
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ce(OH)2Cl and lanthanide-substituted variants as precursors to redox-active CeO2 materials†
Alexander J. A.
Dunn
a,
James W.
Annis
a,
Janet M.
Fisher
b,
David
Thompsett
b and
Richard I.
Walton
 *a
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bJohnson Matthey Technology Centre, Sonning Common, Reading, RG4 9NH, UK
First published on 13th October 2020
Abstract
The cerium(III) hydroxide chloride Ce(OH)2Cl crystallises directly as a polycrystalline powder from a solution of CeCl3·7H2O in poly(ethylene) glycol (Mn = 400) heated at 240 °C and is found to be isostructural with La(OH)2Cl, as determined from high-resolution synchrotron powder X-ray diffraction (P21/m, a = 6.2868(2) Å, b = 3.94950(3) Å, c = 6.8740(3) Å, β = 113.5120(5)°). Replacement of a proportion of the cerium chloride in synthesis by a second lanthanide chloride yields a set of materials Ce1−xLnx(OH)2Cl for Ln = La, Pr, Gd, Tb. For La the maximum value of x is 0.2, with an isotropic expansion of the unit cell, but for the other lanthanides a wider composition range is possible, and the lattice parameters show an isotropic contraction with increasing x. Thermal decomposition of the hydroxide chlorides at 700 °C yields mixed-oxides Ce1−xLnxO2−δ that all have cubic fluorite structures with either expanded (Ln = La, Gd) or contracted (Ln = Pr, Tb) unit cells compared to CeO2. Scanning electron microscopy shows a shape memory effect in crystal morphology upon decomposition, with clusters of anisotropic sub-micron crystallites being seen in the precursor and oxide products. The Pr- and Tb-substituted oxides contain the substituent in a mixture of +3 and +4 oxidation states, as seen by X-ray absorption near edge structure spectroscopy at the lanthanide LIII edges. The mixed oxide materials are examined using temperature programmed reduction in 10%H2 in N2, which reveals redox properties suitable for heterogeneous catalysis, with the Pr-substituted materials showing the greatest reducibility at lower temperature.
1. Introduction
Cerium dioxide is well-known for its widespread applications in catalysis, where it commonly plays the role of a redox-active support, making use of its easy and reversible release and uptake of oxygen.1 This chemistry is exploited in various practical applications, such as the oxygen storage component in three-way catalytic convertors for abatement of pollutants in automotive exhausts,2 and as an oxide-conducting electrolyte in solid oxide fuel cells.3 Emerging applications of ceria in catalysis include in steam reforming,4 soot oxidation,5 water-gas shift,6 oxidation of volatile organics,7 and thermochemical water splitting.8 Beyond heterogeneous catalysis, biomedical applications of ceria are being explored,9 and the material also finds use in UV-shielding,10 as an abrasive agent,11 and in humidity sensors.12The defect chemistry of ceria is crucial in determining its properties,13 as is its crystallite morphology and size,14 particularly on the nanoscale.15 These aspects of fine control of properties of ceria mean that the synthesis method used is of the utmost importance in forming materials with desirable properties.16 The simplest synthesis method is precipitation by increasing the pH of aqueous solutions of cerium salts, such as ammonium cerium(IV) nitrate or cerium(III) nitrate, followed by calcination of the isolated amorphous solid at temperatures of typically 500–700 °C to induce crystallisation.17 Greater control over crystal morphology is obtained by hydrothermal, or more generally solvothermal, routes that allow one-step crystallisation from solutions heated above the boiling point of the solvent.18 Other synthesis routes to ceria fine powders include combustion, with inclusion of a suitable oxidant to induce rapid formation of the oxide product,19 and reverse micelle sol gel techniques, with surfactants to allow confined growth of small particles.20
The thermal decomposition of crystalline precursors provides another convenient approach for formation of ceria, and it has been commonly observed that the crystallite morphology of the ceria product reflects the morphology of the precursor salt.21 This ‘shape memory effect’ has the potential for forming ceria crystallites with unusual surface reactivity, while the inclusion of substituent cations in the precursor salt provides a way of achieving a homogeneous elemental distribution in the oxide. Cerium(III) carboxylates, such as formate,22 and oxalate,23 yield nanostructured ceria at temperatures around 400 °C. The basic carbonate CeCO3(OH) has been widely studied as a precursor and the typical anisotropic morphology of the crystallites permit the formation of particles of CeO2 with unusual morphologies.24 Isovalent substitution of Ce3+ by other trivalent lanthanides to yield Ce1−xLnxCO3(OH) has proved possible,25 and recently cobalt-doping of the precursor was found as a facile route to Co-doped CeO2 nanosheets with electrocatalytic properties.26
The crystalline lanthanide chloride hydroxides, Ln(OH)2Cl, were reported in the 1960s for Ln = La, Pr, Nd, Sm and Gd,27 and their crystal structures determined in these and subsequent works,28 as well as further examples found for other lanthanides.29 An exception is the case of Ce(OH)2Cl, which although was mentioned by Klevtsov et al. in 1969,27c was not structurally characterised until 2017 when Kim et al. prepared the material using a hexamethylenetetramine solution route in the form of powders and as films.30 In this paper we describe a convenient solvothermal route to bulk, polycrystalline samples of Ce(OH)2Cl, and then show how replacement of the cerium for other lanthanides is possible, before studying their thermal decomposition to give substituted cerium oxides, Ce1−xLnxO2−δ, with four examples of substituents chosen to illustrate the scope for substitutional chemistry.
2. Experimental section
CeCl3·7H2O (Sigma-Aldrich, 99.9% purity), LaCl3·7H2O (Alfa Aesar, 99.9% purity), PrCl3·7H2O (Alfa Aesar, 99.9% purity), GdCl3·7H2O (Alfa Aesar, 99.9% purity) and TbCl3·7H2O (Alfa Aesar, 99.999% purity) powders were used as precursor materials. The level of hydration of the metal salts was determined accurately using thermogravimetric analysis. Samples of Ce1−xLnx(OH)2Cl were prepared by solvothermal synthesis from metal salts using Teflon-lined, stainless-steel autoclaves (20 mL). In a typical synthesis, CeCl3·7H2O and LnCl3·7H2O (Ln = La, Pr, Gd and Tb) with desired molar ratio (2 mmol total) were stirred in 8 mL of polyethylene glycol (Mn = 400, Sigma-Aldrich) for 1 h. The reaction mixtures were sealed in autoclaves and then placed in a preheated fan oven at 240 °C for 24 h, before cooling to room temperature. After the reaction, precipitates were obtained that were recovered by suction filtration. All the solid products were washed with deionised water and dried at 70 °C overnight in air before further characterisation.Powder X-ray diffraction (PXRD) was initially performed to assess phase purity using a Siemens D5000 diffractometer operating with Cu Kα1/2 radiation in Bragg–Brentano geometry. Higher resolution PXRD data were recorded at room temperature using a Panalytical X'Pert Pro MPD operating with monochromatic Cu Kα1 radiation and equipped with a PIXcel solid-state detector. For structure solution and refinement of Ce(OH)2Cl PXRD was recorded using the I11 beamline at the Diamond Light Source synchrotron facility (UK) under ambient conditions. A finely-ground sample was mounted in a spinning quartz capillary, and data were collected across a range of 2–92° 2θ with a step size of 0.004° using a wide-angle position sensitive detector based on Mythen-2 Si strip modules. The incident X-ray beam wavelength was 0.82496(10) Å. Data were analysed using the GSAS software visualised using the EXPGUI interface,31 to allow Rietveld refinement of structural models or refinement of lattice parameters using the Le Bail method.
Thermogravimetric analysis (TGA) was performed using a Mettler Toledo TGA/DSC 1 instrument under a constant flow of air (50 mL min−1). Simultaneous differential scanning calorimetry (DSC) curves were also recorded. Data were recorded from room temperature to 1000 °C at a rate of 10 °C min−1. Seven point nitrogen adsorption isotherms were measured on a Micromeritics Tristar Instrument. The samples were pre-treated in flowing nitrogen at 200 °C for 16 h. BET analysis was performed to obtain surface areas.
Scanning electron microscopy was performed using a Zeiss Supra 55-VP field emission scanning electron microscope with applied accelerating voltages within the range of 5–10 kV at an aperture size of around 10 μm. SmartSEM software was used to capture and process images. Aluminium circular SEM stubs (12.5 mm diameter) were covered by adhesive conductive carbon tape. The powdered samples were transferred on to individual SEM stubs. The stubs were placed within a carbon evaporator to coat the sample with carbon particles to reduce charging. Energy dispersive X-ray analysis (EDXA) was used to determine approximate metal ratios and their homogeneity by selectively analysing ∼6 areas of the specimens. Genesis Software was used to collect and quantify the data and process the information into EDXA spectra and the are results quoted as average atomic percentage of the elements identified.
Pr and Tb LIII-edge X-ray absorption spectra were collected on Beamline B18 at Diamond Light Source, UK. The incident energy was selected using a water-cooled, fixed-exit, double-crystal monochromator with Si (111) crystals. On B18 a double toroidal mirror, coated with Cr and Pt, 25 m from the source is used to focus the beam horizontally and vertically onto the sample and a pair of smaller plane mirrors are used for harmonic rejection. X-ray absorption near edge structure (XANES) spectra were normalised to the edge step using the software ATHENA.32 The samples were diluted with polyethylene powder and pressed into a 13 mm diameter pellets, approximately 1 mm thick. The absorption data were collected in transmission mode and the spectra were normalised using the ATHENA software package. The Pr6O11 and Tb4O7 reference materials were supplied by Johnson Matthey Technology Centre, UK.
Temperature programmed reduction (TPR) was measured by thermal conductivity of 10% H2 in N2 (at a flow rate of 30 ml min−1) before and after contact with 0.05–0.10 g sample as a function of temperature. A H2O trap after the sample was used to absorb water created by H2 oxidation. Temperature was increased linearly as a function of time. In order to quantify accurately the hydrogen consumption a known quantity (1 ml) of N2 was injected into the H2/N2 gas stream before the experiment began to create a calibration peak. Quantification of H2 consumption was carried by the integration of TPR profile as a function of time results are presented as the amount of H atoms oxidised per gram of sample, mmol(H) g−1 which can be related to fraction of lanthanide atoms reduced. The error is estimated to be ±5% for the total H2 consumption in these measurements.
3. Results and discussion
The synthesis of Ce(OH)2Cl was achieved using poly(ethylene glycol) with average Mn = 400 as solvent and using CeCl3·7H2O as a reagent at temperatures of 240 °C. Ce(OH)2Cl was mentioned in the 1960s but never structurally characterised until recent work by Kim et al., which appeared during the work we describe here.30 Our sample is a highly crystalline polycrystalline powder and hence we measured synchrotron powder X-ray diffraction in order to refine its structure, based on the structure known for other Ln(OH)2Cl materials. The powder XRD pattern could be indexed and the profile fitted to a monoclinic cell (space group: P21/m), and atomic positions taken from the published structure of Nd(OH)2Cl28c to allow a full structure refinement. The final Rietveld fit is shown in Fig. 1 and crystallographic data are presented in Table 1. Bond valence sums33 give a value of 3.09 for the single cerium site in the unit cell, consistent with the expected +3 oxidation state for cerium. Our structural model for Ce(OH)2Cl is very similar to that deduced by Kim et al.30 and shows that the material is isostructural with La(OH)2Cl and other Ln(OH)2Cl materials.28bFig. 2 shows representations of the crystal structure. The cerium is coordinated to 6 hydroxide ions (Ce–O ∼ 2.5 Å) and 2 chloride ions (Ce–Cl = 3.03 Å), with a further Cl at slightly longer distance (3.12 Å). The immediate coordination environment can therefore be described as capped trigonal prismatic, Fig. 2a. The trigonal prisms share triangular faces to create chains running parallel to the b axis, that are crossed linked by bridging hydroxides to create an extended structure, Fig. 2b. These motifs are held by OH–Cl hydrogen bonds (3.18 Å) in the ac plane, Fig. 2c.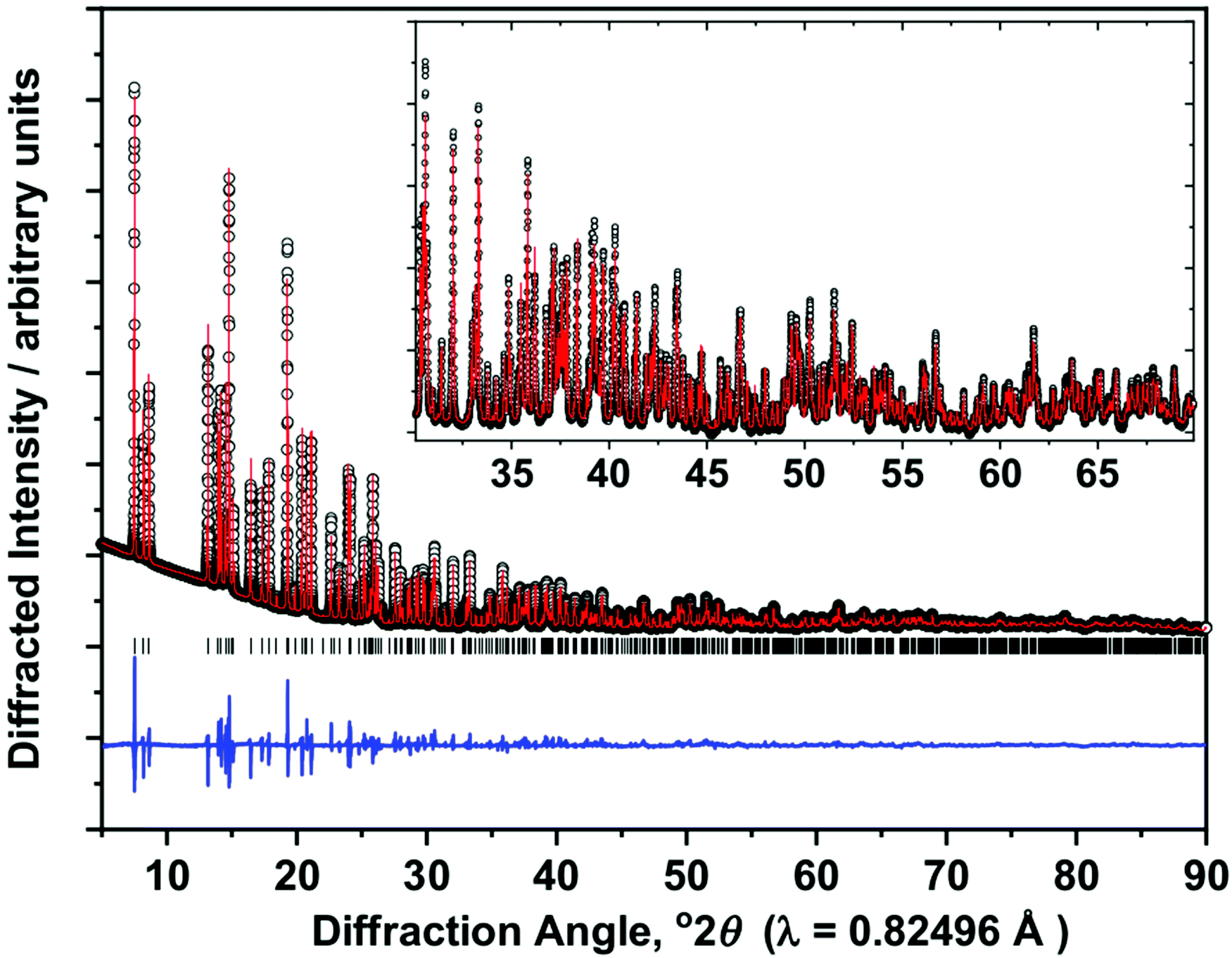 | ||
| Fig. 1 Final Rietveld fit (wRp = 4.66%, GOF = 6.76) of Ce(OH)2Cl. The measured data are the black circles, the red line the fitted profile, the blue line the difference curve and the black ticks the positions of allowed Bragg reflections. See Table 1 for crystal data. | ||
 | ||
| Fig. 2 Views of the crystal structure of Ce(OH)2Cl: (a) the capped trigonal prismatic coordination environment of Ce, (b) columns of face-shared trigonal bipyramids running along b and (c) the ac plane showing hydrogen bond interactions as dotted lines between neighbouring chains. Cerium atoms are grey, oxygen are red and chlorine green and hydrogen atoms are not shown. | ||
| a/Å = 6.2868(2), b/Å = 3.94950(3), c/Å = 6.8740(3), β (°) = 113.5120(5), space group P21/m | |||
|---|---|---|---|
| Site | Wyckoff position | x, y, z | U iso/Å2 |
| Ce | 2e | 0.31183(8), 0.25000, 0.13465(7) | 0.001 |
| Cl | 2e | 0.24658(25), 0.75000, 0.44103(23) | 0.004 |
| O1 | 2e | 0.0970(6), 0.75000, −0.0805(6) | 0.006 |
| O2 | 2e | 0.5718(6), 0.75000, 0.1647(6) | 0.009 |
As with Ce(OH)2Cl the powder XRD patterns of the Ce1−xLax(OH)2Cl materials can be indexed and the profile fitted to a monoclinic cell (space group: P21/m). A composition range 0.1 ≤ x ≤ 0.2 was found and increasing the lanthanum content to x = 0.3 or above led to phase segregation, with an unidentified byproduct. Indeed when LaCl3·7H2O is used solely in the solvothermal reaction the product formed is the same unidentified phase. In contrast to Ce1−xLax(OH)2Cl, there is a greater capability to incorporate praseodymium, gadolinium and terbium into the monoclinic structure, and indeed the pure lanathanide end members Pr(OH)2Cl, Gd(OH)2Cl and Tb(OH)2Cl also proved possible to synthesise by the same method (ESI†). Fig. 3 summarises the fitted lattice parameters, as determined by Pawley fits of measured powder diffraction patterns, of the materials Ce1−xLnx(OH)2Cl for Ln = La, Pr, Gd, Tb. We studied cerium-rich materials since our aim to later use the materials as precursors to substituted cerium oxides so materials with up to x = 0.5 were prepared, except for La, where up to the maximum value of x = 0.2 were produced. It can be seen that for the Pr, Gd and Tb materials there is an almost linear contraction of all lattice parameters, and hence unit cell volume, with increased replacement of cerium, consistent with the smaller ionic radii of the trivalent substituents34 and implying an isotropic response of the crystal structure to the elemental substitution. Only for the case of La does its larger radius lead to an expansion of the unit cell volume. The elemental composition of the materials was confirmed using EDXA on the SEM (ESI†), which gave good agreement with the expected values from the reagent ratios used in the synthesis.
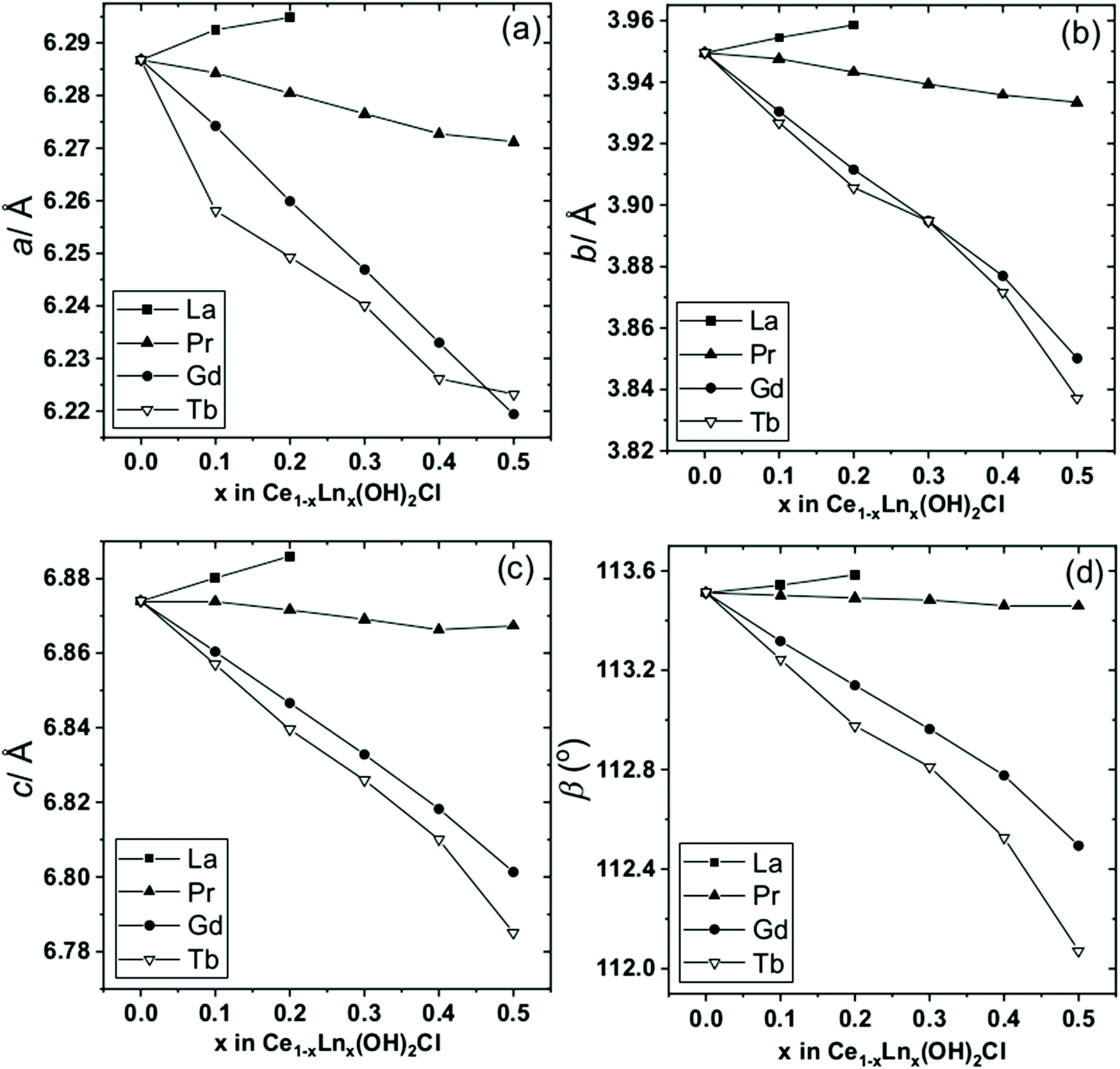 | ||
| Fig. 3 Plots of monoclinic lattice parameters of Ce1−xLnx(OH)2Cl for Ln = La, Pr, Gd, Tb: (a) a, (b) b, (c) c and (d) β. The error bars are smaller than the data points. | ||
If the lanthanide chloride precursors were replaced by lanthanide nitrates, the products of the solvothermal reactions were lanthanide carbonate hydroxides Ce1−xLnxCO3OH (ESI†). Cerium carbonate hydroxide has been studied previously as a precursor to cerium dioxides, so instead we focussed on studying the hydroxide chloride phase, along with its new substituted variants. We used thermogravimetric analysis to define the temperature of decomposition of the Ce1−xLax(OH)2Cl materials, Fig. 4. The parent Ce(OH)2Cl decomposes in air with a total mass loss of 21.54% to 800 °C. This is larger than the release of hydroxide and chloride and oxidation of Ce3+ to Ce4+ to give CeO2 as the final product (expected mass loss 17.9%), but the TGA trace shows a low temperature mass loss below 200 °C of at least 2.5% that is likely to be due to loss of residual solvent at the surface of the crystallites. The TGA traces for all the Ce1−xLax(OH)2Cl materials show a similar behaviour to give Ce1−xLnxO2−δ: Fig. 4 shows the materials with the highest Ln content, and others are plotted in the ESI.† The oxide product is confirmed by XRD analysis (see below), but note that the oxygen content of the oxide product will depend upon the amount of substituent and also its oxidation state. The substituted materials all show a higher temperature of decomposition by at least 50 °C, and also show a more complex decomposition, with several inflections observed in the mass loss. It has been previously reported that the end members Ln(OH)2Cl Ln = La, Pr, Nd, Sn, and Gd, all decompose to give LnOCl before forming Ln2O3 products.27c The exception is Ce(OH)2Cl and indeed the phase CeOCl is known to require rather different conditions for its formation and is unstable in excess oxygen,35 and also does not form on decomposition of CeCl3·7H2O, for example.36 For the Ce1−xLax(OH)2Cl materials it is possible that decomposition takes place via LnOCl intermediate phases. Note that the final mass loss is not easy to interpret, notwithstanding it does not plateau at the highest temperature studied, since the oxygen content of the product will depend upon the oxidation state of the lanthanide present, which may not be +3 for Pr and Tb (see below for further discussion of this), and also the oxygen content may be different at elevated temperature compared to after cooling to ambient conditions.
 | ||
| Fig. 4 Thermogravimetric analysis of Ce1−xLnx(OH)2Cl for Ln = La, Pr, Gd, Tb normalised to show the major mass loss above 250 °C. The inset shows the full trace for the parent Ce(OH)2Cl showing solvent loss at lower temperatures. | ||
Guided by the TGA analysis, a temperature of 700 °C was used to decompose the Ce1−xLnx(OH)2Cl materials to investigate the formation of mixed-metal oxide materials. The precursors were heated until a constant mass was reached, and a time of 5 hours was found to be suitable; note that this is considerably longer than the TGA experiment to ensure that all volatile products were removed. Fig. 5 shows scanning electron microscopy image of representative samples before and after decomposition (see ESI† for further images).
 | ||
| Fig. 5 SEM images of Ce1−xLnx(OH)2Cl as-made (left) and after heating at 700 °C (right) heating for (a) and (b) Ce(OH)2Cl, (c) and (d) Ln = La, x = 0.2, (e) and (f) Ln = Pr, x = 0.5, (g) and (h) Ln = Gd, x = 0.5, and (i) and (j) Ln = Tb, x = 0.5. | ||
All of the Ce1−xLnx(OH)2Cl materials form as agglomerates in which the primary particles are anisotropic forms with submicron dimensions and but overall spherical secondary particles of a few microns in size are found. There is little effect on crystal form on addition of the substituent lanthanide, nor on the amount of the substituent (see ESI†). Upon thermal treatment and collapse to the oxides, the particle morphology is maintained. Such a ‘shape memory effect’ has been observed upon decomposition of other cerium salt precursors into CeO2, such as cerium formate,22 cerium oxalate,23 and cerium basic carbonate.24
Powder XRD of the oxides, produced on decomposition of the Ce1−xLnx(OH)2Cl precursors, shows a fluorite-like CeO2 structure is found for all the materials, with no evidence of phase separation nor the presence of any crystalline chloride-containing byproducts. Fig. 6 shows the patterns of the materials with the highest lanthanide concentration, with other patterns presented in ESI.† Full pattern fitting provides the cubic lattice parameter, while analysis of the angle-dependent peak broadening using the Williamson–Hall method (ESI†) yields an estimate of crystallite domain size and also a strain-broadening parameter, Table 2.
 | ||
| Fig. 6 Profile fits of Ce1−xLnxO2−δ oxides formed by thermal decomposition of Ce1−xLnx(OH)2Cl materials: (a) CeO2, (b) Ce0.8La0.2O2−δ, (c) Ce0.8Pr0.2O2−δ, (d) Ce0.8Gd0.2O2−δ, (e) Ce0.8Tb0.2O2−δ. | ||
| Sample | a (Å) | Cε | L (Å) |
|---|---|---|---|
| CeO2 | 5.4104(2) | 0.0110(3) | 223(1) |
| Ce0.9La0.1O2−δ | 5.4371(3) | 0.01418(6) | 139(1) |
| Ce0.8La0.2O2−δ | 5.4664(4) | 0.015(1) | 113(6) |
| Ce0.9Pr0.1O2−δ | 5.4163(2) | 0.0141(3) | 176(5) |
| Ce0.8Pr0.2O2−δ | 5.4178(2) | 0.0178(3) | 167(4) |
| Ce0.7Pr0.3O2−δ | 5.4171(2) | 0.0201(1) | 176(2) |
| Ce0.6Pr0.4O2−δ | 5.4130(4) | 0.0225(6) | 245(2) |
| Ce0.5Pr0.5O2−δ | 5.4099(6) | 0.0199(1) | 223(1) |
| Ce0.9Gd0.1O2−δ | 5.4181(2) | 0.0113(3) | 174(3) |
| Ce0.8Gd0.2O2−δ | 5.4246(3) | 0.0113(3) | 232(6) |
| Ce0.7Gd0.3O2−δ | 5.4296(2) | 0.0167(2) | 373(13) |
| Ce0.6Gd0.4O2−δ | 5.4360(3) | 0.0218(2) | 679(28) |
| Ce0.5Gd0.5O2−δ | 5.4378(6) | 0.0281(1) | 1240(98) |
| Ce0.9Tb0.1O2−δ | 5.4003(2) | 0.0047(5) | 135(4) |
| Ce0.8Tb0.2O2−δ | 5.3922(4) | 0.0065(5) | 148(5) |
| Ce0.7Tb0.3O2−δ | 5.3832(2) | 0.01826(6) | 296(2) |
| Ce0.6Tb0.4O2−δ | 5.3735(4) | 0.0224(2) | 385(17) |
| Ce0.5Tb0.5O2−δ | 5.3616(2) | 0.01888(4) | 508(5) |
Fig. 7 shows the evolution of lattice parameters with level of lanthanide substitution. This shows an expansion of the unit cell for Ln = La, which is consistent with the larger eight-coordinate ionic radius of La3+ (1.16 Å) compared to Ce4+ (0.97 Å) and a similar effect, although lesser, is seen also for Ln = Gd due to the only slightly larger radius of Gd3+ (1.053 Å) compared to Ce4+.34 The upper limit of La inclusion, x = 0.2, was dictated by the amount included in the Ce1−xLax(OH)2Cl precursor phase, as noted above. Previous work has also found a limit to the amount of La3+ that can be included in CeO2 which is highly dependent on the synthesis methods, and is typically up to 40–70%,37 although in cubic nanocrystals prepared by a solvothermal route, much lower concentrations of La were achieved of 7.5 mol%.38 The case of Pr is more complex, with a small expansion of the unit at the lower levels of substitution and a small contraction at higher levels of substitution, but overall the lattice parameter does not change significantly. Although Pr3+ (1.126 Å) is larger than Ce4+ the presence of Pr4+, which is slightly smaller (0.96 Å) is a distinct possibility, thus it is likely that the Ce1−xPrxO2−δ contain mixtures of Pr3+ and Pr4+ (see below for further analysis of this). In the case of Tb-substitution, a linear contraction of the unit cell is seen with increasing Tb content despite the larger size of Tb3+ (1.04 Å), which would imply the presence of some of the smaller Tb4+ (0.88 Å).
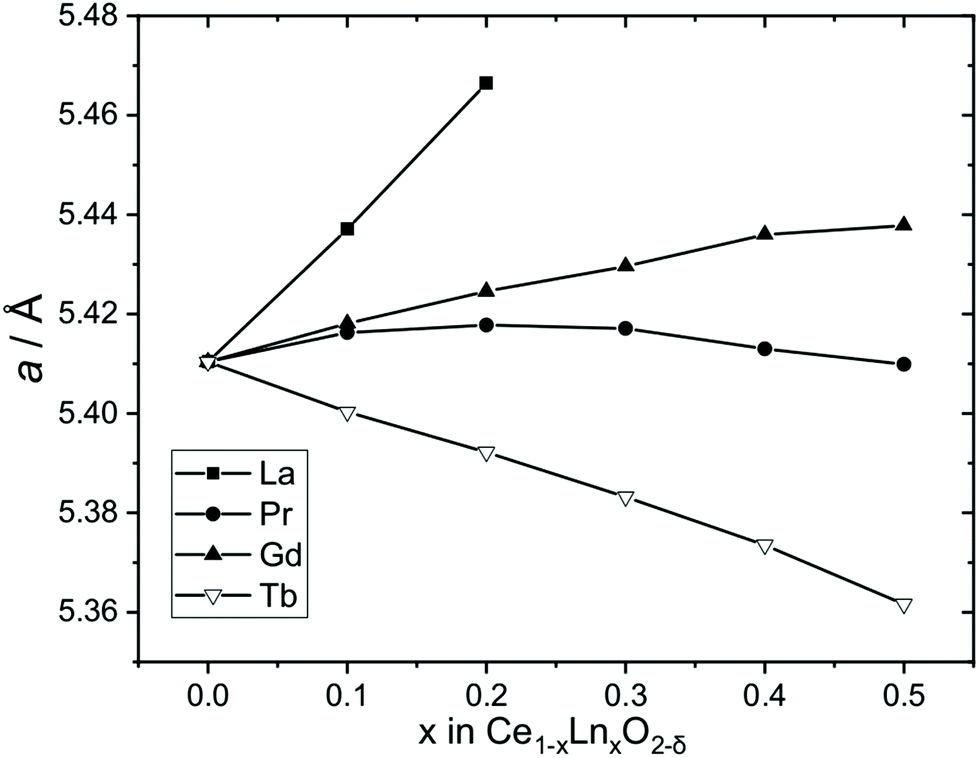 | ||
| Fig. 7 Cubic lattice parameters of Ce1−xLnxO2−δ oxides formed by thermal decomposition of Ce1−xLnx(OH)2Cl materials. The error bars are smaller than the data points (see Table 2). | ||
The Williamson–Hall analysis, Table 2, reveals that most samples have a greater strain parameter than the pure CeO2 prepared by the same method. The increased strain is consistent with the size mismatch between the substituent lanthanide and cerium cations. The only exception is the Ce1−xTbxO2−δ materials with low levels of substitution and for these there is a closer match in ionic radii (see above). The effect of substituent lanthanide on lattice strain has been observed previously using the Williamson–Hall method: for example, Kumar et al., studied Eu-CeO2 produced by coprecipitation and found increased strain with Eu content,39 while Jamshidijam et al. found that different lanthanide cations gave different lattice strains.40 For our materials, the Williamson–Hall analysis yields average crystallite sizes for all the materials in the range 10–130 nm, which is consistent with the primary particles seen by SEM, as mentioned above.
To investigate the oxidation state of Pr and Tb in the oxide materials, we used XANES spectroscopy at the lanthanide LIII edges, Fig. 8. The LIII-edges of these lanthanides show characteristic spectra in the +3 and +4 oxidation states. For the +3 oxidation a strong white line is seen that corresponds to the electron transition 2p3/2 → (4f1)5d, while for the +4 oxidation state the main absorption is shifted to higher energy and a characteristic double feature is observed that corresponds to the 2p3/2 → (4fL)5d and 2p3/2 → (4f0)5d transitions.41 These features provide a fingerprint of the oxidation state.42 For the Ce1−xPrxO2−δ materials the spectra are all shifted to higher energies than Pr(OH)2Cl, which contains solely Pr3+, and resemble that of the mixed-valent oxide Pr6O11 (Pr3+2Pr4+4O11), with the x = 0.5 material likely to have a higher proportion of Pr4+. A similar conclusion can be made for the Ce1−xTbxO2−δ materials, with a shift in the Tb LIII absorption edge to higher energy from Tb(OH)2Cl and the characteristic double feature seen as in Tb4O7 (Tb3+2Tb4+2O7). It can be concluded that the Tb–Ce oxides contain a higher proportion of the +3 oxidation state than the +4, but as with the Pr materials, the proportion changes with composition of the oxide.
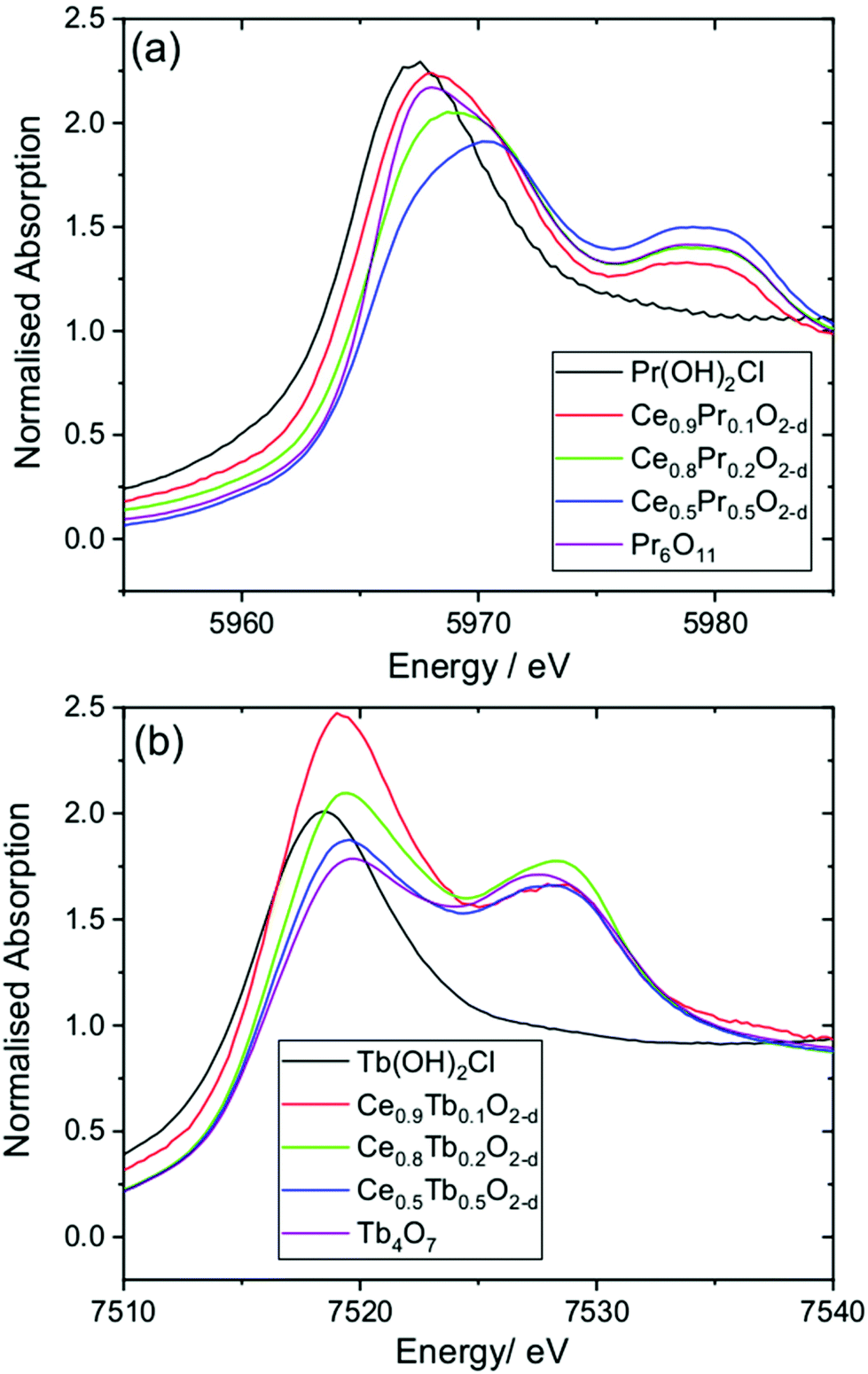 | ||
| Fig. 8 XANES spectra measured at (a) the Pr LIII-edge and (b) the Tb LIII-edge from Ce1−xLnxO2−δ oxides and reference materials. | ||
The results from XANES are consistent with the powder XRD analysis discussed above. The presence of a majority of Pr4+, but some Pr3+, explains the small contraction of the unit cell with increasing Pr content. Similar results have been seen previously for Pr-substituted ceria prepared by calcination methods.43 For Tb the +3 oxidation predominates, but there is a proportion of Tb4+.43d
Temperature programmed reduction of the oxide materials was performed to assess their redox properties, Fig. 9. This is a standard measurement made to examine the ease of release of oxygen from the solids and provides an indication of their potential use in heterogeneous catalysis. To make a comparison, we studied Ce1−xLnxO2−δ materials with x = 0.2. Table 3 summarise the analysis of the results, with the percentage of lanthanide reduced and the temperature of the maximum TPR response. Two TPR runs are shown: these were acquired from the same sample with a mild oxidation between the first and second.
 | ||
| Fig. 9 Temperature programmed reduction of CeO2 and Ce1−xLnxO2−δ measured under 10% H2 in N2 for (a) first cycle TPR1 and (b) second cycle TPR2 for (i) CeO2, (ii) Ce1−xLaxO2−δ, (iii) Ce1−xPrxO2−δ, (vi) Ce1−xGdxO2−δ, (v) Ce1−xTbxO2−δ. | ||
| Sample | S BET/m2 g−1 | First TPR | Second TPR | ||
|---|---|---|---|---|---|
| Total Ln reduced (%) | T max (°C) | Total Ln reduced (%) | T max (°C) | ||
| CeO2 | 93.3 | 48.1 | 836 | 38.4 | 837 |
| Ce0.8La0.2O2−δ | 91.7 | 20.2 | 569 | 36.4 | 630 |
| Ce0.8Pr0.2O2−δ | 69.6 | 56.0 | 463 | 79.2 | 542 |
| Ce0.8Gd0.2O2−δ | 63.7 | 31.4 | 555 | 33.8 | 742 |
| Ce0.8Tb0.2O2−δ | 45.4 | 45.6 | 773 | 44.6 | 295 |
The first TPR response from the CeO2 sample is characteristic of small particle size ceria, with a low temperature feature at around 500 °C, corresponding to surface reduction followed by a high temperature feature at ∼820 °C that represents bulk reduction.44 The second TPR of the ceria shows loss of the surface reduction so that only the bulk reduction remains: this would be consistent with an annealing of the material. The La-substituted oxide shows a lower temperature of reduction than the pure ceria, and no distinction between a surface and bulk reduction, at least on the first cycle: this is consistent with a greater lattice strain, which conceivably lowers the activation for reduction, but overall there is less lanthanide that can be reduced owing to the fixed valence state of La. The TPR response is similar to other reported La-CeO2 materials.45 The Gd-substituted oxide shows a very similar response to the La analogue, both in terms of the extent of reduction and the form of the TPR traces. The Pr and Tb materials show the best performance, and this is to be anticipated with the presence of the substituents in the +4 oxidation state, which themselves add to the total reducibility, along with an ionic size mismatch that leads to lattice strain and a lower activation for reduction, evident by a lower temperature of peak reduction. This is particularly evident for the Pr-containing sample, which would be consistent with a greater proportion of the +4 oxidation state of Pr, as seen by XANES (see above). The TPR traces we have reported are very similar to similar compositions prepared by other methods, particularly the low temperature reduction characteristic of Pr-CeO2.46 It is already established that Pr-substitution in ceria provides the lowest temperature reduction, amongst the lanthanides, followed by Tb-substitution.47 It should be noted that for all materials there is no evidence for phase separation after the two cycles of TPR: powder XRD shows that the average fluorite structure is maintained in all cases (ESI†). It is also noteworthy that the surface areas of the samples show some variation, Table 2, and the poorer reducibility of the Tb-substituted oxide may in part be due to its lower surface area. The contrast between the Pr- and Gd-substituted materials is particularly evident when surface area is considered, since the materials have similar values but markedly different redox behaviour.
4. Conclusions
The solvothermal synthesis of Ce(OH)2Cl provides a convenient precursor route to CeO2 with thermal decomposition providing the shape-memory formation of agglomerates of submicron crystallites. Substitution of cerium by various lanthanides is possible in Ce(OH)2Cl and four examples are provided that lead to substituted ceria materials with redox properties that match those of similar compositions prepared by other methods. The results will add to the repertoire of methods for the preparation of materials for heterogeneous catalysis applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AJAD and JWA thank Johnson Matthey and the Royal Society, respectively, for funding their PhD positions. RIW also thanks the Royal Society for an Industry Fellowship in collaboration with Johnson Matthey. We thank Diamond Light Source for provision of beamtime for XANES as part of the Energy Materials Block Allocation Group SP14239, and we are grateful to Professor Alan Chadwick and Dr Giannantonio Cibin for help with data aquisition. The I11 beamtime was obtained through the Diamond Light Source Block Allocation Group award “Oxford/Warwick Solid State Chemistry BAG to probe composition–structure–property relationships in solids” (EE18786) and we thank Mr Gabriel Clarke for assistance with measuring the data. Some of the equipment used in this work was provided by the University of Warwick's Research Technology Platforms.References
- (a) A. Trovarelli, Catal. Rev. Sci. Eng., 1996, 38, 439–520 CrossRef CAS; (b) T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS.
- A. F. Diwell, R. R. Rajaram, H. A. Shaw and T. J. Truex, in Stud. Surf. Sci. Catal, ed. A. Crucq, Elsevier, 1991, vol. 71, pp. 139–152 Search PubMed.
- N. Jaiswal, K. Tanwar, R. Suman, D. Kumar, S. Upadhyay and O. Parkash, J. Alloys Compd., 2019, 781, 984–1005 CrossRef CAS.
- I. I. Soykal, H. Sohn, D. Singh, J. T. Miller and U. S. Ozkan, ACS Catal., 2014, 4, 585–592 CrossRef CAS.
- S. Liu, X. Wu, D. Weng and R. Ran, J. Rare Earths, 2015, 33, 567–590 CrossRef CAS.
- R. J. Gorte and S. Zhao, Catal. Today, 2005, 104, 18–24 CrossRef CAS.
- Q. Wang, K. L. Yeung and M. A. Bañares, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.05.016.
- A. Le Gal, S. Abanades, N. Bion, T. Le Mercier and V. Harlé, Energy Fuels, 2013, 27, 6068–6078 CrossRef CAS.
- (a) A. B. Shcherbakov, V. K. Ivanov, N. M. Zholobak, O. S. Ivanova, E. Y. Krysanov, A. E. Baranchikov, N. Y. Spivak and Y. D. Tretyakov, Biophysics, 2011, 56, 987–1004 CrossRef; (b) E. Casals, M. Zeng, M. Parra-Robert, G. Fernández-Varo, M. Morales-Ruiz, W. Jiménez, V. Puntes and G. Casals, Small, 2020, 16, 1907322 CrossRef CAS.
- (a) S. Yabe, M. Yamashita, S. Momose, K. Tahira, S. Yoshida, R. Li, S. Yin and T. Sato, Int. J. Inorg. Mater., 2001, 3, 1003–1008 CrossRef CAS; (b) S. Yabe and T. Sato, J. Solid State Chem., 2003, 171, 7–11 CrossRef CAS.
- X. Feng, D. C. Sayle, Z. L. Wang, M. S. Paras, B. Santora, A. C. Sutorik, T. X. T. Sayle, Y. Yang, Y. Ding, X. Wang and Y. S. Her, Science, 2006, 312, 1504–1508 CrossRef CAS.
- X. Q. Fu, C. Wang, H. C. Yu, Y. G. Wang and T. H. Wang, Nanotechnology, 2007, 18, 145503 CrossRef.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- Y. X. Gao, W. D. Wang, S. J. Chang and W. X. Huang, ChemCatChem, 2013, 5, 3610–3620 CrossRef CAS.
- (a) S. S. Lee, H. G. Zhu, E. Q. Contreras, A. Prakash, H. L. Puppala and V. L. Colvin, Chem. Mater., 2012, 24, 424–432 CrossRef CAS; (b) C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 RSC.
- (a) Z. A. Qiao, Z. L. Wu and S. Dai, ChemSusChem, 2013, 6, 1821–1833 CrossRef CAS; (b) M. Lykaki, E. Pachatouridou, E. Iliopoulou, S. A. C. Carabineiro and M. Konsolakis, RSC Adv., 2017, 7, 6160–6169 RSC; (c) A. Trovarelli and J. Llorca, ACS Catal., 2017, 7, 4716–4735 CrossRef CAS; (d) A. Dhall and W. Self, Antioxidants, 2018, 7, 97 CrossRef.
- (a) H. Y. Chang and H. I. Chen, J. Cryst. Growth, 2005, 283, 457–468 CrossRef CAS; (b) M. Kurian and C. Kunjachan, Int. Nano Lett., 2014, 4, 73–80 CrossRef CAS.
- (a) C. S. Riccardi, R. C. Lima, M. L. dos Santos, P. R. Bueno, J. A. Varela and E. Longo, Solid State Ionics, 2009, 180, 288–291 CrossRef CAS; (b) S. Hosokawa, K. Shimamura and M. Inoue, Mater. Res. Bull., 2011, 46, 1928–1932 CrossRef CAS; (c) R. I. Walton, Prog. Cryst. Growth Charact. Mater., 2011, 57, 93–108 CrossRef CAS; (d) J. Zdravkovic, B. Simovic, A. Golubovic, D. Poleti, I. Veljkovic, M. Scepanovic and G. Brankovic, Ceram. Int., 2015, 41, 1970–1979 CrossRef CAS.
- C. Matei, D. Berger, S. Stoleriu, D. Neagu and E. Stefan, J. Optoelectron. Adv. Mater., 2010, 12, 1524–1529 CAS.
- S. Masson, P. Holliman, M. Kalaji and P. Kluson, J. Mater. Chem., 2009, 19, 3517–3522 RSC.
- (a) G. A. M. Hussein, J. Anal. Appl. Pyrolysis, 1996, 37, 111–149 CrossRef CAS; (b) A. A. Titov, M. A. Klimenko, E. G. Goryacheva, N. L. Opolchenova, N. N. Stepareva and N. P. Sokolova, Inorg. Mater., 2008, 44, 1101–1104 CrossRef CAS.
- (a) J. J. Wei, Z. J. Yang and Y. Z. Yang, CrystEngComm, 2011, 13, 2418–2424 RSC; (b) E. R. Lopez-Mena, C. R. Michel, A. H. Martinez-Preciado and A. Elias-Zuniga, Nanoscale Res. Lett., 2017, 12, 169 CrossRef; (c) B. B. Bokhonov, A. A. Matvienko, K. B. Gerasimov and D. V. Dudina, Ceram. Int., 2019, 45, 19684–19688 CrossRef CAS.
- (a) G. J. Zhang, Z. R. Shen, M. Liu, C. H. Guo, P. C. Sun, Z. Y. Yuan, B. H. Li, D. T. Ding and T. H. Chen, J. Phys. Chem. B, 2006, 110, 25782–25790 CrossRef CAS; (b) H. Miyazaki, J. I. Kato, N. Sakamoto, N. Wakiya, T. Ota and H. Suzuki, Adv. Appl. Ceram., 2010, 109, 123–127 CrossRef CAS; (c) Y. N. Li, X. H. Wu, W. W. Wu, K. T. Wang, L. Q. Qin, S. Liao and Y. X. Wen, J. Therm. Anal. Calorim., 2014, 117, 499–506 CrossRef CAS; (d) D. V. Maslennikov, A. A. Matvienko, A. A. Sidelnikov and S. A. Chizhik, Mater. Today: Proc., 2017, 4, 11495–11499 CAS; (e) V. Tyrpekl, P. Markova, M. Dopita, P. Brazda and M. A. Vacca, Inorg. Chem., 2019, 58, 10111–10118 CrossRef CAS.
- (a) Z. T. Guo, F. L. Du, G. C. Li and Z. L. Cui, Inorg. Chem., 2006, 45, 4167–4169 CrossRef CAS; (b) K. Li and P. S. Zhao, Mater. Res. Bull., 2010, 45, 243–246 CrossRef CAS; (c) M. Y. Cui, J. X. He, N. P. Lu, Y. Y. Zheng, W. J. Dong, W. H. Tang, B. Y. Chen and C. R. Li, Mater. Chem. Phys., 2010, 121, 314–319 CrossRef CAS; (d) C. R. Li, Q. T. Sun, N. P. Lu, B. Y. Chen and W. J. Dong, J. Cryst. Growth, 2012, 343, 95–100 CrossRef CAS; (e) F. M. Meng, J. F. Gong, Z. H. Fan, H. J. Li and J. T. Yuan, Ceram. Int., 2016, 42, 4700–4708 CrossRef CAS; (f) J. Ederer, M. Stastny, M. Dosek, J. Henych and P. Janos, RSC Adv., 2019, 9, 32058–32065 RSC.
- (a) M. Jobbagy, C. Sorbello and E. E. Sileo, J. Phys. Chem. C, 2009, 113, 10853–10857 CrossRef CAS; (b) C. Sorbello and M. Jobbágy, Cryst. Growth Des., 2017, 17, 5660–5666 CrossRef CAS.
- S. H. Jiang, R. Y. Zhang, H. X. Liu, Y. Rao, Y. N. Yu, S. Chen, Q. Yue, Y. N. Zhang and Y. J. Kang, J. Am. Chem. Soc., 2020, 142, 6461–6466 CrossRef CAS.
- (a) F. L. Carter and S. Levinson, Inorg. Chem., 1969, 8, 2788–2791 CrossRef CAS; (b) R. F. Klevtsova and L. A. Glinskaya, J. Struct. Chem., 1969, 10, 408–410 CrossRef; (c) P. V. Klevtsov, V. M. Bembel and Z. A. Grankina, J. Struct. Chem., 1969, 10, 543–547 CrossRef.
- (a) T. N. Tarkhova, N. N. Mironov and I. A. Grishin, J. Struct. Chem., 1970, 11, 515–516 CrossRef; (b) T. N. Tarkhova, I. A. Grishin and N. N. Mironov, Zh. Neorg. Khim., 1970, 15, 1340–1341 Search PubMed; (c) V. I. Bukin, Dokl. Akad. Nauk SSSR, 1972, 207, 1332–1335 CAS; (d) R. A. Zehnder, D. L. Clark, B. L. Scott, R. J. Donohoe, P. D. Palmer, W. H. Runde and D. E. Hobart, Inorg. Chem., 2010, 49, 4781–4790 CrossRef CAS.
- P. V. Klevtsov, T. G. Lysenina and L. Y. Kharckenko, J. Struct. Chem., 1973, 14, 76–79 CrossRef.
- H. Kim, M. Kim and S.-H. Byeon, J. Mater. Chem. C, 2017, 5, 444–451 RSC.
- A. C. Larson and R. B. V. Dreele, Los Alamos National Laboratory Report LAUR 86-748, 2000.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Y.-P. Lan, H. Y. Sohn, A. Murali, J. Li and C. Chen, Appl. Phys. A, 2018, 124, 702 CrossRef.
- W. W. Wendlandt, J. Inorg. Nucl. Chem., 1957, 5, 118–122 CrossRef CAS.
- S. A. Liang, E. Broitman, Y. N. Wang, A. M. Cao and G. Veser, J. Mater. Sci., 2011, 46, 2928–2937 CrossRef CAS.
- D. Loche, L. M. Morgan, A. Casu, G. Mountjoy, C. O'Regan, A. Corrias and A. Falqui, RSC Adv., 2019, 9, 6745–6751 RSC.
- A. Kumar, S. Babu, A. S. Karakoti, A. Schulte and S. Seal, Langmuir, 2009, 25, 10998–11007 CrossRef CAS.
- M. Jamshidijam, R. V. Mangalaraja, A. Akbari-Fakhrabadi, S. Ananthakumar and S. H. Chan, Powder Technol., 2014, 253, 304–310 CrossRef CAS.
- (a) A. Bianconi, A. Marcelli, H. Dexpert, R. Karnatak, A. Kotani, T. Jo and J. Petiau, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 806–812 CrossRef CAS; (b) A. V. Soldatov, T. S. Ivanchenko, S. Della Longa, A. Kotani, Y. Iwamoto and A. Bianconi, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 5074–5080 CrossRef CAS.
- Y. Takahashi, H. Sakami and M. Nomura, Anal. Chim. Acta, 2002, 468, 345–354 CrossRef CAS.
- (a) Y. R. Wang, T. Mori, J. G. Li and T. Ikegami, J. Am. Ceram. Soc., 2002, 85, 3105–3107 CrossRef CAS; (b) L. Yan, X. Xing, R. Yu, L. Qiao, J. Chen, J. Deng and G. Liu, Scr. Mater., 2007, 56, 301–304 CrossRef CAS; (c) A. C. Cabral, L. S. Cavalcante, R. C. Deus, E. Longo, A. Z. Simoes and F. Moura, Ceram. Int., 2014, 40, 4445–4453 CrossRef CAS; (d) M. Coduri, M. Scavini, M. Brunelli, E. Pedrazzin and P. Masala, Solid State Ionics, 2014, 268, 150–155 CrossRef CAS.
- M. Boaro, M. Vicario, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2003, 77, 407–417 CrossRef CAS.
- G. M. Mullen, E. J. Evans, B. C. Siegert, N. R. Miller, B. K. Rosselet, I. Sabzevari, A. Brush, Z. Duan and C. B. Mullins, React. Chem. Eng., 2018, 3, 75–85 RSC.
- (a) M.-F. Luo, Z.-L. Yan and L.-Y. Jin, J. Mol. Catal. A: Chem., 2006, 260, 157–162 CrossRef CAS; (b) M. A. Małecka, L. Kępiński and W. Miśta, Appl. Catal., B, 2007, 74, 290–298 CrossRef; (c) K. Krishna, A. Bueno-López, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2007, 75, 189–200 CrossRef CAS; (d) K. Ahn, D. S. Yoo, D. H. Prasad, H. W. Lee, Y. C. Chung and J. H. Lee, Chem. Mater., 2012, 24, 4261–4267 CrossRef CAS.
- Y. Sohn, Appl. Sci. Convergence Technol., 2019, 28, 35–40 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt03435e |
| This journal is © The Royal Society of Chemistry 2020 |