
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphinoborane interception at magnesium by borane-assisted phosphine-borane dehydrogenation†
Louis J.
Morris
a,
Nasir A.
Rajabi
a,
Michael S.
Hill
 *a,
Ian
Manners
*b,
Claire L.
McMullin
*a and
Mary F.
Mahon
a
*a,
Ian
Manners
*b,
Claire L.
McMullin
*a and
Mary F.
Mahon
a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bath.ac.uk
bDepartment of Chemistry, University of Victoria, Victoria, BC V8P 5C2, Canada. E-mail: imanners@uvic.ca
First published on 7th October 2020
Abstract
Reactions of B(C6F5)3 with the β-diketiminato (BDI) alkaline-earth phosphidoborane complexes, 1a [(BDI)Ca(H3B·PPh2)] and 1b [(BDI)Mg(H3B·PPh2)]2 (BDI = [HC{C(CH3)N(2,6-iPr-C6H3)}2]−) result in the formation of phosphinodiboronate complexes 4a [(BDI)Ca(η6-toluene){H3B·PPh2·B(C6F5)3}] and 4b [(BDI)Mg{H3B·PPh2·B(C6F5)3}]. Calcium complex 4a is stable in aromatic solvents at room temperature and does not display well-defined onward reactivity at elevated temperatures. Magnesium complex 4b undergoes a room temperature transformation to provide the known hydridoborate derivative 3b [(BDI)Mg{HB(C6F5)3}] and an N,P,N’-ligated species, 5 [{HC(C(CH3)N(2,6-iPr-C6H3))2(H2BPPh2)}Mg{H3B·PPh2·B(C6F5)3}] that results from interception of the putative phosphinoborane, H2B = PPh2, by the BDI ligand backbone following B(C6F5)3-mediated hydride abstraction. NMR spectroscopic investigations were supported by DFT calculations, which suggested a mechanism involving B(C6F5)3 migration and hydride abstraction within the coordination sphere of magnesium. Interception of H2B = PPh2 by B(C6F5)3 is proposed to stabilise this species, whilst activating it towards ligand-centred nucleophilic attack. The significant stabilisation energy calculated for the Ca-π(toluene) interaction in 4a accounts for the contrasting outcomes between the two Ae-elements. The crystal structures of compounds 4a and 5 are presented and discussed.
Introduction
Polyphosphinoboranes are an emerging class of solution-processable inorganic materials with contrasting properties to their formally isoelectronic all-carbon analogues. Whereas most polyolefins are highly flammable, polyphosphinoboranes, for example, display flame retardant properties1,2 and have seen use as lithographic resists.2–4 Although their synthesis, via the catalytic dehydrogenation-polymerisation of phosphine-borane complexes, was first achieved some 20 years ago, catalysts remain almost exclusively based on iron, ruthenium, rhodium or iridium.2–11 A metal-free stoichiometric approach was described by Manners, Scheer, and co-workers, who demonstrated the metal-free head-to-tail polymerisation of tert-butylphosphinoborane, which was generated in situ from the trimethylamine-stabilised monomer (Scheme 1a).12 More recently, Manners and co-workers have reported that treatment with stoichiometric quantities of cyclic alkyl amino carbenes (CAACs) induces the dehydrogenative coupling of phosphine-boranes to provide access to primary- and, previously unprecedented, secondary polyphosphinoboranes (Scheme 1b).13 Of particular relevance to the current work is the B(C6F5)3 (BCF)-catalysed dehydropolymerisation of phosphine-borane and phenylphosphine-borane reported by Denis et al. in 2003 (Scheme 1c).14 This process was proposed to proceed via iterative borane transfer and dehydrocoupling steps involving a Brønsted-acidic phosphine-borane complex, (C6F5)3B·PPhH2, as the active species. The obtained polymers were, however, of modest molecular weight and accompanied by a significant fraction of low molecular-weight oligomeric species.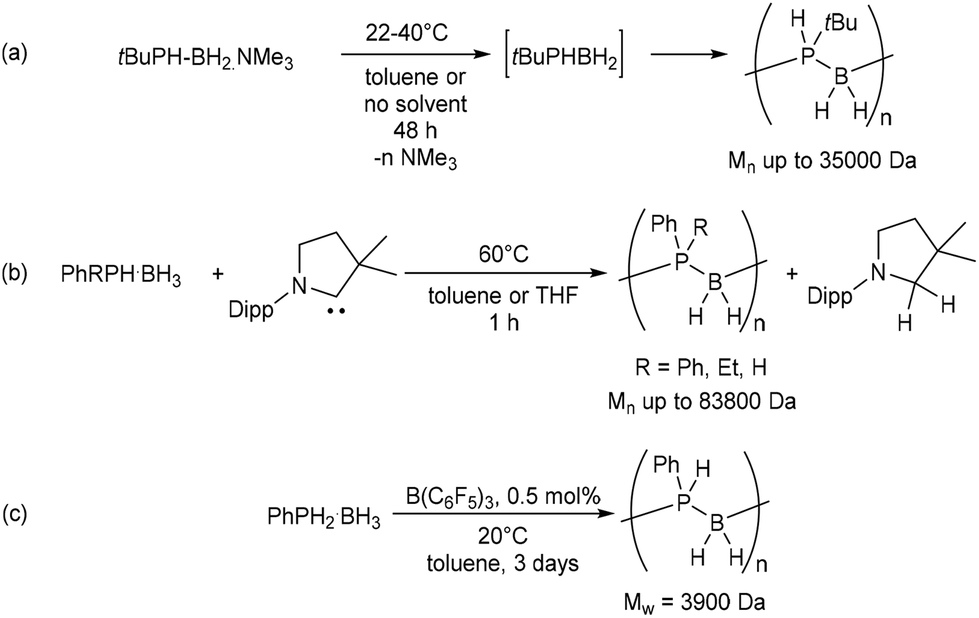 | ||
| Scheme 1 (a) Metal-free head-to-tail polymerisation of tert-butylphosphinoborane.12 (b) Cyclic alkyl amino carbene (CAAC)-mediated polymerisation of phosphine-boranes.13 (c) BCF-catalysed dehydropolymerisation of phenylphosphine-borane.14Mn and Mw values determined by GPC relative to polystyrene standards and, in the case of bimodal distributions, refer to the polymeric fraction only. | ||
Our own interest in this topic is motivated by the use of alkaline-earth (Ae) elements as non-toxic and inexpensive catalysts for the synthesis of inorganic polymers.15,16 Following previous investigations into Ae-mediated catalytic and stoichiometric amine-borane dehydrogenation and dehydrocoupling,17–26 we recently reported a series of β-diketiminate (BDI) supported alkaline-earth (Ae) phosphidoborane complexes, which are prepared by exposing diphenylphosphine-borane to readily accessible BDI-Ae hydride, alkyl, or amide precursors (Scheme 2a).27 Attempts to achieve complete phosphine-borane dehydrogenation were unsuccessful, however, and the use of super-stoichiometric quantities of phosphine-borane resulted in BH3 transfer to provide the phosphinodiboronate complex 2 and uncomplexed diphenylphosphine (Scheme 2a). The preference for these systems to undergo BH3 transfer rather than hydride-elimination was rationalised to be a consequence of the charge-dense Lewis acidic Ae-centre in conjunction with the soft phosphorus-centred Lewis base. In order to address this shortcoming, inspiration was drawn from the previously observed BCF-mediated hydride abstraction of [(BDI)Ae] amidoborane complexes (Scheme 2b).28,29 This transformation yielded hydridoborate derivatives 3a–c with concomitant formation of the cyclic diborazane, (Me2NBH2)2. It was anticipated that analogous BCF addition to phosphidoborane complexes 1a and 1b would provide a similar outcome, thus yielding oligomerisation products of the unsaturated diphenylphosphinoborane monomer.
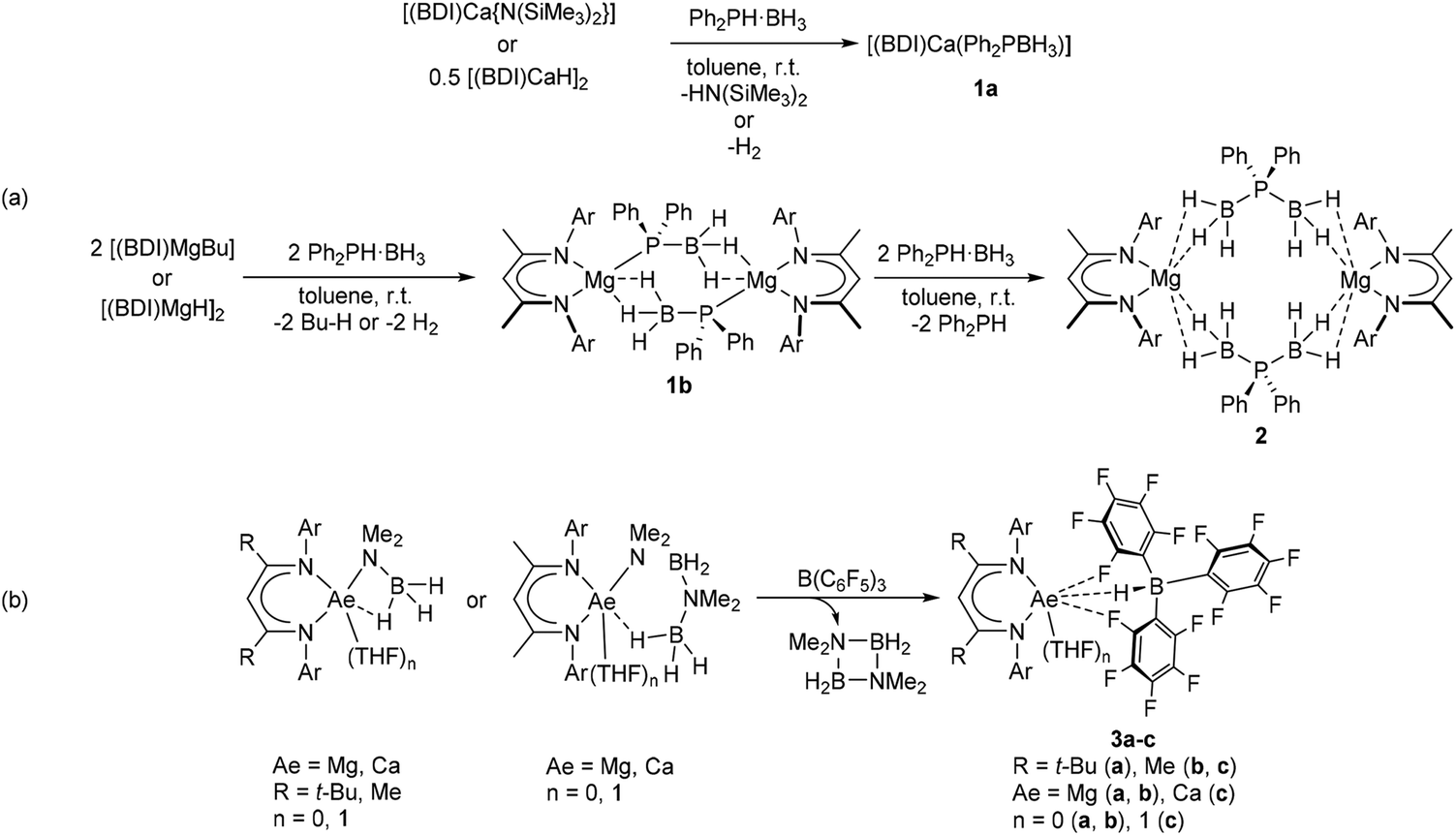 | ||
| Scheme 2 (a) Synthesis and reactivity of Ae-phosphidoborane complexes, 1a and 1b.27 (b) BCF-promoted hydride abstraction of Ae-amidoborane complexes.28,29 Ar = 2,6-di-isopropylphenyl. | ||
Results and discussion
Addition of an equimolar quantity of BCF to a d8-toluene solution of the calcium complex 1a resulted in quantitative conversion to a new BDI-containing species, 4a (Scheme 3), whose γ-CH proton resonated at δ 4.85 ppm. Counter to expectation, the resultant 11B NMR spectrum comprised two broadened signals at δ −11.0 and −31.2 ppm and no resonance corresponding to the anticipated [HB(C6F5)3]− anion was observed. Similarly, the 31P{1H} NMR spectrum showed no resonances corresponding to oligophosphinoboranes;7 instead, a single broad resonance centred at δ −1.43 ppm was detected. Slow evaporation of a saturated toluene solution of the crude product at room temperature yielded single crystals, X-ray diffraction analysis of which provided the solid-state structure of the phosphinodiboronate complex, 4a (Fig. 1), in which the boron-bound hydrogens were located and refined without restraints. Solutions of compound 4a in d8-toluene or C6D6 were stable at room temperature and attempts to induce further reactivity by heating to 60 °C resulted in partial decomposition to the homoleptic complex [(BDI)2Ca]30 and other unidentified species.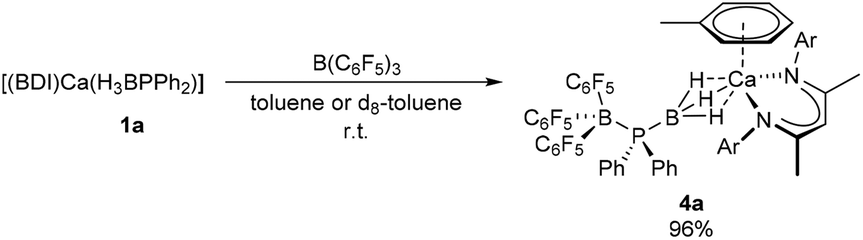 | ||
| Scheme 3 Synthesis of compound 4a. Isolated yield shown, quantitative spectroscopic yield determined by NMR spectroscopy. | ||
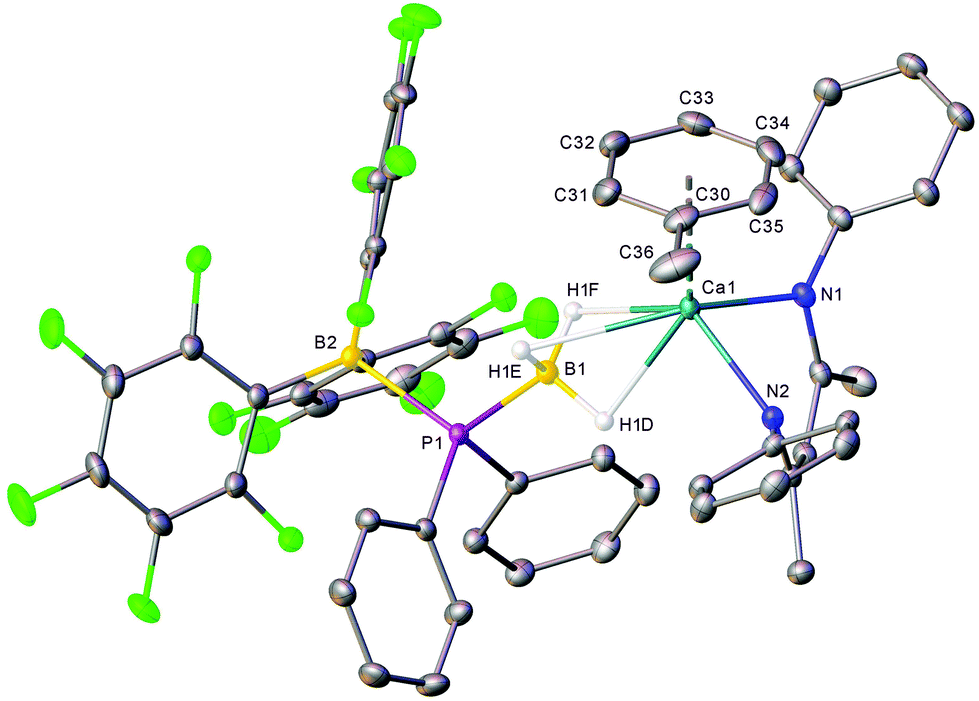 | ||
| Fig. 1 X-ray crystal structure of compound 4a. Thermal ellipsoids displayed at the 30% probability level with iso-propyl groups and hydrogen atoms omitted except those bound to boron. Selected bond lengths (Å) and angles (°): Ca1–N1 2.3147(14), Ca1–N2 2.2904(14), Ca1–C30 3.1365(19), Ca1–C31 3.044(2), Ca1–C32 2.905(2), Ca1–C33 2.843(2), Ca1–C34 2.9055(19), Ca1–C35 3.0416(19), Ca1–(C30–C35 centroid) 2.6391(10), Ca1–(C30–C35 centroid plane) 2.6215(11), P1–C37 1.8343(17), P1–C43 1.8382(16), P1–B1 1.9469(18), P1–B2 2.1368(18), C49–B2 1.640(2), C55–B2 1.640(2), C61–B2 1.639(2), C30–C31 1.388(3), C30–C35 1.396(3), C30–C36 1.502(3), C31–C32 1.389(3), C32–C33 1.381(3), C33–C34 1.379(3), C34–C35 1.383(3), N1–Ca1–(C30–C35 centroid) 110.05(5), N1–Ca1–(C30–C35 normal) 105.58(7), N2–Ca1–(C30–C35 centroid) 109.50(4), N2–Ca1–(C30–C35 centroid) 114.48(7), N2–Ca1–N1 84.76(5), C30–C31–C32 120.7(2), C33–C32–C31 120.3(2), C34–C33–C32 119.8(2), C33–C34–C35 119.9(2), C34–C35–C30 121.2(2), C37–P1–C43 103.82(7), C37–P1–B1 105.99(8), C37–P1–B2 109.52(7), C43–P1–B1 106.29(8), C43–P1–B2 114.17(7), B1–P1–B2 116.06(7), C49–B2–P1 110.52(11), C49–B2–C55 111.11(13), C55–B2–P1 104.30(10), C61–B2–P1 102.97(11), C61–B2–C49 114.13(13), C61–B2–C55 113.02(13). | ||
The BDI-complexed calcium centre of 4a is further coordinated by threefold B-μ2-H-Ca binding of a [(C6F5)3B·PPh2·BH3]− anion and its coordination sphere is completed by an asymmetric η6 interaction with the π-system of a neutral molecule of toluene. The toluene ligand is labile in solution such that when crystals of 4a were dissolved in C6D6, one equivalent of free toluene could be discriminated in the resulting 1H NMR spectrum (Fig. S1†). The phosphinodiboronate anion displays near-perfect tetrahedral geometry at P1, a slightly flattened tetrahedron at B2 and essentially identical structural parameters to those of Lancaster and co-workers’ lithium salt, [{(C6F5)3B·PPh2·BH3}Li(OEt2)3], which was prepared by an analogous route.31 The contrasting P1–B1 (1.9469(18) Å) and P1–B2 (2.1368(18) Å) bond lengths may be rationalised on simple steric grounds.
Although toluene adducts of barium are well established, the comparable ability of magnesium and calcium centres to bind neutral arenes has only been established very recently. Charge-separated [(BDI)Ae]+[WCA]− species (WCA = weakly coordinating anion: [B(C6F5)4]− or [Al{OC(CF3)3}4] −) coordinate arenes via η2,3 or 6 interactions to give complexes of the type [(BDI)Ae·arene]+[WCA]−.32–34 Compound 4a provides, however, the first crystallographic characterisation of an unsupported interaction between a formally charge neutral calcium centre and an arene. The accommodation of the toluene ligand at the pseudo-tetrahedral metal-centre (for a space-filling representation of the crystal structure, see ESI, Fig. S24†) is facilitated by the disposition of the calcium centre, which projects some 1.3821(18) Å out of the mean plane defined by the BDI ligand. Ca1 is located 2.6391(10) Å from the toluene centroid but is asymmetrically coordinated with Ca–C distances ranging from 2.843(2) Å (Ca1–C33) to 3.1365(19) Å (Ca1–C30). The average Ca–C distance of 2.979 Å is, thus, only slightly longer than the comparable measurement in the charge separated cationic component of [(BDI)Ca·C6H6]+[Al{OC(CF3)3}4]− (ca. 2.93 Å).33
Immediate assessment of an analogous reaction performed in C6D6 with the dimeric magnesium complex 1b was also indicative of the effectively instantaneous and quantitative generation of a phosphinodiboronate complex (4b, Scheme 4). The constitution of compound 4b was diagnosed by the emergence of a single new BDI ligand environment in its 1H NMR spectrum and the appearance of a broad signal centred at δ 0.7 ppm in the corresponding 31P{1H} NMR spectrum. In addition to the expected resonances at δ −10.4 and −36.7 ppm, however, a doublet signal at δ −22.0 was observed to grow into the 11B NMR spectrum over the course of 16 hours. While this latter resonance, and the appearance of a further BDI-ligated species in the 1H NMR spectrum, was readily attributed to the formation of the hydridoborate derivative, [(BDI)Mg{HB(C6F5)3}] (3b),28,29 this solution instability frustrated all attempts to isolate and structurally characterise compound 4b.
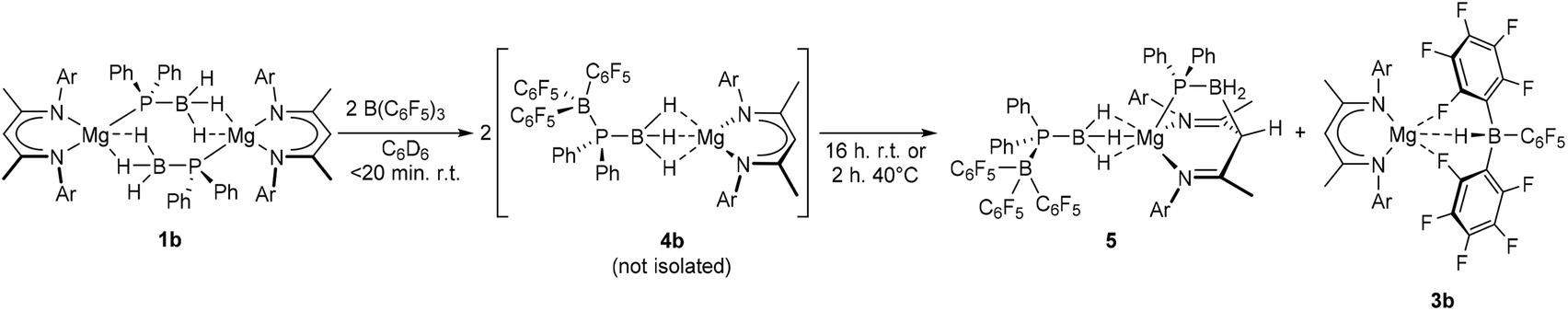 | ||
| Scheme 4 Reaction of compound 1b with BCF to provide compounds 5 and 3bvia4b. Quantitative spectroscopic yields. Ar = 2,6-di-isopropylphenyl. | ||
Although the production of 3b is indicative of the successful abstraction of hydride from the phosphinodiboronate anion of 4b, no signals corresponding to the expected oligomerisation products of [Ph2P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2] could be detected.7 Rather, in addition to the doublet signal of [H(BC6F5)3]− at δ −22.0 ppm, the ultimate 11B NMR spectrum comprised three additional broad resonances at δ −10.2, −24.7 and −35.3 ppm and the 31P{1H} NMR spectrum a broad multiplet at δ −7.2 ppm and a singlet at δ −42.7 ppm. Similarly, two differentiated C6F5 environments were discriminated by 19F NMR spectroscopy, while the 1H NMR spectrum evidenced a further, less symmetrical BDI-coordinated species (5), whose quantitative evolution was simultaneous to that of 3b and took place over the course of 16 hours at room temperature, or two hours at 40 °C. Compound 5 was most clearly characterised by a broad pseudo-doublet at δ 3.05 ppm, which emerged with a similar ratio of 1H NMR signal intensities to those assigned to 3b. These spectroscopic features were eventually rationalised by X-ray diffraction analysis of single crystals of compound 5 (Fig. 2), obtained after removal of solvent and fractional crystallisation of the reaction products from a hexane/toluene solvent system. As for 4a, the hydrogen atoms attached to boron were located and refined freely.
BH2] could be detected.7 Rather, in addition to the doublet signal of [H(BC6F5)3]− at δ −22.0 ppm, the ultimate 11B NMR spectrum comprised three additional broad resonances at δ −10.2, −24.7 and −35.3 ppm and the 31P{1H} NMR spectrum a broad multiplet at δ −7.2 ppm and a singlet at δ −42.7 ppm. Similarly, two differentiated C6F5 environments were discriminated by 19F NMR spectroscopy, while the 1H NMR spectrum evidenced a further, less symmetrical BDI-coordinated species (5), whose quantitative evolution was simultaneous to that of 3b and took place over the course of 16 hours at room temperature, or two hours at 40 °C. Compound 5 was most clearly characterised by a broad pseudo-doublet at δ 3.05 ppm, which emerged with a similar ratio of 1H NMR signal intensities to those assigned to 3b. These spectroscopic features were eventually rationalised by X-ray diffraction analysis of single crystals of compound 5 (Fig. 2), obtained after removal of solvent and fractional crystallisation of the reaction products from a hexane/toluene solvent system. As for 4a, the hydrogen atoms attached to boron were located and refined freely.
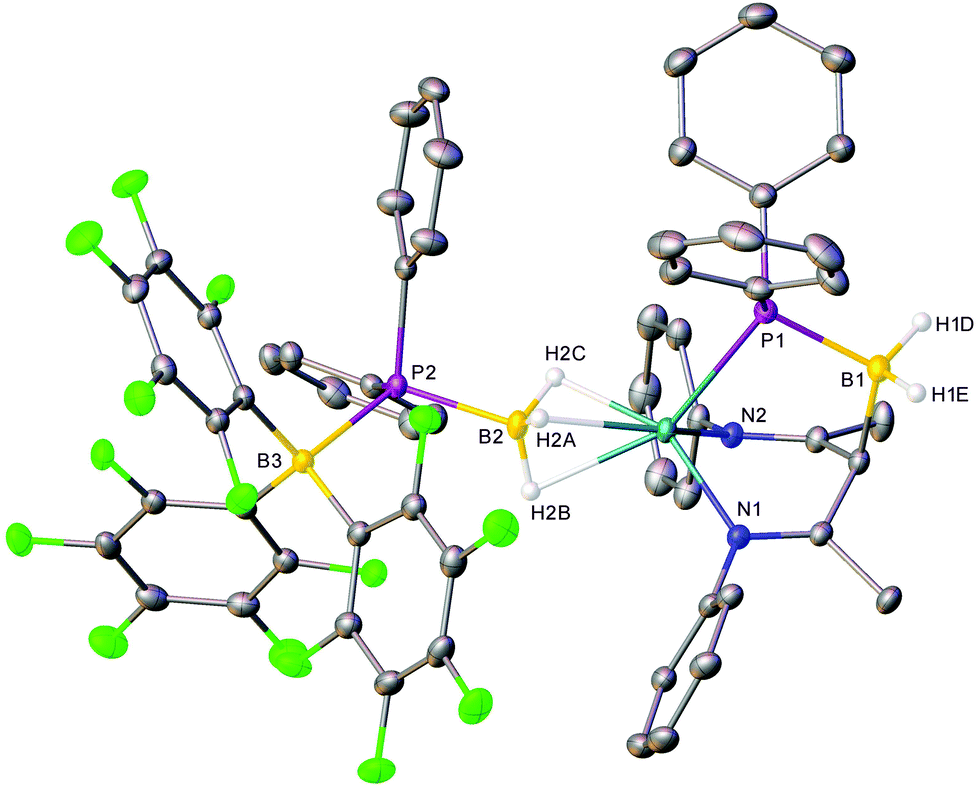 | ||
| Fig. 2 X-ray crystal structure of compound 5. Thermal ellipsoids are shown at the 30% probability level. Hydrogen atoms except for H3 and those bound to boron are omitted for clarity, as are iso-propyl substituents. Selected bond lengths (Å) and angles (°): P1–Mg1 2.5701(6), P1–C30 1.8389(17), P1–C36 1.8358(17), P1–B1 1.9791(18), P2–C42 1.8403(15), P2–C48 1.8339(16), P2–B2 1.9251(17), P2–B3 2.1100(16), Mg1–N1 2.0986(13), Mg1–N2 2.1057(13), N1–C2 1.2950(19), N2–C4 1.293(2), C1–C2 1.502(2), C2–C3 1.494(2), C3–C4 1.488(2), C3–B1 1.726(2), C54–B3 1.649(2), C60–B3 1.636(2), C66–B3 1.655(2), C4–C5 1.504(2), C30–P1–Mg1 121.60(6), C30–P1–B1 111.06(8), C36–P1–Mg1 119.50(6), C36–P1–C30 97.43(7), C36–P1–B1 112.27(8), B1–P1–Mg1 95.60(6), C42–P2–B2 102.12(8), C42–P2–B3 117.18(7), C48–P2–C42 104.20(7), C48–P2–B2 106.70(8), C48–P2–B3 110.48(7), B2–P2–B3 115.06(7), N1–Mg1–P1 94.86(4), N1–Mg1–N2 92.73(5), N2–Mg1–P1 96.01(4), N1–C2–C1 123.85(14), N1–C2–C3 122.85(13), C3–C2–C1 113.27(13), C2–C3–B1 110.54(12), C4–C3–C2 119.41(13), C4–C3–B1 112.13(13), N2–C4–C3 122.49(13), N2–C4–C5 123.50(14), C3–C4–C5 114.00(13), C3–B1–P1 110.30(10), C54–B3–P2 110.68(10), C54–B3–C66 109.84(12), C60–B3–P2 103.54(10), C60–B3–C54 113.11(12), C60–B3–C66 112.61(12), C66–B3–P2 106.73(9), Mg1–B2–P2 163.63(10). | ||
Compound 5 consists of a mononuclear magnesium species ligated by a tripodal N,P,N’-donor ligand and displays close μ-H-Mg contacts to the same [(C6F5)3B·PPh2·BH3]− anion present in the X-ray crystal structure of 4a. The former anionic moiety is postulated to result from nucleophilic attack of the BDI γ-methine carbon centre upon the unsaturated phosphinoborane monomer, [Ph2PBH2], which is generated within the coordination sphere of magnesium by BCF-mediated hydride abstraction from [(C6F5)3B·PPh2·BH3]− or [Ph2P·BH3]−. Although Weller and co-workers’ cationic rhodium complex [Cp*Rh{tBu2P·BH2·PMe3}][BArF4], was similarly derived by interception of a phosphinoborane monomer generated by in situ phosphine-borane dehydrogenation,35 compound 5 is, to the best of our knowledge, the first example of a phosphinoborane trapped within the coordination sphere of a main-group metal. Other kinetically stabilised transition-metal phosphinoborane complexes have been described by the groups of Scheer36,37 and Bourissou,38 but were prepared by non-dehydrogenative metathesis and ligand exchange routes. Harder's calcium borylamide complex [(BDI)Ca(DippNBH2)] bears comparison to compound 5 as an Ae-coordinated dehydrogenation product of a group 13–15 complex.25
Although examples of similar BDI γ-C–B bond formation are limited to a recent report by Jones and co-workers,39 the formation of 5 is clearly also reminiscent of the well documented reactivity of [(BDI)Ae]+ and [(BDI)AlMe]+ cations towards other unsaturated small molecules such as alkynes or CO2.29,40,41 Consistent with the formulation of the tripodal anion as a bis-imine unit, the N1–C2 (1.2950(19) Å) and N2–C4 (1.293(2) Å) bond lengths of compound 5 are significantly shorter than those observed in unperturbed BDI anions. The geometry about C3 is also effectively tetrahedral, with X–C3–X bond angles in the range of 110.5–119.4°. The B1–P1 bond (1.9791(18) Å) is significantly longer than those typical of BP double bonds (1.763–1.913 Å),42–47 and even exceeds the dative P–BH3 bonds of secondary phosphine-boranes (1.91–1.94 Å), and is thus best described as an elongated single bond, consistent with the tetrahedral geometries at both phosphorus and boron.4,48–51 The C3–B1 bond (1.726(2) Å) is also elongated in comparison to the analogous γ-C–B bonds of Jones’ compounds (1.680(2)–1.697(3) Å).39 The [(C6F5)3B·PPh2·BH3]− anion, however, remains geometrically similar to that of compound 4a.
Several potential mechanisms were assessed theoretically by density functional theory (DFT) studies (BP86-D3(BJ)-benzene/BS2//BP86/BS1) of the reaction between 1b and BCF that culminates in the formation of 3b and 5. Initial calculations addressed the direct abstraction of a hydride from the dimeric phosphidoborane, complex 1b (designated as pathway I; see ESI, Fig. S27†). This process ensues by disruption of a B-μ2-H-Mg interaction and the formation of a B-μ2-H-(BCF) intermediate via a free energy barrier of 26.8 kcal mol−1. Although subsequent hydride abstraction, Ph2PBH2 elimination and interception steps were found to be kinetically facile and thermodynamically viable, this computed pathway (I) was discounted as it does not invoke the formation of the experimentally observed product, 4b.
Incorporation of 4b into the mechanism necessarily requires insertion of a BCF molecule into one of the Mg–P bonds of the dimeric starting complex 1b to form species A at +6.7 kcal mol−1 (Fig. 3). Consideration of this requirement also raises a number of possibilities. Formation of a dative P → B bond significantly stabilises A to give intermediate B at −14.2 kcal mol−1. Subsequent addition of a second molecule of BCF to B results in the cleavage of the Mg–P bond and yields two molecules of 4b at ΔGbnz = −15.5 kcal mol−1. Alternatively, the second Mg–P bond in B can dissociate with the endergonic production of one molecule of 4b and one molecule of the monomeric phosphidoborane intermediate, C (ΔGbnz = +6.8 kcal mol−1, left hand side of both Fig. 4a and b). The calculated molecular structure of 4b (see ESI, Fig. S31 and Table S5†) exhibits near co-planarity of the Mg centre with the BDI ligand, resulting in a sterically protected environment that obviates the possibility of a stable Mg-arene solvent interaction, of the type observed for the analogous calcium complex, 4a. The DFT-optimised structure of an η2-benzene adduct of 4b (4b′) was found to reside 22.1 kcal mol−1 higher in energy than the unsolvated complex (see ESI, Fig. S25†).
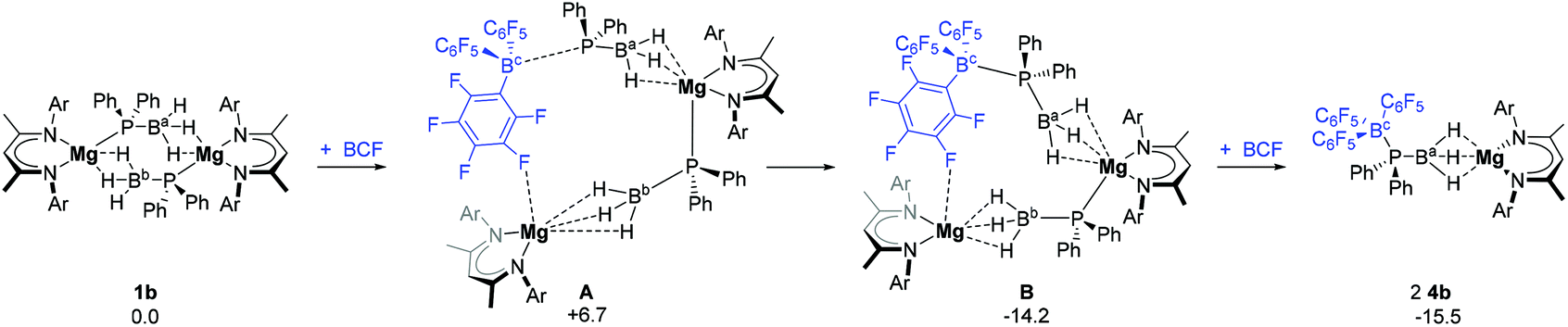 | ||
| Fig. 3 DFT computed free energies (BP86-D3(BJ)-benzene/BS2//BP86/BS1), in kcal mol−1, for the reaction of 1b with BCF to form two molecules of 4b. Ar = 2,6-di-isopropylphenyl. | ||
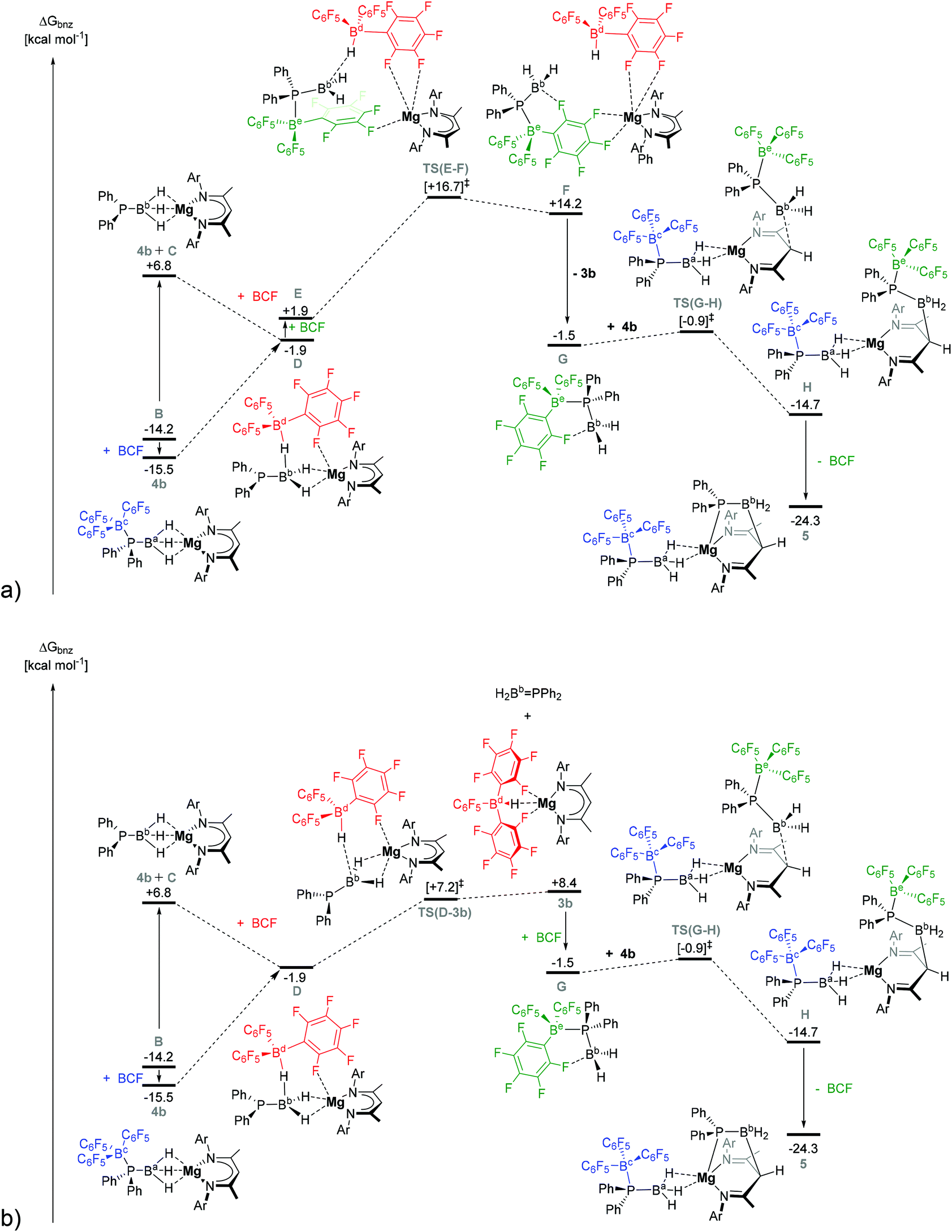 | ||
| Fig. 4 DFT calculated free energy profile (BP86-D3(BJ)-benzene/BS2//BP86/BS1), in kcal mol−1, for the reaction of 4b with BCF by (a) pathway II, and (b) pathway III. Ar = 2,6-di-isopropylphenyl. | ||
C is relatively unencumbered, and one of its three boron-bound hydrides can be readily intercepted by BCF to generate intermediate D, ΔGbnz = −1.9 kcal mol−1 (left hand side of both Fig. 4a and b). D may also be formed directly from 4b by migration of the BCF moiety from the phosphorus centre to hydride of Ba. This was computed to occur with the complete dissociation of BCF from P. Once D has formed, however, the mechanism for formation of 3b (and 5) can diverge along two viable alternative trajectories, designated as pathways II and III, both of which invoke a significant degree of commonality (Fig. 4a and b).
The DFT-calculated Köhn–Sham frontier molecular orbitals of D (see ESI, Table S4†) reveal that the HOMO possesses significant P-lone pair character. As a result, D can either be intercepted by a further molecule of BCF in a marginally endergonic process to provide E, ΔΔGbnz = 3.8 kcal mol−1 (Fig. 4a). Subsequent progression along pathway II invokes hydride abstraction via transition state TS(E–F) at ΔGbnz = +16.7 kcal mol−1 to yield F at ΔGbnz = +14.2 kcal mol−1 in which a {(BDI)Mg}+ cation is loosely bound to a neutral BCF-stabilised phosphinoborane monomer (G) and a [HB(C6F5)3]− anion via μ-F interactions. G can then dissociate from F to provide the observed product 3b (ΔGbnz(3b + GE) = −1.5 kcal mol−1), the DFT-optimised structure of which is in good agreement with the previously published X-ray crystal structure of the magnesium hydridoborate product.28
Alternatively, the direct abstraction of a hydride from D can proceed via pathway III (Fig. 4b) and TS(D-3b) at ΔGbnz = +7.2 kcal mol−1. The B–H distances in TS(D-3b) clearly show near-complete hydride transfer to the flattened tetrahedral boron centre, whilst the unsaturated H2B = PPh2 fragment remains bound to magnesium. Minor adjustments to the [HB(C6F5)3]− anion result in the endergonic elimination of free H2Bb = PPh2 to provide 3b at 23.9 kcal mol−1 above 4b (ΔGbnz = +8.4 kcal mol−1). Interestingly, H2Bb = PPh2 can also be readily intercepted by a free BCF molecule to generate G, ΔGbnz = −1.5 kcal mol−1, such that, from this point onwards, the course of the reaction to produce compound 5 is completely common to both computed pathways II and III.
The DFT calculated Köhn–Sham frontier molecular orbitals of free H2B = PPh2 displayed significant contributions from the π- and π*-molecular orbitals of the P–B bond (see ESI, Table S4†). In contrast, formation of the BCF-complex G results in de-population of the π-orbital and elongation of the P–B bond by 0.08 Å to 1.96 Å. This bond length is comparable to P–B single bonds observed in crystallographically characterised Lewis base- and Lewis acid–base stabilised phosphanylboranes.12,14,36,37,52–54 Whilst the calculated existence of an ortho-F–B donor–acceptor interaction in G provides some degree of a “push–pull” stabilisation (F–B distance = 1.73 Å), the electron-withdrawing effect of the phosphorus-complexed BCF moiety enhances the electrophilicity at boron. The calculated HOMO of 4b (−5.17 eV) involves a relatively exposed π-system delocalised across the BDI-ligand backbone. NBO charge analysis shows that the γ-methine carbon of the BDI ligand exhibits a significant negative charge (−0.41) and can, hence, act as a nucleophile, attacking the electrophilic boron centre of the BCF-complex GviaTS(G–H) at ΔGbnz = −0.9 kcal mol−1 to give H at ΔGbnz = −14.7 kcal mol−1. Although dissociation of BCF is computed to be endergonic by +17.0 kcal mol−1 with respect to H, intramolecular formation of a P–Mg dative bond is thermodynamically favoured. As such, stepwise P–B cleavage and Mg—P bond formation is exergonic, affording compound 5 at ΔGbnz = −24.3 kcal mol−1. The free energy barrier to 3b and 5 formation from 4b in pathway II is, therefore, computed to be 31.2 kcal mol−1 (via intermediates E and F), whilst pathway III (via3b) is computed to be 23.9 kcal mol−1, the latter being 7.3 kcal mol−1 lower in free energy and most consistent with the reaction conditions.
Most known Ae-mediated catalytic processes have been shown to follow a trend of increasing reactivity on descending the group from magnesium to barium, as a result of increasing polarisability, electropositivity and ionic radius.55–57 In this context, the apparent inertness of compound 4a towards formation of calcium analogues of 3b and 5 is intriguing. To this end, the mechanistic pathway for 4a formation and its onward reaction to the hypothetical calcium complexes 3c′ and 5′ was also calculated (see ESI, Fig. S29 and S30†). Viable pathways for BCF-mediated hydride abstraction from the toluene-free intermediate 4a′ displayed similarities to pathways II and III for magnesium. Intermediate 4a′ was, however, located at −4.7 kcal mol−1 with respect to 1a: some 26.7 kcal mol−1 higher in energy than the crystallographically characterised η6-toluene complex 4a (ΔGtol = −31.4 kcal mol−1). This latter species, therefore, may be viewed as an energetic sink, which precludes onward reactivity analogous to that observed for its magnesium analogue 4b such that transformation of 4a to 3c′ + 5′ is both kinetically unrealistic with a suggested free energy barrier of 37.9 kcal mol−1 and being almost thermoneutral.
Conclusion
In conclusion, addition of B(C6F5)3 to the alkaline earth phosphidoborane complexes 1a and 1b initially results in P → B adduct formation to provide the monomeric phosphinodiboronate species, 4a and 4b. In the solid state, the calcium complex 4a features an η6-coordinated toluene molecule which, although labile in solution, was calculated to stabilise the compound towards onward reactivity. In contrast, arene solvent-coordination to the spectroscopically-detected magnesium analogue 4b was calculated to be endergonic, and this species transforms into the known [HB(C6F5)3]− derivative 3b and the N,P,N′-ligated species 5. Crystallographic characterisation of 5 revealed it to be the product of H2B = PPh2 interception by the nucleophilic BDI ligand backbone. Computational assessment of these processes suggests a mechanism involving BCF dissociation and subsequent hydride abstraction to generate the putative intermediate, H2B = PPh2. Key to the thermodynamic viability of the process are μ-F-Mg interactions; complexation of H2B = PPh2 by BCF was calculated to generate an electrophilic intermediate containing a P → B dative bond and a stabilising ortho-F-BH2 interaction. These studies demonstrate the potential for cooperative main-group mediated reactivity in the dehydrogenation of phosphine-borane adducts. Whilst non-innocence of the BDI ligand utilised herein is a clear obstacle towards the use of this system in catalytic phosphine-borane dehydropolymerisation, these results will inform future efforts to develop improved and inexpensive catalysts for the synthesis of useful inorganic polymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for financial support through the Centre for Doctoral Training in Catalysis (EP/L016443/1) and research grant EP/R020752/1. I. M. thanks the University of Bristol for support and the Canadian Government for a C150 Research Chair. This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath.References
- A. B. Burg and R. I. Wagner, J. Am. Chem. Soc., 1953, 75, 3872–3877 CrossRef.
- J. R. Turner, D. A. Resendiz-Lara, T. Jurca, A. Schäfer, J. R. Vance, L. Beckett, G. R. Whittell, R. A. Musgrave, H. A. Sparkes and I. Manners, Macromol. Chem. Phys., 2017, 218, 1700120 CrossRef.
- A. Schäfer, T. Jurca, J. Turner, J. R. Vance, K. Lee, V. A. Du, M. F. Haddow, G. R. Whittell and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 4836–4841 CrossRef.
- T. L. Clark, J. M. Rodezno, S. B. Clendenning, S. Aouba, P. M. Brodersen, A. J. Lough, H. E. Ruda and I. Manners, Chem. – Eur. J., 2005, 11, 4526–4534 CrossRef CAS.
- U. S. D. Paul, H. Braunschweig and U. Radius, Chem. Commun., 2016, 52, 8573–8576 RSC.
- N. T. Coles, M. F. Mahon and R. L. Webster, Organometallics, 2017, 36, 2262–2268 CrossRef CAS.
- H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669–6678 CrossRef CAS.
- D. Han, F. Anke, M. Trose and T. Beweries, Coord. Chem. Rev., 2019, 380, 260–286 CrossRef CAS.
- S. Pandey, P. Lönnecke and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2014, 2456–2465 CrossRef CAS.
- H. Cavaye, F. Clegg, P. J. Gould, M. K. Ladyman, T. Temple and E. Dossi, Macromolecules, 2017, 50, 9239–9248 CrossRef CAS.
- M. A. Huertos and A. S. Weller, Chem. Sci., 2013, 4, 1881–1888 RSC.
- C. Marquardt, T. Jurca, K. C. Schwan, A. Stauber, A. V. Virovets, G. R. Whittell, I. Manners and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13782–13786 CrossRef CAS.
- N. L. Oldroyd, S. S. Chitnis, V. T. Annibale, M. I. Arz, H. A. Sparkes and I. Manners, Nat. Commun., 2019, 10, 1370 CrossRef.
- J. M. Denis, H. Forintos, H. Szelke, L. Toupet, T. N. Pham, P. J. Madec and A. C. Gaumont, Chem. Commun., 2003, 54–55 RSC.
- L. J. Morris, M. S. Hill, M. F. Mahon, I. Manners, F. S. McMenamy and G. R. Whittell, Chem. – Eur. J., 2020, 26, 2954–2966 CrossRef CAS.
- L. J. Morris, G. R. Whittell, J.-C. Eloi, M. F. Mahon, F. Marken, I. Manners and M. S. Hill, Organometallics, 2019, 38, 3629–3648 CrossRef CAS.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem. – Eur. J., 2010, 16, 8508–8515 CrossRef CAS.
- P. Bellham, M. D. Anker, M. S. Hill, G. Kociok-Köhn and M. F. Mahon, Dalton Trans., 2016, 45, 13969–13978 RSC.
- P. Bellham, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2015, 44, 12078–12081 RSC.
- P. Bellham, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 5716–5721 CrossRef CAS.
- S. Harder, J. Spielmann and B. Tobey, Chem. – Eur. J., 2012, 18, 1984–1991 CrossRef CAS.
- J. Spielmann and S. Harder, Dalton Trans., 2011, 40, 8314–8319 RSC.
- J. Spielmann, D. F. J. Piesik and S. Harder, Chem. – Eur. J., 2010, 16, 8307–8318 CrossRef CAS.
- J. Spielmann, M. Bolte and S. Harder, Chem. Commun., 2009, 6934 RSC.
- J. Spielmann and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064–5065 CrossRef CAS.
- J. Spielmann, G. Jansen, H. Bandmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 6290–6295 CrossRef.
- L. J. Morris, M. S. Hill, M. F. Mahon, I. Manners and B. O. Patrick, Organometallics, 2020 DOI:10.1021/acs.organomet.0c00008.
- M. D. Anker, M. Arrowsmith, R. L. Arrowsmith, M. S. Hill and M. F. Mahon, Inorg. Chem., 2017, 56, 5976–5983 CrossRef CAS.
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC.
- S. Harder, Organometallics, 2002, 21, 3782–3787 CrossRef CAS.
- A. M. Fuller, A. J. Mountford, M. L. Scott, S. J. Coles, P. N. Horton, D. L. Hughes, M. B. Hursthouse and S. J. Lancaster, Inorg. Chem., 2009, 48, 11474–11482 CrossRef CAS.
- J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685–8688 RSC.
- L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684–12693 RSC.
- A. Friedrich, J. Pahl, H. Elsen and S. Harder, Dalton Trans., 2019, 48, 5560–5568 RSC.
- T. N. Hooper, A. S. Weller, N. A. Beattie and S. A. Macgregor, Chem. Sci., 2016, 7, 2414–2426 RSC.
- K. C. Schwan, A. Y. Timoskin, M. Zabel and M. Scheer, Chem. – Eur. J., 2006, 12, 4900–4908 CrossRef CAS.
- U. Vogel, P. Hoemensch, K. C. Schwan, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2003, 9, 515–519 CrossRef CAS.
- A. Amgoune, S. Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2012, 134, 6560–6563 CrossRef CAS.
- D. D. L. Jones, A. J. R. Matthews and C. Jones, Dalton Trans., 2019, 48, 5785–5792 RSC.
- J. Pahl, T. E. Stennett, M. Volland, D. M. Guldi and S. Harder, Chem. – Eur. J., 2019, 25, 2025–2034 CrossRef CAS.
- T. E. Stennett, J. Pahl, H. S. Zijlstra, F. W. Seidel and S. Harder, Organometallics, 2016, 35, 207–217 CrossRef CAS.
- A. N. Price and M. J. Cowley, Chem. – Eur. J., 2016, 22, 6248–6252 CrossRef CAS.
- S. J. Geier, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2011, 50, 336–344 CrossRef CAS.
- E. Rivard, W. A. Merrill, J. C. Fettinger and P. P. Power, Chem. Commun., 2006, 3800–3802 RSC.
- R. A. Bartlett, H. V. R. Dias, X. D. Feng and P. P. Power, J. Am. Chem. Soc., 1989, 111, 1306–1311 CrossRef CAS.
- R. A. Bartlett, X. D. Feng and P. P. Power, J. Am. Chem. Soc., 1986, 108, 6817–6819 CrossRef CAS.
- X. D. Feng, M. M. Olmstead and P. P. Power, Inorg. Chem., 1986, 25, 4615–4616 CrossRef CAS.
- K. Izod, J. M. Watson, W. Clegg and R. W. Harrington, Inorg. Chem., 2013, 52, 1466–1475 CrossRef CAS.
- E. M. Pelczar, E. A. Nytko, M. A. Zhuravel, J. M. Smith, D. S. Glueck, R. Sommer, C. D. Incarvito and A. L. Rheingold, Polyhedron, 2002, 21, 2409–2419 CrossRef CAS.
- H. Dorn, E. Vejzovic, A. J. Lough and I. Manners, Inorg. Chem., 2001, 40, 4327–4331 CrossRef CAS.
- M. W. Day, B. Mohr and R. H. Grubbs, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 3106–3108 CrossRef.
- A. Adolf, U. Vogel, M. Zabel, A. Y. Timoshkin and M. Scheer, Eur. J. Inorg. Chem., 2008, 3482–3492 CAS.
- A. Adolf, M. Zabel and M. Scheer, Eur. J. Inorg. Chem., 2007, 2136–2143 CrossRef CAS.
- A. Stauber, T. Jurca, C. Marquardt, M. Fleischmann, M. Seidl, G. R. Whittell, I. Manners and M. Scheer, Eur. J. Inorg. Chem., 2016, 2684–2687 CrossRef CAS.
- A. G. M. Barrett, C. Brinkmann, M. R. Crimmin, M. S. Hill, P. Hunt and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 12906–12907 CrossRef CAS.
- C. Bellini, V. Dorcet, J. F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 4564–4583 CrossRef CAS.
- B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray diffraction analysis of compounds 4a and 5, details for the computational analysis and atomic coordinates of computed structures. CCDC 2024757 and 2024758. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03415k |
| This journal is © The Royal Society of Chemistry 2020 |