
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCytotoxic (cis,cis-1,3,5-triaminocyclohexane)ruthenium(II)-diphosphine complexes; evidence for covalent binding and intercalation with DNA†
Dan E.
Wise
a,
Aimee J.
Gamble
b,
Sham W.
Arkawazi‡
 b,
Paul H.
Walton
b,
M. Carmen
Galan
a,
Michael P.
O'Hagan
a,
Karen G.
Hogg
c,
Joanne L.
Marrison
c,
Peter J.
O'Toole
c,
Hazel A.
Sparkes
a,
Jason M.
Lynam
*b and
Paul G.
Pringle
*a
b,
Paul H.
Walton
b,
M. Carmen
Galan
a,
Michael P.
O'Hagan
a,
Karen G.
Hogg
c,
Joanne L.
Marrison
c,
Peter J.
O'Toole
c,
Hazel A.
Sparkes
a,
Jason M.
Lynam
*b and
Paul G.
Pringle
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: paul.pringle@bristol.ac.uk
bDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: jason.lynam@york.ac.uk
cImaging and Cytometry Laboratory, Bioscience Technology Facility, Department of Biology, University of York, UK
First published on 30th September 2020
Abstract
We report cytotoxic ruthenium(II) complexes of the general formula [RuCl(cis-tach)(diphosphine)]+ (cis-tach = cis–cis-1,3,5-triaminocyclohexane) that have been characterised by 1H, 13C and 31P{1H} NMR spectroscopy, mass spectrometry, X-ray crystallography and elemental analysis. The kinetics of aquation and stability of the active species have been studied, showing that the chlorido ligand is substituted by water at 298 K with first order rate constants of 10−2–10−3 s−1, ideal for potential clinical use as anti-tumour agents. Strong interactions with biologically relevant duplex and quadruplex DNA models correlate with the activity observed with A549, A2780 and 293T cell lines, and the degree of activity was found to be sensitive to the chelating diphosphine ligand. A label-free ptychographic cell imaging technique recorded cell death processes over 4 days. The Ru(II) cis-tach diphosphine complexes exhibit anti-proliferative effects, in some cases outperforming cisplatin and other cytotoxic ruthenium complexes.
Introduction
Ruthenium complexes have potential as alternatives to platinum-based chemotherapy in the treatment of cancers. Compounds A–G in Fig. 1 show the structural diversity of Ru complexes whose anti-cancer activity has been investigated.1 The ruthenium(III) complexes, NAMI-A (A) and KP1019 (B) have undergone clinical trials.2–6 More recently, Sadler7–11 and Dyson12–17 have reported piano-stool (η6-arene)ruthenium(II) compounds that are cytotoxic in vitro and in vivo. Notably, the chelate complex [RuCl(η6-biphenyl)(H2NCH2CH2NH2)]PF6 (C) has been shown to target DNA directly, with the DNA-complex adduct stabilised by hydrogen bonds between the diamine ligand and the O6 of guanine.18 It has been shown that [RuCl2(p-cymene)(PTA)] (D) is active in vivo against secondary metastases.13 In addition, due to their tunable photophysical properties, many Ru(II) polypyridyl complexes have been developed for use in photodynamic therapy (PDT) and photochemotherapy (PCT).19–22 The first example of this class of complexes to have entered human clinical trials was TLD1433 (E) which contains a terthienyl chromophore.19,23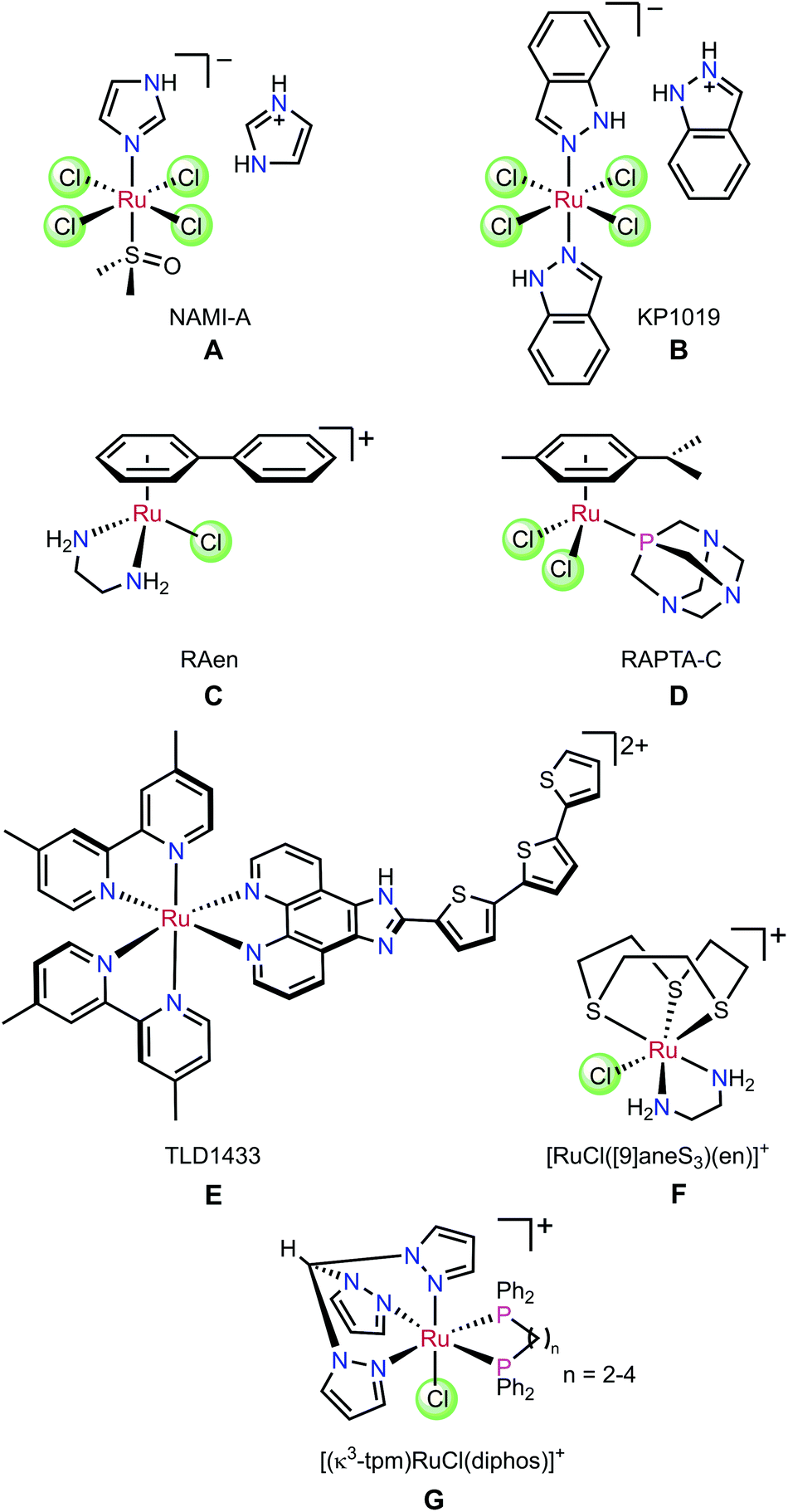 | ||
| Fig. 1 Cytotoxic Ru complexes. | ||
The seminal work on the anti-cancer properties of (η6-arene)Ru complexes has spurred the investigation of many coordination complex analogues of organometallic piano-stool complexes. For example, Alessio et al. replaced the arene with [9]aneS3 to give complex F with minimal loss of biological activity compared to its organometallic analogues.24 Furthermore, Ru-diphosphine complexes such as [(κ3-tpm)RuCl(diphos)]PF6 (G) showed activity in vitro.25 However, despite the variety of facially capping ligands available which could be used to modulate activity, this aspect of the complexes has received far less attention than modification of the other ancillary ligands on the metal.26–28 From a biological perspective, the narrow range of face-capping ligands that have been used limits the rate and extent of the substitution of the halido ligands by water. This rate is known to correlate with in vitro activity,8,29,30 and expanding its range is thus a critical factor in maximising the clinical potential of ruthenium complexes in the treatment of cancer.
The ligand cis-tach (cis–cis-1,3,5-triaminocyclohexane) forms face-capping complexes with many transition metals including ruthenium(II).31–35 The labile complex [RuCl(dmso-S)2(cis-tach)]Cl (1) is the precursor to N,N-chelate complexes 2a–c and P,P-chelate complexes 3a–f (Scheme 1) that have been previously reported.35 The influence of the cis-tach ligand was evaluated by comparison of the structural data with those for (η6-arene)Ru complexes. It has previously been demonstrated35,36 that cis-tach is a strong σ-donor, as would be expected due to the three nitrogen atoms coordinated to the metal. For instance, reaction of the DMSO complex 1 with diamines yields dicationic complexes in which one coordinated DMSO is retained, whereas 1 with diphosphines gave complexes with one chlorido ligand retained (Scheme 1). This difference in behaviour was rationalised on the basis of the different bonding characteristics of diamines and diphosphines. The electron-richness of the Ru created by the cis-tach, is augmented by the diamine σ-donors which leads to strong π-back-donation to the DMSO ligand, strengthening the Ru–S interaction. By contrast, diphosphines, which are better π-acceptors than DMSO, favour the coordination of chloride, which is presumed to be a π-donor. Importantly, for the use of these complexes as anti-cancer agents, it was reasoned that the Ru(cis-tach)(diphos) moiety may promote the rapid aquation of the Ru–Cl bond which might then result in enhanced in vitro activity.31,37
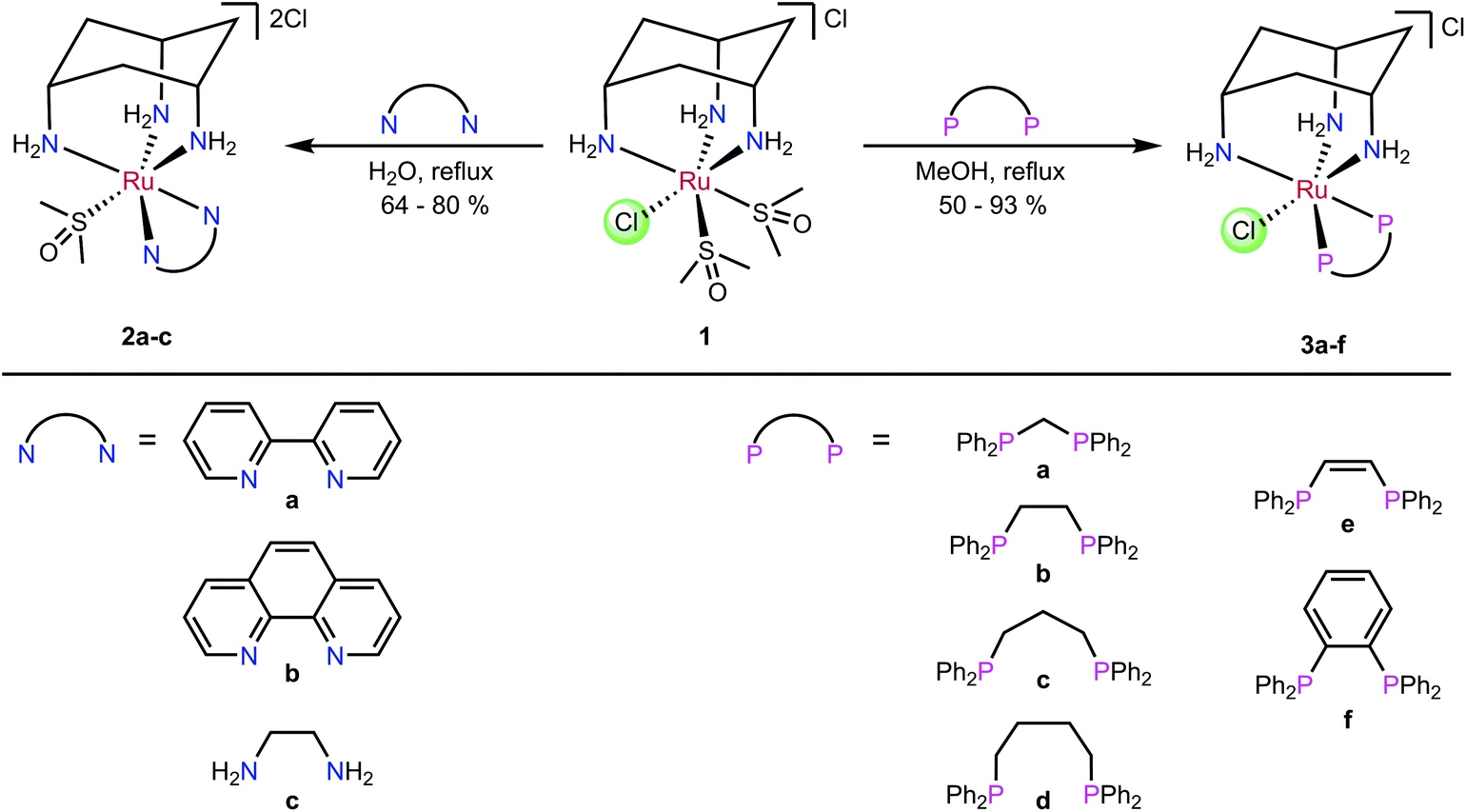 | ||
| Scheme 1 Synthesis of complexes 2a–c and 3a–f.35 | ||
In addition to their potential as effective anti-cancer agents, Ru complexes of cis-tach have other features that make them attractive for medicinal chemistry. For instance, the cationic Ru cis-tach complexes are readily prepared as chloride salts, obviating the use of the toxic PF6− anion in potential pharmaceuticals.38 The NH2 groups of the cis-tach ligand enhance the water solubility of the complexes and, moreover, may strengthen any binding to DNA through hydrogen-bonding interactions, in a similar manner to the DNA binding with [RuCl(η6-biphenyl)(en)]PF6 (C).8,9 Finally, the cyclohexane ring provides a hydrophobic face to the complex, giving steric protection to the hydrophilic metal centre.
It is in this context that we now report a detailed investigation of the in vitro activity of ruthenium cis-tach complexes. It is shown that a range of diphosphine derivatives exhibit activity against three tumour cell lines, in some cases with potency exceeding that of cisplatin or established anti-cancer ruthenium complexes. The extended aromatic backbones of the new diphosphines L1–L3 (Fig. 2) are shown to allow detailed insight into the nature of the biological interactions with their Ru-complexes via a range of physical inorganic and biological measurements including UV/visible, fluorescence and NMR spectroscopy as well as label-free cellular imaging techniques.
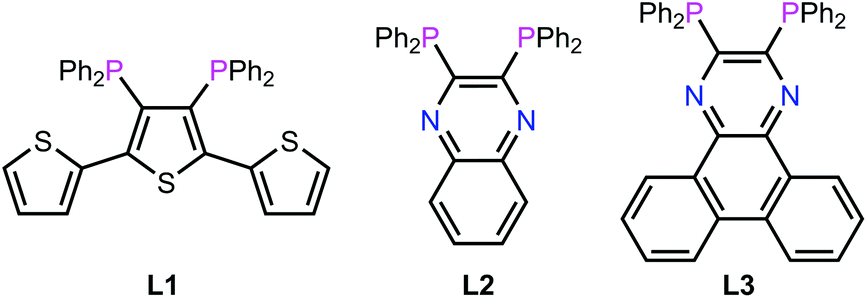 | ||
| Fig. 2 Chemical structures of L1–L3 designed for this work. | ||
Results and discussion
The ruthenium(II) precursor [RuCl(dmso-S)2(cis-tach)]Cl (1) and the (cis-tach)Ru complexes containing N,N-chelates (2a–c) and P,P-chelates (3a–f) were synthesised by previously reported methods (Scheme 1).35 Our initial biological investigations described herein focused on these complexes. Notably, for the purposes of this study, the P,P-chelates 3a–f are freely soluble in water up to millimolar concentrations, well in excess of that needed for therapy.Synthesis of diphosphine ligands and complexes
The ligands L1–L3 were made by the routes shown in Scheme 2. The novel terthiophene diphosphine L1 was prepared by route (i) from tetrabromothiophene using Pd-catalysed cross-coupling.45 It was necessary to install the diphenylphosphine groups in L1 sequentially to avoid the formation of a complex mixture of products. The quinoxaline diphosphine ligand, L2 was prepared by route (ii) following a modified literature procedure.46 The novel extended quinoxaline diphosphine ligand, L3 was prepared by route (iii) from its dichloro precursor (obtained from treatment of 1,4-dihydrodibenzo[f,h]quinoxaline-2,3-dione with PCl3 in DMF, see ESI†).47 The ruthenium complexes 3g–i were prepared in an analogous way to 3a–f (route (iv)); their formation was monitored by 31P{1H} NMR spectroscopy.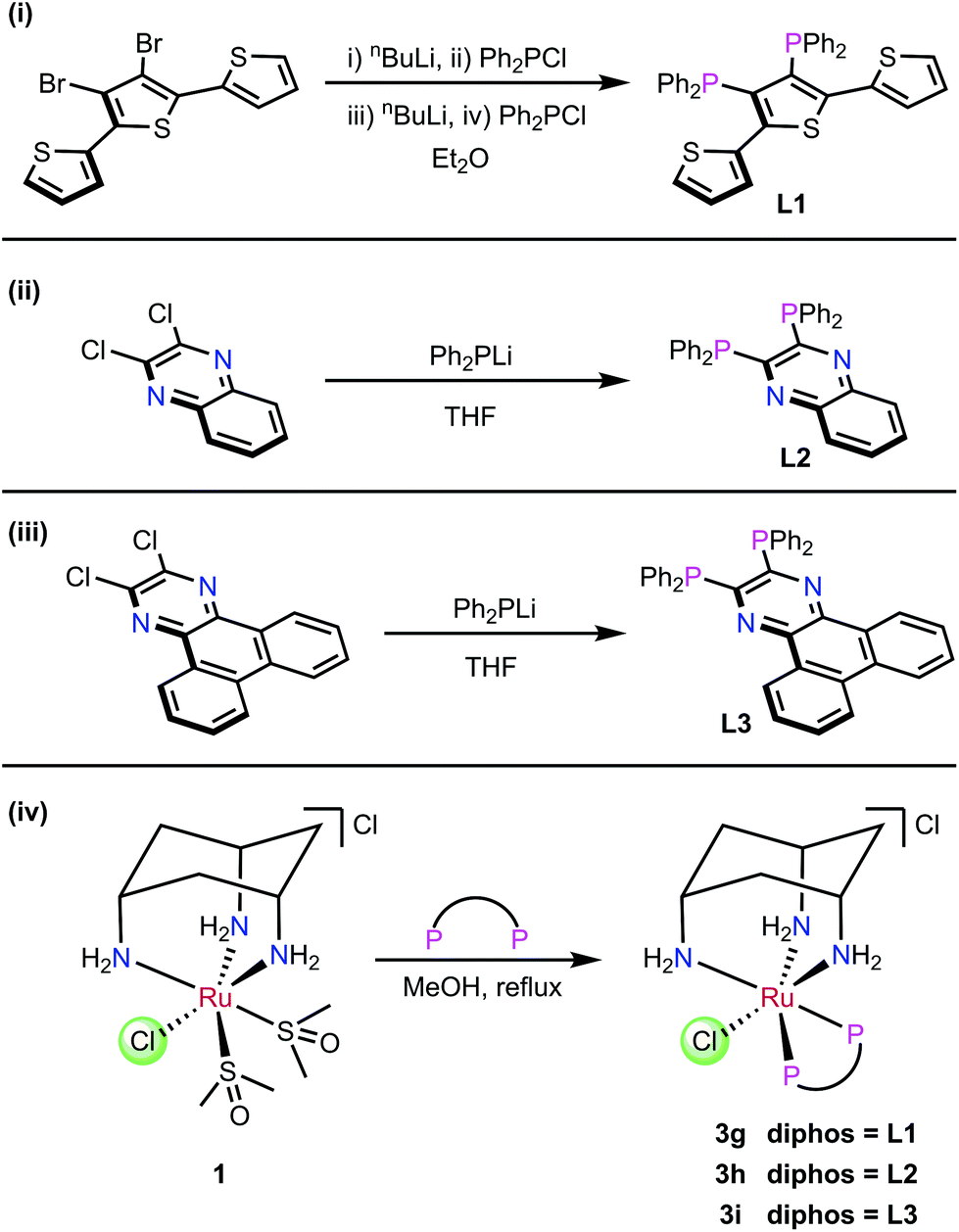 | ||
| Scheme 2 (i)–(iii) Synthesis of ligands L1–L3. (iv) Synthesis of Ru complexes 3g–i. | ||
The X-ray crystal structure of [3h]PF6 (Fig. 3) demonstrates that the addition of the larger, planar aromatic quinoxaline diphosphine ligand L2 does not significantly alter the geometry of the (cis-tach)Ru complex, as shown by the overlap with the phenylene diphosphine analogue (3f) illustrated in Fig. 3. The cis-tach ligand adopts the expected κ3-coordination mode and there are intramolecular interactions detected between the N(4)H2 and the centroids of the phenyl rings of the PPh2 groups. In addition to the lipophilic cyclohexane ring, the PPh2 groups provide further hydrophobicity to the complex and have the potential to interact with biomolecules (see below).
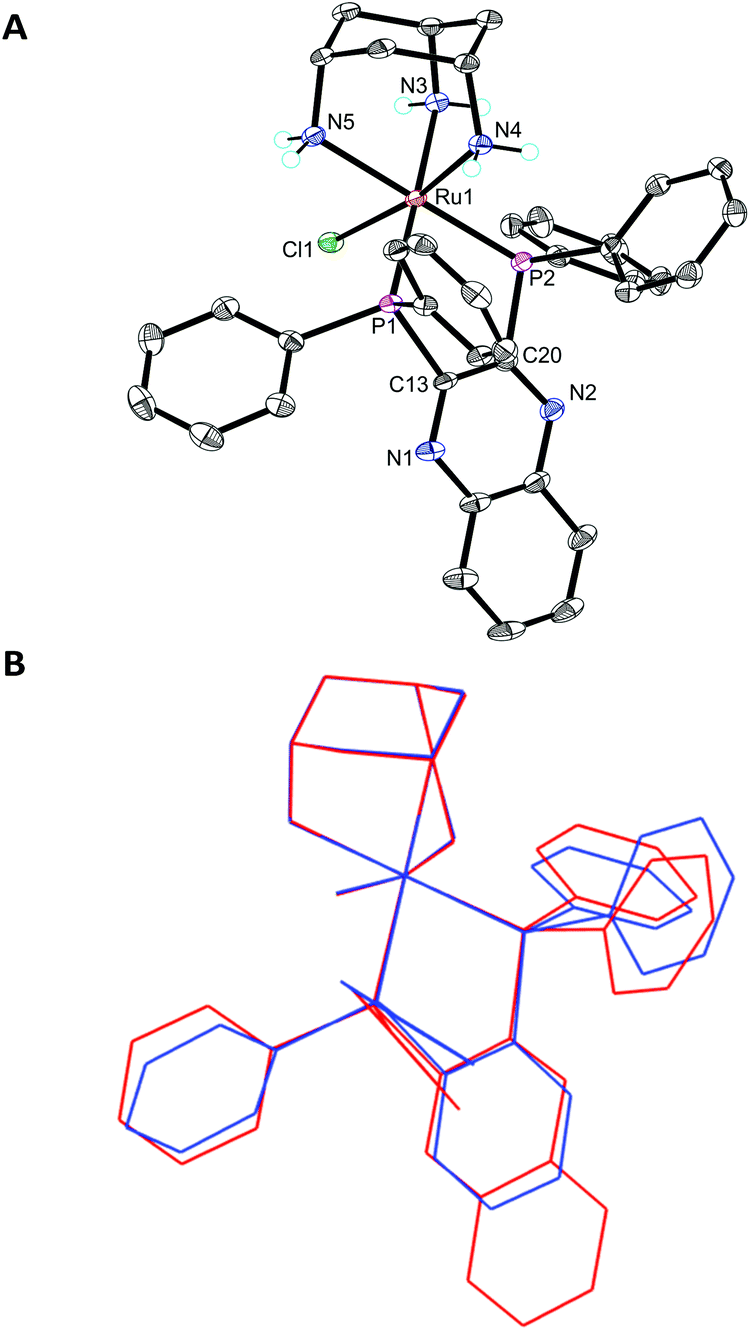 | ||
| Fig. 3 ORTEP diagram of [3h]PF6 (A) and overlap with [3f]PF6 (B), ellipsoids are shown at the 50% probability level and hydrogen atoms (except for the –NH2 groups) and PF6− are omitted for clarity. Selected bond lengths (Å) and angles (°) for [3h]PF6: Ru1–Cl1 2.4335(6), Ru1–P1 2.2857(7), Ru1–P2 2.2654(6), Ru1–N3 2.179(2), Ru1–N4 2.130(2), Ru1–N5 2.182(2); P2–Ru1–P1 84.43(2), N4–Ru1–Cl1 170.90(6), P1–Ru1–Cl1 90.11(2), P2–Ru1–Cl1 93.92(2). | ||
Inhibition of the proliferation of A549 and A2780 cells
The in vitro growth inhibition was determined by MTT assay in two cell lines: A549 (human lung adenocarcinoma) and A2780 (human ovarian adenocarcinoma). The Ru complex of bipy (2a) in concentrations up to 300 μM, did not inhibit the growth of tumour cells and were therefore considered inactive.Ruthenium mono-phosphine complexes containing an η6-arene, have previously been shown11,15,39–42 to be active against cancer cells. We now report clear antiproliferative activity with the P,P-chelates 3a–f as shown in Fig. 4 and some trends can be discerned from the data. (1) The activity generally increases with increasing chelate ring size: 3a < 3b < 3c ≃ 3d; complexes 3c and 3d are over twice as active as cisplatin against the A549 cell line and are equipotent to cisplatin against the A2780 cell line. (2) Complex 3b is over twice as active as complex 3e against both cell lines; although both 3b and 3e are 5-membered chelates, the more active chelate 3b has a less rigid backbone. (3) Complex 3f is significantly more active (by factors of ca. 10 and 7 against the two cell lines) than the ostensibly similar complex 3e. Although both 3e and 3f are rigid, 5-membered chelates, the phenylene backbone in 3f will make the complex more lipophilic. It was also speculated that the intercalating potential of the planar aromatic backbone present in 3f may also be a contributing factor in its higher activity than 3e.
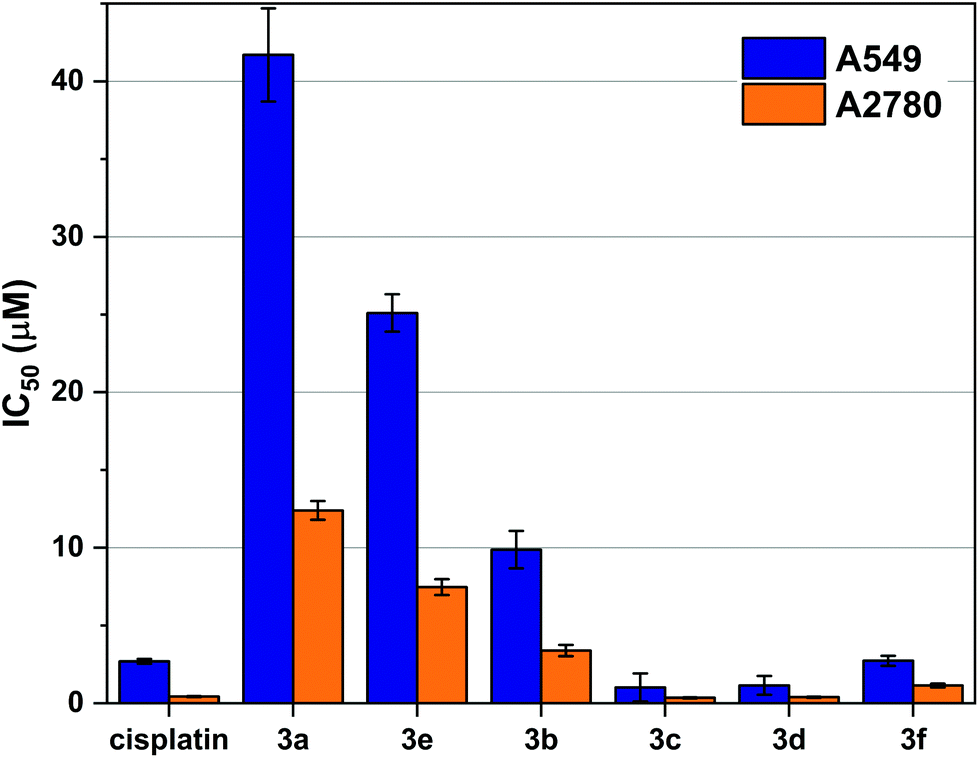 | ||
| Fig. 4 Cell viability data in A549 and A2780 cells treated with cisplatin and 3a–f to show influence of ligand backbone on cytotoxicity. Antiproliferative activities were determined by MTT assay and dose response curves are given in Fig. S5 and 6.† The IC50 calculated is the concentration of drug required for 50% growth inhibition over a 72-hour period. The error bars represent one standard deviation from three independent experiments. | ||
To explore this hypothesis further, the (cis-tach)Ru complexes 3g–i containing diphosphines with extended aromatic surfaces: terthiophenyl diphosphine (L1), quinoxaline diphosphine (L2) and dibenzo[f,h]quinoxaline diphosphine (L3) (see Fig. 2 and Scheme 2) were tested. It was postulated that these novel complexes might exhibit dual-function cytotoxicity by covalently binding to biomolecules and by intercalation with DNA in a similar way to the functioning of the cytotoxic Pt complex phenanthriplatin.43,44 If this were the case, it was reasoned that 3g–i would be expected to be more active than the first-generation Ru cis-tach complexes 3a–f.
We assessed the antiproliferative activity of 3g–i against A549 cells by a 72-hour MTT assay and found that the complexes were comparable in activity to the most active tested P,P-chelate complexes 3c–d with IC50 values of 1.83 ± 0.66 μM for 3g, 11.81 ± 1.23 μM for 3h, and 5.06 ± 1.01 μM for 3i. Taking into account the experimental errors inherent in the MTT assays, the difference between the activity of 3g and 3c–d is not statistically significant. Encouraged by the activities of these more lipophilic derivatives, we sought to understand their interactions with a variety of biomolecules and study their activity in vitro. The MTT data for complexes 3g–i (Table S2†) demonstrated that their activity was comparable to the most active (cis-tach)Ru complexes. However, detailed time course studies using in situ cellular imaging revealed further details of the behaviour of the complexes and indicated that 3g–i are significantly more active against A549 and 293T cells when compared to cisplatin and 3f (see below).
Two potential modes of action of the Ru complexes 3a–i were investigated. Firstly, aquation of the Ru–Cl bond to the labile Ru–OH2 complex followed by covalent interactions with nucleosides; and secondly, the ability of the complexes to interact with DNA by non-covalent interactions.
Aquation of Ru–Cl complexes
The aquation products of 3b, 3c and 3h were characterised by 31P{1H} NMR spectroscopy and ESI-MS. The 31P{1H} NMR spectra for complexes recorded in water at pH 7.4 consisted of two singlet resonances, with one corresponding to the starting chlorido complex, and the other to the aquated (water or hydroxo) complex (Scheme 3). The assignment of each resonance was made by monitoring the changes upon addition of sodium chloride to the aqueous solution (Fig. 5). The high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) data for 3b, 3c and 3h recorded in 75% H2O/25% MeOH supported the formation of the aquated complexes with the molecular ion mass and isotope patterns corresponding to the ion [Ru–Cl + OH]+ in all cases.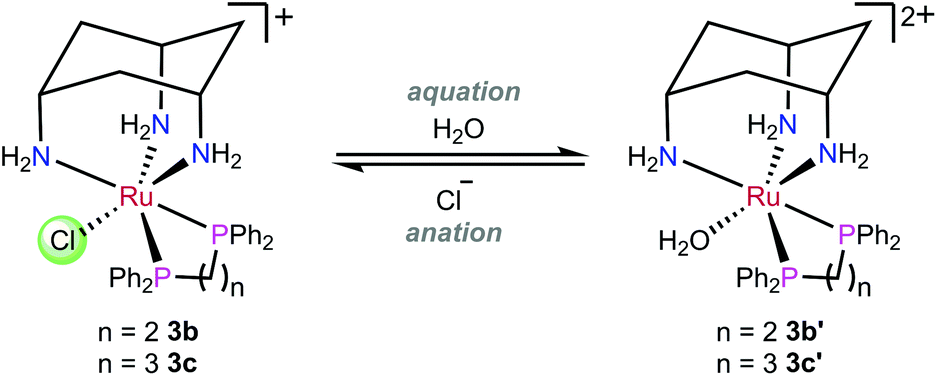 | ||
| Scheme 3 Representative aquation/anation for 3b and 3c. | ||
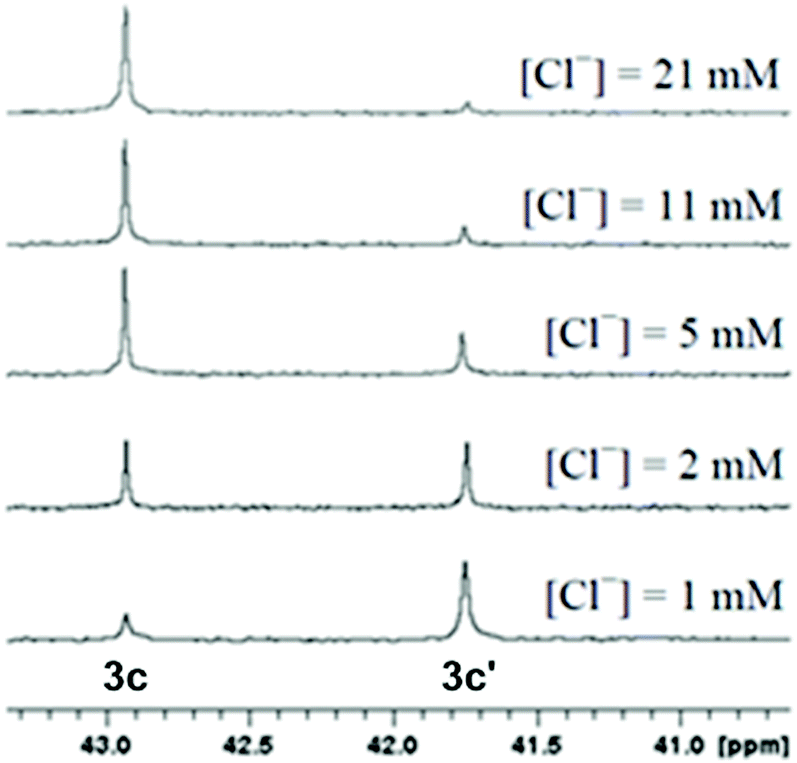 | ||
| Fig. 5 Stacked 31P{1H} NMR spectra of a 500 μM solution (pH 7.4) of 3c with various chloride concentrations to assign the resonances corresponding to the Ru chlorido (3c) and Ru aqua (3c′) complexes. | ||
Kinetics of aquation
The kinetics of aquation and anation of the ruthenium cis-tach complexes 3b, 3f and 3h were investigated by UV/Visible spectroscopy and compared to [RuCl(η6-bip)(en)]+ (C in Fig. 1).29 The time-evolution difference spectra for each complex are shown in Fig. S1.† The presence of isosbestic points in each spectrum suggests that the aquation process involves a single-step mechanism in the formation of the aqua derivative from the chlorido complex. The time dependence for the absorbance of each complex followed first order kinetics (Table 1). The rate of aquation of 3b at physiological temperature (310 K) corresponds to a half-life of 33 s. Therefore, the rate of aquation is not a significant factor in the in vitro activity of the complex as aquation occurs rapidly in comparison to cell proliferation (typically 24 h).| Complex | T/K | k/10−3 s−1 | t ½/s |
|---|---|---|---|
| a Measurements for the aquation of 3b, 3c, 3f and 3h (300 μM) in aqueous solution buffered at pH 7.4 (10 mM sodium phosphate). | |||
| 3b | 288 | 2.09 ± 0.02 | 331 ± 3 |
| 3b | 293 | 3.60 ± 0.08 | 192 ± 5 |
| 3b | 298 | 6.55 ± 0.06 | 106 ± 1 |
| 3b | 303 | 10.7 ± 0.20 | 65 ± 1 |
| 3b | 310 | 21.0 ± 0.70 | 33 ± 1 |
| 3c | 298 | 63.9 ± 6.0 | 10 ± 2 |
| 3f | 298 | 2.23 ± 0.12 | 311 ± 17 |
| 3h | 298 | 1.02 ± 0.01 | 679 ± 3 |
The rate constants for the aquation of 3b (6.55 ± 0.06 × 10−3 s−1) and 3c (63.9 ± 6.0 × 10−3 s−1) at 298 K are approximately 5 and 15 times faster than the η6-biphenyl complex [RuCl(η6-bip)(en)]+ (C) (1.28 × 10−3 s−1) respectively. This difference could be attributed to a weakening of the Ru–Cl bond, as shown by its lengthening in 3b (2.4431(14) Å) and 3c (2.4404(4) Å) when compared to RAen complex C (2.405(6) Å), due to the large trans-effect of the nitrogen donors in the cis-tach ligand.
The reaction rates for aquation and anation at different temperatures allowed for the determination of the Arrhenius activation energy (Ea), activation enthalpy (ΔH‡) and activation entropy (ΔS‡). The Ea and ΔH‡ for the aquation reaction are comparable to those reported for complex C (Table 2), consistent with the fundamental mechanistic steps in the aquation process being the same. Both 3b and 3c were found to be stable for the duration of a typical 72 h MTT assay experiment. Furthermore, over a two-week period at 37 °C, the 1H NMR spectrum of 3b in 10% D2O/90% H2O did not change. Since the rate of aquation of the Ru–Cl is rapid, the biological activity is likely more dependent on the binding to biomolecules once this aquation step has taken place.
| Complex | E a/kJ mol−1 | ΔH‡/kJ mol−1 | ΔS‡/J K−1 mol−1 |
|---|---|---|---|
| a Arrhenius activation energy (Ea), activation enthalpy (ΔH‡) and activation entropy (ΔS‡) for the aquation and anation of 3b and 3b’ at pH 7.4. b Complex C is shown in Fig. 1; values taken from ref. 29. | |||
| 3b | 79.8 ± 0.7 | 77.3 ± 0.7 | −27.6 ± 4.8 |
| 3b′ | 84.9 ± 1.0 | 82.4 ± 1.0 | 24.4 ± 3.4 |
C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
75.6 ± 0.6 | 73.1 ± 0.6 | −55.7 ± 2.0 |
|
C′b |
76.7 ± 1.3 | 74.1 ± 1.3 | −13.6 ± 4.5 |
Determination of the pKa of the Ru–OH2
The pKa of metal aqua complexes can form the basis of structure–activity relationships, and it has been noted that Ru complexes with higher pKa values have exhibited greater potency in vitro.48 The UV/Visible spectra of 3b and 3c were recorded at pH intervals between 2 and 12 at 298 K in order to determine the pKa values for the coordinated water ligands. The data were fitted to the Henderson–Hasselbalch equation which gave pKa values of 10.80 ± 0.06 and 10.42 ± 0.15 for 3b and 3c respectively. The 1H NMR pH titrations, performed in H2O with 1.6% CD3OD to provide an NMR lock signal, resulted in pKa values of 10.85 ± 0.02 and 10.54 ± 0.02 for 3b and 3c respectively and were therefore in good agreement with the UV/Visible spectroscopy determinations (Fig. S2†).The acid-dissociation constants for these complexes are higher than those commonly obtained for (η6-arene)ruthenium(II) complexes, including [Ru(OH2)(η6-biphenyl)(en)]2+ (7.71 ± 0.01), [Ru(OH2)(η6-tha)(en)]2+ (8.01 ± 0.03, tha = tetrahydroanthracene), and [RuCl(OH2)(η6-C6H6)(PTA)]2+ (9.2 ± 0.03). The highest pKa values reported are obtained for O,O-chelates, such as [Ru(OH2)(η6-p-cymene)(malonate)]+ (9.23 ± 0.02) and [Ru(OH2)(η6-p-cymene)(acac)]+ (9.41 ± 0.01).8,10,48,49 The conclusion drawn from the pKa measurements of complexes 3b and 3c is that the deprotonated forms are physiologically inaccessible by more than 3 pH units and aquation therefore affords exclusively the aqua complex.
One likely step in the mechanism of the antiproliferative effect of the Ru complexes 3a–i is through coordination of a biomolecule to the site on Ru initially occupied by the chlorido ligand. The Ru complexes share a structural feature with cisplatin – a nitrogen donor trans to a chlorido ligand and have favourable aquation kinetics. Additionally, the amine groups of the cis-tach ligand are located cis to the chlorido ligand and may have a role in strengthening interactions with a bound molecule through hydrogen bonding. As a result of this reasoning, the series of DNA binding experiments described below were carried out.
Calculated speciation in biological environments
The kinetic analysis shows that the rate of aquation for (cis-tach)Ru complexes is rapid at physiological temperature, reaching equilibrium within minutes. It is of interest to understand the extent to which these complexes are aquated outside and within a cell and to predict the composition of species present in each environment. In order to understand the physiological significance of the aquation/anation equilibrium constants, we have predicted the distribution of chlorido and aqua species for the blood, cytoplasm and cell nucleus based on the reported chloride concentrations (Table S1†).29,50The equilibrium constant, K, for the aquation of 3b (K = 30.6 ± 1.7 × 10−3 M) is significantly greater than for 3c (K = 5.9 ± 0.1 × 10−3 M) and results in a considerable difference in speciation (Fig. S3 and Table S1†). The two equilibrium constants span those of the RAen complexes. The equilibrium constant for the aquation of 3b may result in the formation of the aqua species in the blood, cytoplasm, and cell nucleus, leading to a greater possibility for deactivation reactions to occur. For example, cisplatin is deactivated by glutathione binding prior to reaching the nucleus51 and a similar process could account for the 10-fold reduction in activity of 3b compared to 3c.
The combination of a smaller equilibrium constant for aquation and a lower pKa of the aqua species results in a favourable proportion of 3c′ formed under different physiological environments. It is predicted for 3c that a lower proportion of aqua species would be present in the blood, but a high proportion in the cell nucleus compared to RAen complexes. Therefore, the proportion of aqua species for 3c is greater than for many other Ru complexes that have been evaluated for anti-cancer activity (Table S1†). This produces a good balance between protection of the complex outside the cell and a higher degree of activation inside the cell. As demonstrated by the kinetic study, the rate at which the complex is aquated once inside the cell is very rapid and is not a factor in the intracellular speciation.
Interactions of 3b and 3c with nucleosides
It has been shown that [RuCl(η6-arene)(en)]+ forms strong covalent adducts with DNA, with a preference for the N7 of guanine residues.52–54 These interactions are strengthened by hydrogen bonds between the en ligand and the O6 of an adjacent guanine residue. The reaction of 3b with 9-ethylguanine (EtG) (see ESI†) resulted in a new guanine containing species in the 1H NMR spectrum, evident from a new H8 resonance (Δδ(H8) = –1.93 ppm, Fig. S4†). This is consistent with guanine binding at the N7 position to give [Ru(EtG-N7)(dppe)(cis-tach)]2+.Substitution of the chorido ligand with a guanine derivative was demonstrated by the reaction of 3b with guanosine (Guo) after incubation at 37 °C for 24 h in water. The solution was diluted to 0.1 mM with 50% methanol in water and the ESI mass spectrum recorded. An ion with mass and isotope pattern corresponding to [M–Cl + Guo–H]+ (30%) and [M−Cl + Guo]2+ (30%) was observed at m/z 911.1 and 456.2 respectively along with chlorido (100%) and hydroxy (30%) species. In contrast to 3b, the reaction of 3c with EtG did not produce an observable adduct in the 1H NMR spectrum after 24 h at 37 °C. Therefore, it is plausible that coordination to 3c of the N7 of guanine may not be involved in the mechanism by which this complex inhibits proliferation.
Binding of Ru to CT-DNA
The intercalating fluorescent dye ethidium bromide (EB) was used in a competition assay with complexes 3b, 3f, 3g, 3h, 3i as well as the known DNA intercalator [Ru(bpy)2(DPPZ)]2+ (DPPZ = dipyrido[3,2-a:2′,3′-c]phenazine) as a benchmark (see Fig. 6).55 A solution of 50 μM CT-DNA and 5 μM EB ([CT-DNA]/[EB] = 10:1) was prepared and the EB-CT-DNA adduct was subjected to titration with Ru complexes. The quenching constants (KSV) were calculated according to the Stern–Volmer (SV) equation.56–58where I0 and I are the emission intensity in the absence and presence of quencher complex respectively, KSV is the Stern–Volmer quenching constant and [Q] is the quencher concentration. From these plots, the apparent binding constants (Kapp) were calculated using: KEB[EB] = Kapp[complex], where KEB = 1 × 107 M−1, [EB] = 5 μM, and [complex] is the concentration of Ru complex which gave a 50% reduction of the initial emission intensity of EB.
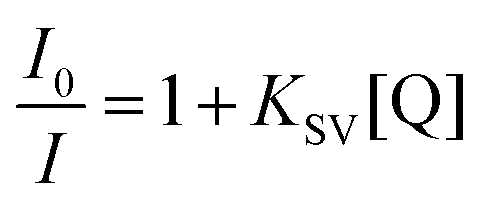
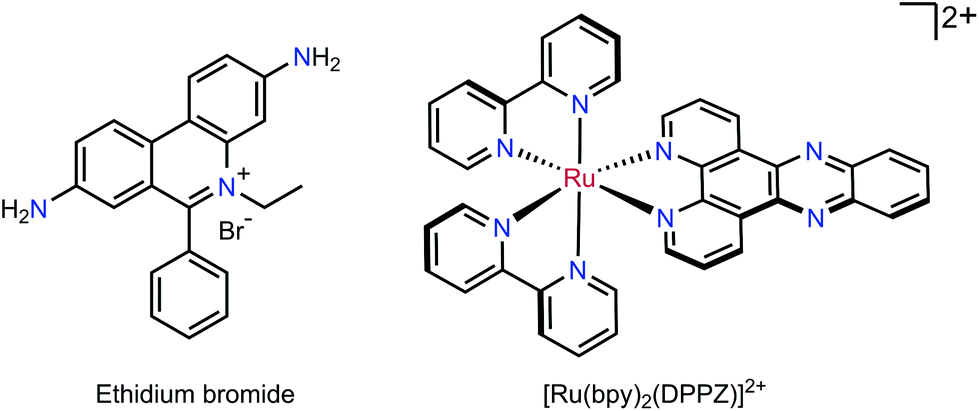 | ||
| Fig. 6 Chemical structures of the DNA intercalating dye ethidium bromide and the known intercalating complex [Ru(bpy)2(DPPZ)]2+. | ||
For titrations with complexes 3g, 3h, 3i, a gradual decrease in emission was observed (Fig. 7 and Fig. S7†) implying that these complexes outcompete the EB to interact with DNA. The same titration was performed with 3b and 3f but no appreciable decrease in emission was observed for the dppe complex indicating little intercalative interaction is present (Fig. S7†).
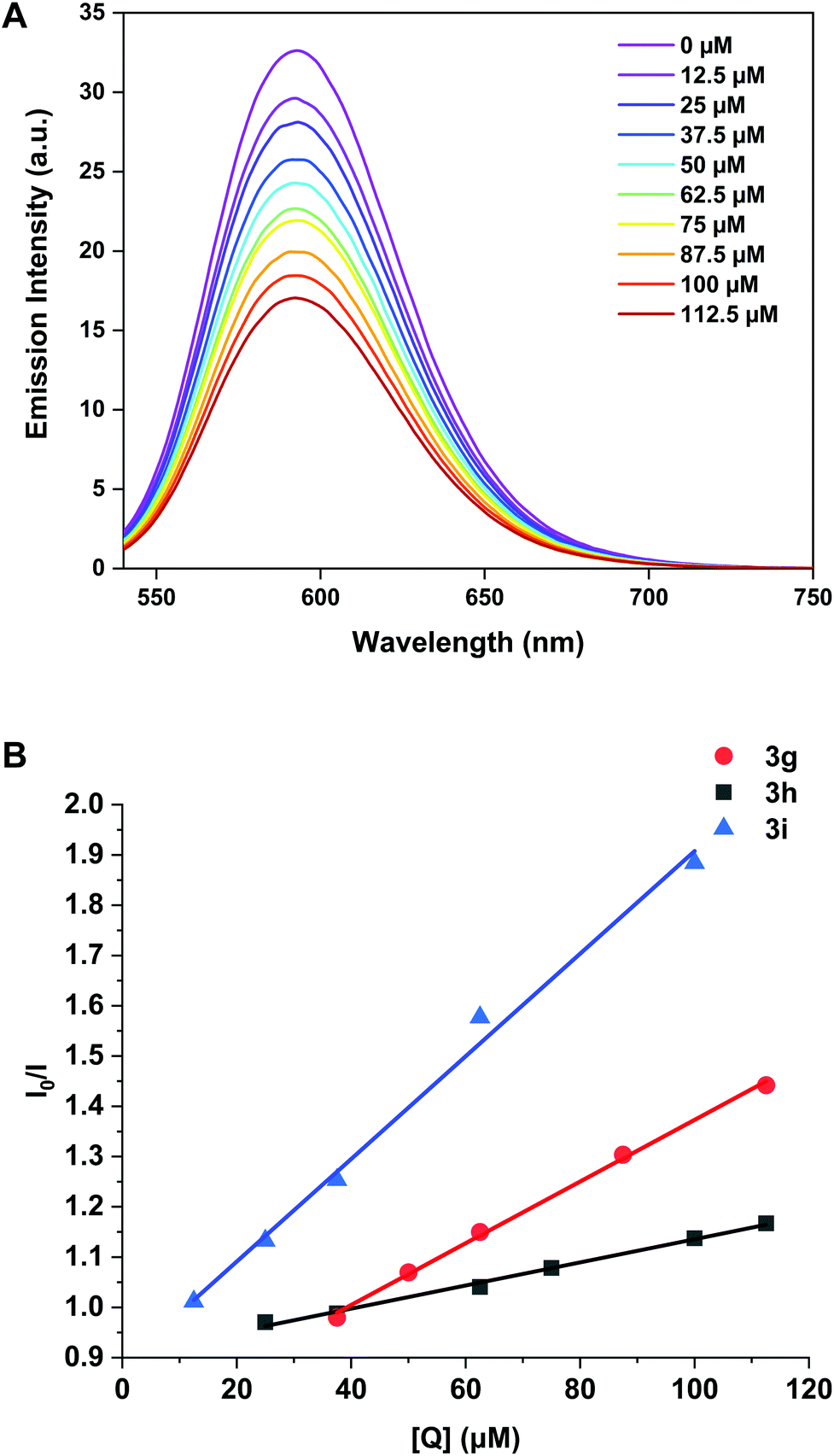 | ||
| Fig. 7 (A) Emission spectra of CT-DNA (50 μM) and EB (5 μM) competition assay with 3h (0–112.5 μM). (B) Stern–Volmer plots EB-CT-DNA vs. the concentration of 3g, 3h and 3i. | ||
For 3f, the decrease was so small that a binding constant could not be calculated and therefore intercalation is not considered a viable mode of action for this complex. The binding constants (Kapp) for 3g, 3h and 3i are 3.07 ± 0.07 × 105 M−1, 1.16 ± 0.04 × 105 M−1 and 5.11 ± 0.25 × 105 M−1 respectively (Table 3), indicating that increasing the aromatic surface of the ligand backbone (3h to 3i) gave a five-fold increase in binding affinity.
These binding constants are comparable, and in some cases superior to, rigid dinuclear (η6-arene)Ru complexes previously reported.59 The apparent binding constant of the known DNA intercalator [Ru(bpy)2(DPPZ)]2+ was calculated as 1.75 ± 0.02 × 106 M−1, only one order of magnitude higher than the (cis-tach)Ru complexes.
Interactions with plasmid DNA
Gel electrophoretic mobility shift assays (GEMSA) were used to study the interactions of 3b and 3c with plasmid DNA. From 1H NMR spectroscopy and mass spectrometry experiments, 9-ethylguanine (EtG) coordination was observed with 3b, whereas 3c showed no reaction with EtG after 24 h at 37 °C (see ESI†). This result was mirrored in the gel electrophoresis assays with pUC18 DNA (Fig. 8), where 3c did not alter the mobility of either the open coil (OC) or super-coiled (CCC) forms of the plasmid under the conditions employed. However, 3b did alter the mobility of the OC and CCC forms of pUC18 DNA at concentrations above 25% bpe (base pair equivalents), where a small shift was observed with increasing concentration (lanes 5–7, Fig. 8). These results suggest that binding to the N7 of guanine is not involved in the mechanism of action for 3c. It is evident that 3b and 3c (lanes 3–7, Fig. 8) have different reactivity with plasmid DNA from cisplatin (lane 8, Fig. 8), indicating that these complexes may have different cellular targets that result in cytotoxicity.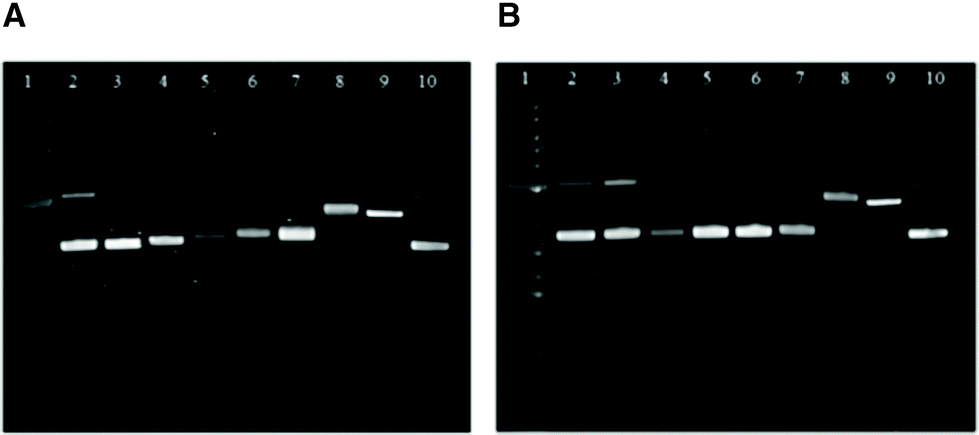 | ||
| Fig. 8 GEMSA of 3b (A) and 3c (B) with pUC18 plasmid DNA after 20 h at 37 °C. Lanes: molecular marker (1); pUC18 (2 and 10); pUC18+ 3b or 3c (% bpe), 2.5 (3), 10 (4), 25 (5), 50 (6), 100 (7); pUC18+ 10% bpe cisplatin (8); pUC18 linearised by single cut with SmaI (9). bpe = base pair equivalents. | ||
Binding to G-quadruplex DNA
G-quadruplexes are four-stranded DNA secondary structures that form from guanine-rich sequences (Fig. 9). They attract particular attention as an anticancer target, owing to the occurrence of quadruplex-forming motifs at chromosome telomeres and in the promoter sequences of several oncogenes, e.g. the c-myc gene.60–62 These structures can adopt a variety of topologies characterised by the relative orientations (parallel/antiparallel) of the DNA strands in the folded structure.63–65 Furthermore, the planar G-tetrads provide additional opportunities for stacking and intercalative interactions with complexes, since the dimensions are significantly larger than those of a classical Watson–Crick base pair.66 These DNA sequences can hypothetically be targeted with high-selectivity in order to reduce or eliminate the off-target effects resulting from indiscriminate binding to duplex DNA.60,66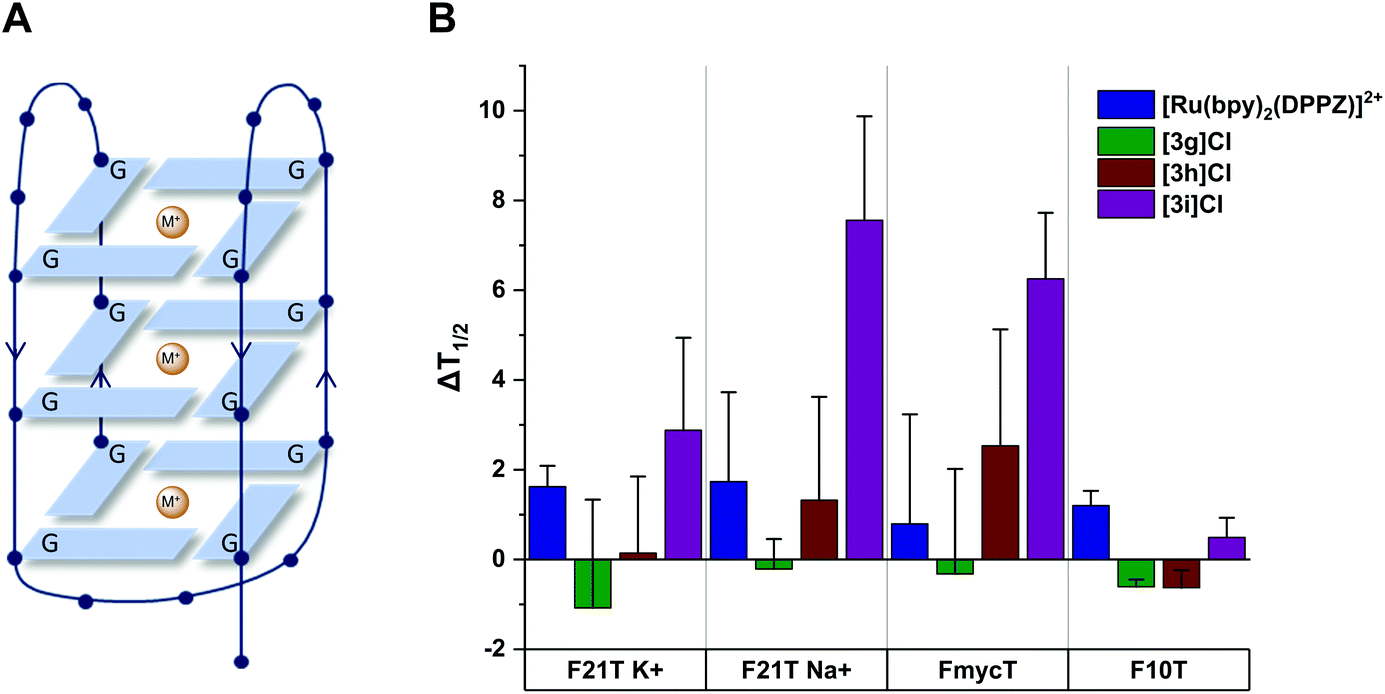 | ||
| Fig. 9 (A) Schematic representation of a DNA G-quadruplex. The folding of the oligonucleotide into a four-stranded structure creates a stacked arrangement of G-tetrads, each formed by the square-planar assembly of four guanine (G) residues. The overall topology is determined by the relative orientation of the neighbouring strands (indicated by arrows) and stabilized by co-ordination to metal ions (M+). (B) Average ΔT1/2 for quadruplex (F21T and FmycT) and duplex (F10T) DNA after treatment with Ru complexes (1 μM). Error bars show standard deviations from four experiments. | ||
This therapeutic hypothesis has led to many groups designing G-quadruplex binders as potential anticancer agents67–70 and many metal complexes are known to bind G-quadruplexes effectively through covalent and non-covalent interactions.71 For example, Liu et al. found that ruthenium polypyridyl complexes containing 4idip (4-indoleimidazo[4,5-f][1,10]phenanthroline) ligands were able to selectively stabilise the human telomeric G-quadruplex structure.68,69
As a result of the intercalating ability of 3g–i indicated by the EB assays, we investigated the ability of the complexes to stabilise G-quadruplex DNA and duplex DNA structures. The extent of stabilisation was quantified by performing a fluorescence resonance energy transfer (FRET) assay initially reported by De Cian et al.72 (see ESI†). The change in DNA melting temperature (ΔT1/2) induced by a Ru complex compared to that of the oligonucleotide in the absence of complex provides an indication of the capacity of the complex to stabilise the G-quadruplex structure. We chose to investigate three models of G-quadruplex DNA and one of duplex DNA (see Fig. 9). The human telomeric sequence (F21T) was studied in potassium- and sodium-containing buffer owing to the known influence of the metal ion on the polymorphism of this sequence.73,74 The G-quadruplex sequence found in the c-myc oncogene promoter (FmycT) was selected as a model of this anticancer target.
The results of the FRET experiments are shown in Fig. 9 and representative raw data in the ESI (Fig. S8†). Complexes 3g and 3h did not induce any appreciable stabilisation of quadruplex DNA (ΔT1/2 < 3 °C at 1 μM complex) but 3i did stabilise F21T (ΔT1/2 = + 7.5 ± 2.3 °C) in sodium-containing buffer. Additionally, 3i was selective for quadruplex DNA structures, stabilising the quadruplex sequence FmycT (ΔT1/2 = + 6.2 ± 1.5 °C) whilst stabilisation of the duplex sequence F10T (Fig. 9) was negligible. Meanwhile, the same complex did not significantly stabilize F21T in K+-rich buffer (ΔT1/2 < 3 °C), suggesting that as well as G4/duplex selectivity, the complex can also discriminate between different G-quadruplex topologies to some extent. As a control, the well-known DNA intercalator complex [Ru(bpy)2(DPPZ)]2+ did not significantly stabilise quadruplex DNA (ΔT1/2 = + 1.6 ± 0.4 °C for F21T) or duplex DNA (ΔT1/2 = + 1.2 ± 0.3 °C for F10T) as previously reported.67
The dibenzo[f,h]quinoxaline moiety in 3i provides a large aromatic surface that may selectively stabilise G-quadruplex DNA through preferential association with the large G-tetrads over intercalation with Watson–Crick base pairs, akin to the 4idip examples previously reported. Complex 3i showed stronger binding to quadruplex DNA than 3g and 3h in experiments with quadruplex DNA as well as CT-DNA as previously shown. This interesting discovery that Ru cis-tach complexes interact with quadruplex DNA warrants further study.
LiveCyte cell imaging
The viability of A549 cells and 293T cells was assessed using Livecyte (Phasefocus Ltd) label-free time-lapse microscopy.75,76 LiveCyte cell imaging does not require the cell to be labelled with antibodies or cellular dyes; the cells are unperturbed and the imaging process is not toxic to the cells; the cellular changes measured are reported with confidence to be associated with the presence of the compounds. Conventional light microscope imaging requires the cells to be labelled with a DNA binding or cytoplasmic dye and requires higher energy lasers, compared to the LiveCyte: both the labelling and the imaging light source in conventional light microscopy may be cytotoxic over prolonged time and needs to be considered when interpreting results.This technique enables quantification of the total cellular dry mass as an indicator of cell death and growth. For A549 and 293T cells, the effect of treatment with cisplatin and 3i is shown at 36 and 72-hour time-points (Fig. 10 and 11). For 0-hour time points and compounds 3f–h see Fig. S11–13.† The MTT assay is an endpoint colourmetric readout of cell viability and does not provide any visual characteristics of the cells state. In this preliminary study, integrated image analysis software (Livecyte Cell Analysis Toolbox) was used to extract real-time changes in morphology and dry mass of each cell over time. The summed mass of the cellular components excluding water (known as dry mass, see Fig. S9 and 10†) was calculated and for each treated population of cells used as a measure of the combined growth and proliferation. For A549 cells (Fig. 10D), the reduction in dry mass is greatest for 3g and 3i followed by the slightly less active derivative 3h (which shows comparable results to cisplatin), and 3f gave the smallest decrease in dry mass of those tested. This difference in dry mass reduction between cisplatin and 3f is notable as the MTT assay derived IC50 values are within error of each other (IC50 = 2.7 μM). For 293T cells (Fig. 11D), this effect is more pronounced with 3f causing significantly less reduction in dry mass than cisplatin.
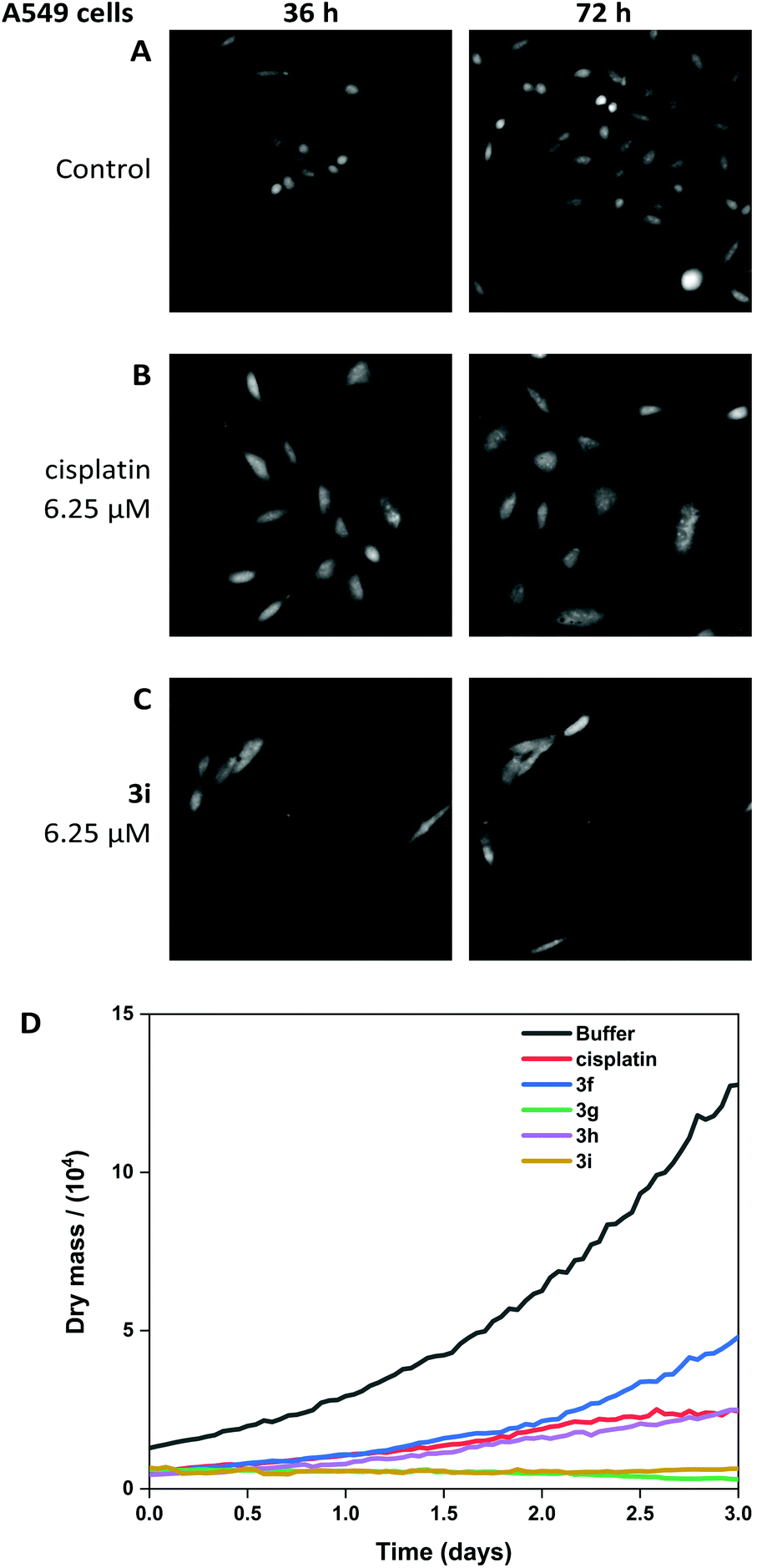 | ||
| Fig. 10 Livecyte time-lapse images of A549 control cells (A) and after treatment with 6.25 μM cisplatin (B) and 6.25 μM 3i (C). (D) Dry mass plot to show growth inhibition when treated with cisplatin, 3f, 3g, 3h, 3i (all at 6.25 μM). | ||
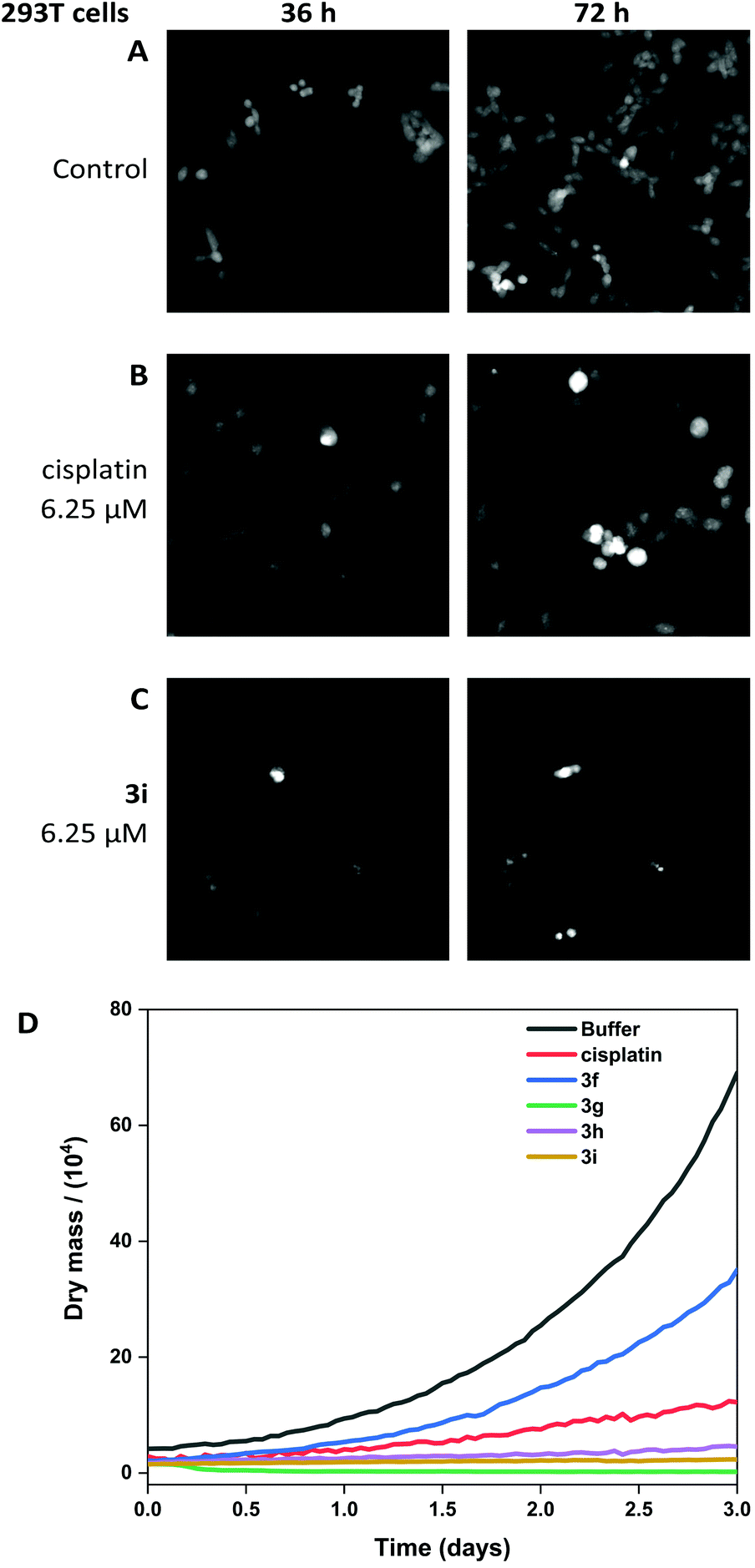 | ||
| Fig. 11 Livecyte time-lapse images of 293T control cells (A) and after treatment with 6.25 μM cisplatin (B) and 6.25 μM 3i (C). (D) Dry mass plot to show growth inhibition when treated with cisplatin, 3f, 3g, 3h, 3i (all at 6.25 μM). | ||
The other (cis-tach)Ru complexes 3g–i caused significant cell death at a concentration of 6.25 μM as is evident from the cell images and dry mass curves, and this greater activity is consistent with 3g–i exhibiting cytotoxicity by multiple mechanisms.
Conclusions
In summary, the compounds presented here are the first reported examples of cytotoxic ruthenium(II) cis-tach complexes. The complex [RuCl(dmso-S)2(cis-tach)]Cl (1) is obtained in high yield and is a very useful precursor for the synthesis of a range of ruthenium(II) derivatives. Complexes 3a–i which contain chelating diphosphine ligands are highly active in vitro against A549, A2780 and 293T cancer cell lines. In particular, complexes with flexible aliphatic backbones (3c and 3d) or planar aromatic backbones (3f–i) are the most cytotoxic, with activity in the A549 cell line more than twice that of cisplatin, and activity in the A2780 cell line equipotent to the clinical drug. The pKa values for 3b–c are significantly higher that most (arene)Ru complexes which, coupled with favourable aquation kinetics, enables high levels of the active Ru–OH2 complex in the cell nucleus.New analogues with planar aromatic backbones have been shown to intercalate strongly with CT-DNA models, and, in the case of 3i also selectively stabilise G-quadruplex DNA over duplex DNA. The anti-proliferative effect has been monitored by LiveCyte, label-free, time-lapse imaging and stark differences are observed between the phenylene derivative 3f and the extended aromatic derivatives (3g and 3i). Overall, these preliminary biological studies suggest that (cis-tach)Ru diphosphine complexes exhibit a dual-action cytotoxic effect, targeting cellular DNA by intercalation, as well as by modes of action involving covalent binding with DNA. This robust, water-soluble molecular architecture could be further developed to produce next generation ruthenium chemotherapeutic agents. Further cell studies exploiting the tunability of phosphine ligands that result in targeted metallodrugs are ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC (EP/L015366/1) and the University of Bristol, for a PhD studentship (DEW). We thank the EPSRC and the University of York (studentship to AJG), University of Garmian (studentship to SWA) and the University of York Research Priming Fund for financial support.References
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549–1560 CrossRef CAS.
- A. Bijelic, S. Theiner, B. K. Keppler and A. Rompel, J. Med. Chem., 2016, 59, 5894–5903 CrossRef CAS.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201–214 CrossRef CAS.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodivers., 2008, 5, 2140–2155 CrossRef CAS.
- R. E. Morris, R. E. Aird, P. Del Socorro Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS.
- R. Fernández, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2004, 10, 5173–5179 CrossRef.
- A. Habtemariam, M. Melchart, R. Fernández, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2007, 13, 2601–2613 CrossRef CAS.
- Q. Wu, L.-Y. Liu, S. Li, F.-X. Wang, J. Li, Y. Qian, Z. Su, Z.-W. Mao, P. J. Sadler and H.-K. Liu, J. Inorg. Biochem., 2018, 189, 30–39 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 15, 1396–1397 RSC.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS.
- H. A. Wee and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 20, 4003–4018 Search PubMed.
- C. Scolaro, A. B. Chaplin, C. G. Hartinger, A. Bergamo, M. Cocchietto, B. K. Keppler, G. Sava and P. J. Dyson, Dalton Trans., 2007, 5065–5072 RSC.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Dröge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 CrossRef.
- A. Bergamo, P. J. Dyson and G. Sava, Coord. Chem. Rev., 2018, 360, 17–33 CrossRef CAS.
- S. M. Guichard, R. Else, E. Reid, B. Zeitlin, R. Aird, M. Muir, M. Dodds, H. Fiebig, P. J. Sadler and D. I. Jodrell, Biochem. Pharmacol., 2006, 71, 408–415 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS.
- A. K. Renfrew, J. Karges, R. Scopelliti, F. D. Bobbink, P. Nowak-Sliwinska, G. Gasser and P. J. Dyson, ChemBioChem, 2019, 20, 2876–2882 CrossRef CAS.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS.
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji and H. Chao, Coord. Chem. Rev., 2018, 363, 17–28 CrossRef CAS.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, Inorg. Chem., 2014, 53, 4548–4559 CrossRef CAS.
- B. Serli, E. Zangrando, T. Gianferrara, C. Scolaro, P. J. Dyson, A. Bergamo and E. Alessio, Eur. J. Inorg. Chem., 2005, 17, 3423–3434 CrossRef.
- J. M. Walker, A. McEwan, R. Pycko, M. L. Tassotto, C. Gottardo, J. Th'ng, R. Wang and G. J. Spivak, Eur. J. Inorg. Chem., 2009, 4629–4633 CrossRef CAS.
- M. M. Haghdoost, G. Golbaghi, M. Létourneau, S. A. Patten and A. Castonguay, Eur. J. Med. Chem., 2017, 132, 282–293 CrossRef CAS.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- F. Wang, H. Chen, S. Parsons, I. D. H. Oswald, J. E. Davidson and P. J. Sadler, Chem. – Eur. J., 2003, 9, 5810–5820 CrossRef CAS.
- H. Huang, P. Zhang, Y. Chen, L. Ji and H. Chao, Dalton Trans., 2015, 44, 15602–15610 RSC.
- B. Greener, L. Cronin, G. D. Wilson and P. H. Walton, J. Chem. Soc., Dalton Trans., 1996, 401–403 RSC.
- B. Greener, S. P. Foxon and P. H. Walton, New J. Chem., 2000, 24, 269–273 RSC.
- B. Greener, M. H. Moore and P. H. Walton, Chem. Commun., 1996, 27–28 RSC.
- L. E. Erickson, D. J. Cook, G. D. Evans, J. E. Sarneski, P. J. Okarma and A. D. Sabatelli, Inorg. Chem., 1990, 29, 1958–1967 CrossRef CAS.
- A. J. Gamble, J. M. Lynam, R. J. Thatcher, P. H. Walton and A. C. Whitwood, Inorg. Chem., 2013, 52, 4517–4527 CrossRef CAS.
- P. Ebrahimpour, M. F. Haddow and D. F. Wass, Inorg. Chem., 2013, 52, 3765–3771 CrossRef CAS.
- T. Kobayashi, S. Tobita, M. Kobayashi, T. Imajyo, M. Chikira, M. Yashiro and Y. Fujii, J. Inorg. Biochem., 2007, 101, 348–361 CrossRef CAS.
- K. G. Moodley, Der Pharma Chem., 2019, 11, 1–19 CAS.
- K. J. Kilpin, S. M. Cammack, C. M. Clavel and P. J. Dyson, Dalton Trans., 2013, 42, 2008–2014 RSC.
- A. Habtemariam, M. Melchart, R. Fernández, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS.
- P. J. Dyson and F. Marchetti, Dalton Trans., 2017, 46, 11973–12366 RSC.
- J. Zhao, D. Zhang, W. Hua, W. Li, G. Xu and S. Gou, Organometallics, 2018, 37, 441–447 CrossRef CAS.
- G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987–11992 CrossRef CAS.
- A. Hucke, G. Y. Park, O. B. Bauer, G. Beyer, C. Köppen, D. Zeeh, C. A. Wehe, M. Sperling, R. Schröter, M. Kantauskaitè, Y. Hagos, U. Karst, S. J. Lippard and G. Ciarimboli, Front. Chem., 2018, 6, 180 CrossRef.
- D. T. Túng, D. T. Tuân, N. Rasool, A. Villinger, H. Reinke, C. Fischer and P. Langer, Adv. Synth. Catal., 2009, 351, 1595–1609 CrossRef.
- S. W. Hunt, L. Yang, X. Wang and M. G. Richmond, J. Organomet. Chem., 2011, 696, 1432–1440 CrossRef CAS.
- Y. Miura, H. Chiba, R. Katoono, H. Kawai, K. Fujiwara, S. Suzuki, K. Okada and T. Suzuki, Tetrahedron Lett., 2012, 53, 6561–6564 CrossRef CAS.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- C. Gossens, A. Dorcier, P. J. Dyson and U. Rothlisberger, Organometallics, 2007, 26, 3969–3975 CrossRef CAS.
- M. Jennerwein and P. A. Andrews, Drug Metab. Dispos., 1995, 23, 178–184 CAS.
- A. K. Godwin, A. Meister, P. J. O'Dwyer, C. S. Huang, T. C. Hamilton and M. E. Anderson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3070–3074 CrossRef CAS.
- C. Gossens, I. Tavernelli and U. Rothlisberger, J. Am. Chem. Soc., 2008, 130, 10921–10928 CrossRef CAS.
- H. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173–186 CrossRef CAS.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS.
- A. Banerjee, J. Singh and D. Dasgupta, J. Fluoresc., 2013, 23, 745–752 CrossRef CAS.
- T. Topală, A. Bodoki, L. Oprean and R. Oprean, Farmacia, 2014, 62, 1049–1061 Search PubMed.
- C. V. Kumar and E. H. Asuncion, J. Am. Chem. Soc., 1993, 115, 8547–8553 CrossRef CAS.
- A. E. Friedman, C. V. Kumar, N. J. Turro and J. K. Barton, Nucleic Acids Res., 1991, 19, 2595–2602 CrossRef CAS.
- Q. Wu, L. Y. Liu, S. Li, F. X. Wang, J. Li, Y. Qian, Z. Su, Z. W. Mao, P. J. Sadler and H. K. Liu, J. Inorg. Biochem., 2018, 189, 30–39 CrossRef CAS.
- S. Neidle, J. Med. Chem., 2016, 59, 5987–6011 CrossRef CAS.
- S. Asamitsu, S. Obata, Z. Yu, T. Bando and H. Sugiyama, Molecules, 2019, 24, 429 CrossRef.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS.
- M. P. O'Hagan, J. C. Morales and M. C. Galan, Eur. J. Org. Chem., 2019, 4995–5017 CrossRef.
- J. Kypr, I. Kejnovská, D. Renčiuk and M. Vorlíčková, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS.
- J. Dai, M. Carver and D. Yang, Biochimie, 2008, 90, 1172–1183 CrossRef CAS.
- A. De Cian, E. DeLemos, J. L. Mergny, M. P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856–1857 CrossRef CAS.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem. – Eur. J., 2006, 12, 4611–4619 CrossRef CAS.
- Q. Yu, Y. Liu, J. Zhang, F. Yang, D. Sun, D. Liu, Y. Zhou and J. Liu, Metallomics, 2013, 5, 222–231 RSC.
- Q. Yu, Y. Liu, C. Wang, D. Sun, X. Yang, Y. Liu and J. Liu, PLoS One, 2012, 7, 1–13 CrossRef.
- M. P. O'Hagan, S. Haldar, M. Duchi, T. A. A. Oliver, A. J. Mulholland, J. C. Morales and M. C. Galan, Angew. Chem., 2019, 58, 4334–4338 CrossRef.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS.
- A. De Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M. P. Teulade-Fichou, L. Lacroix and J. L. Mergny, Methods, 2007, 42, 183–195 CrossRef CAS.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef CAS.
- J. Marrison, L. Räty, P. Marriott and P. O'Toole, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- R. Kasprowicz, R. Suman and P. O'Toole, Int. J. Biochem. Cell Biol., 2017, 84, 89–95 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, characterisation, X-ray crystallography and detailed assay procedures. Table S3, CCDC 1959465. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02612c |
| ‡ Current address, Department of Chemistry, University of Garmian, Kurdistan Region, Iraq. |
| This journal is © The Royal Society of Chemistry 2020 |