
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular O- vs. N-coordination of pyridylidene amide ligands to iron determines activity in alcohol oxidation catalysis†
Dide G. A.
Verhoeven
and
Martin
Albrecht
 *
*
Departement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch
First published on 2nd November 2020
Abstract
A family of polydentate pyridine-substituted pyridylidene amide (PYA) complexes bound to iron(II) was developed. The variation of the coordination set from NN-bidentate PYA to tridentate pincer-type pyPYA2 systems (pyPYA2 = 2,6-bis(PYA)pyridine) had a large influence on the binding mode to iron(II), including a change from the N- to rare O-coordination of the PYA site and a concomitant shift of the predominant ligand resonance structure. These binding mode variations invoke changes in the reactivity of the complexes, which were probed in the peroxide-mediated oxidation of 1-phenylethanol to acetophenone. A comparison with uncomplexed FeCl2 indicated that bidentate NN coordination is unstable and presumably leads to the dissociation of FeCl2. In contrast, the tridentate ligand binding is robust. Remarkably, the tridentate PYA pincer coordination inhibits catalytic activity in the NNN binding mode, while the ONO coordination greatly enhances catalytic performance. Under optimized conditions, the bis-ligated ONO pincer iron complex [Fe(pyPYA2)2][2PF6] reaches full conversion within one hour (0.5 mol% catalyst loading) and under dilute conditions turnover numbers over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (0.005 mol% catalyst loading).
000 (0.005 mol% catalyst loading).
Introduction
Neutral N-donor ligands are ubiquitous in homogeneous catalysis, including imines, amines, pyridines, and as a more recent addition, pyridylidene amide (PYA) ligands.1,2 This latter scaffold has been emerging as a promising class of ligands with strong σ-donating properties. Analysis of PYA's σ-donor strength has been shown to resemble that of common C-coordinating N-heterocyclic carbene (NHC) ligands, making them unique nitrogen-based ligands for homogeneous catalysis.2–5 PYAs consist of a deprotonated amide core bound to an N-alkylated pyridinium ring (Scheme 1). These ligands constitute an overall neutral donor system which is comprised of multiple limiting resonance structures including the neutral imine structure A and zwitterionic structures B and C.2,6 Whereas evidence for resonance structures A and B have been obtained, for example from crystallographic and NMR data,6–9 structure C has solely been proposed.10 As a direct result of these multiple resonance structures, the ligand is electronically flexible in its donor properties to metal centers: it can alter between L-type bonding through resonance form A and X-type metal coordination through resonance form B, and therefore adapt to the influence and requirements of the metal center. Pyridine substituted PYA ligands (Scheme 1, R′ = 2-pyridine) are related to the more common pyridine-bis-carboxamide ligands.11 However, the formally neutral nature of the PYA ligands and their electronically flexible behavior impart unique properties when compared to the more frequently used dianionic and electronically static pyridine bis-carboxamide systems. For example, the CN-coordinating PYA ligand in the complex [Ir(PhPYA)(Cp*)L] adapts its binding mode from a larger contribution of resonance form A when L = Cl to predominantly form B in the solvento complex (L = MeCN) to compensate for the differently charged ligands L (Fig. 1).12 Owing to this electronic flexibility, the PYA ligands can stabilize multiple oxidation states on a metal center and their use has brought attractive results to precious metal complexes in homogeneous catalysis (Fig. 2), such as Pd(pyPYA)Cl2 for cross coupling reactions,2 Ir(PhPYA)(Cp*)Cl for water oxidation13 and [Ru(bisPYA)(p-cym)Cl]PF6 for olefin oxidation.14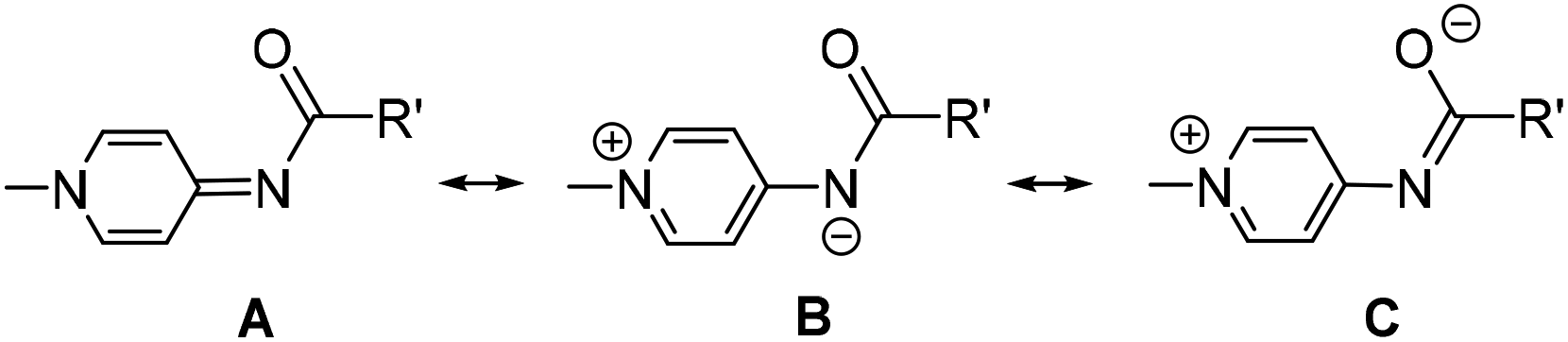 | ||
| Scheme 1 Resonance structures of a generalized PYA ligand. | ||
 | ||
| Fig. 1 Selected literature precedents of PYA complexes: [Pd(pyPYAR)Cl2] (R = Me, benzyl), [Ir(PhPYA)(Cp*)L] (L = Cl, MeCN), and [Ru(bisPYA)(p-cym)Cl]PF6. | ||
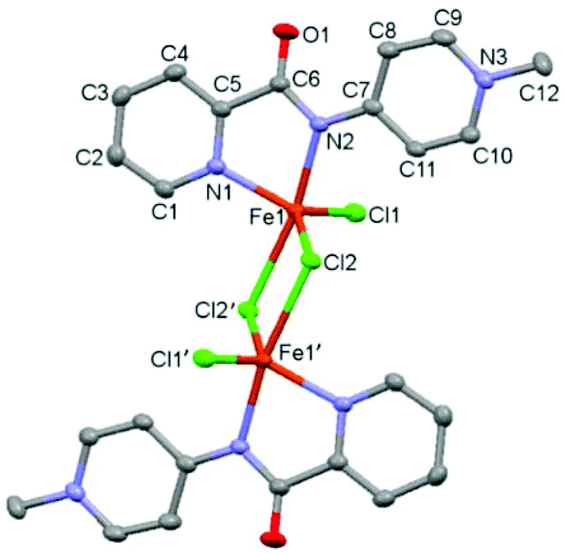 | ||
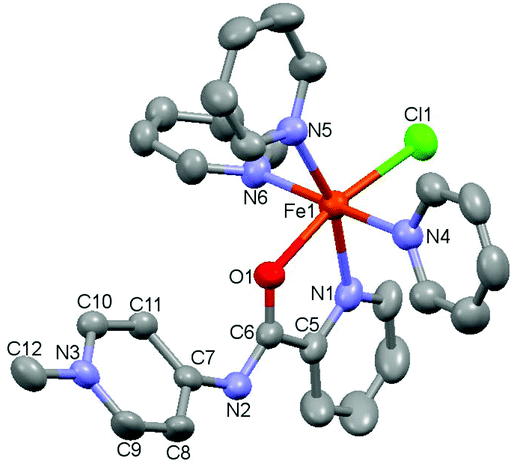
| Fig. 2 ORTEP representation of complex 2 (50% probability ellipsoids; hydrogen atoms omitted for clarity). Selected bond distances in Å: Fe1–N1: 2.1142(16), Fe1–N2: 2.1639(15), O1–C6: 1.229(2), N2–C6: 1.361(2), N2–C7: 1.378(2), N3–C9: 1.346(3), N3–C10: 1.354(2), N3–C12: 1.476(2), C5–C6: 1.511(3), C7–C8: 1.418(3), C7–C11: 1.411(3), C8–C9: 1.362(3), C10–C11: 1.365(3). Bond angle N1–Fe1–N2: 78.19(6)°. | ||
Their N-coordination and donor flexibility make PYA ligands particularly attractive for hard and Earth-abundant first row transition metals, offering the opportunity to stabilize catalytic intermediates of different oxidation states. For example, multidentate nitrogen donor ligands bound to iron have provided attractive catalytic performance for C–H oxidation reactions,15–17 olefin polymerization,18–20 and olefin oxidation.21 Considering these facts, it is remarkable that no PYA ligand has been coordinated to iron. Here, we report the coordination of polydentate PYA ligands to iron and their catalytic activity in oxidation catalysis. Changes in the ligand structure greatly impact the stability, coordination mode, and catalytic activity of the complexes and reveal evidence of the rare O-coordination of these PYA ligands.
Results and discussion
The potentially NN-bidentate coordinating pyridine-substituted PYA ligand (1I) was synthesized following previously reported protocols.2,9 Subsequent anion exchange with NH4PF6 in water yielded 1PF6 (Scheme 2). The coordination of 1PF6 to FeCl2 was accomplished by a stepwise process including first ligand deprotonation with a stoichiometric amount of LiHMDS as a strong non-nucleophilic base, followed by the addition of the metal precursor, which afforded complex 2 after precipitation with Et2O. 1H NMR analysis in CD3CN showed signals ranging from −50 to +73 ppm which are consistent with a paramagnetic compound (Fig. S5†). Magnetic susceptibility measurements performed using the Evans method gave a μeff of 4.8 for each iron unit indicating a high spin configuration of the d6 metal with S = 2.22 In line with this finding, no signal was obtained in the X-band EPR spectrum (solid state and DMSO solution at RT and 77 K). Cyclic voltammetry measurements in MeCN indicate the presence of two oxidative waves at Epa = 1.16 and 2.01 V (vs. Fc+/Fc, Fig. S16 and Table S1†). Both oxidations were irreversible under the employed conditions, presumably due to the loss of a chloride ligand or complex decomposition. Additionally, stability tests by 1H NMR spectroscopy indicated that the complex is air and moisture sensitive. Exposure of the complex to air directly results in the formation of the free ligand and the disappearance of the paramagnetic signals, both in the solid state and in solution.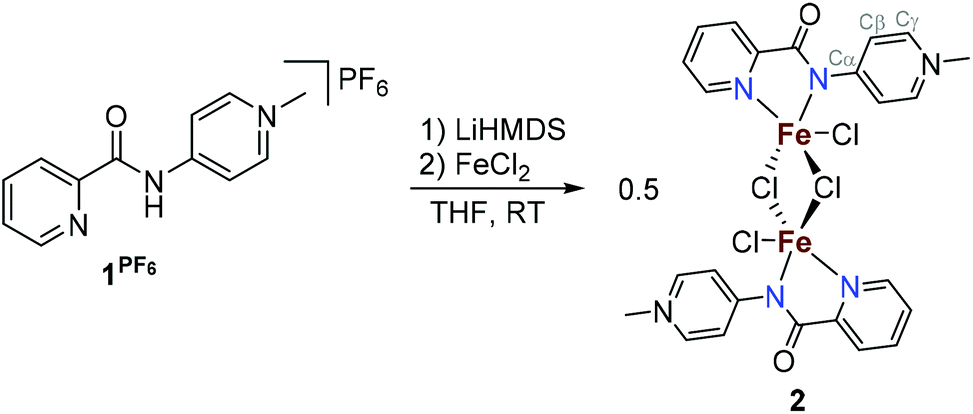 | ||
| Scheme 2 Synthesis of 2. Cα, Cβ and Cγ are defined in 2. | ||
Single crystals suitable for X-ray diffraction analysis were grown by the slow evaporation of a MeCN solution under inert conditions, and identified complex 2 as a dimeric structure (Fig. 2).23 Each asymmetric unit in the crystal consists of an iron center bound to two nitrogen atoms of the bidentate chelating pyridyl-PYA ligand, and a terminal as well as two bridging chlorido ligands. The C–C bond lengths in the PYA pyridyl ring show bond length alteration, since the Cα–Cβ bond is elongated compared to the Cβ–Cγ bond with an average difference of 0.051 Å (Table S4†), which indicates some contribution of the diene resonance form A (cf.Scheme 1).
In an attempt to break this dimer, we explored the addition of stoichiometric amounts of co-ligands, such as 1,2-bis(diphenylphosphino)ethane (dppe), 2,2′-bipyridine (bpy) or ethylenediamine (en), though no reaction was observed. However, stirring the dimer with stoichiometric quantities of NEt4I in THF/pyridine in a 4:1 ratio for several hours and the subsequent crystallization of the mixture revealed the formation of small quantities of monomeric complex 3 as a highly air-sensitive complex.24 The molecular structure of 3 was determined by X-ray diffraction analysis and consists of an octahedral FeII center bound to chloride, three pyridine ligands, and the κ2(N,O)-coordinated PYA ligand, implying a rearrangement of the N-binding mode of the amide moiety to the untypical O-coordination mode (Fig. 3).25 N-coordination of the PYA site instead of O-coordination induces significant changes in the amide unit, with elongation of the C6–O1 bond in 3 compared to 2 (1.272(3) vs. 1.229(2) Å) along with a shortening of C6–N2 bond from 1.378(2) Å in 2 to 1.308(3) Å in 3. These bond length changes reflect a more pronounced contribution of the +N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O− amide resonance form and indicate the increased relevance of resonance structure C, featuring the formal negative charge located at the oxygen atom (cf.Scheme 1). Moreover, complex 3 reveals partial double bond localization in the pyridyl heterocycle. The Cα–Cβ bonds are elongated compared to the Cβ–Cγ bonds with an average difference of 0.048 Å (Table S5†). This difference is similar to that observed in complex 2 and suggests that either C–C bond length analysis is not a useful probe to distinguish the contributions of different resonance structures or that other resonance structures contribute as well; for example, a diene-type structure with the positive and negative charge located on the amide unit (N+C(R)–O−).
C–O− amide resonance form and indicate the increased relevance of resonance structure C, featuring the formal negative charge located at the oxygen atom (cf.Scheme 1). Moreover, complex 3 reveals partial double bond localization in the pyridyl heterocycle. The Cα–Cβ bonds are elongated compared to the Cβ–Cγ bonds with an average difference of 0.048 Å (Table S5†). This difference is similar to that observed in complex 2 and suggests that either C–C bond length analysis is not a useful probe to distinguish the contributions of different resonance structures or that other resonance structures contribute as well; for example, a diene-type structure with the positive and negative charge located on the amide unit (N+C(R)–O−).
 | ||
| Fig. 3 ORTEP representation of complex 3 (50% probability ellipsoids; hydrogen atoms and iodide counterions omitted for clarity). Selected bond distances in Å: Fe1–Cl1: 2.3914(8), Fe1–O1: 2.0927(18), Fe1–N1: 2.189(2), C5–C6: 1.504(4), C7–C8: 1.400(4), C7–C11: 1.405(4), O1–C6: 1.272(3), C8–C9: 1.355(4), C10–C11: 1.354(4), N1–C5: 1.344(4), N2–C6: 1.308(3), N2–C7: 1.377(4), N3–C9: 1.348(4), N3–C10: 1.344(3), N3–C12: 1.474(4). Bond angle O1–Fe1–N1: 75.74(8)°. | ||
Pincer PYA ligands
To increase the stability of the iron PYA complexes, the chelation of the ligand was extended to a tridentate coordinating pincer-type system comprising a central pyridine unit with two PYA arms derived either from para or ortho aminopyridine (4PF6 and 5PF6, Scheme 3).26–28 Complexation of 4PF6 was performed by the deprotonation of the ligand precursor in the presence of Na2CO3 in DMF, followed by filtration into a DMF solution of FeCl2, which induced the precipitation of complex 6 (Scheme 3). The neutral iron(II) complex is paramagnetic as evident from the 1H NMR spectra with shifts in the range of 0 to 73 ppm and from the magnetic susceptibility of μeff = 4.9 (Evans method), consistent with an S = 2 ground state (Fig. S8†). Complex 6 shows two quasi reversible and presumably metal-centered redox processes in cyclic voltammetry (Fig. S17†)29 at E1/2 = −0.46 and −0.31 V (vs. Fc+/Fc in DMF). Single crystals suitable for crystallographic analysis were grown by slow vapor diffusion of Et2O into a concentrated solution of the complex in DMSO. The molecular structure of 6 reveals an iron center bound to a κ3(N,N,N)-coordinated PYA pincer ligand and two chloride ligands, resulting in a five-coordinate trigonal bipyramidal geometry.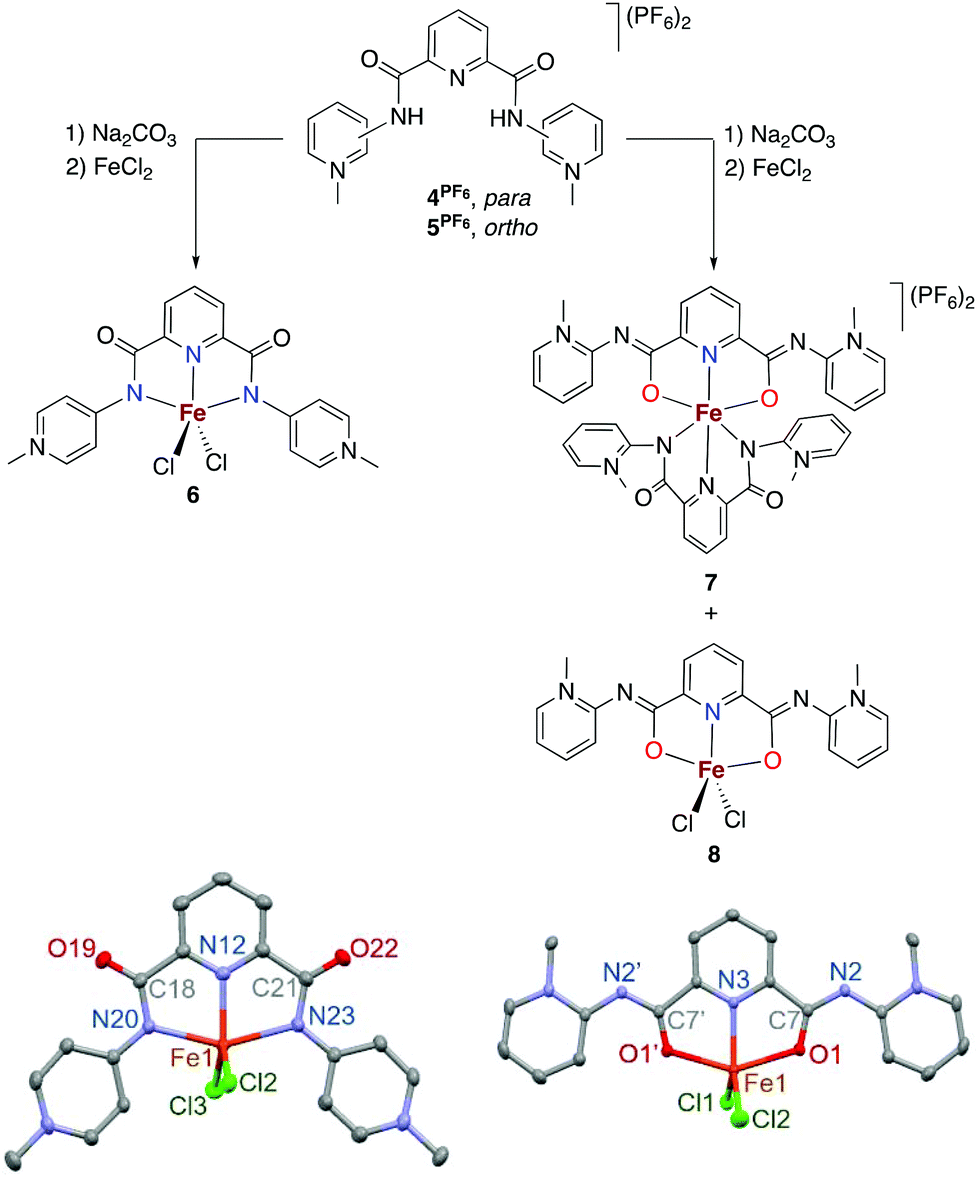 | ||
| Scheme 3 Synthesis (top) and ORTEP plots (bottom) of complexes 6–8 and ORTEP plots of 6 and 8 shown at 50% thermal ellipsoids, hydrogen atoms and co-crystallized DMSO molecules are omitted for clarity. Selected bond distances in Å: 6: Fe1–N12: 2.0696(14), Fe1–N20: 2.2476(14), Fe1–N23: 2.2559(14), O19–C18: 1.231(2), O22–C21: 1.233(2), N20–C18: 1.363(2), N23–C21: 1.357(2). 8: Fe1–N3: 2.109(2), Fe1–O1: 2.1797(12), O1–C7: 1.259(2), N2–C7: 1.329(2). | ||
In contrast, the complexation of the ortho-PYA pincer system 5PF6 by the same procedure resulted in the precipitation of bis-pincer iron(II) complex 7. The purple powder is only sparingly soluble in common solvents, including MeCN and DMF, hampering its recrystallization. A 1H NMR analysis in DMSO-d6 showed that 7 is paramagnetic with shifts ranging from 0 to 70 ppm and that the complex has only limited stability in this solvent (Fig. S9†).30 A CV analysis of 7 reveals oxidation at higher potential compared to that of 6 with E1/2 = −0.25 and −0.05 V (vs. Fc+/Fc in MeCN). The formation of 7 was further supported by HRMS and elemental analysis. Notably, the DMF supernatant from the synthesis of 7 contained traces of a different complex, which crystallized as the mono-pincer complex 8 (Fig. S19†). This complex might be an intermediate in the formation of the bis-pincer complex, which is also the favored product in relation to CoII and NiII chemistry.28 In analogy to these cobalt and nickel complexes, the ortho-PYA pincer ligand in the iron(II) complex 8 features a rare ONO-tridentate bonding mode, presumably because of a combination of effects including steric shielding of the amide nitrogen by the ortho-substitution, electronic factors associated with the relatively weak donor properties of N-coordinating ortho PYA ligands, as well as the hardness of the 3d metal center.31 While the attempts to crystallize complex 7 have failed so far, we speculate that at least one of the pyPYA2 ligands is bound in the ONO-coordination mode based on the structure of 8 and the analogy to other first-row transition metal complexes with this ligand.28
Complexes 6 and 8 are structurally related to anionic pyridine bis-carboxamide complexes such as [FeIII(2,6-bis(N-phenylcarbamoyl)pyridine)2][NEt4].32 Generally, these doubly anionic ligands coordinate through the nitrogen atoms, but steric congestion indicated that O-coordination is also feasible.33,34 Bond length analyses in the N-bound para-PYA unit in 6 revealed an average difference of 0.052 Å between Cα–Cβ and Cβ–Cγ, suggesting similar double bond localization as noted in complexes 2 and 3 (Table S6†). Remarkably, this bond length alteration is just slightly less pronounced in the O-bound ortho-PYA heterocycle of complex 8 (Δd 0.034 Å; Table S7†). In addition, the amide CO bond is only moderately elongated at 1.259(2) Å (cf. 1.232(3) in 6 and 1.272(3) in 3), while the exocylic CPYA–Namid bond is shortened to 1.353(2) Å. Together with the contraction of the adjacent C–N bond to 1.329(2) Å (cf. 1.360(5) Å in 6), these data indicate relevant contributions of resonance structures A and C for complex 8.
Alcohol oxidation catalysis
We have used catalytic alcohol oxidation as a probe to evaluate the effect of the different ligand coordination modes (Table 1 and Table S2†). Initially, the reactivity of complex 2 was explored in the catalytic oxidation of 1-phenyl ethanol to acetophenone as a model reaction.35–39 Under standard conditions, viz. 0.5 mol% of the dimeric iron complex 2, 1.5 eq. t-butyl hydroperoxide (TBHP), 50 °C in MeCN, complex 2 cleanly converted the alcohol to acetophenone and reached full conversion after 24 h, slightly slower than when using only FeCl2 as the catalyst precursor (Table 1, entries 1,2). Substituting TBHP by 30% aq. H2O2 suppressed product formation, which is likely caused by the sensitivity of complex 2 towards water. Under mild conditions (0.1 mol% catalyst at 25 °C), complex 2 and FeCl2 have comparable reactivity, reaching a product yield of 18 ± 1% after 2 days (entries 6, 7; Fig. S18†). This similar activity suggests the formation of an identical catalytically active species when starting from 2 or FeCl2, i.e., catalyst activation of complex 2 presumably involves ligand dissociation and the release of FeCl2 as an active entity.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Fe] | Fe loading (mol%) | Temp. (°C) | Conversion (time) | |
| a General conditions: iron complex (7 μmol, 1 mol%), MeCN (0.5 mL), 1-phenylethanol (0.7 mmol), TBHP (1.05 mmol, 5 M in decane), Ar atmosphere, yields determined by 1H NMR spectroscopy (Fig. S10†); quantities of substrate and catalyst adjusted for runs with lower catalyst loading, see experimental for details. b 0.5 mol% of the dimeric complex was used to keep [Fe] at 1 mol%. c 2 equiv. diphenylamine added at the start of the reaction. d TBHP was added in two portions, 1 equiv. at t = 0 and 1 equiv. at t = 30 min. e 1.5 equiv. TBHP added in two portions, 1 equiv. at t = 0 and 0.5 equiv. at t = 20 h. | |||||
| 1 | FeCl2 | 1 | 50 | 45 (2 h) | 81 (24 h) |
| 2b | 2 | 1 | 50 | 33 (2 h) | >99 (24 h) |
| 3 | 6 | 1 | 50 | 11 (2 h) | 48 (24 h) |
| 4 | 7 | 1 | 50 | 47 (2 h) | >99 (24 h) |
| 5 | 7 | 0.5 | 50 | 42 (2 h) | >99 (24 h) |
| 6 | FeCl2 | 0.1 | 25 | 17 (2 d) | |
| 7b | 2 | 0.1 | 25 | 18 (2 d) | |
| 8 | 6 | 0.1 | 25 | 6 (2 d) | |
| 9 | 7 | 0.1 | 25 | 67 (2 d) | |
| 10 | 7 | 0.05 | 25 | 58 (2 d) | |
| 11c | 7 + Ph2NH | 0.5 | 50 | <3 (1 h) | <3 (24 h) |
| 12d | 7 | 0.5 | 80 | 98 (1 h) | |
| 13d | FeCl2 | 1 | 80 | 50 (1 h) | |
| 14 | 5PF6 | 0 | 80 | 1 (1 h) | |
| 15 | none | 0 | 80 | <5 (1 h) | |
| 16e | 7 | 0.005 | 80 | 92 (24 h) | 98 (48 h) |
In contrast, the pincer type complexes 6 and 7 showed diverging reactivities. Complex 6 with an N,N,N-coordinated PYA pincer ligand only yielded 48% product after 24 h (entry 3, cf. >80% for FeCl2 and complex 2). However, complex 7 reached full conversion after 24 h and resulted in a higher yield at 2 h when compared to 2 (47% vs. 33%, entry 4).40 Even at a catalyst loading of 0.5 mol%, 7 accomplishes full conversion after 24 h (entry 5). Comparing the performance at lower catalyst loading and milder temperatures, viz. 0.1 mol% and 25 °C, demonstrates the distinct activity of complex 7.41 Under these conditions, FeCl2 and complex 2 reached 17 and 18% yield, respectively, after 2 days (entries 6, 7) and complex 6 achieved a modest 6% (entry 8); yet, complex 7 accelerated product formation and afforded an appreciable 67% yield within the same time span (entry 9). Conversions of almost 60% were accomplished at even a lower catalyst loading of 0.05 mol% (entry 10),42 corresponding to turnover numbers (TONs) of 1960. These runs also indicate a considerable rate enhancement compared to FeCl2.
These catalytic experiments suggest that the bidentate ligand system is not binding strong enough and leads to catalytic activity that is commensurate with ligand dissociation (cf. identical activity of FeCl2). Unlike the bidentate system, the tridentate pincer-type coordination has a much more profound impact on complex integrity and catalytic activity (see also Table S2† for various control experiments). The NNN coordination mode of the para-PYA pincer ligand in 6 inhibits catalytic activity significantly, suggesting a tight bonding of the Fe center within the pincer ligand scaffold, while the bis-pincer coordination pattern in complex 7 imparts a substantially enhanced catalytic performance, presumably after the loss of one of the pincer ligands. Even though steric effects cannot be excluded, we surmise that the electronic implication of the PYA resonance structure C with partially anionic oxygen donors beneficially influences the electronic configuration at the iron center to stabilize critical transition states during metal oxidation. In particular, higher oxidation species generated in the reaction of TBHP with the FeII complex are expected to be better stabilized by the ONO-pincer coordination mode due to the high oxophilicity of high-valence iron. This model is supported by catalytic runs in the presence of diphenylamine as an oxygen radical trap aimed at probing the formation of tBuOO˙ or tBuO˙ radicals (entry 11).21,35 Under these conditions, product yield dropped to <3% after 24 h, indicating the relevance of oxygen radicals for enabling catalytic turnover and hence indirectly supporting a mechanistic model involving the intermediates with Fe in spin-unpaired higher oxidation states.
Since the ortho-PYA pincer iron complex 7 showed the best results throughout, further optimization of the catalytic reaction was performed with this complex. Increasing the reaction temperature to 80 °C resulted in a higher reaction rate and a 91% product yield within 2 h. However, full conversion was not reached due to the consumption of the oxidant as observed by the 1H NMR analysis. Adjustment of the protocol therefore included batchwise additions of 1 equiv. TBHP at the onset and again after 30 min. This portioned addition afforded 98% conversion after only 1 h using 0.5 mol% of complex 7 (entry 12), compared to a 50% yield with 1 mol% FeCl2 (entry 13), and <5% with only the ligand or blank reactions (entries 14, 15, entries S1–S8 in Table S2†). The catalytic species shows excellent stability. A catalyst loading as low as 0.005 mol% gave a 92% yield after 24 h and full conversion after 48 h, resulting in 20000 TONs (entry 16).43 Such high TONs are unusual for iron complexes and are assumed to be a direct consequence of the integrity of the catalytic species due to the rigid tridentate coordination of the PYA pincer ligand combined with the beneficial electronic impact of the ONO donor motif.
The scope of the bis-pincer iron complex 7 in oxidative catalysis was explored with a small variety of distinct substrates (Table 2).44 Diphenylmethanol was oxidized quantitatively to benzophenone, indicating that sterically more bulky substrates are converted equally well as 1-phenylethanol (entry 1). In contrast, primary alcohols are not selectively transformed to the corresponding aldehyde. For example, benzyl alcohol produces benzaldehyde in reasonable selectivity initially (32% conversion, 78% selectivity to benzaldehyde after 2 h, entry 2) and longer reaction times led to full conversion to a mixture of products, however, with only traces of benzaldehyde. Aliphatic alcohols such as cyclohexanol and 4-phenyl-2-butanol were also converted, though yields were only moderate (75% and 53%, respectively) and did not improve upon prolonging the reaction time beyond 5 h (entries 3, 4). Thioethers such as thioanisole were oxidized with high selectivity towards the sulfoxide product with only minute overoxidation to the corresponding sulfone product even after extended reaction periods (98/2 ratio after 20 h, entry 5). Notably, oxidation of pyridine to its N-oxide does not proceed (entry 6), indicating selective oxygen transfer with complex 7 to sulfur but not to nitrogen. This selectivity was further evaluated in the oxidation of 1-(2-pyridyl)ethanol, which afforded exclusively and quantitatively 2-acetylpyridine, demonstrating high preference for alcohol over nitrogen oxidation and no catalyst inhibition by the pyridine functionality (entry 7).
| Entry | Substrate | Product | % conversion (time) | |
|---|---|---|---|---|
|
a General conditions: iron complex (3 μmol, 0.5 mol%), MeCN (0.5 mL), substrate (0.7 mmol), 1 eq. TBHP (0.7 mmol, 5 M in decane) and a second eq. TBHP after 30 min, Ar atmosphere, conversions determined by GC analysis or 1H NMR spectroscopy.
b Spectroscopic yield of benzaldehyde.
c Sulfoxide/sulfone ratio 98:2 independent of reaction time.
|
||||
| 1 |
|
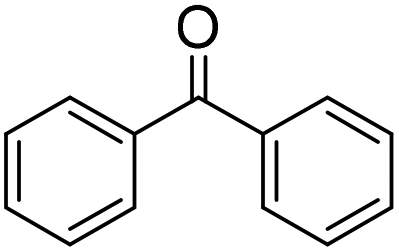
|
>99 (1 h) | |
| 2 |
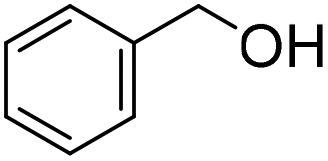
|
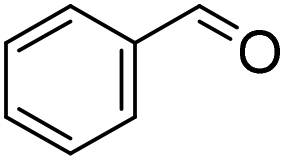
|
32 (1 h) (25%)b | 98 (24 h) (3%)b |
| 3 |
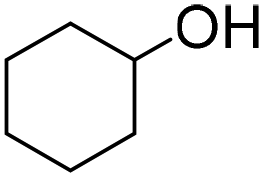
|
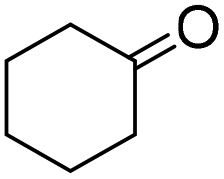
|
69 (3 h) | 75 (24 h) |
| 75 (5 h) | ||||
| 4 |
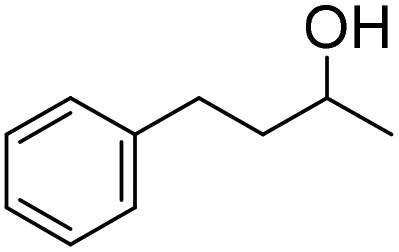
|
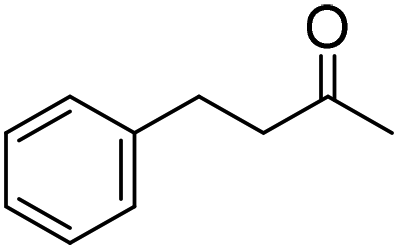
|
51 (5 h) | 53 (24 h) |
| 5 |
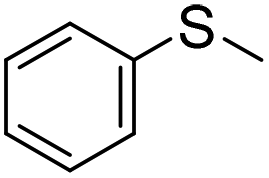
|
|
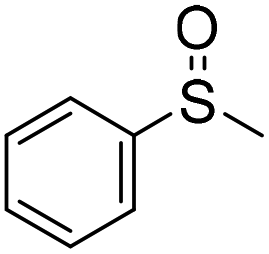
57 (2 h)c | 72 (20 h)c |
| 6 |
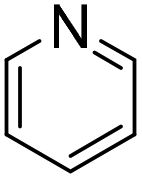
|
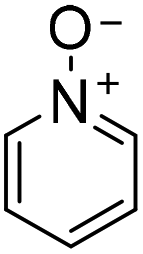
|
0 (1 h) | 0 (24 h) |
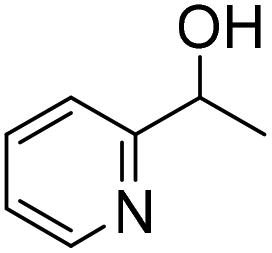
| 7 |
|

|
>99 (1 h) | |
Conclusions
In summary, we present the synthesis of the first iron-based PYA complexes having an NN(N) or ONO coordination. The molecular structures reveal the connection between resonance structures and binding mode: the NN(N) coordinating structures have an increased resonance contribution of the diene form, while the zwitterionic resonance structure with a negative charge on the oxygen center is predominant in the ONO complexes. Peroxide-mediated alcohol oxidation provided a useful probe for the integrity of the complexes. While NN-bidentate coordination is too unstable and leads to complex decomposition, NNN-tridentate binding results in more rigid iron coordination, yet also to catalytic inhibition. In contrast, ONO-tridentate PYA pincer coordination has a positive effect on the stability of the iron complex and its catalytic activity, and this ligand imparts high product yields and excellent TONs. Catalytic oxidation additionally proceeds with sterically more demanding substrates, aliphatic alcohols, and in oxygen transfer oxidation to form sulfoxide, yet leaves imines unaffected. This work highlights the unique coordination flexibility of PYA ligands and their potential to induce attractive catalytic performance with Earth-abundant first row transition metals. Future work will focus on extending the catalytic scope of these iron complexes to other oxidative transformations.Experimental section
General considerations
All reagents were commercially available and used as received unless specified differently. When specified, reactions were run in an MBRAUN LABmaster 130 argon-filled glovebox. When using inert conditions, the chemicals and materials were dried and degassed prior to use. THF, MeCN, hexanes, and Et2O were taken from a solvent purification system (SPS), stored over molecular sieves for at least 2 days, degassed with N2 gas bubbling for 30 min, placed in the glovebox and stored over fresh molecular sieves prior to use. FeCl2, ZnCl2, NEt4I, and Na2CO3 were dried under vacuum for 1–2 days before placing them in the glovebox. Mesitylene and 1-phenyl ethanol were placed on molecular sieves in a Schlenk flask, degassed with N2 bubbling for 30 min, and used under inert N2 conditions. CD3CN and DMSO-d6 were placed in the glovebox directly, and dried further over molecular sieves. Compounds 1I,94PF6,27 and 5PF6 (ref. 28) were synthesized following literature procedures. 1H, 13C, 19F, and 31P NMR spectra were recorded at 25 °C using a Bruker spectrometer operating at 300, 75, 282, and 121 MHz, respectively. 1H and 13C NMR chemical shifts are reported in ppm relative to SiMe4 by using the residual solvent resonance as the internal standard. 31P NMR chemical shifts are externally referenced to 85% aqueous H3PO4. 19F NMR chemical shifts are externally referenced to CFCl3. The purity of the bulk samples of the complexes has been established by NMR spectroscopy, and when possible by elemental analysis. The elemental analyses were performed at the Department of Chemistry and Biochemistry (DCB), University of Bern, Microanalytic Laboratory using a Thermo Scientific Flash 2000 CHNS-O elemental analyzer. High-resolution mass spectrometry was carried out using a Thermo Scientific LTQ Orbitrap XL (ESI-TOF) by the mass spectroscopy group of the DCB. Cyclic voltammograms were recorded using an Autolab PGSTAT101 from Metrohm in the specified solvent solutions: 10 mL solvent, 1 mM sample, and 100 mM tetrabutylammonium hexafluorophosphate (nBu4N)PF6 as supporting electrolyte. Solutions were taken under argon from the glovebox. Redox potentials were measured using a glassy carbon working electrode, Ag/AgCl reference electrode (SSCE) and Pt-wire auxiliary electrode and are tabulated versus a ferrocene internal standard.45 IR spectra were recorded using a Jasco FT/IR 4700 Fourier transform infrared spectrometer.Syntheses
:1 mixture of DMSO-d6/DMSO, resulting in a μeff: 4.9, S = 2. Due to the high sensitivity of the compound, no satisfactory elemental analysis was performed.
:5 mixture of DMSO-d6/DMSO, resulting in a μeff: 4.9, S = 2.
Representative catalytic procedures
In an argon filled glovebox, a 5 mL Teflon capped microwave reaction vial was loaded with the catalyst (1 mol%, 7 μmol), closed and transferred to a fume hood. While stirring, MeCN (0.5 mL), the substrate (1-phenylethanol, 0.7 mmol), and oxidant (TBHP 5 M in decane, 1.05 mmol) were added consecutively by (micro)syringes. The vial was placed in a heating bath. On set times, 0.05 mL samples were taken using a 1 mL syringe, diluted in CD3CN (0.4 mL), filtered through a cotton pad, and analyzed by 1H NMR spectroscopy. When using an NMR standard, the reaction was performed without sampling. Mesitylene (0.5 eq.) was added at the end of the reaction to avoid interference, and a sample was taken and analyzed by NMR spectroscopy as described above. The amounts were adjusted as follows when using lower catalyst loadings: for 0.1 mol% runs: 4 μmol catalyst in 3 mL MeCN with 4 mmol 1-phenylethanol and 6 mmol TBHP; for 0.005 mol% run: 2 μmol catalyst in 3 mL of MeCN with 20 mmol 1-phenylethanol and 2 × 20 mmol TBHP.43Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge generous financial support from the European Research Council (CoG 615653), the Seal of Excellence Fund of the University of Bern (SELF fellowship to D. G. A. V.), and the Swiss National Science Foundation (200020_182663). We thank Fabio Notter for synthetic assistance and the group of Chemical Crystallography of the University of Bern for X-ray analysis of all reported structures.Notes and references
- A. Togni and L. M. Venanzi, Nitrogen Donors in Organometallic Chemistry and Homogeneous Catalysis, Angew. Chem., Int. Ed. Engl., 1994, 33, 497–526 CrossRef.
- P. D. W. Boyd, L. J. Wright and M. Zafar, Extending the Range of Neutral N-Donor Ligands Available for Metal Catalysts: N-[1-Alkylpyridin-4(1H)-ylidene]amides in Palladium-Catalyzed Cross-Coupling Reactions, Inorg. Chem., 2011, 50, 10522–10524 CrossRef CAS.
- M. E. Doster and S. A. Johnson, Selective C–F Bond Activation of Tetrafluorobenzenes by Nickel(0) with a Nitrogen Donor Analogous to N-Heterocyclic Carbenes, Angew. Chem., Int. Ed., 2009, 48, 2185–2187 CrossRef CAS.
- Q. Shi, R. J. Thatcher, J. Slattery, P. S. Sauari, A. C. Whitwood, P. C. McGowan and R. E. Douthwaite, Synthesis, Coordination Chemistry and Bonding of Strong N-Donor Ligands Incorporating the 1H-Pyridin-(2E)-Ylidene (PYE) Motif, Chem. – Eur. J., 2009, 15, 11346–11360 CrossRef CAS.
- R. J. Thatcher, D. G. Johnson, J. M. Slattery and R. E. Douthwaite, Charged Behaviour from Neutral Ligands: Synthesis and Properties of N-Heterocyclic Pseudo-amides, Chem. – Eur. J., 2012, 18, 4329–4336 CrossRef CAS.
- V. Leigh, D. J. Carleton, J. Olguin, H. Mueller-Bunz, L. J. Wright and M. Albrecht, Solvent-Dependent Switch of Ligand Donor Ability and Catalytic Activity of Ruthenium(II) Complexes Containing Pyridinylidene Amide (PYA) N–Heterocyclic Carbene Hybrid Ligands, Inorg. Chem., 2014, 53, 8054–8060 CrossRef CAS.
- C.-H. Chien, M.-K. Leung, J.-K. Su, G.-H. Li, Y.-H. Liu and Y. Wang, Substituent Effects on Pyrid-2-yl Ureas toward Intramolecular Hydrogen Bonding and Cytosine Complexation, J. Org. Chem., 2004, 69, 1866–1871 CrossRef CAS.
- K. F. Donnelly, C. Segarra, L.-X. Shao, R. Suen, H. Müller-Bunz and M. Albrecht, Adaptive N–Mesoionic Ligands Anchored to a Triazolylidene for Ruthenium-Mediated (De)Hydrogenation Catalysis, Organometallics, 2015, 34, 4076–4084 CrossRef CAS.
- M. Navarro, V. Rosar, T. Montini, B. Milani and M. Albrecht, Olefin Dimerization and Isomerization Catalyzed by Pyridylidene Amide Palladium Complexes, Organometallics, 2018, 37, 3619–3630 CrossRef CAS.
- . DFT computational investigation to NN vs. NO binding: C. M. A. Muller, M. V. Babak, M. Kubanik, M. Hanif, S. M. F. Jamieson, A. G. Hartinger and L. J. Wright, Pt(II) pyridinium amidate (PYA) complexes: Preparation and in vitro anticancer activity studies, Inorg. Chim. Acta, 2016, 450, 124–130 CrossRef CAS.
- (a) O. Belda and C. Moberg, Bispyridylamides-coordination chemistry and applications in catalytic reactions, Coord. Chem. Rev., 2005, 249, 727–740 CrossRef CAS; (b) A. Rajput and R. Mukherjee, Coordination chemistry with pyridine/pyrazine amide ligands. Some noteworthy results, Coord. Chem. Rev., 2013, 257, 350–368 CrossRef CAS.
- M. Navarro, M. Li, H. Müller-Bunz, S. Bernhard and M. Albrecht, Donor-Flexible Nitrogen Ligands for Efficient Iridium-Catalyzed Water Oxidation Catalysis, Chem. – Eur. J., 2016, 22, 6740–6745 CrossRef CAS.
- M. Navarro, C. A. Smith, M. Li, S. Bernhard and M. Albrecht, Optimization of Synthetically Versatile Pyridylidene Amide Ligands for Efficient Iridium-Catalyzed Water Oxidation, Chem. – Eur. J., 2018, 24, 6386–6398 CrossRef CAS.
- K. Salzmann, C. Segarra and M. Albrecht, Donor-flexible bis-pyridine amide ligands for highly efficient ruthenium-catalyzed olefin oxidation, Angew. Chem., Int. Ed., 2020, 59, 8932–8936 CrossRef CAS.
- (a) M. S. Chen and M. C. A. White, Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis, Science, 2007, 318, 783–787 CrossRef CAS; (b) M. Cianfanelli, G. Olivio, M. Milan, R. J. M. Klein Gebbink, X. Ribas, M. Bietti and M. Costas, Enantioselective C–H Lactonization of Unactivated Methylenes Directed by Carboxylic Acids, J. Am. Chem. Soc., 2020, 142, 1584–1593 CrossRef CAS.
- C. Kim, K. Chen, J. Kim and L. Que Jr., Stereospecific Alkane Hydroxylation with H2O2 Catalyzed by an Iron(II)-Tris(2-pyridylmethyl)amine Complex, J. Am. Chem. Soc., 1997, 119, 5964–5965 CrossRef CAS.
- P. Talsi and K. P. Bryliakov, Chemo- and stereoselective C–H oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef.
- Z. Guan, Metal Catalysts for Olefin Polymerization, Top Organomet Chem., Springer-Verlag Berlin Heidelberg, 2009, vol. 26, ISBN 978-3-540-87750-9 Search PubMed.
- G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Novel olefin polymerization catalysts based on iron and cobalt, Chem. Commun., 1998, 849–850 RSC.
- M. W. Bouwkamp, E. Lobkovsky and P. J. Chirik, Bis(imino)pyridine Iron(II) Alkyl Cations for Olefin Polymerization, J. Am. Chem. Soc., 2005, 127, 9660–9661 CrossRef CAS.
- A. Gonzalez-de-Castro and J. Xiao, Green and Efficient: Iron-Catalyzed Selective Oxidation of Olefins to Carbonyls with O2, J. Am. Chem. Soc., 2015, 137, 8206–8218 CrossRef CAS.
- . The solution state magnetic moment of 2 was determined per iron center, indicating that the dimeric complex either dissociates to monomeric Fe(L)Cl2 complexes in solution, or that the Fe centers in the dimeric complex have a very weak magnetic coupling if any at all.
- . Crystallographic data of the complexes reported in this paper have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication numbers 2001555 (2), 2001556 (3), 2001557 (6), 2001558 (7). Details of data collection, structure solution, and refinement are compiled in the ESI.†.
- . Yields were low due to crystallization of the product. The product is sensitive to vacuum, as it leads to gradual decomposition.
- . O-coordination has been observed in neutral pyridine carboxamides without N-deprotonation, see: W. Jacob and R. Mukherjee, Synthesis, structure, and properties of monomeric Fe(II), Co(II), and Ni(II) complexes of neutral N-(aryl)-2-pyridinecarboxamides, Inorg. Chim. Acta, 2006, 359(14), 4565–4573 CrossRef CAS.
- A. Dorazco-González, H. Höpfl, F. Medrano and A. K. Yatsimirsky, Recognition of Anions and Neutral Guests by Dicationic Pyridine-2,6-dicarboxamide Receptors, J. Org. Chem., 2010, 75, 2259–2273 CrossRef.
- (a) P. Melle, Y. Manoharan and M. Albrecht, Modular Pincer-type Pyridylidene Amide Ruthenium(II) Complexes for Efficient Transfer Hydrogenation Catalysis, Inorg. Chem., 2018, 57, 11761–11774 CrossRef CAS; (b) P. Melle and M. Albrecht, Ruthenium Complexes with PYA Pincer Ligands for Catalytic Transfer Hydrogenation of Challenging Substrates, Chimia, 2019, 73, 299–303 CrossRef CAS.
- P. Melle, N. Segaud and M. Albrecht, Ambidentate bonding and electrochemical implications of pincer-type pyridylidene amide ligands in complexes of nickel, cobalt and zinc, Dalton Trans., 2020, 49, 12662–12673 RSC.
- . Due to low solubility of 6 in MeCN, and reactivity of 7 with DMF, the complexes could not be analyzed in the same solvent, which limits a direct comparison of the oxidation potentials.
- . Low solubility of the complex hampered the accurate measurement of its magnetic susceptibility by Evans method.
- . This shielding seems to be more relevant for first-row transition metals with small ionic radii, since larger metal ions such as Ir(III), Ru(II), and Pd(II) were previously shown to coordinate to the amide nitrogen also in ortho-PYA complexes, see ref. 9, 12 and 28, while amide O-coordination was shown with Co and Ni, see ref. 28.
- M. Ray, D. Ghosh, Z. Shirin and R. Mukherjee, Highly Stabilized Low-Spin Iron(III) and Cobalt(III) Complexes of a Tridentate Bis-Amide Ligand 2,6-Bis(N-phenylcarbamoyl)pyridine. Novel Nonmacrocyclic Tetraamido-N Coordination and Two Unusually Short Metal-Pyridine Bonds, Inorg. Chem., 1997, 36, 3568–3572 CrossRef CAS.
- (a) G. L. Guillet, J. B. Gordon, G. N. Di Francesco, M. W. Calkins, E. Čižmár, K. A. Abboud, M. W. Meisel, R. García-Serres and L. J. Murray, A Family of Tri- and Dimetallic Pyridine Dicarboxamide Cryptates: Unusual O,N,O–Coordination and Facile Access to Secondary Coordination Sphere Hydrogen Bonding Interactions, Inorg. Chem., 2015, 54, 2691–2704 CrossRef CAS; (b) . For a related example with Ru, see also: J.-C. Waslike, G. Wu, X. Bu, G. Kehr and G. Erker, Ruthenium carbene Complexes Featuring a Tridentate Pincer-Type Ligand, Organometallics, 2005, 17, 4289–4297 CrossRef.
- T. C. Harrop, L. A. Tyler, M. M. Olmstead and P. K. Mascharak, Change in the Spin State of an FeIII Center upon One N-to-O Switch in the Coordination of a 2,6-Pyridinedicarboxamido Unit: The Effect of Methyl Thioether and Methyl Ether Appendages at the Ligand Periphery, Eur. J. Inorg. Chem., 2003, 475–481 CrossRef CAS.
- M. F. Pinto, B. d. P. Cardoso, S. Barroso, A. M. Martins and B. Royo, Chelating bis-N-heterocyclic carbene complexes of iron(II) containing bipyridyl ligands as catalyst precursors for oxidation of alcohols, Dalton Trans., 2016, 45, 13541–13546 RSC.
- E. Balogh-Hergovich and G. Speier, Catalytic oxidation of alcohols to carbonyl compounds with hydrogen peroxide using dinuclear iron complexes, J. Mol. Catal. A: Chem., 2005, 230, 79–83 CrossRef CAS.
- S. Tanaka, Y. Kon, T. Nakashima and K. Sato, Chemoselective hydrogen peroxide oxidation of allylic and benzylic alcohols under mild reaction conditions catalyzed by simple iron-picolinate complexes, RSC Adv., 2014, 4, 37674–37678 RSC.
- M. Sutradhar, T. Roy Barman, A. J. L. Pombeiro and L. M. D. R. S. Martins, Cu(II) and Fe(III) Complexes Derived from N-Acetylpyrazine-2-Carbohydrazide as Efficient Catalysts Towards Neat Microwave Assisted Oxidation of Alcohols, Catalysts, 2019, 9, 1053 CrossRef CAS.
- M. Lenze and E. B. Bauer, Chemoselective, iron(II)-catalyzed oxidation of a variety of secondary alcohols over primary alcohols utilizing H2O2 as the oxidant, Chem. Commun., 2013, 49, 5889–5891 RSC.
- . While 7 is only sparingly soluble in DMF, the purple suspension turns to a bright orange homogeneous solution upon addition of the oxidant.
- . The reactivity of 7 is related to the Fe(L)Cl2 complex reported by Ghosh and coworkers, although the catalysis reported herein vastly exceeds the earlier reported alcohol oxidation activity. N. Tyagi, O. Singh and K. Ghosh, Non-heme iron(III) complex with tridentate ligand: Synthesis, structures and catalytic oxidations of alkanes, Catal. Commun., 2017, 95, 83–87 CrossRef CAS.
- . Complex 7 reached full conversion after 9 days at 0.05 mol% catalyst loading while 0.1 mol% FeCl2, 2 and 6 reached 37%, 43% and 23% conversion, respectively after the same time. These data underline the high activity of complex 7, although deterioration of the oxidant and background oxidations due to oxygen from the atmosphere cannot be excluded in these long-term experiments.
- . Note that in the run with 0.005 mol% catalyst the major solvent is decane and the minor solvent is MeCN (ratio of 8:3), yet the reaction still goes to completion in this apolar solvent mixture.
- S.-I. Murahashi and N. Komiya, New types of catalytic oxidations in organic synthesis, Catal. Today, 1998, 41, 339–349 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and electrochemical data, catalytic and crystallographic details. CCDC 2001555–2001558. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D0DT02818E |
| This journal is © The Royal Society of Chemistry 2020 |