
Exceptional performance of hierarchical Ni–Fe oxyhydroxide@NiFe alloy nanowire array electrocatalysts for large current density water splitting†
Caiwu
Liang
a,
Peichao
Zou
a,
Adeela
Nairan
a,
Yongqi
Zhang
b,
Jiaxing
Liu
a,
Kangwei
Liu
a,
Shengyu
Hu
a,
Feiyu
Kang
a,
Hong Jin
Fan
 *b and
Cheng
Yang
*a
*b and
Cheng
Yang
*a
aDivision of Energy and Environment, Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China. E-mail: yang.cheng@sz.tsinghua.edu.cn
bSchool of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: fanhj@ntu.edu.sg
First published on 8th October 2019
Abstract
Water electrolysis represents a promising sustainable hydrogen production technology. However, in practical application which requires extremely large current densities (>500 mA cm−2), the oxygen evolution reaction (OER) becomes unstable and kinetically sluggish, which is a major hurdle to large-scale hydrogen production. Herein, we report an exceptionally active and binder-free NiFe nanowire array based OER electrode that allows durable water splitting at current densities up to 1000 mA cm−2 up to 120 hours. Specifically, NiFe oxyhydroxide (shell)–anchored NiFe alloy nanowire (core) arrays are prepared via a magnetic-field-assisted chemical deposition method. The ultrathin (1–5 nm) and amorphous NiFe oxyhydroxide is in situ formed on the NiFe alloy nanowire surface, which is identified as an intrinsically highly active phase for the OER. Additionally, the fine geometry of the hierarchical electrode can substantially improve charge and mass (reactants and oxygen bubbles) transfer. In an alkaline electrolyte, this OER electrode can yield current densities of 500 and 1000 mA cm−2 stably over 120 hours at overpotentials of only 248 mV and 258 mV respectively, which are dramatically lower than any recently reported overpotentials. Notably, the integrated alkaline electrolyzer (with pure Ni nanowires as HER electrode) is demonstrated to reach the current density of 1000 mA cm−2 with super low voltage of 1.76 V, outperforming the state-of-the-art industrial catalysts. Our result may represent a critical step towards an industrial electrolyzer for large-scale hydrogen production by water splitting.
Broader contextHydrogen is the energy carrier with the most potential in the future due its cleanness and high energy density, which could play an essential role in solving the global energy crisis and greenhouse issues. Compared to the present major hydrogen production method of steam reformation of fossil fuels, water electrolysis with electricity generated by renewable energy sources such as waste heat, wind, and solar power is widely considered as a sustainable and cost-effective technology for producing high purity hydrogen on a large scale in the future. At present, only a small quantity of hydrogen (<5% of global hydrogen production) is produced by industrial water electrolysis, due to the poor efficiency of both the OER and HER. In particular, in practical applications, the OER process should be energetically catalyzed over a long period at extremely high current densities (≥500 mA cm−2) with overpotentials ≤300 mV. But most reported OER catalysts cannot meet this industrial standard, making it the major bottleneck for large scale production of hydrogen. Here, we report an exceptionally active and durable OER catalyst based on low-cost NiFe materials, which can yield current densities of 500 and 1000 mA cm−2 at overpotentials of only 248 and 258 mV, respectively, in an alkaline electrolyte, with an excellent durability over 120 hours. The fabrication method is also facile and scalable, which is via a magnetic-field-assisted chemical deposition approach. This work may pave a new way for practical and economic production of hydrogen by water splitting. |
Introduction
Splitting water to produce high purity hydrogen provides a sustainable alternative for the conversion and storage of renewable electricity sources, such as wave, wind, and solar power.1,2 To reach a practical level of energy conversion efficiency, the current state-of-the-art technologies mainly involve noble metal based electrocatalysts in proton exchange membrane (PEM) water electrolysis (e.g., Pt/C for the hydrogen evolution reaction (HER) and IrO2 for the oxygen evolution reaction (OER)), so as to lower the overpotential of both the cathodic and anodic sides.3 However, the scarcity and high cost of noble metals severely hinders this technology being widely adopted. Recently, the low-cost alternative alkaline water electrolysis, based on non-noble metal catalysts (e.g., Ni-, Co-, and Fe-based electrocatalysts), has been widely investigated for hydrogen production; but the energy conversion efficiency needs to be substantially improved. In particular, the sluggish OER, which involves multiple proton/electron-coupled steps, has become the bottleneck of water splitting.4,5 Researchers have devoted enormous efforts to developing earth-abundant OER electrocatalysts for alkaline water electrolysis, including transition metal oxides,6 hydroxides,7 sulfides,8,9 phosphides,10 nitrides11 and carbon nanomaterials.12 Among them, NiFe-based catalysts (mainly NiFe oxides and hydroxides) have been investigated extensively due to their ever-known highest activity in alkaline media.13,14 Recently, several effective strategies have been reported to further enhance the OER activity of NiFe-based electrocatalysts, including designing morphologies which can expose more active sites,15 optimizing the Ni/Fe composition and creating defects to regulate the electronic structure,16 and integrating with carbon materials to enhance electron transfer ability.7 Some available studies have demonstrated substantial improvements in OER activity for NiFe-based electrocatalysts. For example, a very low overpotential of ∼191 mV to reach 10 mA cm−2 was achieved with an amorphous FeCoW oxyhydroxide electrocatalyst.17 However, in terms of the requirement for industrial application, OER electrocatalysts are supposed to afford extremely large current densities (≥500 mA cm−2) with overpotentials ≤300 mV over a long period;18,19 the state-of-the-art non-noble metal electrocatalysts are still facing huge challenges for practical water splitting.Currently, to evaluate the catalytic activity of materials, most reported OER catalysts are fabricated into powders, which are bonded onto the current collector surfaces (e.g., glassy carbon) by electrical insulating binding agents such as Nafion,20,21 and tested at relatively small current densities (e.g., <20 mA cm−2). However, utilizing an insulating binder deteriorates the charge transfer within the electrode, and the stacking of powder based electrocatalysts may baffle the ion transfer channels and impede the release of the as-formed oxygen bubbles. As a result, powdery electrocatalysts require high overpotentials at large current densities and are easy to peel off when suffering from serious stress generated by violent oxygen bubbling. Therefore, the poor charge and mass (ion and bubbles) transfer within the electrode of the powder-based electrocatalyst hinders their application in large-current-density water splitting. Recent studies revealed that self-supported and binder-free architectures such as innovative nanoarrays can be an efficient electrode to improve the charge and mass transfer ability during the OER.22,23 For instance, Zou et al. have designed a Ni3S2 nanosheet array supported on nickel foam as a conductive substrate and coated it with amorphous NiFe hydroxide film as a catalyst, which showed overpotentials of ∼370 and ∼479 mV at large current densities of 500 and 1000 mA cm−2, respectively.24 However, to meet the industrial standard, it's still critically challenging to improve the performance of OER electrocatalysts. The challenges in designing catalysts for practical applications primarily stem from the rigorous requirement of electron and mass transfer efficiency at large current densities. Specifically, rationally designing the morphology and architecture of the whole catalytic electrode can elevate the electron transfer efficiency, ion diffusion rate, reaction kinetics, exposure of active sites and mechanical stability, and thus has a profound impact on its OER performance at large current densities.
To address these key challenges, herein we developed robust NixFe1−x alloy (core)–ultrathin amorphous oxyhydroxide (shell) nanowire arrays (denoted as NixFe1−x–AHNAs) via a one-step chemical deposition method with the assistance of uniform magnetic field. Due to the nanoscale confinement effect, the in situ formed NiFe oxyhydroxide layer on the surface of the NiFe alloy nanowire has an ultrathin structure (1–5 nm), which is far thinner than those of NiFe oxyhydroxide prepared via electrodeposition, hydrothermal or chemical deposition methods, reducing the transfer resistance of electrons within catalysts and enhancing the charge transfer ability. Additionally, the self-supported hierarchical nanowire array structure is identified to substantially reduce the contact region between bubbles and the electrode, which facilitates bubble release and ion transfer. With a simultaneous improvement of both charge/mass transfer ability and mechanical stability by elaborate electrode design, we achieve the record lowest overpotentials of 248 and 258 mV to reach large current densities of 500 and 1000 mA cm−2 with small Tafel slopes of 34.7 mV dec−1 and excellent stability over 120 hours. Furthermore, the Ni0.8Fe0.2–AHNA(+)//Ni nanowire array(−) alkaline water electrolyzer demonstrates a low cell voltage of 1.76 V at an industrial grade large current density of 1000 mA cm−2, far surpassing the performance of current industrial catalysts,25 which require 1.8–2.40 V for 200–400 mA cm−2.
Results and discussion
2.1 Synthesis and characterization of the NixFe1−x–AHNAs
NixFe1−x–AHNAs were prepared for the first time by a modified one-step chemical-deposition method under uniform electromagnetic field (see details in Experimental section), which is analogous to our group's previously reported method.26 A uniform magnetic field here was used to render alignment of the reduced ferromagnetic NixFe1−x nanoparticles on the nickel foam substrate and allow them to coalesce with each other (Fig. 1a). To optimize catalytic activity, electrodes with different Ni/Fe ratios were prepared by simply tuning the molar ratio of the Ni precursor over the Fe one, and were named as Ni1Fe0, Ni0.88Fe0.12, Ni0.85Fe0.15, Ni0.8Fe0.2, Ni0.68Fe0.32 and Ni0.62Fe0.38–AHNA according to the inductively coupled plasma-atomic emission spectrometry (ICP-AES) analysis (Table S1, ESI†).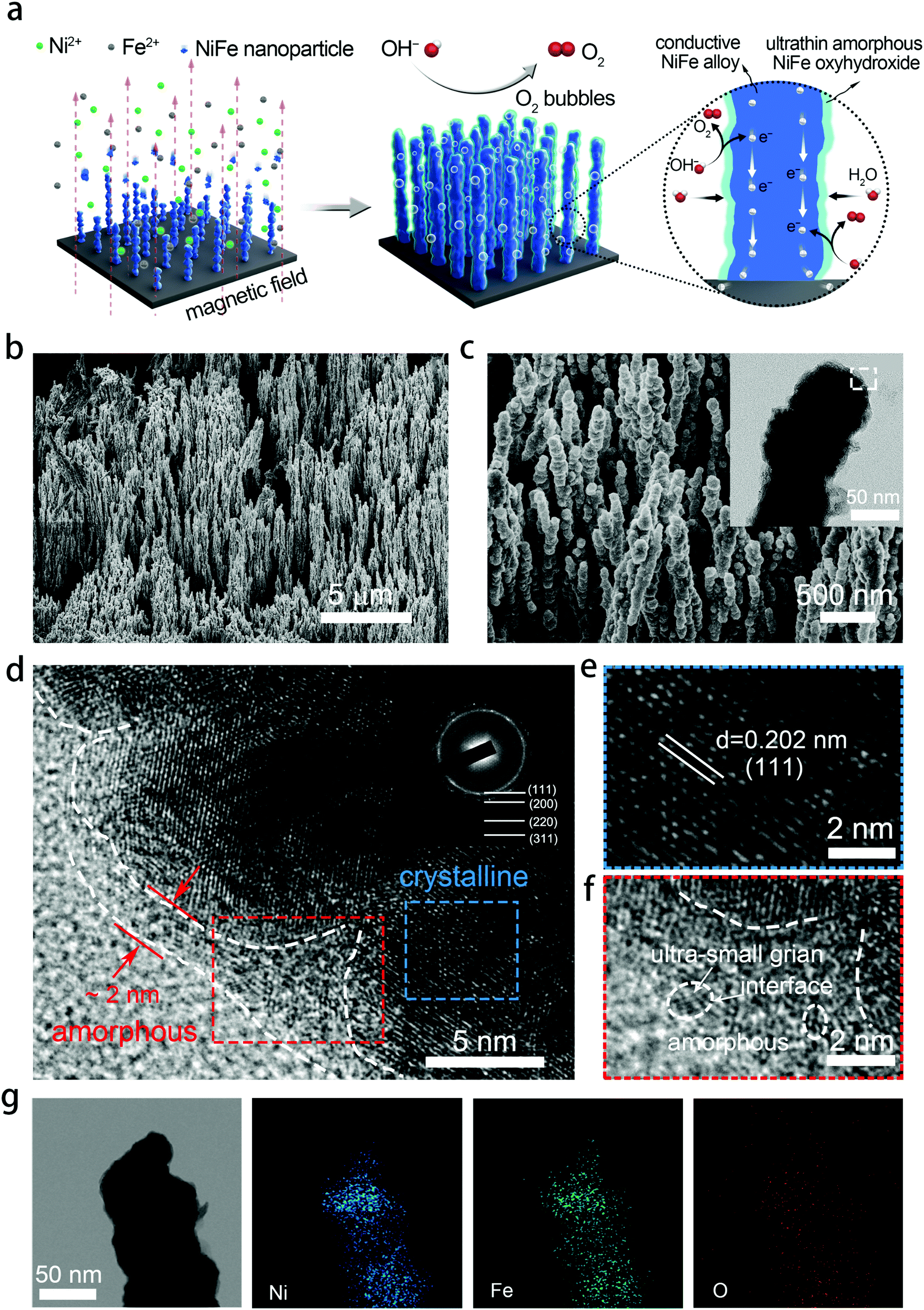 | ||
| Fig. 1 Morphological and structural characterization of NixFe1−x–AHNAs. (a) Schematic of the synthesis of the NixFe1−x–AHNA nanowire array and its catalytic function for the OER. (b and c) SEM images of NixFe1−x–AHNAs at different magnifications. The inset of (c) is a low magnification TEM of a single nanowire. (d–f) High resolution transmission electron microscopy (HRTEM) images of NixFe1−x–AHNAs. Inset of (d) is the SAED pattern. The images (e) and (f) are enlarged views of the selected area in (d). (g) EDX mapping images of Ni, Fe and O elements. | ||
Scanning electron microscopy (SEM) images show that a large number of NixFe1−x nanowires are highly oriented and vertically rooted on the nickel foam with distinctive gaps between nanowires (Fig. 1b, c, and Fig. S1, ESI†). Transmission electron microscopy (TEM) images show that NixFe1−x nanowires with a convex surface are composed of nanoparticles (the inset of Fig. 1c and Fig. S2, ESI†), which well matches the growth mechanism of ferromagnetic metal nanowires under a magnetic field.27,28 Without the induction of a magnetic field, the NiFe nanoparticles cannot grow into nanowires, but randomly stick onto the nickel foam to form agglomerates (Fig. S3, ESI†). The diameter of the nanowires is about 70–130 nm, as can be seen in the statistical result of the SEM images (Fig. S4, ESI†). Such a nanowire array structure can absorb a liquid phase electrolyte onto the electrode surface because of the strong capillary forces.29,30 As a result, it reduces gas–solid interface friction and promotes the release of oxygen bubbles from the electrocatalyst surface, which is crucial for the OER under large current densities. In addition, the well-aligned feature of NixFe1−x–AHNAs reduces the stacking of catalysts, thus exposing more catalytic active sites. The specific surface area of the NixFe1−x–AHNAs was also investigated with Brunauer–Emmett–Teller (BET) measurement, and the value was shown to be 7.4 m2 g−1 (Fig. S5, ESI†), which is far higher than that of nickel foam (<1 m2 g−1). Fig. 1d and Fig. S6 (ESI†) show the high-resolution transmission electron microscopy (HR-TEM) images of NixFe1−x-AHNA. As can be seen, the nanowire consists of well-defined crystalline phase in the core part, and an ultrathin layer (1–5 nm) of amorphous nano-domains on the surface. The crystalline core and amorphous shell were further investigated, as shown in Fig. 1e and f. The well-resolved lattice fringes with spacings of 0.202 nm in Fig. 1e correspond to the (111) plane of NixFe1−x metal alloy, while the absence of ordered lattice fringes in Fig. 1f indicates an amorphous structure. In addition, ultra-small crystalline domains on the nanowire surface, and boundaries between ultra-small crystalline domains and amorphous domains can be observed (Fig. S6, ESI†). Additionally, the selected area electron diffraction (SAED) pattern was performed to confirm the crystal structure of the NixFe1−x-AHNA. As shown in the inset of Fig. 1c, all the well-defined rings can be indexed into the (111), (200), (220) and (311) planes of NixFe1−x metal alloy. Furthermore, Fe, Ni and O elements are homogeneously distributed over the nanowire even at very high magnification, according to the energy dispersive X-ray (EDX) spectrum (Fig. 1g). The Ni/Fe atomic ratio in the NixFe1−x nanowire was determined to be 4.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S7, ESI†), which well matches with the ICP-AES result of 4.1:1 (Table S1, ESI†). The similar Ni/Fe ratio between a single nanowire and nanowire arrays also suggests that both Ni and Fe elements are distributed uniformly in each nanowire. Notably, as the core part was confirmed to be NixFe1−x metal alloy, the oxygen element here may come from the amorphous phase on the surface, which could be NiFe oxide or NiFe hydroxide due to the inevitable oxidation of the NixFe1−x alloy nanowires under the alkaline preparation conditions. It is believed that the core part of the nanowire has a crystalline metallic phase and thus is a good electron conductor. On the nanowire surface, the under-coordinated metal atoms in the amorphous domain and the amorphous/crystalline boundaries are beneficial for facilitating the binding of OH− and catalytic activity.17,18 It is noteworthy that the ultrathin thickness of the amorphous phase here ensures the high efficiency of electron transfer from the surface to the core.
:1 (Fig. S7, ESI†), which well matches with the ICP-AES result of 4.1:1 (Table S1, ESI†). The similar Ni/Fe ratio between a single nanowire and nanowire arrays also suggests that both Ni and Fe elements are distributed uniformly in each nanowire. Notably, as the core part was confirmed to be NixFe1−x metal alloy, the oxygen element here may come from the amorphous phase on the surface, which could be NiFe oxide or NiFe hydroxide due to the inevitable oxidation of the NixFe1−x alloy nanowires under the alkaline preparation conditions. It is believed that the core part of the nanowire has a crystalline metallic phase and thus is a good electron conductor. On the nanowire surface, the under-coordinated metal atoms in the amorphous domain and the amorphous/crystalline boundaries are beneficial for facilitating the binding of OH− and catalytic activity.17,18 It is noteworthy that the ultrathin thickness of the amorphous phase here ensures the high efficiency of electron transfer from the surface to the core.
The crystalline NixFe1−x alloy core–amorphous shell structure was further confirmed by X-ray diffraction (XRD) (Fig. 2a). For the bare Ni nanowire arrays (Ni1Fe0–AHNA), three strong diffraction peaks located at 44.5°, 51.9°, and 76.4° can be observed, which are indexed to the (111), (200) and (220) planes of the face-centered cubic (fcc) Ni (JCPDS No. 04-0850), respectively. The NixFe1−x nanowires have similar diffraction peaks to those of bare Ni, with slight shift toward lower diffraction degrees, and no obvious peak from bare Fe or metal oxide/hydroxide can be observed. Moreover, when the Fe content increases from 0 to 0.38, the 2θ of (111) shifts from 22.28° to 22.07°, and the calculated average lattice parameter increases from 3.52 Å to 3.55 Å. (Fig. S8, ESI†). This could be attributed to the lattice expansion due to the site-to-site lattice distortions caused by the replacement of Ni atoms with Fe. Additionally, diffraction peaks of NixFe1−x nanowires are broader than those of bare Ni, which may be due to the distortion of the lattice and the underlying peak of the amorphous surface.
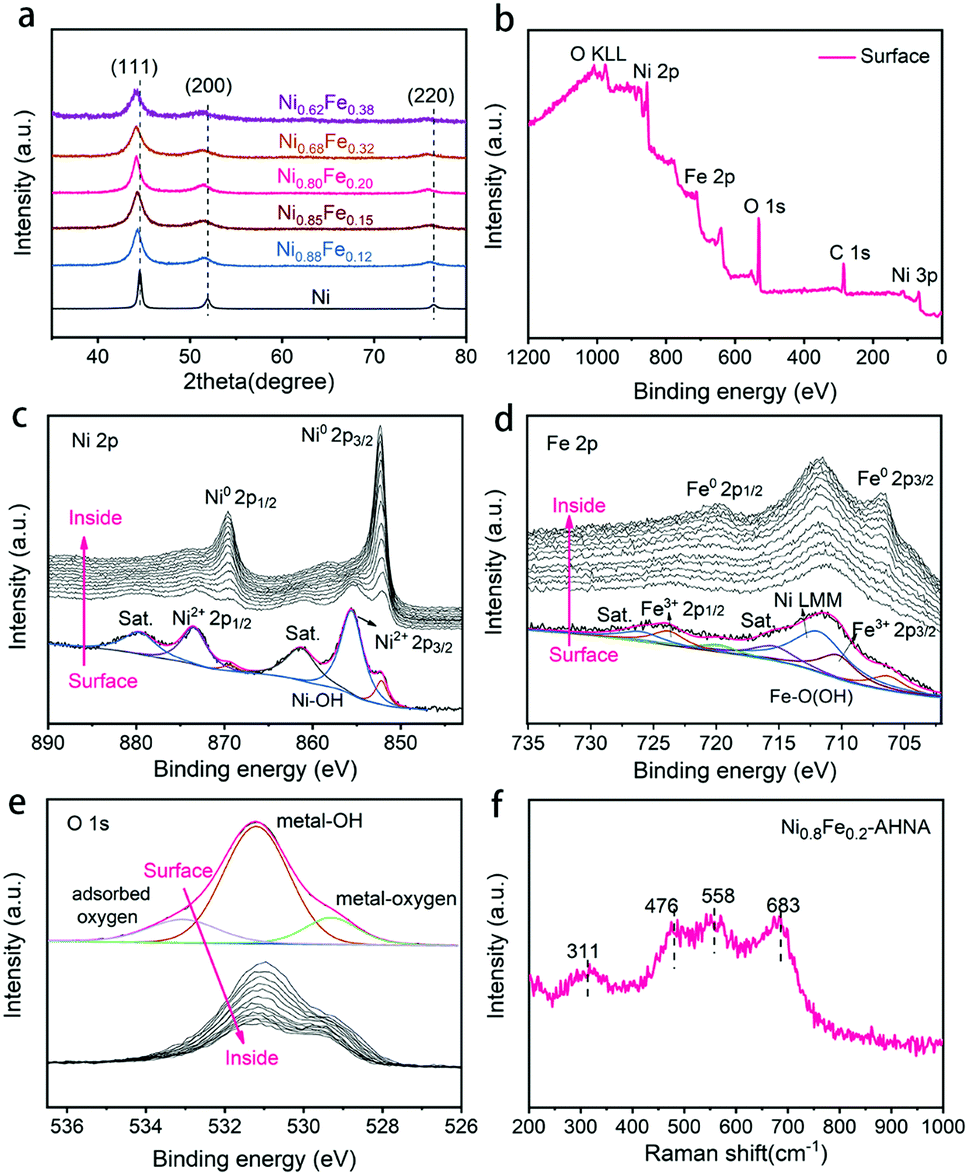 | ||
| Fig. 2 (a) XRD pattern of different NixFe1−x–AHNAs. (b–e) XPS analysis of NixFe1−x–AHNAs: (b) wide-scanning XPS spectrum (surface); (c) Ni 2p XPS depth profile; (d) Fe 2p XPS depth profile; (e) O 1s XPS depth profile. (f) Raman spectra of the Ni0.8Fe0.2–AHNA sample. | ||
X-ray photoelectron spectroscopy (XPS) was conducted to further understand the chemical composition and oxidation state of the NixFe1−x–AHNAs surface. Specifically, the wide-scanning XPS spectrum of the NixFe1−x–AHNA samples reveals the co-existence of Ni, Fe, and O elements on the nanowire surface (Fig. 2b). The fine-scanning Ni 2p spectrum from the nanowire surface (Fig. 2c), consisting of two spin–orbit doublets of Ni 2p3/2 (855.5 eV), Ni 2p1/2 (873.5 eV) and two satellites (abbreviated as “sat.”), can be indexed as Ni(OH)2; while the minor peaks at 852.2 eV and 869.3 eV are assigned to metallic Ni.27,31 Similarly, the deconvoluted peaks located at 710.6 eV and 723.8 eV and two satellites in the Fe 2p profile from the nanowire surface can be attributed to FeOOH, and the peaks located at 706.6 eV and 719.8 eV are assigned to metallic Fe.32 The broad peak at around 711.5 eV can be attributed to an Auger peak of Auger Ni LMM (Fig. 2d).33 Notably, the significant weakening of recognizable signals of metallic Ni or Fe and the emergence of strong signals of Ni2+/3+ and Fe3+ is due to the formation of a thin amorphous layer on the surface of the nanowires. After removing the surface oxide by argon ion beam etching, the signals of metallic Ni and metallic Fe come to emerge gradually, with the disappearance of oxidation state signals of Ni and Fe. Correspondingly, in the O 1s spectrum (Fig. 2e), the intensity of the peaks located at 529.3 eV (typical metal–oxygen bonds),34 531.2 eV (referring to the oxygen in the hydroxide group)35 and 533.0 eV (chemisorbed molecular water)32 decreases dramatically with etching time. The depth profile results indicate the existence of oxidized Ni and Fe and a hydroxide group on the surface of the nanowires and metallic Ni and Fe in the core.
The Raman spectra further confirm the amorphous surface of the nanowires (Fig. 2f), where four evident broad peaks instead of narrow ones can be observed, among which the bands at around 476 and 558 cm−1 can be assigned to the eg bending and A1g stretching vibrations of Ni–O in disordered Ni(OH)2 clusters, as previous studies have reported that the disordered Ni(OH)2 exhibits broadened and positively shifted Ni–O vibration peaks.24,36,37 In addition, the Raman bands at 311 and 683 cm−1 may originate from disordered FeOOH clusters.38,39 Combining the TEM, XPS and Raman analytical results, the amorphous layer on the surface of NixFe1−x alloy nanowires can be finally defined as NixFe1−x oxyhydroxide, which results from the inevitable oxidation of the nanowire under alkaline preparation conditions.
2.2 Electrocatalytic properties of the electrodes for the OER
The as-prepared electrodes were directly applied to a typical three electrode cell as the working electrode in a 1 M KOH electrolyte for OER electrocatalysis. In order to avoid signal overlap between Ni2+/Ni3+ oxidation and the OER, polarization curves were recorded from high initial potential to low potential at a scan rate of 5 mV s−1 (see Experiment section for details). A commercial IrO2 catalyst loaded on nickel foam and bare nickel foam samples were also measured as controls. As illustrated in Fig. 3a, the NixFe1−x–AHNAs show remarkably enhanced catalytic activity compared to other electrodes employed. Among them, Ni0.8Fe0.2–AHNA shows the lowest overpotential of 190 mV to deliver 10 mA cmgeometric−2, which is 110 mV lower than that of the IrO2 catalyst (300 mV). Here, the optimized Ni/Fe atomic ratio (0.8:0.2) also agrees with those of previously-reported NiFe-based OER catalysts.16,36 In particular, at the overpotential of 260 mV, the Ni0.8Fe0.2–AHNA catalyst exhibits a current density up to 1078 mA cm−2, which is 1232-fold higher than the benchmark IrO2 catalyst, demonstrating a huge enhancement of the catalytic activity toward the OER. The OER performance of a Ni0.8Fe0.2–AHNA catalyst is also far better than the Ni0.8Fe0.2 alloy fabricated without the induction of magnetic field (Fig. S9, ESI†). Additionally, we observed a very small Tafel slope of 21.3 to 41.9 mV dec−1 in low overpotential ranges (Fig. 3b), which are smaller than those of control catalysts, i.e., IrO2 (43.3 mV dec−1), Ni nanowire array (44.7 mV dec−1), and nickel foam (121.8 mV dec−1), indicating a fast kinetics during the OER. All of the above experimental observations suggest that the catalytic performance of Ni0.8Fe0.2–AHNA in 1 M KOH solution is superior to most of the previously reported catalysts, which even outperforms the presently most active NiFe–LDH (double layered hydroxide) catalyst (∼200 mV to reach 10 mA cmgeometric−2).40,41 Additionally, the overpotential to yield 1000 mA cm−2 is substantially lower than that of any of the non-noble metal OER catalysts, including recently reported state-of-the-art electrocatalysts such as NiFe–LDH/Cu nanowire arrays (315 mV),22 Ni–Fe–OH@Ni3S2/NF (469 mV),24 FeP/Ni2P (293 mV),42 and [(Ni,Fe)OOH] (289 mV),43etc. Specifically, we compared the OER activity of Ni0.8Fe0.2–AHNA with many other available OER catalysts (Fig. 3c, d and Table S2, ESI†). Clearly, our catalyst requires a low overpotential of 200 mV to achieve 20 mA cmgeometric−2, and can generate the largest current density (1078 mA cmgeometric−2) at 260 mV overpotential, indicating its great potential to drive a water splitting cell at large current densities with low electric energy consumption. It is understandable that for the same catalyst species, increasing mass loading is beneficial to improve the current density normalized by geometric area at the same overpotential, which is practically meaningful for water electrolysis devices.44 For a more comprehensive comparison, we also listed the corresponding mass loading and the substrate of these catalysts in Table S2 (ESI†).
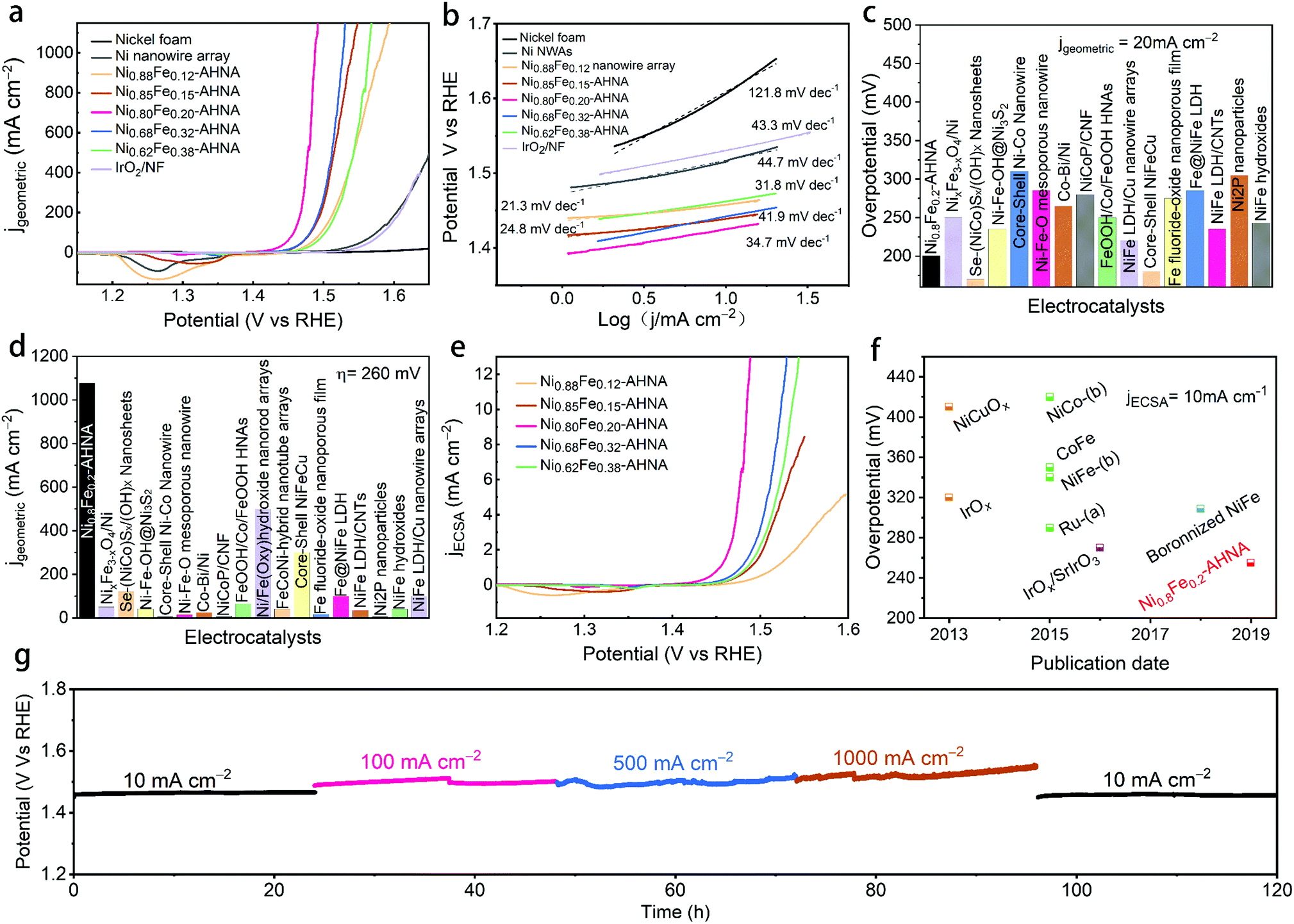 | ||
| Fig. 3 Electrocatalytic oxygen evolution reaction. (a) The polarization curves recorded on different catalysts of different electrodes, (b) the corresponding Tafel plots, (c) comparison of the overpotentials required to reach the current density of 20 mA cm−2 among our catalyst and available reported OER catalysts, (d) comparison of the current densities delivered at 260 mV among our catalyst and available reported OER catalysts. (e) Polarization curves of Ni0.8Fe0.2–AHNAs based on ECSA. (f) Comparison of intrinsic catalytic activities towards the OER with the literature. The currents are normalized to ECSA. (g) Chronopotentiometric curves of Ni0.8Fe0.2–AHNAs in a 1 M KOH electrolyte at various current densities. | ||
Nevertheless, the intrinsic catalytic activity of the catalyst can be more fundamental for increasing the water splitting performance. Here we investigated the intrinsic catalytic activity of different NixFe1−x–AHNAs catalysts towards the OER. The intrinsic catalytic activities are evaluated by the overpotentials to reach a certain current density which was normalized to the catalyst's effective electrochemical surface area (ECSA), rather than the geometric surface area. The ECSA values were estimated using an electrochemical capacitance measurement method (Fig. S10, ESI†). As shown in Fig. 3e, with the increase of Fe concentration, the normalized polarization curves of NixFe1−x–AHNAs show significant improvement of OER catalytic performance first with subsequent degradation. Among all samples, Ni0.8Fe0.2–AHNA shows the highest intrinsic catalytic activity, which affords 1 mA cmECSA−2 at an overpotential of 234 mV, lower than those of Ni0.88Fe0.12–AHNA (297 mV), Ni0.62Fe0.38–AHNA (268 mV), Ni0.62Fe0.38–AHNA (257 mV) and Ni0.85Fe0.15–AHNA (264 mV). Electrochemical impedance spectroscopy (EIS) measurement shows that Ni0.8Fe0.2–AHNA exhibits the smallest charge transfer resistance (Rct, the radius of semicircle in high frequency region) among all investigated samples, further confirming its high catalytic activity (Fig. S11, ESI†). It has been reported that the electronic structure of NiOOH varies with Fe content and thus affects the OER activity. Here, we also found that the Ni redox waves shift toward higher potential in the LSV curves (Fig. 3e and Fig. S12, ESI†) with increasing Fe content, and the binding energies of Ni and Fe 2p also shift when the Fe content varies in the NixFe1−x–AHNA samples (Fig. S13, ESI†). These indicate the change in the electronic structure of different NixFe1−x–AHNAs. Therefore, it is believed that the Ni0.8Fe0.2–AHNA showing the highest intrinsic catalytic activity is due to the most favourable electronic configuration among all samples. Notably, the Ni0.8Fe0.2–AHNA shows a significantly low overpotential of 255 mV to reach a current density of 10 mA cmECSA−2, while IrO2 and RuO2 require overpotentials of 320 mV and 290 mV to give the same current density, respectively, as shown in Fig. 3f and Table S3 (ESI†). Even the most recently reported state-of-the-art non-noble metal OER catalysts, such as Ni-based bi-/multi-metallic hydr(oxy)oxides and boronized NiFe alloy,45–47 still require overpotentials higher than 300 mV to reach the same current density. Therefore, our Ni0.8Fe0.2–AHNA has an outstanding intrinsic catalytic activity for the OER under alkaline conditions.
Electrochemical stability is another key index to evaluate the electrocatalytic performance. For practical application, the catalyst must survive over a long period under high-current-density operation conditions. Here, we probed the long-term electrochemical stability of the Ni0.8Fe0.2–AHNA catalyst by testing at five constant current densities (10, 100, 500, 1000 and 10 mA cmgeometric−2) for a total of 120 hours, and we found that the real-time potential presents negligible increase during continuous long-time operation (Fig. 3g), suggesting excellent durability. These results confirm the superior durability of the Ni0.8Fe0.2–AHNA catalyst for the OER in an alkaline electrolyte.
2.3 Enhanced charge and mass transfers of NixFe1−x–AHNAs
To further understand the origin of high catalytic activity, compositional and structural evolutions of Ni0.8Fe0.2–AHNA after the OER were investigated by SEM, TEM, XPS, XRD and Raman. Specifically, SEM and TEM images (Fig. 4a) show that the nanowire structure can be well maintained after long-term electrocatalysis, and ultrathin nanosheets are uniformly formed on the surface of the nanowires. The HR-TEM image (Fig. 4b) reveals that the core part of the nanowires remains conductive NiFe alloy phase while the nanosheets are NiFe oxyhydroxide, which was further confirmed by the XRD pattern (Fig. S14, ESI†). This core–shell composition was further verified by the XPS and Raman data (Fig. S15 and S16, ESI†). Furthermore, according to the energy dispersive X-ray (EDX) spectrum (Fig. S17, ESI†), Fe, Ni and O elements are still homogeneously distributed over the nanowire after the OER test. Notably, no oxyhydroxide could be observed deep inside the nanowire, further confirming the uniform coverage of oxyhydroxide only on the nanowire surface; this is because NiFe LDH is densely covered on the nanowire surface and is stable under alkaline conditions, thus protecting the NiFe alloy nanowire against further corrosion in the alkaline and oxidative circumstance.48,49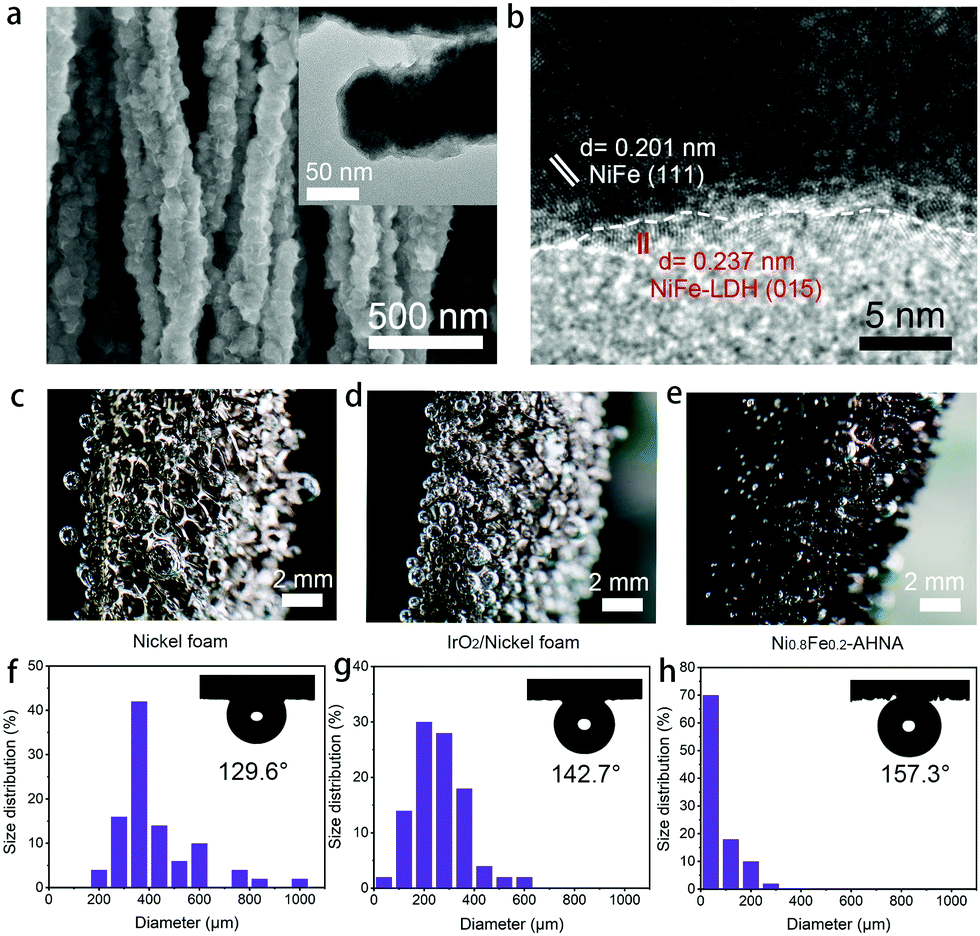 | ||
| Fig. 4 (a) SEM image of Ni0.8Fe0.2–AHNA after the OER. The inset is the TEM image of the nanowire surface. (b) HR-TEM image of Ni0.8Fe0.2–AHNA after the OER. (c–e) Digital photos demonstrating the oxygen bubbles on the surface of nickel foam, IrO2/nickel foam and Ni0.8Fe0.2–AHNA during the OER process. (f–h) The corresponding size distribution statistics of releasing bubbles for fifty bubbles. The insets are the corresponding photos of the bubble/catalyst contact angles under the electrolyte. | ||
There have been a few reports showing that after long-term OER electrocatalysis the surface species can finally transform into oxyhydroxides,16,50 and there is no exception for our one. Additionally, the morphological and compositional stability of the sample surface after long-term service is of key importance. Herein, the in situ formed NiFe oxyhydroxide nanosheets during the OER process are ultrafine (<5 nm) in both the lateral and vertical directions. These NiFe oxyhydroxides are different from the ones prepared via electrodeposition, hydrothermal or chemical deposition methods in previous reports,22,49 which generally have a large lateral size (e.g., several hundred nanometers or even micrometers), though the vertical thickness may be below 10 nm. It is reasonable to believe that due to the nanoscale confinement effect of NiFe nanowires with a small diameter of 70–130 nm, the mass supply and valid area for NiFe oxyhydroxide growth are limited, thus greatly limiting the grain size of the in situ formed NiFe oxyhydroxides. Therefore, the small lateral size and ultrathin thickness of these NiFe oxyhydroxides can excellently reduce the transport resistance of electrons from the surface catalyst to the conductive core, and thus can substantially enhance the charge transfer during the OER.
During the OER, oxygen bubbles are bound to generate at the interface between the electrocatalyst surface and electrolyte, which will severely hinder the contact of active sites and reactants. Thus, bubble releasing manners become very critical for evaluating the mass transfer within the OER electrode. We captured the digital photography of bubble releasing behaviour during a galvanostatic scan at the current density of 10 mA cm−2 (Fig. 4c–e) and counted the size of fifty releasing gas bubbles for each sample (Fig. 4g and h). As can be seen, oxygen bubbles firmly adhere to the surface of bare nickel foam and IrO2/nickel foam and grow to very large sizes (i.e., larger than 800 μm), while leaving easily from the surface of Ni0.8Fe0.2–AHNA with a small size (i.e., smaller than 100 μm). The average sizes of the released bubbles from Ni0.8Fe0.2–AHNA, IrO2/nickel foam and bare nickel foam are calculated to be 76.2 μm, 252.6 μm and 420.2 μm, respectively, which proves the much faster re-exposure of catalytic sites to the surrounding electrolyte for Ni0.8Fe0.2–AHNA. Furthermore, the under-electrolyte surface wettability of the as-prepared electrodes was investigated by bubble contact angle measurements in 1 M KOH (the inset of Fig. 4f–h). The bubble contact angle of Ni0.8Fe0.2–AHNA reaches 157.3°, while the bubble contact angles of nickel foam and IrO2/nickel foam are 129.6° and 142.7° respectively, indicating the remarkable electrolyte wettability of Ni0.8Fe0.2–AHNA. According to the solid–liquid–gas interface theory, structures with roughness at both micro- and nanoscale can reduce the contact region between the bubbles and electrode, thus leading to a low interfacial adhesion of bubbles and facilitating the wettability toward the electrolyte.29,51 Indeed, the quick release of gas bubbles on Ni0.8Fe0.2–AHNA verifies that this architecture is conducive to enhancing the mass transfer and facilitating the reaction kinetics.
It is important to clarify that the encouraging catalytic activity is not mainly attributed to the large surface area of the nanostructure electrocatalyst, as the Ni0.8Fe0.2–AHNA also exhibits very low overpotential of 255 mV to reach a current density of 10 mA cmECSA−2. We believe that the remarkable catalytic performance of our Ni0.8Fe0.2–AHNA can be mainly attributed to the enhancement of charge and mass transfer by our unique design of the whole electrode, which are: (i) the abundant defects in amorphous and low crystallinity phase NiFe oxyhydroxide promote the reaction kinetics and the optimized Ni/Fe ratio provides a favourable electronic structure; (ii) the self-supported hierarchical nanowire array structure substantially reduces the contact region between the bubbles and electrode, which facilitates the bubble release and reactant transfer; (iii) the super small size of in situ transformed NiFe oxyhydroxide in both lateral and vertical directions increases the number of active sites and reduces the electron transport resistance from the surface catalyst to the conductive core; (iv) directly growing the Ni0.8Fe0.2 nanowire catalyst on conductive substrates without polymer binders and the strong adhesion of in situ formed NiFe oxyhydroxide with NiFe alloy nanowires both reduce the interface contact resistance and benefit their mechanical and electrocatalytic stabilities.
2.4 Evaluation of the electrocatalytic overall water splitting performance
To move further toward industrial applications, it is desirable to investigate full-cell water splitting, which simultaneously converts water into oxygen at the anode and hydrogen at the cathode. We further evaluated the HER performance of NixFe1−x–AHNA catalysts with different Ni/Fe ratios. As shown in Fig. S18 (ESI†), with the increase of Fe concentration, the HER catalytic performance of the nanowire arrays shows significant degradation. The bare Ni nanowire array shows the highest activity toward the HER, which affords 10 mA cmgeometric−2 and 1000 mA cmgeometric−2 at low overpotentials of 21 mV and 207 mV, surpassing most of the reported non-noble metal HER catalysts (Table S4, ESI†). EIS results further confirm its high catalytic activity (Fig. S19, ESI†). Finally, we paired a Ni0.8Fe0.2–AHNA OER electrocatalyst with the Ni nanowire array HER electrocatalyst to construct an alkaline electrolyzer in 1 M KOH. The steady-state potential polarization curves of this Ni0.8Fe0.2–AHNA(+)//Ni nanowire array(−) water alkaline electrolyzer are shown in Fig. 5a. Remarkably, the overall-water-splitting activity of this electrolyzer is far superior to the IrO2(+)//Pt(−) and stainless-steel(+)//Ni foam(−) couples. At room temperature, our electrolyzer requires only a cell voltage of 1.41 V to achieve a water-splitting current density of 10 mA cmgeometric−2, which is substantially lower than that of the coupled benchmarking IrO2–Pt catalysts (1.57 V) and standard Ni and stainless-steel pair (1.73 V). This result is also superior to those of most ever-reported water electrolyzer cells, which generally require cell voltages higher than 1.50 V to deliver the same current density of 10 mA cm−2 (Fig. 5c and Table S5, ESI†). In particular, the Ni0.8Fe0.2–AHNA(+)//Ni nanowire array(−) alkaline water electrolyzer can generate extremely high current densities of 100, 500, and 1000 mA cmgeometric−2 at very low cell voltages of 1.55, 1.70, and 1.76 V, respectively. This outperforms the standard Ni and stainless-steel pair used in industrial alkaline electrolyzers by 368 mV, 420 mV and 486 mV, respectively at room temperature. For a rough estimation, when 1 kg H2 is generated at the current density of 500 mA cm−2, our Ni0.8Fe0.2–AHNA(+)//Ni nanowire array(−) alkaline water electrolyzer can save 11.16 kWh electricity compared to the standard Ni//stainless-steel pair (see calculation details in the ESI†). The above results show that our catalyst performs excellently at both low and high current densities. In contrast, most previously reported cells can only deliver below 100 mA cmgeometric−2 even at 1.7 V, as shown in Fig. 5d. As for the electrochemical durability, it is remarkable that this electrolyzer can sustain an overall-water-splitting performance with negligible decay for over 24 hours when operating at constant current densities of 20, 100, and 500 mA cmgeometric−2 (Fig. 5b). In addition, the faradaic efficiency for H2 and O2 generation by this electrolyzer was also evaluated (Fig. S20, ESI†). H2 and O2 with a volume ratio close to 2:1 can be observed, and the amount of measured H2 and O2 matches well with the calculated results, indicating a nearly 100% faradaic efficiency for both the HER and OER processes during water electrolysis. Overall, these electrochemical results imply that our alkaline electrolyzer has great potential for scale-up industrial implementation of hydrogen production with high efficiency and low cost.
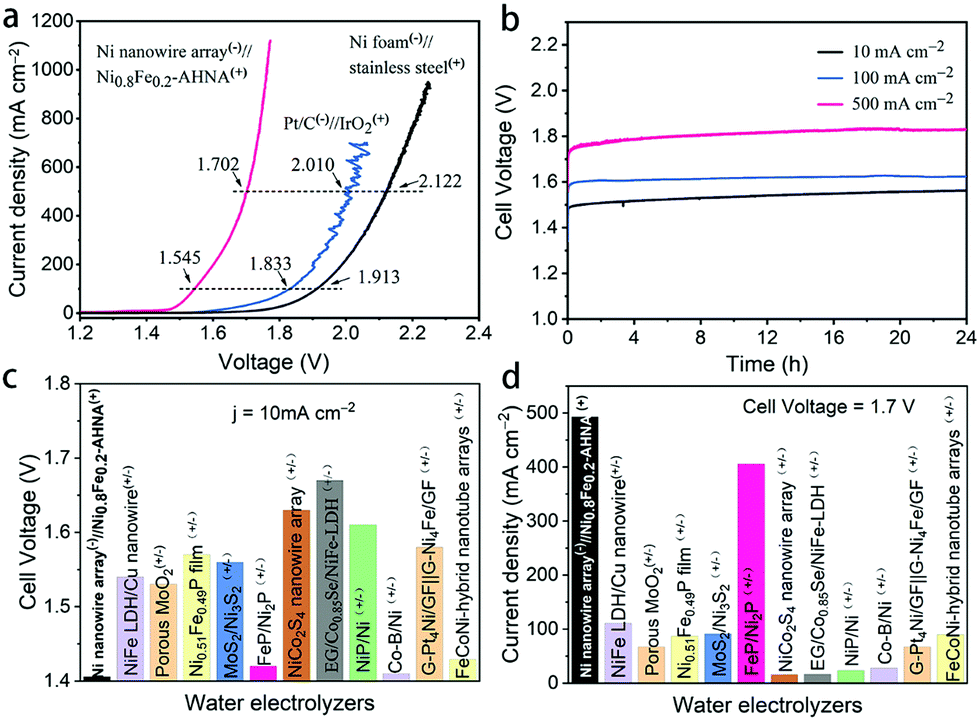 | ||
| Fig. 5 Overall water splitting. (a) Comparison of the overall water splitting activities of our designed electrolyzer to the IrO2(+)//Pt(−) and stainless steel(+)//Ni foam(−) electrolyzers at 1 M KOH. (b) Durability tests of this electrolyzer at different current densities in 1 M KOH, (c) Comparison of the cell voltages to achieve 10 mA cm−2 among different water alkaline electrolyzers. (d) Comparison of the current densities at 1.7 V for this electrolyzer with currently available robust electrolyzers in 1 M KOH. | ||
Conclusions and outlook
In summary, we have demonstrated a hierarchically structured NixFe1−x–AHNA with an adjustable ratio of Ni/Fe for highly efficient OER electrocatalysis under alkaline conditions. Benefiting from a series of advantages, including intrinsically high activity of the in situ formed amorphous NixFe1−x oxyhydroxides, large surface areas of the nanowire forests, extremely low charge transfer resistance and efficient release of gas bubbles, the optimized Ni0.8Fe0.2–AHNA catalyst can deliver current densities of 500 and 1000 mA cmgeometric−2 for over 120 hours at record low overpotentials of 248 and 258 mV, respectively. In particular, the optimized Ni0.8Fe0.2–AHNA outperforms the state-of-the-art IrO2 catalyst in 1 M KOH. Notably, pure nickel nanowire arrays prepared through the same magnetic-field-assisted method also show excellent HER electrocatalyst activity. Hence, we have constructed a Ni0.8Fe0.2–AHNA(+)//Ni nanowire array(−) alkaline water electrolyzer, all being inexpensive materials, which exhibits super-low voltages of 1.41 and 1.76 V for current densities of 10 and 1000 mA cm−2, respectively.This work demonstrates that we can rationally design the electrode structure of common non-noble NiFe materials and achieve an exceptionally high OER/HER performance to meet the industrial standard of alkaline water electrolysis. Looking forward to the next step, we are optimistic that the electrode performance can be further improved with higher activity and longer lifespan through modulating the electronic structure by introducing extra elements (such as metal Co, Mo and W or even non-metal N, S and P), and optimizing the diameter, length and density of nanowires. Hence, we believe our work is meaningful towards practical and economic production of hydrogen by water splitting.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01N111), Shenzhen Geim Graphene Center, the National Natural Science Foundation of China (Project No. 51578310 & 21950410512), Guangdong Province Science and Technology Department (Project No. 2015A030306010 & 2017A030313279), and the Shenzhen Government (Project No. JSGG20160607161911452). H. J. Fan acknowledges the financial support from an AME Individual Research Grant (Grant number: A1983c0026), Agency for Science, Technology, and Research (A*STAR).References
- M. Wang, Z. Wang, X. Gong and Z. Guo, Renewable Sustainable Energy Rev., 2014, 29, 573–588 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS.
- H. Sun, Y. Lian, C. Yang, L. Xiong, P. Qi, Q. Mu, X. Zhao, J. Guo, Z. Deng and Y. Peng, Energy Environ. Sci., 2018, 11, 2363–2371 RSC.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS.
- Z. Ma, Q. Zhao, J. Li, B. Tang, Z. Zhang and X. Wang, Electrochim. Acta, 2018, 260, 82–91 CrossRef CAS.
- Z. Ma, H. Meng, M. Wang, B. Tang, J. Li and X. Wang, ChemElectroChem, 2018, 5, 335–342 CrossRef CAS.
- X. Y. Yu, Y. Feng, B. Y. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 RSC.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef CAS.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. Gong and H. J. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. Garcia de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- R. D. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- H. Zhou, F. Yu, J. Sun, R. He, S. Chen, C.-W. Chu and Z. Ren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5607–5611 CrossRef CAS PubMed.
- B. Wang, C. Tang, H. F. Wang, X. Chen, R. Cao and Q. Zhang, Adv. Mater., 2018, e1805658, DOI:10.1002/adma.201805658.
- Y. Zhou, Z. Wang, Z. Pan, L. Liu, J. Xi, X. Luo and Y. Shen, Adv. Mater., 2019, 31, e1806769 CrossRef PubMed.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- H. Meng, W. Zhang, Z. Ma, F. Zhang, B. Tang, J. Li and X. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 2430–2441 CrossRef CAS.
- X. Zou, Y. Liu, G. D. Li, Y. Wu, D. P. Liu, W. Li, H. W. Li, D. Wang, Y. Zhang and X. Zou, Adv. Mater., 2017, 29, 1700404 CrossRef PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- C. Xu, Z. Li, C. Yang, P. Zou, B. Xie, Z. Lin, Z. Zhang, B. Li, F. Kang and C. P. Wong, Adv. Mater., 2016, 28, 4105–4110 CrossRef CAS.
- P. Zou, J. Li, Y. Zhang, C. Liang, C. Yang and H. J. Fan, Nano Energy, 2018, 51, 349–357 CrossRef CAS.
- M. Kawamori, S. Yagi and E. Matsubara, J. Electrochem. Soc., 2011, 158, E79–E83 CrossRef CAS.
- Y. Luo, L. Tang, U. Khan, Q. Yu, H. M. Cheng, X. Zou and B. Liu, Nat. Commun., 2019, 10, 269 CrossRef.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- Y. Wang, C. Xie, Z. Zhang, D. Liu, R. Chen and S. Wang, Adv. Funct. Mater., 2018, 28, 1703363 CrossRef.
- T. T. H. Hoang and A. A. Gewirth, ACS Catal., 2016, 6, 1159–1164 CrossRef CAS.
- J. Huang, J. Han, R. Wang, Y. Zhang, X. Wang, X. Zhang, Z. Zhang, Y. Zhang, B. Song and S. Jin, ACS Energy Lett., 2018, 3, 1698–1707 CrossRef CAS.
- C. Dong, T. Kou, H. Gao, Z. Peng and Z. Zhang, Adv. Energy Mater., 2018, 8, 1701347 CrossRef.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS.
- B. J. Trzesniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef CAS PubMed.
- W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan, G. Qian, J. Zou, A. Du and X. Yao, Adv. Mater., 2017, 29, 1700017 CrossRef.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef.
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2858–2864 RSC.
- S. Sun, H. Li and Z. J. Xu, Joule, 2018, 2, 1024–1027 CrossRef.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS.
- F.-F. Guo, Y. Wu, H. Chen, Y. Liu, L. Yang, X. Ai and X. Zou, Energy Environ. Sci., 2019, 12, 684–692 RSC.
- D. E. Hall, J. Electrochem. Soc., 1982, 129, 310–315 CrossRef CAS.
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G. D. Li, X. Zou and J. S. Chen, Nat. Commun., 2018, 9, 2609 CrossRef.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Wang, J. Luo, X. Lei and L. Jiang, Adv. Mater., 2014, 26, 2683–2687 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02388g |
| This journal is © The Royal Society of Chemistry 2020 |