
Heterointerface engineering for enhancing the electrochemical performance of solid oxide cells
Chenhuan
Zhao†
 a,
Yifeng
Li†
a,
Wenqiang
Zhang
a,
Yun
Zheng
ab,
Xiaoming
Lou
a,
Bo
Yu
*a,
Jing
Chen
*a,
Yan
Chen
*c,
Meilin
Liu
*d and
Jianchen
Wang
a
a,
Yifeng
Li†
a,
Wenqiang
Zhang
a,
Yun
Zheng
ab,
Xiaoming
Lou
a,
Bo
Yu
*a,
Jing
Chen
*a,
Yan
Chen
*c,
Meilin
Liu
*d and
Jianchen
Wang
a
aInstitute of Nuclear and New Energy Technology (INET), Collaborative Innovation Center of Advanced Nuclear Energy Technology, Tsinghua University, 30 Shuang’qing Road, Beijing 100084, P. R. China. E-mail: cassy_yu@mail.tsinghua.edu.cn; jingxia@mail.tsinghua.edu.cn
bDepartment of Chemical Engineering, University of Waterloo, Waterloo, ON, Canada
cGuangzhou Key Laboratory for Surface Chemistry of Energy Materials, New Energy Institute, School of Environment and Energy, South China University of Technology, Guangzhou 510006, China. E-mail: escheny@scut.edu.cn
dSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0245, USA. E-mail: meilin.liu@mse.gatech.edu; Tel: +1-404-894-6114
First published on 13th September 2019
Abstract
Solid oxide cells (SOCs) have the potential to be the most efficient energy storage and conversion systems. To minimize energy loss due to charge and mass transport associated with the operation of SOC systems at intermediate temperatures, electrodes and electrolytes containing different types of heterointerfaces have been designed, fabricated, and tested under various conditions. While heterointerfaces can significantly enhance not only the ionic and/or electronic conductivity but also the electrocatalytic activity and stability of SOC components, as predicted by theoretical calculations and demonstrated by experimental results, the mechanisms of these enhancements are yet to be fully understood. In this review, we start with an overview of the techniques for fabrication of heterointerfaces with controlled composition, structure, and morphology. Then, the latest developments in performance enhancement of SOCs with heterointerfaces are summarized, including boosting the ionic conductivity of heterostructured electrolytes (oxygen ion conductors and proton conductors) and increasing the electrocatalytic activity and durability of heterostructured electrodes (oxygen electrodes and fuel electrodes). Subsequently, we will highlight the unique attributes of heterointerfaces in the enhancement of the SOC performance and provide important insights into the mechanisms of performance enhancement in order to establish the scientific basis for rational design of better electrolyte and electrode materials. Finally, the remaining challenges in design and fabrication of novel materials for advanced solid-state electrochemical systems will be discussed, together with possible strategies to overcome these critical issues, new research directions, and future perspectives.
 Chenhuan Zhao | Chenhuan Zhao is now studying for a PhD degree from the Institute of Nuclear and New Energy Technology (INET) at Tsinghua University, China. She received her BS degree in the Department of Chemical Engineering from Tsinghua University in 2014. Her research interests include energy storage and conversion using solid oxide cells, especially for heterostructured oxygen electrodes with high activity. |
 Yifeng Li | Yifeng Li is currently studying for a PhD degree from the Institute of Nuclear and New Energy Technology (INET) at Tsinghua University, China. He received his BS degree in the Department of Chemical Engineering at Tsinghua University in 2015. His research interests include electrochemical energy storage and conversion, especially the research of high temperature electrolysis of CO2/H2O to produce sustainable fuels using solid oxide electrolytic cells (SOECs), as well as the degradation issues of SOEC electrode materials. |
 Bo Yu | Dr Bo Yu is an Associate Professor and PhD Supervisor of Tsinghua University, China. She received her PhD from Tsinghua University in 2004 and joined Nuclear Science & Engineering at Massachusetts Institute of Technology as a visiting researcher in 2012. Dr Yu has been responsible for the research and development of nuclear hydrogen or syngas production at Institute of Nuclear and New Energy Technology since 2005. She has published more than 100 articles and holds over 30 patents. Her research interests are electrochemical energy storage and conversion with a focus on high temperature electrolysis of CO2/H2O through SOEC technologies. |
 Jing Chen | Dr Jing Chen, as a professor, works at Institute of Nuclear and New Energy Technology (INET), Tsinghua University, China. He received his BS degree and PhD degree in Chemical Engineering from Tsinghua University in 1992 and 1996, respectively. His research area is chemical engineering for advanced energy systems, especially hydrogen production and utilization via high temperature solid oxide cell systems. |
 Yan Chen | Yan Chen is a Professor at the School of Environment and Energy at the South China University of Technology. She received her BS and ME from Peking University, and PhD from the Massachusetts Institute of Technology. Prior to her current position, she worked as a postdoctoral fellow at MIT. Her research focuses on rational design of new materials to enable high performance, economical energy devices based on understanding and controlling the surface and interface properties under extreme conditions (electric polarization, reactive environment, radiation etc.). She is also working on the application of ion beam implantation in low dimensional materials for energy and electronics. |
 Meilin Liu | Meilin Liu is the B. Mifflin Hood Chair, Regents' Professor, and Associate Chair of the School of Materials Science and Engineering at the Georgia Institute of Technology, Atlanta, Georgia, USA. He received his BS from the South China University of Technology and both MS and PhD from the University of California at Berkeley. His research interests include design, fabrication, in situ/operando characterization, and simulation of membranes, thin films, and nanostructured electrodes in devices for energy storage and conversion, aiming at achieving rational design of materials and structures with unique functionalities. |
Broader contextLow-cost and efficient energy storage technologies are the enabler for the implementation of intermittent renewable energy technologies. Solid oxide cells (SOCs), when operated in reversible mode, have the potential to be the most efficient option for large-scale energy storage and power generation. However, broader commercialization of SOC technology hinges on breakthroughs in material development for high performance and long operational life. Heterostructured materials have demonstrated significantly enhanced charge and mass transport and electro-catalytic activity, especially at relatively low temperatures. This review provides a timely and comprehensive update on the latest advancements in the development of heterostructured SOC components. The methodology and engineering strategies are valuable not only to SOC technology but also to a wide range of energy applications for a clean and sustainable energy future, including photo/electrochemical water splitting and solar thermal CO2 reduction. |
1. Introduction
1.1 Overview of solid oxide cells
Rapid growth of energy consumption in the fields of industrial production, transportation, and electricity generation has posed unprecedented challenges to sustainable growth. In addition, the use of fossil fuels discharges large amounts of CO2 and polluting gases that cause global warming and serious environmental pollution. To meet future energy demands and reduce CO2 emissions, many renewable and environmentally friendly energy sources (such as solar, geothermal, wind, and biomass) have been extensively investigated. However, the deployment of these renewable resources is severely hindered by their intermittent nature, leading to urgent demands for efficient energy storage and CO2 conversion systems. Due to their high efficiency and the capability to meet the requirement for large scale energy storage and conversion, solid oxide cells (SOCs) have attracted global attention.1–9Fig. 1 shows the working principle of SOCs for electric energy storage and conversion, together with a number of potential applications such as distributed power generation, electrical vehicles, and smart grids.10,11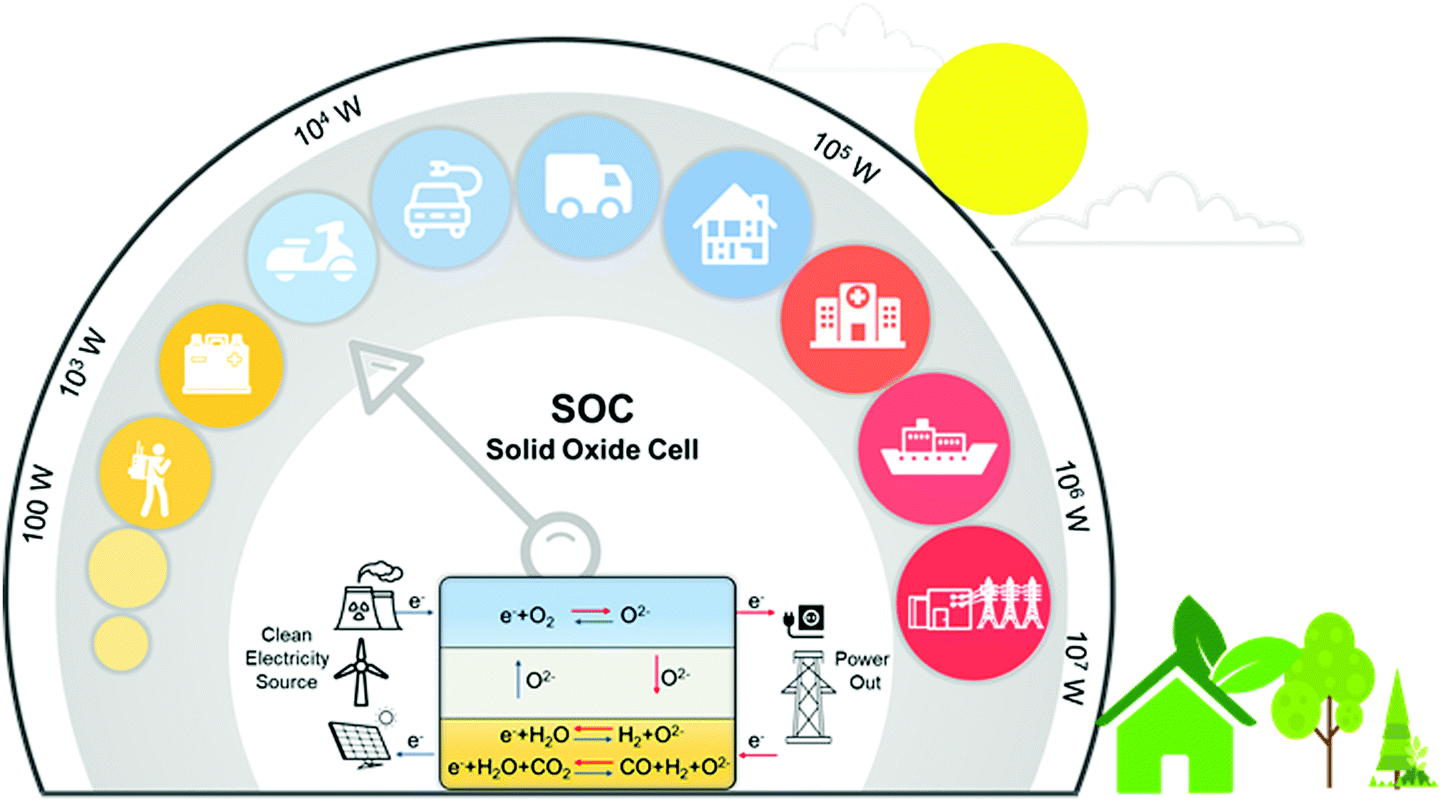 | ||
| Fig. 1 Working principles and representative applications of SOCs. | ||
A SOC consists of a dense electrolyte, an oxygen electrode, and a fuel electrode. During operation, the oxygen electrode provides active sites for the oxygen reduction or evolution reaction (ORR/OER)12–14 whereas the fuel electrode provides active sites for oxidation of fuel (e.g., H2, CH4, and CO) or production of fuel (e.g., electrolysis of H2O and CO2). Both electrodes must provide not only a sufficient number of active sites for the electrode reactions but also proper pathways for rapid transport of the species involved in the electrode reactions (such as ions, electrons, and gas molecules). The electrolyte in an SOC facilitates ion migration (oxygen ions, protons, or both) between the oxygen and the fuel electrodes while blocking the transport of electrons and gas molecules to avoid the short circuiting of the cell. SOCs can operate as a solid oxide fuel cell (SOFC) or as a solid oxide electrolysis cell (SOEC) as needed.15–18 SOFCs can convert chemical fuels directly to electricity with high energy efficiency, high energy and power densities, and fuel flexibility.3,4,7,19,20 SOECs can store “excess” electric energy in the form of chemical fuel (H2, CO, syngas, or other renewable fuels) through the electrolysis of H2O and/or CO221–24 while maintaining similar advantages such as high energy conversion efficiencies, high production rates, fast power cycling, and low manufacturing costs.
Despite these numerous advantages and broad application prospects, the large-scale application of SOC systems is severely inhibited by the high operating temperature (e.g., 800 to 1000 °C).18,25 Although such high operating temperatures can significantly accelerate the reaction kinetics and ionic conductivity, they can lead to detrimental interactions between cell components, leading to poor durability and high costs of utilization.3,26,27 Therefore, to develop more efficient, durable, and economically feasible SOC systems, the SOC operating temperature must be reduced to about 500 °C while maintaining sufficiently high performance.28–30 As the operating temperature is reduced, however, the cell performance will decrease exponentially due to slower oxygen reduction/evolution (ORR/OER) reaction kinetics on the oxygen electrode,31–35 lower ionic conductivity of the electrolyte,36 and increased polarization resistance of the fuel electrode.37 Therefore, new materials or novel structures for SOC components need to be designed to enable high ionic conductivity in the electrolytes, high electrocatalytic activity at the electrodes, and good compatibility with other cell components under operating conditions.38,39
1.2 Heterointerface engineering
Both experimental results and theoretical calculations have shown that heterointerfaces have the potential to significantly enhance the ionic and/or electronic conductivity, electrocatalytic activity, and stability of electrolytes and electrodes in SOCs.40–45 For example, Su et al.46 reported that the ionic conductivity of vertically aligned nanocomposite GDC/YSZ heterostructured electrolytes (0.96 S cm−1) was 2 times higher than that of the YSZ single phase at 600 °C. Furthermore, Sase et al.47 used 18O isotope exchange with secondary ion mass spectrometry (SIMS) to clearly demonstrate ∼103 times faster oxygen exchange kinetics at the heterointerface of (La,Sr)CoO3/(La,Sr)2CoO4 (LSC113/LSC214) with PLD-layered films. Crumlin et al.48 decorated (La,Sr)CoO3 thin films with epitaxial growth of (La,Sr)2CoO4 using PLD and achieved a ∼1–3 orders of magnitude enhancement in the ORR activity. In addition, Sun et al.49 reported that La0.3Sr0.6Ce0.1Ni0.1Ti0.9O3−δ (LSCNT) fuel electrodes with Ni–Ce exsoluted particles presented outstanding activities with a peak power density of 600 mW cm−2 and were resistant to coking for more than 80 h in a CH4 atmosphere.Overall, these results demonstrate that heterointerface engineering is a promising approach to enhancing the SOC component performance at intermediate temperatures. Such enhancements can be attributed to many factors, including lattice strain, dislocation, cation segregation, changes in electronic structure, and expansion of triple phase boundaries (TPBs) (Fig. 2).4,50,51 For example, research into electrolytes52,53 and oxygen electrodes54,55 suggested that tensile strain at heterointerfaces can accelerate ionic transport by decreasing the migration barrier of charge carriers. In addition, results showed that the electronic structure near the heterointerface of LSC113/LSC214 oxygen electrodes can be different from their single-phase counterpart, leading to strongly enhanced ORR kinetics.56 The presence of cation segregation near the interfacial region was reported to provide more active sites for the ORR/OER.57,58 Furthermore, Ni/metal oxide interfaces of fuel electrodes have been reported to have higher coking resistance by facilitating water adsorption and water-mediated carbon removal.59,60 To guide the rational design of high performance SOCs, it is imperative to fully understand the role of heterointerfaces in each of the key cell components: the electrolyte, the oxygen electrode, and the fuel electrode.
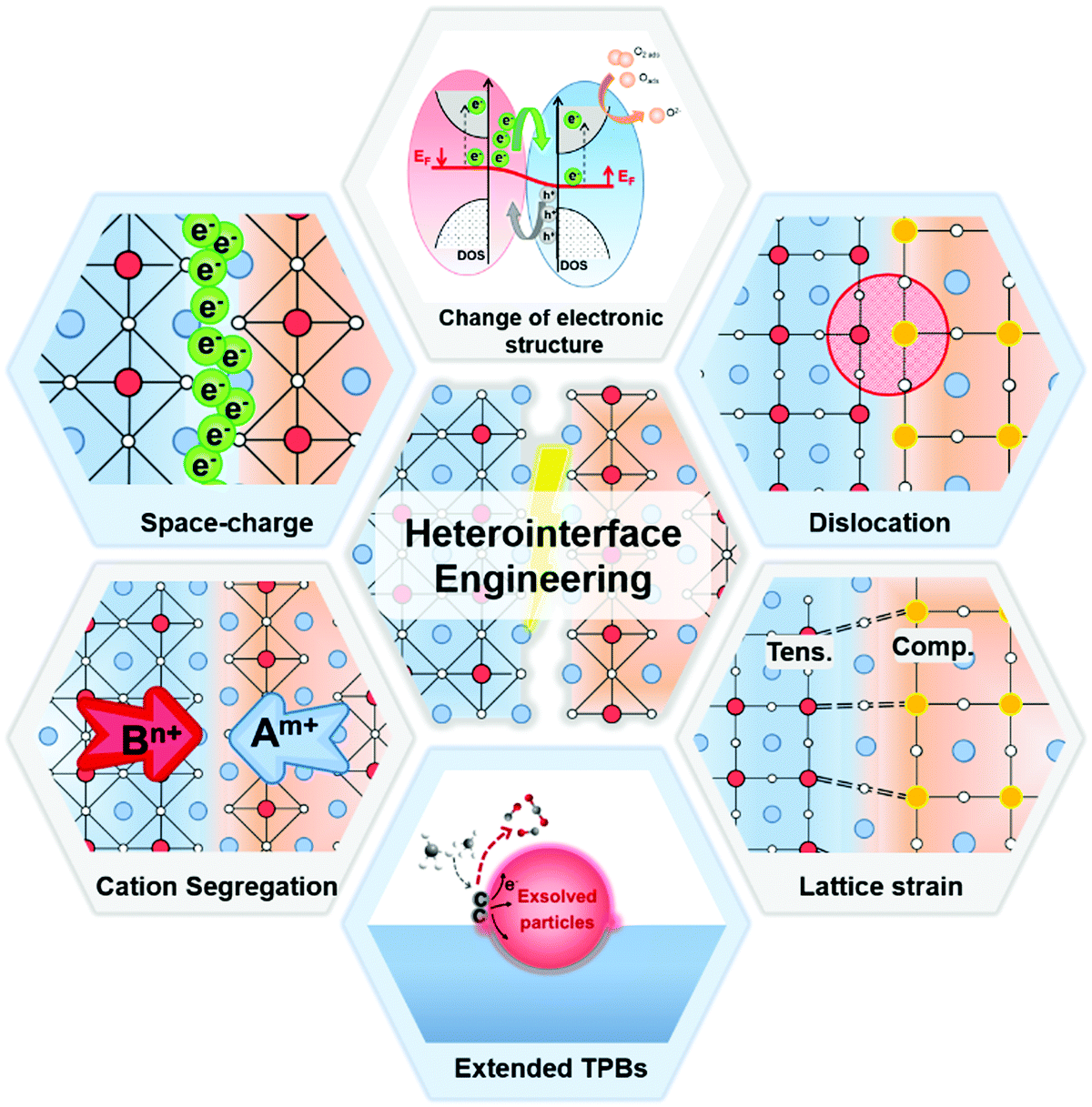 | ||
| Fig. 2 The possible mechanisms for the performance enhancement in heterostructured SOC components. | ||
It is noted that the factors that affect the properties of heterostructured SOC components (Fig. 2) are often inter-related. For examples, small lattice mismatches at a heterointerface may be readily accommodated by local lattice strains;52,53 when the lattice mismatches are sufficiently large, however, the formation of dislocations may become energetically favorable to relieve local strain and stabilize interfacial structures. Furthermore, it has been proven that lattice strain may influence defect formation energies and migration barriers,54,55 thus altering the distribution of charge carriers near heterointerfaces or in the space charge region (e.g., cation segregation at the interfacial region57,58). The presence of or a change in lattice strain, defects (e.g., oxygen vacancies or dislocations), and redistribution (accumulation or depletion) of charge carriers in space-charge regions can induce electron transfer and influence the electronic structure56 (or the energy landscape) of the interface, which determines the fundamental properties of the heterointerfaces. However, the specific correlations among these factors are yet to be fully understood.
This review aims to provide a comprehensive and in-depth overview of the impacts of heterointerfaces on the ionic transport, catalytic activity, and stability of heterostructured materials, providing important insights into the mechanisms of performance enhancement and establishing a scientific basis for the rational design of better electrolyte and electrode materials. The fundamentals of heterointerfaces are reviewed in Section 2, including basic concepts, structural characteristics, and controlled fabrication processes. Subsequently, applications of heterostructures in electrolytes, oxygen electrodes and fuel electrodes of SOCs are highlighted, along with possible mechanisms for the performance enhancement in those heterostructured SOC components, including lattice strain, dislocation, cation segregation, modified electronic structure, and extended TBPs at heterointerfaces. In addition, new research directions, future perspectives, existing challenges and possible solutions for the rational design of novel materials for advanced solid-state electrochemical systems involving heterostructures will be summarized. Overall, this review will provide a useful guide for the rational design of novel materials with unique functionalities for high-performance SOCs. The knowledge and methodology illustrated in this review are also suitable for the development of novel catalysts for other energy storage and conversation systems.
2. Fundamentals of heterointerfaces for SOCs
This section will provide a brief introduction to the basic concepts of heterointerfaces along with representative structures and synthesis techniques of oxide heterostructures used in SOCs. After a brief discussion of the physical properties of heterojunctions, we will classify heterointerfaces into different categories according to their structures. Then, several approaches for synthesis of heterointerfaces will be discussed, including atomic layer deposition, infiltration, screen printing, etc.2.1 Basic concepts of heterojunctions
When two dissimilar materials are brought into contact, a heterointerface is formed. The difference in lattice constant of the two dissimilar materials will result in strain near the heterojunction and the difference in Fermi energy of the two materials will lead to electron transfer across the heterointerface, inducing a built-in electric field and a space charge region. Similarly, the difference in mobile ion concentrations of the two materials causes ion transfer across the heterointerface and a space charge region. The unique characteristics of heterojunctions include local accumulation (or depletion) of charge carriers, enhanced (or decreased) charge carrier mobility, quantum effects, and other extraordinary effects not observed in single phase materials.61With technological developments and in-depth studies of heterojunctions, the definition and research of heterostructures have largely expanded. The extraordinary properties associated with heterointerfaces include high electrical conductivity,36,62 enhanced thermoelectric effects, boosted catalytic activity,63 super-wettability,64,65 and induced magnetization.66–69 The first study on the enhancement of ionic conductivity in composite materials was based on inorganic salt/Al2O3 and AgI/AgBr heterointerfaces in the 1980s.70 Subsequently, significantly enhanced ion conduction was also found in multi-layered CaF2/BaF236,71 and YSZ/STO epitaxial heterostructures.72 In addition, high-mobility electron gases at the heterointerface of LaAlO3/SrTiO3 were investigated by Ohtomo et al.73,74 and Nakagawa et al.75, which demonstrated extremely high carrier mobility exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1 as a result of the built-in polarity discontinuity at the interface. These remarkable enhancements in conductivity and activity inspired the application of heterostructured oxides in SOCs.
000 cm2 V−1 s−1 as a result of the built-in polarity discontinuity at the interface. These remarkable enhancements in conductivity and activity inspired the application of heterostructured oxides in SOCs.
2.2 Structures of heterointerfaces
Based on the structural characteristics, heterointerfaces in SOCs can be classified into several categories, including (a) multilayers, (b) vertically aligned nanocomposites (VAN), (c) composites, and (d) skeleton/surface coating (Fig. 3). Due to the significantly different structures, these heterointerfaces display large variations in local composition, lattice strain, electronic structure, and other properties.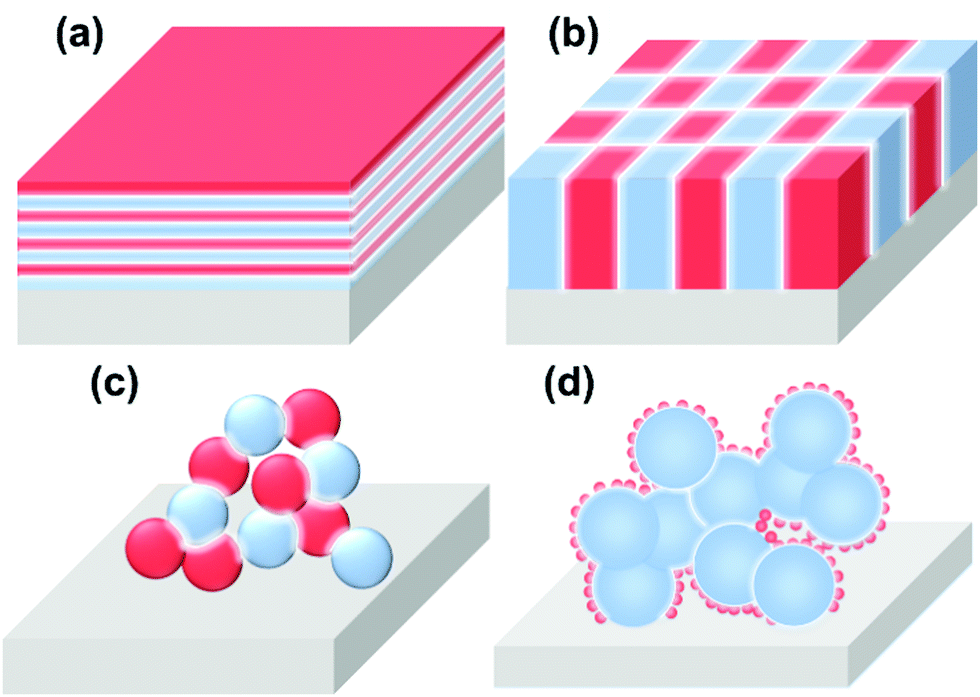 | ||
| Fig. 3 Schematics of the heterogeneous structures of component materials in SOCs. (a) Multilayer structure, (b) vertically aligned nanocomposite (VAN) structure, (c) composite structure, (d) skeleton/surface coating structure. | ||
(a) The multilayer structure is a classical model for theoretical investigations in which heterointerfaces between two adjacent layers are parallel to the surface. Here, well-ordered interface structure enables the precise control of interfacial properties such as local compositions, lattice strains, electronic structures, etc. And in particular, the interfacial properties can play a dominate role in determining the functionality of the multilayer when the thickness of each layer approaches several nanometers.
(b) The VAN structure is another widely used model system for the investigation of the role of interfaces in the functionality of materials. Compared with the multilayer structure model, the unique advantage of the VAN structure is the maximum exposure of the active heterointerface. Furthermore, the interfaces in VAN structures are either directly exposed to gas (as electrodes) or along the direction of ion flow (as electrolytes), enabling full utilization of the heterointerfaces in SOCs. In addition, a VAN structure can sustain higher interfacial strain compared with a multilayer structure.
(c) Composite structures are often used in practical solid oxide cells; the heterointerface in this structure is random and the actual contact of the two phases is complicated. It is difficult to quantify the interfacial region between the two phases.
(d) The skeleton/surface coating structure is composed of a porous backbone decorated with a thin-film coating, which is also widely used in practical SOCs. In this structure, the backbones (or the materials serving as the mechanical support) generally possess good ionic or mixed conductivity, whereas the coating materials are generally highly active catalysts.
2.3 Fabrication of heterostructured materials or devices
Tremendous techniques have been developed for fabrication of materials containing heterointerfaces with specific structures and compositioins,76 including pulse laser deposition (PLD),77–81 molecular beam epitaxy (MBE),36,82,83 atomic layer deposition (ALD),84–88 electrophoretic deposition (EPD),89 infiltration,90–94 freeze drying tape casting,92,95–98 screen printing,99,100etc. In this review, advanced atomic layer deposition techniques are emphasized for the precise fabrication of high-quality oxide heterostructures.To be specific, PLD can be used to fabricate complex heterostructured thin films with desired stoichiometric ratios, in which a high-power pulsed laser beam is produced by an excimer laser (e.g. KrF) and focused inside a vacuum chamber through a lens to a target (Fig. 4a).103 Here, the target material is evaporated as a plasma plume in a high vacuum or in the presence of a background gas (e.g. oxygen of various concentrations) and subsequently deposited onto a substrate. And by optimizing operating conditions such as temperature, oxygen partial pressure, laser energy, etc., thin films with well-ordered crystal lattice units and specific orientations can be prepared. In addition, the use of PLD can produce various heterostructured oxide morphologies including bilayer,106 ML107 and VAN77 structures. For example, Schichtel et al.108 fabricated YSZ|Sc2O3 multilayers using PLD to study strain effects on the ionic conductivity.
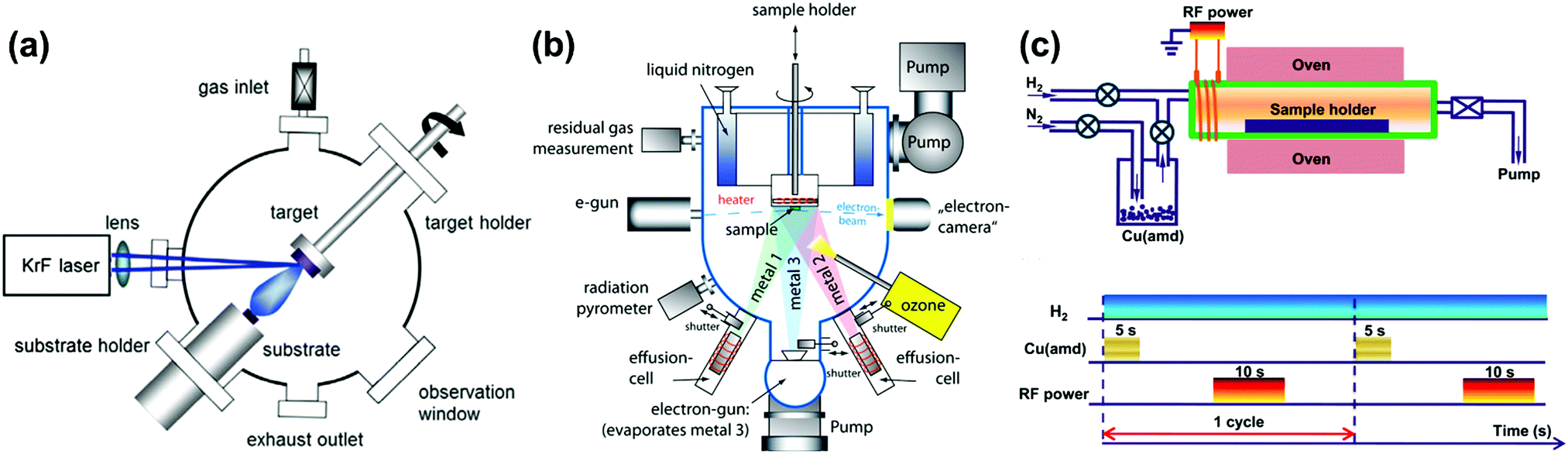 | ||
| Fig. 4 Schematics of a (a) PLD system,103 (b) MBE system104 and (c) up: tubular ALD reactor, down: layout of a plasma-enhanced ALD cycle for Cu deposition.105 Reproduced from ref. 103–105 with permission from RSC, copyright 2016; Baiutti et al; license Beilstein-Institute (https://www.beilstein-journals.org/bjnano/articles/5/70), copyright 2014 and American Chemical Society, 2015, respectively. | ||
The main advantage of the MBE method (Fig. 4b) is the excellent control of oxide growth as a result of the single-element fluxes, minimized kinetic energies and slow kinetics.104,109,110 The difficulty of the MBE method, however, is the inability to maintain desired cationic stoichiometry during growth, which requires cooperation with an in situ monitoring auxiliary system, including a quartz crystal microbalance (QCM), real-time absorption spectroscopy (AAS), reflection high-energy electron diffraction (RHEED) etc. Furthermore, applications of MBE and PLD are also limited by their expensive deposition systems.
In addition, ALD is applied in layer-by-layer structure fabrication, which is based on the gas–solid reaction between the atmosphere and a substrate (Fig. 4c).105,111 ALD can produce conformal and pinhole-free coatings of porous structures and is usually used in the fabrication of binary or ternary oxides.112,113 For example, high quality films of many single oxides (e.g., MgO,114 NiO115 and CuO116) can be readily coated onto perovskite LSCF backbones using ALD.
3. Heterointerfaces in electrolytes of SOC devices
The reduction of operating temperatures increases the need for superior alternatives with high-conductivity electrolytes at intermediate temperatures. Here, heterostructured electrolyte materials with extraordinary ionic conductivities have been shown to be promising in SOC applications at intermediate temperatures. However, the corresponding mechanisms need to be further verified. The impact of heterointerfaces on the ionic conductivity and the mechanisms of such impacts will be introduced in this section.Electrolytes are key components of fuel cells, which largely determine SOC electrode compositions.3,88,121,122 To improve the performance of SOCs at intermediate temperatures, the electrolyte materials should meet the following requirements: (1) high ionic conductivity and negligible electronic conductivity under operating conditions; (2) good stability in various atmospheres over a wide range of temperature; (3) a small mismatch in the thermal expansion coefficient with electrodes and other cell components, (4) negligible interactions with electrodes and interconnect materials under operating and processing conditions, and (5) adequate mechanical strength or integrity.
High ionic conductivity of SOC electrolytes requires a sufficient concentration of mobile charge carriers with high mobility. Based on the types of ionic charge carriers, SOC electrolytes can be classified into oxygen ion conductors, proton conductors, and mixed ion conductors. To date, tremendous efforts have been contributed to the development of high-performance electrolyte materials with oxygen ion conductivity,123,124 including doped zirconia,125,126 doped ceria,125,127,128 lanthanum gallate-based oxides,129,130 and bioxide based materials.131,132 The activation energy for O2− transport in oxygen ion conductors is usually in the range of 50–60 kJ mol−1,133,134 indicating significantly lower ionic conductivity at reduced temperatures.
In addition, since the discovery of alkali earth doped cerates (ACeO3, A = Sr and Ba) with perovskite structures as promising proton conductors,135,136 researchers have devoted much attention to these materials.137–141 The general formula of perovskite type proton conductors can be expressed as AB1−xMxO3−δ, in which M denotes a trivalent dopant and δ represents the oxygen deficiency per unit cell.139,142 Here, the mechanisms of proton conduction include proton transfer, structural reorganization, diffusion motion of extended moieties, etc.135,136,139,143
3.1 Performance of electrolytes with heterointerfaces
Heterostructured electrolytes have attracted great attention due to the enhancement of ionic conductivities by the heterointerfaces, especially at low or intermediate temperatures. Representative mechanisms of enhanced ionic conductivities through heterointerfaces are highlighted in Fig. 5 and more detailed information is summarized in Table 1. Heterogeneous structures of various architectures and morphologies have been designed and fabricated to enhance the ionic conductivity, including multilayers,36,72,144–151 vertically aligned nanocomposites,78,152–154 and composites of randomly distributed phases.155,156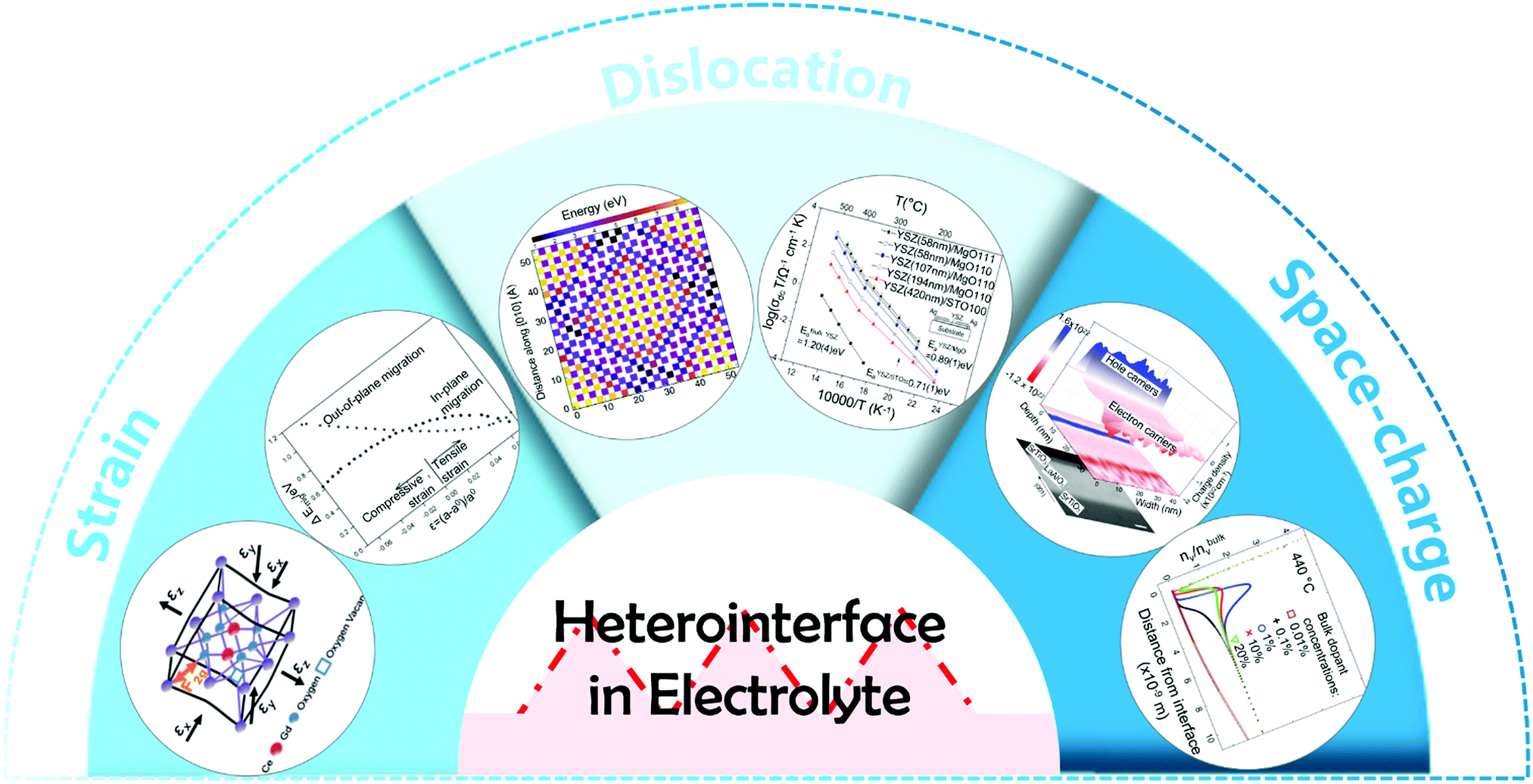 | ||
| Fig. 5 Schematic of the functional principles for heterostructured electrolytes. Insets: (left) Schematic view illustrating the distortion of the unit cell under lattice strain;145 activation energies for vacancy migration in CeO2 biaxially strained along [100] and [010];157 oxygen vacancy formation energies at the MgO layer in a TiO2-terminated SrTiO3 interface;158 lateral ionic conductivity versus inverse temperature for YSZ films deposited on different substrates and with film thicknesses in the range 420 to 58 nm;159 charge distribution in the STO/LAO/STO heterostructure;160 normalized vacancy concentration as a function of distance from the GB interface at 440 °C161,162 (right). Reproduced from ref. 145 and 157–161 with permission from RSC, copyright 2017; RSC, copyright 2012; Springer Nature, copyright 2014; WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2010; Springer Nature, copyright 2018 and RSC, copyright 2015, respectively. | ||
| Material | Geometry | T (°C) | Relative increase in conductivity | Mechanism | Ref. |
|---|---|---|---|---|---|
| GdCeO/MgO | Bilayer | 500–800 | ×3 | Heat treatment that releases strain energy and removes dislocation | 148 |
| YSZ/MgO | Bilayer | 400–800 | ×102.5 | Lattice mismatch strain | 43 |
| YSZ/MgO YSZ/STO | Bilayer | 150–500 | ×103 | Combination of mismatched dislocation density and elastic strain | 159 |
| Ce0.9Gd0.1O2−δ/STO | Bilayer | 450–900 | ×10–102 | Compressive strain | 163 |
| YSZ/Al2O3 | Bilayer | 27–377 | ×100.5–1.5 | Tensile strain and edge dislocation | 164 |
| BZY/NGO | Bilayer | 550–600 | ×102 | Dislocation | 144 |
| Ce0.8Gd0.2O2−δ/Si3N4 | Bilayer | 300–500 | ×10 | Strain | 165 |
| GDC/Si3N4/Si | Bilayer | 327 | ×102 | Strain | 145 |
| YSZ/LAO, NGO, MgO, Al2O3 | Bilayer | 300–500 | ×10−2 | Film texture and grain boundary density | 166 |
| SDC/STO/BZO/MgO | Bilayer | 600 | ×3 | Strain | 146 |
| YSZ/Y2O3, Sc2O3, Lu2O3 | Multilayer | 610 | ×2–10 | Strain | 167 |
| YSZ/CZO | Multilayer | 650 | Almost 0 | Strain | 168 |
| GDC/ZrO2 | Multilayer | 377 | 18 (compared to YSZ single crystal) | Strain and dislocation | 169 |
| CSZ/Al2O3 | Multilayer | 350–700 | ×102 | Structural disorder/mismatch | 150 |
| YSZ/STO | Multilayer | 84–258 | ×108 | Atomic reconstruction | 72 |
| YSZ/Y2O3 | Multilayer | 350–700 | ×10 | Dislocation | 170 |
| YSZ/STO | Multilayer | 357–531 | — | p-Type conductivity | 171 |
| YSZ/STO | Multilayer | 100–500 | ×105 compared to YSZ/Al2O3 | Cation interdiffusion | 172 |
| (all electronic cond.) | |||||
| Ce0.8Sm0.2O2−δ (SDC)/YSZ | Multilayer | 400–800 | ×10 compared to SDC | Tensile strain | 173 |
| ×10 compared to YSZ | |||||
| YSZ/STO | Multilayer | Room T–500 | ×102 | Cation interdiffusion | 174 |
| Sc2O3/YSZ | Multilayer | 420–780 | ×105 total | Strain | 108 |
| YSZ/CeO2 | Multilayer | 400–700 | Almost 0 | Strain | 125 |
| ZrO2/STO | Multilayer | — | Fluorite not stable for ε > 0.05 | Strain | 151 |
| SrTiO3/YSZ/SrTiO3 | Multilayer | 227 | Almost ×103 | Defect redistribution | 175 |
| YSZ/Gd2Zr2O7 | Multilayer | 250–475 | ×102 | Tensile strain | 176 |
| YSZ/Y2O3 | Multilayer | 520 | ×102 | Strain | 177 |
| Sc2O3/YSZ | Multilayer | 420–780 | ×105 total | Strain | 108 |
| CeO2/Y2O3, La2O3, Gd2O3 | Multilayer | 610–1000 | ×10 | Lattice mismatch strain | 149 |
| GDC/YSZ | VAN | 600 | ×2 | Strain and fast ionic transport rates | 46 |
| YSZ/SDC, STO | VAN | 360 | ×102 | Strain and structurally mismatched vertical interface | 152 |
| SrTiO3/SDC | VAN | 400 | ×10 | High crystallinity nanopillars | 153 |
| YSZ/STO | Composite | 127–727 | ×101.5 @ 1000 K ×103.5 @ 400 K | Lattice strain | 53 |
| SDC/LiNaCO3 | Composite | 300–650 | ×10 | Surface properties and electrolyte thickness | 156 |
| CeO2/M2O3 | Composite | 227 | ×104 | Strain | 157 |
| YSZ/STO | Composite | 200–600 | ×10−2 | Defect formation and transport, p-type conductivity | 178 |
| YSZ | Calculation | 137–927 | ×10 | Strain | 179 |
| BZY/MgO | Calculation | 300–700 | Almost 0 | Segregation tendency, but saturated | 180 |
| ZrO2/Cr2O3 | Calculation | 527–927 | — | Defect redistribution | 181 |
Many multilayer structured oxides have been reported to show high conductivity and low activation energies due to the presence of heterointerfaces. For example, Azad et al.169 synthesized a multilayer structure consisting of Gd2O3-doped CeO2 and ZrO2 films grown on Al2O3(0001) using MBE and reported that the conductivity of the 10-layer GDC/ZrO2 heterostructure was 2.9 × 10−4 S cm−1 at 650 K in air, which was an ∼18 times enhancement as compared with YSZ single crystals. In another example, Peters et al.150 found that in CSZ (ZrO2 + 8.7 mol% CaO)/Al2O3 multilayer systems with coherent interfaces, the activation energy for ionic conduction was decreased from 1.47 eV to 0.96 eV as the number of CSZ layers increased from 1 to 5. There was a two orders of magnitude enhancement in the total conductivity when the individual CSZ layers got thinner from 0.78 μm to 40 nm (Fig. 6a and b). Sillassen et al.159 deposited YSZ thin films onto MgO(110)(111) substrates and observed remarkable conductivity enhancements; the ionic conductivity of the resulting YSZ thin film was increased to 1 S cm−1 at 500 °C, nearly 3.5 orders of magnitude higher than that of bulk YSZ.
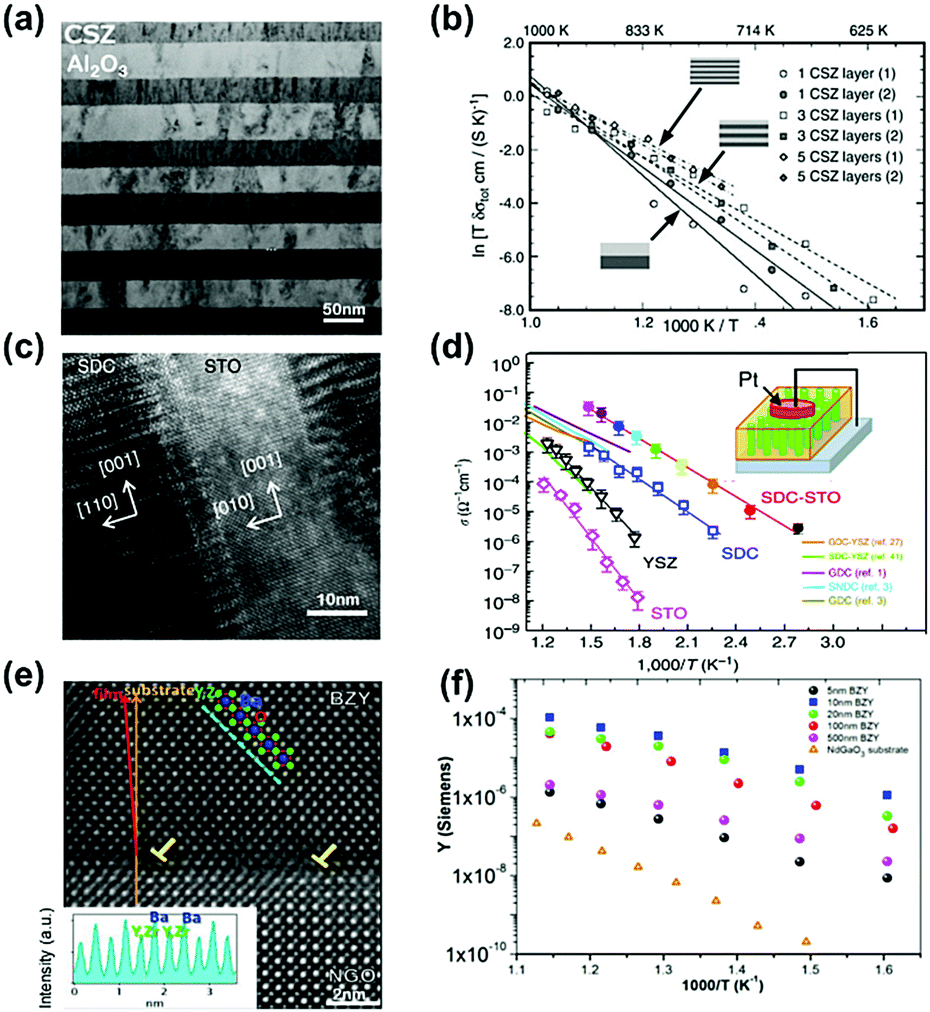 | ||
| Fig. 6 (a) TEM micrograph of a CSZ/Al2O3 multilayer system after heat treatment at 800 °C for ∼100 h. (b) Arrhenius plot of the total conductivities (σtot) measured on different CSZ/Al2O3 multilayer systems. Reprinted with permission from ref. 150. Copyright 2007 Elsevier. (c) High-resolution TEM image of a vertical SDC–STO interface in a cross-sectional view. Scale bar, 10 nm. (d) Temperature dependence of the real part of the ac conductivity (σac′) measured in the nanoscaffold SDC–STO with comparisons with the single phase thin film and literature data.153 Reprinted with permission from ref. 153. Copyright 2015 Springer Nature. (e) Z-STEM image of the BZY thin film and its interface with the NGO substrate, inset: Z-dependent intensity profile of the cations taken along the dashed line in the (−1 0 1) direction. (f) Arrhenius plot of the conductance for the whole measured set of film thicknesses.144 Reprinted with permission from ref. 144. Copyright 2014 AIP publishing. | ||
Compared with multilayer structures, vertically aligned nanocomposite (VAN) heterostructure films can potentially accommodate larger strain near the interface, creating fast ionic diffusion paths along the vertical interface.78,152 For example, Yang et al.153 prepared highly crystalline micrometer-thick vertical nanocolumns on a Nb:STO substrate using PLD (Fig. 6c) and reported that this SDC–STO VAN structure exhibited significantly enhanced oxygen ion conductivities, approaching 3 × 10−2 S cm−1 at 400 °C as recorded using scanning probe microscopy measurements. In contrast, the conductivity of SDC, YSZ, and STO single phase thin films is on the order of 10−3, 10−4, and 10−6 S cm−1, respectively, under the same conditions (Fig. 6d). Here, the activation energy for oxygen migration in the SDC–STO VAN structure (0.65 ± 0.02 eV) was also slightly lower than that of the SDC film (0.70 ± 0.04 eV). Lee et al.152 also demonstrated that a double-layered vertical YSZ–STO/SDC–STO heterostructured film prepared by PLD can provide a high ionic conductivity of ∼10−2 S cm−1 at 635 K, which was over 2 orders of magnitude higher than that of YSZ films (∼6 × 10−5 S cm−1). In addition, Su et al.46 reported that in GDC/YSZ VAN systems, two-phase strain coupling can improve the ionic conductivity along the vertical heterointerface. The ionic conductivity at 600 °C of the GDC/YSZ VAN thin film (0.96 S cm−1) was about 2 times higher than that of single phase YSZ thin films (0.41 S cm−1) and slightly larger than that of the GDC/YSZ multilayers (0.82 S cm−1), as determined from impedance spectroscopy. As a result, the corresponding single cell (Ni–YSZ|YSZ/GDC/VAN|LSC) exhibited peak power densities (PPD) of 0.488, 0.694 and 0.883 W cm−2 at 700, 750 and 800 °C, respectively.
Similarly, heterostructured proton conductors have been reported to exhibit higher proton conductivity than those of the corresponding single phases. For example, Foglietti et al.144 reported that the conductivity of a 10 nm thick film was about 2 orders of magnitude larger than that of a 500 nm thick film (Fig. 6e and f) and the average in-plane conductivity of the 10 nm thick film was 20 S cm−1 at 550–600 °C, as demonstrated by a series of strained BaZr0.8Y0.2O3−δ (BYZ) thin films of different thicknesses deposited on NdGaO3 (NGO) substrates using PLD. The enhancement in proton conductivity was attributed to fast ionic transport induced by dislocations along the heterointerfaces between dissimilar materials. In another example, Yang et al.182 characterized the electrochemical activities and transport properties of BaZr0.8Y0.2O3−δ (BZY)/NdGaO3 (NGO) heterointerfaces using electrochemical strain microscopy (ESM), demonstrating that the mismatch dislocation network reduced the activation energy (0.1 eV for the 20 nm thick film) and induced a 2D transport phenomenon, which will be discussed in detail in Section 3.2.2.
3.2 Impacts of heterointerfaces on the ionic conductivity
Due to the extraordinary effects of heterointerfaces in various electrolyte systems, great efforts have been devoted to gaining a deeper understanding of the detailed mechanisms of ionic conductivity enhancement,183 including the effect of strain, defects (such as dislocations), and the space-charge region near the heterointerfaces.Tensile elastic strain is generally believed to accelerate ionic transport by decreasing the energy barrier of migration or by reducing the activation enthalpy for charge-carrier migration.52,53,157,187 For example, Kushima et al.53 investigated changes in oxygen ion migration barriers in YSZ under biaxial lattice strain using DFT and the nudged elastic band (NEB) method. They found that the oxygen migration pathways may diversify and coexist depending on the local distribution of defects such as vacancies and dopant cations along the migration path. Here, the researchers reported that at lower stress levels, tensile strain can expand the migration space, increase the distance between cations (Δ(Zr–Zr)) among the migration path in this configuration and weaken O–C bonds (Fig. 7), all of which can decrease vacancy migration barriers and exponentially increase the oxygen diffusivity. Specifically, under 4% biaxial tensile strain, a maximum 6.8 × 103 times diffusivity enhancement was obtained at 400 K for YSZ layers.
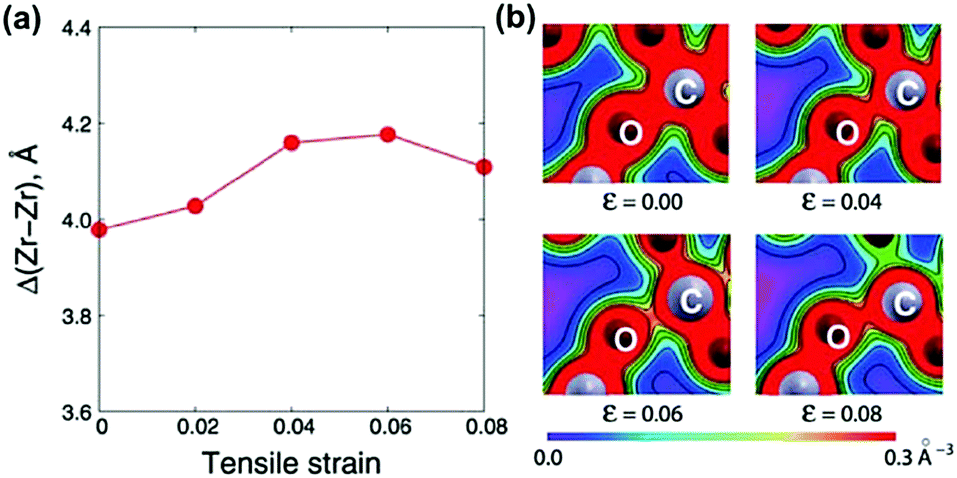 | ||
| Fig. 7 (a) Change in the cation–cation distance Δ(Zr–Zr) as a function of tensile strain from 0.00 to 0.08; and (b) simulated bond strengths for O–C in the migration process under biaxial lattice strain. Reprinted with permission from ref. 53. Copyright 2010 the RSC. | ||
Furthermore, Souza et al.157 used quantitative equations to describe the impacts of stress on the activation enthalpy and ionic conductivity in fluorite structured solid solutions such as ZrO2–M2O3 and CeO2–M2O3. Here, the researchers used static atomistic simulations to calculate the energy change along oxygen vacancy migrations in strained CeO2 based on empirical pair-potentials (EPP) and evaluated the correlation between conductivity and strain using the equations:
 | (1) |
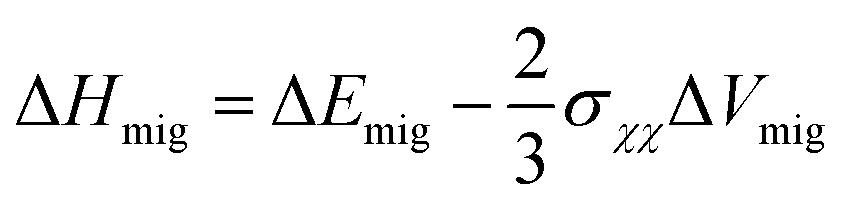 | (2) |
Based on the model described by eqn (1) and (2), Korte et al.167 further established a computational model by simulating elastically deformable crystallites. And based on grain boundary dependent strain relaxation in solid electrolyte films, their equation can adequately approximate the relationship between the total conductivity and structural parameters when the lattice mismatches are below 10%:
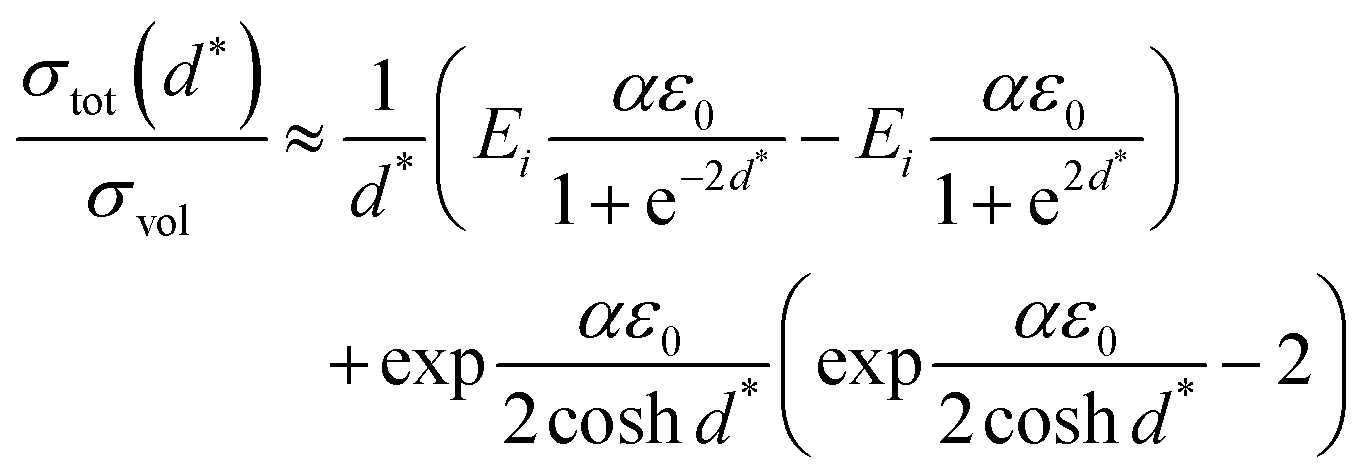 | (3) |
Subsequent experimental measurements were also consistent with these theoretical models and highlighted the dependence of ionic conductivity on lattice strain.108,159,189 Schichtel et al.108,189 systematically investigated the influence of interfacial stain effects at semi-coherent and coherent heterointerfaces such as YSZ(111)/RE2O3(111)/Al2O3(0001) and Al2O3(0001)/Sc2O3(111)/YSZ(111) on ionic transport along the interface. In these studies, the researchers reported that the conductivity decreased with increasing interfacial density and attributed this to the compressive strain in YSZ at the interface. This was also in agreement with model (3). Furthermore, Sillassen et al.159 suggested that eqn (3) more accurately explained strain effects on continuous, flat and coherent interfaces (or with slight dislocations).
Overall, interfacial tensile strain generally increases the ionic conductivity and decreases ionic migration barriers whereas compressive strain has the opposite effect. However, lattice mismatch for heterointerfaces can lead to tensile strain on one side and compressive strain on the other, in which heterointerfaces coupled with two opposite effects simultaneously can face uncertainty and complexity. Therefore, the aim of heterointerface engineering is to maximize positive effects through the optimal design of the morphology and selection of material systems.
 | ||
| Fig. 8 (a) HRTEM micrograph of a Sc2O3/YSZ heterointerface with a regular network of dislocations (white marks).108 Reprinted from ref. 108 with permission from RSC, copyright 2013. (b) HRTEM micrograph of a YSZ/MgO interface with mismatch dislocations marked by T symbols. The dotted rectangle exhibits a Burgers circuit in which the difference between atomic steps on each side represents the quantity of dislocations in each of the two orientations.159 Reprinted from ref. 159 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2010. | ||
Recent studies also suggested that dislocations can critically impact the ionic transport properties.158,159,182,191–193 For example, Sillassen et al.159 found that the conductivity of YSZ/MgO systems with 58 nm-thick YSZ films can be enhanced by almost 3.5 orders of magnitude with a large lattice mismatch of −18.01% between the YSZ and MgO lattices. They assumed that the dislocation network along the semi-coherent YSZ/MgO interface (Fig. 8b) may play a role of fast transport pathways for oxygen diffusion.
In another study on the MgO/SiTiO3 system, Dholabhai et al.158 used atomistic simulations with LAMMPS and proposed that dislocation networks in the TiO2-terminated interface can reduce oxygen vacancy formation energies and accelerate ionic transport. Here, the researchers reported that oxygen vacancies preferred to form along dislocations lines with lower formation energies, which was similar to those in metallic nanocomposites.194 They also suggested that TiO2-terminated interfaces can exhibit analogous pipe diffusion mechanisms to metallic nanocomposites,195 which were responsible for remarkable ionic conductivities. In comparison, SrO-terminated interfaces possessed oxygen vacancies that were concentrated on terraces and were inhibited from migrating.
Based on TEM analysis and calculation, Foglietti et al.144 suggested that the enhanced in-plane proton conductivity was due likely to the high densities of defects, particularly dislocations, at the interface of the film. Similarly, Yang et al.182 proposed that the 2D dislocation network near the interfacial region can facilitate the proton conductivity by lowering the activation energies (Fig. 9a). STEM characterization identified mismatch dislocations spaced −10 unit cells apart at the BZY–NGO interface. Here, the atomic arrangements were highly disturbed with more oxygen vacancies along the dislocation network, allowing the redistribution of defects to induce charge imbalance and attract proton incorporation near the interface. In addition, they reported that the local distortion of oxygen octahedra can reduce the trapping effect, allowing the 2D dislocation network at the BZY/NGO interface to provide easily accessible sites to anchor H+ and decrease activation energies.
 | ||
| Fig. 9 (a) Schematic of the dislocation network at the interface of a BZY thin film and NGO substrate based on STEM characterization results.182 (b) Map of oxygen vacancy formation energies around one dislocation core.197 Reprinted with permission from ref. 182 and 197, both with permission from American Chemical Society, 2015. | ||
Despite many studies reporting positive impacts of dislocations on the ionic conductivity, some have also reported negative impacts. For example, Sun et al.196 used atomic simulations and found that strain fields can enrich the concentration of trivalent dopant cations and oxygen defects near the 1/2 〈110〉 {100} edge dislocations in CeO2 with depletion at the other edge. In the enriched zone, strong dopant–vacancy and vacancy–vacancy interactions can diminish oxide ion diffusion, whereas in the depleted zone, the lack of charge carriers can decrease the conductivity. In another study, Marrocchelli et al.197 found that the redistribution of defects resulted in overlapping electrostatic fields surrounding the dislocation cores with positive charge, which further influenced the electrical and ionic conductivity. And although the oxygen vacancy formation energies were lower than 2 eV at sites close to the dislocation cores of 〈100〉 {011} as compared with the SrTiO3 bulk (Fig. 9b), these researchers did not find any significant enhancement in the oxide ion mobility. Similarly, Schichtel et al.108 reported the negligible role of dislocations in the ionic conductivity along the heterointerface of Al2O3(0001)/Sc2O3(111)/YSZ(111). Yep et al.198 also reported that edge dislocations induced by kinking on the (220) slip plane near the YSZ/quartz interface can inhibit oxygen ion migration along the interfacial region. Overall, the complex role of dislocations in the ionic diffusion kinetics is still not well understood and requires further systematic research.
mol%-doped YSZ can be as narrow as 1 Å at 500 °C according to the Gouy–Chapman conditions. The space-charge effect in undoped conductors or conductors with low dopant concentrations can be described using the Poisson–Boltzmann equation:201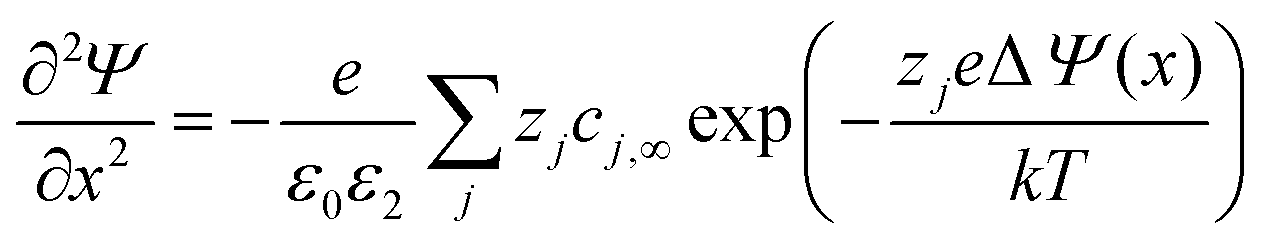 | (4) |
The positive impact of space-charge effects on the ionic conductivity at interfacial regions has been highlighted in several studies.43,204,205 For example, Kosacki et al.43 proposed that due to space-charge effects, the ionic conductivity of 1.6 nm-thick YSZ/MgO interface layers can reach ∼2 S cm−1 at 700 °C, which was 3 orders of magnitude larger than that of the lattice. In another example, Karthikeyan et al.205 reported that in YSZ/MgO, the ionic conductivity increased as the film thickness decreased. They also found that the high activation energy indicated that the space-charge effect at the interface and grain boundary dominated the conductivity enhancements. Despite these promising results, however, a straightforward relationship between the space-charge effect and ionic enhancement still needs to be further investigated. Thus far, there were only a few reports about tuning the space-charge potential and defining the vacancy accumulation regions clearly.206–208 For example, Gäbel et al.207 reported that a nanocrystalline GDC thin film on a MgO substrate can exhibit relatively high ionic conductivities (2.7 × 10−4 S m−1 S at 700 °C in 10−5 bar O2) with a low space-charge potential of 0.19 ± 0.05 V, in which the researchers attributed this to the low depletion of oxygen vacancies at the grain boundaries.
Alternatively, there were studies demonstrating that the impacts of space-charge effects on conductivities are complicated. Some researchers found that the positively charged grain boundary cores can block the migration of oxygen vacancies, significantly influencing the electronic and ionic conductivities.209,210 For example, Adepalli et al.211 reported that dislocation-induced space-charge fields can enhance the n-type conductivity of SrTiO3 by 50 times at 10−5 atm p(O2) at 650 °C and decrease the p-type conductivity by 50 times and reduce the oxygen diffusion coefficient by three orders of magnitude at high oxygen pressures. In another study, Lupetin et al.212 reported that in SrTiO3, a reduction of the grain size to 30 nm can bring out a 3 orders of magnitude enhancement of the n-type conductivity and a significant decrease in the p-type conductivity and oxygen vacancy conductivity. The researchers attributed such decreases to increased space-charge effects near the interface as the particle size decreases. Similar effects have also been found for nanocrystalline ceria.213
Furthermore, Mebane et al.161 combined the Poisson–Boltzmann with the Cahn–Hilliard model to estimate space-charge effects in solid solutions with wide dopant levels. The ‘Poisson–Cahn’ theory is based on local and non-local chemical interactions and can yield activity coefficients for point defects. And in the case of heterostructured CeO2–Gd2O3, the model successfully predicted defect behaviors near the grain boundary over the entire concentration range. In addition, the model, included existing conductivity considering local dopant concentrations, covered various conditions of solid solutions, and finally gave a comprehensive prediction of both bulk and grain boundary conductivities.
In this section, we have discussed the role of heterointerfaces in electrolyte materials for SOCs. In summary, heterointerfaces can greatly influence the transfer properties of charge carriers (such as oxygen ions and protons) via various mechanisms including lattice strain, dislocation and space-charge effects. Actually, the evolution of the local lattice structure, cation arrangement, and electronic structure induced by heterointerfaces may not only affect the mass transfer, but also have a more profound influence on the primitive steps of electrode reactions, which will be discussed in detail in the next sections.
4. Heterointerfaces in oxygen electrodes of SOC devices
Due to sluggish ORR/OER activities at intermediate temperatures, the development of oxygen electrodes with high catalytic activity and stability is essential for high performance SOCs.214–218 To address this, heterointerface engineering has recently emerged as a promising approach to optimize the oxygen electrode performance. This section will provide an overview of oxygen electrodes with heterointerfaces and the mechanisms behind the impact of the interface on the ORR/OER kinetics.ORR/OER processes on mixed ionic and electronic conductor (MIEC) electrode materials involve several elementary steps. 215 The detailed ORR process includes the following steps (Fig. 10):
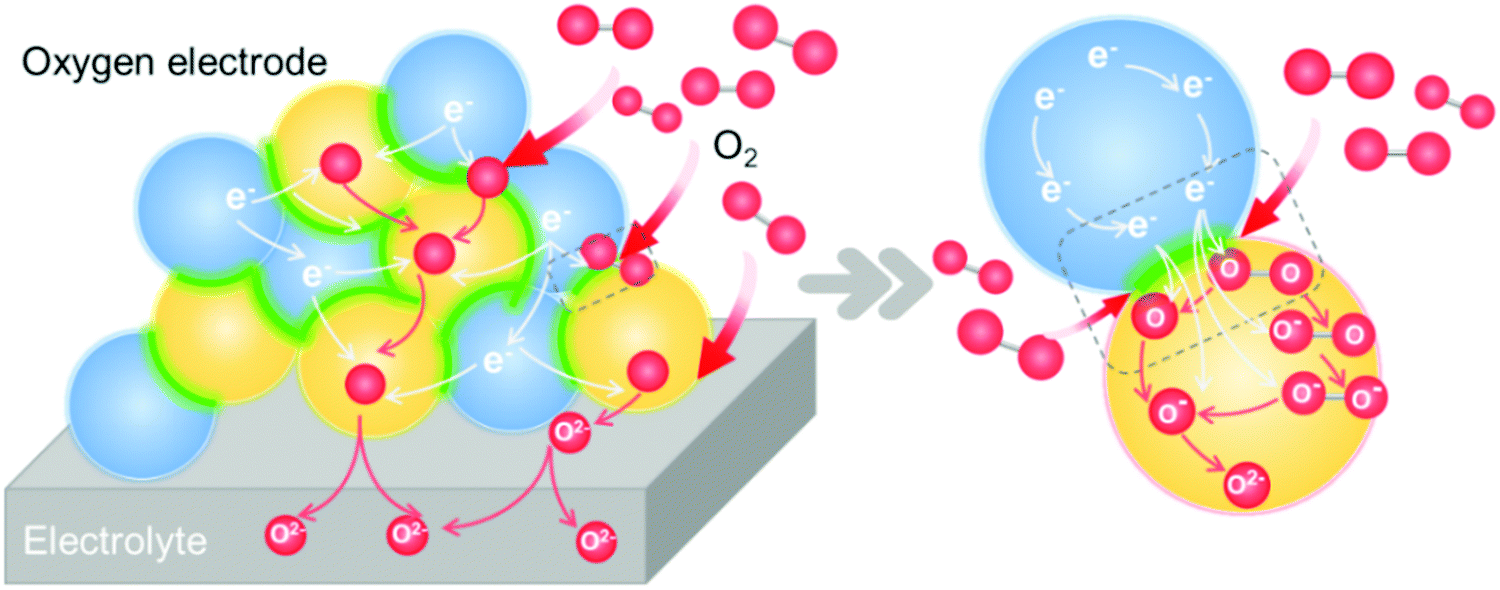 | ||
| Fig. 10 Schematic of the elementary steps of an oxygen reduction reaction process on a heterostructured oxygen electrode. | ||
① Gas diffusion, O2(gas) → O2(near surface);
② Adsorption, O2(near surface) → O2(ads);
③ Dissociation, O2(ads) → 2O(ads);
④ Electron transport, O2(ads) + e− → O2−(ads), O2−(ads) + e− → O22−(ads), O22−(ads) → 2O−(ads), O(ads) + e− → O−(ads), O−(ads) + e− → O2−(ads);
⑤ 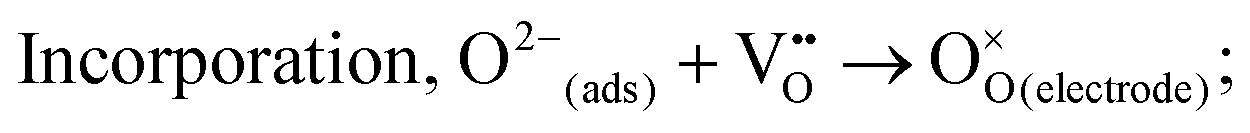
⑥ Diffusion of O2− to the electrode/electrolyte interface;
⑦ O2− transport across the electrode/electrolyte interface.
Many of the elementary reaction steps above involve the transportation of ionic and electronic species. Therefore, optimal ionic and electronic conductivities as well as surface exchange coefficients are desirable for oxygen electrodes. Furthermore, oxygen electrode materials should meet additional requirements such as high chemical stability, compatibility with electrolyte materials, thermodynamic properties matching other materials, etc.
4.1 Performance of SOC oxygen electrodes with heterointerfaces
Many theoretical and experimental studies have demonstrated that heterogeneous structures can exhibit much faster oxygen surface exchange and diffusion kinetics compared with the single-phase counterparts (Fig. 10). To be specific, oxygen electrodes with heterointerfaces that exhibit high performance include (La,Sr)MnO3(LSM)/LaNiO3,218 LSM/LaAlO3,192 Er0.4Bi0.6O3−δ/La0.8Sr0.2MnO3−δ,219 La0.85Sr0.15MnO3/La0.6Sr0.4Co0.2Fe0.8O3−δ,220 Ba0.5Sr0.5TiO3/La0.67Sr0.33MnO3,221 LSC113/LSC214,48,222 Nd0.8Sr1.2CoO4±δ (NSC214)/Nd0.5Sr0.5CoO3−δ (NSC113),223 SrCo0.2Fe0.6Ni0.2O3 (SCFN)/La0.8Sr0.2Co0.2Fe0.8O3−δ (LSCF),224 La0.6Sr0.4Co0.2Fe0.8O3−δ/CeO2,225etc. and are summarized in Table 2.| Material | Geometry | T (°C) | Performance | Mechanism | Ref. |
|---|---|---|---|---|---|
| La0.8Sr0.2CoO3−δ/LaSrCoO4+δ | Multilayer | 550 | k q (from EIS) ∼103–104 enhancement | Strain, space-charge effects and an increase in electronic structure or oxygen vacancies | 48 |
| La0.8Sr0.2CoO3−δ/YSZ | Multilayer | 510 | Surface exchange coefficient ∼10 times | Sr decoration | 226 |
| (La0.5Sr0.5)2CoO4±δ/La0.6Sr0.4CoO3−δ | Multilayer | 550 | k q ∼ 3 orders of magnitude enhancement | Heterointerface | 222 |
| SrTi1−xFeO3/YSZ | Multilayer | 345 | Band gap increased from 2.5 ± 0.5 eV to 3.6 ± 0.6 eV | Surface electronic structure | 227 |
| La1−xSrxCoO3−δ/SrTiO3 | Multilayer | 280–475 | Tensile strain with faster surface exchange (∼4 times) and diffusion (∼10 times) | Strain | 228 |
| La1−xSrxCoO3−δ/LaAlO3 | |||||
| La0.8Sr0.2CoO3−δ/La0.8Sr0.2MnO3−δ | Multilayer | 550 | k q (cm s−1) (from EIS) ∼10–102 enhancement | Mn substitution (cation diffusion) | 79 |
| La0.8Sr0.2CoO3/(La0.5Sr0.5)2CoO4 | Multilayer | 250 | Energy gap decreased from 1.4 eV (LSC113) and 2.6 eV (LSC214) to 1.0 eV (LSC113/214) | Electron donation and transfer of oxygen vacancies across the heterointerface | 229 |
| (La,Sr)2CoO4±δ/La1−xSrxMO3−δ | Multilayer | 550 | Surface exchange coefficients enhanced by 102 times | Sr segregation | 230 |
| SrCoOx/(LaAlO3–(SrAl0.5Ta0.5O3)0.7) or SrTiO3 or DyScO3 or GdScO3 or KTaO3) | Multilayer computation | 300 | Oxygen activation energy barriers decreased by ∼30% | Strain | 231 |
| La0.6Sr0.4CoO3−δ/Ce0.9Gd0.1O1.95 | Multilayer | 700 | Slight increase in 18O ratio | Extended TPB | 232 |
| La0.8Sr0.2MnO3/SrTiO3 | Multilayer | 600 | Oxygen ion diffusion enhanced by 103 | Dislocation and strain | 192 |
| La0.8Sr0.2MnO3/LaAlO3 | |||||
| La0.8Sr0.2CoO3−δ/Nd2NiO4+δ | Multilayer | 300–415 | Oxygen exchange kinetics are much slower | Charge transfer | 233 |
| La0.6Sr0.4Co0.2Fe0.8O3/PrxCe1−xO2 | Multilayer | 600 | PrO2/LSCF: nearly 6 times faster | Inducing higher oxygen vacancy concentrations | 234 |
| CeO2/LSC: slight enhancement | |||||
| La0.6Sr0.4CoO3−δ/LaSrCoO4±δ | Multilayer | 600–850 | k q ∼ 5–10 times that of the single phase | Sr enrichment at the interfacial region and stabilized against detrimental Sr segregation | 106 |
| Nd0.5Sr0.5CoO3−δ/Nd0.8Sr1.2CoO4±δ | Multilayer | 500 | k q ∼ 102–103 times that of the single phase | Enriched Sr concentration and decrease in the valence state of Co | 107 |
| La0.65Sr0.35MnO3/SrTi0.2Fe0.8O3 | Multilayer computation | — | Bandgap values (1 and 2 eV for LSM and STF) decreased to 0.3 and 1 eV for LSM and STF | Electronic structure, strain, defects | 235 |
| La0.8Sr0.2CoO3/(La0.5Sr0.5)2CoO4 | Multilayer | 300 | 103 times increase in charge transfer rates to oxygen | Electronic structure | 56 |
| La0.8Sr0.2CoO3/HfO2 decoration | Multilayer | 530 | k q ∼ 30 times enhancement | Optimum surface oxygen vacancy concentration | 236 |
| La0.6Sr0.4CoO3−δ/Co2O3 decoration | Multilayer | 450 | Surface exchange resistance reduction by 13% per laser pulse | Reactivation of LSC surface | |
| La0.8Sr0.2CoO3−δ/LaSrCoO4+δ | VAN | 320–400 | k q ∼ 10 times enhancement | Electronic activation and more stable cation composition at the interface | 77 |
| La0.6Sr0.4CoO3−δ/LaSrCoO4+δ | Composite | 500 | Exchange coefficient k* (cm s−1) (from SIMS) enhanced by 103 | Fast oxygen incorporation paths along the heterointerface boundary | 216 |
| La0.6Sr0.4CoO3−δ/LaSrCoO4+δ | Composite | 500 | Exchange coefficient σe (S cm−2) (from EIS) 10–101.5 | Heterointerface enhanced the oxygen catalytic reaction | 237 |
| Fe2O3–LaCePrOx | Composite | 400–600 | Ionic conductivity increased by 10 times | Heterogeneity and interfacial conduction effects | 238 |
| SP Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125/DP Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125 | Composite | 550–700 | ASR ∼3 times lower | Cooperative DP/SP effects | 239 |
| Er0.4Bi1.6O3−δB/La0.8Sr0.2MnO3−δ | Composite | 550–650 | R p/5 | Particle sizes | 219 |
| La0.6Sr0.4Fe0.8Co0.2O3−δ/Gd0.2Ce0.8O1.9−δ–ZrO2 | Skeleton/surface coating | 800 | ∼25% degradation rates after overcoating in 1100 hours of operation | Synergistic function of porosity, mixed conductivity and suppressed Sr enrichment | 240 |
| La0.6Sr0.4CoO3−δ/ZrO2 | Skeleton/surface coating | 700 | Lower polarization resistances and 19 times slower degradation rates over 4000 h | Porosity, mixed conductivity and suppressed Sr enrichment | 241 |
| La0.58Sr0.4Co0.2Fe0.8O3−δ/GDC | Skeleton/surface coating | 900 | k chem ∼ 6 times higher than pure LSCF | Facilitating dissociative adsorption of O2 | 242 |
| La0.6Sr0.4CoO3−δ/Al2O3 | Skeleton/surface coating | 800 | Power output reduced from 1.62 W cm2 to 0.742 W cm2 | Blocking effect of excessive Al2O3 | 243 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ/MgO | Skeleton/surface coating | 650 | ASR reducing from 0.49 Ω cm2 to 0.31 Ω cm2 | Promotion of charge transfer process | 114 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ/CuO | Skeleton/surface coating | 750 | k chem ∼ 9.3 × 10−5 cm s−1, 3.5 times enhancement | Additional reactions on both the CuO surface and the LSCF–CuO-gas boundaries (3PBs) | 116 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ/NiO | Skeleton/surface coating | 800 | k q ∼ 6.9 × 10−4 cm s−1, 20 times enhancement | Promotion of oxygen incorporation process | 115 |
| (La,Sr)2FeO4−δ/La0.8Sr0.2FeO3−δ | Skeleton/surface coating | 650–800 | ORR activity enhanced by 10 times | Higher catalytic activity of LSF214 and mismatch between LSF214 and LSF113 | 244 |
| La0.8Sr0.2MnO3/Ba0.5Sr0.5Co0.8Fe0.2O3−δ | Skeleton/surface coating | 650–750 | Electrode resistances of ∼0.101, 0.049 and 0.023 Ω cm2 at 650, 700 and 750 °C respectively | Material intrinsic properties and fabrication method | 245 |
| La0.84Sr0.16MnO3−δ/Bi1.4Er0.6O3 | Skeleton/surface coating | 750 | 10–102 ionic conductivity | Effective TPB extension | 246 |
| PrSrCoMnO6−δ/La0.6Sr0.4Co0.2Fe0.8O3−δ | Skeleton/surface coating | 750 | R p of 0.093 Ω cm2 after running for 500 h with a performance enhancement of 61.7% as compared with blank LSCF | Material intrinsic properties and fabrication method | 247 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ/La0.85Sr0.15MnO3±δ | Skeleton/surface coating | 700 | Power density of 655 mW cm−2 at 700 °C | Formation of LSM(C) phase | 248 |
| (La0.6Sr0.4)0.995Co0.2Fe0.8O3−δ/La2NiO4+δ | Skeleton/surface coating | 750 | 67% increase in peak power density, a low degradation rate of 0.39% for ∼500 h | Cation segregation of LSCF and favorable acceptance by LNO of Sr/Co doping. | 249 |
| La2NiO4+δ/Ba0.5Sr0.5Co0.8Fe0.2O3−δ | Skeleton/surface coating | 600 | ASR decreased to 0.078 Ω cm2 | Shell increased TPB | 250 |
| Complete resistivity to CO2 attacks at IT | |||||
| Er0.4Bi1.6O3/La0.76Sr0.19MnO3+δ | Skeleton/surface coating | 550–750 | Power density of 162 W cm−2; excellent stability in SOFC, SOEC and reversible SOC operating models for over 200 h | Fast oxygen ion migration at the heterointerface | 6 |
| (La,Sr)CoO3−δ/(La,Sr)2CoO4+δ | Computation | 500 | 400 times faster oxygen incorporation kinetics | Anisotropy and strain | 51 |
| La0.8Sr0.2CoO3−δ/(La0.5Sr0.5)2CoO4−δ | Computation | 500–600 | Oxygen vacancy concentrations enhanced by 102–102.5 | Sr segregation | 223 |
| (La1−ySry)2CoO4±δ/La1−xSrxCoO3−δ | Computation | — | Anomalous Sr segregation | Cation diffusion | 57 |
| LaCoO3/LaAlO3 | Computation | — | (100) oriented film possessed better OER performance | Spin-state transition of Co | 40 |
Based on the compositions, heterostructured oxygen electrodes can be sorted into four categories: (1) perovskite/Ruddlesden–Popper (ABO3/A2BO4), (2) perovskites with different cations (ABO3/A′B′O3), (3) perovskite/simple metal oxides, and (4) others, which will be discussed respectively as follows.
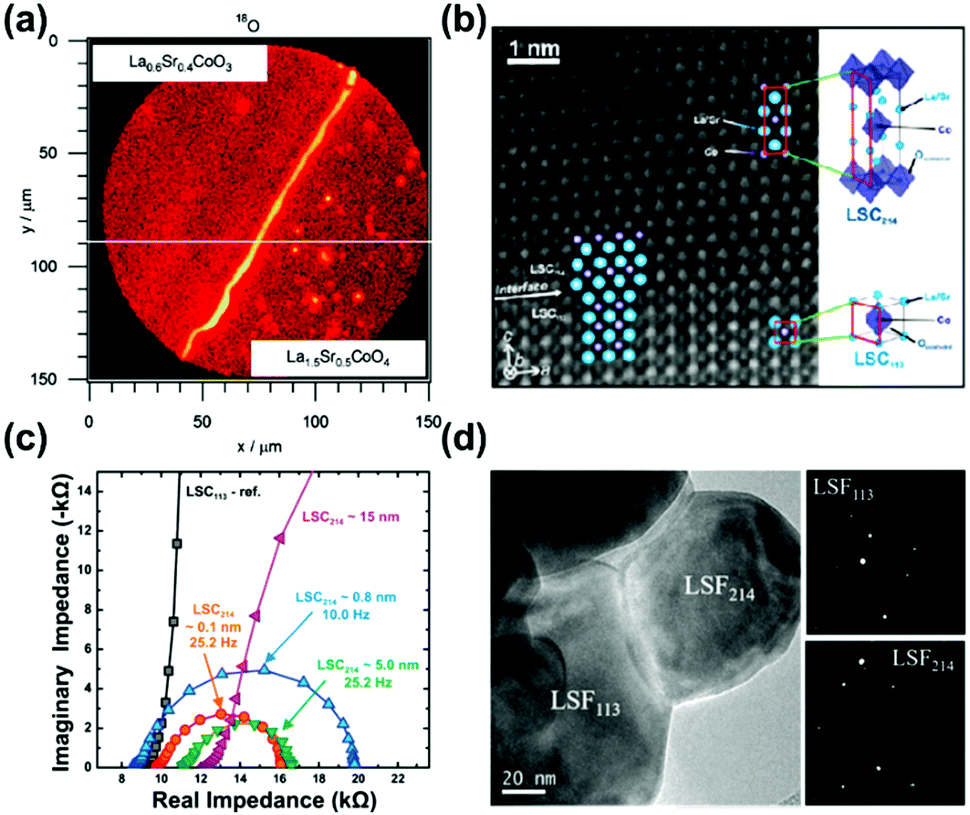 | ||
| Fig. 11 (a) Mapping profile of 18O signals by SIMS at the surface above LSC113/LSC214/CGO after oxygen isotope exchange.47 (b) HAAADF STEM micrograph of the LSC113/LSC214 interface;48 (c) EIS results of the microelectrodes (∼200 μm) for LSC113 films with ∼0.1, ∼0.8, ∼5 and ∼15 nm LSC214 surface decoration and the LSC113 reference at 550 °C at 1% p(O2).48 (d) TEM image and selected area electron diffraction of a LSC113/LSF214 particle.244 Reproduced from ref. 47, 48 and 244 with permission from the Electrochemical Society, copyright 2008; and American Chemical Society publications, copyright 2010 and 2017, respectively. | ||
The ultra-fast oxygen exchange kinetics near the interface of LSC113/214 was confirmed by Crumlin et al.48 using epitaxial La0.8Sr0.2CoO3−δ (LSC113) thin films with partial decoration of (La0.5Sr0.5)2CoO4±δ (LSC214) (Fig. 11b). Here, the researchers reported that the LSC214 decorated LSC113 presented a 3–4 orders of magnitude enhancement in the surface exchange coefficient (kq) compared with the LSC113 single phase reference, reaching as high as ∼3 × 10−6 cm s−1 in 1 atm O2 at 550 °C (Fig. 11c). Furthermore, Lee et al.230 compared the exchange coefficients for LSC113/LSC214 and LSCF113/LSC214 heterostructures and reported that in 1 atm O2 at 550 °C, both LSCF113/LSC214 (∼7 × 10−8 cm s−1) and LSC113/LSC214 (∼2 × 10−7 cm s−1) exhibited significantly enhanced kq values relative to the LSCF113 reference (∼3 × 10−8 cm s−1) and the LSC113 reference (∼5 × 10−9 cm s−1), respectively.
Recently, Zhao et al.214 also reported that bilayered LSC113/LSC214 was able to maintain superior ORR activities at 873–1173 K relative to single-phase LSC113. At 973 K in air, kq of LSC113/LSC214 was 2.5 × 10−5 cm s−1, which was 10 times higher than that of the LSC113 reference. Moreover, in another study conducted by the same group, Zheng et al.11 fabricated a series of Nd0.5Sr0.5CoO3−δ/Nd0.8Sr1.2CoO4±δ multilayers with the same total thickness and different numbers of interfaces. By conducting 18O isotope exchange experiments (97.1% 18O isotope enriched, 773 K, 15 s) and utilizing SIMS, the researchers found that the 18O− signal strongly concentrated along the NSC214/NSC113 interface. With the increased layers of the heterostructure, kq showed a significant enhancement. Specifically, the bilayer NSC214/113 film (∼5.5 × 10−8 cm s−1) exhibited a ∼41 times higher kq value than that of the NSC113 reference (∼1.3 × 10−9 cm s−1), whereas the 18-layered NSC214/113 film (∼2.9 × 10−7 cm s−1) exhibited a 165 times enhancement.
Hong et al.244 used the infiltration method to fabricate a heterostructured (La,Sr)2FeO4−δ (LSF214)–La0.8Sr0.2FeO3−δ (LSF113) electrode (Fig. 11d). After repeated infiltration of LSF214 onto a LSF113 skeleton (4 times), numerous LSF214 nano-islands were generated, and its chemical surface exchange coefficient (kchem) increased from 1 × 10−5 cm s−1 to 4 × 10−4 cm s−1 accordingly, as calculated by ECR experiments at 750 °C.
Aside from bilayer or multilayer structures, Ma et al.77 also fabricated VAN structured LSC113/LSC214 oxygen electrodes that were coherently grown onto STO(001) and YSZ/GDC(001) substrates using PLD. Results from high resolution STEM-EDX verified the separation of LSC113 and LSC214 columns in near-vertical orientation with a ∼300 nm average size of each column. Here, the ASR of the VAN thin-film electrode (∼2 × 104 Ω cm2) was only ∼10% of LSC113 (∼1.6 × 105 Ω cm2) and LSC214 (∼3.5 × 105 Ω cm2) single phase electrodes in air at 320 °C.
ABO3/A2BO4 electrodes with different elemental composition of the ABO3 and A2BO4 components have also been reported to exhibit enhanced performance. For example, Zhang et al.249 demonstrated that the infiltration of La2NiO4+δ (LNO) into (La0.6Sr0.4)0.995Co0.2Fe0.8O3−δ (LSCF) can decrease the ASR value from 1.34 Ω cm2 to 0.042 Ω cm2 at 750 °C with the activation energy changing from 1.38 eV to 1.06 eV. Here, the resulting cell with the LSCF–LNO cathode demonstrated a 67% increase in peak power density (697 mW cm−2) relative to the cell with LSCF at 750 °C. And after long-term operation at a constant current density of 250 mA cm−2, the degradation rate was reported to be as low as 0.39% after 500 hours’ testing.
 | ||
| Fig. 12 (a) kq of LSC82 with a thin LSM82 coating (0.9 nm) and the LSC82 reference as a function of annealing time.79 (b) Comparison of the electrochemical activities of LSCF decorated with CeO2, PCO and PrO2.234 (c) Schematics of a LSCF backbone and conformal, dense PNM decoration as well as exsoluted PrOx nanoparticles.256 (d) Typical I–V–P curves and (e) long-term stability performance (at a constant cell voltage of 0.7 V) for Ni–YSZ anode supported cells with different cathodes: bare LSCF, PNM-, PrOx- and hybrid PNM–PrOx catalyst-decorated LSCFs with 3% humidified H2 as the fuel and air as the oxidant at 750 °C.256 Reproduced from ref. 79, 234 and 256 with permission from American Chemical Society, copyright 2014; American Chemical Society, copyright 2018 and RSC, copyright 2017, respectively. | ||
Lynch et al.248 also reported that the electrochemical properties of porous La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) cathodes can be enhanced through a uniform La0.85Sr0.15MnO3±δ (LSM) decorating layer using the infiltration method. The current densities improved from 0.7 A cm−2 to 1.1 A cm−2, resulting in enhanced peak power densities (655 mW cm−2 at 700 °C) in 200 hours’ operation. Results from TEM and convergent beam electron diffraction (CBED) for the aged oxygen electrodes showed that the LSCF skeleton retained its perovskite structure after operation, and the LSM surface coatings exhibited a loss in crystallinity. In another example, Zhu et al.245 fabricated a (Ba,Sr)(Co,Fe)O3 porous cathode and decorated it with a (La,Sr)MnO3 nanofilm through solution impregnation. The polarization resistance of the LSM-decorated BSCF was found to be 0.023 Ω cm2 at 750 °C in air, which was ∼20% lower than that of the undecorated cathode. The researchers also reported that the LSMO layer on the BSCF backbone prevented inherent electrode poisoning by CO2, H2O and SO2 in air, resulting in considerable stability enhancements. Moreover, Wang et al.252 reported that the infiltration of perovskite SrCo0.9Nb0.1O3−δ (SCN10) onto LSCF can lead to an 89.4% reduction in ASR, indicating enhanced ORR/OER kinetics.
Tsvetkov et al.236 conducted systematic research into the decoration of less reducible cations on perovskite LSC, including V2O5, Nb2O5, TiO2, ZrO2, HfO2 and Al2O3, and concluded that related metal oxide decoration can improve the surface exchange kinetics with the exception of V2O5. A ‘volcano’ relation between kq and the oxygen vacancy formation enthalpy (ΔHVf(MeOx)) was proposed in which optimal kq enhancement (∼30 times) was achieved through HfO2 addition after 54 h in air at 530 °C.
Furthermore, Rupp et al.253 used in situ impedance spectroscopy during pulsed laser deposition (IPLD) to investigate the electrochemical activity of LSC during the deposition of well-defined monolayer fractions of Sr-, Co- and La-oxides. Here, the researchers reported that Co2O3 decoration using one laser pulse reduced the surface exchange resistance (Rsurfexch) by 13%, whereas SrO decoration using one laser pulse (∼13 pmol Sr) increased Rsurfexch by 42 ± 6%. The researchers in this study attributed this difference to the fact that Co decoration can induce more active sites whereas Sr decoration can deactivate LSC surfaces.
The decoration of Zr-based and Ce-based oxides onto perovskite surfaces has also been reported to improve the reaction activity and operation stability of oxygen electrodes.240–242 For example, Gong et al.241 decorated LSC surfaces with less electrocatalytically active ZrO2 using atomic layer deposition (ALD) and reported that the ZrO2 overcoating gradually became porous and mixed conducting after thermal exposure, resulting in elevated ORR properties. In addition, the researchers reported that the ZrO2 coating prevented LSC nanoparticles from agglomerating and suppressed Sr enrichment at the surface during long-term operation. As a result, the nanostructured LSC–ZrO2 electrode exhibited significantly lower polarization resistance and a 19 times slower degradation rate over 4000 h at 700 °C in air as compared with pristine LSC. In another study, Gong et al.240 reported that ZrO2 coatings can reduce the degradation rate of porous LSCF–GDC to ∼25% as compared with undecorated samples during 1100 hours of operation at 800 °C. Saher et al.242 also reported that LSCF skeletons with a porous nanoparticulate GDC layer can exhibit a high surface exchange coefficient (kchem) of ∼3 × 10−4 m s−1 at 900 °C, which was 6 times higher than that of pure LSCF. Here, the researchers suggested that the decoration of GDC particles facilitated the dissociative adsorption of O2, leading to enhanced activities for the heterostructure.
Dispersed MgO,114 NiO115 and CuO116 particles have also been highlighted as promising synergistic catalysts of LSCF electrodes. For example, Yang et al.114 reported that by infiltrating MgO particles onto LSCF skeletons, the ASR of the oxygen electrode can be reduced from 0.49 Ω cm2 to 0.31 Ω cm2 at 650 °C. In addition, researchers reported that the introduction of CuO can greatly improve the surface exchange kinetics of LSCF with kchem rising from 2.6 × 10−5 cm s−1 to 9.3 × 10−5 cm s−1 at 750 °C.116 Furthermore, coatings on cathodes can also enhance the cell performance, in which cells with Ni–YSZ|YSZ/SDC|LSCF–NiO can exhibit a high peak power density of 1.031 W cm−2 at 800 °C, which is ∼1.5 times higher than that of single cells with bare LSCF as the cathode.115
Despite these promising results, however, there are also studies highlighting the fact that surface decorations with simple metal oxides may reduce the electrochemical performance. For example, Kim et al.243 modified La0.6Sr0.4CoO3-based electrodes with a Al2O3 layer using ALD and reported that excessive Al2O3 coating on LSC-based electrodes can negatively affect the power output (reduced from 1.62 to 0.742 W cm2 at 800 °C). In addition, there are also controversial evaluations concerning CoOx and SrO coatings,254,255 in which the results suggest that the surface modification process needs to be rationally controlled. Here, proper amounts of decoration can activate electrode surfaces whereas excessive coating may further block active sites and degrade the performance.
Furthermore, Chen et al.256 demonstrated that a hybrid catalyst coating consisting of a conformal PrNi0.5Mn0.5O3 (PNM) thin film and exsoluted PrOx nanoparticles on LSCF obtained using a one-step infiltration process can significantly improve the ORR kinetics and durability (Fig. 12c). The heterostructured cathode reduced the polarization resistance to ∼0.022 Ω cm2 at 750 °C, which was 1/6 that of bare LSCF cathodes. Here, the researchers reported that in full cell operation, anode-supported cells with the hybrid catalyst-coated LSCF cathode exhibited an extraordinary peak power density of ∼1.21 W cm−2 (Fig. 12d) and excellent durability at 0.7 V for 500 h (Fig. 12e). And if applied in BaZr0.1Ce0.7Y0.1Yb0.1O3-based protonic fuel cells, electrodes combining PNM with exsoluted PrOx particles can exhibit excellent electrocatalytic activity with ASRs as low as 0.052 Ω cm2 at 700 °C and operation stability under 0.7 V for ∼500 h.257 In another study, a PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) nanofibre-based cathode with PrOx nanoparticles exhibited high activity for oxygen reduction. The PPD of PBSCF–PrOx|SDC|Ni–BZCYYb reached as high as 0.37 W cm−2 at 500 °C with nearly dry methane as the fuel and air as the oxidant.258
4.2 The role of heterointerfaces in oxygen electrodes
Heterostructured oxygen electrodes demonstrate strongly enhanced performance and in the following section the mechanisms proposed in the literature regarding the effects of heterointerfaces on the oxygen electrode performance are summarized,48,229 such as strain effects,48,51,231 electronic structure reconstructions,40,56,77,200,259 cation diffusion79,223,230,260–262 and extended TPBs.4,48,232,235,263,264 These mechanisms will be discussed in detail respectively in the following sections.Researchers have proposed that tensile strain can generally increase the free spacing in crystal lattices and decrease bond energies and weaken interactions between ions,53 thus facilitating the formation of oxygen vacancies.231,267 For example, Petrie et al.231 reported that the amount of oxygen deficiency in strained SrCoO3−x can increase with x changing from 0 to 0.25 as the tensile strain increases from +1% to +4.2% (Fig. 13a). Here, the researchers used DFT and proposed that tensile strain can decrease the electronic hybridization between Co 3d and O 2p and further weaken Co–O bonds, leading to a greater intercalation formation enthalpy (Hi) and more thermodynamic oxygen instability in the tensile strained SrCoOx. The researchers also reported that a 2% tensile strain can reduce the activation energy barrier (Ea) by ∼30% and facilitate the process of oxygen diffusion. Such an increase in Hi and decrease in Ea will both result in an oxygen deficient state and further influence the electrochemical properties.
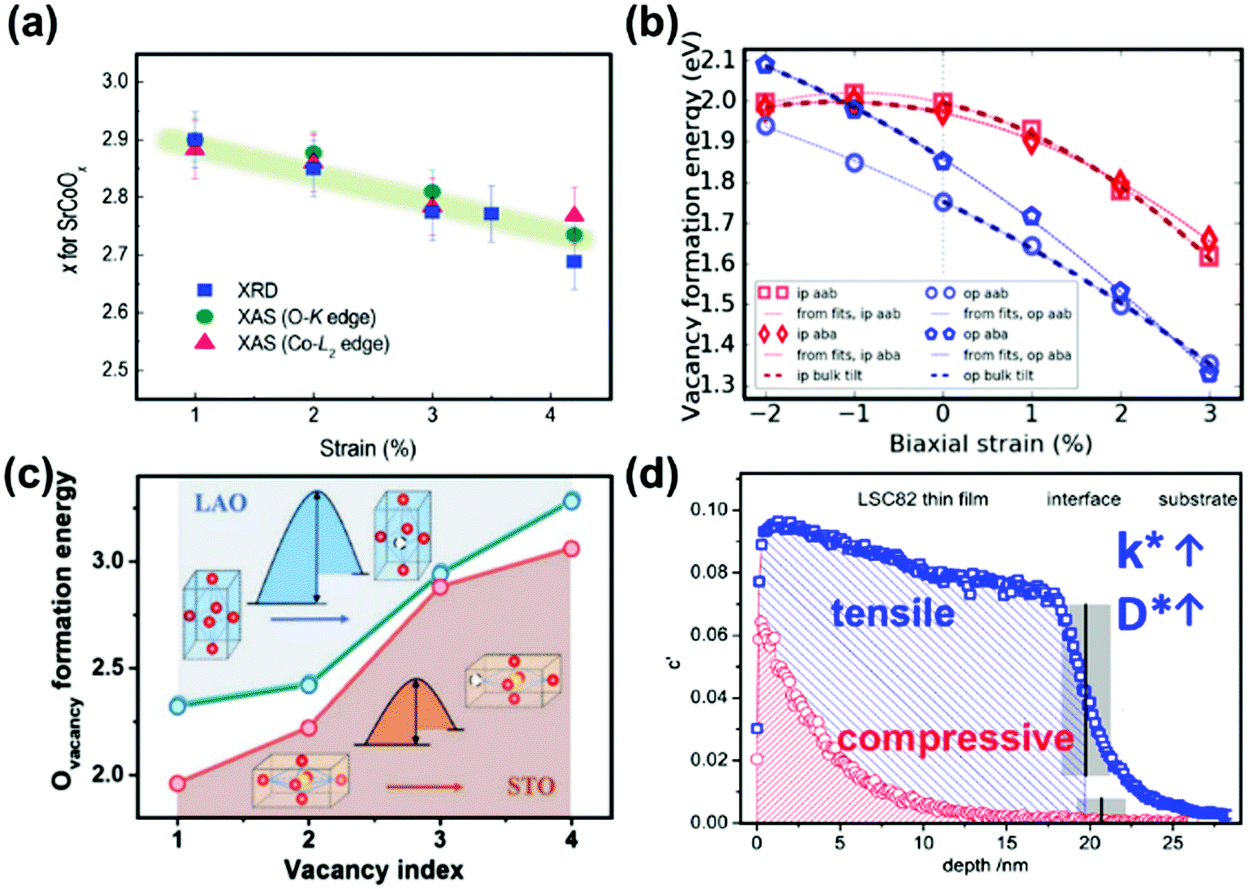 | ||
| Fig. 13 (a) Oxygen deficiency in tensile strained P-SCO determined by XRD and O-K and Co-L2 edge XAS measurements.231 (b) Vacancy formation energies of LSM as a function of strain using an oxygen chemical potential for 1173 K and 0.1 atm pO2.267 (c) Comparison of calculated Ovacancy formation energies for LSC/STO and LSC/LAO with different oxygen vacancy concentrations. The insets exhibit the smaller oxygen vacancy formation energies for LSC/STO.268 (d) Depth profiles of 18O− distributions for tensile strained (100) LSC82 thin films on STO and compressive strained LSC82 on LAO after oxygen isotope exchange at 400 °C for 5 min. Reproduced from ref. 228, 231, 267 and 268 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2016; Elsevier, copyright 2017; WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2019 and American Chemical Society, copyright 2013 (https://doi.org/10.1021/nn305987x), respectively. | ||
In another example, Morgan et al.267 used ab initio calculations to develop a quantitative model for the evaluation of strain impacts on vacancy formation energies (Fig. 13b) and proposed that tensile strain can increase lattice volumes, which caused decreased vacancy formation energies. By systemically studying strained LaMnO3, La0.75Sr0.25MnO3, LaFeO3 and La0.75Sr0.25FeO3, the researchers inferred that the vacancy formation energies for most oxides generally declined under tensile strain; a 1% increase in the biaxial strain was estimated to correspond to a ∼30–100 meV decrease in the vacancy formation energy.
Recently, Chen et al.268,269 investigated the critical impact of lattice strain on the oxygen defect chemistry and OER activity of LSC in liquid solutions through a combination of DFT calculations and experimental measurements. Here, the DFT results showed that the oxygen vacancy formation energy of tensile LSC/STO was smaller than that of LSC/LAO (Fig. 13c). As a result, LSC/STO contained more lattice oxygen vacancies than LSC/LAO. These calculations were also consistent with the larger δ values observed in HRXRD and the increased Co2+ signals in XPS characterization of LSC/STO.
Han et al.51 suggested that strain effects can influence not only the formation of oxygen vacancies, but also the incorporation and transportation of oxygen defects. By using DFT+U calculations, the researchers proved that lattice strain near the LSC113/LSC214 heterointerfacial region can enhance the concentration and mobility of oxygen vacancies, leading to highly accelerated oxygen dissociation and incorporation kinetics.265,270 Here, the researchers reported that the tensile strain in LSC113(001) (+1.9%) reduced elastic interactions along the oxygen migration paths and facilitated both oxygen adsorption and oxygen vacancy formation in neighboring regions. In addition, the researchers also reported that the tensile strain on LSC113 in the (110) direction can lead to more available spacing among crystal lattices for oxygen migration, minimizing energy barriers. Furthermore, calculations in this study demonstrated that lattice strain can contribute to 3 × 102 times of conductivity enhancement at 500 °C (considered both in LSC113 and LSC214 together).
There were also some studies indicating that strain effects on electronic structures can greatly influence the charge transfer kinetics in ORR/OER processes. For example, Cai et al.259 studied the dependence of surface electronic structures on strain states in epitaxial La1−xSrxCoO3−δ (LSC) thin films with STO and LAO substrates. Results from in situ STM/STS revealed an electronic structural transformation from a semiconducting state with an energy gap of 0.8–1.5 eV at room temperature to a metallic-like state with no energy gaps at 200–300 °C. The researchers also found that in tensile strained LSC, the electronic density around the Fermi level was strengthened following this transition, facilitating charge transfer in the ORR. And by collaborating with Kubicek et al.,228 these researchers further demonstrated that the surface exchange coefficient (k*) and diffusion coefficient (D*) of the tensile strain of LSC–STO were ∼4 and ∼10 times larger than that of the compressive strain of LSC–LAO, respectively (Fig. 13d).
In a recent review article, Hwang et al.271 discussed the impacts of lattice strain on the electronic structure and electrocatalytic activity of perovskite oxides. They proposed that lattice strain can change the level of octahedral rotation and structural deviation as well as the bond length of metal-oxides, thus modifying the oxide electronic structures through 3d orbital occupancy and M 3d–O 2p orbital overlap.272,273 As the energy gap between M 3d and O 2p centers was considered as a quantitative descriptor of the electronic changes, the lattice strain may therefore have broad effects on not only the surface reactivity such as the oxygen adsorption and dissociation energy, but also the bulk defect energetics such as the oxygen vacancy formation and migration energy.
Zhang et al.249 used TEM/EDAX to observe Sr, Fe and Co signal peaks on LNO nanoparticles in LNO-infiltrated LSCF cathodes after long term testing, revealing the occurrence of the cation segregation phenomenon (Fig. 14a). Here, the researchers reported that the segregation of highly-active cobaltic oxides accelerated oxygen surface exchange and improved the electrocatalytic activity. Furthermore, the Sr diffusion increased the concentration of electron holes and enhanced the charge transport process. These resulted in a 67% increase in power output over long-term operation.
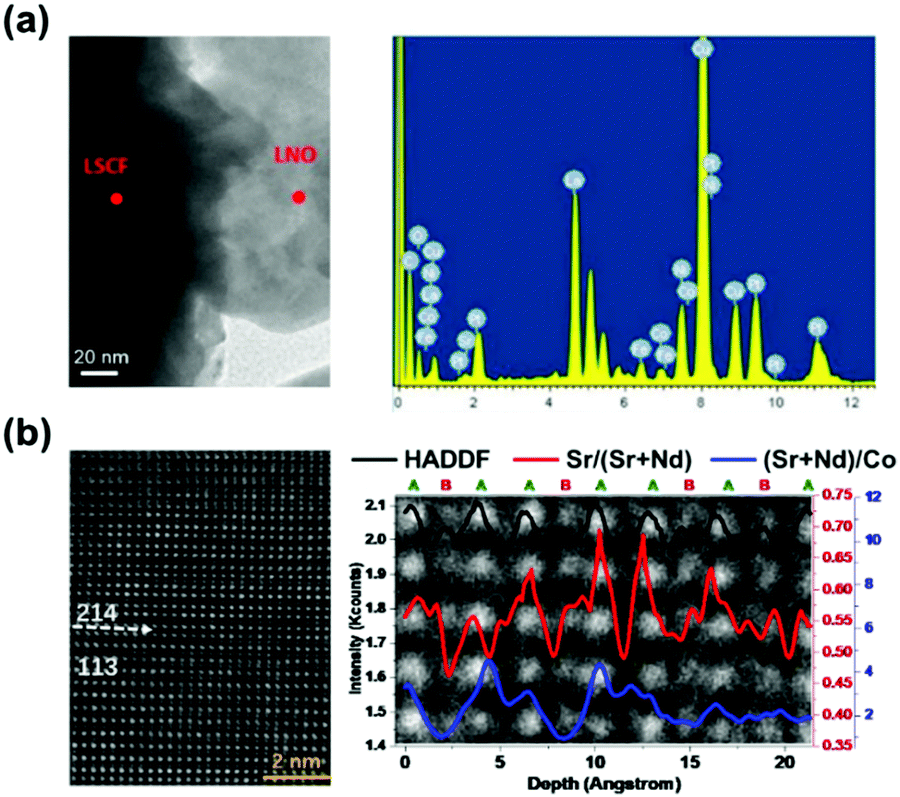 | ||
| Fig. 14 (a) TEM image and EDAX spectra of LNO-infiltrated LSCF particles after 1000 h operation.249 (b) HADDF-STEM micrograph of the NSC113/NSC214 interface of the NSC214/113-17L sample and ratios of Sr/(Sr + Nd) and (Sr + Nd)/Co along the (001) direction.107 Reproduced from ref. 107 and 249 with permission from Elsevier, copyright 2014 and 2018, respectively. | ||
Researchers have attempted to uncover the driving forces of cation segregation at heterointerfaces. For example, Zheng et al.107 used HADDF-STEM to obtain direct evidence of cation segregation in the interfacial region of NSC113/214. According to their results, the stacking sequence of atoms along the c-axis was in the order of perovskite (Nd0.5Sr0.5CoO3−δ) and Ruddlesden–Popper (Nd0.8Sr1.2CoO4±δ), and the (Sr + Nd)/Co ratio conformed to the stoichiometry, respectively. However, the Sr/(Sr + Nd) ratio was found to be enhanced obviously at the interfacial region, revealing that Sr strongly segregated near the NSC113/214 interface (Fig. 14b). The researchers also used DFT calculations to reveal that E(SrNd) (the energy required to replace a surface Nd3+ cation with a lattice Sr2+ cation) decreased significantly at the interfacial region (−0.53 eV), indicating a strong thermodynamic trend that can drive Sr from the perovskite bulk to the interface.
Gadre et al.223 used DFT simulations to calculate cation diffusion at the LSC113/LSC214 heterointerface and reported that Sr tended to migrate to the interfacial region thermodynamically. Moreover, the heterointerface can stabilize high dopant Sr concentrations at the interface and suppress the formation of secondary passive particles (such as SrO). In addition, the researchers reported that the increased concentration of Sr can induce the formation of oxygen vacancies, leading to enhanced electrocatalytic activities. In addition, combining COBRA with ab initio calculations, Feng et al.57 provided atomic-scale structural information on the LSC113/LSC214 heterointerface, in which the signals at (0,0,0) and (0.5,0.5,0) (Fig. 15a and b) exhibited apparent Sr segregation at the interface and near the surface of LSC214. Here, the low SrLa substitution energy in the heterogeneous region was believed to be responsible for Sr segregation. Using ab initio calculations, Lee et al.230 found that the SrLa substitution energy in the LSCF113/LSC214 heterogeneous region was much lower than that of LSC113/214 (Fig. 15c), which was consistent with the weaker Sr interdiffusion trend of LSCF113/LSC214 as observed by AES.278
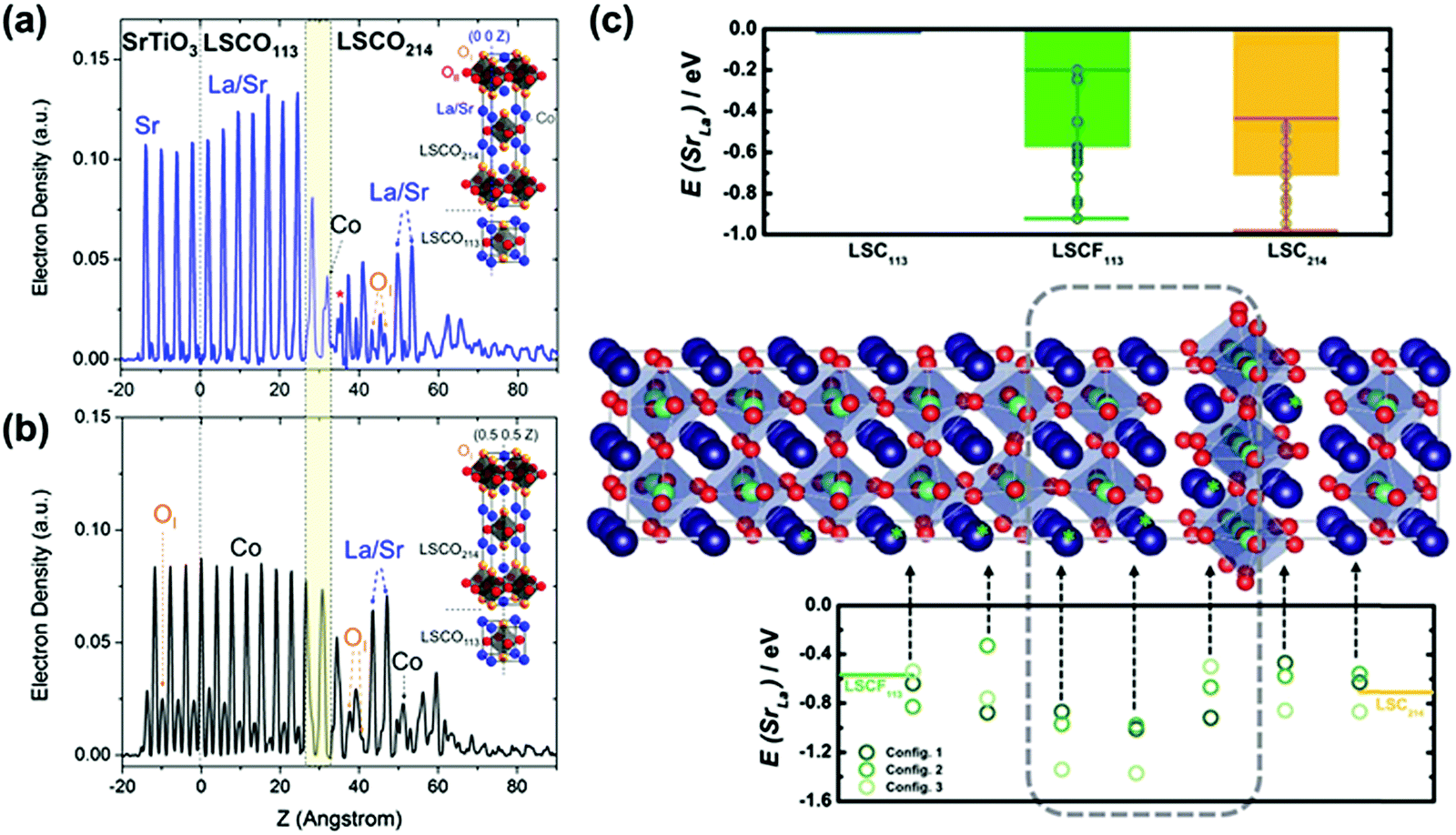 | ||
| Fig. 15 Electron densities of as-deposited LSCO214/LSCO113/STO thin films along: (a) the (0 0 Z) and (b) (0.5, 0.5, Z) lines using COBRA.57 (c) Up: SrLa substitution energies in bulk LSC214, LSC113 and LSCF113 calculated by DFT simulations. Middle: heterostructure model used in DFT simulations. La/Sr (dark blue), Fe/Co (light blue, center of the octahedra) and O (red). Bottom: SrLa substitution energies of each layer near the heterointerfacial region.230 Reproduced from ref. 57 and 230 with permission from American Chemical Society, copyright 2014 (https://pubs.acs.org/doi/10.1021/jz500293d) and RSC, copyright 2015, respectively. | ||
In addition, Tsvetkov et al.236 proposed that the phase separation of Sr-rich particles was detrimental to the ORR kinetics and that the decoration of less reducible cations ( , Me = V5+, Nb5+, Ti4+, Zr4+, Hf4+ and Al3+) onto perovskite LSC can balance Sr segregation with the ORR kinetics. Here, the researchers suggested that
, Me = V5+, Nb5+, Ti4+, Zr4+, Hf4+ and Al3+) onto perovskite LSC can balance Sr segregation with the ORR kinetics. Here, the researchers suggested that  decoration can reduce the oxygen vacancy concentration acting as reactive sites, while the
decoration can reduce the oxygen vacancy concentration acting as reactive sites, while the  coatings can increase the Co oxidation state and decrease the electrostatic attraction of Sr enrichment onto the surface, further suppressing harmful phase separation and stabilizing the surface composition. Based on the results of APXPS/XAS, the researchers summarized that these two factors resulted in a ‘volcano’ relationship between kq and ΔHVf(MeOx). In another study, Rupp et al.253 reported that SrO decoration onto LSC severely deactivated the LSC thin film, whereas deposited Co3O4 re-activated the surface. Here, the researchers used IPLD and found that deposited Sr cations can migrate and attach to highly active sites of inhomogeneous surfaces, hindering ionic and electronic transport in the ORR process,279 leading to the deactivation of active sites. Alternatively, Co3O4 decoration can induce highly active sites on surfaces.280
coatings can increase the Co oxidation state and decrease the electrostatic attraction of Sr enrichment onto the surface, further suppressing harmful phase separation and stabilizing the surface composition. Based on the results of APXPS/XAS, the researchers summarized that these two factors resulted in a ‘volcano’ relationship between kq and ΔHVf(MeOx). In another study, Rupp et al.253 reported that SrO decoration onto LSC severely deactivated the LSC thin film, whereas deposited Co3O4 re-activated the surface. Here, the researchers used IPLD and found that deposited Sr cations can migrate and attach to highly active sites of inhomogeneous surfaces, hindering ionic and electronic transport in the ORR process,279 leading to the deactivation of active sites. Alternatively, Co3O4 decoration can induce highly active sites on surfaces.280
Surface Sr segregation and separation are major causes of the degradation of Sr containing electrodes. It has been demonstrated that the deposition of a secondary phase to form a heterostructure can effectively suppress the phase separation of Sr. For example, Wen et al.281 proposed that ZrO2 coatings fabricated using ALD can suppress excess Sr segregation in LSC epitaxial films and improve the stability of oxygen electrodes. Here, the researchers suggested that the existence of cation exchange between Zr and Co near the interface can reduce the electrostatic force of surface Sr segregation, thus improving the thermal and electrochemical stability. In another study, Gong et al.241 confirmed that ZrO2 overcoats can optimize nanostructured LSC surfaces by preventing agglomeration and suppressing Sr segregation in long term operation. Similarly, the suppression of surface Sr segregation using surface coatings was also reported for LSM-decorated LSCF,41 Pr0.2Ce0.8O2/CeO2-decorated LSCF,234,282 PSM/PSCM-decorated LSCF,247,283etc.
By combining focused ion beam (FIB) milling235 with in situ scanning tunneling microscopy and spectroscopy (STM/STS), Chen et al.56 discovered that LSC214 in a LSC113/214 multilayer (ML) can be electronically activated. This electronic activation was highlighted by the smaller bandgap and a reversible transition from a semiconductor state to a metallic-like state with the temperature increasing to ∼250 °C (Fig. 16a). Here, the researchers reported that in the LSC113/214 interface, electron injection from LSC113 to LSC214 can result in band alignment and a decreased distance between the Fermi level and the conducting band of LSC214. The reduced distance can exponentially increase the quantity of available electrons and accelerate the kinetics of electron transfer from the electrode surface to adsorptive oxygen (Fig. 16b). Furthermore, Ma et al.77 reported that VAN-structured LSC113/214 exhibited similar electronic structure changes to ML LSC113/214. The energy bandgap of VAN was 1.5 ± 0.2 eV at room temperature, while metallic-like behavior with no energy gap was found above 250 °C. Here, the researchers proposed that electronic activation and stable surface composition can lead to enhanced ORR activity. In a recent study, Chen et al.80 used HRXRD (high resolution X-ray diffraction) and HAXPES (hard X-ray photoelectron spectroscopy) to probe the distribution of oxygen defects across the heterointerfaces of LSC113/LSC214 and LSC113/LNO214. Here, larger concentrations of oxygen vacancies were found in the LSC113 of LSC113/LSC214 than in the LSC113 of LSC113/LNO214 in a reducing environment, while both the LSC214 and LNO214 layers in the heterostructured samples were found to be more reduced compared with the respective single counterparts (Fig. 16c).
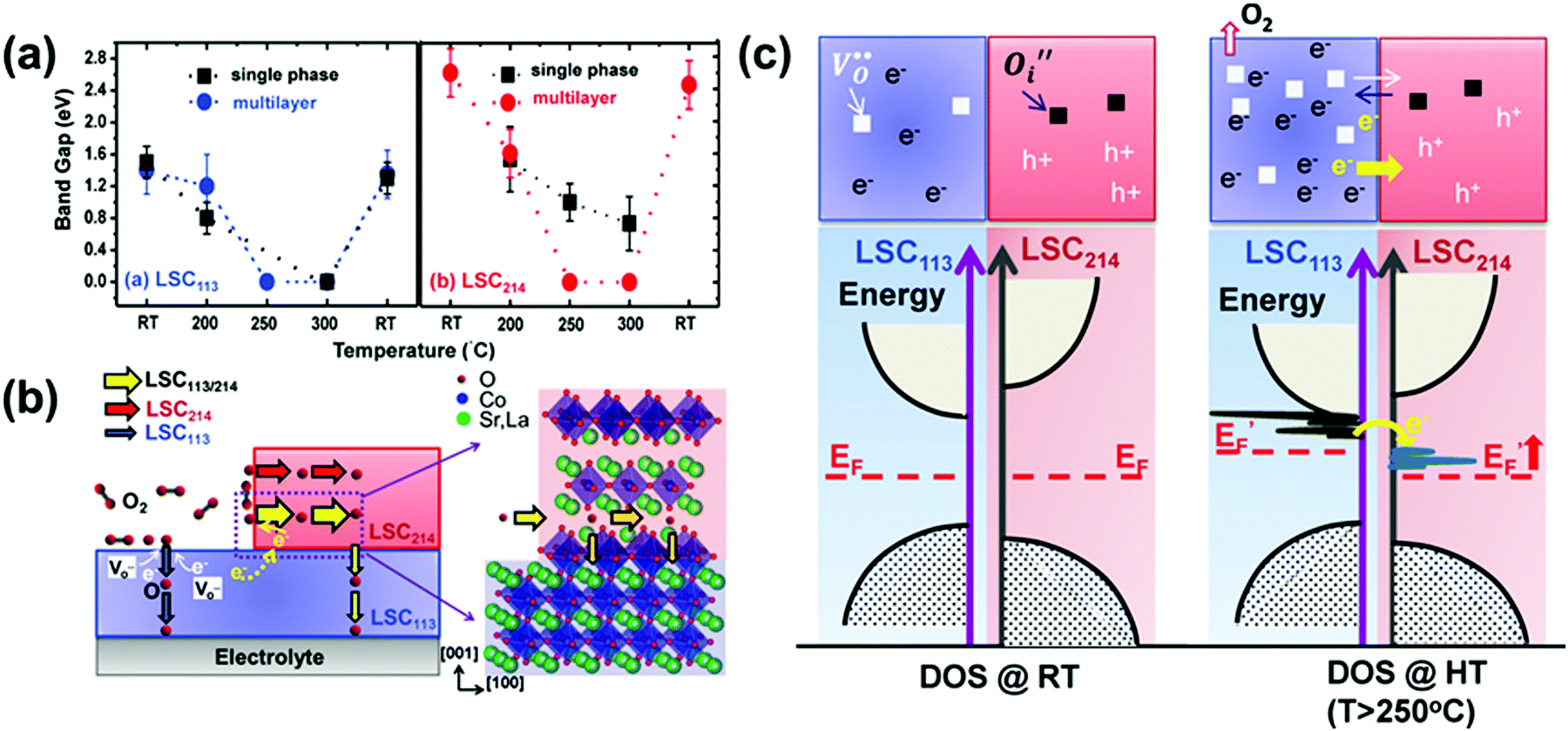 | ||
| Fig. 16 (a) Transformation of apparent energy gaps in single phase LSC113 and LSC214 as well as multilayered LSC113/LSC214 at different temperatures.56 (b) Origin of the enhanced ORR activity at heterostructured LSC113/LSC214 layers.56 (c) Illustrations of how the LSC113/LSC214 and LSC113/LNO214 interfaces induce the formation of local electric fields and influence local oxygen defect equilibria; (left) the misalignment of Fermi levels in LSC113 and LSC214 (LNO214) single phases after the loss of oxygen through annealing at high temperatures, (right) the alignment of Fermi levels in LSC113 and LSC214 (LNO214) heterostructures after annealing through the creation of local electric fields.80 Reproduced from ref. 56 and 80 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2013 and American Chemical Society, copyright 2018, respectively. | ||
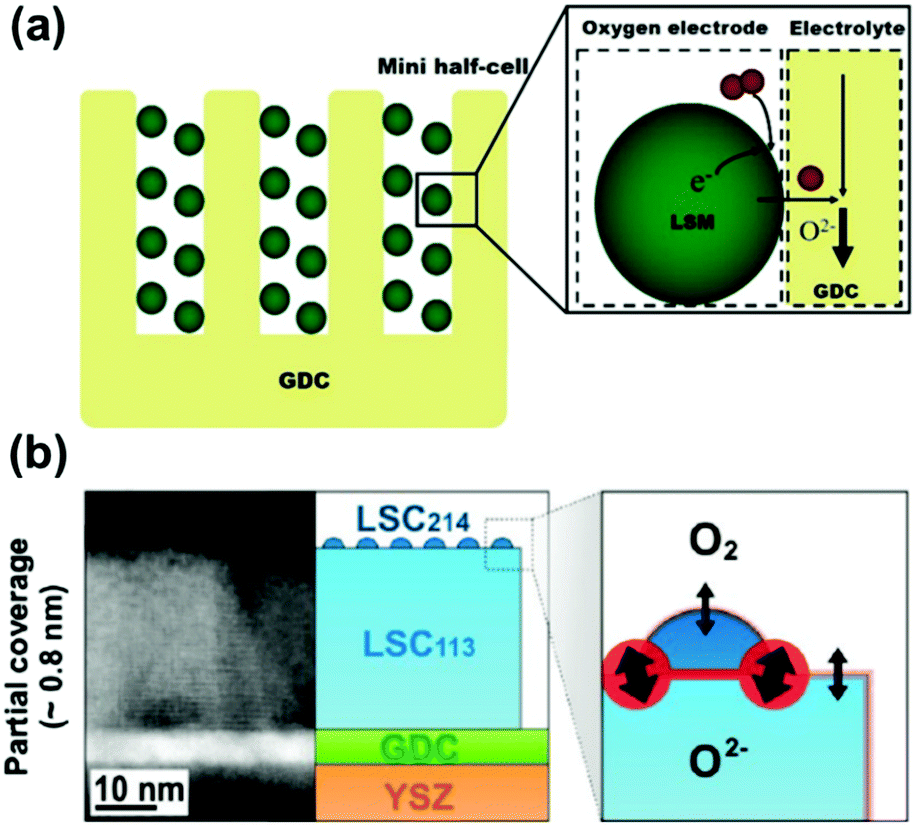 | ||
| Fig. 17 (a) Illustration of an idealized model of an integrated miniature half-cell with sufficient TPB; (b) TEM image and schematic of a LSC113 thin film with LSC214 particle decoration.48 Reproduced from ref. 48 with permission from American Chemical Society, copyright 2010. | ||
Crumlin et al.48 reported high ORR activities on the heterointerface of nano-island LSC214 (La0.5Sr0.5)2CoO4±δ decorated LSC113 (La0.8Sr0.2CoO3−δ) films, in which they proposed that the dispersive nano-island LSC214 can extend the TPB, leading to excellent ORR activity (Fig. 17b). Wan et al.288 also proposed that the oxygen reduction kinetics of PrBaCo2O5+δ–Sm0.2Ce0.8O1.9 (PBC–SDC) composites can be enhanced through the introduction of 3PB (PBC–SDC-gas), in which the polarization resistances of the oxygen electrode decreased from 0.281 Ω cm2 (for bare PBC) to 0.107 Ω cm2 (for the composite with 60 wt% SDC) at 700 °C. Here, the researchers suggested that the 3PB accounted for more than 80% of active sites in the oxygen incorporation processes.
5. Heterointerfaces in fuel electrode of SOC devices
SOC fuel electrodes can provide active sites for fuel oxidation or CO2/H2O decomposition depending on the operating modes. High ionic conductivity, electronic conductivity, and catalytic activity are essential characteristics. In addition, high resistance to sulfur poisoning and carbon coking are also necessary because SOC fuel electrodes may be fed with H2, H2S, CO and hydrocarbon containing fuels such as syngas, biogas and gasification reformate from hydrocarbons.289–291 Currently, nickel/yttria-stabilized zirconia (Ni–YSZ) has been the most widely used fuel electrode material for SOCs due to its high catalytic activity, operational reliability and acceptable cost.292 However, the long-term durability of Ni–YSZ is inadequate due to inherent redox instability and serious carbon deposition during operation. To address this, studies have recently highlighted the introduction of secondary phases into Ni–YSZ electrodes to form heterostructures as a potential approach to enhance the electrochemical performance and suppress surface coke deposition.59,293,294 In addition, alternatives to Ni–YSZ electrode materials, including various perovskite-based heterostructured oxides, have been developed and investigated, many of which have shown considerable catalytic activity and stability.258,295,296 Furthermore, studies have shown that treating these perovskite-based electrode materials under reducing conditions can further lead to the generation of metallic nanoparticles (known as in situ exsolution) on oxide matrixes and the formation of unique oxide–metal heterostructures. Different from the skeleton/surface coating structure fabricated by conventional approaches (such as infiltration), the concentration of oxygen vacancies can promote the catalytic activity of CO2 electrolysis and ionic conductivity, while the strong bonding of the exsolved metal particles to the parent phase can greatly enhance the stability of the heterostructures under fuel cell operating conditions (Fig. 18).4,297,298 | ||
| Fig. 18 Comparison of heterostructured fuel electrodes fabricated by infiltration and exsolution. | ||
Overall, the development of high-performance heterointerfaces is not only a promising strategy to enhance the electrode performance but can also greatly extend the scope of selection for new electrode materials. Therefore, recent progress in the utilization of heterostructured oxides as high performance fuel electrodes will be summarized in this section to provide direction for the research of more efficient and economical fuel electrode materials to further improve the SOC performance and stability.
5.1 Ni/oxide heterostructures in Ni/YSZ fuel electrodes
Conventional Ni/YSZ electrodes are troubled by surface coking in carbon-containing fuels. To address this, efforts have been made to modify the surface chemistry in which researchers report that extrinsic decoration with simple oxides (BaO, CaO, MgO, etc.) can impact the fuel electrode performance.59,293,294,299,300 For example, finely distributed nanoparticle oxide coatings have been recently deposited onto anode surfaces to generate oxide/metal heterostructures through various surface engineering techniques, including infiltration,301 evaporation deposition,293 carbonate decomposition,59etc. They have shown promise in the efficient enhancement of the electrochemical catalytic properties and the simultaneous extension of lifespans for Ni/YSZ electrodes if applied in hydrocarbon atmospheres.In one example, Yang et al.293 synthesized a novel BaO–Ni/YSZ anode with a finely distributed Ni/BaO heterointerface using vapor deposition and subsequent reduction and tested a single cell (BaO–Ni/YSZ|YSZ/SDC|LSCF) in CO. Here the researchers achieved a typical PPD of ∼0.70 W cm−2 at 750 °C, which was more than twice that of conventional Ni–YSZ anodes.301,302 Furthermore, the cell in this study provided stable power outputs for ∼100 h with 500 mA cm−2 in wet CO, demonstrating superior coking resistance. In another example, Tao et al.60 found that the BaO/Ni interface can stabilize cathode surfaces against coking in hydrocarbon fuels, in which the peak power density of their oxide cell (Ni–BaZr0.1Ce0.7Y0.2O3−δ (BZCY)–SDC|SDC|LSCF–SDC) reached 570 mA cm−2 at 700 °C in methane in 150 minutes of operation with a low electrode resistance of 0.136 Ω cm2. Likewise, Islam et al.303 reported that 5 wt% BaO loading on Ni/YSZ through infiltration increased the peak power density by ∼40% at 1073 K in 100% CH4.
Researchers have studied the mechanisms of coking resistance properties provided by extrinsic metal oxide decoration.59,60,293 For example, Yang et al.293 proposed that under operating conditions, carbon can be reversibly generated and removed at Ni surfaces through 5 primitive steps, including: (1) CO adsorption onto Ni due to a high adsorption energy (−2.6 eV) and the water-gas shift reaction; (2) carbon deposition on the surface of Ni catalysts due to the dehydrogenation of hydrocarbon fuels or CO disproportionation; (3) strong H2O adsorption on the BaO sites of BaO/Ni(111), leading to barrierless O–H bond cleavage surrounding BaO; (4) the reaction between dissociated OH and adsorbed C on Ni surfaces to form an intermediate COH, which dissociates to CO and H; and (5) CO and H diffusion to the TPB and electrochemical oxidation to CO2 and H2O.
Alternatively, Li et al.59 have studied the origin of enhanced coking resistance through BaO decoration on Ni surfaces using in situ Raman spectroscopy. They found the formation of a thin Ba(OH)2 layer on Ni surfaces during operation in wet hydrocarbon fuels, which further enabled the conversion of deposited coke to CO or CO2 rapidly. Here, the possible chemical reactions occurring near the interface can be summarized as follows:
BaO + H2O ⇔ Ba(OH)2; Cad + Ba(OH)2 → BaO + CO + H2; CO + Ba(OH)2 → BaO + CO2 + H2; BaO + CO2 → BaCO3.
This mechanism was supported by the rapid decrease in the –OH intensity accompanied by the gradually increasing –CO3 signal as measured by in situ Raman spectroscopy (Fig. 19).59 Likewise, Tao et al.60 proposed that the BaO/Ni interface can accelerate water adsorption and enhance the reaction of water-mediated carbon removal, whereas fast oxygen ion transportation and generated steam at the anode can promote hydrocarbon reformation to enhance the performance and stability.
 | ||
| Fig. 19 (a) Integrated peak intensities of the carbon G-bands for bare Ni and BaO–Ni as functions of exposure time in wet C3H8 at 450 °C. (b) Collection of time-resolved Raman spectra on Ni powders and (c) on BaO modified Ni powders. Reproduced from ref. 59 with permission from American Chemical Society, copyright 2015. | ||
Despite enhanced surface stability, however, surface decoration needs to be precisely controlled to avoid negative effects such as the formation of insulating alkaline oxides that can block active sites. For instance, the covering of TPB active regions with BaO particles can increase electrode polarization resistances and partly lower power densities.59,294,304,305 To address this, Islam et al.303 recently developed a novel fabrication technique combining microwave irradiation with vapor deposition to control the distribution and quantity of deposited nanosized BaO particles to ensure that additive components were only deposited separately on Ni surfaces.
In addition to BaO, decoration with alkaline earth metal oxides such as MgO and CaO has also been reported to enhance the electrode stability through similar mechanisms. For example, Qu et al.294 modified Ni–SDC anodes using CaO nanoparticles and achieved only slight degradations in performance during operation in a wet CH4 atmosphere for 70 h at 650 °C whereas an unmodified Ni–SDC-based cell failed to generate power within 1.2 h in the same conditions. Hua et al.299 also infiltrated Ni–YSZ cermet anodes with MgO additives and reported an Rp of 0.93 Ω cm2 after 48 h operation, which was 31% lower than that of a NiO infiltrated anode. Furthermore, Lay et al.300 proposed that a moderate amount of MgO decoration on the Ni–YSZ surface can lead to accelerated steam gasification reactions and enable less generation of surface carbon fibers in CH4 atmospheres.
5.2 Fluorite/perovskite heterostructures in Ni/YSZ fuel electrodes
Many perovskite structured oxides have been shown to possess considerable conductivities and superior coking tolerances in application as electrolytes or oxygen electrodes. However, these materials possess limited application as fuel electrodes due to inherent structural instabilities in reducing atmospheres. Despite this, some nanosized perovskites have been shown to remain stable under reducing conditions and currently investigations aimed at introducing such perovskite nanoparticles onto SOC fuel electrode surfaces to form fluorite/perovskite heterostructures and enhance the stability against surface side reactions are being conducted.258,295,296,306Vincent et al.295 fabricated a YSZ/La0.4Sr0.5Ba0.1TiO3 anode through infiltration and achieved a peak power density in the single cell of 200 mW cm−2 at 850 °C in H2/H2S (5000 ppm). Moreover, Xu et al.296 fabricated a CeO2/La0.3Sr0.7Ti0.3Fe0.7O3−δ composite anode and reported polarization resistances as low as 0.151 Ω cm2 at 850 °C in a CO atmosphere. More recently, Chen et al.258 designed a Ni–BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb)/SDC hybrid SOFC anode (Fig. 20) and reported that the corresponding cell was able to operate in nearly dry CH4 fuel (with ∼3.5% H2O) for more than 550 h at only 500 °C without coke formation.
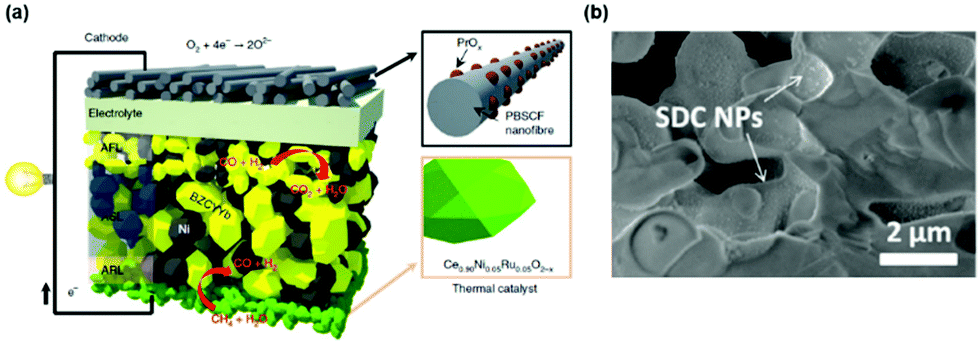 | ||
| Fig. 20 (a) Schematic of a single cell with yellow and grey grains representing BZCYYb and Ni phases. (b) SEM of the anode after long term operation for ∼500 h in nearly dry CH4, indicating excellent coking tolerance. Reproduced from ref. 258 with permission from Springer Nature, copyright 2018. | ||
In another example, Hua et al.307 reported that decorating Ni/YSZ anodes with PrBaMn2O5+δ nanoparticles can significantly enhance the catalytic activity and durability, leading to a 1.35 W cm−2 peak power density in a biogas atmosphere (containing CH4, CO2 and small amounts of H2S) at 800 °C as well as negligible performance degradation after 100 h of testing. Here, the researchers suggested that through infiltrating the Ni/YSZ surfaces with perovskite oxides, the finely distributed perovskites with high catalytic activity can act as micro-anodes that can increase the TPB area largely and enhance the processing capacity of the anode for converting hydrocarbon fuels (Fig. 21). Furthermore, Irvine et al.4 suggested that the heterogeneous and nanosized decoration of perovskite oxides such as SrTiO3 and LaCrO3 on fuel electrode surfaces can improve the long-term stability, resulting from the superior stability over a wide range of environments and moderate electronic conductivities. This mechanism appears to also be applicable in perovskite-modified Ni/YSZ electrode systems as well.
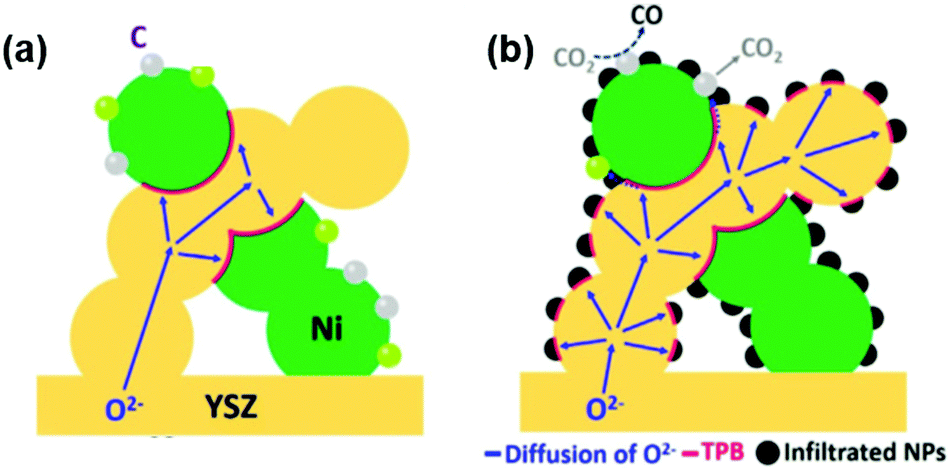 | ||
| Fig. 21 Comparison of Ni/YSZ anodes before (a) and after (b) PrBaMn2O5+δ infiltration. The infiltrated perovskite nanoparticles resisted carbon species deposition on the Ni/YSZ surfaces and enhanced carbon removal. Reproduced from ref. 307 with permission from Elsevier, copyright 2017. | ||
5.3 In situ assembly of oxide–metal heterostructures through exsolution
To improve the stability of Ni-based electrodes in carbon-containing environments, many perovskite-structured materials have been studied as potential alternatives to conventional Ni/YSZ fuel electrodes. B-site cations in perovskite lattices can be partly reduced in reducing conditions and generate numerous finely distributed metallic nanoparticles, thus forming numerous perovskite–metal heterointerfaces, which is defined as B-cation exsolution.4,7 These in situ exsolved metal normally are strong bonded to the oxide skeleton, leading to facilitated charge and mass transfer processes and high stability of the materials during operation.308Self-assembled metallic nanoparticles can normally exhibit higher conductivities and catalytic activities than perovskite bulk phases, leading to significant enhancements in fuel electrode electrochemical activity.4,309–312 For example, Yang et al.311 proposed that exsolved NiO particles on SrFe0.85Ti0.1Ni0.05O3−δ (STFNi) electrode surfaces can significantly reduce the ASR from ∼0.4 to ∼0.15 Ω cm2 at 700 °C, in which the anode-supported cell with NiO–STFNi as the fuel electrode can exhibit high peak power densities of 1650, 1420 and 1100 mW cm−2 at 750, 700 and 650 °C respectively. Tsekouras et al.312 further compared La0.4Sr0.4Ni0.06Ti0.94O2.94 (LSNT) with Ni0 exsolution, La0.4Sr0.4Fe0.06Ti0.94O2.97 (LSFT) with Fe0 exsolution and La0.4Sr0.4TiO3 (LST) and found that the exsolution trend of Ni0 was generally larger than Fe0 exsolution. Here, electrolysis cells operating in 47% H2O/53% N2 at 900 °C at −1.5 V with the above cathodes showed reduced HTSE onset potentials (EHTSE) from −1.19 V for the LST cathode to 0.98 V and 0.63 V for the Fe0 and Ni0 exsoluted cathodes, respectively, with Rp decreasing from 1.7 to 1.5 and 1.2 Ω cm2. Likewise, Li et al.309 fabricated a perovskite-structured (La0.2Sr0.8)(Ti0.9Mn0.1)O3−δ (LSTM) skeleton with exsolved Ni nanocrystals on the surface and reported a ∼50% enhancement in catalytic activity for CO2 electrolysis as compared with a bare electrode. The high catalytic activity for CO2 electrolysis is likely associated with the high oxygen vacancy concentration.298 The exsolution of metal particles is often accompanied by formation and aggregation of oxygen vacancies near the interface. Theoretical calculations indicated that the exsolved Ni clusters and oxygen vacancies could synergistically weaken the C–O1 bond, thus activating the adsorbed CO2 molecule and enhancing the CO2 reduction. In addition, the increase in the oxygen vacancy concentration enhances the ionic conductivity of the fuel electrode through a vacancy-mediated mechanism.
Many studies have also confirmed that the surface composition and electrochemical performance of these exsolved particle systems experience no significant decline after many redox cycles, demonstrating considerable operational stability.7,314–317 For example, Liu et al.318 used a double-layered perovskite ((Pr0.4Sr0.6)3(Fe0.85Mo0.15)2O7) with exsolved Co/Fe metallic nanoparticles and found that no graphite was generated during polarization for 100 h of CO2 electrolysis, indicating excellent anti-coking properties. Furthermore, Song et al.319 fabricated a self-assembled dual-phase structure (R-LS0.6FCN) consisting of both SP and RP (SLF214 + LFO113) layers with exsolved Fe–Cu bimetal nanofibers and applied it as the fuel electrode of a SOFC. Here, the researchers reported that the peak power density of the solid oxide cell using R-LS0.6FCN as the anode yielded 1.03 W cm−2 at 850 °C in H2/3% H2O, which was higher than that of a similar single cell with Ni–Fe/SDC cermet as the anode. These researchers also reported that there was no obvious performance degradation after operation in H2/50 ppm H2S or syngas at 700 °C for 100 h, demonstrating high durability against coking/sulfur deposition. Here, such high stabilities were attributed to the formation of nanofiber networks without agglomeration and to the construction of an SP/RP heterointerface at the anode. More studies about the coking resistance capacity of perovskite-based fuel electrodes with exsolved metals are summarized in Table 3.
| Exsolved metal | Perovskite precursor | Temperature (°C) | Atmosphere | Cell configuration | Performance | Ref. |
|---|---|---|---|---|---|---|
| Fe | La0.6Sr0.3Cr0.85Fe0.15O3−δ (LSCFe) | 800 | CH4 | LSCFe|YSZ–YSZ–LSM|YSZ | PPD ∼ 330 mW cm−2 | 320 |
| Co | Pr0.5Ba0.5Mn0.9Co0.1O2.89 (PBMCo) | 900 | Syngas | PBMCo|GDC–GDC–YSZ–GDC–LSM|GDC | PPD ∼ 900 mW cm−2 | 321 |
| Ni | La0.52Sr0.28Ni0.06Ti0.94O3 (LSNT) | 800 | 20% CH4/H2 | Slight carbon fiber growth after 4 h exposure | 313 | |
| La0.9Mn0.8Ni0.2O3 (LMN) | 700 | CH4/CO2 | Negligible change for 24 h | 322 | ||
| Cu | La0.5Sr0.5Fe0.8Cu0.15Nb0.05O3−δ (LSFCNb) | 800 | Wet syngas | LSFCNb–SDC–ScSZ–SDC–LSFCNb | PPD ∼ 640 mW cm−2; 9.4% degradation rate for 100 h at 0.7 V | 323 |
| Co–Fe alloys | (Pr0.4Sr0.6)3(Fe0.85Nb0.15)2O7 (RP-PSFN) | 800 | Wet C3H8 | RP-PSFN–CFA|LSGM|BSCF | Stable operation for 100 h under 400 mA cm−2 | 324 |
| Pr0.8Sr1.2(Co,Fe)0.8Nb0.2O4+δ (K-PSCFN) | 850 | C3H8 | K-PSCFN–CFA|LSGM|P-PSCFN | PPD ∼ 940 mW cm−2; negligible degradation during 26 cycles of redox testing | 325 | |
| La0.5Ba0.5Mn0.8Fe0.1Co0.1O3−δ | 850 | Wet CH4 | CoFe–LBMF|LDC–LSGM|BSCF | PPD ∼ 503 mW cm−2; stable power output for 200 h | 326 | |
| La0.3Sr0.7Cr0.3Fe0.6Co0.1O3−δ (LSCrFeCo10) | 850 | C3H8/H2O | LSCrFeCo10–GDC|LDC–LSGM|LSCF–GDC | PPD ∼ 700 mW cm−2 | 327 | |
| (Pr0.4Sr0.6)0.3(Fe0.85Mo0.15)2O7 (DLP-PSFM) | 750 | C2H6 | Stable operation for 100 h under 650 mA cm−2 | 328 | ||
| Fe–Ni alloys | Sr2FeMo0.65Ni0.35O6−δ (SFMNi) | 850 | Wet CH4 | SFMNi–LDC|LSGM|LSCF | PPD ∼ 500 mW cm−2, 24 h of stability under 200 mA cm−2 | 329 |
| (La0.7Sr0.3)(Cr0.85Ni0.1125Fe0.0375)O3−δ (LSCNi-Fe) | 850 | 5000 ppm H2S-syngas | LSCNi-Fe|YSZ|YSZ–LSM/YSZ | PPD ∼ 400 mW cm−2 and a 1300 mA cm−2 maximum current density | 330 | |
| La0.5Sr0.5Fe0.8Ni0.2O3−δ (LSFN) | 725 | Wet syngas | LSFN–SDC|ScSZ|SDC–LSFCNb | Stable operation for 20 h at 0.7 V | 331 | |
| Fe–Pb alloys | La0.8Sr0.2Fe0.9Nb0.1Pd0.04O3−δ (LSFNP) | 800 | Dry C3H8 | LSFNP|SDC|NBCCo–SDC | 25 h operation under 80 mA cm−2 | 332 |
| Fe–Re alloys | LaRexFe0.8O-La3ReO8 (x < 0.2) (LRF) | 860 | CH4/CO2 | No significant carbon deposition after 70 h operation | 333 | |
| Fe–Cu alloys | (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9(Cu1−xFex)0.1O3−δ | 850 | Dry CH4 | Cu0.5Fe0.5–LSCM|LSGM |LSM | PPD ∼ 40 mW cm−2, negligible degradation after 100 h operation and 10 redox cycles | 334 |
| Ni–Ce alloys | La0.3Sr0.6Ce0.1Ni0.1Ti0.9O3−δ (LSCNT) | 900 | CH4 | LSCNT–YSZ|YSZ|LSM–YSZ | PPD ∼ 600 mW cm−2, no degradation for 80 h under 0.5 V | 49 |
| Ni–Cu alloys | (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9(Ni1−xCux)0.1O3−δ (LSCM-Ni1−xCux) | 800 | CH4/CO2 | Symmetric cell | 300 h stability at 0.9 V | 335 |
Alternatively, Neagu et al.313 proposed that the high stability of perovskite–metal heterosystems resulted from a combination of size effects and formation of robust interfaces, in which for a specific La0.52Sr0.28Ni0.06Ti0.94O3 system with in situ exsolved Ni metal (Fig. 22a), after operation in a 20% CH4/H2 environment at 800 °C for 4 h, there was nearly no carbon fiber on the exsolved Ni particles with sizes of 25–60 nm, while carbon growth was found on exsolved Ni particles of ∼80 nm in size. For comparison, there was obvious carbon fiber accumulation on La0.4Sr0.4TiO3 anodes infiltrated by 20 nm Ni particles. TEM micrographs indicated that exsolved nanoparticles were embedded at a considerable depth in the parent perovskite oxide support (Fig. 22b), resulting in a robust interface between the perovskite oxide and the exsolved metal particles. The TEM micrographs and electron diffraction revealed that the exsolved metallic particles could grow epitaxially from the parent perovskite oxide in a specific orientation (perovskite(111), Ni(111)), forming a well-ordered structure across the interface with good stability (Fig. 22c and d). In addition, the cation diffusion across the interface between the metal particles and the parent oxide greatly increases the adhesion of the metal particles to the oxide phases. The strong bonding between the exsolved metal particles and the parent perovskite oxide plays a vital role in preventing particle uplifting and subsequent carbon fiber growth, resulting in high coking resistance and stable catalytic activity in a carbon-containing atmosphere.
 | ||
| Fig. 22 (a) Schematic of the possible mechanism of B-site exsolution in a perovskite-structured La0.52Sr0.28Ni0.06Ti0.94O3 model system. (b) HRTEM image of the interfacial region between the perovskite matrix and exsolved Ni nanoparticles. (c) HRTEM micrographs at the perovskite–metal interface, demonstrating the epitaxic atomic planes and orientations. (d) Atomic arrangement at the Ni–La0.52Sr0.28Ni0.06Ti0.94O3 interface according to structure characterization. Reproduced with permission from ref. 313, copyright 2015 Macmillan Publishers Limited. | ||
The driving force for B-site cation exsolution was also investigated by several groups.4,310,336–339 According to Irvine et al.,4,310,313 the exsolution of B-site cations is dynamically driven through a combination of chemical and electrochemical reduction and generally initiates at surface defect sites. Specifically, in lattices experiencing reducing conditions, the reduction of B-site transition metals and the formation of surface defects (i.e. oxygen vacancies) become more favorable, collectively leading to metal nucleation at the surface and the lowering of nucleation barriers through crystal defects.30 More recently, Han et al.336 successfully controlled B-site exsolution in La0.2Sr0.7Ni0.1Ti0.9O3−δ (LSNT) thin films through lattice strain control, resulting in a high degree of Ni exsolution with more than 1100 particles μm−2 and tiny particle sizes of ∼5 nm in diameter. Here, thermodynamic analysis indicated that the small atomic diffusion coefficients of the exsoluted Ni under both tensile and compressive strain were much lower compared to conventionally deposited Ni, indicating a small thermodynamic potential of particle coarsening and agglomeration. Furthermore, the researchers suggested that due to the small size of the exsoluted Ni and strong interactions between the embedded Ni and perovskites, carbon fibers grew in “base growth” without particle growth. The key factor was deduced to be the mismatch-strain relaxation energy (ΔGmr), which can reduce nuclei sizes and nucleation barriers and simultaneously quicken nucleation rates, leading to smaller exsolution particles with higher densities.
Alternatively, Kwon et al.337,338 explained B-site exsolution in terms of thermodynamics, in which, based on density functional simulations, the researchers proposed that in a PrBa(Mn,T)2O5+δ (T = Fe, Co and/or Ni) double-perovskite model system, substituted cations such as Co2+ and Ni2+ possess lower exsolution energies than the host cation Mn2+ (−0.55 and −0.50 eV vs. −0.47 eV) and demonstrate more favorable thermodynamic trends. In addition, Co/Ni doped PrBa(Mn,T)2O5+δ exhibits a lower surface oxygen vacancy-formation energy than undoped samples (2.46 and 2.85 eV vs. 2.97 eV), which can also assist in stabilizing the self-grown metallic phase.
6. Summary, challenges and possible research directions
SOCs are promising and environmentally friendly devices that can achieve efficient conversion between electricity and chemical energy. Tremendous efforts involving the understanding and control of heterointerfaces have been undertaken to improve the performance of SOC materials at intermediate temperatures. For heterostructured electrolytes, significantly enhanced ionic conductivities are attributed to lattice strain, dislocation and space-charge effects. Due to the presence of lattice strain, cation segregation, modified electronic structures and expanded TBPs, many heterostructured oxygen electrodes were reported to show outstanding ORR/OER activities. Decorating suitable secondary oxide decoration on the surface presents an effective approach to improve both the reaction activity and long-term stability of oxygen electrodes. For fuel electrodes, different types of heterostructure, particularly oxide skeletons with exsolved metal nanoparticles, have been reported to exhibit good fuel oxidation activity and high resistance to sulfur poisoning and carbon coking. Despite many successful studies about improving the performance of SOC components through heterointerface engineering, there are still many challenges to overcome, which require further systematic investigation.First, precise control of the composition and microstructure of heterointerfaces is necessary for the rational design of novel electrode or electrolyte materials via interface engineering. It has been reported that the atomic structure of the heterointerface such as local strain and dislocations can profoundly impact the ionic conductivity and/or the electrocatalytic activity of the heterostructure. The use of thin film techniques such as PLD, ALD and MBE presents unique advantages in controlling the atomic structure of the heterointerfaces, which makes them idea tools for synthesis of novel heterostructured SOC components. The challenges that need to be overcome are the high price of the equipment and the slow growth rates of these thin film techniques.
Secondly, advanced characterization techniques, particular the ones with in situ measurement capability, are needed to further reveal the role of heterointerfaces in determining the ionic conductivity and electrocatalytic activity of the heterostructure. Especially, it is necessary to probe how the interface evolves under the working conditions of SOCs, such as high temperature, a reactive gas environment and electrical bias. There have been several successful attempts to use in situ STM, XPS, and SIMS to investigate the evolution of the electronic structure, cation valence and ion distribution at the interfacial region at elevated temperature or under bias.11,56,233
Thirdly, although several factors have been proposed to explain the impact of heterointerfaces, an overall theoretical model that describes the role of heterointerfaces in determining the transport properties and catalytic activity of SOC components is still lacking. Advanced theoretical modelling and simulation need to be carried out to be combined with experimental results to uncover the effects of heterointerfaces, hence providing guidance for the rational design of novel materials.
Finally, besides improving the performance of each component of SOCs, system level optimization is also necessary. The compatibility of SOC materials with other parts of the system such as interconnection materials needs to be tested. The solid oxide cell and stack design and the thermal management of the whole system are also critical for the long stability of devices.
While SOC components are taken as examples in this review to introduce the influence of heterointerfaces on the conductivity, electrochemical activity and stability, the mechanism of the performance improvement and methodology for controllable fabrication of interfaces are applicable to other energy conversion systems for a wide range of applications such as photo/electrochemical water splitting, solar thermal CO2 reduction, and lithium ion batteries.
Nomenclature and acronyms
| ALD | Atomic layer deposition |
| ASR | Area surface resistance |
| BSCF | Ba1−xSrxCo1−yFeyO3−δ |
| BZCY | BaZr0.4Ce0.4Y0.2O3 |
| BZO | BaZrO3 |
| BZY | BaZrxY1−xO3−y |
| CFA | Co–Fe bi-metallic alloy |
| COBRA | Coherent Bragg rod analysis |
| CSZ | ZrO2 + CaO |
| DFT | Density functional theory |
| DP-BSCFW | Double perovskite Ba0.5Sr0.5(Co0.7Fe0.3)0.687W0.312O3−δ |
| EC | Electron conduction |
| EELS | Electron energy loss spectroscopy |
| EPD | Electrophoretic deposition |
| HAXPES | Hard X-ray photoelectron spectroscopy |
| HRXRD | High resolution X-ray diffraction |
| HRTEM | High resolution transmission electron microscope |
| HTPC | High-temperature proton conductor |
| IC | Ionic conduction |
| LAO | LaAlO3 |
| LCP | LaCePrOx |
| LNO | LaNiO3±δ |
| LSC113 | La1−xSrxCoO3±δ |
| LSC214 | La2−xSrxCoO4±δ |
| LSCF | La1−xSrxCoyFe1−yO3±δ |
| LSCM | La1−xSrxCoyMn1−yO3±δ |
| LSCMC | (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9Cu0.1O3−δ |
| LSCMN | (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9Ni0.1O3−δ |
| LSGM | LaxSr1−xGayMg1−yO3−δ |
| LSF | La1−xSrxFeO3±δ |
| LSM | La1−xSrxMnO3±δ |
| LSZG | Li13.9Sr0.1Zn(GeO4)4 |
| MBE | Molecular beam epitaxy |
| MIEC | Mixed ionic and electronic conductor |
| ML | Multilayer |
| NEXAFS | Near edge X-ray absorption fine structure |
| NGO | NdGaO3 |
| NSC113 | Nd1−xSrxCoO3±δ |
| OER | Oxygen evolution reaction |
| ORR | Oxygen reduction reaction |
| PLD | Pulsed laser deposition |
| PPD | Peak power density |
| PSCM | PrSrCoMnO6−δ |
| RP | Ruddlesden–Popper |
| RP-PSFN | RP type layered (Pr0.4Sr0.6)3(Fe0.85Nb0.15)2O7 |
| SCDC | Sm/Ca co-doped ceria |
| SCFN | SrCo0.2Fe0.6Ni0.2O3 |
| SDC | Samarium doped ceria |
| SIMS | Secondary-ion mass spectrometry |
| SOC | Solid oxide cell |
| SOEC | Solid oxide electrolysis cell |
| SOFC | Solid oxide fuel cell |
| SP-BSCFW | Single perovskite Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125O3−δ |
| SSC–GDC | Sm0.5Sr0.5CoO3–Gd0.1Ce0.9O2−δ |
| STEM | Scanning transmission electron microscope |
| STF | SrTi0.2Fe0.8O3 |
| STM/STS | Scanning tunnelling microscopy/spectroscopy |
| STO | SrTiO3 |
| TEM | Transmission electron microscope |
| TOF-SIMS | Time of flight secondary ion mass spectroscopy |
| TPB/3PB | Triple phase boundaries |
| VAN | Vertical aligned nanocomposite |
| XANES | X-ray absorption near-edge structure |
| XAS | X-ray absorption spectroscopy |
| XPS | X-ray photoemission spectroscopy |
| YBCO | (Y,Ca)Ba2Cu3O7 |
| YSZ | Yttira-stabilized zirconia |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 91645126, 21273128, 11975102 and 21677162), the ‘Tsinghua University Initiative Scientific Research Program’ (2018Z05JZY010), the ‘Program for Changjiang Scholars and Innovative Research Team in University’ (IRT13026), Guangdong Innovative and Entrepreneurial Research Team Program (No. 2014ZT05N200), Phillips 66 Research Center, and the ‘Tsinghua-MIT-Cambridge’ Low Carbon Energy University Alliance Seed Fund Program (201LC004). All of the above financial support is gratefully acknowledged.References
- S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265–267 CrossRef CAS PubMed.
- Z. Shao, S. M. Haile, J. Ahn, P. D. Ronney, Z. Zhan and S. A. Barnett, Nature, 2005, 435, 795–798 CrossRef CAS PubMed.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 15014 CrossRef CAS.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- J. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- D. Burnat, R. Kontic, L. Holzer, P. Steiger, D. Ferri and A. Heel, J. Mater. Chem. A, 2016, 4, 11939–11948 RSC.
- Y. Zhang, R. Knibbe, J. Sunarso, Y. Zhong, W. Zhou, Z. Shao and Z. Zhu, Adv. Mater., 2017, 29, 1700132 CrossRef.
- S. H. Jensen, C. Graves, M. Mogensen, C. Wendel, R. Braun, G. Hughes, Z. Gao and S. A. Barnett, Energy Environ. Sci., 2015, 8, 2471–2479 RSC.
- Y. Zheng, W. Zhang, Y. Li, J. Chen, B. Yu, J. Wang, L. Zhang and J. Zhang, Nano Energy, 2017, 40, 512–539 CrossRef CAS.
- Y. Chen, S. Yoo, Y. Choi, J. H. Kim, Y. Ding, K. Pei, R. Murphy, Y. Zhang, B. Zhao, W. Zhang, H. Chen, Y. Chen, W. Yuan, C. Yang and M. Liu, Energy Environ. Sci., 2018, 11, 2458–2466 RSC.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- A. M. Saranya, A. Morata, D. Pla, M. Burriel, F. Chiabrera, I. Garbayo, A. Hornés, J. A. Kilner and A. Tarancón, Chem. Mater., 2018, 3, 5621–5629 CrossRef PubMed.
- M. A. Laguna-Bercero, H. Monzón, A. Larrea and V. M. Orera, J. Mater. Chem. A, 2016, 4, 1446–1453 RSC.
- J. T. S. Irvine and S. Tao, Nat. Mater., 2003, 2, 320–323 CrossRef PubMed.
- D. M. Bastidas, S. Tao and J. T. S. Irvine, J. Mater. Chem., 2006, 16, 1603–1605 RSC.
- N. Ai, N. Li, S. He, Y. Cheng, M. Saunders, K. Chen, T. Zhang and S. P. Jiang, J. Mater. Chem. A, 2017, 5, 12149–12157 RSC.
- T. Li, M. F. Rabuni, L. Kleiminger, B. Wang, G. H. Kelsall, U. W. Hartley and K. Li, Energy Environ. Sci., 2016, 9, 3682–3686 RSC.
- C. C. Duan, D. Hook, Y. C. Chen, J. H. Tong and R. O'Hayre, Energy Environ. Sci., 2017, 10, 176–182 RSC.
- W. Zhang, B. Yu and J. Xu, Int. J. Hydrogen Energy, 2012, 37, 837–842 CrossRef CAS.
- B. Yu, W. Zhang, J. Xu, J. Chen, X. Luo and K. Stephan, Int. J. Hydrogen Energy, 2012, 37, 12074–12080 CrossRef CAS.
- L. Chen, F. Chen and C. Xia, Energy Environ. Sci., 2014, 7, 4018–4022 RSC.
- Y. Li, Y. Li, Y. Wan, Y. Xie, J. Zhu, H. Pan, X. Zheng and C. Xia, Adv. Energy Mater., 2019, 9, 1803156 CrossRef.
- J. Tallgren, O. Himanen and M. Noponen, ECS Trans., 2017, 78, 3103–3111 CrossRef CAS.
- M. Kramer, I. H. Stairs, R. N. Manchester, M. A. McLaughlin, A. G. Lyne, R. D. Ferdman, M. Burgay, D. R. Lorimer, A. Possenti, N. D’Amico, J. M. Sarkissian, G. B. Hobbs, J. E. Reynolds, P. C. C. Freire and F. Camilo, Science, 2006, 314, 97–102 CrossRef CAS.
- T. Hibino, A. Hashimoto, T. Inoue, J. Tokuno, S. Yoshida and M. Sano, Science, 2000, 288, 2031–2033 CrossRef CAS.
- Y. Zhou, H. Wu, T. Luo, J. Wang, Y. Shi, C. Xia, S. Wang and Z. Zhan, Adv. Energy Mater., 2015, 5, 1500375 CrossRef.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 162–1644 Search PubMed.
- Y. Li, W. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. Liu, Chem. Soc. Rev., 2017, 46, 6345–6378 RSC.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. Mcphail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- J. Fleig, Annu. Rev. Mater. Res., 2003, 33, 361–382 CrossRef CAS.
- F. Santoni, D. M. Silva Mosqueda, D. Pumiglia, E. Viceconti, B. Conti, C. Boigues Muñoz, B. Bosio, S. Ulgiati and S. J. McPhail, J. Power Sources, 2017, 370, 36–44 CrossRef CAS.
- D. Heidari, S. Javadpour and S. H. Chan, Energy Convers. Manage., 2017, 136, 78–84 CrossRef CAS.
- C. Zhang and K. Huang, J. Power Sources, 2017, 342, 419–426 CrossRef CAS.
- N. Sata, K. Eberman, K. Eberl and J. Maier, Nature, 2000, 408, 946–949 CrossRef CAS.
- P. Boldrin, E. Ruiz-Trejo, J. Mermelstein, J. M. Bermúdez Menéndez, T. Ramírez Reina and N. P. Brandon, Chem. Rev., 2016, 116, 13633–13684 CrossRef CAS.
- S. Zhang, H. Wang, M. Y. Lu, A. Zhang, L. V. Mogni, Q. Liu, C. Li, C. Li and S. A. Barnett, Energy Environ. Sci., 2018, 11, 1870–1879 RSC.
- Y. Lin, S. Fang, D. Su, K. S. Brinkman and F. Chen, Nat. Commun., 2015, 6, 6824 CrossRef CAS PubMed.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chem., 2017, 3, 812–821 CAS.
- M. Först, A. D. Caviglia, R. Scherwitzl, R. Mankowsky, P. Zubko, V. Khanna, H. Bromberger, S. B. Wilkins, Y. D. Chuang, W. S. Lee, W. F. Schlotter, J. J. Turner, G. L. Dakovski, M. P. Minitti, J. Robinson, S. R. Clark, D. Jaksch, J. M. Triscone, J. P. Hill, S. S. Dhesi and A. Cavalleri, Nat. Mater., 2015, 14, 883–888 CrossRef.
- R. Yoshimi, A. Tsukazaki, K. Kikutake, J. G. Checkelsky, K. S. Takahashi, M. Kawasaki and Y. Tokura, Nat. Mater., 2014, 13, 253–257 CrossRef CAS.
- I. Kosacki, C. Rouleau, P. Becher, J. Bentley and D. Lowndes, Solid State Ionics, 2005, 176, 1319–1326 CrossRef CAS.
- Z. Gao, V. Y. Zenou, D. Kennouche, L. Marks and S. A. Barnett, J. Mater. Chem. A, 2015, 3, 9955–9964 RSC.
- Y. Ling, L. Chen, B. Lin, W. Yu, T. T. Isimjan, L. Zhao and X. Liu, RSC Adv., 2015, 5, 17000–17006 RSC.
- Q. Su, D. Yoon, A. Chen, F. Khatkhatay, A. Manthiram and H. Wang, J. Power Sources, 2013, 242, 455–463 CrossRef CAS.
- M. Sase, F. Hermes, K. Yashiro, K. Sato, J. Mizusaki, T. Kawada, N. Sakai and H. Yokokawa, J. Electrochem. Soc., 2008, 155, B793–B797 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, S. Ahn, G. J. aO, D. N. Leonard, A. Borisevich, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, J. Phys. Chem. Lett., 2010, 1, 3149–3155 CrossRef CAS.
- Y. Sun, X. Zhou, Y. Zeng, B. S. Amirkhiz, M. Wang, L. Zhang, B. Hua, J. Li, J. Li and J. Luo, J. Mater. Chem. A, 2015, 3, 22830–22838 RSC.
- Z. Cai, Y. Kuru, J. W. Han, Y. Chen and B. Yildiz, J. Am. Chem. Soc., 2011, 133, 17696–17704 CrossRef CAS.
- J. W. Han and B. Yildiz, Energy Environ. Sci., 2012, 5, 8598–8607 RSC.
- C. Leon, J. Santamaria and B. A. Boukamp, MRS Bull., 2013, 38, 1056–1063 CrossRef CAS.
- A. Kushima and B. Yildiz, J. Mater. Chem., 2010, 20, 4809–4819 RSC.
- B. Yildiz, MRS Bull., 2014, 39, 147–156 CrossRef CAS.
- K. Wen, W. Lv and W. He, J. Mater. Chem. A, 2015, 3, 20031–20050 RSC.
- Y. Chen, Z. Cai, Y. Kuru, W. Ma, H. L. Tuller and B. Yildiz, Adv. Energy Mater., 2013, 3, 1221–1229 CrossRef CAS.
- Z. Feng, Y. Yacoby, M. J. Gadre, Y. Lee, W. T. Hong, H. Zhou, M. D. Biegalski, H. M. Christen, S. B. Adler, D. Morgan and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1027–1034 CrossRef CAS.
- G. J. aO, R. F. Savinell and Y. Shao-Horn, J. Electrochem. Soc., 2009, 156, B771–B781 CrossRef.
- X. Li, M. Liu, S. Y. Lai, D. Ding, M. Gong, J. Lee, K. S. Blinn, Y. Bu, Z. Wang, L. A. Bottomley, F. M. Alamgir and M. Liu, Chem. Mater., 2015, 27, 822–828 CrossRef CAS.
- Z. Tao, G. Hou, N. Xu, Q. Zhang and H. Ding, Electrochim. Acta, 2014, 150, 55–61 CrossRef CAS.
- Y. Chen, S. Huang, X. Ji, K. Adepalli, K. Yin, X. Ling, X. Wang, J. Xue, M. Dresselhaus, J. Kong and B. Yildiz, ACS Nano, 2018, 12, 2569–2579 CrossRef CAS.
- J. Garcia-Barriocanal, F. Y. Bruno, A. Rivera-Calzada, Z. Sefrioui, N. M. Nemes, M. Garcia-Hernández, J. Rubio-Zuazo, G. R. Castro, M. Varela, S. J. Pennycook, C. Leon and J. Santamaria, Adv. Mater., 2010, 5, 627–632 CrossRef.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS.
- X. Feng, L. Feng, M. Jin, J. Zhai and D. Zhu, J. Am. Chem. Soc., 2004, 126, 62–63 CrossRef CAS.
- H. S. Lim, D. Kwak, D. Y. Lee, S. G. Lee and K. Cho, J. Am. Chem. Soc., 2007, 129, 4128–4129 CrossRef CAS.
- S. Singh, J. T. Haraldsen, J. Xiong, E. M. Choi, P. Lu, D. Yi, X. Wen, J. Liu, H. Wang, Z. Bi, P. Yu, M. R. Fitzsimmons, J. L. MacManus-Driscoll, R. Ramesh, A. V. Balatsky, J.-X. Zhu and Q. X. Jia, Phys. Rev. Lett., 2014, 113, 047204 CrossRef.
- C. Ran, X. Jiang, N. Qian and B. Arne, Phys. Rev. Lett., 2014, 113, 057601 CrossRef.
- M. Li, Q. Song, W. Zhao, J. A. Garlow, T. Liu, L. Wu, Y. Zhu, J. S. Moodera, M. H. W. Chan, G. Chen and C. Chang, Phys. Rev. B, 2017, 96, 201301 CrossRef.
- M. Liu and L. Jiang, Sci. China Mater., 2016, 59, 239–246 CrossRef CAS.
- M. Joachim, J. Phys. Chem. Solids, 1985, 46, 309–320 CrossRef.
- X. X. Guo, I. Matei, J. S. Lee and J. Maier, Appl. Phys. Lett., 2007, 91, 103102 CrossRef.
- J. Garcia-Barriocanal, A. Rivera-Calzada, M. Varela, Z. Sefrioui, E. Iborra, C. Leon, S. J. Pennycook and J. Santamaria, Science, 2008, 321, 676–680 CrossRef CAS.
- A. Ohtomo, Nature, 2002, 419, 378–380 CrossRef CAS.
- A. Ohtomo and H. Y. Hwang, Nature, 2004, 441, 423 CrossRef.
- N. Nakagawa, H. Y. Hwang and D. A. Muller, Nat. Mater., 2012, 5, 204–209 CrossRef.
- L. S. Mahmud, A. Muchtar and M. R. Somalu, Renewable Sustainable Energy Rev., 2017, 72, 105–116 CrossRef CAS.
- W. Ma, J. J. Kim, N. Tsvetkov, T. Daio, Y. Kuru, Z. Cai, Y. Chen, K. Sasaki, H. L. Tuller and B. Yildiz, J. Mater. Chem. A, 2015, 3, 207–219 RSC.
- M. G. Blamire, H. Yang, R. Yu, J. L. MacManus-Driscoll, H. Wang, Q. Jia, P. Zerrer, A. Fouchet and J. Yoon, Nat. Mater., 2008, 7, 314–320 CrossRef.
- D. Lee, Y. L. Lee, A. Grimaud, W. T. Hong, M. D. Biegalski, D. Morgan and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 14326–14334 CrossRef CAS.
- Y. Chen, D. D. Fong, F. W. Herbert, J. Rault, J. P. Rueff, T. Nikolai and B. Yildiz, Chem. Mater., 2018, 30, 3359–3371 CrossRef CAS.
- A. Evans, A. Bieberle-Hutter, J. L. M. Rupp and L. J. Gauckler, J. Power Sources, 2009, 194, 119–129 CrossRef CAS.
- D. Kuo, C. J. Eom, J. K. Kawasaki, G. Petretto, J. N. Nelson, G. Hautier, E. J. Crumlin, K. M. Shen, D. G. Schlom and J. Suntivich, J. Phys. Chem. C, 2018, 122, 4359–4364 CrossRef CAS.
- K. H. L. Zhang, Y. Du, P. V. Sushko, M. E. Bowden, V. Shutthanandan, S. Sallis, L. F. J. Piper and S. A. Chambers, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 155129 CrossRef.
- H. Kim, H.-B.-R. Lee and W. J. Maeng, Thin Solid Films, 2009, 517, 2563–2580 CrossRef CAS.
- N. Cheng, Y. Shao, J. Liu and X. Sun, Nano Energy, 2016, 29, 220–242 CrossRef CAS.
- Y. Chen, J. Wang, X. Meng, Y. Zhong, R. Li, X. Sun, S. Ye and S. Knights, Int. J. Hydrogen Energy, 2011, 36, 11092–11805 Search PubMed.
- G. Y. Cho, Y. Kim, S. W. Hong, W. Yu, Y. Kim and S. W. Cha, Nanotechnology, 2018, 29, 345401 CrossRef.
- P. Su, C. Chao, J. H. Shim, R. Fasching and F. B. Prinz, Nano Lett., 2008, 8, 2289–2292 CrossRef CAS PubMed.
- L. Besra and M. Liu, Prog. Mater. Sci., 2007, 52, 1–61 CrossRef CAS.
- A. J. Samson, M. Sogaard, P. Hjalmarsson, J. Hjelm, N. Bonanos, S. P. V. Foghmoes and T. Ramos, Fuel Cells, 2013, 13, 511–519 CrossRef CAS.
- Y. Xiang, S. Lu and S. P. Jiang, Chem. Soc. Rev., 2012, 41, 7291–7321 RSC.
- Y. Chen, Q. Liu, Z. Yang, F. Chen and M. Han, RSC Adv., 2012, 2, 12118–12121 RSC.
- Y. Choi, S. Choi, H. Y. Jeong, M. Liu, B. Kim and G. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 17352–17357 CrossRef CAS.
- T. E. Burye and J. D. Nicholas, J. Power Sources, 2015, 276, 54–61 CrossRef CAS.
- Y. Chen, Y. Lin, Y. Zhang, S. Wang, D. Su, Z. Yang, M. Han and F. Chen, Nano Energy, 2014, 8, 25–33 CrossRef CAS.
- Y. Chen, Y. Zhang, G. Xiao, Z. Yang, M. Han and F. Chen, ChemElectroChem, 2015, 2, 672–678 CrossRef CAS.
- Y. Chen, Y. Zhang, J. Baker, P. Majumdar, Z. Yang, M. Han and F. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 5130–5136 CrossRef CAS PubMed.
- R. Yan, Y. Chen, Y. Lin and F. Chen, Electrochim. Acta, 2016, 217, 187–194 CrossRef CAS.
- D. Dang, B. Zhao, D. Chen, M. D. Ben, C. Qu, S. Dai, X. Zeng, J. Liu, Y. Lu, S. Liao and M. Liu, Energy Storage Mater., 2018, 12, 79–84 CrossRef.
- J. Piao, K. Sun, N. Zhang and S. Xu, J. Power Sources, 2008, 175, 288–295 CrossRef CAS.
- I. Garbayo, F. Baiutti, A. Morata and A. Tarancón, J. Eur. Ceram. Soc., 2019, 39, 101–114 CrossRef CAS.
- Z. Huang, Ariando, W. X. Renshaw, A. Rusydi, J. Chen, H. Yang and T. Venkatesan, Adv. Mater., 2018, 30, 1802439 CrossRef.
- Z. Yang and J. Hao, J. Mater. Chem. C, 2016, 4, 8859–8878 RSC.
- F. Baiutti, G. Christiani and G. Logvenov, Beilstein J. Nanotechnol., 2014, 5, 596–602 CrossRef.
- Z. Guo, H. Li, Q. Chen, L. Sang, L. Yang, Z. Liu and X. Wang, Chem. Mater., 2015, 27, 5988–5996 CrossRef CAS.
- C. Zhao, X. Liu, W. Zhang, Y. Zheng, Y. Li, B. Yu, J. Wang and J. Chen, Int. J. Hydrogen Energy, 2019, 44, 19102–19112 CrossRef CAS.
- Y. Zheng, Y. Li, T. Wu, W. Zhang, J. Zhu, Z. Li, J. Chen, B. Yu, J. Wang and J. Zhang, Nano Energy, 2018, 51, 711–720 CrossRef CAS.
- N. Schichtel, C. Korte, D. Hesse, N. Zakharov, B. Butz, D. Gerthsen and J. Janek, Phys. Chem. Chem. Phys., 2010, 12, 14596 RSC.
- M. Brahlek, A. S. Gupta, J. Lapano, J. Roth, H. T. Zhang, L. Zhang, R. Haislmaier and R. Engel-Herbert, Adv. Funct. Mater., 2017, 28, 1702772 CrossRef.
- X. Yuan, L. Tang, S. Liu, P. Wang, Z. Chen, C. Zhang, Y. Liu, W. Wang, Y. Zou and C. Liu, Nano Lett., 2015, 15, 3571 CrossRef CAS.
- R. Zhao and X. Wang, Chem. Mater., 2018, 31, 445–453 CrossRef.
- X. Meng, X. Wang, D. Geng, C. Ozgit-Akgun, N. Schneider and J. W. Elam, Mater. Horiz., 2017, 4, 133–154 RSC.
- J. Wang, Z. Guo, X. Wei and X. Wang, Chem. – Eur. J., 2018 DOI:10.1002/chem.201803327.
- Y. Yang, M. Li, Y. Ren, Y. Li and C. Xia, Int. J. Hydrogen Energy, 2018, 43, 3797–3802 CrossRef CAS.
- M. Nadeem, B. Hu and C. Xia, Int. J. Hydrogen Energy, 2018, 43, 8079–8087 CrossRef CAS.
- T. Hong, K. Brinkman and C. Xia, J. Power Sources, 2016, 329, 281–289 CrossRef CAS.
- T. Wu, W. Zhang, Y. Li, Y. Zheng, B. Yu, J. Chen and X. Sun, Adv. Energy Mater., 2018, 8, 1802203 CrossRef.
- D. Ding, X. Li, S. Y. Lai, K. Gerdes and M. Liu, Energy Environ. Sci., 2014, 7, 552–575 RSC.
- F. Liang, J. Chen, S. P. Jiang, B. Chi, J. Pu and L. Jian, Electrochem. Commun., 2009, 11, 1048–1051 CrossRef CAS.
- K. Yashiro, T. Takamura, M. Sase, F. Hermes, K. Sato, T. Kawada and J. Mizusaki, ECS Trans., 2007, 7, 1287–1292 Search PubMed.
- M. Tsuchiya, B. Lai and S. Ramanathan, Nat. Nanotechnol., 2011, 6, 282–286 CrossRef CAS.
- S. M. Haile, D. A. Boysen, C. R. I. Chisholm and R. B. Merle, Nature, 2001, 910–913 CrossRef CAS.
- V. Kharton, F. Marques and A. Atkinson, Solid State Ionics, 2004, 174, 135–149 CrossRef CAS.
- T. Ishihara, Bull. Chem. Soc. Jpn., 2006, 79, 1155–1166 CrossRef CAS.
- D. Pergolesi, E. Fabbri, S. N. Cook, V. Roddatis, E. Traversa and J. A. Kilner, ACS Nano, 2012, 6, 10524–10534 CrossRef CAS.
- I. Kosacki, C. M. Rouleau, P. F. Becher, J. Bentley and D. H. Lowndes, Electrochem. Solid-State Lett., 2004, 12, A459–A461 CrossRef.
- L. Fan, C. Wang, M. Chen and B. Zhu, J. Power Sources, 2013, 234, 154–174 CrossRef CAS.
- K. Lee, K. Ahn, Y. Chung, J. Lee and H. Yoo, Solid State Ionics, 2012, 229, 45–53 CrossRef CAS.
- Y. Wu and S. Chen, Int. J. Hydrogen Energy, 2017, 42, 27284–27297 CrossRef CAS.
- F. Bozza, R. Polini and E. Traversa, Electrochem. Commun., 2009, 11, 1680–1683 CrossRef CAS.
- N. M. Sammes, G. A. Tomsett, H. Nafe and F. Aldinger, J. Eur. Ceram. Soc., 1999, 19, 1801–1826 CrossRef CAS.
- Y. Mishima, H. Mitsuyasu, M. Ohtaki and K. Eguchi, J. Electrochem. Soc., 1998, 145, 1004–1007 CrossRef CAS.
- J. B. Goodenough, Annu. Rev. Mater. Res., 2003, 33, 91–128 CrossRef CAS.
- N. P. Brandon, S. Skinner and B. C. H. Steele, Annu. Rev. Mater. Res., 2003, 331, 183–213 CrossRef.
- H. Iwahara, H. Uchida, K. Ono and K. Ogaki, J. Electrochem. Soc., 1988, 135, 369–375 CrossRef.
- H. Iwahara, T. Esaka, H. Uchida and N. Maeda, Solid State Ionics, 1981, 3, 359–363 CrossRef.
- Z. Zhu, E. Guo, Z. Wei and H. Wang, J. Power Sources, 2018, 373, 132–138 CrossRef CAS.
- K. Xie, R. Yan, X. Chen, S. Wang, Y. Jiang, X. Liu and G. Meng, J. Alloys Compd., 2009, 473, 323–329 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355 RSC.
- S. Sun and Z. Cheng, J. Electrochem. Soc., 2017, 10, F3104–F3113 CrossRef.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. OHayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- L. Bi, S. Boulfrad and E. Travers, Chem. Soc. Rev., 2014, 43, 8255–8270 RSC.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T. Lim and G. Kim, Nano Energy, 2018, 44, 121–126 CrossRef CAS.
- V. Foglietti, N. Yang, A. Tebano, C. Aruta, E. Di Bartolomeo, S. Licoccia, C. Cantoni and G. Balestrino, Appl. Phys. Lett., 2014, 104, 81612 CrossRef.
- Y. Shi, I. Garbayo, P. Muralt and J. L. M. Rupp, J. Mater. Chem. A, 2017, 5, 3900–3908 RSC.
- A. Fluri, D. Pergolesi, V. Roddatis, A. Wokaun and T. Lippert, Nat. Commun., 2016, 7, 10692 CrossRef CAS.
- D. S. Aidhy, B. Liu, Y. Zhang and W. J. Weber, J. Phys. Chem. C, 2014, 118, 30139–30144 CrossRef CAS.
- L. Chen, C. L. Chen, X. Chen, W. Donner, S. W. Liu, Y. Lin, D. X. Huang and A. J. Jacobson, Appl. Phys. Lett., 2003, 83, 4737–4739 CrossRef CAS.
- W. Shen, J. Jiang and J. L. Hertz, J. Phys. Chem. C, 2014, 118, 22904–22912 CrossRef CAS.
- A. Peters, C. Korte, D. Hesse, N. Zakharov and J. Janek, Solid State Ionics, 2007, 178, 67–76 CrossRef CAS.
- W. L. Cheah and M. W. Finnis, J. Mater. Sci., 2012, 47, 1631–1640 CrossRef CAS.
- S. Lee, W. Zhang, F. Khatkhatay, H. Wang, Q. Jia and J. L. MacManus-Driscoll, Nano Lett., 2015, 15, 7362–7369 CrossRef.
- S. M. Yang, S. Lee, J. Jian, W. Zhang, P. Lu, Q. Jia, H. Wang, T. W. Noh, S. V. Kalinin and J. L. MacManus-Driscll, Nat. Commun., 2015, 6, 8588 CrossRef CAS.
- X. B. Liao, Y. Ni, H. Yang and L. H. He, Appl. Phys. Lett., 2013, 103, 141903 CrossRef.
- T. He, J. Feng, J. Ru, Y. Feng, R. Lian and J. Yang, ACS Nano, 2019, 13, 830–838 CrossRef CAS PubMed.
- C. Xia, Y. Li, Y. Tian, Q. Liu, Z. Wang, L. Jia, Y. Zhao and Y. Li, J. Power Sources, 2010, 195, 3149–3154 CrossRef CAS.
- R. A. De Souza, A. Ramadan and S. Hörner, Energy Environ. Sci., 2012, 5, 5445–5453 RSC.
- P. P. Dholabhai, G. Pilania, J. A. Aguiar, A. Misra and B. P. Uberuaga, Nat. Commun., 2014, 5, 5043 CrossRef CAS.
- M. Sillassen, P. Eklund, N. Pryds, E. Johnson, U. Helmersson and J. Bøttiger, Adv. Funct. Mater., 2010, 20, 2071–2076 CrossRef CAS.
- H. Lee, N. Campbell, J. Lee, T. J. Asel, T. R. Paudel, H. Zhou, J. W. Lee, B. Noesges, J. Seo, B. Park, L. J. Brillson, S. H. Oh, E. Y. Tsymbal, M. S. Rzchowski and C. B. Eom, Nat. Mater., 2018, 17, 231–236 CrossRef CAS.
- D. S. Mebane and R. A. De Souza, Energy Environ. Sci., 2015, 8, 2935–2940 RSC.
- R. A. De Souza and E. C. Dickey, Philos. Trans. R. Soc., A, 2019, 377, 20180430 CrossRef CAS PubMed.
- K. M. Kant, V. Esposito and N. Pryds, Appl. Phys. Lett., 2010, 97, 143110 CrossRef.
- J. Jiang, X. Hu, W. Shen, C. Ni and J. L. Hertz, Appl. Phys. Lett., 2013, 102, 143901 CrossRef.
- Y. Shi, A. H. Bork, S. Schweiger and J. L. M. Rupp, Nat. Mater., 2015, 14, 721–728 CrossRef CAS PubMed.
- G. F. Harrington, A. Cavallaro, D. W. Mccomb, S. J. Skinner and J. A. Kilner, Phys. Chem. Chem. Phys., 2017, 19, 14319–14336 RSC.
- C. Korte, J. Keppner, A. Peters, N. Schichtel, H. Aydin and J. Janek, Phys. Chem. Chem. Phys., 2014, 16, 24575–24591 RSC.
- W. Shen and J. L. Hertz, J. Mater. Chem. A, 2015, 3, 2378–2386 RSC.
- S. Azad, O. A. Marina, C. M. Wang, L. Saraf, V. Shutthanandan, D. E. McCready, A. El-Azab, J. E. Jaffe, M. H. Engelhard, C. H. F. Peden and S. Thevuthasan, Appl. Phys. Lett., 2005, 86, 131906 CrossRef.
- C. Korte, A. Peters, J. Janek, D. Hesse and N. Zakharov, Phys. Chem. Chem. Phys., 2008, 10, 4623 RSC.
- X. Guo, Science, 2009, 324, 465 CrossRef CAS PubMed.
- A. Cavallaro, M. Burriel, J. Roqueta and E. Al, Solid State Ionics, 2010, 181, 592–601 CrossRef CAS.
- S. Sanna, V. Esposito, A. Tebano, S. Licoccia, E. Traversa and G. Balestrino, Small, 2010, 6, 1863–1867 CrossRef CAS PubMed.
- A. Cavallaro, M. Burriel, J. Roqueta, A. Apostolidis, A. Bernardi, A. Tarancón, R. Srinivasan, S. N. Cook, H. L. Fraser, J. A. Kilner, D. W. McComb and J. Santisoa, Solid State Ionics, 2010, 181, 592–601 CrossRef CAS.
- R. A. De Souza and A. H. H. Ramadan, Phys. Chem. Chem. Phys., 2013, 15, 4505–4509 RSC.
- B. Li, J. Zhang, T. Kaspar, V. Shutthanandan, R. C. Ewing and J. Lian, Phys. Chem. Chem. Phys., 2013, 15, 1296–1301 RSC.
- H. Aydin, C. Korte, M. Rohnke and J. Janek, Phys. Chem. Chem. Phys., 2013, 15, 1944–1955 RSC.
- E. Ruiz-Trejo, K. Thyden, N. Bonanos and M. B. Mogensen, Solid State Ionics, 2016, 288, 82–87 CrossRef CAS.
- W. Lv, N. Feng, Y. Niu, F. Yang, K. Wen, M. Zou, Y. Han, J. Zhao and W. He, Solid State Ionics, 2016, 289, 168–172 CrossRef CAS.
- J. M. Polfus, T. Norby and R. Bredesen, Solid State Ionics, 2016, 297, 77–81 CrossRef CAS.
- J. Yang, M. Youssef and B. Yildiz, Phys. Chem. Chem. Phys., 2017, 19, 3869–3883 RSC.
- N. Yang, C. Cantoni, V. Foglietti, A. Tebano, A. Belianinov, E. Strelcov, S. Jesse, D. Di Castro, E. Di Bartolomeo, S. Licoccia, S. V. Kalinin, G. Balestrino and C. Aruta, Nano Lett., 2015, 15, 2343–2349 CrossRef CAS PubMed.
- E. Fabbri, D. Pergolesi and T. Enrico, Sci. Technol. Adv. Mater., 2010, 11, 54503 CrossRef PubMed.
- C. Korte, N. Schichtel, D. Hesse and J. Janek, Monatsh. Chem., 2009, 140, 1069–1080 CrossRef CAS PubMed.
- M. Zou, F. Yang, K. Wen, W. Lv, M. Waqas and W. He, Int. J. Hydrogen Energy, 2016, 41, 22254–22259 CrossRef CAS.
- J. L. M. Rupp, E. Fabbri, D. Marrocchelli, J. Han, D. Chen, E. Traversa, H. L. Tuller and B. Yildiz, Adv. Funct. Mater., 2014, 24, 1562–1574 CrossRef CAS.
- H. Jalili, J. W. Han, Y. Kuru, Z. Cai and B. Yildiz, J. Phys. Chem. Lett., 2011, 2, 801–807 CrossRef CAS.
- N. Schichtel, C. Korte, D. Hesse and J. Janek, Phys. Chem. Chem. Phys., 2009, 11, 3043–3048 RSC.
- T. Nakamura, K. Yashiro, K. Sato and J. Mizusaki, Phys. Chem. Chem. Phys., 2009, 11, 3055 RSC.
- T. Petrişor, A. Meledin, A. Boulle, R. B. Moş, M. S. Gabor, L. Ciontea and T. Petrişor, Appl. Surf. Sci., 2018, 433, 668–673 CrossRef.
- S. Krzysztof, S. Wolfgang, B. Gustav and W. Rainer, Nat. Mater., 2006, 5, 312 CrossRef PubMed.
- E. Navickas, Y. Chen, Q. Lu, W. Wallisch, T. M. Huber, J. Bernardi, M. Stöger-Pollach, G. Friedbacher, H. Hutter, B. Yildiz and J. Fleig, ACS Nano, 2018, 11, 11475–11487 CrossRef PubMed.
- C. Chang, M. Chu, H. Jeng, S. Cheng, J. Lin, J. Yang and C. Chen, Nat. Commun., 2014, 5, 3522–3530 CrossRef PubMed.
- M. J. Demkowicz, A. Misra and A. Caro, Curr. Opin. Solid State Mater. Sci., 2012, 16, 101–108 CrossRef CAS.
- L. Marc, D. Gerhard, A. Eduard and B. T. John, Science, 2008, 319, 1646–1649 CrossRef PubMed.
- L. Sun, D. Marrocchelli and B. Yildiz, Nat. Commun., 2015, 6, 6294 CrossRef CAS PubMed.
- D. Marrocchelli, L. Sun and B. Yildiz, J. Am. Chem. Soc., 2015, 137, 4735–4748 CrossRef CAS PubMed.
- T. Yeh, R. Lin and J. Cherng, Thin Solid Films, 2015, 574, 66–70 CrossRef CAS.
- A. Ohtomo and H. Hwang, Nature, 2004, 427, 423–426 CrossRef CAS PubMed.
- E. Dagotto, Science, 2007, 318, 1076–1077 CrossRef CAS PubMed.
- J. Maier, Prog. Solid State Chem., 1995, 23, 171–263 CrossRef CAS.
- G. Gregori, R. Merkle and J. Maier, Prog. Mater. Sci., 2017, 89, 252–305 CrossRef CAS.
- M. Shirpour, B. Rahmati, W. Sigle, P. A. V. Aken, R. Merkle and J. Maier, J. Phys. Chem. C, 2012, 116, 2453–2461 CrossRef CAS.
- D. Lee, S. H. Baek, T. H. Kim, J. G. Yoon, C. M. Folkman, C. B. Eom and T. W. Noh, Phys. Rev. B, 2011, 84, 125305 CrossRef.
- A. Karthikeyan, C. L. Chang and S. Ramanathan, Appl. Phys. Lett., 2006, 89, 1123–1127 CrossRef.
- W. J. Bowman, J. Zhu, R. Sharma and P. A. Crozier, Solid State Ionics, 2015, 272, 9–17 CrossRef CAS.
- M. C. Göbel, G. Gregori and J. Maier, J. Phys. Chem. C, 2013, 117, 22560–22568 CrossRef.
- H. Li, F. L. Souza and R. H. R. Castro, J. Eur. Ceram. Soc., 2018, 38, 1750–1759 CrossRef CAS.
- J. M. Polfus, T. Kazuaki, O. Fumiyasu, T. Isao and H. Reidar, Phys. Chem. Chem. Phys., 2012, 14, 12339–12346 RSC.
- X. Guo, W. Sigle and J. Maier, J. Am. Ceram. Soc., 2010, 86, 77–87 CrossRef.
- K. K. Adepalli, J. Yang, J. Maier, H. L. Tuller and B. Yildiz, Adv. Funct. Mater., 2017, 27, 1700243 CrossRef.
- P. Lupetin, G. Gregori and J. Maier, Angew. Chem., Int. Ed., 2010, 49, 10123–10126 CrossRef CAS PubMed.
- A. Tschöpe, E. Sommer and R. Birringer, Solid State Ionics, 2001, 139, 267–280 CrossRef.
- W. Tong, W. Zhang, B. Yu and J. Chen, Int. J. Hydrogen Energy, 2017, 42, 29900–29910 CrossRef.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- T. Wu, B. Yu, W. Zhang, J. Chen and S. Zhao, RSC Adv., 2016, 6, 68379–68387 RSC.
- S. Liu, W. Zhang, Y. Li and B. Yu, RSC Adv., 2017, 7, 16332–16340 RSC.
- S. Liu, B. Yu, W. Zhang, Y. Zhai and J. Chen, Int. J. Hydrogen Energy, 2016, 41, 15952–15959 CrossRef CAS.
- S. J. Kim, A. M. Dayaghi, K. J. Kim and G. M. Choi, J. Power Sources, 2017, 344, 218–222 CrossRef CAS.
- L. Luo, Y. Lin, J. Shi, L. Cheng, Y. Wu and L. Sun, J. Inorg. Mater., 2016, 7, 756–760 Search PubMed.
- J. Miao, H. Y. Tian, X. Y. Zhou, K. H. Pang and Y. Wang, J. Appl. Phys., 2007, 101, 84101 CrossRef.
- E. J. Crumlin, S. Ahn, D. Lee, E. Mutoro, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, J. Electrochem. Soc., 2012, 159, F219–F225 CrossRef CAS.
- Y. Zheng, Y. Li, T. Wu, C. Zhao, W. Zhang, J. Zhu, Z. Li, J. Chen, J. Wang, B. Yu and J. Zhang, Nano Energy, 2019, 62, 521–529 CrossRef CAS.
- K. Świerczek, Solid State Ionics, 2011, 192, 486–490 CrossRef.
- W. Zhang, H. Wang, K. Guan, Z. Wei, X. Zhang, J. Meng, X. Liu and J. Meng, ACS Appl. Mater. Interfaces, 2019, 11, 26830–26841 CrossRef CAS PubMed.
- E. Mutoro, E. J. Crumlin, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 3689–3696 RSC.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tulle and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979–7988 RSC.
- M. Kubicek, Z. Cai, W. Ma, B. Yildiz, H. Hutter and J. Fleig, ACS Nano, 2013, 7, 3276–3286 CrossRef CAS PubMed.
- N. Tsvetkov, Y. Chen and B. Yildiz, J. Mater. Chem. A, 2014, 2, 14690–14695 RSC.
- D. Lee, Y. L. Lee, W. T. Hong, M. D. Biegalski, D. Morgan and Y. Shao-Horn, J. Mater. Chem. A, 2015, 3, 2144–2157 RSC.
- J. R. Petrie, C. Mitra, H. Jeen, W. S. Choi, T. L. Meyer, F. A. Reboredo, J. W. Freeland, G. Eres and H. N. Lee, Adv. Funct. Mater., 2016, 26, 1564–1570 CrossRef CAS.
- Y. Fujimaki, K. Mizuno, Y. Kimura, T. Nakamura, K. D. Bagarinao, K. Yamaji, K. Yashiro, T. Kawada, F. Iguchi, H. Yugami and K. Amezawa, ECS Trans., 2017, 78, 847–853 CrossRef CAS.
- C. Lenser, Q. Lu, E. Crumlin, H. Bluhm and B. Yildiz, J. Phys. Chem. C, 2018, 122, 4841–4848 CrossRef CAS.
- H. Chen, Z. Guo, L. A. Zhang, Y. Li, F. Li, Y. Zhang, Y. Chen, X. Wang, B. Yu, J. Shi, J. Liu, C. Yang, S. Cheng, Y. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 39785–39793 CrossRef CAS PubMed.
- Y. Kuru, H. Jalili, Z. Cai, B. Yildiz and H. L. Tuller, Adv. Mater., 2011, 23, 4543–4548 CrossRef CAS PubMed.
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Nat. Mater., 2016, 15, 1010–1016 CrossRef CAS PubMed.
- M. Sase, K. Sato and K. Yashiro, Electrochem. Solid-State Lett., 2009, 9, B135–B137 Search PubMed.
- C. Xia, Y. Cai, Y. Ma, B. Wang, W. Zhang, M. Karlsson, Y. Wu and B. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 20748–20755 CrossRef CAS PubMed.
- J. F. Shin, W. Xu, M. Zanella, K. Dawson, S. N. Savvin, J. B. Claridge and M. J. Rosseinsky, Nat. Energy, 2017, 2, 16214 CrossRef CAS.
- Y. Gong, R. L. Patel, X. Liang, D. Palacio, X. Song, J. B. Goodenough and K. Huang, Chem. Mater., 2013, 25, 4224–4231 CrossRef CAS.
- Y. Gong, D. Palacio, X. Song, R. L. Patel, X. Liang, X. Zhao, J. B. Goodenough and K. Huang, Nano Lett., 2013, 13, 4340–4345 CrossRef CAS PubMed.
- A. Samreen, S. Saher, S. Ali, H. Seema and A. Qamar, Int. J. Hydrogen Energy, 2019, 44, 6223–6228 CrossRef CAS.
- E. Kim, H. Jung, K. An, J. Park, J. Lee, I. Hwang, J. Kim, M. Lee, Y. Kwon and J. Hwang, Ceram. Int., 2014, 40, 7817–7822 CrossRef CAS.
- T. Hong, M. Zhao and K. Brinkman, ACS Appl. Mater. Interfaces, 2017, 9, 8659–8668 CrossRef CAS PubMed.
- X. Zhu, H. Xia, Y. Li and Z. Lü, Mater. Lett., 2015, 161, 549–553 CrossRef CAS.
- J. Li, S. Wang, Z. Wang, R. Liu, T. Wen and Z. Wen, J. Power Sources, 2009, 194, 625–630 CrossRef CAS.
- D. Ding, M. Liu, Z. Liu, X. Li, K. Blinn, X. Zhu and M. Liu, Adv. Energy Mater., 2013, 3, 1149–1154 CrossRef CAS.
- M. E. Lynch, Y. Lei, W. T. Qin, J. J. Choi, M. F. Liu, K. Blinn and M. L. Liu, Energy Environ. Sci., 2011, 4, 2249–2258 RSC.
- X. Zhang, Z. Hui and X. Liu, J. Power Sources, 2014, 269, 412–417 CrossRef CAS.
- W. Zhou, F. Liang, Z. Shao and Z. Zhu, Sci. Rep., 2012, 2, 327 CrossRef PubMed.
- S. Stammler, R. Merkle, B. Stuhlhofer, G. Logvenov, K. Hahn, P. A. van Aken and J. Maier, Solid State Ionics, 2017, 303, 172–180 CrossRef.
- J. Wang, T. Yang, Y. Wen, Y. Zhang, C. Sun and K. Huang, J. Power Sources, 2019, 414, 24–30 CrossRef CAS.
- G. M. Rupp, A. K. Opitz, A. Nenning, A. Limbeck and J. Fleig, Nat. Mater., 2017, 16, 640–645 CrossRef CAS PubMed.
- H. J. Choi, K. Bae, D. Y. Jang, J. W. Kim and J. H. Shim, J. Electrochem. Soc., 2015, 162, F622–F626 CrossRef CAS.
- A. Karimaghaloo, J. Koo, H. Kang, S. A. Song, J. H. Shim and M. H. Lee, Int. J. Pr. Eng. Man-GT, 2019, 6, 611–628 Search PubMed.
- Y. Chen, Y. Chen, D. Ding, Y. Ding, Y. Choi, L. Zhang, S. Yoo, D. Chen, B. DeGlee, H. Xu, Q. Lu, B. Zhao, G. Vardar, J. Wang, H. Bluhm, E. J. Crumlin, C. Yang, J. Liu, B. Yildiz and M. Liu, Energy Environ. Sci., 2017, 10, 964–971 RSC.
- Y. Chen, S. Yoo, K. Pei, D. Chen, L. Zhang, B. DeGlee, R. Murphy, B. Zhao, Y. Zhang, Y. Chen and M. Liu, Adv. Funct. Mater., 2018, 28, 1704907 CrossRef.
- Y. Chen, B. DeGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS.
- J. Chakhalian, J. W. Freeland, H. U. Habermeier, G. Cristiani, G. Khaliullin, M. V. Veenendaal and B. Keimer, Science, 2007, 318, 1114–1117 CrossRef CAS PubMed.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- F. S. Baumann, J. Fleig, M. Konuma, U. Starke, H. U. Habermeier and J. Maier, J. Electrochem. Soc., 2005, 152, A2074–A2079 CrossRef CAS.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- S. N. Stephen, K. Rainer, V. John and A. B. Dawn, ACS Nano, 2013, 7, 6330–6336 CrossRef PubMed.
- W. C. Chueh, Y. Hao, W. Jung and S. M. Haile, Nat. Mater., 2012, 11, 155–161 CrossRef CAS PubMed.
- J. W. Han and B. Yildiz, J. Mater. Chem., 2011, 21, 11899–18983 Search PubMed.
- B. Koo, H. Kwon, Y. Kim, H. G. Seo, J. W. Han and W. Jung, Energy Environ. Sci., 2018, 11, 71–77 RSC.
- T. Mayeshiba and D. Morgan, Solid State Ionics, 2017, 311, 105–117 CrossRef CAS.
- X. Liu, L. Zhang, Y. Zheng, Z. Guo, Y. Zhu, H. Chen, F. Li, P. Liu, B. Yu, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Sci., 2019, 1801898, DOI:10.1002/advs.201801898.
- F. Li, Y. Li, H. Chen, H. Li, Y. Zheng, Y. Zhang, B. Yu, X. Wang, J. Liu, C. Yang, Y. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 36926–36932 CrossRef CAS PubMed.
- Y. A. Mastrikov, R. Merkle, E. Heifets, E. A. Kotomin and J. Maier, J. Phys. Chem. C, 2010, 114, 3017–3027 CrossRef CAS.
- J. Hwang, Z. Feng, N. Charles, X. R. Wang, D. Lee, K. A. Stoerzinger, S. Muy, R. R. Rao, D. Lee, R. Jacobs, D. Morgan and Y. Shao-Horn, Mater. Today, 2019 10.1039/C4RA11973H.
- D. Pesquera, G. Herranz, A. Barla, E. Pellegrin, F. Bondino, E. Magnano, F. Sánchez and J. Fontcuberta, Nat. Commun., 2012, 3, 1189 CrossRef CAS PubMed.
- J. M. Rondinelli and N. A. Spaldin, Adv. Mater., 2011, 23, 3363–3381 CrossRef CAS PubMed.
- W. C. Jung and H. L. Tuller, Energy Environ. Sci., 2012, 5, 5370–5378 RSC.
- Y. Chen, H. Téllez, M. Burriel, F. Yang, N. Tsvetkov, Z. Cai, D. W. McComb, J. A. Kilner and B. Yildiz, Chem. Mater., 2015, 27, 5436–5450 CrossRef CAS.
- S. He, K. F. Chen, M. Saunders, Z. Quadir, S. W. Tao, J. T. S. Irvine., C. Q. Cui and S. P. Jiang, Solid State Ionics, 2018, 325, 176–188 CrossRef CAS.
- Y. Li, W. Zhang, T. Wu, Y. Zheng, J. Chen, B. Yu, J. Zhu and M. Liu, Adv. Energy Mater., 2018, 8, 1801893 CrossRef.
- Y. L. Lee, J. Kleis, J. Rossmeisl, S. H. Yang and D. Morgan, Energy Environ. Sci., 2011, 4, 3966–3970 RSC.
- M. B. Mogensen, C. Chatzichristodoulou, C. Graves, K. V. Hansen, K. K. Hansen, A. Hauch, T. Jacobsen and K. Norrman, ECS Trans., 2016, 72, 93–103 CrossRef CAS.
- G. M. Rupp, H. Téllez, J. Druce, A. Limbeck, T. Ishihara, J. Kilner and J. Fleig, J. Mater. Chem. A, 2015, 3, 22759–22769 RSC.
- Y. Wen, T. Yang, D. Lee, H. N. Lee, E. J. Crumlin and K. Huang, J. Mater. Chem. A, 2018, 6, 24378–24388 RSC.
- H. Ding, A. Virkar, M. Liu and F. Liu, Phys. Chem. Chem. Phys., 2013, 15, 489–496 RSC.
- D. Ding, M. Liu and M. Liu, ECS Trans., 2013, 57, 1801–1810 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 9, 1383–1385 CrossRef PubMed.
- P. Bo, C. Gang, W. Tao, J. Zhou, J. Guo, Y. Cheng and W. Kai, J. Power Sources, 2012, 201, 174–178 CrossRef.
- A. Yamada, Y. Suzuki, K. Saka, M. Uehara, D. Mori, R. Kanno, T. Kiguchi, F. Mauvy and J. C. Grenier, Adv. Mater., 2010, 20, 4124–4128 CrossRef.
- K. Chen, N. Ai and S. P. Jiang, Int. J. Hydrogen Energy, 2014, 39, 10349–10358 CrossRef CAS.
- Y. Wan, B. Hu and C. Xia, Electrochim. Acta, 2017, 252, 171–179 CrossRef CAS.
- Y. Song, W. Wang, L. Ge, X. Xu, Z. Zhang, P. S. B. Julião, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1700337 CrossRef PubMed.
- S. Cui, J. Li, X. Zhou, G. Wang, J. Luo, K. T. Chuang, Y. Bai and L. Qiao, J. Mater. Chem. A, 2013, 1, 9689–9696 RSC.
- B. Hua, N. Yan, M. Li, Y. Q. Zhang, Y. F. Sun, J. Li, T. Etsell, P. Sarkar, K. Chuang and J. L. Luo, Energy Environ. Sci., 2016, 9, 207–215 RSC.
- W. Wang, C. Su, Y. Wu, R. Ran and Z. Shao, Chem. Rev., 2013, 113, 8104–8151 CrossRef CAS PubMed.
- L. Yang, Y. Choi, W. Qin, H. Chen, K. Blinn, M. Liu, P. Liu, J. Bai, T. A. Tyson and M. Liu, Nat. Commun., 2011, 2, 357 CrossRef PubMed.
- J. Qu, W. Wang, Y. Chen, X. Deng and Z. Shao, Appl. Energy, 2016, 164, 563–571 CrossRef CAS.
- A. L. Vincent, A. R. Hanifi, J. Luo, K. T. Chuang, A. R. Sanger, T. H. Etsell and P. Sarkar, J. Power Sources, 2012, 215, 301–306 CrossRef CAS.
- J. Xu, X. Zhou, X. Dong, L. Pan and K. Sun, Int. J. Hydrogen Energy, 2017, 42, 15632–15640 CrossRef CAS.
- J. Zhou, T. H. Shin, C. Ni, G. Chen, K. Wu, Y. Cheng and J. T. S. Irvine, Chem. Mater., 2016, 28, 2981–2993 CrossRef CAS.
- L. Ye, M. Zhang, P. Huang, G. Guo, M. Hong, C. Li, J. T. S. Irvine and K. Xie, Nat. Commun., 2017, 8, 14785 CrossRef CAS PubMed.
- B. Hua, W. Zhang, L. Meng, W. Xin, C. Bo, P. Jian and L. Jian, J. Power Sources, 2014, 247, 170–177 CrossRef CAS.
- E. Lay, C. Metcalfe and O. Kesler, J. Power Sources, 2012, 218, 237–243 CrossRef CAS.
- M. Asamoto, S. Miyake, K. Sugihara and H. Yahiro, Electrochem. Commun., 2009, 11, 1508–1511 CrossRef CAS.
- D. L. Rosa, A. Sin, M. L. Faro, G. Monforte, V. Antonucci and A. S. Aricò, J. Power Sources, 2009, 193, 160–164 CrossRef.
- S. Islam and J. M. Hill, J. Mater. Chem. A, 2014, 2, 1922–1929 RSC.
- M. D. McIntyre, J. D. Kirtley, A. Singh, S. Islam, J. M. Hill and R. A. Walker, J. Phys. Chem. C, 2015, 119, 7637–7647 CrossRef CAS.
- A. Singh and J. M. Hill, J. Power Sources, 2012, 214, 185–194 CrossRef CAS.
- W. Yue, Y. Li, Y. Zheng, T. Wu, C. Zhao, J. Zhao, G. Geng, W. Zhang, J. Chen, J. Zhu and B. Yu, Nano Energy, 2019, 62, 64–78 CrossRef CAS.
- B. Hua, M. Li, Y. Sun, Y. Zhang, N. Yan, J. Li, T. Etsell, P. Sarkar and J. Luo, Appl. Catal., B, 2017, 200, 174–181 CrossRef CAS.
- W. Wang, L. Gan, J. P. Lemmon, F. Chen, J. T. S. Irvine and K. Xie, Nat. Commun., 2019, 10, 1550 CrossRef PubMed.
- Y. Li, K. Xie, S. Chen, H. Li, Y. Zhang and Y. Wu, Electrochim. Acta, 2015, 153, 325–333 CrossRef CAS.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- G. Yang, W. Zhou, M. Liu and Z. Shao, ACS Appl. Mater. Interfaces, 2016, 8, 35308–35314 CrossRef CAS PubMed.
- G. Tsekouras, D. Neagu and J. T. S. Irvine, Energy Environ. Sci., 2012, 6, 256–266 RSC.
- D. Neagu, T. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- S. Liu, Q. Liu and J. Luo, ACS Catal., 2016, 6, 6219–6228 CrossRef CAS.
- H. Wei, K. Xie, J. Zhang, Y. Zhang, Y. Wang, Y. Qin, J. Cui, J. Yan and Y. Wu, Sci. Rep., 2015, 4, 5156 CrossRef PubMed.
- K. Xie, J. Zhang, S. Xu, B. Ding, G. Wu, T. Xie and Y. Wu, Fuel Cells, 2014, 14, 1036–1045 CrossRef CAS.
- W. Qi, C. Ruan, G. Wu, Y. Zhang, Y. Wang, K. Xie and Y. Wu, Int. J. Hydrogen Energy, 2014, 39, 5485–5496 CrossRef CAS.
- S. Liu, Q. Liu and J. Luo, J. Mater. Chem. A, 2016, 4, 17521–17528 RSC.
- Y. Song, Y. Yin, L. Li, Z. Chen and Z. Ma, Chem. Commun., 2018, 54, 12341–12344 RSC.
- Y. Sun, J. Li, Y. Zeng, B. S. Amirkhiz, M. Wang, Y. Behnamian and J. Luo, J. Mater. Chem. A, 2015, 3, 11048–11056 RSC.
- Y. Sun, Y. Zhang, J. Chen, J. Li, Y. Zhu, Y. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J. Luo, Nano Lett., 2016, 16, 5303–5309 CrossRef CAS PubMed.
- T. Wei, L. Jia, H. Zheng, B. Chi, J. Pu and J. Li, Appl. Catal., A, 2018, 564, 199–207 CrossRef CAS.
- N. Zhou, Y. Yin, Z. Chen, Y. Song, J. Yin, D. Zhou and Z. Ma, J. Electrochem. Soc., 2018, 165, F629–F634 CrossRef CAS.
- C. Yang, J. Li, Y. Lin, J. Liu, F. Chen and M. Liu, Nano Energy, 2015, 11, 704–710 CrossRef CAS.
- C. Yang, Z. Yang, C. Jin, G. Xiao, F. Chen and M. Han, Adv. Mater., 2012, 24, 1439–1443 CrossRef CAS PubMed.
- N. Hou, T. Yao, P. Li, X. Yao, T. Gan, L. Fan, J. Wang, X. Zhi, Y. Zhao and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 6995–7005 CrossRef CAS PubMed.
- K. Lai and A. Manthiram, Chem. Mater., 2018, 30, 2515–2525 CrossRef CAS.
- S. Liu, K. T. Chuang and J. Luo, ACS Catal., 2016, 6, 760–768 CrossRef CAS.
- Z. Du, H. Zhao, S. Yi, Q. Xia, Y. Gong, Y. Zhang, X. Cheng, Y. Li, L. Gu and K. Świerczek, ACS Nano, 2016, 10, 8660–8669 CrossRef CAS PubMed.
- Y. Sun, J. Li, L. Cui, B. Hua, S. Cui, J. Li and J. Luo, Nanoscale, 2015, 7, 11173–11181 RSC.
- Z. Wang, Y. Yin, Y. Yu, Y. Song, Z. Ma and J. Yin, Int. J. Hydrogen Energy, 2018, 43, 10440–10447 CrossRef CAS.
- J. Li, B. Wei, Z. Cao, X. Yue, Y. Zhang and Z. Lü, ChemSusChem, 2018, 11, 254–263 CrossRef CAS PubMed.
- D. Zubenko, S. Singh and B. A. Rosen, Appl. Catal., B, 2017, 209, 711–719 CrossRef CAS.
- W. Wang, C. Zhu, K. Xie and L. Gan, J. Power Sources, 2018, 406, 1–6 CrossRef CAS.
- J. Lu, C. Zhu, C. Pan, W. Lin, J. P. Lemmon, F. Chen, C. Li and K. Xie, Sci. Adv., 2018, 4, 5100 CrossRef PubMed.
- H. Han, J. Park, S. Y. Nam, K. J. Kim, G. M. Choi, S. S. P. Parkin, H. M. Jang and J. T. S. Irvine, Nat. Commun., 2019, 10, 1471 CrossRef PubMed.
- O. Kwon, K. Kim, S. Joo, Y. J. Hu and G. Kim, J. Mater. Chem. A, 2018, 6, 15947–15953 RSC.
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y. W. Ju, J. W. Han and G. Kim, Nat. Commun., 2017, 8, 15967 CrossRef CAS PubMed.
- B. H. Park and G. M. Choi, Solid State Ionics, 2014, 262, 345–348 CrossRef CAS.
Footnote |
| † These two authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2020 |