
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rigid anionic Janus bis(NHC) – new opportunities in NHC chemistry†
Nabila Rauf
Naz
a,
Gregor
Schnakenburg
a,
Zsolt
Kelemen
b,
Dalma
Gál
b,
László
Nyulászi
 *b,
René T.
Boeré
*c and
Rainer
Streubel
*a
*b,
René T.
Boeré
*c and
Rainer
Streubel
*a
aInstitut für Anorganische Chemie der Rheinischen Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Strasse 1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn.de; Web: http://anorganik.chemie.uni-bonn.de/akstreubel
bDepartment of Inorganic and Analytical Chemistry and MTA-BME Computation Driven Chemistry Research Group, Budapest University of Technology and Economics, Szt Gellert ter 4, 1111 Budapest, Hungary. E-mail: nyulaszi@mail.bme.hu
cDepartment of chemistry and biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, AB T1K3M4, Canada. E-mail: boere@uleth.ca
First published on 10th December 2020
Abstract
A phosphanido-type bridged bis(imidazolium) salt, readily prepared in two steps via reductive deselenization of a tricyclic 1,4-diphosphinine diselone, affords access to a novel anionic P-functional tricyclic bis(NHC) via deprotonation. The former also offers a P-functionalization/deprotonation sequence to access the first mixed P-substituted tricyclic bis(NHCs), as well as coordination of the phosphorus centers to rhodium(I) fragments.
Introduction
Owing to the wide range of structural and property modifications of N-heterocyclic carbenes (NHCs), they have become potent ligands in organometallic chemistry and catalysis.1,2 It has been reported that the incorporation of an anionic functionality confers higher stability to resulting NHC complexes compared to related neutral donor substituents.3 Backbone-functionalized NHCs having anionic heteroatom substituents especially facilitate π-electron interactions with the heterocyclic ring, which can lead to electronic tuning of the donor properties of the carbene.4 Only a small number of NHCs of type I possessing an anionic low-coordinate moiety such as enolate,5 borate,6 amido7 and phosphanido,8 have been re-ported (Fig. 1). Some of these enabled an additional and/or competing metal binding site, resulting in ligand polytopicity or ambidenticity in bimetallic coordination.9,10 The anionic bis-(NHC) II has been reported, obtained via reduction of a bis-(imidazol-2-thione-4-yl)phosphane using a large excess of potassium metal;11 but II could neither be isolated nor structurally confirmed. | ||
| Fig. 1 Anionic mono and bis(NHCs) I, II and rigid Janus bis(NHCs) III–V (R,R1,R2 = organic substituents). | ||
A new ambidentate Janus-type ligand combining a carbene and an anionic imidate centers within the same heterocyclic framework was reported by Lavigne et al., appears to be suitable for the directed construction of a variety of homo-and/or heteropolymetallic complexes.3c In 2017, a dianionic bis(maloNHC)3d was reported as a bridging ligand to construct zwitterionic complexes by Tapu et al. Furthermore, it served as building block for the preparation of novel organometallic frameworks, not handy with neutral Janus-type bis(NHC)s. Due to their unique electronic properties, these zwitterionic NHC-metal species exhibit potentially valuable advantages such as enhanced catalytic activity and solubility relative to the classical cationic metal complexes of the neutral NHCs.
Recently, we established a new series of tricyclic rigid Janus bis(NHCs) III, tuned by PRn moieties in different phosphorus oxidation states, and reported on their use in coinage metal(I) complex chemistry.12 Thereafter, continuing efforts have been made to establish anionic low-coordinate P-linked bis(NHCs) in order to achieve tuneable electronic communication and redox activity.
Herein, we report on the synthesis of a stable anionic Janus-type tricyclic bis(NHC) IV having a P-localized charge, and its use in main group and transition metal chemistry to access, e.g., bis(NHCs) V, having a mixed P-substitution pattern.
Results and discussion
Synthesis of anionic bis(NHC) 4via reductive deselenization
Following our recent synthetic protocol,13 the new tricyclic 1,4 diphosphinine diselone 1 (Scheme 1) was synthesized via mild reduction of the P–Cl substituted 1,4-dihydro-1,4-diphosphi-nine12 precursor and, finally, isolated as a deep violet solid. According to TD-DFT calculations, the colour of this electron-delocalized compound could be attributed to a HOMO–LUMO transition (details in the ESI†). Diselone 1 has been treated with 2 equivalents of trifluoromethyl methylsulfonate (MeOTf) in dichloromethane to afford the doubly Se-methylated salt 2 which was isolated as a yellow solid and fully characterized, including single crystal X-ray diffraction analysis (Fig. 2).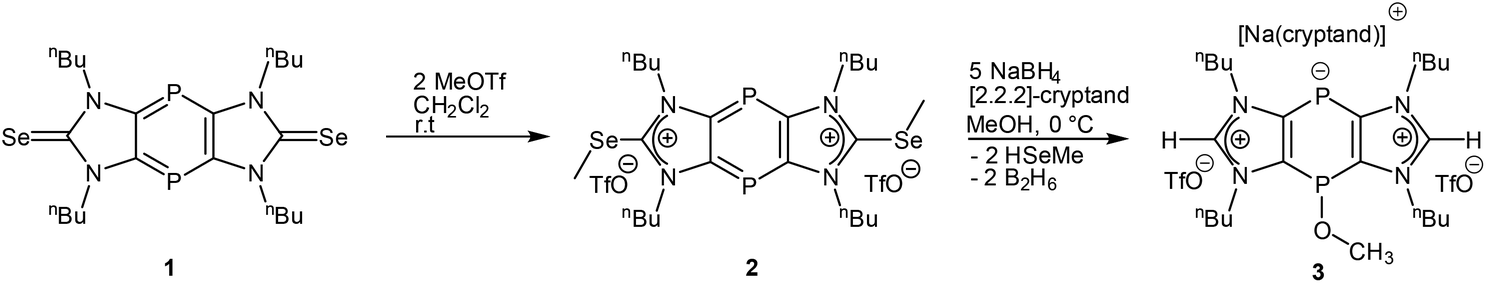 | ||
| Scheme 1 Synthesis of doubly Se-methylated salt 2 and bis(imidazolium) salt 3 starting from tricyclic diselone 1. | ||
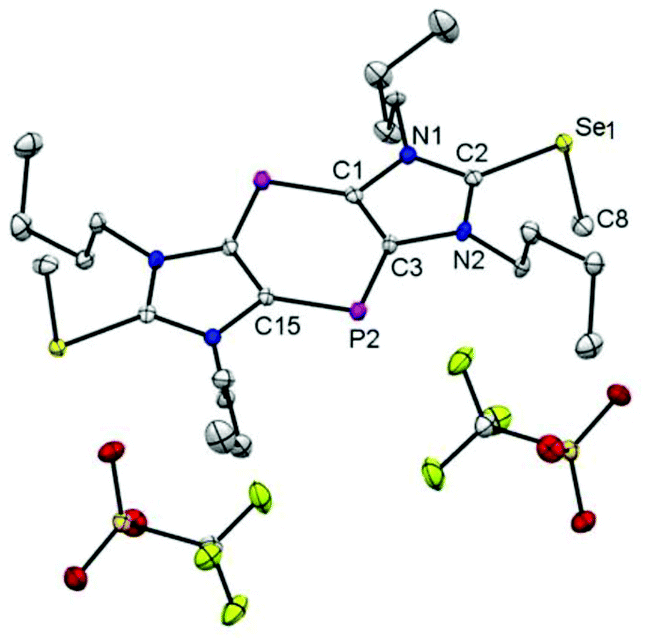 | ||
| Fig. 2 Molecular structure of compound 2 (ellipsoids at the 50% probability-substituted level); hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C2–Se1 1.896(4), Se1–C8 1.952(4), C2–N2 1.339(5), N2–C3 1.397(5), C3–P2 1.736(4), C1–C3 1.412(6); N1–C2–N2 108.8(3), C3–P2–C15 96.3(2). | ||
The centrosymmetric molecular structure of 2 has a C2–Se1 bond distance of 1.896(4) Å that is slightly elongated compared to 1.8240(16) Å for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se in the precursor 1 (ESI†), but remains significantly shorter than the 1.952(4) Å Se–C8 bond to the methyl group.
Se in the precursor 1 (ESI†), but remains significantly shorter than the 1.952(4) Å Se–C8 bond to the methyl group.
Doubly Se-methylated salt 2 was subjected to reductive deselenization with NaBH4 in the presence of [2.2.2]-cryptand in methanol to afford (somewhat surprisingly) bis(imidazolium) salt 3 (Scheme 1) with an anionic phosphorus centre. Salt 3, obtained in pure form via extraction with dichloromethane followed by washing with diethylether, showed two resonance signals in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in the 31P NMR spectrum at 20.1 ppm (POMe) and −67.3 ppm (anionic P), but no 3J(P,P) coupling (Table 1). Further confirmation for 3 was obtained from NMR and MS experiments as well as elemental analysis. DFT calculations, performed on N-Me model compounds,14 reveal that the aro-matic character of the middle ring in 3′ is lower (NICS(0) = −4.9) than in 1′14 ((NICS(0) = −8.1) or 2′ (NICS(0) = −10.0); nevertheless some – apparently hyperconjugative – cyclic conjugation is still operative. In contrast, the outer rings retain high aromatic character (NICS(0) = −11.1).
:1 ratio in the 31P NMR spectrum at 20.1 ppm (POMe) and −67.3 ppm (anionic P), but no 3J(P,P) coupling (Table 1). Further confirmation for 3 was obtained from NMR and MS experiments as well as elemental analysis. DFT calculations, performed on N-Me model compounds,14 reveal that the aro-matic character of the middle ring in 3′ is lower (NICS(0) = −4.9) than in 1′14 ((NICS(0) = −8.1) or 2′ (NICS(0) = −10.0); nevertheless some – apparently hyperconjugative – cyclic conjugation is still operative. In contrast, the outer rings retain high aromatic character (NICS(0) = −11.1).
:0.3) and 6cis/trans (1:0.3)
| δ(31P)/ppm (CD2Cl2) | δ(13C)/ppm (CD2Cl2)b | |
|---|---|---|
|
a In case of 4 and 6cis/trans (1:0.4) THF-d8.
b
C
2 carbon.
|
||
| 3 | 20.1 (s), −67.3 (s) | 137.3 (d, 3JP,C = 4.5 Hz) |
| 4 | 25.2 (s), −74.8 (s) | 208.8 (br) |
| 5cis/trans | δ = −71.58 (d, 3JP,H = 5.2 Hz), −66.23 (d, 3JP,H = 4.9 Hz), 39.57 (br), 43.7 (br) | 142.4 (br), 143.37 (br) |
| 6cis/trans | δ = −74.0 (d, 2JP,H = 4.8 Hz), −68.6 (d, 2JP,H = 3.7 Hz), 37.2(d, 3JP,H = 4.6 Hz), 41.3 (d, 3JP,H = 3.8 Hz) | 224.2, 223.8, (t, 3JP,C = 2.7 Hz) |
To access the first example of an anionic P-functional bis(NHC), deprotonation of the bis(imidazolium) salt 3 was performed in THF using two equivalents of KHMDS (Scheme 2). After extraction with THF/diethyl ether (1:1.5), compound 4 was isolated as a dark orange solid which has two resonances in its 31P NMR spectrum at 25.2 ppm (P-OMe) and −74.8 ppm (anionic P). The 1H-NMR spectrum confirms the absence of the C2–H proton and the 13C{1H} NMR spectrum a broad resonance at 208.8 ppm assigned to the C2 atom of dicarbene 4. The proposed constitution of 4 is also supported by HR-MS (negative ESI; exp. 449.2605 vs. calc. 449.2607). In order to establish the stability of the carbene 4, an isodesmic reaction (see ESI†) yields 113.3 kcal mol−1 stabilization for 4′.14 This is very similar to our earlier reported 111.1 kcal mol−1 value12 for III (R:Me, R′:cis-NEt2), indicating that the carbene character in the anionic tri-cycle is virtually unchanged. Compared to 3′, the aromaticity of the middle ring is slightly higher (NICS(0) = −5.5), while that of the outer rings is lower (NICS(0) = −9.0), as is usual for NHCs if compared to imidazolium salts.15
 | ||
| Scheme 2 Synthesis of anionic bis(NHC) 4, PIII/III bis(imidazolium) salts 5cis/trans (1:0.3) and mixed substituted bis(NHCs) 6cis/trans (1:0.3). | ||
Formation of mixed substituted PIII/III bridged bis(NHCs) 6cis/trans
We then targeted to use the P-anionic functionality to access bis(NHCs) with a mixed P-substitution pattern. Therefore, 3 was treated with MeI in diethyl ether at −80 °C which resulted in a clean formation of bis(imidazolium) salt 5cis/trans with an isomeric ratio of 1:0.3 (signals not assigned) (Scheme 2). The 31P{1H} NMR spectrum of the reaction mixture showed two sets of signals for two isomers at δ(P–CH3) = −71.6 (d, 3JP,H = 5.0 Hz), −66.2 (d, 3JP,H = 5.0 Hz) and δ(P–OCH3) = 39.6 (br), 43.7 (br) (ratio 1:0.4). The isolated mixture of 5cis/trans (1:0.3) was subsequently deprotonated using two equiv. of KHMDS in THF to afford the mixed-substituted bis(NHCs) 6cis/trans (ratio 1:0.3).
Clear evidence for the latter came from the 1H NMR spectrum of this mixture, since the former C2–H signal (δ = 9.47 (t, 3JP,H = 3.03 Hz), 9.56 (br)) of 5cis/trans were absent. This was further supported by the 13C{1H} NMR spectrum as the characteristic downfield signals for the C2-nuclei were found (Table 1), revealing the formation of the bis(NHCs) 6cis/trans (ratio 1:0.3). 6trans and 6cis are computed to have high stability (the isodesmic reaction energies are 108.8 for 6′trans14 and 108.6 kcal mol−1 for 6′cis). The aromatic character of the middle ring is significantly lower (NICS(0) = −0.1), indicating that when both phosphorus centres are saturated the central ring loses its aromaticity, whilst that of the outer rings (NICS(0) = −9.7) remains high.
Initial coordination chemistry experiments were undertaken to explore the reactivity of tricyclic bis(imidazolium) salt 3. Thus, reaction with a half equiv. of [Rh(cod)Cl]2 affords exclusively the mono rhodium(I) complex 7 (Scheme 3). The coordination of 3 to the Rh(cod)Cl fragment is confirmed by the 31P NMR spectrum as resonances of 7 appeared at high-field (−70.6 (s, anionic P) and 47.5 (d) ppm) having a rhodium–phosphorus coupling of 1JRh,P = 192.4 Hz, assigned to the neutral phosphorus atom. This coordination mode is surprising since the HOMO of 3′ is located at the dicoordinate (anionic) phosphorus atom, and the involved tricoordinate phosphorus, largely representing HOMO−1, is lower in energy by as much as 1.9 eV (see ESI†). Nevertheless, our calculations on the isomeric complexes of 7′ showed that the favoured coordination site is indeed the tricoordinate phosphorus, 7′ being more stable than the possible isomers (cis and trans) by more than 5 kcal mol−1 (see ESI†). To examine how many Rh(I) fragments could be coordinated to 3, it was then treated with 1.5 equiv. of the rhodium dimer which afforded selectively an isomeric mixture of trinuclear phosphanido complexes 8cis/trans (ratio 0.9:1) (Scheme 3). Upon coordination, two sets of signals are present in the 31P NMR spectrum at 65.0 (d, 1JRh,P = 195.6 Hz) (minor)/64.1 (dd, 1JRh,P = 200.1 Hz, 3JP,P = 5.3 Hz) (major) and −120.3 (t br, 1JRh,P = 126.9 Hz) (major)/−123.4 (t, 1JRh,P = 126.9 Hz) (minor).
 | ||
| Scheme 3 Synthesis of mononuclear rhodium complex 7 and trinuclear rhodium complexes 8cis/trans (1:0.7). | ||
Cyclic voltametric studies supported by DFT calculations
The electrochemical properties of bis(NHCs) 4 and 6cis/trans (1:0.3) were investigated by cyclic voltammetry (CV) in THF (0.2 M [nBu4N][PF6]) at gold ceramic screen printed electrodes (Au CSPE) in an Ar-filled glove box. Voltammetric data were measured on solutions containing 1.0 mM analyte and representative CVs are presented in Fig. 3 (and in greater detail in the ESI†). The observed behaviour of 6cis/trans (Fig. 3b) is reminiscent of dicarbenes III (Rn = PIIINEt2)12 and we therefore start the analysis here.
 | ||
| Fig. 3 CV diagrams (a) of 4, and (b) of 6′cis/trans (scan rate: 200 mV s−1). In each case, the blue trace indicates the solvent/electrolyte background (0.2 M) [nBu4N][PF6] in (THF), and the red that from a 1.0 mM solution of analyte. | ||
There are a series of facile, chemically irreversible oxidations with anodic peak potentials Ea1p = −0.29 V, Ea2p = −0.07 V and Ea3p = +0.65. The processes are nonetheless repeatable and stable to scanning first in anodic or cathodic directions, and continue right up to the anodic potential limit at around +1.0 V. This is consistent with oxidations involving the carbene σ(p)-centred HOMO and HOMO−1 as determined from B3LYP/6-31+G*//M06-2X/6-31+G* computations undertaken on the model structures 6′cis/trans15 (Fig. 4). The more positive first oxidations Ea1p = −0.29 V for 6cis/trans compared to III (Rn = PIIINEt2), for which Ea1p = −0.61 V (ref. 12) reflect the lower lying HOMO energies of −5.93/−5.92 eV of the former versus −5.78/−5.77 eV for the latter; as before, we are not able to identify separate CV processes for the two geometrical isomers that are known to co-exist in solution. The true electrochemical reductions for the two types of PIII dicarbenes both occur at very low potentials (less than −3.5 V) and probably cannot be measured accurately; the shoulder on the main reduction wave in the CVs (e.g. Ec1p) are likely from breakdown products of the IRR oxidations (see ESI† for further explanations).
 | ||
| Fig. 4 FMO topologies and energies for the model (R = CH3) calculated structures of 4′, 6′cis and 6′trans at the B3LYP/6-31+G*//M06-2X/6-31+G*level of theory (PCM solvent model). | ||
In contrast to this established behaviour of the diphosphinine dicarbenes, the CVs measured on 4 are less well defined although the onset of oxidation is definitely lower in potential than in 6cis/trans with Ea5p = −0.74 V (Fig. 3a). The computed HOMO of model system 4′ is very different (Fig. 4) and is essentially localized at the anionic P atom, and, as expected for an anion, is also at a much higher computed energy (−0.89 eV). Experimentally, the onset of oxidation does not have the expected well-separated peak for a first 1e oxidation of such a localized MO, followed by further processes after a considerable gap.
A plausible explanation for the observed behaviour is that 1e oxidation gives P-centred radical bis(NHC) 9 which rapidly dimerizes to give tetrakis NHC 10 (Scheme 4). Related tricyclic dithione P-radicals have been shown to be very short-lived furnishing structurally verified dimers with a P–P bond.16 On this view, the remaining processes such as Ea6p = −0.30 V and Ec1p = −2.5 V recorded in the CVs of 4 (Fig. 3a) are best under-stood as redox processes of such a tetrakis NHC.
 | ||
| Scheme 4 One electron reduction of 4 to neutral 9 with rapid dimerization to 10. | ||
Experimental section
Experimental details and devices
All experiments were done under an argon atmosphere, using common Schlenk techniques and dry solvents. Tetrahydrofuran, n-pentane and diethyl ether were dried over sodium wire/benzophenone and further purified by subsequent distillation. The precursor for 1, the 1,4-dichloro-1,4-dihydro-1,4-diphosphinine, was synthesized using standard protocols.12 All NMR spectra were recorded on a Bruker AX-300 spectrometer (300.1 MHz for 1H, 75.5 MHz for 13C, and 121.5 MHz for 31P) and pollux-500 (500.1 MHz for 1H, 125.75 MHz for 13C, and 500.0 MHz for 31P) spectrometer. The 1H and 13C NMR spectra were referenced to the residual proton resonances and the 13C NMR signals of the deuterated solvents and 31P to 85% H3PO4 as external standard, respectively. Elemental analyses were carried out on a Vario EL gas chromatograph. Melting points were determined in one-side melted off capillaries using a Büchi Type S or a Carl Roth Type MPM-2 apparatus, they are uncorrected Mass spectrometric data were collected on a Kratos MS 50 spectrometer using EI, 70 eV. IR spectra of all compounds were recorded on a Thermo IR spectrometer with an attenuated total reflection (ATR) attachment. The X-ray analyses were performed on a Bruker APEX-II CCD or a Bruker X8-KappaApexII type diffractometer at 100(2) K. The structures were solved by direct methods refined by full-matrix least-squares technique in anisotropic approximation for non-hydrogen atoms using SHELXS97 and SHELXL9717 program packages. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2032731 (1), 2032732 (2).†
Synthesis of compounds
1. To a clear solution of 1,4-bis(diethylamino)-1,4-dihydro-1,4-diphosphinine12 (2.5 g, 3.5 mmol) in dichloromethane, PCl3 (0.61 mL, 6.9 mmol) was added and stirred for 4 hours at −40 °C. The reaction mixture was then warmed to room temperature and tris(n-butyl)phosphane (0.34 mL, 1.4 mmol) was added in a dropwise manner. After 10 minutes stirring, a colour change of the solution from orange to violet was observed. After concentrating the reaction mixture under reduced pressure, the residue was filtered via a silica® bed with diethyl ether and toluene mixture (1:1). It was then dried under reduced pressure (6 × 10−3 mbar and washed with n-pentane (3 × 10 mL) to get rid of the aminophosphane Et2NPCl2. Finally, the solution was con-centrated in vacuo (6 × 10−3 mbar) to get 1 as pure compound. Yield: 1.2 g (2.1 mmol) 61%; violet solid. M.p. 223 °C. 1H NMR (300.1 MHz, C6D6, 25 °C): δ = 0.8 (t, 12H, 3JH,H = 7.3 Hz, NCH2CH2CH2![[M with combining low line]](https://www.rsc.org/images/entities/i_char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/i_char_0065_0332.gif) ), 1.2–1.4 (m, 8H, NCH2CH2
), 1.2–1.4 (m, 8H, NCH2CH2![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2Me), 1.8–1.9 (m, 8H, NCH22CH2Me), 4.4–4.4 (m, 8H, N2CH2CH2Me). 13C{1H} NMR (75.5 MHz, C6D6, 25 °C): δ = 13.55 (s, NCH2CH2CH2), 19.9 (s, NCH2CH22Me), 28.9 (t, 3JP,C = 1.6 Hz, NCH22CH2Me), 48.1 (t, 3JP,C = 4.6 Hz, NCH22CH2Me), 152.2 (t, 1JP,C = 23.1 Hz, P– of the middle ring), 168.32 (br, 2). 31P{1H} NMR (121.5 MHz, C6D6, 25 °C): δ = 78.2 (s). 77Se NMR (57.28 MHz, CDCl3): 179.4 (s). IR [cm−1]:
2Me), 1.8–1.9 (m, 8H, NCH22CH2Me), 4.4–4.4 (m, 8H, N2CH2CH2Me). 13C{1H} NMR (75.5 MHz, C6D6, 25 °C): δ = 13.55 (s, NCH2CH2CH2), 19.9 (s, NCH2CH22Me), 28.9 (t, 3JP,C = 1.6 Hz, NCH22CH2Me), 48.1 (t, 3JP,C = 4.6 Hz, NCH22CH2Me), 152.2 (t, 1JP,C = 23.1 Hz, P– of the middle ring), 168.32 (br, 2). 31P{1H} NMR (121.5 MHz, C6D6, 25 °C): δ = 78.2 (s). 77Se NMR (57.28 MHz, CDCl3): 179.4 (s). IR [cm−1]:  = 2998.1 (v), 2752.8 (m), 2654.0 (m), 1487.0 (s), 1289.1 (s), 1201.5 (s), 1098.2 (m), 964.0 (s). MS (EI, 70 eV): m/z (%) = 578.0 (100) [M]˙+, 498.1 (20) [M − Se]˙+. UV-vis (CH2Cl2): λmax [nm] (abs.): 296 (1.375), 383 (0.156), 554(0.139). EA [%]: theor./exp. C 45.84/45.64, H 6.30/6.46; N 9.72/9.63.
= 2998.1 (v), 2752.8 (m), 2654.0 (m), 1487.0 (s), 1289.1 (s), 1201.5 (s), 1098.2 (m), 964.0 (s). MS (EI, 70 eV): m/z (%) = 578.0 (100) [M]˙+, 498.1 (20) [M − Se]˙+. UV-vis (CH2Cl2): λmax [nm] (abs.): 296 (1.375), 383 (0.156), 554(0.139). EA [%]: theor./exp. C 45.84/45.64, H 6.30/6.46; N 9.72/9.63.
2. 2 equivalents of trifluoromethyl methylsulfonate (0.4 mL, 3.4 mmol) was added to a solution of 1 (1.0 g, 1.7 mmol) in dichloromethane, at room temperature. The reaction mixture was stirred for 2 hours resulting in a color change from violet to light yellow. After concentrating the reaction mixture under reduced pressure (6 × 10−3 mbar), the residue was washed with n-pentane (2 × 5 mL) to get 2 as pure light yellow solid. Yield 1.4 g (1.5 mmol) 91%; light yellow solid. M.p. 96 °C. 1H NMR (300.1 MHz, CDCl3, 25 °C): 1.04 (t, 12H, 3JH,H = 7.4 Hz, NCH2CH2CH2), 1.52–1.62 (m, 8H, NCH2CH22Me), 2.05–2.15 (m, 8H, NCH22CH2Me), 2.8 (s, 6H, Se) 4.89–4.94 (m, 8H, N2CH2CH2Me). 13C{1H} NMR (75.5 MHz, CDCl3, 25 °C): δ = 11.72 (s, Se), 13.44 (s, NCH2CH2CH2), 20.16 (s, NCH2CH22Me), 30.73 (s, NCH22CH2Me), 52.18 (s, NCH22CH2Me), 148.84 (t, 3JP,C = 4.6 Hz, Se–2), 154.94 (t, 1JP,C = 26.1 Hz, P– of the middle ring). 31P NMR (121.5 MHz, CDCl3, 25 °C): δ = 119.95 (s). 77Se NMR (57.28 MHz, CDCl3): 138.89 (s). IR [cm−1]:  = 3009.1 (v), 2992.8 (m), 2954.0 (m), 1623.0 (w), 1529.1 (w), 1461.5 (m), 1236.2 (m), 1201.3 (w), 1075.4 (m), 1032.2 (v), 974.0 (s). Pos. ESI-MS: [C25H42F3N4O3P2SSe2]+ calcd (found) 757.0728 (757.0750). neg. ESI-MS: TfO− theor./exp. 148.9(149.5). EA [%]: theor./exp. C 34.52/34.33, H 4.68/4.71; N 6.19/6.0, S 7.08/6.99.
= 3009.1 (v), 2992.8 (m), 2954.0 (m), 1623.0 (w), 1529.1 (w), 1461.5 (m), 1236.2 (m), 1201.3 (w), 1075.4 (m), 1032.2 (v), 974.0 (s). Pos. ESI-MS: [C25H42F3N4O3P2SSe2]+ calcd (found) 757.0728 (757.0750). neg. ESI-MS: TfO− theor./exp. 148.9(149.5). EA [%]: theor./exp. C 34.52/34.33, H 4.68/4.71; N 6.19/6.0, S 7.08/6.99.
3. To a solution of 2 (1.5 g, 1.6 mmol) in methanol, 5 equivalents of sodium tetrahydridoborate (0.3 g, 8.2 mmol) and one equivalent of [2.2.2]-cryptand was added as solid at 0 °C. The reaction mixture turned to orange-red with strong odour due to a liberation of methylselane (HMeSe). The solution was then concentrated in vacuo (6 × 10−3 mbar) after 30 minutes stirring. Extraction was done with dichloromethane followed by washing with diethyl ether (2 × 5 mL) to get 3 as pure orange red solid. Yield: 1.2 g (1.04 mmol) 65%; red orange solid, M.p. 142 °C 1H NMR (300.1 MHz, CD2Cl2, 25 °C): δ = 1.0 (t, 12H, 3JH,H = 7.3 Hz, NCH2CH2CH2), 1.3–1.5 (m, 8H, NCH2CH22Me), 1.9–2.1 (m, 8H, NCH22CH2Me), 2.4 (d, 3H, 3JP,H = 7.3 Hz, O–) 2.6 (t, 12H, cryptand), 3.6 (t, 12H, cryptand), 3.7 (s, 12H, cryptand), 4.1–4.6 (m, 8H, N2CH2CH2Me), 8.9 (t, 2H, 4JP,H = 1.7 Hz, C2–). 13C{1H} NMR (75.5 MHz, CD2Cl2, 25 °C): δ = 13.2 (s, NCH2CH2CH2), 19.6 (s, NCH2CH22Me), 30.2 (s, NCH22CH2Me), 47.8 (s, N2CH2CH2Me), 67.5 (s, cryptand), 68.5 (s, cryptand), 120.9 (d, 2JP,C = 7.3 Hz, O–3), 121.7 (q, 1JP,F = 321.0 Hz, F3), 137.2 (d, 3JP,C = 4.5 Hz, H–2), 155.5 (ddd, 1/2JP,C = 47.0 Hz, 4/5). 31P NMR (121.5 MHz, CD2Cl2, 25 °C): δ = 20.12 (P–OMe), −67.34 (anion P). IR [cm−1]:  = 2984 (v), 2921.8 (m), 2894.0 (m), 1542.0 (w), 498.1 (w), 1423.5 (m), 1246.2 (m), 1206.3 (w), 1012.4 (m), 968.4 (s). Pos. ESI-MS: m/z (%) = 451.3 (100) [M]+, 399.1 (97) [Na(C18N2H36O6)]˙+. HRMS: [C23H41N4OP2]+ theor./exp. 451.2750 (451.2754). UV/vis (CH2Cl2): λ [nm] (abs.): 346 (0.791). EA [%]: theor./exp. C 44.94/45.12, H 6.75/6.64; N 7.31/6.74, S 5.58/5.68.
= 2984 (v), 2921.8 (m), 2894.0 (m), 1542.0 (w), 498.1 (w), 1423.5 (m), 1246.2 (m), 1206.3 (w), 1012.4 (m), 968.4 (s). Pos. ESI-MS: m/z (%) = 451.3 (100) [M]+, 399.1 (97) [Na(C18N2H36O6)]˙+. HRMS: [C23H41N4OP2]+ theor./exp. 451.2750 (451.2754). UV/vis (CH2Cl2): λ [nm] (abs.): 346 (0.791). EA [%]: theor./exp. C 44.94/45.12, H 6.75/6.64; N 7.31/6.74, S 5.58/5.68.
4. A solution of potassium hexamethyldisilazide (KHMDS) (0.7 g, 3.5 mmol) in 5 mL of THF was added dropwise to a solution of 3 (2 g, 1.7 mmol) in 10 mL of THF at room temperature. After 1 h, all volatiles were removed in vacuo (6 × 10−3 mbar). The residue was washed (twice) with diethyl ether followed by extraction with mixture of THF and diethyl ether (1:1.5) to remove the potassium triflate. After concentrating the extracted solution, the product 4 was obtained as dark orange solid. Yield: 1.1 g (1.3 mmol) 76%; Dark orange. M.p. 207 °C. 1H NMR (500.1 MHz, THF, 25 °C): δ = 0.9 (t, 12H, 3JH,H = 6.5 Hz, NCH2CH2CH2), 1.2–1.3 (m, 8H, NCH2CH22Me), 1.8–1.9 (m, 8H, NCH22CH2Me), 2.1 (d, 3H, 4JP,H = 6.3 Hz, O–) 2.5 (t, 12H, cryptand), 3.5 (t, 12H, cryptand), 3.6 (s, 12H, cryptand), 3.8–4.5 (m, 8H, N2CH2CH2Me). 13C{1H} NMR (75.5 MHz, THF-d8, 25 °C): δ = 13.5 (s, NCH2CH2CH2), 20.1 (s, NCH2CH22Me), 32.3 (s, NCH22CH2Me), 47.5 (s, N2CH2CH2Me), 67.6 (s, cryptand), 70.4 (s, cryptand), 120.2 (d, 2JP,C = 7.5 Hz, O–3), 154.2 (d, 1/2JP,C = 43.9 Hz, 4/5), 215.5 (d, JP,C = 2.7, 2). 31P NMR (500.0 MHz, THF-d8, 25 °C): δ = 25.15 (P–OMe), −74.78 (anion P). IR [cm−1]:  = 3191.2 (w), 3045.0 (m), 2975.7 (m), 1623.4 (m), 1436.3 (m), 1375.3 (m), 1245.7 (s), 1184.8 (s), 1016.5 (s), 968.5 (s). Neg. ESI-MS: m/z (%) = 449.261 (15) [M]+, HRMS: [C23H39N4OP2]− theor./exp. 449.2605 (449.2608). UV/vis (THF): λ [nm] (abs.): 407 (0.517).
= 3191.2 (w), 3045.0 (m), 2975.7 (m), 1623.4 (m), 1436.3 (m), 1375.3 (m), 1245.7 (s), 1184.8 (s), 1016.5 (s), 968.5 (s). Neg. ESI-MS: m/z (%) = 449.261 (15) [M]+, HRMS: [C23H39N4OP2]− theor./exp. 449.2605 (449.2608). UV/vis (THF): λ [nm] (abs.): 407 (0.517).
5cis/trans. To a suspension of 3 (2.5 g, 2.1 mmol) in 50 mL of diethyl ether, methyl iodide (1.35 mL, 2.1 mmol) was added dropwise at −80 °C. The reaction mixture was stirred for 18 hours and warmed to room temperature. All volatiles were removed in vacuo (6 × 10−3 mbar). Residue was extracted with dichloromethane followed by washing (twice) with diethyl ether. The solvent was removed under vacuum (6 × 10−3 mbar) which resulted in a pure colorless liquid. Yield: 1.2 g (1.6 mmol) 75%; colorless liquid. Ratio of two isomers 1:0.3. M.p. – (liquid at r.t.). 1H NMR (300.1 MHz, CD2Cl2, 25 °C): δ = 1.04, 1.06 (t, 12H, 3JH,H = 7.4 Hz, NCH2CH2CH2), 1.41–1.54 (m, 8H, PCH2CH22Me), 1.67 (d, 2JP,H = 5.2 Hz, ![[P with combining low line]](https://www.rsc.org/images/entities/i_char_0050_0332.gif) –), 1.87 (d, 2JP,H = 6.8 Hz,
–), 1.87 (d, 2JP,H = 6.8 Hz, ![[O with combining low line]](https://www.rsc.org/images/entities/i_char_004f_0332.gif) –);1.92–2.11 (m, 8H, NCH22CH2Me, 4.28–4.57 (m, 8H, N2CH2CH2Me), 9.47 (t, 2H, 3JP,H = 3.03 Hz, C2–), 9.56 (brs, C2–) 2nd isomer. 13C{1H} NMR (75.5 MHz, CD2Cl2, 25 °C): δ = 13.08, 13.10 (s, NCH2CH2CH2 of two isomers), 19.52 (br, –), 19.68 (s, NCH2CH22Me), 29.68 (br, NCH22CH2Me), 31.78 (d, 3JP,C = 2.6 Hz, NCH22CH2Me), 32.23 (d, 3JP,C = 2.4 Hz, NCH22CH2Me) (2nd isomer), 49.13 (ddd, 3JP,C = 9.1 Hz, N2CH2CH2Me), 49.96 (ddd, 3JP,C = 8.2 Hz, N2CH2CH2Me) (2nd isomer), 122.84 (q, 1JP,F = 319.5 Hz, F3), 131.54 (d, 2JP,C = 9.5 Hz, O–3), 135.38 (ddd, 1/2JP,C = 3.7 Hz, – of the middle ring), 135.72 (t, 1/2JP,C = 3.0 Hz, P– of the middle ring) (2nd isomer), 142.38 (br, H–2), 143.37 (br, H–2) 2nd isomer. 31P NMR (500.0 MHz, CD2Cl2, 25 °C): δ = −71.58 (d, 3JP,H = 5.2 Hz, P–), −66.23 (d, 3JP,H = 4.9 Hz, P–); 39.57 (br, P–OMe) & 43.7 (br, P–OMe). IR [cm−1]:
–);1.92–2.11 (m, 8H, NCH22CH2Me, 4.28–4.57 (m, 8H, N2CH2CH2Me), 9.47 (t, 2H, 3JP,H = 3.03 Hz, C2–), 9.56 (brs, C2–) 2nd isomer. 13C{1H} NMR (75.5 MHz, CD2Cl2, 25 °C): δ = 13.08, 13.10 (s, NCH2CH2CH2 of two isomers), 19.52 (br, –), 19.68 (s, NCH2CH22Me), 29.68 (br, NCH22CH2Me), 31.78 (d, 3JP,C = 2.6 Hz, NCH22CH2Me), 32.23 (d, 3JP,C = 2.4 Hz, NCH22CH2Me) (2nd isomer), 49.13 (ddd, 3JP,C = 9.1 Hz, N2CH2CH2Me), 49.96 (ddd, 3JP,C = 8.2 Hz, N2CH2CH2Me) (2nd isomer), 122.84 (q, 1JP,F = 319.5 Hz, F3), 131.54 (d, 2JP,C = 9.5 Hz, O–3), 135.38 (ddd, 1/2JP,C = 3.7 Hz, – of the middle ring), 135.72 (t, 1/2JP,C = 3.0 Hz, P– of the middle ring) (2nd isomer), 142.38 (br, H–2), 143.37 (br, H–2) 2nd isomer. 31P NMR (500.0 MHz, CD2Cl2, 25 °C): δ = −71.58 (d, 3JP,H = 5.2 Hz, P–), −66.23 (d, 3JP,H = 4.9 Hz, P–); 39.57 (br, P–OMe) & 43.7 (br, P–OMe). IR [cm−1]:  = 3204.7 (m), 3145 (m), 2975.5 (w), 2768.8 (m), 1534.3 (w), 1445.3 (s), 1317.7 (m), 1206.8 (m), 1046.9 (m), 1009.5 (s), 921.5 (s). Pos. ESI-MS: m/z (%) = 615.251 (54) [M − TfO]+; HRMS: [C25H44F3N4O4P2S]+ theor./exp. 615.2505 (615.2511). EA [%]: theor./exp. C 40.84/40.53, H 5.80/5.93, N 7.33/7.36.
= 3204.7 (m), 3145 (m), 2975.5 (w), 2768.8 (m), 1534.3 (w), 1445.3 (s), 1317.7 (m), 1206.8 (m), 1046.9 (m), 1009.5 (s), 921.5 (s). Pos. ESI-MS: m/z (%) = 615.251 (54) [M − TfO]+; HRMS: [C25H44F3N4O4P2S]+ theor./exp. 615.2505 (615.2511). EA [%]: theor./exp. C 40.84/40.53, H 5.80/5.93, N 7.33/7.36.
6cis/trans. A solution of potassium hexamethyldisilazide (KHMDS) (1.04 g, 5.2 mmol) in 5 mL of THF was added dropwise to a solution of 5cis/trans (2.0 g, 2.6 mmol) in 10 mL of THF at room temperature. After 1 h, all volatiles were removed in vacuo (6 × 10−3 mbar). Residue was extracted with diethyl ether to remove potassium triflate using filtering cannulation. After concentrating the extracted solution, the product 6cis/trans was obtained as yellow liquid. Yield: 0.92 g (1.9 mmol) 76%; (ratio of two isomers 1:0.30). M.p. – (liquid at r.t.) 1H NMR (500.1 MHz, THF-d8, 25 °C): δ = 0.8, 1.1 (t, 12H, 3JH,H = 7.1 Hz, NCH2CH2CH2), 1.2 (d, 2JP,H = 5.3 Hz, –), 1.2–1.4 (m, 8H, PCH2CH22Me), 1.9–2.1 (m, 8H, NCH22CH2Me), 2.7 (d, 2JP,H = 7.2 Hz, –), 3.9–4.2 (m, 8H, N2CH2CH2Me), 4.3–4.5 (m, 8H, N2CH2CH2Me; 2nd isomer). 13C{1H} NMR (125.75 MHz, THF-d8, 25 °C): δ = 12.9, 12.8 (s, NCH2CH2CH2 of two isomers), 19.9 (s, –), 23.7 (s, NCH2CH22Me), 25.7 (s, NCH22CH2Me), 32.8 (d, 3JP,C = 2.1 Hz, NCH22CH2Me), 32.9 (d, 3JP,C = 2.2 Hz, NCH22CH2Me; 2nd isomer), 48.5 (ddd, 3JP,C = 9.6 Hz, N2CH2CH2Me), 49.6 (ddd, 3JP,C = 8.3 Hz, N2CH2CH2Me; 2nd isomer), 118.5 (d, 2JP,C = 9.2 Hz, O–3), 131.4 (br, – of the middle ring), 132.2 (d, 1/2JP,C = 2.5 Hz, – of the middle ring; 2nd isomer), 223.4 ((t, 3JP,C = 2.7 Hz, 2), 224.2 (t, 3JP,C = 2.7 Hz, 2; 2nd isomer). 31P NMR (500 MHz, THF-d8, 25 °C): δ = −74.0 (d, 3JP,H = 4.8 Hz, –), −68.6 (d, 3JP,H = 3.7 Hz, –); 41.3 (d, 3JP,H = 3.8 Hz) & 37.2 (d, 3JP,H = 4.6 Hz). IR [cm−1]:  = 2992.2 (m), 2962.5 (m), 2842.2 (w), 1501.2 (w), 1472.4 (m), 1415.8 (s), 1367.3 (s), 1146.0 (s), 1052.1 (m), 986.5 (m). Pos. ESI-MS: m/z (%) = 465.290 (31) [M + H]˙+ [C24H43ON4P2]1+ theor./exp. 465.2907 (465.2909). UV/vis (CH2Cl2): λ [nm] (abs.): 347 (0.124).
= 2992.2 (m), 2962.5 (m), 2842.2 (w), 1501.2 (w), 1472.4 (m), 1415.8 (s), 1367.3 (s), 1146.0 (s), 1052.1 (m), 986.5 (m). Pos. ESI-MS: m/z (%) = 465.290 (31) [M + H]˙+ [C24H43ON4P2]1+ theor./exp. 465.2907 (465.2909). UV/vis (CH2Cl2): λ [nm] (abs.): 347 (0.124).
7. To a solution of 3 (2.0 g, 1.7 mmol) in dichloromethane, [Rh(cod)Cl]2 (0.43 g, 0.87 mmol) was added as solid at ambient temperature. Reaction mixture was stirred for 6 hours, at which point volatiles were removed in vacuo (6 × 10−3 mbar). Residue was washed (twice) with diethyl ether and subsequent drying in vacuo (6 × 10−3 mbar) resulted in an orange solid. Yield: 2.1 g (1.5 mmol) 88%; Orange solid; M.p. 82 °C. 1H NMR (300.1 MHz, CD2Cl2, 25 °C): δ = 0.99 (t, 12H, 3JH,H = 7.2 Hz, NCH2CH2CH2), 1.26–1.57 (m, 8H, NCH2CH22Me), 1.92 (m, 4H, cod), 1.99–2.21 (m, 8H, NCH22CH2Me), 2.36 (m, 4H, cod), 2.56 (d, 8H, 3JP,H = 10.2 Hz, O–3), 3.68 (m, 2H, cod), 4.06–4.21 (m, 8H, N2CH2CH2Me), 5.37 (m, 2H, cod), 8.99 (brs, C2–). 13C{1H} NMR (125.75 MHz, CD2Cl2, 25 °C): δ = 13.46 (s, NCH2CH2CH2), 19.95 (s, NCH2CH22Me), 28.52 (s, cod), 30.14 (br, NCH22CH2Me), 33.27 (s, cod), 49.48 ((d, 3JP,C = 7.2 Hz, N2CH2CH2Me), 69.6 (br, cod), 73.21 (d, 1JRh,C = 11.2 Hz, cod), 122.13 (q, 1JP,F = 322.8 Hz, F3), 108.68 (d, 2JP,C = 11.1 Hz, O–3), 155.96 (br, P– of the middle ring), 156.35 (d, 1/2JP,C = 44.2 Hz, P– of the middle ring), 137.96 (br, H–2). 31P NMR (500.0 MHz, CD2Cl2, 25 °C): δ = −70.56 (s), 47.45 (d, 1JRh,P = 188.2 Hz). IR [cm−1]:  = 2975.2 (m), 2931.4 (m), 2840.1 (w), 1511.4 (s), 1480.4 (m), 1398.1 (m), 1247.8 (s), 1175.5 (m), 1129.8 (m), 1007.1 (s), 910.2 (s). Neg. ESI-MS: m/z (%) = 995.151 (29) [M]˙+. HRMS: [C33H53ClF3N4O7P2RhS2F6]+ theor./exp. 995.1473 (995.1494). UV/vis (CH2Cl2): λ [nm] (abs.): 299 (0.924). EA [%]: theor./exp. C 43.89/43.89, H 6.43/6.73, N 6.02/76.03, S 4.59/4.34.
= 2975.2 (m), 2931.4 (m), 2840.1 (w), 1511.4 (s), 1480.4 (m), 1398.1 (m), 1247.8 (s), 1175.5 (m), 1129.8 (m), 1007.1 (s), 910.2 (s). Neg. ESI-MS: m/z (%) = 995.151 (29) [M]˙+. HRMS: [C33H53ClF3N4O7P2RhS2F6]+ theor./exp. 995.1473 (995.1494). UV/vis (CH2Cl2): λ [nm] (abs.): 299 (0.924). EA [%]: theor./exp. C 43.89/43.89, H 6.43/6.73, N 6.02/76.03, S 4.59/4.34.
8cis/trans. To a solution of 3 (1.5 g, 1.3 mmol) in dichloromethane, 1.5 equivalent of [Rh(cod)Cl]2 (0.97 g, 1.9 mmol) was added as solid at ambient temperature. Reaction mixture was stirred at ambient temperature for 12 hours. Solvent was then removed in vacuo (6 × 10−3 mbar) and the residue washed (twice) with diethyl ether. Subsequent drying in vacuo (6 × 10−3 mbar) resulted in a dark orange solid. Yield: 1.7 g (0.9 mmol) 69%; dark orange solid; (ratio of two isomers 1:0.7). M.p. 102 °C. 1H NMR (500.1 MHz, CD2Cl2, 25 °C): δ = 1.1–1.2 (t, 12H, 3JH,H = 7.0 Hz, NCH2CH2CH2), 1.5–1.7 (m, 8H, NCH2CH22Me), 2.2–2.4 (m, 24H, cod), 2.5–2.6 (m, 8H, NCH22CH2Me), 2.9, 3.0 (d, 3H, 3JP,H = 12.4 Hz, O–3), 3.9 (m, 6H, cod), 4.2 (br, 8H, N2CH2CH2Me), 5.2 (m, 6H, cod), 9.5 (brs, C2–), 9.7 (brs, C2–). 13C{1H} NMR (125.75 MHz, CD2Cl2, 25 °C): δ = 13.5, 13.8 (s, NCH2CH2CH2) two isomers, 19.9, 20.1 (s, NCH2CH22Me) two isomers, 28.4 (s, cod), 31.3 (br, NCH22CH2Me), 32.7 (s, cod), 49.9 (d, 3JP,C = 6.3 Hz, N2CH2CH2Me), 49.9 (br, N2CH2CH2Me), 72.0 (d, 1JRh,C = 13.0 Hz, cod), 72.6 (d, 1JRh,C = 13.0 Hz, cod), 73.21 (d, 1JRh,C = 13.1 Hz, cod), 74.2 (d, 1JRh,C = 13.2 Hz, cod), 74.6 (d, 1JRh,C = 13.0 Hz, cod), 75.4 (d, 1JRh,C = 13.1 Hz, cod), 113.4 (d, 2JP,C = 9.5 Hz, O–3), 121.1 (q, 1JP,F = 332.7 Hz, F3), 131.9 (br, – of the middle ring), 133.3 (d, 1/2JP,C = 45.2 Hz, – of the middle ring), 141.9 (br, H–2), 142.4 (br, H–2) 2nd isomer. 31P NMR (500.0 MHz, CD2Cl2, 25 °C): δ = 65.0 (d, 1JRh,P = 195.6 Hz) (minor)/64.1 (dd, 1JRh,P = 200.1 Hz, 3JP,P = 5.3 Hz) (major) and −120.3 (t br, 1JRh,P = 126.9 Hz) (major)/−123.4 (t, 1JRh,P = 126.9 Hz) (minor). IR [cm−1]: 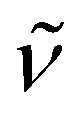 = 2984.1 (m), 2971.0 (m), 2861.8 (w), 1545.0 (s), 1491.7 (m), 1421.1 (m), 1327.8 (s), 1265.6 (m), 1129.8 (w), 1069.1 (s), 978.0 (s). Pos. ESI-MS: m/z (%) = 1153.205 (36) [M − Cl − 2TfO]˙+. HRMS: [C47H76Cl2N4OP2Rh3]˙+ theor./exp. 1153.2031 (1153.2043). UV/vis (CH2Cl2): λ [nm] (abs.): 386 (0.216). EA [%]: theor./exp. C 42.61/41.38, H 6.03/6.04, N 4.45/4.22, S 3.40/3.62.
= 2984.1 (m), 2971.0 (m), 2861.8 (w), 1545.0 (s), 1491.7 (m), 1421.1 (m), 1327.8 (s), 1265.6 (m), 1129.8 (w), 1069.1 (s), 978.0 (s). Pos. ESI-MS: m/z (%) = 1153.205 (36) [M − Cl − 2TfO]˙+. HRMS: [C47H76Cl2N4OP2Rh3]˙+ theor./exp. 1153.2031 (1153.2043). UV/vis (CH2Cl2): λ [nm] (abs.): 386 (0.216). EA [%]: theor./exp. C 42.61/41.38, H 6.03/6.04, N 4.45/4.22, S 3.40/3.62.
Conclusion
The first example of an anionic P-bridged tricyclic bis(NHC) was obtained via an unexpected reductive deselenization combined with a subsequent deprotonation of the bis(imidazolium) salt. Reaction of the latter with MeI as electrophile resulted finally in the first example of a neutral P-functional bis(NHCs) with a mixed substitution pattern. Initial studies of the coordination properties of the tricyclic bis(imidazolium) salt revealed a pre-ference of the neutral P-ligands site over the anionic phosphorus centre. Detailed electrochemical studies of the anionic bis(NHC) showed multiple, closely spaced oxidation processes owing, most probably, to the formation of a short lived P-centred radical that yields a new intermediate, a tetrakis(NHC), having a P–P bond linkage.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the University of Bonn for financial support. The reinvitation grants for R. T. B. and L. N. from the Alexander von Humboldt Foundation are acknowledged. Z. K. is grateful for the general support of Hungarian Academy of Science under the Premium Postdoctoral Research Program 2019.Notes and references
- (a) For some books, see: N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006 Search PubMed (b) S. D. Gonzalez, N-Heterocyclic, Carbenes: From Laboratory Curiosities To Efficient Synthetic Tools, Royal Society of Chemistry, Cambridge, U.K., 2011 Search PubMed.
- (a) F. E. Hahn and M. C. Jancke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS; (b) K. J. Cavell, Dalton Trans., 2008, 6676 RSC; (c) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (d) D. Bourissou, O. Guerret, F. Gabbaı and G. Bertrand, Chem. Rev., 2000, 100, 32 CrossRef; (e) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS; (f) A. Prades, E. Peris and M. Alcarazo, Organometallics, 2012, 31, 4623 CrossRef CAS; (g) H. Valdes, M. Poyatos and E. Peris, Organometallics, 2015, 34, 1725 CrossRef CAS.
- (a) S. Conde-Guadano, M. Hanton, R. P. Tooze, A. A. Dano-poulos and P. Braunstein, Dalton Trans., 2012, 41, 12558 RSC; (b) S. Conde-Guadano, A. A. Danopoulos, R. Pattacini, M. Hanton and R. P. Tooze, Organometallics, 2012, 31, 1643 CrossRef CAS; (c) N. Vujkovic, V. Cesar, N. Lugan and G. Lavigne, Chem. – Eur. J., 2011, 17, 13151 CrossRef CAS; (d) A. Carter, A. Mason, M. A. Baker, D. G. Bettler, A. Changas, C. D. McMillen and D. Tapu, Organometallics, 2017, 36, 1867 CrossRef CAS.
- D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, Chem. – Asian J., 2011, 6, 1099 CrossRef CAS.
- L. Benhamou, V. César, H. Gornitzka, N. Lugan and G. Lavigne, Chem. Commun., 2009, 4720 RSC.
- S. Kronig, E. Theuergarten, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 3240 CrossRef CAS.
- A. A. Danopoulos, K. Y. Monakhov and P. Braunstein, Chem. – Eur. J., 2013, 19, 450 CrossRef CAS.
- P. K. Majhi, G. Schnakenburg, Z. Kelemen, L. Nyulaszi, D. P. Gates and R. Streubel, Angew. Chem., Int. Ed., 2013, 52, 10080 CrossRef CAS.
- (a) N. Vujkovic, V. César, N. Lugan and G. Lavigne, Chem. – Eur. J., 2011, 17, 13151 CrossRef CAS; (b) D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, Chem. – Asian J., 2011, 6, 1099 CrossRef CAS.
- D. M. Khramov, A. J. Boydston and C. W. Bielawski, Angew. Chem., Int. Ed., 2006, 45, 6186 CrossRef CAS.
- P. K. Majhi, A. Koner, G. Schnakenburg, Z. Kelemen, L. Nyulászi and R. Streubel, Eur. J. Inorg. Chem., 2016, 3559 CrossRef CAS.
- (a) N. R. Naz, G. Schnakenburg, A. Mikeházi, Z. Kelemen, L. Nyulászi, R. T. Boeré and R. Streubel, Chem. Commun., 2020, 56, 2646 RSC; (b) for a recent account on bis(NHCs), see: S. Ibáñez, M. Poyatos and E. Peris, Acc. Chem. Res., 2020, 53, 1401 CrossRef.
- A. Koner, G. Pfeifer, Z. Kelemen, G. Schnakenburg, L. Nyulászi, T. Sasamori and R. Streubel, Angew. Chem., Int. Ed., 2017, 56, 9231 CrossRef CAS.
- 1′, 2′, 3′, 4′, 5′cis/trans, 6′cis/trans calculated compounds with methyl substituents at N and for ions without counter ions; see also ref. 12a and 13.
- Z. Kelemen, R. Streubel and L. Nyulászi, RSC Adv., 2015, 5, 41795 RSC.
- I. Begum, G. Schnakenburg, Z. Kelemen, L. Nyulászi, R. T. Boeré and R. Streubel, Chem. Commun., 2018, 54, 13555 RSC.
- (a) SHELXS-97: G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef; (b) G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2032731 and 2032732. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03915b |
| This journal is © The Royal Society of Chemistry 2021 |