
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of zinc di(O-2,2-dimethylpentan-3-yl dithiocarbonates) bearing pyridine or tetramethylethylenediamine coligands and investigation of their thermal conversion mechanisms towards nanocrystalline zinc sulfide†
Efthymia
Vakalopoulou‡
 a,
Christine
Buchmaier‡
a,
Andreas
Pein
a,
Robert
Saf
a,
Roland C.
Fischer
b,
Ana
Torvisco
b,
Fernando
Warchomicka
c,
Thomas
Rath
*a and
Gregor
Trimmel
a
a,
Christine
Buchmaier‡
a,
Andreas
Pein
a,
Robert
Saf
a,
Roland C.
Fischer
b,
Ana
Torvisco
b,
Fernando
Warchomicka
c,
Thomas
Rath
*a and
Gregor
Trimmel
a
aInstitute for Chemistry and Technology of Materials (ICTM), NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at
bInstitute of Inorganic Chemistry, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
cInstitute of Materials Science, Joining and Forming, Graz University of Technology, Kopernikusgasse 24, 8010, Graz, Austria
First published on 14th October 2020
Abstract
Metal xanthates are versatile single source precursors for the preparation of various metal sulfides. In this study, we present the synthesis of the two novel zinc xanthate complexes bis(O-2,2-dimethylpentan-3-yl-dithiocarbonato)(N,N,N′,N′-tetramethylethylenediamine)zinc(II) and bis(O-2,2-dimethylpentan-3-yl-dithiocarbonato)(pyridine)zinc(II). A thorough investigation of these compounds revealed distinct differences in their structural and thermal properties. While in the complex containing the chelating tetramethylethylenediamine, the xanthate groups coordinate in a monodentate way, they are bidentally coordinated to the zinc atom in the pyridine containing complex. Both compounds show a two-step thermal decomposition with an onset temperature of 151 °C and 156 °C for the tetramethylethylenediamine and pyridine containing complex, respectively. Moreover, different mechanisms are revealed for the two phases of the decomposition based on high resolution mass spectrometry investigations. By the thermal conversion process nanocrystalline zinc sulfide is produced and the coligand significantly influences its primary crystallite size, which is 4.4 nm using the tetramethylethylenediamine and 11.4 nm using the pyridine containing complex for samples prepared at a temperature of 400 °C.
Introduction
Zinc sulfide (ZnS) is a wide band gap metal chalcogenide, which belongs to the II–VI compound semiconductor family. It can form two types of crystal structures, the sphalerite/zinc blende (cubic) and the wurtzite (hexagonal) phase.1 The cubic structure is stable at low temperature and in bulk ZnS it is converted to the high temperature stable hexagonal phase at temperatures above 1000 °C, however, for nanocrystalline ZnS, a transformation from cubic to hexagonal phase has been observed already at much lower temperatures starting at 250 °C.2–4 ZnS in both crystal structures has wide band gaps of ∼3.54 eV and ∼3.90 eV for the cubic and hexagonal phase, respectively.1 Due to its interesting luminescence and optoelectronic properties, ZnS is used in a broad spectrum of applications including light-emitting diodes (LEDs),5,6 photoluminescence devices,7,8 solar cells,9,10 sensors,11–13 and catalysis.14,15The synthesis routes towards ZnS nanocrystals and thin films are manifold and versatile. ZnS nanostructures can be prepared in form of 3D (particle), 2D (thin film), 1D (wire, rod, tube, sheet, belt) as well as 0D structures (quantum dot).16 Generally, metal chalcogenides can be synthesized via several routes, such as solution-based reactions, hydrothermal/solvothermal methods, microwave and sonochemical methods as well as chemical and physical vapor deposition-based methods.17–20 In addition, methods using single-source precursors (SSPs) gain increasing attention for preparing metal chalcogenides.21–23 The method is based on the idea of using compounds containing already a metal sulfur bond. The precursor molecule is then converted into the metal sulfide by a chemical reaction, in most cases initiated by a thermal treatment. Metal dithiocarbonates, also known as metal xanthates, belong to this class of materials and their use for the preparation of a wide range of metal sulfides, for instance, CdS,24,25 ZnS,26,27 NiS,28 SnS,29 PbS,30 Ga2S3,31 Bi2S3,32 CuInS2,33,34 CuSbS2,35 and Cu2ZnSnS4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 is already reported. A further beneficial property of metal xanthates as precursors is that the conversion can take place via thermal treatment in solution,37,38 solid state reactions in a matrix,39,40 solvent-free melt reactions41–43 or aerosol assisted chemical vapour deposition (AA-CVD) processes.44,45 Additionally, by mixing different ratios of metal xanthates, doped metal sulfides with various transition metals can be resulted and several studies showed that such systems are very useful for controlling the optical, electrical and crystalline properties.46–48 The metal xanthates are generally stable at room temperature and their decomposition typically occurs thermally via a mechanism known as Chugaev elimination.24 However, it is also possible to decompose them chemically with the use of primary amines at room temperature49 as well as photochemically with UV irradiation.50 An interesting advantage of metal xanthates is the ability to alter their properties such as solubility and decomposition behaviour by modifying the organic side chain or by adding coligands such as pyridine, which are known as metal xanthate complexes.24 As a result, by designing these compounds, a better control on the stoichiometry and morphology of the final nanostructures can be achieved, which is of great importance for their applications. Furthermore, several studies reported in literature show that features of the final metal sulfide nanomaterials such as the size, phase and shape, can be controlled by the nature of the capping agents51 or the type of the alkyl chain.52,53
36 is already reported. A further beneficial property of metal xanthates as precursors is that the conversion can take place via thermal treatment in solution,37,38 solid state reactions in a matrix,39,40 solvent-free melt reactions41–43 or aerosol assisted chemical vapour deposition (AA-CVD) processes.44,45 Additionally, by mixing different ratios of metal xanthates, doped metal sulfides with various transition metals can be resulted and several studies showed that such systems are very useful for controlling the optical, electrical and crystalline properties.46–48 The metal xanthates are generally stable at room temperature and their decomposition typically occurs thermally via a mechanism known as Chugaev elimination.24 However, it is also possible to decompose them chemically with the use of primary amines at room temperature49 as well as photochemically with UV irradiation.50 An interesting advantage of metal xanthates is the ability to alter their properties such as solubility and decomposition behaviour by modifying the organic side chain or by adding coligands such as pyridine, which are known as metal xanthate complexes.24 As a result, by designing these compounds, a better control on the stoichiometry and morphology of the final nanostructures can be achieved, which is of great importance for their applications. Furthermore, several studies reported in literature show that features of the final metal sulfide nanomaterials such as the size, phase and shape, can be controlled by the nature of the capping agents51 or the type of the alkyl chain.52,53
In this contribution, we report on the synthesis and crystal structure of two new zinc xanthate complexes bearing 2,2-dimethylpentan-3-yl alkyl moieties and additional either pyridine or N,N,N′,N′-tetramethylethylenediamine (TMEDA), as coligand. In addition, we extend our study to the investigation of the thermal decomposition of these compounds. In particular, it is expected that the coligands will influence the formation of the nanocrystalline ZnS by changing the thermal decomposition pathway. Therefore, a special focus is set on the detailed analysis of the thermal decomposition by mass spectrometry in order to shine light of the underlying reaction mechanisms.
Results and discussion
Synthesis and spectroscopic characterization
The synthesis of zinc(II) O-2,2-dimethylpentan-3-yl dithiocarbonates (ZnHep) was performed according to previous reports.34,54 ZnHep was then used as a starting material to synthesize the two complexes bis(O-2,2-dimethylpentan-3-yl-dithiocarbonato)(N,N,N′,N′-tetramethylethylenediamine)zinc(II) (ZnHepTMEDA) and bis(O-2,2-dimethylpentan-3-yl-dithiocarbonato)(pyridine)zinc(II) (ZnHepPyr). The chemical structures of the compounds and a scheme of the synthetic procedure are shown in Scheme 1. The zinc xanthate complexes contain a bulky, branched alkyl group, which provides the compound good solubility in nonpolar solvents such as chloroform, toluene and dichlorobenzene. Moreover, the modification of the ZnHep with two amine ligands, pyridine and TMEDA, is expected to influence the thermal decomposition of the compounds and the formation of ZnS particles.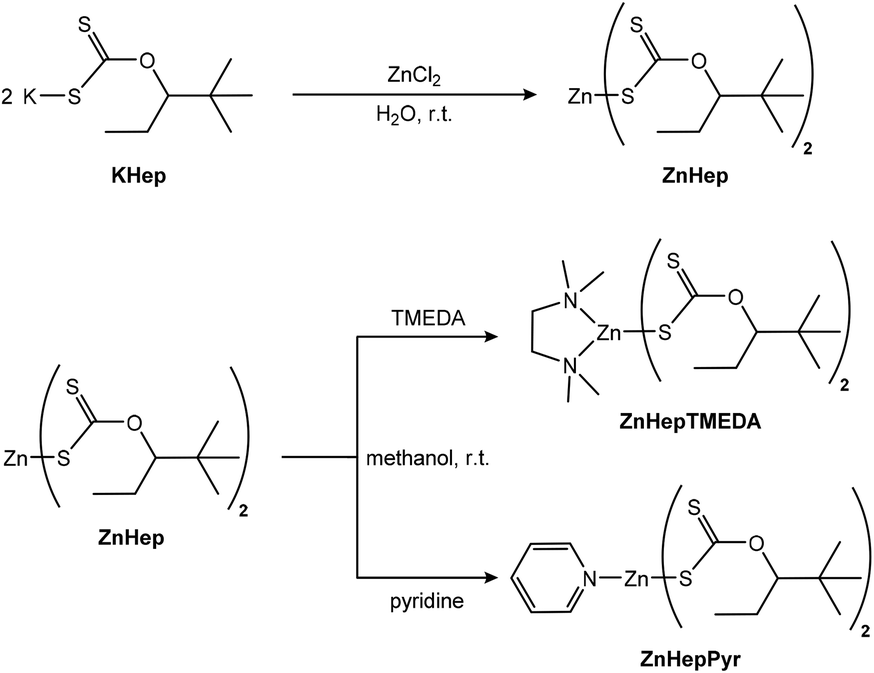 | ||
| Scheme 1 Synthesis routes towards ZnHep, ZnHepTMEDA and ZnHepPyr. | ||
The obtained white powders are stable under ambient conditions and pure according to the NMR analysis. Furthermore, the 13C-NMR spectra (Fig. S1†) reveal a characteristic chemical shift at 229.1 ppm (ZnHep), 228.7 ppm (ZnHepPyr) and 225.9 ppm (ZnHepTMEDA), which corresponds to the dithiocarbonate group (S2COR). The presence of the additional ligand causes a change of the chemical shift. The coordination of additional donating nitrogen ligands to the metal center leads to an increased electron density on the zinc and thus to weaker zinc–sulfur bonds. Therefore, also the adjacent carbon has a higher electron density and the carbon signals show in both cases an upfield shift. Additionally, the 1H-NMR (Fig. S2†) resonance signals of TMEDA (δ = 2.68, 2.58 ppm) and pyridine (δ = 8.94, 8.00, 7.59 ppm) ligands connected to the zinc metal show a downfield shift compared to the free TMEDA (δ = 2.38, 2.24 ppm)55 and pyridine (δ = 8.52, 7.55, 7.16 ppm).56 The latter is a strong indication that the nitrogen atoms of the amine ligands coordinate with the metal atom. Moreover, the quantitative analysis of the 1H-NMR spectrum of ZnHepPyr shows that only one pyridine ligand is bound to the zinc atom.
The IR spectra of the zinc xanthate and its complexes are presented in Fig. 1 and fit well with the suggested structures. The spectra show the typical xanthate bands at approximately 1200 cm−1, 1140 cm−1 and 1040 cm−1. The bands are assigned to the asymmetric and symmetric stretching vibrations of COC and (S)CO and the asymmetric stretching vibrations of SCS and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S.57,58 A list of all peaks and their assignment can be found in Table S1 (ESI†).
S.57,58 A list of all peaks and their assignment can be found in Table S1 (ESI†).
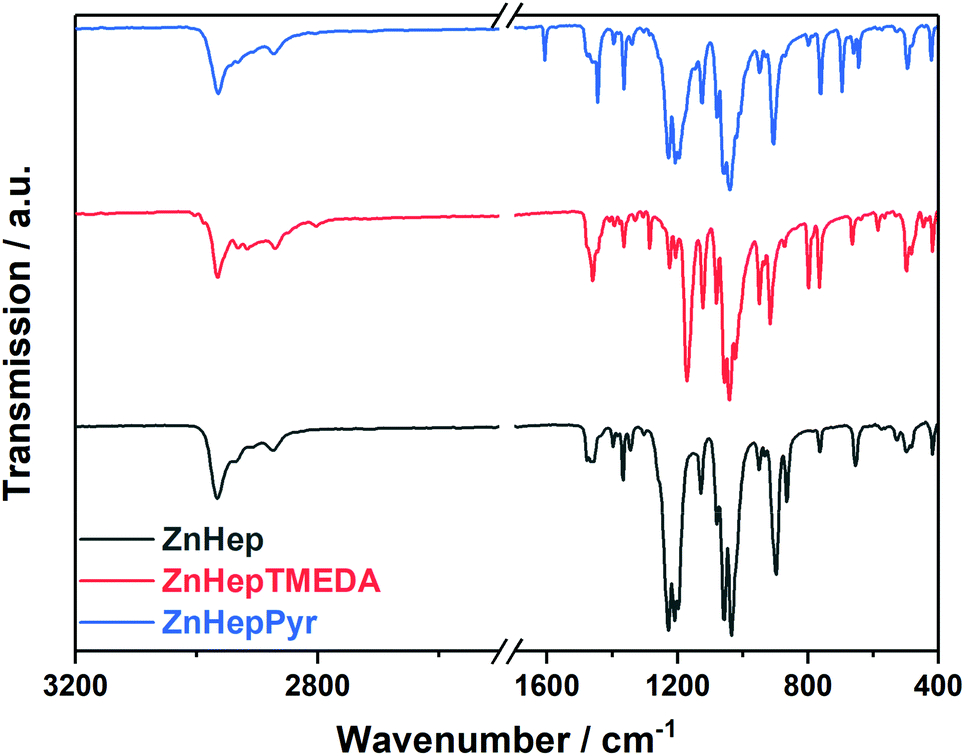 | ||
| Fig. 1 FT-IR spectra of zinc xanthate (ZnHep) and the complexes ZnHepTMEDA and ZnHepPyr. The spectra are shifted vertically for better visibility. | ||
In addition, the spectra prove the presence of the amine ligands. More specifically, the ZnHepPyr complex shows three additional bands at 1608 cm−1, 1483 cm−1 and 1444 cm−1, which can be assigned to plane vibrations of CC and CN of the pyridine ligand.56 On the contrary, ZnHepTMEDA displays bands at 2801 cm−1, which correspond to the symmetric stretching vibrations of CH and CH3 (N–CH3, N–CH2), at 1461 cm−1 due to the symmetrical CH deformation vibration, as well as at 1285 cm−1, which is assigned to the C–N stretching vibration. By comparing the position of the characteristic IR bands of the free TMEDA ligand (1454 cm−1, 1267 cm−1)55 with these of the ZnHepTMEDA complex, it is obvious that there is a slight shift to higher wavenumbers, which originates from the coordination of the nitrogen atoms to the metal.
The position of the asymmetric stretching vibration of the COC bond of the metal xanthate can provide essential information about the metal–sulfur bond, as well as the coordination of the xanthate ligand on the metal atom. A bidentate coordination of the xanthate ligand on the metal atom causes an increased electron flow from the sulfur atoms to the metal, leading to a strong pπ–pπ oxygen–carbon bond and thus to a shift of the asymmetric COC stretching vibration (νas COC) to higher wavenumbers.59,60 A comparison among the zinc xanthates regarding the asymmetric vibration of COC, shows this band in the ZnHepTMEDA sample at lower wavenumbers (1171 cm−1) than for ZnHep (1210 cm−1) and ZnHepPyr (1208 cm−1). This could be probably attributed to the different coordination of the xanthate ligands on the metal atom. In the case of ZnHepTMEDA, a monodentate coordination of the xanthate ligand is expected due to steric and electronic effects of the bidentate coordinated diamine ligand TMEDA.61 In ZnHep57 and ZnHepPyr,60 we assume a bidentate coordination. More details considering this subject will be discussed later in the crystal structure analysis.
Structural properties based on single crystal X-ray diffraction
Furthermore, single-crystal X-ray diffraction analysis of the complexes ZnHepTMEDA and ZnHepPyr was performed and the crystal structures are presented in Fig. 2. It was not possible to obtain suitable single crystals of ZnHep. The analysis revealed comprehensive information regarding the coordination of the amine and xanthate ligands on the metal atom. The N-donor ligands coordinate differently on the metal atom and consequently change the coordination of the xanthate ligands.60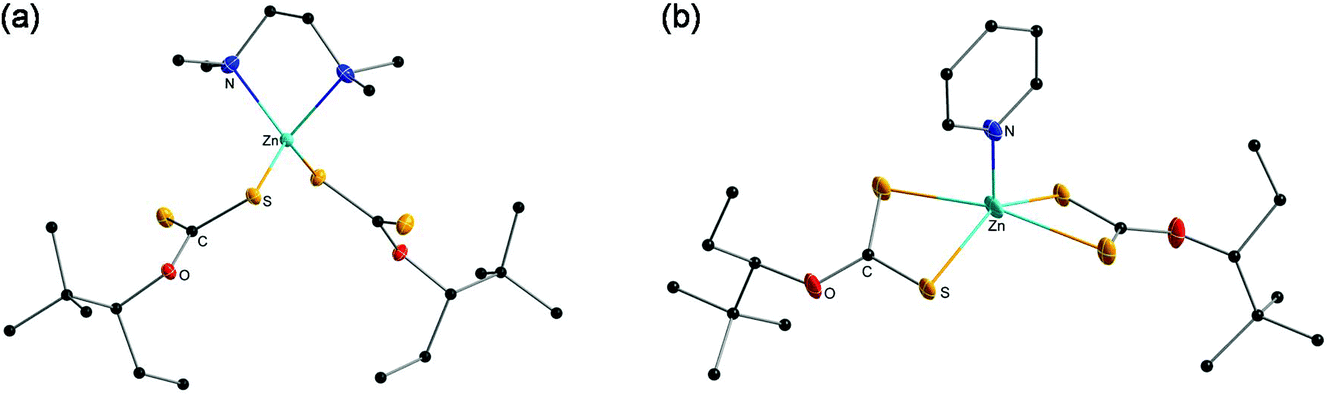 | ||
| Fig. 2 Crystal structure of the zinc xanthate complexes (a) ZnHepTMEDA and (b) ZnHepPyr. All non-carbon atoms shown as 30% shaded ellipsoids. Hydrogen atoms removed for clarity. | ||
In the case of ZnHepTMEDA, the zinc atom is tetrahedrally coordinated with both nitrogen atoms of the non-planar diamine ligand TMEDA and one sulfur atom from each of the two symmetrical xanthate ligands, confirming the monodentate character of the xanthate ligands, which has already been indicated by the IR spectral analysis. The Zn–N and Zn–S bonds show lengths of 2.136(2) Å and 2.304(6) Å and are very close to the values of the similar Zn(S2COEt)2TMEDA complex reported in the literature (Zn–N 2.147; Zn–S 2.304). The coordinated sulfur has a single bond with the carbon with a length of 1.716(2) Å, while the non-coordinated sulfur is bound to the carbon by a double bond and has a length of 1.663(2) Å; both values are in accordance with typical values found for C–S and CS bonds in literature.62 Further selected bond lengths and angles are summarized in Tables 1 and 2, respectively.
| Selected bond lengths (Å) | ||
|---|---|---|
| ZnHepTMEDA | ZnHepPyr | |
| Symmetry code: (i) −x + 1, y, −z + 1/2. | ||
| Zn1–N1 | 2.136(2) | 2.020(3) |
| Zn1–N1i | 2.136(2) | — |
| Zn1–S1 | — | 2.3296(9) |
| Zn1–S1i | — | 2.3296(9) |
| Zn1–S2 | 2.3014(6) | 2.6368(11) |
| Zn1–S2i | 2.3014(6) | 2.6368(11) |
| S1–C1 | 1.716(2) | 1.717(3) |
| S2–C1 | 1.663(2) | 1.685(2) |
| O1–C1 | 1.337(3) | 1.323(3) |
| Selected angles (°) | ||
|---|---|---|
| ZnHepTMEDA | ZnHepPyr | |
| Symmetry code: (i) −x + 1, y, −z + 1/2. | ||
| N1i–Zn1–N1 | 84.63(12) | — |
| N1i–Zn1–S2 | 114.45(6) | — |
| N1–Zn–S2i | 114.45(6) | — |
| N1–Zn1–S2 | 104.97(6) | 97.34(2) |
| N1i–Zn–S2i | 104.97(6) | — |
| S1–Zn–S2i | — | 101.10(3) |
| S1i–Zn–S2 | — | 101.10(3) |
| S1i–Zn–S2i | — | 72.93(3) |
| S1–Zn1–S2 | — | 72.93(3) |
| C1–S1–Zn1 | 99.95(8) | 87.48(8) |
| C1–S2–Zn1 | — | 78.53(9) |
| S1–C1–S2 | 125.27(14) | 121.06(14) |
| O1–C1–S1 | 109.96(16) | 113.4(2) |
| O1–C1–S2 | 124.77(17) | 125.3(2) |
| C1–O1–C2 | 122.23(17) | 122.4(2) |
In contrast to ZnHepTMEDA, the zinc atom of ZnHepPyr is square pyramidally coordinated by the nitrogen atom of the pyridine ligand and both sulfur atoms of each of the xanthate ligands. A stronger Zn–S bond with a bond length of 2.3296(9) Å and a weaker Zn–S bond with 2.6368(11) Å on the zinc atom coordinate the two symmetrical, bidentate xanthate ligands. The values fit with those found by Raston et al. for the Zn(S2COEt)2(C5H5N) complex (Zn–S, 2.294 Å, 2.748 Å).63 The Zn–N bonds have a length of 2.020(3) Å and are slightly shorter than in the case of ZnHepTMEDA, where both nitrogen atoms are coordinated on the metal. With bond lengths of 1.685(2) Å and 1.717 (3) Å, both sulfur–carbon bonds are approximately in the same range as in the ZnHepTMEDA complex.
In addition to resulting in different metal coordination environments, the use of the bidentate donor TMEDA and monodentate pyridine also affect the types of hydrogen bonds present in the compounds and their crystal packing in the extended solid state. Both ZnHepTMEDA and ZnHepPyr display intra- and intermolecular hydrogen bonds through the sulfur atoms (C–H⋯S) (Table S2†). In both compounds, nearly identical intramolecular hydrogen bonds occur from both methylene hydrogens of each S2COEttBu ligand and a sulfur atom of the same S2COEttBu moiety, despite their different coordination environments. The values for these intramolecular C–H⋯S hydrogen bonds are 2.52 Å and with an angle of 114° for ZnHepTMEDA through the uncoordinated sulfur atom and 2.54 Å and with an angle of 115° for ZnHepPyr. This type of intramolecular hydrogen bonding was also observed in a Ni(S2COEttBu)2 analog.28 However, in the case of ZnHepTMEDA, several other intramolecular hydrogen bonds (2.84–3.02 Å) are observed from the methyl groups of the TMEDA base and both sulfur atoms (Table S2†). Hydrogens from the methyl groups of the TMEDA base are also involved in intermolecular hydrogen bonds with the uncoordinated sulfur atom from the S2COEttBu ligand of a neighboring molecule (2.93 Å, 151°) propagating 2D sheets (Fig. S3†). For ZnHepPyr, hydrogen atoms from the pyridine base are also involved in intermolecular hydrogen bonds between sulfur atoms from the S2COEttBu ligand of neighboring molecules (2.96 Å, 134° and 3.02 Å, 127°). Additionally, hydrogens from the tBu groups also afford intermolecular hydrogen bonds (2.96 Å, 150°) resulting in 2D sheets (Fig. S4†).
Investigation of the thermal decomposition
The thermal decomposition behavior of the zinc xanthate and complexes was investigated by thermogravimetric measurements (TGA, see Fig. 3). The analysis shows almost no difference in the onset temperature of the decomposition (Td, determined at 5% mass loss). The decomposition proceeds in two steps for all the compounds, as it is even clearer from differential thermogravimetric (DTG) plots. However, the temperature plateau between the two decomposition steps is very small implying the initiation of the second decomposition before the termination of the first step. Additionally, it is obvious that the ligands slightly influence the decomposition. ZnHepPyr decomposes at approximately 5 °C higher than the other both compounds, but the reaction is faster than in the other cases. Although the exact mechanism cannot be deduced from these measurements, the sharp weight loss steps are in accordance with the expectation of a swift chemical reaction, i.e. that the Chugaev elimination reaction takes place. The experimental mass losses of 77.1, 82.6 and 81.8% are well in line with the theoretical calculations for the formation of ZnS (Table 3). Any deviation can be attributed to measurement uncertainties.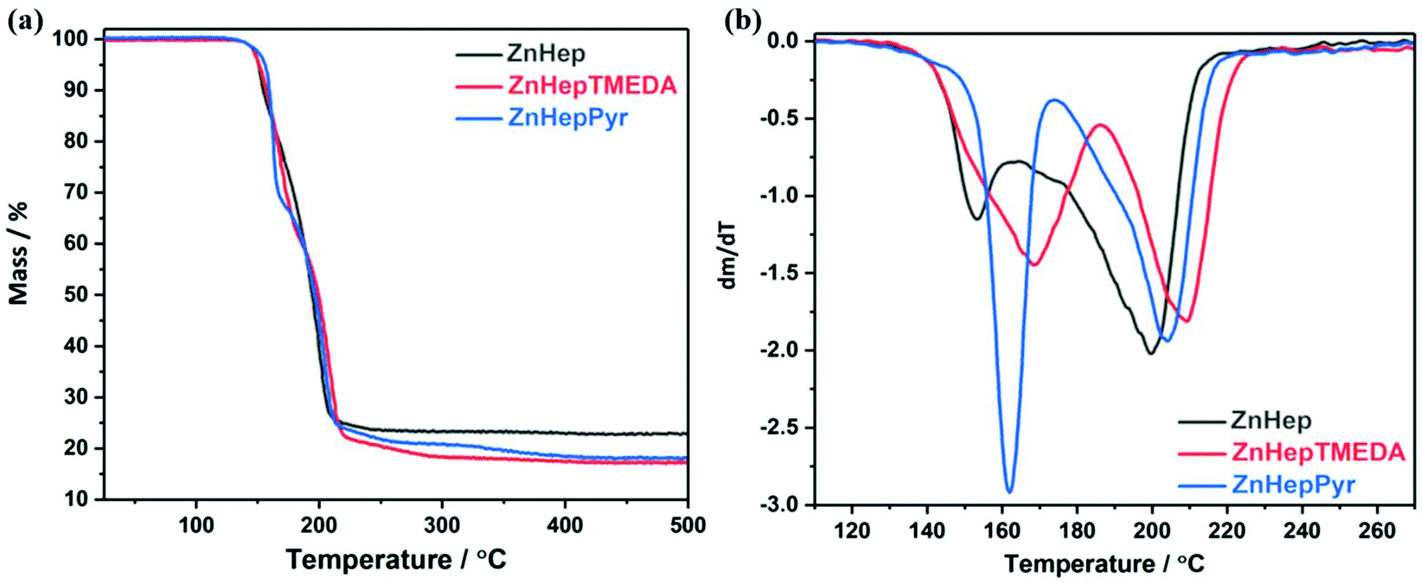 | ||
| Fig. 3 (a) TGA and (b) DTG plots of the zinc xanthate (ZnHep) and complexes (ZnHepTMEDA, ZnHepPyr). | ||
| Compound | Theoretical mass loss (%) | Observed mass loss (%) | Decomposition temperature Td (°C) |
|---|---|---|---|
| ZnHep | 78.2 (ZnS) | 77.2 | 150.7 |
| ZnHepTMEDA | 82.7 (ZnS) | 82.6 | 151.3 |
| ZnHepPyr | 81.5 (ZnS) | 81.4 | 156.0 |
Additionally, the thermal behavior of the zinc xanthates and potassium xanthate (KHep), respectively, was investigated by means of electron impact ionization (EI) mass spectrometry (MS) with direct insertion (DI) of samples. In comparison to common TGA-MS – where samples are heated under atmospheric pressure – DI/EI offers the possibility for analogous experiments under high vacuum conditions. Consequently, species with reduced volatility can be evaporated and detected too.
As starting point, KHep was investigated to gather more information concerning the thermal behavior of the ligand. First, it is important to note that no ions were observed that indicated (partial) evaporation of KHep. Mass spectra showing various ions of volatile thermal decomposition products of KHep after approximately 20% and 80% thermal decomposition, respectively, are depicted in Fig. S5a and S5b in the ESI.† The following products were identified by accurate mass analysis (interpretations as well as calculated exact mass data are given in the caption of Fig. S5,† compare with data shown in the spectra), as well as by comparison with library spectra (Wiley/NIST):
(i) 4,4-dimethyl-2-pentene and (ii) COS
(iii) 2,2-dimethyl-3-pentanol and (iv) CS2
The formation of an alcohol and CS2 in addition to the products of the Chugaev elimination (alkene and COS) was also already found by Gilman et al. as an alternative competing mechanism for the pyrolysis of organic xanthates.64
The extracted ion chromatograms of the signals assigned to 4,4-dimethyl-2-pentene and COS, respectively, showed quite the same time/temperature dependence, what is assigned to simultaneous formation of these two products according to a Chugaev mechanism. They were the first observable decomposition products and continuously formed during the whole thermal process. Chromatograms of ions typical for 2,2-dimethyl-3-pentanol and CS2, respectively, also showed rather equal time/temperature dependent development. However, a comparison with the chromatograms of the Chugaev products clearly indicated that the formation of these products starts a bit later and intensifies significantly later on during the thermal decomposition process. Comparison of the relative intensities of corresponding ions in Fig. S5a and S5b† show that too. A possible explanation would be that KSH – which is a product from the Chugaev reaction – donates protons for the direct formation of 2,2-dimethyl-3-pentanol, and that subsequently CS2 is formed via thermal decomposition of an intermediate like potassium trithiocarbonate or that protonation leads to the formation of the corresponding free xanthic acid which is thermally unstable and also decomposes to 2,2-dimethyl-3-pentanol and CS2.
During DI/EI-MS of the zinc xanthates, first volatile species were generally observed at temperatures a bit below the onset temperature for mass loss in TGA. The thermal decomposition of ZnHep appeared comparable to KHep, but some distinct differences were observed. First, the molecular ion M+ of ZnHep (C16H30O2S4Zn, m/zcalc = 446.0420 Da; m/zfound = 446.0423 Da) as well as characteristic fragment ions (e.g. [M − Xanthate]+, C8H15OS2Zn, m/zcalc = 254.9856 Da, m/zfound = 254.9866 Da) were detected (Fig. 4). This unambiguously proves the structure of ZnHep as well as partial evaporation under high vacuum conditions. Signals below 120 Da corresponded well to those observed during the investigation of KHep (compare Fig. S5 and 4†). Normalized extracted ion chromatograms of selected ions indicate that the thermal procedure – when using a heating rate of 20 °C min−1 (Fig. 4b) – starts with simultaneous formation of 2,2-dimethyl-3-pentanol and CS2, respectively. Next, evaporation of some undecomposed ZnHep intensifies and reaches its maximum shortly before the also quite well matching formation of the Chugaev products. At a higher heating rate, 100 °C min−1, the sequence of the decomposition products is the same, but they are more visible (Fig. 4c). This decomposition sequence generally differs from the thermal behavior of KHep, where the decomposition started with development of the Chugaev products. However, these DI/EI data clearly indicate “steps”/“a sequence” in the thermal behavior of ZnHep, with significant overlap of individual processes. Note, that TGA data also showed two not clearly separated steps during the thermal decomposition of ZnHep. Keeping in mind that the TGA experiments were performed close to atmospheric pressure – where ZnHep will not evaporate, where transfer of heat and removal of volatile decomposition products will be different, etc. – DI/EI-MS and TGA fit quite satisfactory.
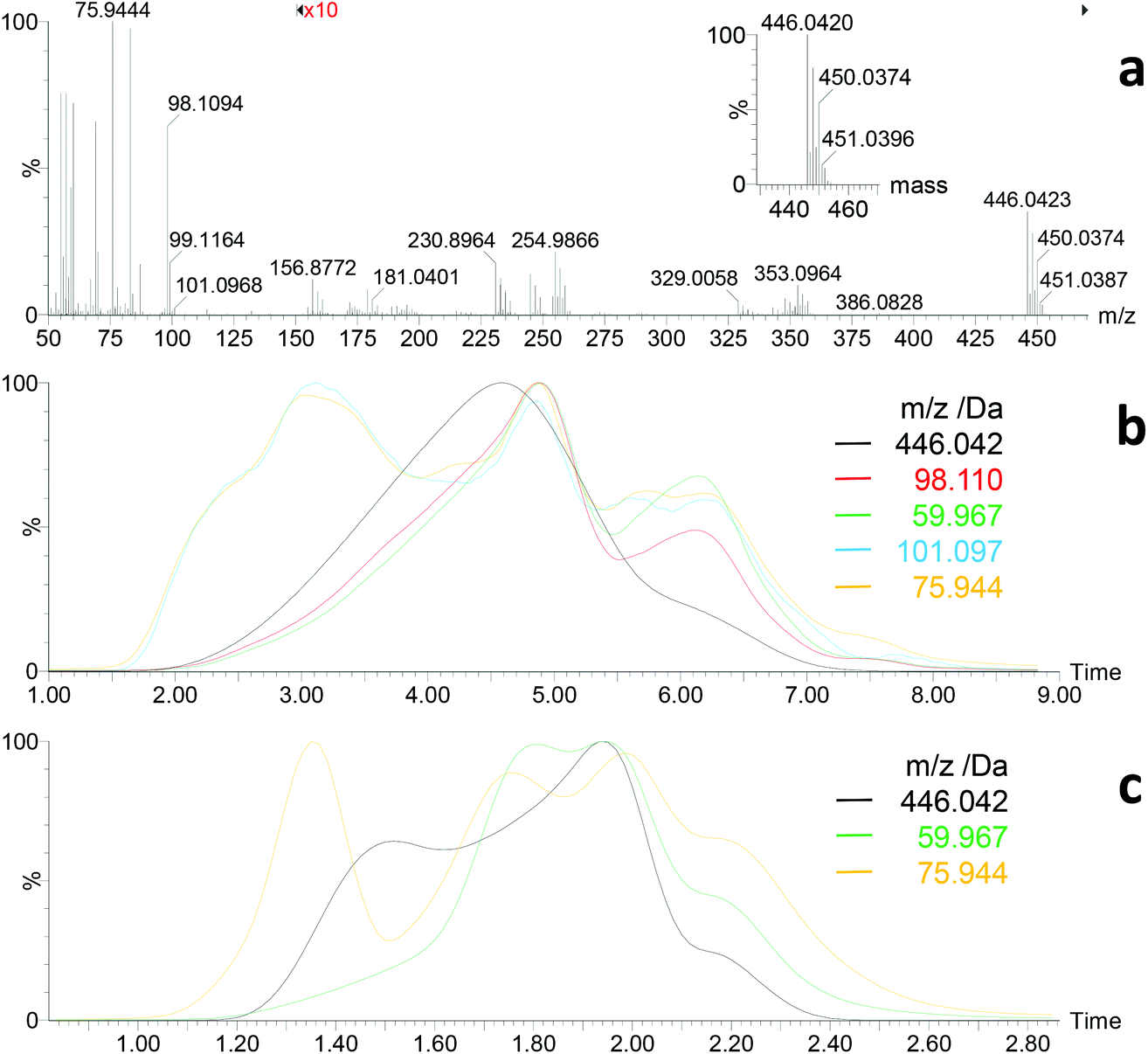 | ||
| Fig. 4 DI/EI mass spectrometry of ZnHep. (a) Mass spectrum after approx. 30–40% thermal decomposition/evaporation showing M+ of ZnHep at m/z = 446.0423 Da (inset: calculated isotope pattern for C16H30O2S4Zn), fragment ions (e.g. [M − CHOS2]+, C15H29OS2Zn, m/zcalc = 353.0951 Da; [M − Xanthate]+, C8H15OS2Zn, m/zcalc = 254.9856 Da), as well as xanthate decomposition products in the low mass region (compare Fig. S5†). (b) Normalized extracted ion chromatograms at a heating rate of 20 and (c) 100 °C min−1, respectively (M+ of ZnHep, 446.042 Da; M+ of 4,4-dimethyl-2-pentene, 98.110 Da; M+ of COS, 59.967 Da; [M − CH3]+ of 2,2-dimetyl-3-pentanol, 101.097 Da; M+ of CS2, 75.994 Da). | ||
DI/EI data of ZnHepTMEDA basically showed (i) early intensifying evaporation of TMEDA, (ii) partial evaporation of complexed zinc, and again (iii) thermal degradation leading to COS, CS2, etc. In detail, the behavior turned out to be a bit more complex. The highest mass observed was at m/z = 446.0424 Da (Fig. S6a†). This corresponds well to [ZnHepTMEDA − TMEDA]+ = ZnHep+ (C16H30O2S4Zn+, m/zcalc = 446.0420 Da). Thus, the molecular ion M+ of ZnHepTMEDA (C22H46N2O2S4Zn, m/zcalc = 562.1733 Da) was not detected. This happens frequently in DI/EI-MS due to intense fragmentation. But ion chromatograms of selected masses (Fig. S6b and S6c;†m/z = 446.042 (ZnHep+) and 116.131 Da (M+ of TMEDA), respectively) suggest, that thermal decomposition of ZnHepTMEDA to ZnHep and TMEDA (as well as other decomposition products) is faster than simple evaporation, and that this is the reason why the M+ was not detected. A large fraction of TMEDA evaporates quite early, but significant amounts were continuously found until the thermal process was completed, especially when lower heating rates were used (Fig. S6b†). The evaporation of ZnHep starts and intensifies a bit later and drops quite rapidly after reaching a maximum. The evaporation of TMEDA shows an additional maximum in the regime where the release of ZnHep already decreases. This is assigned to evaporation of TMEDA temporarily bonded to non-volatile decomposition products containing zinc. Concerning the development of decomposition products of the xanthate ligand (COS, CS2, etc.), it is important to note that a rather strong dependence on the heating rate was observed. Using fast heating rates, the “sequence” for the development of these products was rather comparable to what was observed for pure ZnHep – “CS2 intensifies first” (compare Fig. 4 and Fig. S6c†). In contrast to that, when ZnHepTMEDA is heated slower, COS and CS2 are developed rather simultaneously (Fig. S6b†). Hence, the presence of TMEDA seems to have an impact on the thermal decomposition of the xanthate groups too. Together with the observation that fractions of TMEDA remain in the samples quite long, this can of course have an impact on the formation process of the zinc sulfide.
The DI/EI-MS results obtained for ZnHepPyr were in principle rather comparable (Fig. S7†). Again, the molecular ion was not detected. Data indicate thermally triggered decomposition to ZnHep and pyridine as the first step. The release of pyridine starts earlier and intensified faster in comparison to the development of TMEDA during heating of ZnHepTMEDA. This fits well to what has to be expected for the monodentate in comparison to the bidentate ligand. Nevertheless, again significant amounts of the N-donor ligand were found until the thermal process was completed. The influence of the heating rate on the “sequence” of the formation of degradation products from the xanthate ligands (COS, CS2) was less pronounced.
Conversion to nanocrystalline ZnS
In a further step, the compounds were used as single-source precursors (SSPs) for the synthesis of ZnS. The conversion reaction was performed at 300 °C and 400 °C under inert conditions (N2 atmosphere) because the presence of the oxygen at high temperatures causes the production of ZnO/ZnS mixtures.65,66 X-ray diffraction measurements were performed in order to gain information regarding their crystallinity and phase composition. The obtained data show that the chosen temperature has a significant influence on the primary crystallite size, as well as the phase composition. According to the TGA results, the main decomposition of the compounds is completed at 250–300 °C. The XRD patterns (Fig. 5) prove the formation of ZnS by exhibiting the three main diffraction peaks of cubic ZnS at 28.6 (111), 47.7 (220) and 56.5° 2θ (311) (reference pattern for cubic ZnS: COD ID: 1100043). Furthermore, the broad diffraction peaks at 28.6° 2θ reveal minor shoulders at approx. 27.1 and 30.6° 2θ, which indicates the presence of small amounts of the hexagonal wurtzite phase of ZnS.67 In the diffraction patterns of the samples prepared from the ZnHep and ZnHepPyr precursors at 400 °C, the shoulder at 26.8° 2θ is most pronounced and even visible as an additional minor diffraction peak. A mixed cubic and hexagonal stacking is not uncommon for ZnS nanocrystals and has been already found in other studies dealing with ZnS37,68 and isostructural CdS.24,69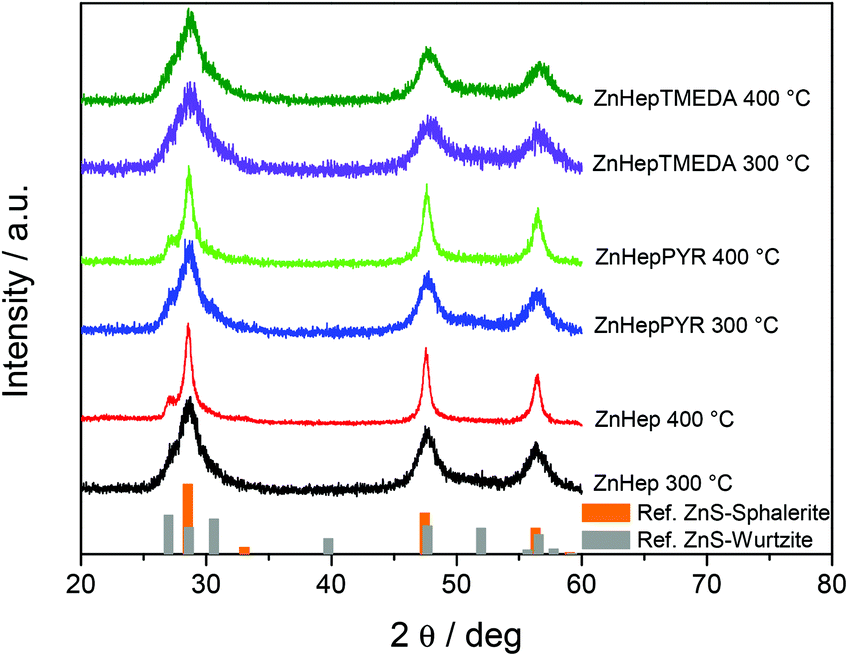 | ||
| Fig. 5 X-ray diffraction patterns of the ZnS powders obtained by decomposition of the zinc xanthate precursors ZnHep, ZnHepTMEDA and ZnHepPyr, at 300 and 400 °C with reference patterns for cubic sphalerite ZnS (COD ID: 1100043) and hexagonal wurtzite ZnS (COD ID: 9008878). The diffraction patterns are shifted vertically for better visibility. | ||
The broad diffraction peaks signify the formation of nanocrystalline ZnS. As it was expected, in all cases the primary crystallite size (estimated via the Scherrer formula) is larger in the ZnS samples prepared at 400 °C (see Table 4). The primary crystallite sizes of ZnS synthesized at 300 °C show minor differences, but a slight decrease from ZnHep to ZnHepPyr to ZnHepTMEDA is observed. The ZnS samples prepared at 400 °C exhibit bigger differences in their crystallite sizes, however, the same trend, a decrease in primary crystallite size from ZnHep to ZnHepPyr to ZnHepTMEDA is also observed, while the ZnS nanocrystals synthesized from the ZnHepTMEDA precursor have a distinctly smaller size compared to the other samples. These differences in crystallite size originate from the fact that minor amounts of the coligands are not removed at the first steps of the decomposition (according to the MS results), but remain temporarily bound to zinc-containing species and thereby act as capping ligands in the zinc sulfide nanocrystal formation. Thus, the ZnS nanocrystal growth is less pronounced in case of ZnHepTMEDA and ZnHepPyr, compared to ZnHep. As a result, the ZnHep gives the biggest crystals at both temperatures. In case of ZnHepPyr, some pyridine is bonded on zinc products leading at smaller crystals at 300 °C, but due to its monodental character, it is cleaved at higher temperatures, leading to crystal sizes similar to ZnHep. Additionally, the TMEDA ligand seems to bond more steadily on zinc products due to its bidental character, even at higher temperatures, leading to significantly smaller crystals at both temperatures compared to the other two compounds. Moreover, the lattice constants of the final ZnS powders are slightly influenced by the presence of a coligand. The ZnHep precursor produces ZnS with a lattice constant of 5.412 Å, whereas a small decrease is observed for ZnHepPyr (5.403 Å) and a more significant decrease is found for ZnHepTMEDA with a lattice constant of 5.387 Å. These differences are in accordance with the changes monitored among different primary crystallite sizes, as it has been described in previous work.70
| Compound | Primary crystallite size at 300 °C (nm) | Primary crystallite size at 400 °C (nm) |
|---|---|---|
| ZnHep | 4.9 | 14.1 |
| ZnHepTMEDA | 3.1 | 4.4 |
| ZnHepPyr | 4.7 | 11.4 |
The morphology of the final ZnS powders was observed by field emission scanning electron microscopy (FESEM). Fig. 6 shows the images of the ZnS powders resulted from the different zinc xanthate precursors after annealing at 400 °C, which reveal that the starting material influences the morphology of the final ZnS samples. The presence of a ligand on the zinc xanthate results in a material that exhibits less compactness compared to the ligand-free precursor, which gives a highly dense material. On the other hand, the type of the ligand also affects the morphology. The pyridine ligand leads to ZnS that consists of slightly packed spherical clusters, while the TMEDA containing zinc xanthate provides a material, which exhibits a smoother surface with high porosity.
 | ||
| Fig. 6 FESEM images at two different magnifications of the ZnS powders derived from (a, d) ZnHep, (b, e) ZnHepPyr and (c, f) ZnHepTMEDA, after decomposition at 400 °C under inert conditions. | ||
Conclusion
In this study, two new zinc xanthate complexes of zinc(II) O-2,2-dimethylpentan-3-yl-dithiocarbonate (ZnHep) with two amine coligands, TMEDA and pyridine (ZnHepTMEDA and ZnHepPyr, respectively) were prepared, characterized and their use as single source precursor for ZnS was demonstrated. The crystal structures revealed that in the case of ZnHepTMEDA zinc is fourfold coordinated in a tetrahedral arrangement with both xanthate ligands bound monodentally. In contrast to that, ZnHepPyr shows a fivefold, square pyramidal coordination of zinc with the two chelating xanthate units forming the square and pyridine on the top. However, the thermal decomposition is only slightly influenced by the additional ligand and all three compounds lead to ZnS at temperatures below 300 °C. Mass spectrometry indicates that the decomposition of the xanthates takes place via the Chugaev elimination leading to COS and the corresponding alkene as well as via protonation of the xanthate leading to the formation of CS2 and the corresponding alcohol. Additionally, the ligands pyridine as well as TMEDA already split off at very low temperatures and are thus influencing the decomposition, however, in both cases, some of the ligands remain till rather high temperatures in the material, indicating that they might act as coordinative ligands during the growth of the ZnS nanocrystals. This is supported by the XRD results, as the primary crystallite size is dependent on the precursor, leading to smaller crystallites if pyridine or TMEDA are present.These findings reveal that the use of coligands clearly induces changes in the decomposition pathway of metal xanthates as well as in the structure of the resulting metal sulfides. Thus, further research should be directed to elaborate the correlation between the structure of the ligand and the thermal reactions, which would then enable the design of new metal xanthate compounds with a better control and tunability of the final metal sulfide thin films.
Experimental section
All chemicals and solvents were used as purchased without any additional purification: chloroform (≥99.2%, VWR), methanol (VWR), diethyl ether (≥99.5%, Sigma-Aldrich), ethanol (≥99.8%, Sigma-Aldrich), acetone (≥99%, Sigma-Aldrich), deuterium oxide (Eurisotop), deuterated chloroform (99.8 atom % D, 0.03% (v/v) TMS, Eurisotop), potassium tert-butoxide (≥98%, Sigma-Aldrich), 2,2-dimethyl-3-pentanol (≥97%, Sigma-Aldrich), carbon disulfide (≥99.9%, Sigma-Aldrich), zinc chloride (≥98%, Sigma-Aldrich), pyridine (≥99%, Sigma-Aldrich), N,N,N′,N′-tetramethylethylenediamine (≥98%, Merck).Potassium O-2,2-dimethylpentan-3-yl-dithiocarbonate (1)
Potassium O-2,2-dimethylpentan-3-yl dithiocarbonate was synthesized for further use in the metal xanthates synthesis. The compound was synthesized according to a previous procedure.71 Potassium tert-butoxide (1.0 equiv.) was dissolved in dry THF under N2 flow. The solution was then cooled down to 0 °C and the 2,2-dimethyl-3-pentanol (1.1 equiv.) was slowly added. After several minutes stirring, CS2 (1.1 equiv.) was added dropwise. The reaction mixture was stirred for 5 h under N2 at 0 °C. Following, the product was precipitated by adding diethyl ether and collected by filtration. For recrystallization, the dry product was dissolved in acetone and precipitated in diethyl ether. The yellowish solid was collected by filtration and dried under vacuum.
1
H-NMR (300 MHz, D2O): δ = 5.38–5.34 (m, 1H, CH), 1.80–1.55 (m, 2H, CH2), 0.93–0.86 (m, 12H, 4 × CH3) ppm. 13C-NMR (75 MHz, D2O): δ = 234.3 (CS2O), 93.8 (CH), 35.1 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CH3)), 25.4 (C(H3)), 22.9 (CH2), 10.4 (CH2-H3) ppm.
(CH3)), 25.4 (C(H3)), 22.9 (CH2), 10.4 (CH2-H3) ppm.
Zinc(II) O-2,2-dimethylpentan-3-yl-dithiocarbonate (ZnHep) (2)
The compound was synthesized following an adapted procedure reported previously.34,54 An aqueous solution of compound (1) (2.1 equiv.) was slowly added to an aqueous solution of zinc(II) chloride (1.0 equiv.) under ambient conditions. The reaction mixture was stirred for 2 h. The white solid was filtered off, washed with plenty of distilled water and dried under vacuum. For recrystallization, the dry product was dissolved in chloroform and precipitated by a water/methanol mixture. The pure white powder was collected and dried under vacuum. 1H-NMR (300 MHz, CDCl3): δ = 5.30 (t, 1H, CH), 1.97–1.89 (m, 2H, CH2), 1.48–0.95 (m, 12H, 4 × CH3) ppm. 13C-NMR (76 MHz, CDCl3): δ = 229.1 (CS2O), 86.0 (CH), 49.4 ((CH3)), 30.2 (C(H3)), 29.9 (CH2), 21.5 (CH2-H3) ppm. FT-IR (cm−1): see Table S1.†
Bis(O-2,2-dimethylpentan-3-yl-dithiocarbonato)(N,N,N′,N′-tetramethylethylenediamine)zinc(II) (ZnHepTMEDA) (3)
Compound (2) (1.0 equiv.) was dissolved in methanol and stirred for some minutes at room temperature. N,N,N′,N′-Tetramethylethylenediamine (TMEDA) (1.2 equiv.) was then added and the reaction mixture was stirred for 2 h. The white precipitate was collected by filtration, washed with distilled water and dried under vacuum. For recrystallization, the dry product was dissolved in chloroform and precipitated by water/methanol mixture. The pure white powder was collected and dried under vacuum. 1H-NMR (300 MHz, CDCl3): δ = 5.46 (t, 1H, CH), 2.68 (s, 4H, 2 × CH2, TMEDA), 2.58 (s, 6H, 2 × CH3, TMEDA), 1.95–1.87 (m, 2H, CH2), 1.39–0.93 (m, 12H, 4 × CH3). 13C-NMR (76 MHz, CDCl3): δ = 225.9 (CS2O), 81.2 (CH), 57.0 (CH2, TMEDA), 49.6 ((CH3)), 46.9 (CH3, TMEDA), 30.3 (C(H3)), 30.0 (CH2), 21.6 (CH2-H3). FT-IR (cm−1): see Table S1.†
Bis(O-2,2-dimethylpentan-3-yl-dithiocarbonato)(pyridine)zinc(II) (ZnHepPyr) (4)
Compound (2) (1.0 eqiv) was dissolved in methanol and stirred for some minutes at room temperature. Pyridine (1.2 equiv.) was then added and the reaction mixture was stirred for 2 h. The white precipitate was collected by filtration, washed with distilled water and dried under vacuum. For recrystallization, the dry product was dissolved in chloroform and precipitated by water/methanol mixture. The pure white powder was collected and dried under vacuum. 1H-NMR (300 MHz, CDCl3): δ = 8.94 (d, 2H, 2 × CH, pyridine), 8.00 (t, 1H, CH, pyridine), 7.59 (t, 2H, 2 × CH, pyridine), 5.35 (t, 1H, CH), 1.97–1.90 (m, 2H, CH2), 1.46–0.97 (m, 12H, 4 × CH3). 13C-NMR (76 MHz, CDCl3): δ = 228.7 (CS2O), 149.1 (CH, pyridine), 140.0 (CH, pyridine), 125.5 (CH, pyridine) 83.7 (CH), 49.5 ((CH3)), 30.2 (C(H3)), 30.0 (CH2), 21.5 (CH2-H3). FT-IR (cm−1): see Table S1.†
Conversion of the precursors to nanocrystalline zinc sulfide
The zinc xanthate precursor (300 mg) was placed in glass containers and subsequently inserted into a quartz tube. The tube was placed in a furnace and heated with a rate of 10 °C min−1 to the desired temperature (300 and 400 °C). The samples stayed at the desired temperature for 15 min and then cooled down to room temperature. The whole procedure was performed under N2 flow.NMR spectra were recorded on a Bruker Ultrashield 300 MHz NMR spectrometer (1H-NMR: 300 MHz, 13C NMR: 75 MHz). The solvents used were deuterated chloroform with tetramethylsilane (TMS) (1H NMR: 7.26 ppm, 13C NMR: 77.16 ppm) and deuterium oxide (1H NMR: 4.79 ppm).72 The spectra were evaluated with TopSpin 3.1 from Bruker. The chemical shifts were given in units of the δ scale in parts per million (ppm).
FTIR spectra were measured on a Bruker Alpha FTIR spectrometer in attenuated total reflection (ATR) mode using the ALPHA's Platinum ATR single-reflection diamond ATR module. All spectra were recorded in a range between 4000 and 400 cm−1 with 24 scans and the air as background.
Electron impact (EI; 70 eV, source temperature 250 °C) mass spectra were recorded on an orthogonal time-of-flight mass spectrometer (Waters GCT Premier) equipped with a direct insertion probe (DI). Tiny crystals of the samples were filled in the glass cup used for the DI and rapidly transferred into the vacuum. The acquisition of mass spectra (mass range: 50–800 Da; 1 spectrum per s; resolution: approx. 7500 FWHM) was started immediately. Spectra were continuously acquired while the sample was heated from room temperature to 500 °C (rapidly heated until detection of first volatile species, afterwards with a heating between 20 and 100 °C min−1). Data were processed using MassLynx (version 4.1).
Thermal gravimetric measurements were performed on a Netzsch Jupiter STA 449C thermogravimetric analyzer in aluminium oxide crucibles under helium atmosphere with a flow rate of 50 mL min−1. The operated temperature range was between 20–550 °C with a heating rate of 10 °C min−1.
X-ray diffraction measurements were taken on a RIGAKU MiniFlex 600 with D/Tex Ultra detector operated at 40 kV and 15 mA using CuKα radiation (λ = 1.5418 Å). The measurement was performed on a silicon zero-background sample holder. The diffraction patterns were evaluated with the SmartLabStudioIIx64v4.2.890 software.
For the single-crystal X-ray diffraction measurements, crystals were prepared with an antisolvent method. The purified xanthates were dissolved in chloroform and filtered through a 0.2 μm filter to remove any impurities. The filtered solution was placed in a glass vial and covered with a paraffin film, which was pierced with a needle. The glass vial was placed in a sealable screw vessel, which had previously been filled with a small amount of the antisolvent (ethanol). ZnHepTMEDA crystallized at room temperature, while ZnHepPyr at about 4 °C in the refrigerator.
Scanning electron microscopic images were acquired on a TESCAN MIRA3 Field Emission Scanning Electron Microscope (FESEM) using an In-Beam secondary electron (SE) detector. The microscope was operated at 5 kV, with a working distance of about 4 mm and an electron beam spot size of 4 nm. Samples for FESEM characterisations were coated with a thin carbon layer by sputtering.
All crystals suitable for single crystal X-ray diffractometry were removed from a vial and immediately covered with a layer of silicone oil. A single crystal was selected, mounted on a glass rod on a copper pin, and placed in the cold N2 stream provided by an Oxford Cryosystems cryostream. XRD data collection was performed for compounds ZnHepPyr and ZnHepTMEDA, on a Bruker APEX II diffractometer with use of Mo Kα radiation (λ = 0.71073 Å) and a CCD area detector. Empirical absorption corrections were applied using SADABS or TWINABS.73,74 The structures were solved with use of the intrinsic phasing option in SHELXT and refined by the full-matrix least-squares procedures in SHELXL.75–77 The space group assignments and structural solutions were evaluated using PLATON.78,79 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in calculated positions corresponding to standard bond lengths and angles. Disorder was handled by modelling the occupancies of the individual orientations using free variables to refine the respective occupancy of the affected fragments (PART).80 In some cases, the similarity SAME restraint, the similar-ADP restraint SIMU and the rigid-bond constraints EXYZ and EADP were used in modelling disorder to make the ADP values of the disordered atoms more reasonable. In some cases, the distances between arbitrary atom pairs were restrained to possess the same value using the SADI instruction. In some tough cases of disorder, anisotropic Uij-values of the atoms were restrained (ISOR) to behave more isotropically. In ZnHepTMEDA, disordered positions for the dithiocarbonate group were refined using 90/10 split positions. All crystal structures representations were made with the program Diamond81 with all atoms displayed as 30% ellipsoids. CIF files were edited, validated and formatted either with the program publCIF82 or Olex2.83
Table 5 contains crystallographic data and details of measurements and refinement for compounds ZnHepPyr and ZnHepTMEDA. CCDC 1993703 – (ZnHepTMEDA) and 1993704 – (ZnHepPyr)† contain the supplementary crystallographic data for compounds ZnHepPyr and ZnHepTMEDA, respectively.
| Compound | ZnHepPyr | ZnHepTMEDA |
|---|---|---|
| Mo Kα(λ = 0.71073 Å). R1 = Σ/|Fo| − |Fc|/|Σ|Fd; wR2 = [Σw(Fo2 − F22)2/Σw(Fo2)2]1/2. | ||
| Formula | C21H35NO2S4Zn | C22H46N2O2S4Zn |
| Fw (g mol−1) | 527.11 | 564.22 |
| a (Å) | 23.559(8) | 11.1922(3) |
| b (Å) | 8.682(3) | 11.5319(3) |
| c (Å) | 13.815(5) | 22.9826(6) |
| α (°) | 90 | 90 |
| β (°) | 101.55(2) | 90 |
| γ (°) | 90 | 90 |
| V (Å3) | 2768.5(17) | 2966.30(14) |
| Z | 4 | 4 |
| Crystal size (mm) | 0.20 × 0.15 × 0.05 | 0.34 × 0.27 × 0.22 |
| Crystal habit | Block, colourless | Block, colourless |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | C2/c | Pbcn |
| d calc (Mg m−3) | 1.265 | 1.263 |
| μ (mm−1) | 1.20 | 1.13 |
| T (K) | 100(2) | 100(2) |
| 2θ range (°) | 2.5–31.1 | 3.1–27.9 |
| F(000) | 1112 | 1208 |
| T min, Tmax | 0.622, 0.747 | 0.673, 0.746 |
| R int | 0.073 | 0.041 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 71670, 4577, 3236 |
75890, 3246, 3102 |
| Independent reflections | 4577 | 3246 |
| No. of parameters, restrains | 137, 0 | 178, 36 |
| Δρmax, Δρmin (e Å−3) | 1.37, −0.81 | 0.55, −0.31 |
| R 1, wR2 (all data) | R 1 = 0.0858 | R 1 = 0.0367 |
| wR2 = 0.1571 | wR2 = 0.0728 | |
| R 1, wR2 (>2σ) | R 1 = 0.0544 | R 1 = 0.0349 |
| wR2 = 0.1396 | wR2 = 0.0735 | |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Graz University of Technology for financial support through the Lead Project Porous Materials at Work (LP-03) as well as Josefine Hobisch and Karin Bartl for their support regarding thermogravimetric analysis and mass spectrometric measurements.References
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, Prog. Mater. Sci., 2011, 56, 175–287 CrossRef CAS.
- A. K. Kole and P. Kumbhakar, Results Phys., 2012, 2, 150–155 CrossRef.
- S. B. Qadri, E. F. Skelton, D. Hsu, A. D. Dinsmore, J. Yang, H. F. Gray and B. R. Ratna, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 9191–9193 CrossRef CAS.
- C. S. Tiwary, P. Kumbhakar, A. K. Mitra and K. Chattopadhyay, J. Lumin., 2009, 129, 1366–1370 CrossRef CAS.
- X. Ma, J. Song and Z. Yu, Thin Solid Films, 2011, 519, 5043–5045 CrossRef CAS.
- Y. Xie, C. Geng, Y. Gao, J. G. Liu, Z.-H. Zhang, Y. Zhang, S. Xu and W. Bi, Materials, 2017, 10, 1242 CrossRef.
- F. Montanarella, T. Altantzis, D. Zanaga, F. T. Rabouw, S. Bals, P. Baesjou, D. Vanmaekelbergh and A. van Blaaderen, ACS Nano, 2017, 11, 9136–9142 CrossRef CAS.
- J. Z. Liu, P. X. Yan, G. H. Yue, J. B. Chang, D. M. Qu and R. F. Zhuo, J. Phys. D: Appl. Phys., 2006, 39, 2352–2356 CrossRef CAS.
- A. Goudarzi, A. D. Namghi and C.-S. Ha, RSC Adv., 2014, 4, 59764–59771 RSC.
- X. Yang, B. Chen, J. Chen, Y. Zhang, W. Liu and Y. Sun, Mater. Sci. Semicond. Process, 2018, 74, 309–312 CrossRef CAS.
- V. Kumar, I. Rawal, V. Kumar and P. K. Goyal, Phys. B, 2019, 575, 411690 CrossRef CAS.
- S. Hussain, T. Liu, M. S. Javed, N. Aslam and W. Zeng, Sens. Actuators, B, 2017, 239, 1243–1250 CrossRef CAS.
- Q. An, X. Meng, K. Xiong, Y. Qiu and W. Lin, Nanotechnology, 2017, 28, 105502 CrossRef.
- R. Zhou and M. I. Guzman, J. Phys. Chem. C, 2016, 120, 7349–7357 CrossRef CAS.
- Z. Ye, L. Kong, F. Chen, Z. Chen, Y. Lin and C. Liu, Optik, 2018, 164, 345–354 CrossRef CAS.
- X. Xu, S. Li, J. Chen, S. Cai, Z. Long and X. Fang, Adv. Funct. Mater., 2018, 28, 1802029 CrossRef.
- D. Barreca, A. Gasparotto, C. Maragno, E. Tondello and C. Sada, Chem. Vap. Deposition, 2004, 10, 229–236 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- D. Moore and Z. L. Wang, J. Mater. Chem., 2006, 16, 3898–3905 RSC.
- M. P. Sarma, J. M. Kalita and G. Wary, Mater. Sci. Semicond. Process, 2017, 61, 131–136 CrossRef CAS.
- S. Shen, Y. Zhang, L. Peng, B. Xu, Y. Du, M. Deng, H. Xu and Q. Wang, CrystEngComm, 2011, 13, 4572–4579 RSC.
- D. Barreca, E. Tondello, D. Lydon, T. R. Spalding and M. Fabrizio, Chem. Vap. Deposition, 2003, 9, 93–98 CrossRef CAS.
- I. Jen-La Plante, T. W. Zeid, P. Yang and T. Mokari, J. Mater. Chem., 2010, 20, 6612–6617 RSC.
- N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS.
- A. J. MacLachlan, T. Rath, U. B. Cappel, S. A. Dowland, H. Amenitsch, A.-C. Knall, C. Buchmaier, G. Trimmel, J. Nelson and S. A. Haque, Adv. Funct. Mater., 2015, 25, 409–420 CrossRef CAS.
- D. Barreca, A. Gasparotto, C. Maragno and E. Tondello, J. Electrochem. Soc., 2004, 151, G428 CrossRef CAS.
- P. Selvaganapathi, S. Thirumaran and S. Ciattini, Polyhedron, 2018, 149, 54–65 CrossRef CAS.
- C. Buchmaier, M. Glänzer, A. Torvisco, P. Poelt, K. Wewerka, B. Kunert, K. Gatterer, G. Trimmel and T. Rath, J. Mater. Sci., 2017, 52, 10898–10914 CrossRef CAS.
- M. Al-Shakban, P. D. Matthews, E. A. Lewis, J. Raftery, I. Vitorica-Yrezabal, S. J. Haigh, D. J. Lewis and P. O'Brien, J. Mater. Sci., 2019, 54, 2315–2323 CrossRef CAS.
- P. D. McNaughter, S. A. Saah, M. Akhtar, K. Abdulwahab, M. A. Malik, J. Raftery, J. A. M. Awudza and P. O'Brien, Dalton Trans., 2016, 45, 16345–16353 RSC.
- S. A. Alderhami, D. Collison, D. J. Lewis, P. D. McNaughter, P. O'Brien, B. F. Spencer, I. Vitorica-Yrezabal and G. Whitehead, Dalton Trans., 2019, 48, 15605–15612 RSC.
- V. Kaltenhauser, T. Rath, W. Haas, A. Torvisco, S. K. Müller, B. Friedel, B. Kunert, R. Saf, F. Hofer and G. Trimmel, J. Mater. Chem. C, 2013, 1, 7825–7832 RSC.
- V. Perner, T. Rath, F. Pirolt, O. Glatter, K. Wewerka, I. Letofsky-Papst, P. Zach, M. Hobisch, B. Kunert and G. Trimmel, New J. Chem., 2019, 43, 356–363 RSC.
- T. Rath, M. Edler, W. Haas, A. Fischereder, S. Moscher, A. Schenk, R. Trattnig, M. Sezen, G. Mauthner, A. Pein, D. Meischler, K. Bartl, R. Saf, N. Bansal, S. A. Haque, F. Hofer, E. J. W. List and G. Trimmel, Adv. Energy Mater., 2011, 1, 1046–1050 CrossRef CAS.
- T. Rath, A. J. MacLachlan, M. D. Brown and S. A. Haque, J. Mater. Chem. A, 2015, 3, 24155–24162 RSC.
- M. Al-Shakban, P. D. Matthews, N. Savjani, X. L. Zhong, Y. Wang, M. Missous and P. O'Brien, J. Mater. Sci., 2017, 52, 12761–12771 CrossRef CAS.
- A. S. Pawar, S. Mlowe, S. S. Garje, M. P. Akerman and N. Revaprasadu, Inorg. Chim. Acta, 2017, 463, 7–13 CrossRef CAS.
- A. Fischereder, A. Schenk, T. Rath, W. Haas, S. Delbos, C. Gougaud, N. Naghavi, A. Pateter, R. Saf, D. Schenk, M. Edler, K. Bohnemann, A. Reichmann, B. Chernev, F. Hofer and G. Trimmel, Monatsh. Chem., 2013, 144, 273–283 CrossRef CAS.
- T. Rath, D. Scheunemann, R. Canteri, H. Amenitsch, J. Handl, K. Wewerka, G. Kothleitner, S. Leimgruber, A.-C. Knall and S. A. Haque, J. Mater. Chem. C, 2019, 7, 943–952 RSC.
- T. Rath, V. Kaltenhauser, W. Haas, A. Reichmann, F. Hofer and G. Trimmel, Sol. Energy Mater. Sol. Cells, 2013, 114, 38–42 CrossRef CAS.
- M. Al-Shakban, Z. Xie, N. Savjani, M. A. Malik and P. O'Brien, J. Mater. Sci., 2016, 51, 6166–6172 CrossRef CAS.
- M. D. Khan, G. Murtaza, N. Revaprasadu and P. O'Brien, Dalton Trans., 2018, 47, 8870–8873 RSC.
- M. D. Khan, M. Aamir, M. Sohail, S. Bhoyate, M. Hyatt, R. K. Gupta, M. Sher and N. Revaprasadu, Dalton Trans., 2019, 48, 3714–3722 RSC.
- M. A. Buckingham, A. L. Catherall, M. S. Hill, A. L. Johnson and J. D. Parish, Cryst. Growth Des., 2017, 17, 907–912 CrossRef CAS.
- N. Alam, M. S. Hill, G. Kociok-Köhn, M. Zeller, M. Mazhar and K. C. Molloy, Chem. Mater., 2008, 20, 6157–6162 CrossRef CAS.
- A. A. K. Bakly, B. F. Spencer and P. O'Brien, J. Mater. Sci., 2018, 53, 4360–4370 CrossRef CAS.
- T. Alqahtani, M. D. Khan, D. J. Kelly, S. J. Haigh, D. J. Lewis and P. O'Brien, J. Mater. Chem. C, 2018, 6, 12652–12659 RSC.
- G. B. Shombe, M. D. Khan, A. M. Alenad, J. Choi, T. Ingsel, R. K. Gupta and N. Revaprasadu, Sustainable Energy Fuels, 2020, 4, 5132–5143 RSC.
- C. Buchmaier, T. Rath, F. Pirolt, A.-C. Knall, P. Kaschnitz, O. Glatter, K. Wewerka, F. Hofer, B. Kunert, K. Krenn and G. Trimmel, RSC Adv., 2016, 6, 106120–106129 RSC.
- T. Rath, C. Padeste, M. Vockenhuber, C. Fradler, M. Edler, A. Reichmann, I. Letofsky-Papst, F. Hofer, Y. Ekinci and T. Griesser, J. Mater. Chem. A, 2013, 1, 11135–11140 RSC.
- M. D. Khan, J. Akhtar, M. A. Malik and N. Revaprasadu, ChemistrySelect, 2016, 1, 5982–5989 CrossRef CAS.
- R. Akram, M. D. Khan, C. Zequine, C. Zhao, R. K. Gupta, M. Akhtar, J. Akhtar, M. A. Malik, N. Revaprasadu and M. H. Bhatti, Mater. Sci. Semicond. Process., 2020, 109, 104925 CrossRef CAS.
- G. B. Shombe, M. D. Khan, C. Zequine, C. Zhao, R. K. Gupta and N. Revaprasadu, Sci. Rep., 2020, 10, 3260 CrossRef CAS.
- T. C. Vagvala, S. S. Pandey, S. Krishnamurthy and S. Hayase, Z. Anorg. Allg. Chem., 2016, 642, 134–139 CrossRef CAS.
- M. A. Adam, M. S. Refat, T. Sharshar and Z. K. Heiba, Spectrochim. Acta, Part A, 2012, 95, 458–477 CrossRef.
- X. H. Jiang, W. G. Zhang, Y. Zhong and S. L. Wang, Molecules, 2002, 7, 549–553 CrossRef CAS.
- D. Barreca, A. Gasparotto, C. Maragno, R. Seraglia, E. Tondello, A. Venzo, V. Krishnan and H. Bertagnolli, Appl. Organomet. Chem., 2005, 19, 59–67 CrossRef CAS.
- D. Barreca, A. Gasparotto, C. Maragno, R. Seraglia, E. Tondello, A. Venzo, V. Krishnan and H. Bertagnolli, Appl. Organomet. Chem., 2005, 19, 1002–1009 CrossRef CAS.
- E. P. Cullen, J. Doherty, A. R. Manning, P. McArdle and D. Cunningham, J. Organomet. Chem., 1988, 348, 109–122 CrossRef CAS.
- I. Ara, F. El Bahij and M. Lachkar, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2006, 36, 399–406 CrossRef CAS.
- J. Cusack, M. G. B. Drew and T. R. Spalding, Polyhedron, 2004, 23, 2315–2321 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- C. L. Raston, A. H. White and G. Winter, Aust. J. Chem., 1976, 29, 731–738 CrossRef CAS.
- R. E. Gilman and M. J. Bogdanowicz, Can. J. Chem., 1971, 49, 3362–3366 CrossRef CAS.
- Y.-Z. Yoo, Y. Osaka, T. Fukumura, Z. Jin, M. Kawasaki, H. Koinuma, T. Chikyow, P. Ahmet, A. Setoguchi and S. F. Chichibu, Appl. Phys. Lett., 2001, 78, 616–618 CrossRef CAS.
- J. Liu, Z. Guo, F. Meng, T. Luo, M. Li and J. Liu, Nanotechnology, 2009, 20, 125501 CrossRef.
- S. Biswas and S. Kar, Nanotechnology, 2008, 19, 045710 CrossRef.
- H. Zhang and J. F. Banfield, J. Phys. Chem. C, 2009, 113, 9681–9687 CrossRef CAS.
- T. Di Luccio, A. M. Laera, L. Tapfer, S. Kempter, R. Kraus and B. Nickel, J. Phys. Chem. B, 2006, 110, 12603–12609 CrossRef CAS.
- T. Rath, B. Kunert, R. Resel, G. Fritz-Popovski, R. Saf and G. Trimmel, Inorg. Chem., 2008, 47, 3014–3022 CrossRef CAS.
- A. González-Roura, J. Casas and A. Llebaria, Lipids, 2002, 37, 401–406 CrossRef.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- Bruker APEX2 and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement: A Crystallographer's Guide to SHELXL, Oxford University Press, 2006 Search PubMed.
- H. Putz and K. Brandenburg, Diamond – Crystal and Molecular Structure Visualization, 3.2i, Crystal Impact, Bonn, Germany Search PubMed.
- S. P. Westrip, J. Appl. Crystallogr., 2010, 43, 920–925 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional NMR, crystal packing, and mass spectrometry data. CCDC 1993703 and 1993704. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03065a |
| ‡ Both authors have equally contributed. |
| This journal is © The Royal Society of Chemistry 2020 |