
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing N-, P-, and S-donor leaving groups: an investigation of the chemical and biological properties of ruthenium, rhodium and iridium thiopyridone piano stool complexes†
Sophia
Harringer
 a,
Debora
Wernitznig
a,
Natalie
Gajic
a,
Andreas
Diridl
a,
Dominik
Wenisch
a,
Michaela
Hejl
a,
Michael A.
Jakupec
ab,
Sarah
Theiner
c,
Gunda
Koellensperger
c,
Wolfgang
Kandioller
*ab and
Bernhard K.
Keppler
ab
a,
Debora
Wernitznig
a,
Natalie
Gajic
a,
Andreas
Diridl
a,
Dominik
Wenisch
a,
Michaela
Hejl
a,
Michael A.
Jakupec
ab,
Sarah
Theiner
c,
Gunda
Koellensperger
c,
Wolfgang
Kandioller
*ab and
Bernhard K.
Keppler
ab
aInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Strasse 42, 1090 Vienna, Austria. E-mail: wolfgang.kandioller@univie.ac.at; Tel: +43 1 4277 52609
bResearch Cluster “Translational Cancer Therapy Research”, Waehringer Strasse 42, 1090 Vienna, Austria
cInstitute of Analytical Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Strasse 38, 1090 Vienna, Austria
First published on 2nd November 2020
Abstract
A series of 15 piano-stool complexes featuring either a RuII, RhIII or IrIII metal center, a bidentate thiopyridone ligand, and different leaving groups was synthesized. The leaving groups were selected in order to cover a broad range of different donor atoms. Thus, 1-methylimidazole served as a N-donor, 1,3,5-triaza-7-phosphaadamantane (pta) as a P-donor, and thiourea as a S-donor. Additionally, three complexes featuring different halido leaving groups (Cl, Br, I) were added. Leaving group alterations were carried out with respect to a possible influence on pharmacokinetic and pharmacodynamic parameters, as well as the cytotoxicity of the respective compounds. The complexes were characterized via NMR spectroscopy, X-ray diffraction (where possible), mass spectrometry, and elemental analysis. Cytotoxicity was assessed in 2D cultures of human cancer cell lines by microculture and clonogenic assays as well as in multicellular tumor spheroids. Furthermore, cellular accumulation studies, flow-cytometric apoptosis and ROS assays, DNA plasmid assays, and laser ablation ICP-MS studies for analyzing the distribution in sections of multicellular tumor spheroids were conducted. This work demonstrates the importance of investigating each piano-stool complexes' properties, as the most promising candidates showed advantages over each other in certain tests/assays. Thus, it was not possible to single out one lead compound, but rather a group of complexes with enhanced cytotoxicity and activity.
Introduction
It is widely acknowledged that antitumoral metal complexes offer a vast window of possible modes of action which are inaccessible for purely organic compounds.1 A huge part of this diversity is due to the various oxidation states of the central atom, accompanied by different coordination numbers and geometries, and the possibility of fine-tuning pharmacokinetic and pharmacodynamic properties via ligand variation.2 However, the miraculous rise of Pt(II) drugs that followed the discovery of their antineoplastic properties was compromised by their adverse side effects and resistances.3 While many treatment regimens are still based on these drugs (e.g. cisplatin), research is focused on alternatives in order to overcome these problems.4,5 Various approaches have been tried and brought to light the class of organometallic anticancer complexes, where titanocene dichloride,6,7 budotitane,8 and the tamoxifen-ferrocenyl derivative ferrocifen9 are just a few examples of pioneer drug candidates (Chart 1A–C). However, none of these drugs were pursued after initial clinical trials, as they revealed stability, solubility or formulation issues, or an undesirable toxicity profile.1 Another great hope lies with ruthenium anticancer compounds, as the central atom is accessible in various oxidation states under physiologically relevant conditions, and its complexes feature slow ligand exchange rates.2 Indeed, the first RuIII drug candidates displayed low overall toxicity, and pronounced antitumor or at least antimetastatic potency in preclinical models (e.g. NAMI-A and BOLD-100; Chart 1D and E).10 Both RuII and RuIII compounds may act as prodrugs, but they differ fundamentally in one respect: while the poorly reactive RuIII parent compounds are assumed to be activated by reduction to their more active RuII congeners in vivo, piano-stool complexes already feature the more active RuII species, which is stabilized by its coordination sphere.11,12 Amongst this group of transition metal complexes, RM175 and RAPTA-C (Chart 2, A and B) are two well-studied examples, which highlight the importance for the coordination sphere and slight modifications thereof on the activity profile. RM175 features a biphenyl, a bidentate ethylene diamine (en) ligand, and a chlorido leaving group, and its mode of action is supposedly based on DNA intercalation, as well as DNA and GSH adduct formation.13,14 On the other hand, RAPTA-C's coordination sphere is built of a p-cymene, two monodentate chlorido ligands and a 1,3,5-triaza-7-phosphaadamantane (pta) leaving group. These changes lead to a distinctly different mode of action, where protein adduct formation is the alleged key factor.15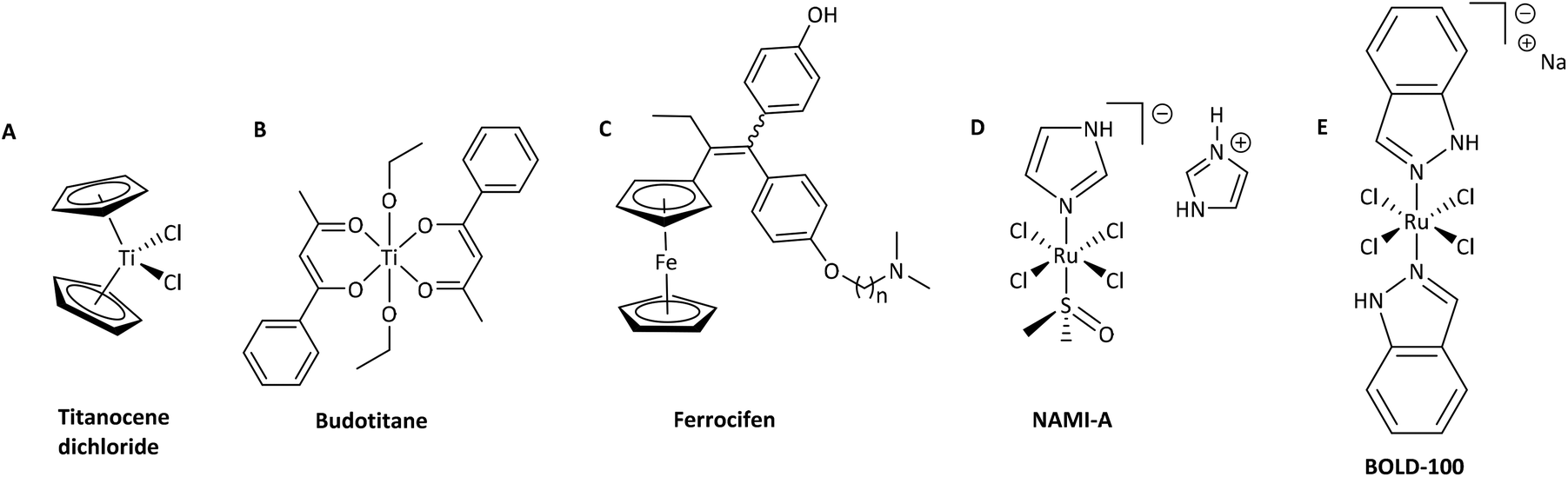 | ||
| Chart 1 Overview of five exemplary transition-metal complexes: titanocene dichloride (A), budotitane (B), ferrocifen (C; where n = 2, 3, 4, 5, 8), NAMI-A (D), and BOLD-100 (E). | ||
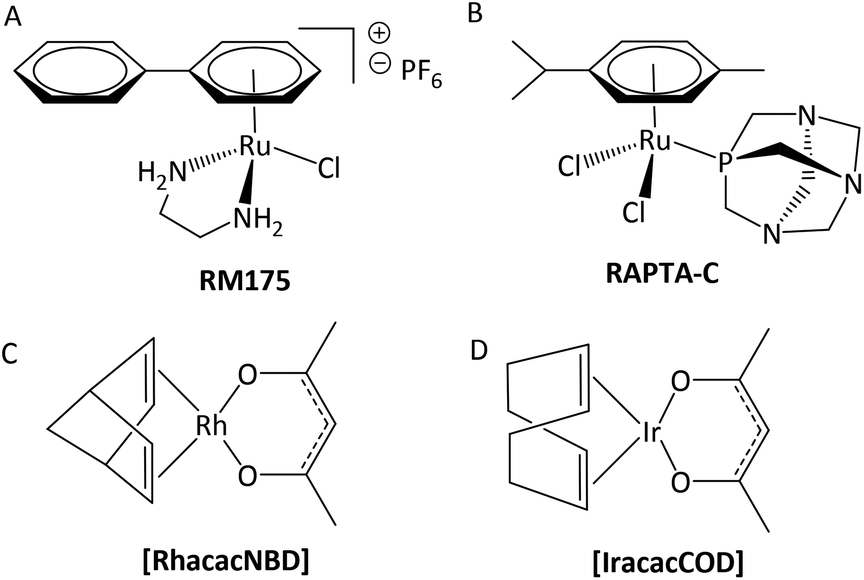 | ||
| Chart 2 Structures of RuII piano-stool complexes which are in advanced preclinical stages of drug development (A and B) and RhI [RhacacNBD] and IrI [IracacCOD] organometallics (C and D) which were evaluated in vivo. | ||
In this context, organometallic Rh and Ir complexes and their possible clinical application are also noteworthy. While Pt and Ru agents are well studied at this point, Rh and especially Ir drugs are still in their infancy. Nevertheless, promising results were obtained with RhI and IrI compounds [RhacacNBD] and [IracacCOD], which cured a 100% of mice bearing Ehrlich ascites carcinoma at sublethal dosages (Chart 2, C and D).16 On the other hand, several RhIII and IrIII congeners of literature-known RuIII drugs failed in biological tests, which highlights the tremendous impact of ligand scaffold and metal center on anticancer properties. For instance, [ImH][trans-{IrCl4(DMSO)(Im)}] and [(DMSO)2H][trans-{IrCl4(DMSO)2}] which are structurally related to NAMI-A, were found to be too inert with regard to their kinetics and poorly active in biological studies.17 Another example is the RhIII complex ([ImH][trans-{RhCl4(Im)2}]), which showed significantly slower ligand exchange reactions and was found to be biologically inactive.18
While many RuII piano-stool compounds and their RhIII and IrIII counterparts feature almost the same ligand sphere, one major difference is the stabilizing arene moiety. While benzene derivatives are employed in the case of RuII and OsII complexes, electron-rich cyclopentadienyl ligands (Cp) are better suited for RhIII and IrIII compounds.19 Apart from that, it is possible to generate libraries of organometallics with slight variations of their coordination sphere, which is an excellent opportunity for in-depth studies of each building block on anticancer activity (e.g. RAPTA-C, OSPTA-C, [Ir(η5-C5Me5)(PTA)Cl2]).20 The findings of the RAPTA studies showed that the Ru compound was most active, followed by OSPTA-C and finally its Ir congener. While anticancer activity was generally very low in the ovarian cancer cell line A2780, RAPTA-C showed the ability to reduce the number and weight of lung metastases in vivo.20,21
Another compound class are thiopyridones based on a thiomaltol scaffold. While thiomaltol organometallics showed considerably low IC50 values in cytotoxicity tests but lacked stability, the exchange of the heterocyclic oxygen for an amine functionality resulted in even lower IC50 values, combined with excellent water solubility and increased stability under biologically relevant conditions.22,23
In this paper, we investigate a set of closely related thiopyridone complexes featuring different metal centers (RuII, RhIII, IrIII), thiopyridone ligands (methyl, benzyl), and leaving groups (1-methylimidazole, pta, and thiourea). The two thiopyridone ligands were chosen based on our previously published results, where benzyl-thiopyridone complexes were the most active representatives in a series of 16 complexes.23 On the other hand, thiodeferiprone (sulfur analogue of deferiprone) was chosen as a negative control as the respective complexes were the least active of the established substance class. This work investigates the impact of the leaving group on stability and biological behavior (e.g. interaction, IC50 values in different cell culture models, cellular accumulation).
Results and discussion
Synthesis and characterization
The complexes of this work were designed in order to gather further insights into the chemical and biological behavior of thiopyridone organometallics via leaving group variation. These organometallics were synthesized with special focus on the influence of this building block on their properties. The introduced monodentate leaving groups feature different donor atoms (e.g. N, P, and S), as well as steric volume, which influences characteristics such as stability under biologically relevant conditions, as well as solubility (depending on the employed counter ion where applicable). For a detailed overview of the chemical structures see Chart 3.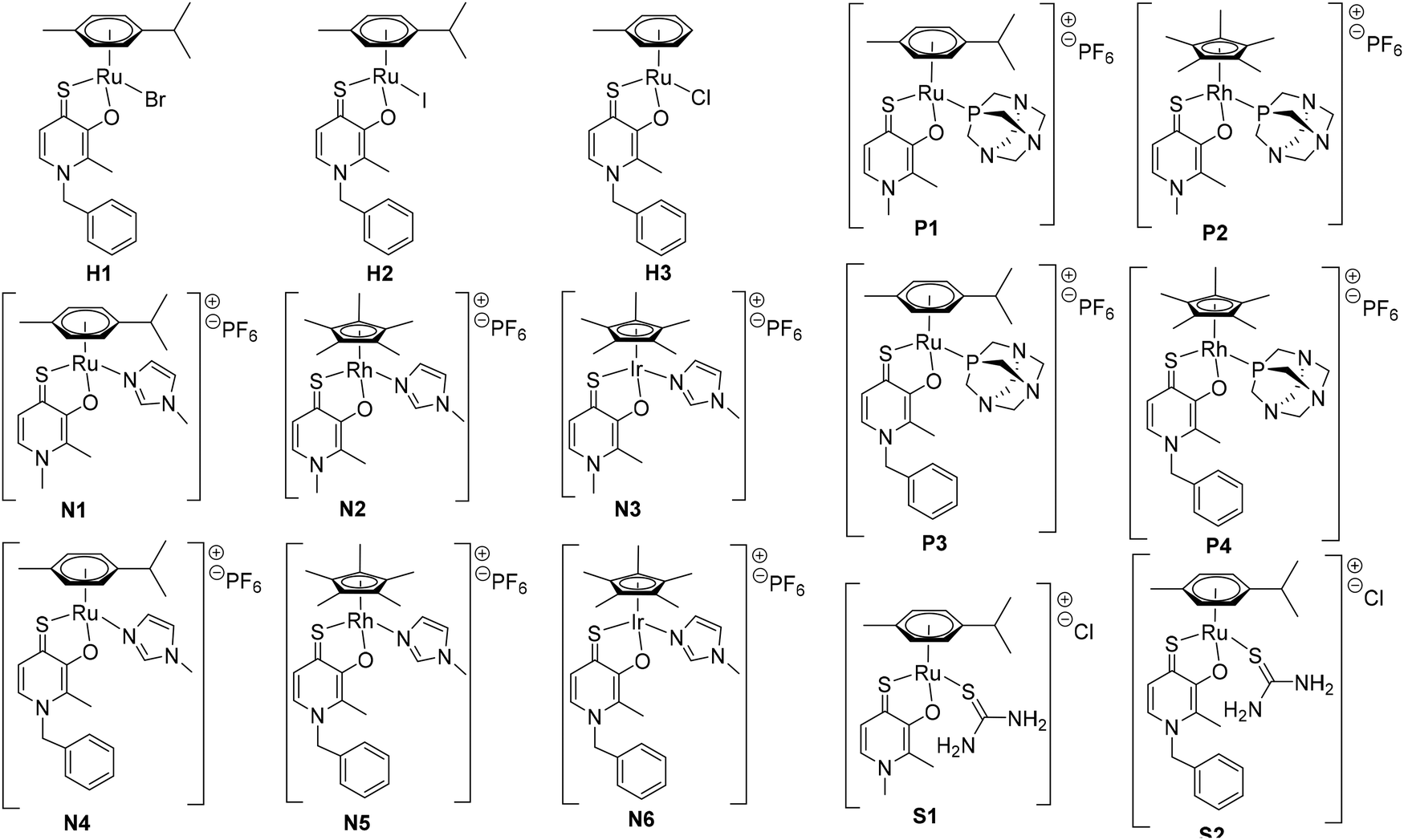 | ||
| Chart 3 Overview of the synthesized thiopyridone organometallics featuring different halides (H1, H2), 1-methylimidazole (N1–N6), pta (P1–P4), or thiourea (S1–S2) as leaving groups, and RuII complex H3 with a toluene arene moiety. | ||
In a first approach the chloride leaving group was exchanged for bromide and iodide (H1, H2), as well as the p-cymene arene for toluene (H3) in order to estimate the impact on dimerization in aqueous solution, which was previously published.23 To gain more detailed insights, well-studied leaving groups such as 1-methylimidazole (N1–N6) and 1,3,5-triaza-7-phosphaadamantane (pta; P1–P4), as well as bioactive building blocks (e.g. thiourea (tu); S1, S2) were employed, resulting in 15 new complexes (Chart 3). The synthetic route started from commercially available maltol, which was reacted with methyl or benzyl amine to give the respective pyridones. In a second step, the pyridone of choice was thionated under inert conditions by use of Lawesson's reagent according to literature.22–24 In the case of organometallics H1–H3 complexation was performed according to standard procedures.25 Thus, the respective ligand was deprotonated with NaOMe by means of Schlenk technique and brought to reaction with the desired dimeric metal precursor. This procedure had to be extended by another step for pta compounds P1–P4 and thiourea derivatives S1, S2. As previously reported, thiopyridone complexes featuring a chloride leaving group undergo dimerization under polar protic conditions.23 This species features a double positive charge. In this state, leaving group exchange seemed unfeasible and therefore, the solvent system was changed from polar protic (MeOH) to polar aprotic (DCM) where the intermediate chloride complex is present in its monomeric form. Subsequently, the desired leaving group (either pta or tu) was added and the mixture was stirred for another 1–24 hours. Noticeably shorter reaction times (1.5–2.5 hours) have been observed for pta complexes, while the exchange for tu took 24 hours. In the case of pta compounds (P1–P4) silver hexafluorophosphate (AgPF6) was added to exchange the counter ion and simplify work up. In the case of 1-methylimidazole piano-stools (N1–N6) it was reported that the above described standard procedure led to product mixtures.22 Therefore, a monomeric 1-methylimidazole metal precursor was synthesized under microwave conditions and used instead of the dimeric precursor.26 Again, the thiopyridone ligand was deprotonated by use of NaOMe, the 1-methylimidazole precursor was added, and after a prolonged reaction time of 24 hours, AgPF6 was added to exchange the counter ion. All products were obtained after filtration and precipitation from DCM/Et2O or DCM/n-hex in poor to good yields (30–83%; Scheme 1). The isolated compounds were characterized by 1H- and 13C-NMR, elemental analysis, ESI-MS measurements, and X-ray diffraction analysis (where possible). Successful complexation was confirmed via1H-NMR spectroscopy due to the vanishing of the ligand's OH peak, as well as shifts of the coordinated leaving group signals. For example, in comparison to free 1-methylimidazole the signals shifted an average of 0.45 ppm downfield upon coordination to IrIII in the spectrum of complex N3 in d4-MeOD. An additional indicator for successful coordination was observed for pta metalorganics (P1–P4), as 1H-NMR signals of free pta and coordinated pta look significantly different. While pta gives a doublet at 4.06 ppm and a singlet at 4.62 ppm, they change to a doublet of doublets at 4.04 ppm and a multiplet from 4.37–4.56 ppm when coordinated to the metal center in RuII methyl pta complex P1 (Fig. 1). This observation can be explained by loss of rotational freedom upon coordination to the sterically more demanding organometallic scaffold. NMR spectra of complexes bearing bromide, and iodide halides as well as a toluene arene (H1–H3) were recorded in deuterated dimethyl sulphoxide (d6-DMSO; Fig. S1–6†) as well as deuterated water (D2O; Fig. S31–36†) in order to investigate if these modifications had any effect on dimerization. According to expectations, dimerization was immediately observed for all three compounds in D2O (dimeric compounds will be annotated with a * henceforth). However, for organometallics featuring either 1-methylimidazole (N1–N6), pta (P1–P4), or thiourea (S1, S2) no dimerization in polar protic solvents can be reported. These findings prove that the affinity of the leaving group's donor atom as well as its bulkiness have considerable impact on the behavior of these compounds in solution. As aqueous solubility for these complexes is lower due to their single positive charge compared to the double positively charged dimers, spectra were recorded in deuterated organic solvents (e.g. d6-DMSO, d4-MeOD or CDCl3; Fig. S7–30†). Counterintuitively, RuII chlorido compounds H1 and H2 showed only two doublets attributable to the p-cymene's aromatic protons, which has been observed for maltol-based complexes before.27 However, the aromatic shifts change drastically when employing sterically more demanding leaving groups (N1–N6, P1–P4, S1, S2) where four distinct signals could be observed. According to literature, this observation can be rationalized by the hindered inversion at the metal center.22 ESI-MS investigations produced [M-L]+ fragments for all complexes, and [M]+ fragments for selected organometallics (P1, P3, P4).
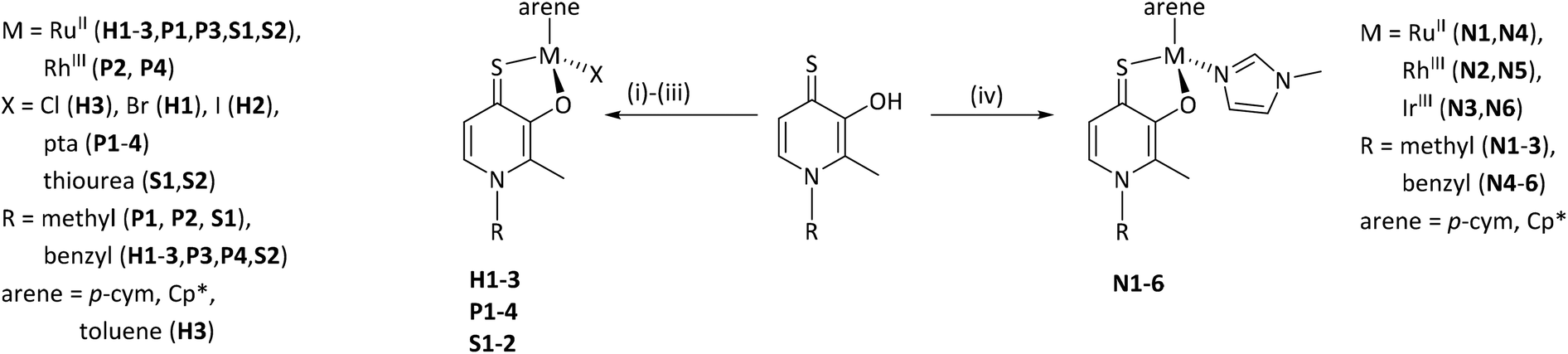 | ||
| Scheme 1 Overview of synthesized complexes. (i) [(p-cym)/(tol)/(Cp*)MCl2]2, NaOMe, MeOH. (ii) [(p-cym)/(Cp*)MCl2]2, NaOMe, MeOH, pta, AgPF6. (iii) [(p-cym)/(Cp*)MCl2]2, NaOMe, MeOH, tu. (iv) [(p-cym)/(Cp*)M(MeIm)2Cl]Cl, NaOMe, MeOH, AgPF6. Charges omitted for clarity. | ||
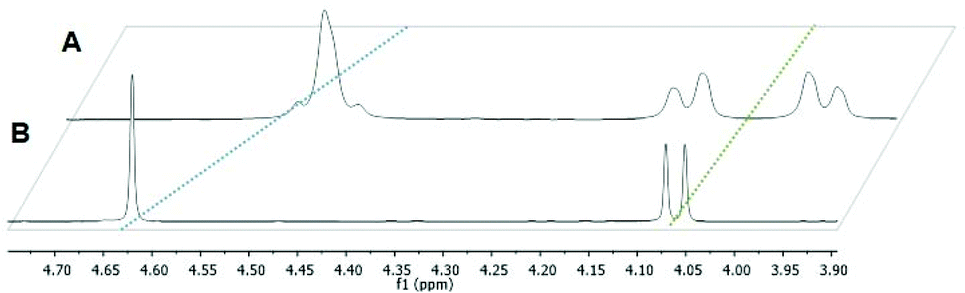 | ||
| Fig. 1 Stacked 1H-NMR spectra of complex P1 (A) and free pta (B) recorded in CDCl3 at 500.10 MHz. | ||
X-ray diffraction analysis
Crystals suitable for X-ray diffraction analysis were obtained for two Ru benzyl dimers (H1*, H2*), 1-methylimidazole (N1–N6), pta (P1–P4), and thiourea complex (S2). Single crystals suitable for diffraction analysis were obtained via vapor diffusion from DCM/Et2O. Their structures are shown in Fig. 2 and Fig. S37–48,† and selected bond lengths and angles are listed in Table 1 and Tables S2–25.† The CCDC numbers can be found in Table S1.† These findings confirm the piano-stool configuration, where the metal center is surrounded by a stabilizing arene moiety constituting the seat, as well as a bidentate thiopyridone ligand and a monodentate leaving group (bromide, iodide, 1-methylimidazole, pta, or thiourea) forming its legs. All 1-methylimidazole complexes (N1, N2, N4–N6) crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The same observations could be made for pta complexes P1, P3 and P4, while Rh methyl compound P2 crystallized in the monoclinic space group P21/c. Thiourea complex S2 crystallized in the monomeric space group P21/n. Selected lattice values for RuII methyl compounds featuring 1-methylimidazole (N1) and pta (P1) leaving groups are listed in Table 1. The Ru–S and Ru–O distances are in the same range for both compounds. On the other hand, the bond length between the metal center and the leaving group increases in the order N1 (2.110 Å) < P1 (2.294 Å). These results are according to expectations, as the nitrogen donor in N1 features a smaller atom radius combined with a higher electronegativity than the phosphorus containing compound P1. The findings for pta compound P1 are in the same range as literature known values for [Ru(η6-p-cymene)Cl2(pta-Me)]Cl [Ru–P: 2.2753(11)].28 Furthermore, the C
. The same observations could be made for pta complexes P1, P3 and P4, while Rh methyl compound P2 crystallized in the monoclinic space group P21/c. Thiourea complex S2 crystallized in the monomeric space group P21/n. Selected lattice values for RuII methyl compounds featuring 1-methylimidazole (N1) and pta (P1) leaving groups are listed in Table 1. The Ru–S and Ru–O distances are in the same range for both compounds. On the other hand, the bond length between the metal center and the leaving group increases in the order N1 (2.110 Å) < P1 (2.294 Å). These results are according to expectations, as the nitrogen donor in N1 features a smaller atom radius combined with a higher electronegativity than the phosphorus containing compound P1. The findings for pta compound P1 are in the same range as literature known values for [Ru(η6-p-cymene)Cl2(pta-Me)]Cl [Ru–P: 2.2753(11)].28 Furthermore, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, C–O, and π-plane distances are approximately the same, indicating that the leaving group variation has no major effects on the remaining complex bonds.
S, C–O, and π-plane distances are approximately the same, indicating that the leaving group variation has no major effects on the remaining complex bonds.
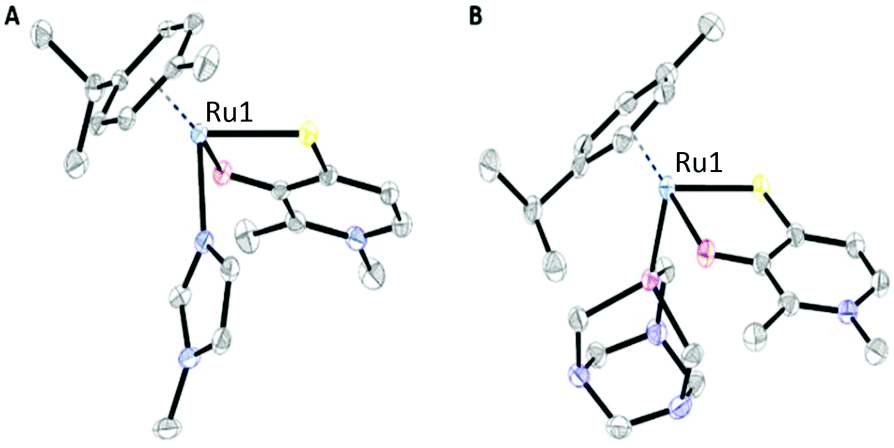 | ||
| Fig. 2 ORTEP views of complex N1 (A) bearing a 1-methylimidazole N-donor leaving group and P1 (B) featuring a pta P-donor. Hydrogen atoms and counter ions were omitted for clarity. | ||
| Compound | N1 | P1 |
|---|---|---|
| RuS [Å] | 2.3724(5) | 2.3737(11) |
| Ru–O [Å] | 2.0655(5) | 2.0895(9) |
| Ru–L [Å] | 2.1098(6) | 2.2945(9) |
| CS [Å] |
1.7346(4) | 1.7381(7) |
| C–O [Å] | 1.3177(3) | 1.3155(5) |
| π-Plane centroid distance [Å] | 1.7108(4) | 1.7259(9) |
Stability investigations
One step in elucidating the mode of action or possible drug candidates is to determine their fate under physiologically relevant conditions. These experiments help in establishing stabilities, delineating differences in aqueous behavior, and identification of the biologically active species. Consequently, the hydrolysis of all compounds was investigated by UV-vis spectrophotometry and incubation in PBS at a pH of 7.2 over 24 hours at 25 °C. RuII complexes featuring bromide (H1) and iodide (H2) leaving groups, as well as a toluene arene (H3) gave almost identical UV spectra with an absorption maximum at 413 nm (H1, H2), or 407 nm (H3) and confirmed stability over 24 hours (Fig. S49A–C†). Comparing the spectra of the 1-methylimidazole bearing group (N1–N6; Fig. S50†) it becomes apparent that RuII and RhIII complexes featuring methyl ligands (N1, N2) and benzyl ligands (N4, N5) give similar results, with absorption bands around 390–400 nm. Additionally, stability over 24 hours could be confirmed for all substances with the slight deviation of RhIII methyl complex N2, which showed decreasing absorption bands due to slow precipitation from solution over time (Fig. S50, A and B†). On the contrary, the respective IrIII 1-methylimidazole congeners revealed distinctly different absorption curves. IrIII complexes N3 featuring a methyl ligand, and N6 featuring a benzyl ligand exhibited an absorption maximum at 373 nm, followed by another maximum at 310 nm (Fig. S50, E and F†). However, RhIII compound N3 showed a slight shift to lower absorption wavelengths over time and isosbestic points at 325 and 370 nm, while the spectrum for complex N6 remained unchanged (Fig. S50, C and D†). The third group consisted of RuII and RhIII organometallics bearing a pta leaving group (P1–P4; Fig. S51†). In this group all spectra look approximately the same with a strong absorption band at 389 nm and stability over 24 hours. Finally, the UV-vis results for thiourea compounds (S1, S2; Fig. S52, A and B†) revealed hydrolysis over time. While RuII compound bearing a methyl ligand (S1) showed only slight shifts to lower wave lengths over a large window of time, its counterpart featuring a benzyl ligand (S2) was stable over 18 hours followed by a drastic drop of the absorption intensity at 400 nm. These findings indicate that S2 is stable during a certain time window, followed by quick deterioration.Cytotoxicity
Several representatives of (thio)maltol-based organometallics have already been tested for their cytotoxic potency in vitro and revealed a great range of IC50 values. While RuII and OsII piano-stool complexes bearing the bidentate O,O-maltol coordination motif were deemed completely inactive,27 the introduction of S,O-ligands brought to light a markedly higher cytotoxicity of the respective compounds. Accordingly, thiomaltol and thiopyridone piano-stool complexes showed moderate to good activity against human lung cancer A549, colon carcinoma SW480, and ovarian teratocarcinoma CH1/PA-1 cell lines.22,23 In order to gather further insights into the cytotoxic behavior of the present monomeric thiopyridone library, the 3-(3,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test was carried out with all 15 complexes in the three cell lines mentioned above. In the MTT assay the capacity of reducing a tetrazolium salt to a formazan product serves as an indicator for cell viability.29 The results are listed in Table 2 and illustrated in Fig. S53–S56.† The first group of three, featuring the dimeric RuII compounds with a benzyl ligand and either bromide (H1*) or iodide (H2*) as leaving group or toluene (H3*) as an arene, showed IC50 values in the same range as cisplatin and their related thiopyridone compounds featuring a chloride leaving group and p-cymene as an arene.23 Therefore, it can be concluded that neither the halogen nor the arene have a significant impact on the cytotoxic potency of these organometallics. The second sub-group featuring 1-methylimidazole as a leaving group (N1–N6) showed the same ligand-dependent trends that were previously reported for dimeric thiopyridone complexes.23 Thus, the RuII compound featuring a methyl ligand (N1) was least active overall, followed by the RhIII (N2) and IrIII (N3) congeners.| IC50a/μM | |||
|---|---|---|---|
| A549 | SW480 | CH1/PA-1 | |
| a IC50 concentrations were determined in three different human cancer cell lines by use of MTT assay with an exposure time of 96 h. | |||
| Me Im | >200 | >200 | >200 |
| pta | — | ~200 | 147 ± 2 |
| Thiourea | >200 | >200 | >200 |
| H1 | 1.8 ± 0.4 | 3.2 ± 0.5 | 1.2 ± 0.2 |
| H2 | 1.4 ± 0.1 | 2.9 ± 0.8 | 1.2 ± 0.1 |
| H3 | 1.4 ± 0.3 | — | 1.0 ± 0.1 |
| N1 | 45 ± 23 | 23 ± 10 | 26 ± 1 |
| N2 | 18 ± 1 | 9.8 ± 3.5 | 15.1 ± 0.4 |
| N3 | 14 ± 6 | 6.0 ± 0.7 | 14 ± 2 |
| N4 | 3.0 ± 1.2 | 3.8 ± 0.3 | 1.2 ± 0.2 |
| N5 | 1.2 ± 0.1 | 0.57 ± 0.01 | 0.53 ± 0.04 |
| N6 | 1.2 ± 0.1 | 0.33 ± 0.04 | 0.46 ± 0.02 |
| P1 | 0.60 ± 0.03 | 1.7 ± 0.4 | 0.96 ± 0.08 |
| P2 | 18 ± 3 | 4.9 ± 0.3 | 3.3 ± 0.7 |
| P3 | — | 20 ± 9 | 1.0 ± 0.1 |
| P4 | 1.3 ± 0.1 | 0.47 ± 0.06 | 0.51 ± 0.04 |
| S1 | 23 ± 1 | 22 ± 4 | 26 ± 2 |
| S2 | 2.0 ± 0.6 | 2.6 ± 0.3 | 1.3 ± 0.1 |
| Cisplatin30 | 1.3 ± 0.4 | 3.5 ± 0.3 | 0.16 ± 0.03 |
| BOLD-100 31 | 156 ± 11 | 88 ± 19 | 62 ± 9 |
When employing a benzyl ligand, IC50 values were significantly lower for all compounds, but again the RuII complex (N4) revealed higher IC50 values than its RhIII (N5) and IrIII (N6) counterparts, which were in the nanomolar range. Generally, it can be seen in 2D settings that all N-donor bearing compounds were least active in the multidrug-resistant A549 cell line, with IC50 values ranging from 1.2 μM to 45 μM. However, they exhibited high activities in partly chemoresistant SW480 and chemosensitive CH1/PA-1 cells where the lowest 50% inhibitory concentrations were observed for IrIII benzyl compound N6 with 0.33 μM and 0.46 μM, respectively. Similar results could be observed for pta complexes (P1–P4). Again, organometallics bearing methyl ligands (P1, P2) were less active than their benzyl containing counterparts (P3, P4) and RhIII complexes were more active than the structurally related RuII complexes. Also the cell line trend was the same for P- and N-donor compounds, with moderate to low activities in A549 cells and activities in the nanomolar range in SW480 and CH1/PA-1 cell lines. The thiourea sub-group only contains two RuII analogues with either a methyl (S1) or a benzyl (S2) ligand. Again, the more active complex is S2 with a benzyl thiopyridone scaffold. In summary, the obtained IC50 values from these 2D MTT assays are similar to those previously reported for their halogen leaving group congeners.23 Furthermore, the same cytotoxicity trends could be observed. Thus, 50% inhibitory concentrations increase in the order Ir < Rh < Ru, and generally benzyl complexes are more active than methyl complexes. Leaving group variation had only a marginal effect in this setting. However, the most active organometallics were Rh compound N5 featuring a benzyl ligand and a 1-methylimidazole leaving group and its Ir congener N6. Additionally, Rh complex P4 featuring a benzyl ligand and a pta leaving group showed activities in the same range. IC50 values are in the nanomolar range for these three complexes in SW480 and CH1/PA-1 cells.
Lipophilicity and cellular accumulation
In order to gather further insights into the influence of the N-, P-, and S-donor leaving groups on the pharmacokinetic properties, cellular accumulation was determined for seven representatives and compared to the values of their chlorido parental compounds (Table 3). These findings are in accordance with expectations, as higher cellular accumulation levels correlate with lower IC50 values in most cases (Fig. 3). This is nicely exemplified by compounds with a 1-methylimidazole group: while Ru methyl complex N1 had the lowest uptake level and the highest IC50 value, accumulation of its structurally related benzyl congener N4 was 8 times as high and the 50% inhibitory concentration 5.5 times lower. Accumulation of Rh benzyl complex N5 was 8.5 times as high as that of N4, associated with an IC50 value in the nanomolar range. The only exception from this trend appears in the pta series. On the one hand, higher intracellular concentrations led to a higher cytotoxic activity for Ru pta P1 featuring a methyl ligand, as well as Rh pta featuring a benzyl ligand (P4). On the other hand, higher uptake of Ru pta complex P3 featuring a benzyl ligand did not increase cytotoxicity. In a series of related structures, miLogP values allow the comparison of relative lipophilicity.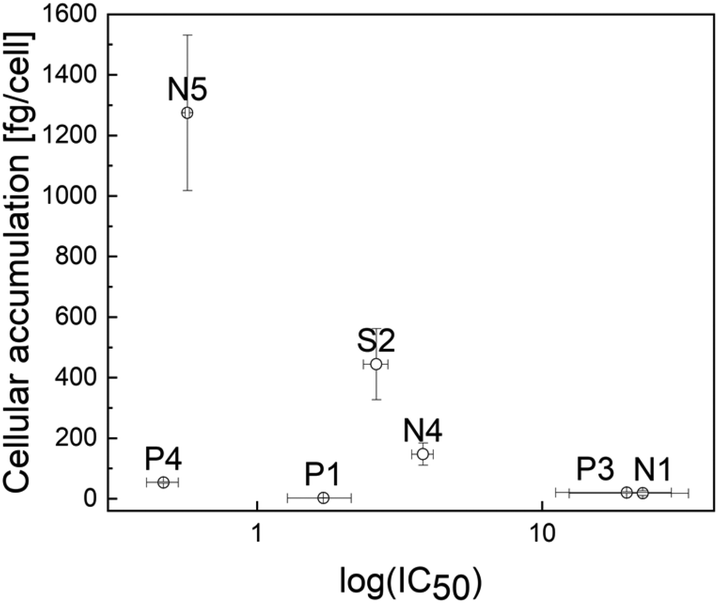 | ||
| Fig. 3 Scatter plot of cellular accumulation versus log(IC50) of compounds N1, N4, N5, P1, P3, P4, and S2. | ||
| Central atom | Cellular accumulationa [fg Ru or Rh per cell] | miLogP (leaving group) | IC50 [μM] | |
|---|---|---|---|---|
| a Cellular accumulation in SW480 cells, determined by inductively coupled plasma mass spectrometry (ICP-MS); exposure time 2 h; c = 50 μM. Abbreviations refer to the respective chlorido parent complexes (e.g. Ru–Me features the same scaffold as N1, and P1, but is equipped with a chlorido leaving group). | ||||
| N1 | RuII | 18 ± 8 | −0.01 | 23 ± 10 |
| N4 | RuII | 147 ± 36 | −0.01 | 3.8 ± 0.3 |
| N5 | RhIII | 1275 ± 257 | −0.01 | 0.57 ± 0.01 |
| P1 | RuII | 2.4 ± 0.8 | −0.67 | 1.7 ± 0.4 |
| P3 | RuII | 20 ± 2 | −0.67 | 20 ± 9 |
| P4 | RhIII | 54 ± 2 | −0.67 | 0.47 ± 0.06 |
| S2 | RuII | 444 ± 118 | −0.46 | 2.6 ± 0.3 |
| Ru–Me 23 | RuII | 21 ± 5 | — | 55 ± 18 |
| Ru–Bz 23 | RuII | 147 ± 54 | — | 3.1 ± 0.2 |
| Rh–Bz 23 | RhIII | 855 ± 66 | — | 0.28 ± 0.02 |
Recently, lipophilicity indices of the parental chlorido compounds have been determined. These experiments showed that compounds bearing a methyl ligand are less lipophilic than their respective benzyl congeners, which was in good accordance with the reported IC50 values and uptake studies.23 In this series of organometallics the molecule scaffold remains the same and is thus assumed to contribute a constant lipophilicity value.
Therefore, changes in lipophilicity can be attributed to the respective leaving group. The predicted miLogP values are in accordance with expectation, as the most lipophilic leaving group is 1-methylimidazole, followed by thiourea and, finally, pta. These values further support cellular accumulation levels, which in the case of the Ru methyl complex bearing a 1-methylimidazole leaving group (N1) was 7 times higher than that of the pta counterpart (P1). A comparable increase in accumulation was observed for Ru benzyl MeIm complex (N4) relative to the pta analogue (P3). In the case of the Rh analogues, the difference was even more pronounced, as the cellular concentration of benzyl 1-methylimidazole organometallic (N5) was 24 times as high as that of the pta compound (P4). Comparing these values to those previously reported for the chlorido parent compounds reveals that leaving group variation has a noticeable impact on this parameter. In this series of seven, both RhIII compounds showed the highest accumulation and cytotoxicity (in the nanomolar range) in SW480 cells. In summary, leaving group variation has a noticeable impact on properties such as lipophilicity, cellular accumulation, reflecting a good correlation between these parameters. While the least lipophilic leaving group was associated with lower cellular accumulation, higher intracellular metal levels could be attributed to a more lipophilic character of the respective organometallic compound.
Cytotoxicity in spheroid tumor models
The cytotoxic behavior of the aforementioned seven compounds was also tested in multicellular spheroids of four different cancer cell lines (A549, CH1/PA-1, HCT-116, as well as HT29). It is possible to get further insights into the cytotoxic behavior of drug candidates, as spheroids better recapitulate properties of solid tumors than 2D monolayer cultures do.32 Therefore, multicellular spheroids were grown and exposed to the complexes for 96 h (Fig. 4 and Fig. S57†). In nearly all cases, cytotoxic activity was noticeably reduced in these 3D cultures (Table 4), which has been reported for structurally related compounds, too (IC50 values up to 183 times higher).33,34 Overall, the most active compound in the sensitive model CH1/PA-1 was Ru methyl complex P1, followed by its benzyl counterpart P3, and finally P4 featuring a RhIII center. It is noteworthy that the 50% inhibitory concentration for P1 was unaltered in A549 cells, and only slightly elevated in CH1/PA-1 cells compared to the 2D results (Table 2). Generally, the majority of complexes were most active in the chemosensitive CH1/PA-1 cell line (Fig. S57†). However, due to the variable patterns, it is not possible to identify one complex with the highest activity in spheroids of all four cell lines based on the obtained data. Further 3D in vitro studies focusing on specific tumor types are required to address the question whether the cytotoxic profile would point to a specific indication.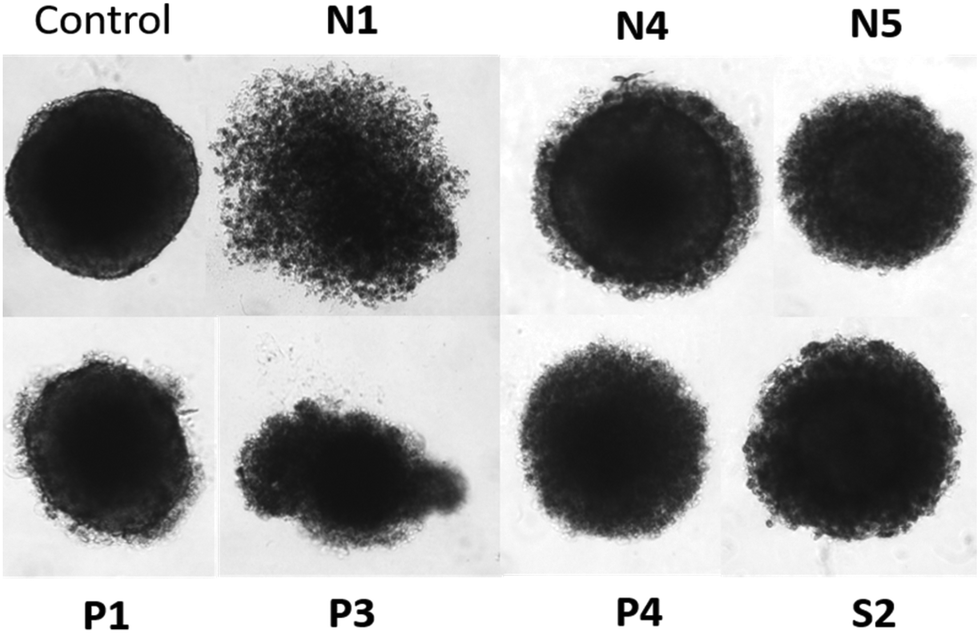 | ||
| Fig. 4 Comparison of untreated HCT-116 spheroids with cells treated at the IC50 concentration of the respective complexes for 96 h. | ||
| IC50/μM | ||||
|---|---|---|---|---|
| A549 | CH1/PA-1 | HCT-116 | HT29 | |
| N1 | >400 | 253 ± 16 | 283 ± 7 | 235 ± 59 |
| N4 | 130 ± 6 | 146 ± 5 | 108 ± 10 | 236 ± 14 |
| N5 | 128 ± 5 | 65 ± 3 | 56 ± 10 | 74 ± 2 |
| P1 | 0.44 ± 0.10 | 1.4 ± 0.6 | 318 ± 8 | >400 |
| P3 | 153 ± 23 | 39 ± 1 | 263 ± 43 | >400 |
| P4 | 245 ± 29 | 71 ± 1 | 17 ± 10 | 175 ± 7 |
| S2 | 139 ± 2 | 117 ± 4 | 242 ± 29 | 293 ± 28 |
Apoptosis induction in a flow cytometry assay
The investigation of cellular apoptosis has gathered interest since the introduction of potent flow cytometry assays.35 In these assays, differences in plasma membrane integrity and permeability are exploited – by combining propidium iodide (PI) and fluorochrome-conjugated annexin-V staining.36–38 The penetration of the PI dye into the nuclei is dependent on the permeabilization of the cell membrane. Viable and early apoptotic cells have intact cell membranes, leading to no PI signal.39 On the other hand, during late apoptotic or necrotic stages, the integrity of their membranes decreases, thus allowing PI to enter the cells and intercalate into DNA, resulting in increased fluorescence signal.36,37,40–42 In this work, the induction of apoptotic cell death by complexes N1, N4, N5, P1, P3, P4 and S2 was investigated via annexin V-FITC and PI double staining of HTC-116 cells and flow cytometric analysis (Fig. 5 and Fig. S58, S59†). All assays were carried out with the IC50 concentrations obtained in Alamar Blue 3D assays and treatment for 96 h. Compound N1 showed the strongest induction of apoptosis with 70% apoptosis after treatment for 48 h. Cell death in HCT-116 spheroids was significantly induced by compounds N1, P4, S2, P3, with the strongest effect observed in N1 treated cells. However, compared to the untreated control no significant induction of apoptosis could be observed for compounds N4, N5, and P1. After treatment with compound N1 most cells were found to be in a late apoptotic state, whereas comparably small percentages were early apoptotic or necrotic (Fig. S58 and S59†). The same pattern was established for complexes P3, P4, and S2 to a lower extent. Necrosis was not induced to a high percentage by the complexes of this work, with the highest necrotic potential of 10% for P4.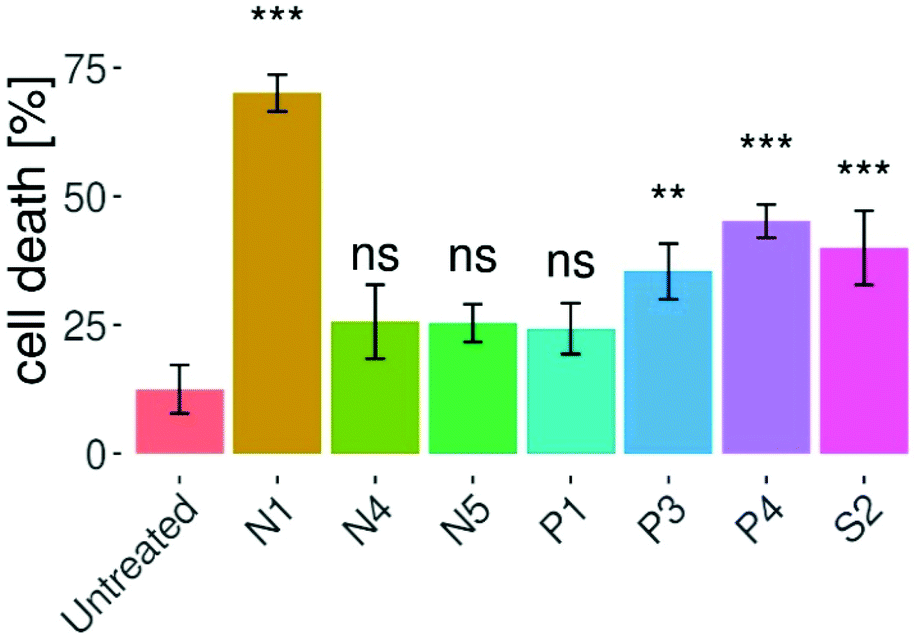 | ||
| Fig. 5 Induction of apoptosis in HCT-116 spheroids after 96 h of treatment with the indicated complexes at the respective 50% inhibitory concentration presented as means ± SDs. Compounds N1, P3, P4, and S2 induced elevated apoptosis compared to the untreated control, indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant). | ||
Apoptosis induction in a confocal microscopy assay
Furthermore, in order to support and better visualize the findings obtained by the flow cytometry assay, treated HCT-116 spheroids were stained with annexin-V and PI and live cell images were obtained by confocal microscopy. The images obtained from a stack of optical sections from the spheroids depict green dots that correspond to annexin V-FITC positive cells (early apoptotic), red dots to PI positive cells (necrotic), and yellow dots to annexin V-FITC/PI double positive (late apoptotic) cells. These findings support the flow cytometry data, where compounds N1 and S2 induced the strongest fluorescence signals (corresponding to a late apoptotic or necrotic stage; Fig. 6 and Fig. S60†). The combination of the Alamar Blue and apoptosis assays (flow cytometry and live cell imaging) highlighted an important aspect in the activity of metal-based drugs. Here, we focused on the ability of the compounds to induce apoptotic cell death according to their respective IC50 values. Even though compound N1 featured a high IC50, it induced the highest apoptosis levels, while other compounds such as N4 and N5 despite having lower IC50 values, were not able to induce significant levels of apoptotic cell death compared to the untreated controls. These results suggest that these compounds might be more cytostatic rather than cytocidal.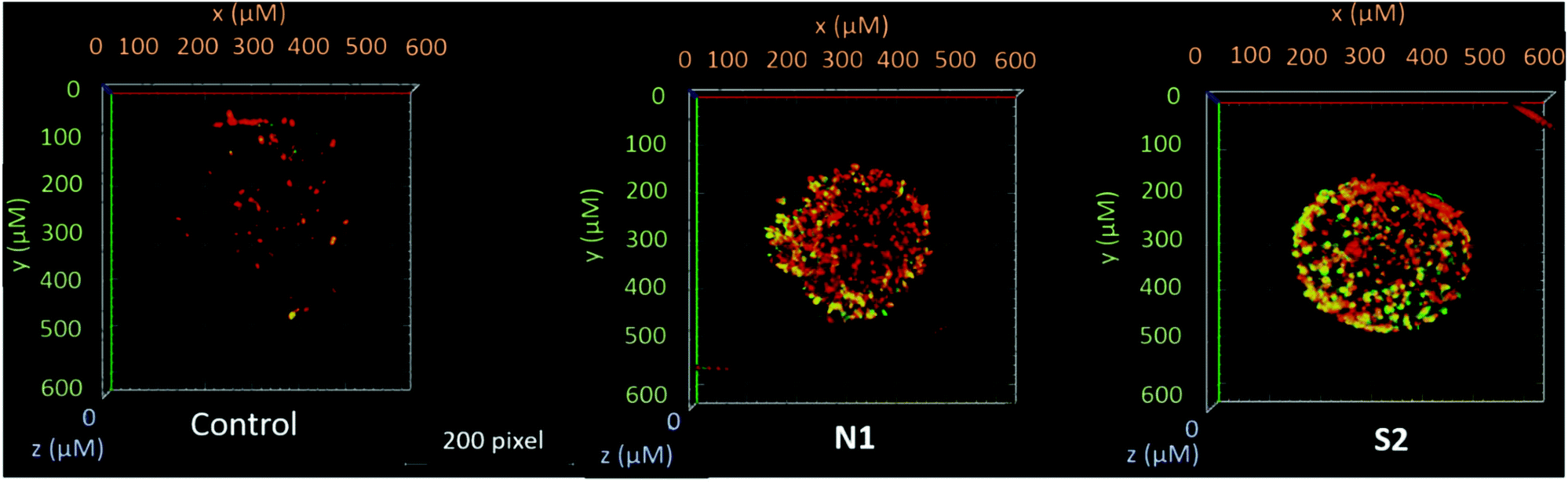 | ||
| Fig. 6 Representative 3D reconstructions of HCT-116 spheroids, untreated or treated with complexes N1 and S2 at their respective 50% inhibitory concentrations after 48 h. Confocal microscope images were obtained from a stack of optical sections from the spheroids. | ||
ROS generation
Oxidative stress can be linked to a set of commonly known diseases such as cancer, Alzheimer's and Parkinson's.43 Under normal conditions, the ratio between reducing and oxidizing agents in cells is tightly balanced in order to maintain the well-functioning cellular machinery. Imbalances between these two groups of oxidants (e.g. reactive oxygen and nitrogen species; ROS/RNS) and anti-oxidants may cause modifications in gene expression, signaling pathways, as well as biomolecules (e.g. DNA, lipids or proteins).44 ROS levels are noticeably elevated in many cancer cells compared to normal cells, which is explained by their augmented energy supply leading to an increased metabolic burden on the electron transport chain.45 Despite the well-known fact that ROS can promote cancer cell survival and even increase cancer cell proliferation, but also DNA damage, it is believed that this imbalance can be exploited in chemotherapy.46 In this way, the cancer cell's buffer capacity may be pushed over a critical limit and ultimately the cell could be destroyed.45 In previous studies, piano-stool complexes featuring a thiomaltol ligand and 1-methylimidazole leaving group only induced slightly increased ROS levels in cancer cells.22 Therefore, seven representatives of this series have been evaluated regarding their capacity of ROS generation (Fig. 7). A flow cytometry CellROX®-based assay was carried out at the IC50 concentration of the respective drug, where HCT-116 cells were treated for 24 h and stained with CellROX® and PI, followed by flow cytometry analysis. Compared to the control, only compounds P1, P3 and S2 induced a significant increase in ROS levels with a maximum of 20% for P1. Contrary, complexes N1, N4, N5, and P4 did not induce significant changes in ROS concentrations compared to the untreated cells.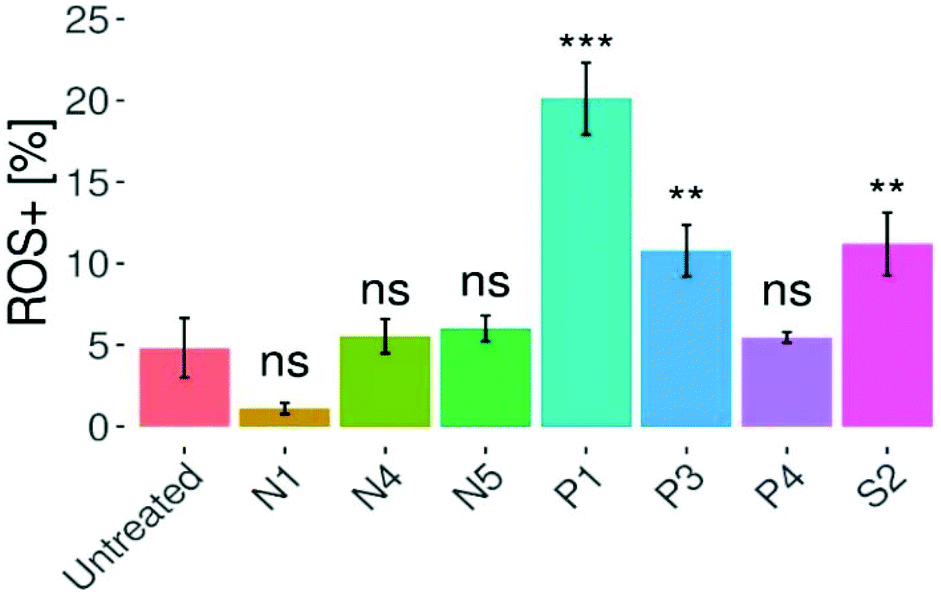 | ||
| Fig. 7 Levels of ROS in HTC-116 cells. The graph shows that compounds P1, P3, and S2 elevate intracellular ROS levels after 24 h of treatment. Columns show mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant). | ||
Clonogenic assay
We employed the colony formation assay in order to assess cell survival and proliferation based on the ability of one cell to grow into a colony.47 Amongst others, Sadler et al.48 as well as Samuelson et al.4 have designed organometallic RuII complexes with the ability to significantly inhibit colony formation. In this experimental setting three different human cancer cell lines (A549, SW480, CH1/PA-1; Fig. 8 and Fig. S61–63†) have been treated with N1, N4, N5, P1, P3, P4, and S2 at the respective IC50 concentration for seven days. Overall, colony formation in A549 cells was markedly inhibited by all complexes except for S2. In CH1/PA-1 cells colony formation was inhibited by P3, and N1. Finally, in SW480 cells P3 showed the highest inhibitory potency. Treatment of these cells with all other compounds lead to a decrease in colonies (except for RhIII complex N5). Compound P3 inhibited colony formation in all three cell lines, which suggests that this compound is able to reduce survival and proliferation under prolonged treatment regimens.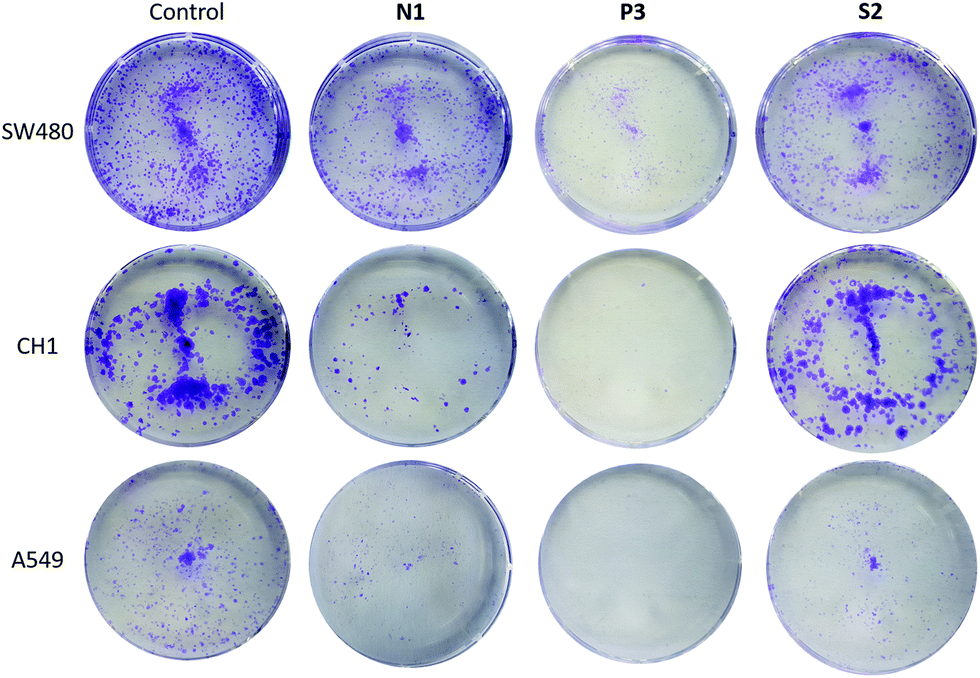 | ||
| Fig. 8 Selected cell colony images of SW480, CH1, and A549 cells. The untreated control group, and cells treated with N1, P3, and S2 for 7 days (from left to right). | ||
DNA plasmid assay
It is known that DNA can adopt different conformations in solution, with their mobility depending on the topology of the probe.49,50 Interaction of test compounds with a supercoiled dsDNA plasmid (e.g. pUC19) may alter its migratory properties in gel electrophoresis in a variety of ways: while in untreated controls most DNA plasmids are present in their supercoiled (SC) form, induction of single- or double-strand breaks leads to the instant adoption of an open-circular (OC) or linear form, respectively; whereas untwisting or bending of the plasmid by adduct formation or intercalation results in gradual convergence of the SC and OC forms.51–53 However, the present piano-stool complexes showed little to no DNA interaction (Fig. S64–S79;† agarose gel pictures Fig. S70†), apart from pta complexes P1 and P3 generating OC levels >10% after 6 h of incubation (Fig. 9 and Fig. S67†). On the other hand, no noticeable interaction could be observed for 1-methylimidazole derivatives N4 and N5. Overall, it can be assumed that DNA is not the primary target of these complexes, which is in accordance with previously published results for various organometallic complexes.15,20,54–58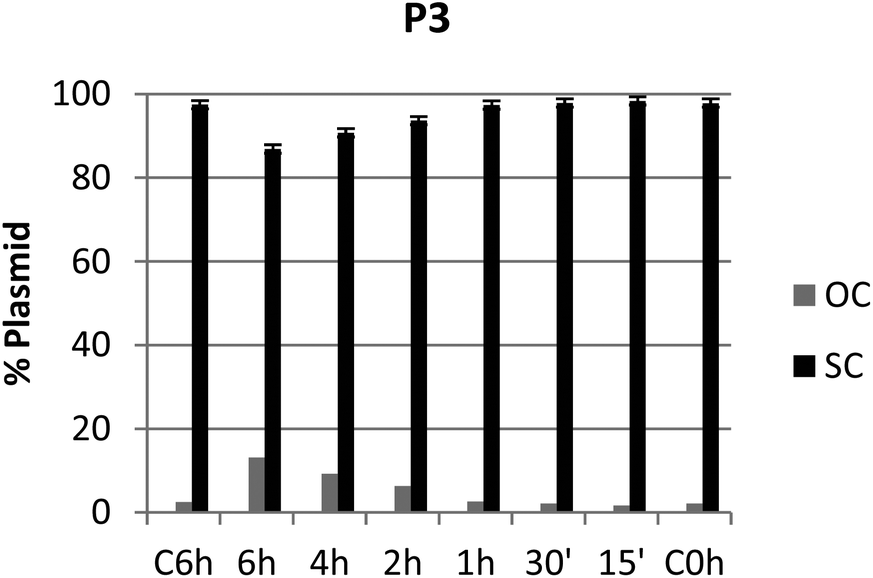 | ||
| Fig. 9 Graph showing the ratio between OC and SC DNA plasmid after 15 min, 30 min, 1 h, 2 h, 4 h, and 6 h of treatment with compound P3 at the respective IC50 inhibitory concentration. | ||
Laser ablation-ICP MS studies of 3D tumor models
Another important and useful tool in the determination of compound distribution within tumors and small organisms is laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS).59–61 In this work, LA-ICP-MS was used to determine the ruthenium and rhodium distribution in colon cancer HCT-116 multicellular tumor spheroids after treatment with the respective compounds (Fig. 10 and Fig. S71†). Tumor spheroids were treated with compounds N1, N4, N5, P1, P3, or S2 at the respective IC50 concentration for 96 h. For compounds N1, N4, N5 and S2 (N4 in Fig. 9 and N1, N5 in Fig. S71†) highest ruthenium or rhodium accumulation was observed in the proliferating cells at the outer rim of the spheroid sections. Significantly lower metal levels were detected in the middle part of the spheroids, which correspond to quiescent cells. This is in accordance with previous LA-ICP-MS studies on platinum compounds in tumor spheroids where highest platinum levels corresponded to proliferating cells, whereas quiescent cells displayed low levels of platinum.59,62 For compounds N1, P1 and P3 pronounced accumulation was observed in the spheroid's inner area corresponding to the necrotic core. Additionally, it is possible to compare these findings to our previously reported LA-ICP-MS results of the accumulation of thiopyridone compounds in tumor spheroids.23 The chlorido congener of complexes N1 and P1 showed higher accumulation in the necrotic core.23 However, the chlorido counterpart to compounds N4, N5, P3 and S2 only penetrated the outer layers of the spheroids.23 In the case of RuII methyl compound N1 introduction of the 1-methylimidazole leaving group decreased the complexes’ ability to migrate into the cell core compared to the chlorido parent molecule (Fig. S71A†).23 However, introduction of a pta leaving group increased ruthenium levels in the spheroid's center for both the methyl P1 and benzyl P3 congener. This highlights an interesting trend where organometallics featuring a pta leaving group (P1, P3) are able to penetrate the tumor core, whereas 1-methylimidazole organometallics remain in the spheroid's outer rims. These findings again highlight the role of the leaving group's influence on the intracellular fate of the herein presented complexes.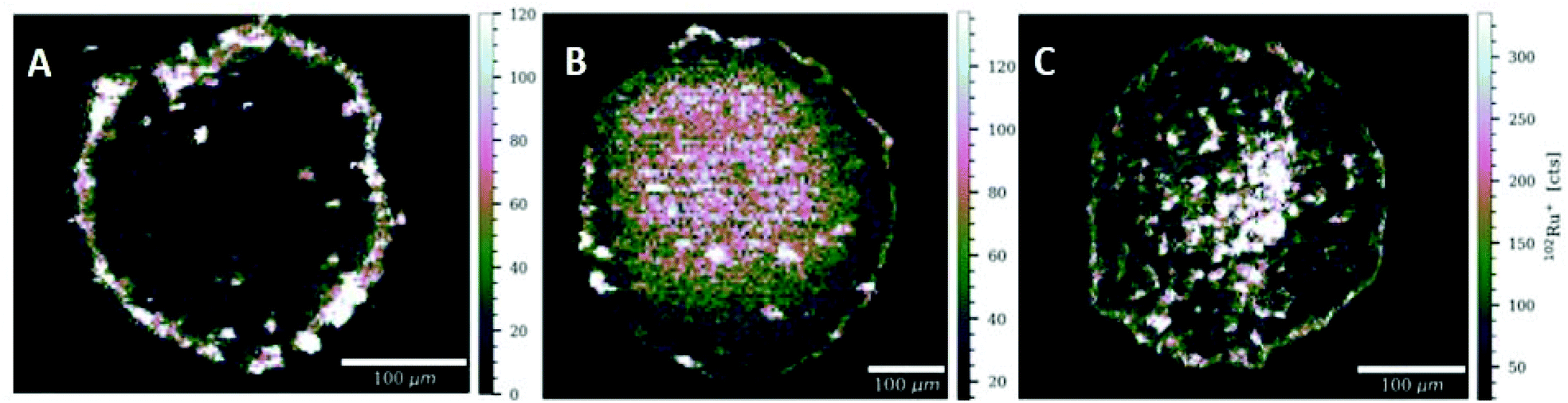 | ||
| Fig. 10 Signal intensity maps of 102Ru+ obtained by LA-ICP-MS analysis of HCT-116 tumor spheroids after treatment with N4 (A), P1 (B), or P3 (C). High resolution LA-ICPMS images were obtained with a pixel size of 5 μm; scale bar 100 μm. | ||
Conclusions
Metal complexes remain at the center of cancer therapy and research into possible future drugs with improved selectivity and efficacy as compared to established PtII drugs is continuing. In the scope of this paper, 15 piano-stool complexes have been synthesized with a special focus on leaving group variation (e.g. 1-methylimidazole, pta, thiourea). Additionally, the small library of this work has been characterized by use of standard techniques and thoroughly investigated via diverse biological assays. The results brought to light that it is not possible to single out one optimal leaving group, but rather representatives of each group had special advantages over the others. These trends were dependent upon various factors, such as the chosen molecule scaffold (e.g. metal center, ligand, leaving group), as well as the treated cell line. Generally, the complexes were mildly to highly cytotoxic, induced apoptosis, partly generated ROS, and inhibited colony formation of cancer cells. However, plasmid assays revealed little to no DNA interactions, which is according to expectations. In detail, complexes P4, N5, and N6 had 50% inhibitory concentrations in the nanomolar range in SW480 and CH1/PA-1 cells. Cellular accumulation studies suggest a connection between complex lipophilicity and permeation into cells, as more lipophilic compounds generally showed elevated cellular accumulation. Cytotoxicity assays in multicellular tumor spheroids confirmed trends from experiments in monolayer cell cultures; however, IC50 values were considerably higher. A notable exception was RuII methyl pta complex P1, which revealed an outstandingly high activity in the nanomolar range in A549 and very low micromolar range in CH1/PA-1 spheroids. Flow-cytometric apoptosis assays showed that compounds N1, P4, S2 and P3 induced apoptosis, which was further supported by confocal microscopy. Furthermore, compounds P1, P3 and S2 generated elevated ROS levels in HCT-116 cells. Colony formation of A549 cells was inhibited by all investigated complexes, while only compound P3 had inhibitory activity in all three tested cell lines (A549, SW480, CH1/PA-1) in the clonogenic assay. Finally, plasmid assays showed little to no overall interaction with cellular DNA. However, highest interaction was observed for RuII benzyl pta complex P3.These results make it impossible to identify one lead drug candidate, as all compounds showed a different activity profile. Thus, this library highlights once more the importance of each part of a piano-stool complex including the leaving group, as well as the thorough investigation of each drug candidate. In order to determine if one candidate's unique set of properties is properly suited for the complex biological environment of a mammalian organism, in vivo tests should be carried out.
Experimental part
Materials and methods
All dimeric metal precursors [Ru(p-cym)Cl2]2,63 [Ru(p-cym)Br2]2,64 [Ru(p-cym)I2]2,64 [Ru(toluene)Cl2]2,65 [Rh(Cp*)Cl2]2,66 and [Ir(Cp*)Cl2]2,66 monomeric MeIm precursors [Ru(p-cym)(MeIm)2Cl]Cl,26 [Rh(Cp*)(MeIm)2Cl]Cl,26 [Ir(Cp*)(MeIm)2Cl]Cl,26 and ligands 2a,67 and 2b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 were prepared according to literature. The solvents used were purchased from commercial sources and dried before use if needed. MeOH (HPLC grade, Fisher), DCM (HPLC grade, Fisher), and silica gel (mesh 40–63 μM) were used for column chromatography. 3-Hydroxy-2-methyl-4H-pyran-4-one (≥99.0%, Sigma-Aldrich), Lawesson's reagent (99%, Acros-Fisher), hydrochloric acid (37%, Acros-Fisher), MeOH (HPLC grade, Fisher), n-hexane (Reag. Ph. Eur. ACS), DCM (stab. with 0.2% EtOH; VWR), toluene (HPLC grade, VWR), α-terpinene (90%, Alfa Aesar), 1,2,3,4,5-pentamethylcyclopentadiene (>93%, TCI Europe), iridium(III) chloride (Johnson Matthey), ruthenium(III) chloride·H2O (Johnson Matthey), rhodium(III) chloride·H2O (Johnson Matthey), sodium methoxide (ca. 95%, Fluka), benzylamine (99%, Sigma-Aldrich), methyl amine (40% in water; Riedel-de-Haen), AgPF6 (>98%, TCI Europe), KBr (99%, Fluka), KI (99%, Alfa Aesar), 1,3,5-triaza-7-phosphaadamantane (97%, Alfa Aesar), thiourea (99%, TCI Europe), 1-methylimidazole (99%, Sigma-Aldrich) were purchased and used as received. HCl and HNO3 were purchased from Sigma–Aldrich in puriss quality. Microwave reactions were carried out on a Biotage® Initiator + system. 1H-, 13C- and 2D-NMR spectra were recorded on a Bruker Avance III™ 500 MHz FT-NMR or Bruker Avance III 600 MHz spectrometers at 500.21/600.25 MHz (1H) or 125.75/150.95 MHz (13C) at 298 K from solutions in deuterated dimethyl sulfoxide (d6-DMSO), methanol (d4-MeOD), chloroform (CDCl3) or water (D2O). CHNS elemental analyses were carried out on a Eurovector EA3000 elemental analyzer in the Microanalytical Laboratory of the University of Vienna. High resolution electronspray ionization mass spectra were recorded on a Bruker Maxis UHR qTOF Mass Spectrometer at the Core Facility for Mass Spectrometry of the University of Vienna (Faculty of Chemistry).
23 were prepared according to literature. The solvents used were purchased from commercial sources and dried before use if needed. MeOH (HPLC grade, Fisher), DCM (HPLC grade, Fisher), and silica gel (mesh 40–63 μM) were used for column chromatography. 3-Hydroxy-2-methyl-4H-pyran-4-one (≥99.0%, Sigma-Aldrich), Lawesson's reagent (99%, Acros-Fisher), hydrochloric acid (37%, Acros-Fisher), MeOH (HPLC grade, Fisher), n-hexane (Reag. Ph. Eur. ACS), DCM (stab. with 0.2% EtOH; VWR), toluene (HPLC grade, VWR), α-terpinene (90%, Alfa Aesar), 1,2,3,4,5-pentamethylcyclopentadiene (>93%, TCI Europe), iridium(III) chloride (Johnson Matthey), ruthenium(III) chloride·H2O (Johnson Matthey), rhodium(III) chloride·H2O (Johnson Matthey), sodium methoxide (ca. 95%, Fluka), benzylamine (99%, Sigma-Aldrich), methyl amine (40% in water; Riedel-de-Haen), AgPF6 (>98%, TCI Europe), KBr (99%, Fluka), KI (99%, Alfa Aesar), 1,3,5-triaza-7-phosphaadamantane (97%, Alfa Aesar), thiourea (99%, TCI Europe), 1-methylimidazole (99%, Sigma-Aldrich) were purchased and used as received. HCl and HNO3 were purchased from Sigma–Aldrich in puriss quality. Microwave reactions were carried out on a Biotage® Initiator + system. 1H-, 13C- and 2D-NMR spectra were recorded on a Bruker Avance III™ 500 MHz FT-NMR or Bruker Avance III 600 MHz spectrometers at 500.21/600.25 MHz (1H) or 125.75/150.95 MHz (13C) at 298 K from solutions in deuterated dimethyl sulfoxide (d6-DMSO), methanol (d4-MeOD), chloroform (CDCl3) or water (D2O). CHNS elemental analyses were carried out on a Eurovector EA3000 elemental analyzer in the Microanalytical Laboratory of the University of Vienna. High resolution electronspray ionization mass spectra were recorded on a Bruker Maxis UHR qTOF Mass Spectrometer at the Core Facility for Mass Spectrometry of the University of Vienna (Faculty of Chemistry).
UV/Vis spectra were recorded using a Hewlett Packard 8452A diode-array spectrophotometer between 200–800 nm with PTP (Peltier Temperature Programmer) and Julabo AWC 100 recirculating cooler. The path length (l) was either 1 or 2 cm.
Single-crystal X-ray diffraction analysis
The X-ray intensity data were measured on a Bruker D8 Venture diffractometer equipped with multilayer monochromator, Mo and Cu K/α INCOATEC micro focus sealed tube and Oxford cooling system. The structures were solved by Direct Methods, Patterson Methods and Intrinsic Phasing. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were inserted at calculated positions and refined with riding model. The following software was used: Bruker SAINT software package68 using a narrow-frame algorithm for frame integration, SADABS69 for absorption correction, OLEX270 for structure solution, refinement, molecular diagrams and graphical user-interface, Shelxle71 for refinement and graphical user-interface SHELXS-201572 for structure solution, SHELXL-201573 for refinement, Platon74 for symmetry check. Experimental data and CCDC-codes experimental data (available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html) can be found in Table S1.† Crystal data, data collection parameters, and structure refinement details are given in Tables S1–25.† Asymmetric unit visualized in Fig. 2 and Fig. S37–48.†Cell culture
CH1/PA-1 (ovarian teratocarcinoma) cells were a gift from L. R. Kelland (CRC Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, UK). A549 (non-small cell lung cancer), SW480, HCT-116 and HT29 (all colorectal carcinoma) cells were kindly provided by the Institute of Cancer Research, Dept. of Medicine I, Medical University of Vienna, Austria. Monolayer cultures were grown in Eagle's minimal essential medium (MEM) supplemented with L-glutamine (4 mM), sodium pyruvate (1 mM) and 1% v/v non-essential amino acid solution (all from Sigma-Aldrich) and 10% v/v heat-inactivated fetal calf serum (FCS, from BioWest) in 75 cm2 flasks (Starlab) at 37 °C under a humidified atmosphere containing 5% CO2 in air.MTT assay
Antiproliferative activity of the compounds was determined with the colorimetric MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay. 1 × 103 CH1/PA-1, 2 × 103 SW480 and 3 × 103 A549 cells were seeded in 100 μL per well into 96-well microculture plates (Starlab). After 24 h, test compounds were dissolved in DMSO (Fisher Scientific), serially diluted in complete MEM not to exceed a final DMSO content of 0.5% v/v and added in 100 μL per well. After 96 h, the drug-containing medium was replaced with 100 μL of RPMI 1640/MTT mixture [6 parts of RPMI 1640 medium (Sigma-Aldrich; supplemented with 10% heat-inactivated fetal bovine serum and 4 mM L-glutamine), 1 part of MTT in phosphate-buffered saline (5 mg mL−1; both from Sigma-Aldrich)]. After incubation for 4 h, the MTT-containing medium was replaced with 150 μL DMSO per well to dissolve the formazan product formed by viable cells. Optical densities at 550 nm (and a reference wavelength of 690 nm) were measured with a microplate reader (ELx808, Bio-Tek). The 50% inhibitory concentrations (IC50) relative to untreated controls were interpolated from concentration-effect curves. At least three independent experiments were performed, with triplicates per concentration level each.Cellular accumulation/ICP-MS
1.8 × 105 SW480 cells per well were seeded into 12-well plates in aliquots of 1 mL supplemented MEM (see above) and incubated at 37 °C for 24 h. Then, cells were exposed for 2 h at 37 °C to 50 μM solutions of the test compounds (containing ≤0.5% DMSO) in fresh 0.5 mL of complete MEM per well upon exchange of the medium. Afterwards, cells were washed three times with 1 mL PBS and lysed with 0.4 mL subboiled HNO3 per well for 1 h at room temperature. Aliquots of 300 μL lysate were diluted with Milli-Q water to a total volume of 8 mL each. Ruthenium and rhodium contents were quantified by inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent 7800 ICP-MS instrument, equipped with an SPS 4 autosampler and a MicroMist nebulizer, at a sample uptake rate of approx. 0.2 mL min−1. Ruthenium, rhodium and indium standards were obtained from CPI International. The instrument was equipped with nickel cones and operated at an RF power of 1550 W, with argon as the plasma gas (15 L min−1), nebulizer gas (1.06 L min−1) and auxiliary gas (0.9 L min−1). The Agilent MassHunter software package (Workstation Software, Version C.01.04, 2018) was used for data processing. Robustness was ensured by tuning the ICP-MS on a daily basis, by using an internal standard (115In) and measuring calibration standards with each measurement. Data from adsorption/desorption blanks were subtracted from the corresponding sample data, and metal content is given relative to the cell number. Results are based on at least three independent experiments, each performed in triplicates.Spheroid formation
For spheroid generation, HCT-116, HT-29, CH1/PA-1 and A549 cells were harvested from culture flasks by trypsinization, resuspended in their respective supplemented medium and seeded in ultra-low attachment round-bottom 96-well plates (Corning) at a density of 500 viable cells per well. Plates were incubated at 37 °C with 5% CO2 for 96 h to allow spheroid formation and then used for the experiments.Cytotoxicity test in spheroids
Test compounds were first dissolved in DMSO, and stock solutions were prepared in the appropriate medium according to the cell line and diluted stepwise to obtain a serial dilution. 100 μL of the respective dilutions were added to each well, and the plates were incubated for 96 h at 37 °C with 5% CO2. 16 h before the end of the incubation time, a 440 μM resazurin sodium salt (Sigma-Aldrich, Austria) in PBS solution was prepared and 20 μL were added to each well. Fluorescence was measured with a Synergy HT reader (BioTek).Clonogenic assay
SW480, CH1/PA-1 and A549 cells were harvested from culture flasks by trypsinization, resuspended in their respective supplemented medium and seeded in 6-well plates at a density of 3000 viable cells per well. Plates were incubated at 37 °C with 5% CO2 for 24 h to allow proper cell attachment to the wells. Cells were treated and incubated with test compounds for 7 days. Plates were washed with PBS and fixed with methanol for 30 min at 4 °C. Cells were washed twice with PBS and stained with a crystal violet solution (Sigma-Aldrich) for 5 min. Plates were washed with tap water to remove excess dye and air dried, and the extent of colony formation was compared with untreated controls.
Spheroid growth
HCT-116, HCT-15 and HT-29 spheroids were treated with test compounds (at their respective IC50s according to the cytotoxicity test) at 37 °C with 5% CO2 for 96 h. Pictures were taken before treatment and after treatment to compare growth among the different treatment groups, by using an Olympus CKX41 microscope and Cell^F.Flow cytometry analysis – apoptosis
After 48 h of treatment with the test compounds, spheroids were collected and pooled. Samples were washed with PBS followed by dissociation with Tryple Express (Gibco, Austria) for 15 min at 37 °C. 500 μL medium containing 10% FCS were added to each tube to stop trypsinization. Samples were centrifuged at 2200g for 3 min and were incubated with anti-annexin-V FITC conjugated antibody (eBioscience) for 15 min followed by nuclei staining using propidium iodide (Sigma-Aldrich) in annexin-V binding buffer solution. Samples were analyzed immediately by using a Guava EasyCyte flow cytometer (Merck/Millipore). The results were analyzed using the FlowJo software. For the confocal imaging approach, spheroids were treated for 48 h with the test compounds and labelled with Annexin-V-FITC Apoptosis Staining/Detection Kit (ab14085 Abcam) according to manufacturer's instructions. Spheroids were analyzed with a confocal microscope (Zeiss LSM 780). Images were processed with the software Zen (Zeiss).Detection of reactive oxygen species (ROS)
After treatment, spheroids were collected and pooled. Samples were washed with PBS followed by dissociation with Tryple Express (Gibco, Austria) for 15 min at 37 °C. 500 μL medium containing 10% FCS were added to each tube to stop trypsinization. Samples were centrifuged at 2200g for 3 min and were incubated with Cellrox® (C10492 Molecular Probes) dye according to manufacturer's instructions. Samples were analyzed immediately by a Guava EasyCyte flow cytometer (Merck/Millipore). The results were analyzed using the FlowJo software.Electrophoretic dsDNA plasmid assay
For the cell-free plasmid DNA interaction assay, 400 ng of the bacterial-derived pUC19 plasmid were incubated for different periods of time (from 15 min to 6 h) with 50 μM of the test compound at 37 °C under continuous shaking. After 6 h, interaction was stopped by cooling samples on ice, 20 μL of the probes were mixed with 4 μL 6× DNA loading dye (Thermo Fisher Scientific), and the obtained solutions were loaded on a 1% agarose gel (in 1× TBE). Electrophoresis was accomplished in 1× TBE-buffer: infiltration of the gel was achieved at 60 V for 5 min and separation of the distinct DNA conformations at 120 V for about 90 min. Then, the gel was stained for 20 min with ethidium bromide in 1× TBE (0.75 μg mL−1) under continuous shaking. Images of the stained gel were taken with a GelDoc-It Imaging System Fusion Fx7 (Vilber Lourmat), and quantitative evaluation was performed with ImageJ software.LA-ICP-TOFMS analysis
Spheroids were collected, pooled and embedded in TissueTek (Sakura). Samples were cut into sections of 5 μm thickness using a Cryostat (Leica) and placed onto Superfrost slides. An Analyte Excite Excimer 193 nm laser ablation system (Teledyne Photon Machines, Bozeman, MT, USA) was coupled to an icpTOF 2R (TOFWERK AG, Thun, Switzerland) TOF-based ICP-MS instrument. The laser ablation system is equipped with a prototype COBALT ablation cell and the aerosol rapid introduction system (ARIS). Through the low-dispersion mixing bulb of the ARIS an Ar make-up gas flow (∼1 L min−1) is introduced into the optimized He carrier gas flow (0.50 L min−1) before entering the plasma. Laser ablation sampling was performed in fixed dosage mode 2, at a repetition rate of 100 Hz and using a square spot size with 10 μm in diameter. The line scans overlapped one another by 5 μm and the used laser ablation parameters resulted in a pixel size of 5 μm. Tumor spheroids were removed quantitatively using a fluence of 1.0 J cm−2. The standard operation mode was used for ICP-TOFMS measurements, which allows the analysis of ions from m/z = 14–256. The integration and read-out rate match the laser ablation repetition rate. The instrument was equipped with a torch injector of 2.5 mm inner diameter and nickel sample and skimmer cones with a skimmer cone insert of 2.8 mm in diameter. A radio frequency power of 1440 W, an auxiliary Ar gas flow rate of ∼0.80 L min−1 and a plasma Ar gas flow rate of 15 L min−1 was used. Data was recorded using TofPilot 1.3.4.0 (TOFWERK AG, Thun, Switzerland). Post-acquisition data processing was performed with Tofware v3.2.0. The data was further processed with HDIP version 1.3.1.1038 (Teledyne Photon Machines, Bozeman, MT, USA).miLogP calculation
Molinspiration (v2018.10) was used to predict the octanol–water partition coefficient logP.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.76 (s, 1H, H4′), 7.32 (s, 1H, H6), 7.13 (s, 1H, H2′), 7.05 (s, 1H, H5), 6.80 (s, 1H, H3′), 5.68 (s, 1H, Hc), 5.57 (s, 1H, Hd), 5.47 (s, 1H, Hd), 5.36 (s, 1H, Hc), 3.78 (s, 3H, H7), 3.64 (s, 3H, H1′), 2.63 (s, 1H, Hf), 2.43 (s, 3H, H1), 2.03 (s, 3H, Ha), 1.18 (d, J = 6 Hz, 6H, Hg) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 166.7 (C3), 160.0 (C4), 139.8 (C4′), 135.2 (C2), 128.9 (C3′), 128.5 (C6), 121.5 (C2′), 120.6 (C5), 101.1 (Ce), 98.5 (Cb), 84.2 (Cc), 83.4 (Cd), 81.4 (Cd), 80.8 (Cc), 43.1 (C1′), 34.1 (C7), 30.6 (Cf), 22.6 (Cg), 22.1 (Cg), 17.6 (C1), 11.9 (Ca) ppm.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.82 (s, 1H, H4′), 7.40 (d, J = 5 Hz, 1H, H6), 7.22 (s, 1H, H2′), 7.09 (s, 1H, H5), 6.91 (s, 1H, H3′), 3.81 (s, 3H, H7), 3.68 (s, 3H, H1′), 2.45 (s, 3H, H1), 1.61 (s, 15H, Cp*) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 166.7 (C3, C4), 139.0 (C4′), 136.8 (C2), 129.4 (C6), 128.2 (C3′), 122.6 (C2′), 121.8 (C5), 79.3 (Cp*), 43.8 (C7), 34.7 (C1′), 12.6 (C1), 8.9 (Cp*) ppm.
1H-NMR (600.25 MHz, d4-MeOD, 25 °C): δ = 7.73 (s, 1H, H4′), 7.30 (s, 1H, H6), 7.16 (s, 1H, H2′), 7.10 (s, 1H, H5), 6.99 (s, 1H, H3′), 3.86 (s, 3H, H7), 3.75 (s, 3H, H1′), 2.57 (s, 3H, H1), 1.72 (s, 15H, Cp*) ppm; 13C-NMR (150.95 MHz, d4-MeOD, 25 °C): δ = 170.0 (C3), 139.7 (C4′), 130.4 (C2), 129.0 (C6, C3′), 122.9 (C2′, C5), 43.9 (C7), 34.5 (C1′), 12.6 (C1), 8.9 (Cp*) ppm.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.66 (s, 1H, H4′), 7.42–7.30 (m, 4H, H6, H10–12), 7.16 (d, J = 6 Hz, 1H, H5), 6.99 (m, 3H, H2′, H9, H13), 6.95 (s, 1H, H3′), 5.61 (s, 1H, Hc), 5.54 (s, 1H, Hc), 5.40–5.33 (m, 4H, Hd, H7), 3.67 (s, 3H, H1′), 2.73–2.63 (m, 1H, Hf), 2.37 (s, 3H, H1), 2.05 (s, 3H, Ha), 1.26–1.21 (m, 6H, Hg) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 167.3 (C3), 161.6 (C4), 139.9 (C4′), 135.3 (C2), 134.4 (C8), 129.1 (C10, C12), 128.9 (C3′), 128.8 (C6), 128.2 (C11), 126.5 (C9, C13), 121.5 (C2′), 121.0 (C5), 101.5 (Ce), 98.4 (Cb), 84.3 (Cc), 83.3 (Cc), 81.5 (Cd), 80.9 (Cd), 58.0 (C7), 34.1 (C1′), 30.6 (Cf), 22.5 (Cg), 22.0 (Ca), 17.7 (Ca), 12.0 (C1) ppm.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.84 (s, 1H, H4′), 7.56 (d, J = 6.4 Hz, 1H, H6), 7.41–7.36 (m, 2H, H10, H12), 7.35–7.31 (m, 1H, H11), 7.25–7.22 (m, 1H, H5), 7.18 (d, J = 6 Hz, 1H, H2′), 7.11–7.07 (m, 2H, H9, H13), 6.92 (s, 1H, H3′), 5.44 (s, 2H, H7), 3.68 (s, 3H, H1′), 2.36 (s, 3H, H1), 1.59 (s, 15H, Cp*) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 167.0 (C3), 160.8 (C4), 138.6 (C4′), 135.2 (C2, C8), 129.1 (C10, C12), 128.7 (C6), 128.2 (C11), 127.6 (C3′), 126.7 (C9, C13), 121.9 (C2′), 121.6 (C5), 58.1 (C7), 33.9 (C1′), 12.1 (C1), 8.5 (Cp*) ppm.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.75 (s, 1H, H4′), 7.65 (s, 1H, H6), 7.41–7.37 (m, 2H, H10, H12), 7.36–7.31 (m, 1H, H11), 7.29 (s, 1H, H5), 7.18 (s, 1H, H2′), 7.13–7.09 (m, 2H, H9, H13), 6.86 (t, J = 1 Hz, 1H, H3′), 5.48 (s, 2H, H7), 3.68 (s, 3H, H1′), 2.40 (s, 3H, H1), 1.66–1.56 (m, 15H, Cp*) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 168.6 (C3), 161.6 (C4), 138.4 (C4′), 135.0 (C2, C8), 129.6 (C10, C12), 129.1 (C6), 128.3 (C11), 127.7 (C3′), 126.8 (C9, C13), 121.7 (C5), 58.1 (C7), 33.8 (C1′), 12.2 (C1), 8.3 (Cp*) ppm.
1H-NMR (500.10 MHz, CDCl3, 25 °C): δ = 7.12 (d, J = 6 Hz, 1H, H6), 7.04 (d, J = 6 Hz, 1H, H5), 5.83 (d, J = 9 Hz, 1H, Hc), 5.80 (d, J = 6 Hz, 1H, Hd), 5.60 (d, J = 6 Hz, 1H, Hd), 5.48 (d, J = 6 Hz, 1H, Hc), 4.56–4.37 (m, 6H, Hpta), 4.04 (dd, J = 70 Hz, 6H, Hpta), 3.83 (s, 3H, H7), 2.59–2.50 (m, 1H, Hf), 2.42 (s, 3H, H1), 2.05 (s, 3H, Ha), 1.24–1.16 (m, 6H, Hg) ppm; 13C-NMR (125.75 MHz, CDCl3, 25 °C): δ = 168.7 (C3), 161.5 (C4), 136.8 (C2), 128.1 (C5), 121.5 (C6), 106.1 (Ce), 99.9 (Cb), 91.5 (Cd), 91.3 (Cc), 88.6 (Cd), 87.2 (Cc), 73.0 (Cpta), 51.3 (Cpta), 51.2 (Cpta), 43.9 (C7), 31.5 (Cf), 23.0 (Cg), 22.7 (Cg), 18.3 (C1), 12.3 (Ca) ppm.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.47 (d, J = 6.5 Hz, 1H, H6), 7.20 (d, J = 6.4 Hz, 1H, H6), 4.48–4.37 (m, 6H, Hpta), 4.10–4.00 (m, 6H, Hpta), 3.85 (s, 3H, H7), 2.40 (s, 3H, H1), 1.67 (s, 15H, Cp*) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 166.5 (C3), 158.0 (C4), 137.5 (C2), 128.8 (C5), 121.0 (C6), 99.7 (Cp*q), 71.9 (Cpta), 48.8 (Cpta), 43.5 (Cpta), 12.2 (C1), 9.0 (Cp*) ppm.
1H-NMR (500.10 MHz, CDCl3, 25 °C): δ = 7.41–7.34 (m, 3H, H6, H10, H12), 7.21 (d, J = 6 Hz, 1H, H5), 7.07–7.04 (m, 3H, H9, H11, H13), 5.88 (d, J = 6 Hz, 1H, Hd), 5.84 (d, J = 6 Hz, 1H, Hc), 5.61 (d, J = 6 Hz, 1H, Hc), 5.51 (d, J = 6 Hz, 1H, Hd), 5.26 (dd, J = 38, 16 Hz, 2H, H7), 4.52 (s, 6H, Hpta), 4.06 (dd, J = 58, 15 Hz, 6H, Hpta), 2.58–2.51 (m, 1H, Hf), 2.34 (s, 3H, H1), 2.05 (s, 3H, Ha), 1.23–1.16 (m, 6H, Hg) ppm; 13C-NMR (125.75 MHz, CDCl3, 25 °C): δ = 169.2 (C3), 162.6 (C4), 136.7 (C2), 133.5 (C8), 129.7 (C10, C12), 129.1 (C6), 127.7 (C11), 126.8 (C9, C13), 121.6 (C5), 106.1 (Ce), 100.1 (Cb), 91.9 (Cd), 91.5 (Cc), 88.9 (Cd), 87.1 (Cc), 72.9 (Cpta), 59.5 (C7), 51.2 (Cpta), 31.5 (Cf), 22.9 (Cg), 22.8 (Cg), 18.3 (C1), 12.3 (Ca) ppm.
1H-NMR (500.10 MHz, d6-DMSO, 25 °C): δ = 7.64 (d, J = 7 Hz, 1H, H6), 7.41–7.37 (m, 2H, H10, H12), 7.36–7.31 (m, 2H, H11, H5), 7.11–7.08 (m, 2H, H9, H13), 5.47 (s, 2H, H7), 4.48–4.38 (m, 6H, Hpta), 4.09–4.00 (m, 6H, Hpta), 2.31 (s, 3H, H1), 1.66 (s, 15H, Cp*) ppm; 13C-NMR (125.75 MHz, d6-DMSO, 25 °C): δ = 167.1 (C3), 159.6 (C4), 136.7 (C2), 135.1 (C8), 129.1 (C10, C12), 129.1 (C6), 128.3 (C11), 126.6 (C9, C13), 121.4 (C5), 99.8 (Cp*), 71.9 (Cpta), 58.3 (C7), 48.9 (Cpta), 48.8 (Cpta), 12.2 (C1), 9.0 (Cp*) ppm.
1H-NMR (500.10 MHz, CDCl3, 25 °C): δ = 8.94 (s, 2H, NH2), 7.15 (d, J = 6 Hz, 1H, H6), 7.07 (s, 2H, NH2), 6.96 (d, J = 6 Hz, 1H, H5), 3.76 (s, 3H, H7), 2.70–2.62 (m, 1H, Hf), 2.38 (s, 3H, H1), 2.12 (s, 3H, Ha), 1.18 (d, J = 7 Hz, 6H, Hg) ppm; 13C-NMR (125.75 MHz, CDCl3, 25 °C): δ = 183.0 (tu), 167.0 (C3), 164.2 (C4), 135.6 (C2), 128.4 (C6), 122.1 (C5), 102.1 (Ce), 100.5 (Cb), 85.9 (Cd), 84.8 (Cc), 83.9 (Cd), 83.0 (Cc), 43.8 (C7), 31.0 (Cg), 18.2 (C1), 12.7 (Ca) ppm.
1H-NMR (500.10 MHz, CDCl3, 25 °C): δ = 8.83 (s, 2H, NH2), 7.40–7.35 (m, 3H, H6, H10, H12), 7.22 (d, J = 7 Hz, 1H, H5), 7.06 (s, 2H, NH2), 7.04–6.98 (m, 3H, H9, H11, H13), 5.57–5.33 (m, 3H, Hc, Hd), 5.28–5.12 (m, 3H, Hd, H7), 2.71–2.63 (m, 1H, Hf), 2.32 (s, 3H, H1), 2.12 (s, 3H, Ha), 1.19 (d, J = 7 Hz, 6H, Hg) ppm; 13C-NMR (125.75 MHz, CDCl3, 25 °C): δ = 183.0 (tu), 167.3 (C3), 165.0 (C4), 135.6 (C2), 133.4 (C8), 129.7 (C10, C12), 129.2 (C6), 128.2 (C11), 126.7 (C9, C13), 122.3 (C5), 101.9 (Ce), 100.7 (Cb), 86.3 (Cd), 84.7 (Cc), 84.0 (Cd), 83.4 (Cc), 59.3 (C7), 31.0 (Cf), 23.0 (Cg), 22.4 (Cg), 18.3 (Ca), 12.6 (C1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Vienna for financial support. Furthermore, we would like to thank Claudia Cseh for her kind support with the evaluation of DNA plasmid assays.Notes and references
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- C. S. Allardyce and P. J. Dyson, Platinum Met. Rev., 2001, 45, 62–69 CAS.
- I. Kostova, Curr. Med. Chem., 2006, 13, 1085–1107 CrossRef CAS.
- R. Mitra, S. Das, S. V. Shinde, S. Sinha, K. Somasundaram and A. G. Samuelson, Chem. – Eur. J., 2012, 18, 12278–12291 CrossRef CAS.
- L. Ma, R. Ma, Z. Wang, S. M. Yiu and G. Zhu, Chem. Commun., 2016, 52, 10735–10738 RSC.
- H. Köpf and P. Köpf-Maier, Angew. Chem., Int. Ed. Engl., 1979, 91, 509–509 Search PubMed.
- M. Guo, Z. Guo and P. Sadler, J. Biol. Inorg. Chem., 2001, 6, 698–707 CrossRef CAS.
- B. K. Keppler, M. R. Berger and M. E. Heim, Cancer Treat. Rev., 1990, 17, 261–277 CrossRef CAS.
- G. Jaouen, S. Top, A. Vessieres, G. Leclercq and M. McGlinchey, Curr. Med. Chem., 2004, 11, 2505–2517 CrossRef CAS.
- M. Hanif, H. Henke, S. M. Meier, S. Martic, M. Labib, W. Kandioller, M. A. Jakupec, V. B. Arion, H. B. Kraatz, B. K. Keppler and C. G. Hartinger, Inorg. Chem., 2010, 49, 7953–7963 CrossRef CAS.
- S. M. Meier, M. Hanif, W. Kandioller, B. K. Keppler and C. G. Hartinger, J. Inorg. Biochem., 2012, 108, 91–95 CrossRef CAS.
- M. Caterino, M. Herrmann, A. Merlino, C. Riccardi, D. Montesarchio, M. A. Mroginski, D. Musumeci, F. Ruffo, L. Paduano, P. Hildebrandt, J. Kozuch and A. Vergara, Inorg. Chem., 2019, 58, 1216–1223 CrossRef CAS.
- R. E. Morris, R. E. Aird, P. del Socorro Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS.
- F. Wang, J. Xu, K. Wu, S. K. Weidt, C. L. MacKay, P. R. R. Langridge-Smith and P. J. Sadler, Dalton Trans., 2013, 42, 3188–3195 RSC.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem. – Eur. J., 2011, 17, 3562–3566 CrossRef CAS.
- T. Giraldi, G. Sava, G. Mestroni, G. Zassinovich and D. Stolfa, Chem.–Biol. Interact., 1978, 22, 231–238 CrossRef CAS.
- L. Messori, J. Inorg. Biochem., 2003, 95, 37–46 CrossRef CAS.
- G. Mestroni, E. Alessio, A. Sessanta o Santi, S. Geremia, A. Bergamo, G. Sava, A. Boccarelli, A. Schettino and M. Coluccia, Inorg. Chim. Acta, 1998, 273, 62–71 CrossRef CAS.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS.
- A. Casini, F. Edafe, M. Erlandsson, L. Gonsalvi, A. Ciancetta, N. Re, A. Ienco, L. Messori, M. Peruzzini and P. J. Dyson, Dalton Trans., 2010, 39, 5556–5563 RSC.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS.
- C. M. Hackl, M. S. Legina, V. Pichler, M. Schmidlehner, A. Roller, O. Dömötör, E. A. Enyedy, M. A. Jakupec, W. Kandioller and B. K. Keppler, Chem. – Eur. J., 2016, 22, 17269–17281 CrossRef CAS.
- S. Harringer, B. Happl, M. Ozenil, C. Kast, M. Hejl, D. Wernitznig, A. A. Legin, A. Schweikert, N. Gajic, A. Roller, G. Koellensperger, M. A. Jakupec, W. Kandioller and B. K. Keppler, Chem. – Eur. J., 2020, 26, 5419–5433 CrossRef CAS.
- M. P. Cava and M. I. Levinson, Tetrahedron, 1985, 41, 5061–5087 CrossRef CAS.
- C. A. Riedl, M. Hejl, M. H. M. Klose, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2018, 47, 4625–4638 RSC.
- M. Schmidlehner, P. S. Kuhn, C. M. Hackl, A. Roller, W. Kandioller and B. K. Keppler, J. Organomet. Chem., 2014, 772, 93–99 CrossRef.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2007, 13, 2601–2613 CrossRef CAS.
- A. Dorcier, P. J. Dyson, C. Gossens, U. Rothlisberger, R. Scopelliti and I. Tavernelli, Organometallics, 2005, 24, 2114–2123 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- V. Pichler, S. M. Valiahdi, M. A. Jakupec, V. B. Arion, M. Galanski and B. K. Keppler, Dalton Trans., 2011, 40, 8187–8192 RSC.
- P. S. Kuhn, V. Pichler, A. Roller, M. Hejl, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2015, 44, 659–668 RSC.
- A. S. Nunes, A. S. Barros, E. C. Costa, A. F. Moreira and I. J. Correia, Biotechnol. Bioeng., 2019, 116, 206–226 CrossRef CAS.
- E. Schreiber-Brynzak, E. Klapproth, C. Unger, I. Lichtscheidl-Schultz, S. Göschl, S. Schweighofer, R. Trondl, H. Dolznig, M. A. Jakupec and B. K. Keppler, Invest. New Drugs, 2015, 33, 835–847 CrossRef CAS.
- H. Geisler, D. Wernitznig, M. Hejl, N. Gajic, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2020, 49, 1393–1397 RSC.
- A. M. Rieger, K. L. Nelson, J. D. Konowalchuk and D. R. Barreda, J. Visualized Exp., 2011, 3–6 Search PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS.
- I. Vermes, C. Haanen and C. Reutelingsperger, J. Immunol. Methods, 2000, 243, 167–190 CrossRef CAS.
- M. Cornelissen, J. Philippé, S. De Sitter and L. De Ridder, Apoptosis, 2002, 7, 41–47 CrossRef CAS.
- Z. Darzynkiewicz, S. Bruno, G. Del Bino, W. Gorczyca, M. A. Hotz, P. Lassota and F. Traganos, Cytometry, 1992, 13, 795–808 CrossRef CAS.
- G. Kroemer, B. Dallaporta and M. Resche-Rigon, Annu. Rev. Physiol., 1998, 60, 619–642 CrossRef CAS.
- G. Denecker, D. Vercammen, W. Declercq and P. Vandenabeele, Cell. Mol. Life Sci., 2001, 58, 356–370 CrossRef CAS.
- L. Faleiro and Y. Lazebnik, J. Cell Biol., 2000, 151, 951–960 CrossRef CAS.
- J. A. Khan, F. Forouhar, X. Tao and L. Tong, Expert Opin. Ther. Targets, 2007, 11, 695–705 CrossRef CAS.
- J. J. Soldevila-Barreda and P. J. Sadler, Curr. Opin. Chem. Biol., 2015, 25, 172–183 CrossRef CAS.
- K. W. Wellington, RSC Adv., 2015, 5, 20309–20338 RSC.
- G.-Y. Liou and P. Storz, Free Radic. Res., 2010, 44, 479–496 CrossRef CAS.
- N. A. P. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- S. Zorbas-Seifried, C. G. Hartinger, K. Meelich, M. Galanski and B. K. Keppler, DNA Repair, 2006, 14817–14825 CAS.
- H. Li, H. Bo, J. Wang, H. Shao and S. Huang, Cytotechnology, 2011, 63, 7–12 CrossRef.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS.
- S. Göschl, H. P. Varbanov, S. Theiner, M. A. Jakupec, M. Galanski and B. K. Keppler, J. Inorg. Biochem., 2016, 160, 264–274 CrossRef.
- S. Göschl, E. Schreiber-Brynzak, V. Pichler, K. Cseh, P. Heffeter, U. Jungwirth, M. A. Jakupec, W. Berger and B. K. Keppler, Metallomics, 2017, 9, 309–322 RSC.
- S. Moon, M. Hanif, M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, ChemPlusChem, 2015, 80, 231–236 CrossRef CAS.
- M. Hanif, A. A. Nazarov, A. Legin, M. Groessl, V. B. Arion, M. A. Jakupec, Y. O. Tsybin, P. J. Dyson, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 1475–1477 RSC.
- M. Zaki, S. Hairat and E. S. Aazam, Scope of organometallic compounds based on transition metal-arene systems as anticancer agents: Starting from the classical paradigm to targeting multiple strategies, Royal Society of Chemistry, 2019, vol. 9 Search PubMed.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599–616 CrossRef CAS.
- S. J. Dougan, A. Habtemariam, S. E. McHale, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11628–11633 CrossRef CAS.
- S. Theiner, S. J. M. Van Malderen, T. Van Acker, A. Legin, B. K. Keppler, F. Vanhaecke and G. Koellensperger, Anal. Chem., 2017, 89, 12641–12645 CrossRef CAS.
- M. Zoriy, A. Matusch, T. Spruss and J. S. Becker, Int. J. Mass Spectrom., 2007, 260, 102–106 CrossRef CAS.
- R. Lobinski, C. Moulin and R. Ortega, Biochimie, 2006, 88, 1591–1604 CrossRef CAS.
- S. Theiner, E. Schreiber-Brynzak, M. A. Jakupec, M. Galanski, G. Koellensperger and B. K. Keppler, Metallomics, 2016, 8, 398–402 RSC.
- S. B. Jensen, S. J. Rodger and M. D. Spicer, J. Organomet. Chem., 1998, 556, 151–158 CrossRef CAS.
- M. G. Mendoza-Ferri, C. G. Hartinger, A. A. Nazarov, R. E. Eichinger, M. A. Jakupec, K. Severin and B. K. Keppler, Organometallics, 2009, 28, 6260–6265 CrossRef CAS.
- L. E. Heim, S. Vallazza, D. van der Waals and M. H. G. Prechtl, Green Chem., 2016, 18, 1469–1474 RSC.
- J. W. Kang, K. Moseley and P. M. Maitiis, J. Am. Chem. Soc., 1969, 91, 5970–5977 CrossRef CAS.
- W. Tejchman, E. Zesławska, K. Zborowski, W. Nitek and M. Zylewski, ARKIVOC, 2015, 2015, 216–230 Search PubMed.
- Bruker SAINT v8.38B Copyright © 2005-2019, Bruker AXS Search PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef.
- G. M. Sheldrick, SHELXS v 2016/4, University of Göttingen, Germany, 2015 Search PubMed.
- G. M. Sheldrick, SHELXL v 2016/4, University of Göttingen, Germany, 2015 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR characterization, crystallographic data, UV-Vis spectra, as well as further data concerning MTT assay, apoptosis assays, clonogenic assay, confocal microscopy, plasmid assay, laser ablation ICP-MS. CCDC 2018340–2018345 and 2018351–2018356. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03165h |
| This journal is © The Royal Society of Chemistry 2020 |