
Protonation-induced self-assembly of bis-phenanthroline macrocycles into nanofibers arrayed with tetrachloroaurate, hexachloroplatinate or phosphomolybdate ions†
 *a
Mitsuhiko
Shionoya
*a
*a
Mitsuhiko
Shionoya
*a
Abstract
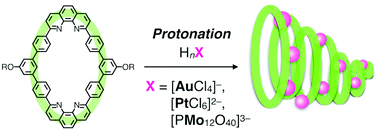
One-dimensional self-assembly of macrocycles is one of the important strategies for constructing fibrous nanomaterials with anisotropic functions such as one-dimensional transport and accumulation of molecules and ions. Herein we report on the synthesis and properties of self-assembled nanofibers using macrocycles to develop a multipurpose template for one-dimensional array of noble metal ions. The nanofibers were prepared by protonation-induced self-assembly of bis-phenanthroline macrocycles, which have enabled the accumulation of some metal-containing anions, such as tetrachloroaurate, hexachloroplatinate and phosphomolybdate. Microscopic observations have demonstrated that the supramolecular nanofibers were reproducibly formed in a similar way, regardless of the structures and charge numbers of the anions. Moreover, the resulting nanofibers, arrayed with several metal ions, were chemically reduced, producing dispersible gold nanoparticles and mixed-valence nanofibers.
 Please wait while we load your content...
Please wait while we load your content...

