
Design of a mixed conductive garnet/Li interface for dendrite-free solid lithium metal batteries†
Hanyu
Huo
 abc,
Yue
Chen
ab,
Ruying
Li
c,
Ning
Zhao
d,
Jing
Luo
c,
João Gustavo
Pereira da Silva
e,
Robert
Mücke
e,
Payam
Kaghazchi
e,
Xiangxin
Guo
*d and
Xueliang
Sun
*c
abc,
Yue
Chen
ab,
Ruying
Li
c,
Ning
Zhao
d,
Jing
Luo
c,
João Gustavo
Pereira da Silva
e,
Robert
Mücke
e,
Payam
Kaghazchi
e,
Xiangxin
Guo
*d and
Xueliang
Sun
*c
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Mechanical and Materials Engineering, University of Western Ontario, Ontario N6A 5B9, Canada. E-mail: xsun9@uwo.ca
dCollege of Physics, Qingdao University, Qingdao 266071, China. E-mail: xxguo@qdu.edu.cn
eForschungszentrum Jülich GmbH, Institute of Energy and Climate Research, Materials Synthesis and Processing (IEK-1), 52425, Jülich, Germany
First published on 27th November 2019
Abstract
Solid-state batteries (SSBs) with metallic lithium (Li) anodes and nonflammable solid-state electrolytes (SSEs) are viewed as the next-generation batteries because of their potential improvement in energy density and guarantee of safety. However, even though the high-density solid garnet SSE pellets exhibit high ionic conductivity, high transference number, and large shear modulus, the unexpectedly serious occurrence of dendrite propagation remains a problem. Herein, a mixed conductive layer (MCL) consisting of electron-conductive nanoparticles embedded in an ion-conductive network is introduced at the interface between the garnet SSE and the Li anode. Such MCL not only leads to the transition from lithiophobicity to lithiophilicity, but also homogenizes the electric-field distribution inside the MCL and relieves the electronic attacks to the garnet. As a result, the Li/MCL/garnet/MCL/Li cells show a critical current density as high as 1.2 mA cm−2 and stable cycling for over 1000 h at 0.1 mA cm−2. The LiCoO2/Li cells with the MCL-protected interface show excellent cycling and rate performance at room temperature. These results demonstrate a rational design for a stable garnet/Li interface and an effective strategy to enable Li metal anodes in SSBs.
Broader contextThe promise to obtain long-lifespan and dendrite-free solid-state batteries (SSBs) with high energy density and high safety is thrilling. As a key component, garnet-type solid-state electrolytes (SSEs) are highly attractive due to their high ionic conductivity at room temperature and high stability against Li metal. However, they are more likely to cause Li dendrite propagation than the conventional liquid electrolytes, leading to rapid short circuiting of SSBs. The relevant mechanism investigation has attracted considerable attention recently. It is generally acknowledged that an ideal SSE/Li interface requires high Li+ conductivity but electronic insulation; in particular, the high electronic conductivity of SSEs was reported as evil for short-circuiting the SSBs. However, our research demonstrates a mixed ion/electron-conductive interface as beneficial for Li dendrite suppression, where electron-conductive nanoparticles are embedded in an ion-conductive network. The built-in electronic pathways can guide a uniform electric field for dendrite-suppressed Li deposition. The SSBs with protected Li anodes show excellent cycling and rate performance in both Li symmetric cells and LiCoO2/Li cells. |
The growing demands of smart electronics and electric vehicles underscore the need for new rechargeable batteries with high energy densities.1 Solid-state batteries (SSBs) are promising candidates. The key materials, solid-state electrolytes (SSEs), have demonstrated the potential to incorporate high-voltage cathodes and high-capacity Li metal anodes, achieving high energy densities for SSBs. In addition, the safety concerns of conventional Li-ion batteries are well addressed by replacing the hazardous liquid electrolytes with nonflammable SSEs in SSBs.2
As a key component in SSBs, various types of SSEs have been studied for decades, including perovskite-type,3 sodium superionic conductor (NASICON)-type,4,5 lithium phosphorus oxynitride (LiPON),6 sulfide-type,7 and garnet-type materials.8 Among them, garnet-type Li7La3Zr2O12 (LLZO) is highly attractive due to its high ionic conductivity at room temperature and the excellent chemical and electrochemical stabilities against Li metal.9,10 The partial substitutions of aliovalent cations such as Ta and Nb for the Zr element of LLZO can further improve the ionic conductivity to over 10−3 S cm−1.11,12 However, LLZO shows an even higher tendency to cause Li dendrite formation than the conventional liquid electrolytes, even though the high Li transference number (∼1), high shear modulus (∼55 GPa), and high relative density (>99%) of LLZO are supposed to suppress Li dendrites.13–15
Although the mechanism of dendrite growth in SSBs remains unclear, LLZO is well acknowledged as lithiophobic, which causes the point contacts between the Li metal and the SSE and initiates the dendrite nucleation at the interface.16,17 Once a tiny dendrite forms, the local electrical field will change rapidly. The Li+ preferentially deposits on the existing dendrite spots and further propagates in the LLZO bulk through the grain boundaries, voids, and other defects, causing short circuiting of the SSBs eventually (Scheme 1).18,19 Therefore, improving the interfacial contact is important to suppress the Li dendrite formation. Various intermediate layers including Al2O3, Nb, Si, and Sn have been introduced between the LLZO and the Li, which indeed enhance the wettability of LLZO with molten Li and thus reduce Li dendrite growth to some extent at low current densities.20–23 However, at a high current density above 1.0 mA cm−2, lithium infiltration still occurs. This indicates that the sole improvement of wettability is not sufficient to thoroughly solve the dendrite problem. Recently, Han et al. revealed that the residual electronic conductivity of LLZO ceramic is the origin of dendrite formation in SSBs using neutron depth profiling (NDP).24 Guo et al. pointed out that the injection of electrons could cause precipitation of Li in polycrystalline garnets.25 They further compared the functionality of various intermediate layers and suggested that an ideal intermediate layer should lead to small interfacial resistance, high ionic conductivity but negligible electronic conductivity, and mechanical stability during repetitive cycles.2
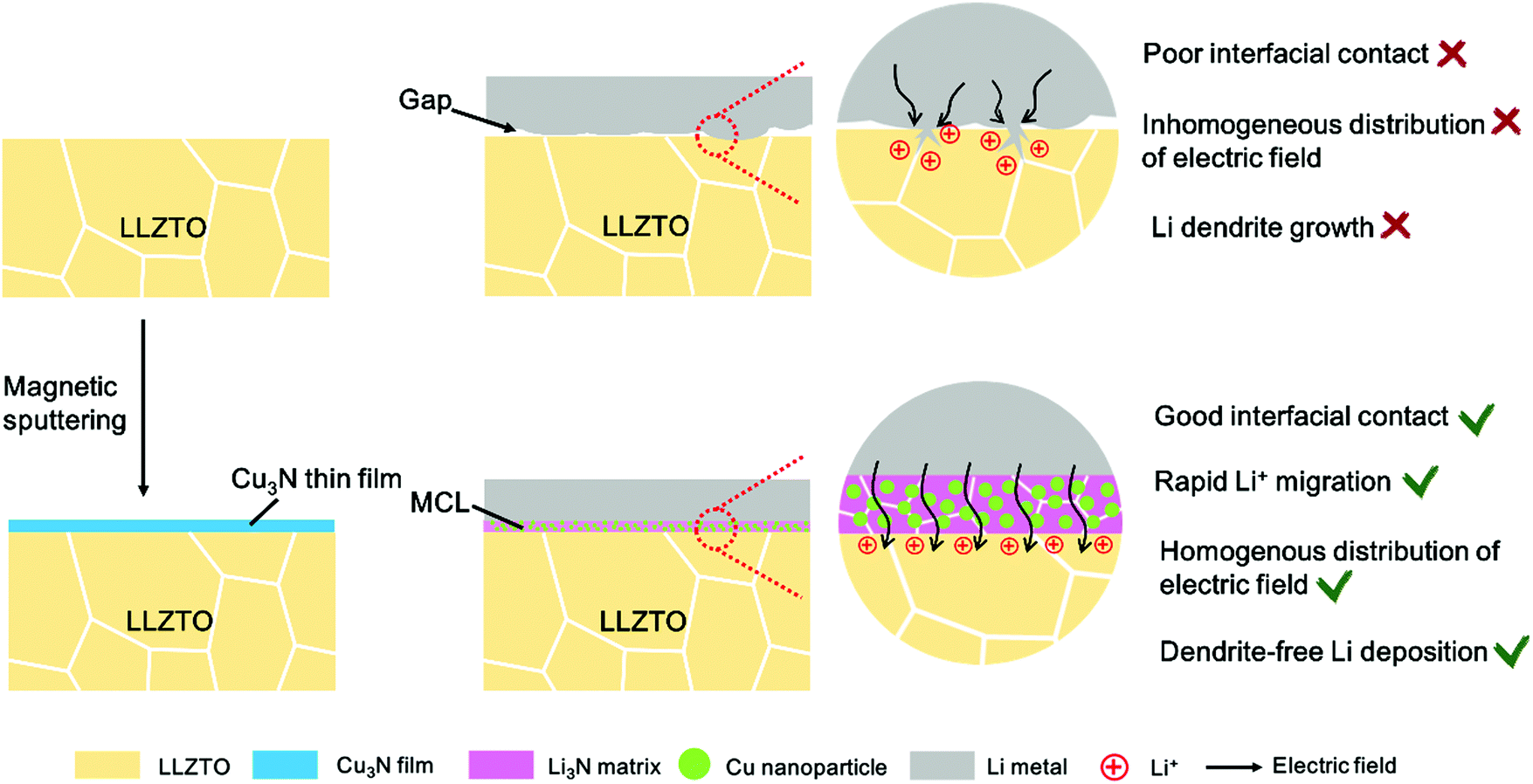 | ||
| Scheme 1 The schematic of the mixed conductive intermediate layer (MCL) protected LLZTO/Li interface for dendrite-free Li metal solid batteries. | ||
It is generally acknowledged that an ideal solid electrolyte interface (SEI) in liquid-based cells and electrode/electrolyte interface in SSBs requires two primary features: high Li+ conductivity and electronic insulation.26 The Li+ conductivity ensures ionic channels at the electrolyte/Li interface, while the electronic insulation is required to prevent undesired side reactions between the electrolytes and the Li metal anodes. However, due to the solid–solid contact, ensuring interfacial wettability between the Li metal and the SSE is crucial in SSBs, unlike in liquid-based cells in which the organic liquid electrolytes can readily cover the Li metal surface. An intermediate layer with ionic conductivity alone showed large interfacial resistance, which could not satisfy the long-term cycles of SSBs so far.21 A mixed ionic/electronic conductive layer (MCL) with built-in electronic conductivity may alleviate the Li+ concentration gradient and level the local current distribution on the Li metal surface, thus leading to homogenous Li deposition.27 The MCL protected Li metal cells with inner electronic channels can render superior cycling performance in liquid-based cells. Zhang et al. reported an armored LiF/Cu-based protective MCL, which renders the cells with reduced impedance and long lifespan.28 Peng et al. also stabilized the electrolyte/Li interface with a MCL based on nanoscale LiF/Ni domains, indicating that the MCLs are good SEI layers to protect the Li metal in liquid-based cells.29 This abundant knowledge of liquid-electrolyte based Li metal batteries can be learned from to address the dendrite issue in SSBs. Therefore, it is worth examining the effect of MCLs on Li dendrite suppression at the interface between Li and garnet, where interfacial contacts and electrochemical kinetics remain challenging.
Herein, an in situ formed Li3N/Cu MCL is proposed to modify the LLZO/Li interface by a facile conversion reaction between a Cu3N thin film and molten Li at 200 °C (Scheme 1). Such an MCL shows a strong wetting interaction with Li metal, substantially decreasing the interfacial resistance from 1138.5 to 83.4 Ω cm2 at 25 °C. In addition, it is more stable than Li alloy layers that may be detached from the garnet pellets after hundreds of cycles. The Li3N as the ionically conducting matrix possesses high Li+ conductivity (close to 10−3 S cm−1) and low energy barriers for Li+ migration (0.007–0.038 eV) at room temperature, which is beneficial for rapid Li+ transport across the interface.30 The uniformly dispersed Cu nanoparticles inside the MCL not only guide a homogeneous electronic distribution to suppress the lithium dendrite nucleation but also serve as a supported matrix to alleviate the volume change in case of a Li-defective state. As a proof of concept, lithium plating–stripping behavior exhibits long-term stability in Li symmetric cells and LiCoO2/Li cells.
As shown in Fig. 1a, the Cu3N film was prepared on a Ta-doped LLZO (LLZTO) pellet by a reactive sputtering method under N2/Ar gas. The LLZTO pellets were prepared by a hot-pressing sintering method according to our previous work.11 All the diffraction peaks in Fig. 1b matched well with the standard pattern of cubic-phase LLZO (PDF#45-0109). The stabilized cubic phase enabled a high ionic conductivity of 1.1 × 10−3 S cm−1 at 25 °C and the relative density of LLZTO was over 99% by hot pressing (Fig. S1, ESI†). The LLZTO pellets were polished to eliminate the Li2CO3 surface contaminants before depositing a Cu3N film. Fig. 1c compares the optical and scanning electron microscope (SEM) images of the LLZTO pellet with or without Cu3N coating. The bare LLZTO surface exhibited clear polishing scratches with exposed grain boundaries. After coating Cu3N on LLZTO, the white LLZTO surface turned yellow and was completely covered. The Cu3N film was also deposited on a glass plate for X-ray diffraction (XRD) analysis to confirm the phase purity. The sharp XRD peak at 40.8° corresponding to Cu3N (111) indicated good crystallinity and strong preferential orientation of the Cu3N film (Fig. 1b).31
 | ||
| Fig. 1 (a) Schematic illustration of the deposition of Cu3N film on the LLZTO ceramic pellet. (b) XRD patterns of LLZTO pellet and Cu3N film on the glass plate. (c) SEM images of LLZTO surfaces with or without Cu3N deposition. (d) TOF-SIMS depth profiles for the LLZTO–Cu3N pellet. (e) TOF-SIMS chemical mappings after sputtering. | ||
Various thicknesses of Cu3N films were deposited by adjusting the sputtering time. An optimal sputtering time of 30 s was selected based on the resulting interfacial resistance (Fig. S2, ESI†). The thickness and homogeneity of the optimized Cu3N film on LLZTO were characterized by the time-of-flight secondary-ion mass spectrometry (TOF-SIMS) technique. The TOF-SIMS depth profiling reveals the evolution of fragments from the specimen as sputtering proceeds in a negative mode. Here, the Cu− and Cu2N− fragments represented the Cu3N layer, while TaO2−, ZrO2− and LaO− fragments indicated the LLZTO underneath. As shown in Fig. 1d, the Cu− and Cu2N− signal intensities remained high over the first 25 s of Cs+ sputtering and then gradually declined. Complementarily, signals of the TaO2−, ZrO2− and LaO− fragments from the LLZTO were absent initially but increased after 25 s of Cs+ sputtering. Evidently, a homogeneous layer of Cu3N was covering the LLZTO pellet, which verifies the SEM results. The thickness of Cu3N was estimated to be 24 nm according to the sputtering rate of 0.96 nm s−1. Fig. 1e shows the TOF-SIMS mappings of Cu−, Cu2N− TaO2−, ZrO2− and LaO− signals after sputtering. A sharp contrast between the Cu3N and LLZTO distributions was observed, where intense TaO2−, ZrO2− and LaO− signals were observed from the sputtered region and strong Cu− and Cu2N− signals across the pristine region. The cross-sectional view of the sputtered volume of LLZTO–Cu3N again confirmed the uniform coverage of Cu3N on LLZTO (Fig. S3, ESI†).
The Cu/Li3N MCL was in situ formed by reacting the Cu3N film with molten Li at 200 °C. It should be noted that a high temperature over 300 °C will lead to the decomposition of Cu3N, releasing N2 gas.31 The conversion reaction of Cu3N to Cu/Li3N MCL on the surface of the LLZTO pellet was confirmed by X-ray photoelectron spectroscopy (XPS). Fig. 2a shows the N 1s, Cu 2p, Li 1s, and Zr 3d XPS spectra of LLZTO–Cu3N and LLZTO-MCL. The peaks at the binding energies of 398.0, 953.0, and 933.2 eV were assigned to N 1s, Cu 2p1/2, and 2p3/2 excitations, respectively, for Cu3N. All these Cu3N related peaks were absent in the XPS spectra of the LLZTO-MCL, indicating a complete reaction. The N 1s peak at 398.0 eV shifted 398.4 eV due to the formation of Li3N.32 Two peaks at 54.1 and 55.0 eV in the Li 1s spectra for the LLZTO–Cu3N pellet can be assigned to the LLZTO substrate and the Li2CO3 surface contaminant, respectively.33 The peak shifting from 54.1 to 54.6 eV could result from the formation of Li3N. The peak slightly shifted from 55.0 to 55.1 eV with enhanced intensity due to the residual Li metal covered on the surface. No Cu signal was detected on LLZTO-MCL, which could be ascribed to the Cu nanoparticles that were covered by the Li3N component. As shown in Fig. S4 (ESI†), the XRD pattern of the formed MCL also confirmed the reaction products of Cu (111) (2θ = 43.2°) and Li3N (2θ = 36.7°, 51.2°, and 52.4°). The Li2CO3 peak was presumably due to the short exposure to air.
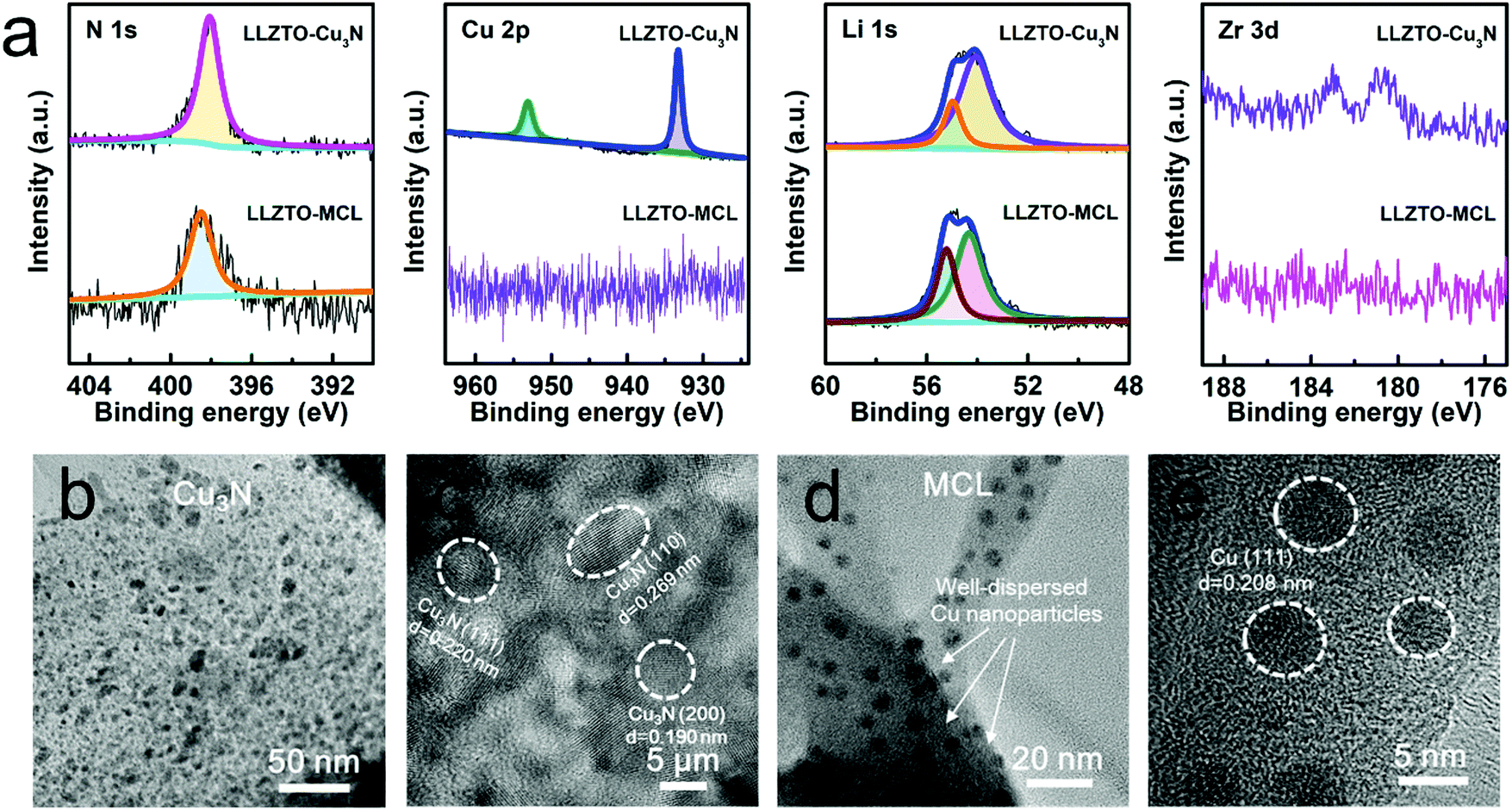 | ||
| Fig. 2 (a) XPS spectra of LLZTO–Cu3N and LLZTO-MCL. (b and c) TEM images of Cu3N at different magnifications. (d and e) TEM images of MCL at different magnifications. | ||
Transmission electron microscopy (TEM) was used to further investigate the microstructure and compositional distribution of the MCL. In agreement with the XRD result, crystalline Cu3N was shown in Fig. 2b and c. The Cu3N film was actually composed of numerous nano-spherical Cu3N crystal grains with the size of 5–10 nm. The inter-planar spacings of 0.220, 0.269 and 0.190 nm corresponded to the (111), (110), and (201) planes, respectively. After reacting with molten Li, the crystalline Cu3N turned into a Li3N network with Cu nanoparticles evenly dispersed inside (Fig. 2d). The inter-planar spacing of 0.208 nm corresponded to the Cu phase as highlighted in Fig. 2e. The uniform dispersion of Cu nanoparticles in the MCL may be beneficial for guiding a homogeneous electronic flux at the LLZTO/Li interface and effectively suppressing Li dendrite nucleation.32
The LLZTO-MCL/Li interface was evaluated by investigating the wettability between LLZTO-MCL and molten Li. As shown in Fig. 3a, the pure LLZTO pellet exhibited a lithiophobic nature with Li metal. Flowable molten Li can easily roll into a liquid sphere on the LLZTO surface, showing a large contact angle. The poor wetting behavior consequently led to micro gaps at the interface, as shown in Fig. 3a. In contrast, an intimate contact was observed between the LLZTO-MCL and the Li metal layer without any micro gaps at the interface (Fig. 3b). This could significantly lower the interfacial resistance and improve the electrochemical performance.
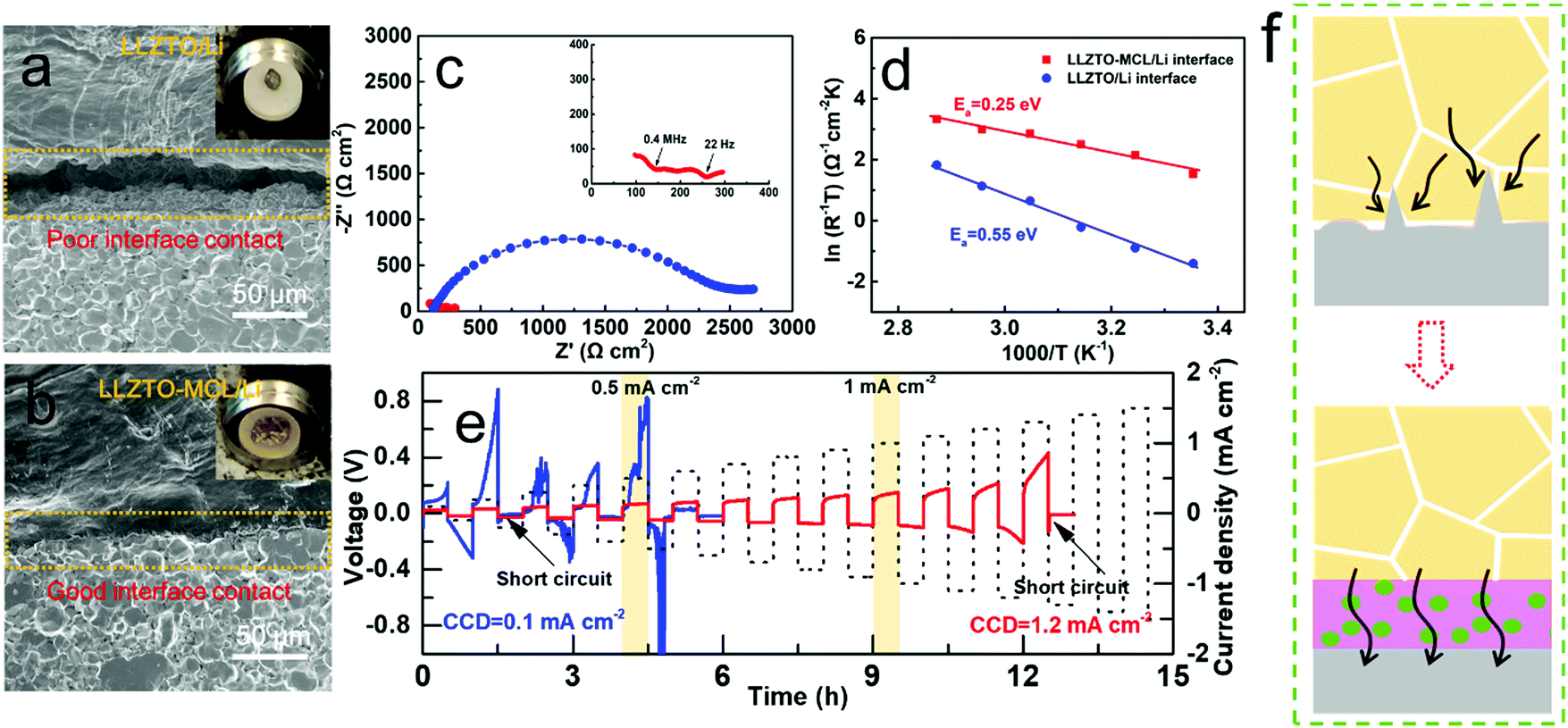 | ||
| Fig. 3 SEM images of (a) the LLZTO/Li interface and (b) the LLZTO-MCL/Li interface. Insets are the corresponding digital images showing the wetting behaviors of molten Li on bare LLZTO or LLZTO-MCL. (c) EIS spectra, (d) Ea, and (e) CCD of the Li/LLZTO/Li and the Li/LLZTO-MCL/Li cells. (f) Mechanism of increased CCD by MCL. | ||
The symmetric cells of Li/LLZTO-MCL/Li and Li/LLZTO/Li were assembled for electrochemical characterization. Electrochemical impedance spectroscopy (EIS) was used to compare the interfacial resistance of cells with or without MCL modification on LLZTO. Fig. 3c shows the Nyquist plots obtained at 25 °C. The Nyquist plot of the Li/LLZTO/Li cell exhibited one huge semicircle ascribed to the large interfacial resistance between LLZTO and Li metal. The initial point of the spectra corresponds to the resistance from the bulk LLZTO pellet. Considering charge transfer across the two Li/LLZTO interfaces in one symmetric cell, the interfacial resistance determined from the semicircle was divided by two to obtain the value of each Li/LLZTO interface. Thus, the resistance of a single LLZTO/Li interface was 1138.5 Ω cm2. Different from the bare LLZTO cell, the Li/LLZTO-MCL/Li symmetric cell exhibited multiple semicircles resulting from the MCL bulk and the MCL/LLZTO interface at high frequency and the MCL/Li interface at low frequency. The overall resistance of the LLZTO-MCL/Li interface due to the MCL modification was 83.4 Ω cm2.21 The significant decrease in interfacial resistance from 1138.5 to 83.4 Ω cm2 could be attributed to the lithiophilic property of MCL by a conversion reaction. The MCL even exhibited a lower interfacial resistance than the pure Li3N layer (175 Ω cm2) reported previously,34 further confirming the advantage of MCL. In addition, the temperature-dependent interfacial resistance evolution was measured from 25 °C to 75 °C, showing good Arrhenius behavior. The activation energy (Ea) of the MCL modified or unmodified interface was calculated by the Arrhenius law, giving a value of 0.25 eV for the LLZTO-MCL/Li interface and 0.55 eV for the LLZTO/Li interface (Fig. 3d and Fig. S5, ESI†). The decreased interfacial Ea can be ascribed to the high ionic conductivity and low energy barrier of Li migration of Li3N in MCL.26 This can be beneficial for rapid Li+ transport across the interface for high rate performance.
Critical current density (CCD) is a key parameter to characterize the stability of the LLZTO/Li interface and the effectiveness of Li dendrite suppression. The applied current density was increased from 0.1 to 1.5 mA cm−2 with a step increase of 0.1 mA cm−2 per hour (0.5 h stripping and 0.5 h plating) at 25 °C. The CCD was defined as the current density at which the cell was short circuited. As shown in Fig. 3e, the CCD of the Li/LLZTO/Li cell was only 0.1 mA cm−2 due to the poor interface, while the CCD of the Li/LLZTO-MCL/Li cell was substantially improved to 1.2 mA cm−2. The voltage profile of the Li/LLZTO-MCL/Li cell remained smooth and stable before the sudden occurrence of short circuiting. The significant improvement of CCD was a result of the combined contributions from the ion-conductive Li3N network and the electron-conductive Cu nanoparticles of the MCL. More specifically, the Li3N matrix provided smooth pathways for rapid Li+ transport, and the well-dispersed Cu nanoparticles helped to guide a uniform electric field for dendrite-suppressed Li deposition (Fig. 3f). To our best knowledge, the CCD of 1.2 mA cm−2 is the highest value obtained in garnet-based SSBs (Table S1, ESI†). Even though many surface modification methods helped to decrease the interfacial resistance, the CCD was still limited by the poor interfacial Li+ conductivity and interfacial stability at high current densities.
In order to further demonstrate the homogenized electric-field distribution induced by the MCL for Li dendrite suppression, calculated current densities of LLZTO/Li dendrite and LLZTO-MCL/Li dendrite are illustrated in Fig. 4. The LLZTO/Li dendrite showed an inhomogeneously distributed current density. The maximum current density was obtained at the tip of the dendrite. While the current density in LLZTO-MCL/Li dendrite was homogeneous, and it was three orders of magnitude lower than that at the tip of Li dendrite in contact with LLZTO. This result showed that the decreased resistivity by MCL leads to a charge smearing in the Li dendrite, which can effectively suppress the needle-like morphology growth of the Li metal.
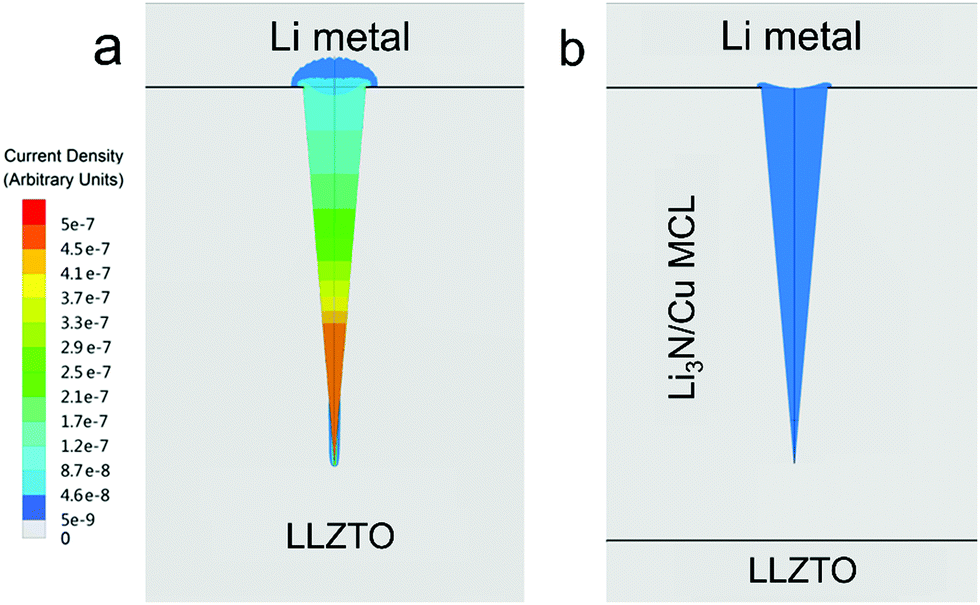 | ||
| Fig. 4 Calculated current densities for (a) LLZTO/Li dendrite and (b) LLZTO-MCL/Li dendrite. | ||
Galvanostatic cycling tests were carried out to evaluate the long-term stability of Li+ transport across the interface. As shown in Fig. 5a, the Li/LLZTO/Li cell readily showed an inclined overpotential up to 0.29 V at the first plating/stripping cycle at 0.1 mA cm−2 (0.05 mA h cm−2), indicating uneven Li deposition and dissolution. A rapid short circuit occurred within 8 cycles. The poor LLZTO/Li contact and the large interfacial resistance induced inhomogeneous current distribution and local hot spots for Li+ flux at the defects, leading to the dendrite growth.12 After disassembling the short-circuited cell and removing the Li metal electrodes by sanding, the Li dendrites grown into the LLZTO pellet were optically visualized as dark spots on the white LLZTO surface (Fig. S6a, ESI†). This was confirmed by SEM (Fig. S6b and c, ESI†). The cross-sectional SEM image along with energy dispersion spectrum (EDS) elemental mappings clearly showed the propagation of Li dendrites along the LLZTO grain boundaries (Fig. 5b and c), which caused short circuiting of the cell. As a note, the SEM specimen was exposed to air briefly during the transfer to the SEM, so the C and O elements were rich at the dendrite locations due to Li2CO3. In great contrast, the Li/LLZTO-MCL/Li cell maintained stable cycling over 1000 h with a smooth overpotential plateau of 30.1 mV at 0.1 mA cm−2 (Fig. 5d). Very similar overall resistances of 312.4 and 323.3 Ω cm2 were maintained before and after cycling, respectively. The optical image showed an all-white LLZTO surface without dark spots of dendrites after sanding (Fig. S7, ESI†). These again indicated the stable interface between the LLZTO-MCL and Li metal anode. Moreover, as shown in Fig. S8 (ESI†), the Li/LLZTO-MCL/Li cells maintained stability over hundreds of hours even with a higher areal capacity of 0.2 mA h cm−2 at various current densities of 0.1, 0.2, and 0.5 mA cm−2. The superior performance of LLZTO-MCL compared to other surface-modified LLZTO was attributed to dendrite-free Li deposition and long-term interfacial stability (Table S1, ESI†).
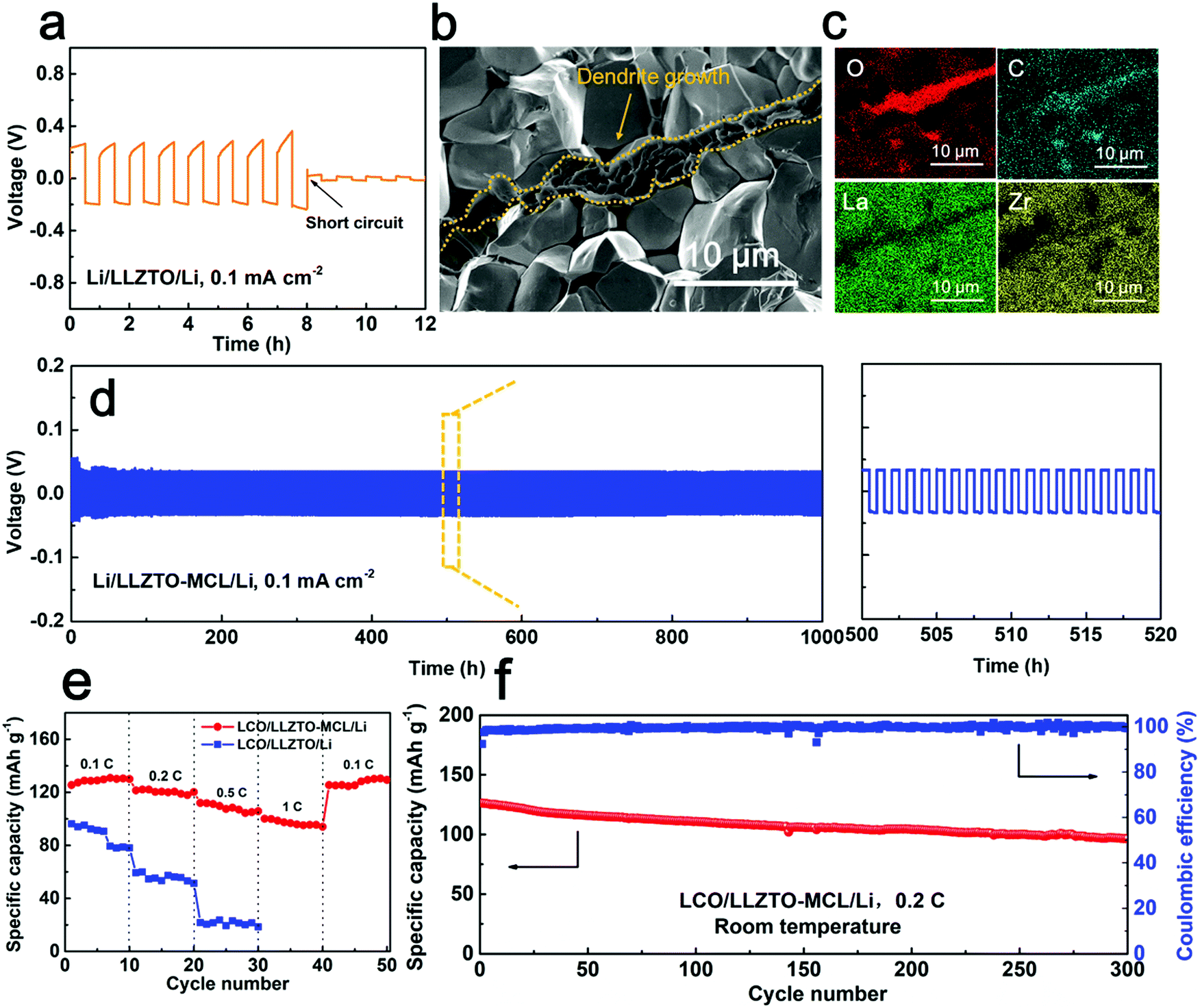 | ||
| Fig. 5 (a) Galvanostatic cycling performance of the Li/LLZTO/Li cell under 0.1 mA at 25 °C. (b) Cross-sectional SEM image and (c) EDS mappings of the LLZTO pellet collected after cell short circuiting. (d) Galvanostatic cycling performance of the Li/LLZTO-MCL/Li cell under 0.1 mA at 25 °C. (e) Rate performance of the Li/LLZTO/LCO and the Li/LLZTO-MCL/LCO cells. (f) Cycle performance of the Li/LLZTO-MCL/LCO cell under 0.2 C at room temperature. | ||
Full SSBs with a Li metal anode and a LiCoO2 (LCO) cathode were constructed using LLZTO-MCL in comparison with bare LLZTO. Fig. S9 (ESI†) shows the schematic configuration of the SSBs. The composite cathode was prepared using ionic liquid as the wetting agent and super P as the conductive additive for room-temperature feasibility.35 In agreement with the reduced interfacial resistance by the MCL modification, the total resistance of the Li/LLZTO-MCL/LCO cell (1029.2 Ω cm2 at 25 °C) was smaller than that of the Li/LLZTO/LCO cell (3354.7 Ω cm2) (Fig. S10, ESI†). Correspondingly, the Li/LLZTO-MCL/LCO cell showed smaller polarizations than the Li/LLZTO/LCO cell at different current rates (Fig. S11, ESI†). The LLZTO-MCL cell delivered an initial specific discharge capacity of 130.0 mA h g−1 with a Coulombic efficiency of 91.3% at 0.1 C. The discharge capacities were 125.3, 115.5 and 104.0 mA h g−1 at 0.2, 0.5 and 1 C, respectively (Fig. 5e). After high-rate cycling, the cell recovered a discharge capacity of 123.4 mA h g−1 at 0.1 C. The high capacity and excellent rate performance could be attributed to the good interfacial contact and high mixed ionic/electronic conductivities at the interface. In contrast, the bare LLZTO cell with a less favorable Li interface delivered a discharge capacity of 88.3 mA h g−1 with a high overpotential at 0.1 C. The discharge capacity decreased to 49.7 and 18.9 mA h g−1 at 0.2 and 0.5 C, respectively (Fig. 5e). Moreover, the SSB with LLZTO-MCL maintained a high capacity retention of 81.1% after 300 cycles under 0.2 C at room temperature (Fig. 5f).
Conclusions
In summary, a Li3N/Cu MCL is constructed at the interface between a Li metal anode and a garnet SSE. The interfacial resistance dramatically decreases from 1138.5 to 83.4 Ω cm2 at 25 °C as the result of the conversion reaction. The Li3N matrix in the MCL provides efficient ion-conducting pathways for Li+, while the well-dispersed Cu nanoparticles guide a homogenous electric field at the interface. The synergistic effect of the external ionic conductivity and internal electronic conductivity relieves the attacks of electrons to the garnet SSE, thus suppressing Li dendrite nucleation. The resulting CCD of the MCL-protected Li symmetrical cell is as high as 1.2 mA cm−2. The cells exhibit stable cycling over 1000 h with a low overpotential of 30.1 mV under 0.1 mA cm−2. The LCO/Li cell with LLZTO-MCL maintains a high capacity retention of 81.1% after 300 cycles under 0.2 C at 25 °C. This superior electrochemical performance clearly indicates the effectiveness of the designed MCL for depressing Li propagation into the garnet SSEs. This work sheds light on the rational design of an excellent interface between the Li metal anode and the SSE, affording the feasibility to obtain long-lifespan and dendrite-free SSBs with high energy density and high safety.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. H. conceived and designed the experimental work and prepared the manuscript; Y. C. did the sputtering and TEM characterizations; R. L. helped with the SEM characterizations; Dr N. Z. fabricated the garnet ceramic pellets. J. L. polished the language. J. G. P. S., R. M., and P. K. did the simulations. X. G., and X. S. supervised the overall project. All authors have given approval to the final version of the manuscript. The authors would like to thank “the National Key R&D Program of China (Grant No. 2018YFB0104300)”, the National Natural Science Foundation of China (Grant No. 51771222, 51532002, 51772314 and 51702346), the “Taishan Scholars Program”, Natural Sciences and Engineering Research Council of Canada (NSERC), Canada Research Chair Program (CRC), and University of Western Ontario.References
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- N. Zhao, W. Khokhar, Z. Bi, C. Shi, X. Guo, L.-Z. Fan and C.-W. Nan, Joule, 2019, 3, 1190–1199 CrossRef CAS.
- Y. Inaguma, L. Q. Chen, M. Itoh, T. Nakamura, T. Uchida, H. Ikuta and M. Wakihara, Solid State Commun., 1993, 86, 689–693 CrossRef CAS.
- J. B. Goodenough, H. Y. P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS.
- Y. Liu, Q. Sun, Y. Zhao, B. Wang, P. Kaghazchi, K. R. Adair, R. Li, C. Zhang, J. Liu and L.-Y. Kuo, ACS Appl. Mater. Interfaces, 2018, 10, 31240–31248 CrossRef CAS PubMed.
- N. J. Dudney, J. B. Bates, R. A. Zuhr, C. F. Luck and J. D. Robertson, Solid State Ionics, 1992, 53, 655–661 CrossRef.
- J. H. Kennedy, S. Sahami, S. W. Shea and Z. M. Zhang, Solid State Ionics, 1986, 18–19, 368–371 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- A. J. Samson, K. Hofstetter, S. Bag and V. Thangadurai, Energy Environ. Sci., 2019, 12, 2957–2975 RSC.
- V. Thangadurai, D. Pinzaru, S. Narayanan and A. K. Baral, J. Phys. Chem. Lett., 2015, 6, 292–299 CrossRef CAS PubMed.
- F. Du, N. Zhao, Y. Li, C. Chen, Z. Liu and X. Guo, J. Power Sources, 2015, 300, 24–28 CrossRef CAS.
- M. He, Z. Cui, C. Chen, Y. Li and X. Guo, J. Mater. Chem. A, 2018, 6, 11463–11470 RSC.
- S. Yu, R. D. Schmidt, R. Garcia-Mendez, E. Herbert, N. J. Dudney, J. B. Wolfenstine, J. Sakamoto and D. J. Siegel, Chem. Mater., 2015, 28, 197–206 CrossRef.
- C. Brissot, M. Rosso, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81, 925–929 CrossRef.
- C.-L. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef CAS PubMed.
- J. Dai, C. Yang, C. Wang, G. Pastel and L. Hu, Adv. Mater., 2018, 30, e1802068 CrossRef PubMed.
- H. Huo, Y. Chen, N. Zhao, X. Lin, J. Luo, X. Yang, Y. Liu, X. Guo and X. Sun, Nano Energy, 2019, 61, 119–125 CrossRef CAS.
- R. H. Basappa, T. Ito and H. Yamada, J. Electrochem. Soc., 2017, 164, A666–A671 CrossRef CAS.
- E. J. Cheng, A. Sharafi and J. Sakamoto, Electrochim. Acta, 2017, 223, 85–91 CrossRef CAS.
- X. Han, Y. Gong, K. K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- N. Zhao, R. Fang, M.-H. He, C. Chen, Y.-Q. Li, Z.-J. Bi and X.-X. Guo, Rare Met., 2018, 37, 473–479 CrossRef CAS.
- W. Luo, Y. Gong, Y. Zhu, K. K. Fu, J. Dai, S. D. Lacey, C. Wang, B. Liu, X. Han, Y. Mo, E. D. Wachsman and L. Hu, J. Am. Chem. Soc., 2016, 138, 12258–12262 CrossRef CAS PubMed.
- C. Wang, H. Xie, L. Zhang, Y. Gong, G. Pastel, J. Dai, B. Liu, E. D. Wachsman and L. Hu, Adv. Energy Mater., 2017, 1701963, DOI:10.1002/aenm.201701963.
- F. D. Han, A. S. Westover, J. Yue, X. L. Fan, F. Wang, M. F. Chi, D. N. Leonard, N. Dudney, H. Wang and C. S. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- X. Xie, J. Xing, D. Hu, H. Gu, C. Chen and X. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 5978–5983 CrossRef CAS PubMed.
- X.-B. Cheng, C. Yan, X.-Q. Zhang, H. Liu and Q. Zhang, ACS Energy Lett., 2018, 3, 1564–1570 CrossRef CAS.
- J. Maier, Solid State Ionics, 1995, 75, 139–145 CrossRef CAS.
- C. Yan, X.-B. Cheng, Y.-X. Yao, X. Shen, B.-Q. Li, W.-J. Li, R. Zhang, J.-Q. Huang, H. Li and Q. Zhang, Adv. Mater., 2018, 1804461, DOI:10.1002/adma.201804461.
- Z. Peng, N. Zhao, Z. Zhang, H. Wan, H. Lin, M. Liu, C. Shen, H. He, X. Guo, J.-G. Zhang and D. Wang, Nano Energy, 2017, 39, 662–672 CrossRef CAS.
- W. Li, G. Wu, C. M. Araújo, R. H. Scheicher, A. Blomqvist, R. Ahuja, Z. Xiong, Y. Feng and P. Chen, Energy Environ. Sci., 2010, 3, 1524–1530 RSC.
- J. Wang, J. Chen, X. Yuan, Z. Wu, B. Miao and P. Yan, J. Cryst. Growth, 2006, 286, 407–412 CrossRef CAS.
- Q. Li, H. Pan, W. Li, Y. Wang, J. Wang, J. Zheng, X. Yu, H. Li and L. Chen, ACS Energy Lett., 2018, 3, 2259–2266 CrossRef CAS.
- Y. Li, X. Chen, A. Dolocan, Z. Cui, S. Xin, L. Xue, H. Xu, K. Park and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 6448–6455 CrossRef CAS PubMed.
- H. Xu, Y. Li, A. Zhou, N. Wu, S. Xin, Z. Li and J. B. Goodenough, Nano Lett., 2018, 18, 7414–7418 CrossRef CAS PubMed.
- Y. Shao, H. Wang, Z. Gong, D. Wang, B. Zheng, J. Zhu, Y. Lu, Y.-S. Hu, X. Guo and H. Li, ACS Energy Lett., 2018, 3, 1212–1218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee01903k |
| This journal is © The Royal Society of Chemistry 2020 |