-S. Hong, J. S. Hwang, K.-W. Jun, J. C. Sur and K.-W. Lee, Deactivation study on a coprecipitated Fe-Cu-K-Al catalyst in CO2 hydrogenation, Appl. Catal., A, 2001, 218, 53–59 CrossRef CAS.
- S.-R. Yan, K.-W. Jun, J.-S. Hong, M.-J. Choi and K.-W. Lee, Promotion effect of Fe–Cu catalyst for the hydrogenation of CO2 and application to slurry reactor, Appl. Catal., A, 2000, 194-195, 63–70 CrossRef CAS.
- M.-D. Lee, J.-F. Lee and C.-S. Chang, Hydrogenation of Carbon Dioxide on Unpromoted and Potassium-Promoted Iron Catalysts, Bull. Chem. Soc. Jpn., 1989, 62, 2756–2758 CrossRef CAS.
- M. K. Gnanamani, W. D. Shafer, D. E. Sparks and B. H. Davis, Fischer–Tropsch synthesis, Catal. Commun., 2011, 12, 936–939 CrossRef CAS.
- Y. Yao, X. Liu, D. Hildebrandt and D. Glasser, Fischer–Tropsch Synthesis Using H 2/CO/CO 2 Syngas Mixtures over an Iron Catalyst, Ind. Eng. Chem. Res., 2011, 50, 11002–11012 CrossRef CAS.
- J.-S. Kim, S. Lee, S.-B. Lee, M.-J. Choi and K.-W. Lee, Performance of catalytic reactors for the hydrogenation of CO2 to hydrocarbons, Catal. Today, 2006, 115, 228–234 CrossRef CAS.
- Y. H. Choi, Y. J. Jang, H. Park, W. Y. Kim, Y. H. Lee, S. H. Choi and J. S. Lee, Carbon dioxide Fischer-Tropsch synthesis: A new path to carbon-neutral fuels, Appl. Catal., B, 2017, 202, 605–610 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- Technologies for Sustainability and Climate Protection. Chemical Processes and Use of CO2, ed. A. Bazzanella and D. Krämer, Seltersdruck & Verlag Lehn GmbH + Co. KG, Selters, 2019 Search PubMed.
- C. V. Olshausen and K. Hauptmeier, in Zukünftige Kraftstoffe, ed. W. Maus, Springer Berlin, Berlin, 1st edn, 2019, pp. 410–432 Search PubMed.
- Sunfire GmbH, Blue Crude, https://www.sunfire.de/de/unternehmen/news/detail/blue-crude-sunfire-produziert-nachhaltigen-erdoelersatz, (accessed April 2020).
- F. V. Vázquez, J. Koponen, V. Ruuskanen, C. Bajamundi, A. Kosonen, P. Simell, J. Ahola, C. Frilund, J. Elfving, M. Reinikainen, N. Heikkinen, J. Kauppinen and P. Piermartini, Power-to-X technology using renewable electricity and carbon dioxide from ambient air: SOLETAIR proof-of-concept and improved process concept, J. CO2 Util., 2018, 28, 235–246 CrossRef.
- Kopernikus Projekt website, Kopernikus-Projekt: P2X, https://www.kopernikus-projekte.de/projekte/p2x, (accessed 10 March 2020).
- Carbon-neutral Fuels from Air and Green Power, https://www.kit.edu/kit/english/pi_2019_107_carbon-neutral-fuels-from-air-and-green-power.php, (accessed 10 March 2020).
- Sunfire GmbH, Breakthrough for Power-to-X: Sunfire puts first co-electolysis into operation and starts scaling, https://www.sunfire.de/en/company/news/detail/breakthrough-for-power-to-x-sunfire-puts-first-co-electrolysis-into-operation-and-starts-scaling, (accessed April 2020).
- Nordic Blue Crude – Transforming energy, saving the Earth, https://nordicbluecrude.no/, (accessed 10 March 2020).
- Sunfire GmbH, Sunfire-Synlink Factsheet, https://www.sunfire.de/files/sunfire/images/content/Produkte_Technologie/factsheets/Sunfire-SynLink_FactSheet.pdf, (accessed April 2020).
- Soletair, Finnish demo plant Soletair produces renewable fuels form sun and air, https://soletair.fi/news/finnish-demo-plant-produces-renewable-fuels/, (accessed 26 February 2020).
- Soletair, Technical Specifications, https://soletair.fi/technical-specifications/renewable-energy/, (accessed 26 February 2020).
- Sunfire GmbH, Rotterdam The Hague Airport initiates study for the production of renewable jet fuel from air, https://www.sunfire.de/en/company/news/detail/rotterdam-the-hague-airport-initiates-study-for-the-production-of-renewable-jet-fuel-from-air, (accessed 2 March 2020).
- J. L. Feimer, P. L. Silveston and R. R. Hudgins, Influence of forced cycling on the Fischer-Tropsch synthesis. Part I. Response to feed concentration step-changes, Can. J. Chem. Eng., 1984, 62, 241–248 CrossRef CAS.
- H. Eilers, PhD thesis, Karlsruher Institut für Technologie, 2018.
- H. Eilers, M. I. González and G. Schaub, Lab-scale experimental studies of Fischer–Tropsch kinetics in a three-phase slurry reactor under transient reaction conditions, Catal. Today, 2016, 275, 164–171 CrossRef CAS.
- H. Gruber, P. Groß, R. Rauch, A. Reichhold, R. Zweiler, C. Aichernig, S. Müller, N. Ataimisch and H. Hofbauer, Fischer-Tropsch products from biomass-derived syngas and renewable hydrogen, Biomass Convers. Biorefin., 2019, 48, 22 Search PubMed.
- F. Habermeyer, Flexibility in renewable fuel production from biomass – the role of electrolysis boosted Fischer–Tropsch synthesis, Lisbon, 2019 Search PubMed.
- M. Rafati, L. Wang, D. C. Dayton, K. Schimmel, V. Kabadi and A. Shahbazi, Techno-economic analysis of production of Fischer-Tropsch liquids via biomass gasification: The effects of Fischer-Tropsch catalysts and natural gas co-feeding, Energy Convers. Manage., 2017, 133, 153–166 CrossRef CAS.
- A. S. Snehesh, H. S. Mukunda, S. Mahapatra and S. Dasappa, Fischer-Tropsch route for the conversion of biomass to liquid fuels - Technical and economic analysis, Energy, 2017, 130, 182–191 CrossRef CAS.
- G. Haarlemmer, G. Boissonnet, E. Peduzzi and P.-A. Setier, Investment and production costs of synthetic fuels – A literature survey, Energy, 2014, 66, 667–676 CrossRef.
- G. N. Choi, S. J. Kramer, S. T. Tam and J. M. Fox, Design/economics of a natural gas based Fischer–Tropsch plant, Houston, 1996 Search PubMed.
- J. H. Gregor, Fischer-Tropsch products as liquid fuels or chemicals, Catal. Lett., 1991, 7, 317–331 CrossRef.
- G. N. Choi, S. J. Kramer, S. S. Tam, J. M. Fox, N. L. Carr and G. R. Wilson, Coal Liquefaction & Solid Fuels Contractors Review Conference, 1997.
- F. G. Albrecht, D. H. König, N. Baucks and R.-U. Dietrich, A standardized methodology for the techno-economic evaluation of alternative fuels – A case study, Fuel, 2017, 194, 511–526 CrossRef CAS.
- IEA, Biofuel and fossil-based transport fuel production cost comparison, https://www.iea.org/data-and-statistics/charts/biofuel-and-fossil-based-transport-fuel-production-cost-comparison-2017, (accessed 9 March 2020).
- Jährliche Verbraucherpreise für Mineralölprodukte, https://www.mwv.de/statistiken/verbraucherpreise/, (accessed 2 March 2020).
- The World Bank, Pink Sheet. annual commodity prices, 2020, https://www.worldbank.org/en/research/commodity-markets, (accessed 2 March 2020).
Footnote |
| † Within the context of this article synthesis gas, which mainly includes CO and H2 will be referred to as ‘syngas’. |
| This journal is © The Royal Society of Chemistry 2020 |