
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dianionic C3-symmetric scorpionate: synthesis and coordination chemistry†
Serhii
Tretiakov
 a,
Johannes A. M.
Damen
a,
Martin
Lutz
b and
Marc-Etienne
Moret
*a
a,
Johannes A. M.
Damen
a,
Martin
Lutz
b and
Marc-Etienne
Moret
*a
aUtrecht University, Organic Chemistry & Catalysis, Debye Institute for Nanomaterials Science, Faculty of Science, 3584 CG Utrecht, The Netherlands. E-mail: M.Moret@uu.nl
bUtrecht University, Crystal and Structural Chemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, 3584 CH Utrecht, The Netherlands
First published on 28th August 2020
Abstract
Introducing charges into ligand systems fine-tunes their electronic properties and influences the solubility of their metal complexes. Herein, we present a synthesis of a dianionic, C3-symmetric ligand combining three anionic N-donors tethered to a positively charged phosphonium center. The tris-skatylmethylphosphonium (TSMP) ligand, isolated in the form of its dipotassium salt TSMPK2, is the first dianionic homoscorpionate capable of metal exchange. The potassium cations in TSMPK2 are exchangeable for other metals, which results in rich coordination chemistry. Thus, the ligand displays a bridging μ2:κ2:κ1 coordination mode with trigonal planar Cu(I) centers in the tetrameric complex [(TSMP)Cu]44−. The κ3 mode is accessed upon addition of 1 equiv. of P(OEt)3 per Cu(I) to yield the tetrahedral monomeric complex [(TSMP)CuP(OEt)3]−. Both Fe(II) and Ni(II) in pyridine give octahedral high-spin κ3 complexes with composition (TSMP)M(Py)3 (M = Fe, Ni). Displacement of three pyridine ligands in (TSMP)Fe(Py)3 for a second equivalent of TSMP gives a high-spin pseudotetrahedral 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex [(TSMP)2Fe]2− with the ligands in κ2 coordination mode. The reduction in coordination number is likely due to electrostatic repulsion of the negatively-charged indolides as well as their weaker π-accepting character as compared to pyridine.
:1 complex [(TSMP)2Fe]2− with the ligands in κ2 coordination mode. The reduction in coordination number is likely due to electrostatic repulsion of the negatively-charged indolides as well as their weaker π-accepting character as compared to pyridine.
Introduction
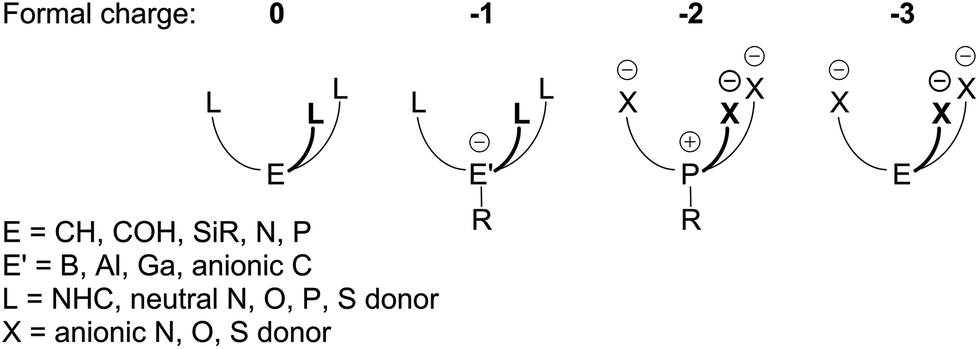
The term “scorpionate” was introduced by Trofimenko1 and refers to tripodal tridentate ligand systems that are able to coordinate to a metal with two identical donor moieties, similarly to the pincers of a scorpion. The third donor moiety rotates forward akin to a scorpion stinger to attack the metal in a fac manner. If it is identical to the first two, a C3-symmetric homoscorpionate complex forms; otherwise, coordination results into a heteroscorpionate complex.Most scorpionates are six-electron donors, which makes them isoelectronic to another common ligand, cyclopentadienyl (Cp). A considerable amount of research was done to compare these two systems.1,2 It is important to point out, however, that while they are isoelectronic, they are not isolobal,3 therefore the extent of such a comparison is limited. Additionally, in some situations, scorpionates are capable of displaying a κ2 coordination mode freeing the third arm for binding to another metal center,1 which sets them apart from Cp ligands.
The first generation of homoscorpionates involved trispyrazolylborates1 and proved to be highly versatile spectator ligands. By modifying the nature, number and position of substituents of the pyrazolyl rings, a wide range of ligands was prepared enabling both electronic and steric properties of a coordinated metal to be fine-tuned. Such systems have found applications in biomimetics,4 catalysis,5 material science6 and production of radiopharmaceuticals.7 Following this success, the definition of scorpionates has been extended to tripodal tridentate systems with other donor groups and bridging atoms. Among the employed donors are imidazole,8 pyridine,9 triazole,10 indole,11 methimazole,12 oxazoline,13 N-heterocyclic carbenes14 and others, and even acyclic donor groups.15 Variation of the bridging atom allowed to further tune electronics and charge of the scorpionate ligands, thus influencing coordination behavior and solubility of their complexes. Reported systems include CH/COH,9 CH3/C6H5-Si,16 N,9a P,8,9,17 [R–P]+,18 P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,9b,19 As,9a AsO,9b [CH3-Al]−,20 [CH3-Ga]−,21 C−,22 Si-,23 Ge−, Sn−,24 Pb−.20
O,9b,19 As,9a AsO,9b [CH3-Al]−,20 [CH3-Ga]−,21 C−,22 Si-,23 Ge−, Sn−,24 Pb−.20
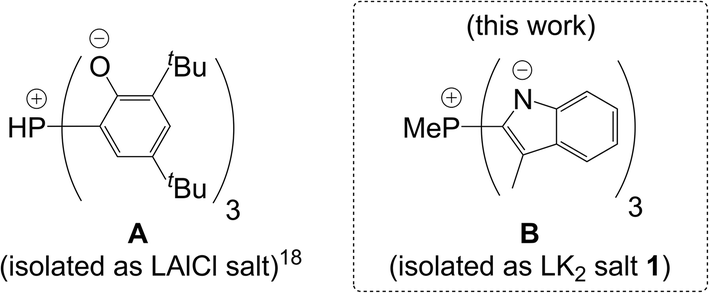
A more general way to classify the existing C3-symmetric scorpionates is by formal charge, which is a combined property of the donor moieties and the bridging atom. Thus, one can differentiate neutral, mono-, di- and trianionic homoscorpionates (Chart 1). Whereas there is a plethora of known neutral8,9,14,16,17 and monoanionic1,10,12,13,20–22,24 homoscorpionates as well as some trianionic examples,11 dianionic ligands are rare with only one reported precedent18 (A in Chart 2). The latter was isolated as a LAlCl complex, and its LAlMe complex could be methylated at phosphorus to afford a methylphosphonium derivative. However strong Al–O bonds would likely preclude the possibility of a subsequent metal exchange and make broader exploration of its coordination chemistry difficult.
 | ||
| Chart 1 C3-Symmetric scorpionates with distinct formal charges. | ||
 | ||
| Chart 2 Dianionic C3-symmetric scorpionates. | ||
Herein, we present the synthesis of a C3-symmetric dianionic ligand B (Chart 2), isolated as its dipotassium salt 1, which fills the aforementioned gap in the assortment of charged scorpionates available for complexation. Taking advantage of the fact that potassium cations are easily exchangeable for other metal ions, we further delve into its coordination chemistry with Cu(I), Fe(II) and Ni(II) salts.
Results and discussion
Ligand synthesis
The dipotassium salt of tris-(2-skatyl)methylphosphonium (1), further abbreviated as TSMPK2, was synthesized as depicted in Scheme 1. In the first step, skatole (2) was treated with di-tert-butyl dicarbonate (Boc2O) in the presence of 4-dimethylaminopyridine (DMAP) to yield the corresponding N-Boc derivative (3). The second position of the indole ring was subsequently lithiated with lithium diisopropylamide (LDA) using N-Boc as a directing group. Without isolation, the lithium salt was quenched with a third of an equivalent of PCl3 to yield tris-2-(N-Boc-skatyl)phosphine (4). The phosphine was then methylated by treatment with an excess (2.0 equiv.) of methyl iodide to form the corresponding methylphosphonium iodide (5). Attempted methylation with an equimolar amount of methyl iodide instead resulted in the incomplete methylation and partial deprotection of the N-Boc-skatole subunits. It is worth pointing out that tris-2-(N-Boc-skatyl)methylphosphonium iodide (5) exists in DCM-d2 solution as a ca. 1.00:0.15 mixture of interexchanging tris-exo and bis-exo-mono-endo rotamers (see ESI section S4.1†).25
 | ||
| Scheme 1 Synthesis of TSMPK2 salt 1. Unless otherwise stated, parentheses underneath reaction arrows indicate isolated yields. | ||
Complete deprotection of the isolated methylphosphonium iodide 5 can be achieved in a 1:1 mixture of dichloromethane and trifluoroacetic acid (TFA). The product, 6, was subjected to ion exchange using the chloride form of the anion exchange resin Amberlite® IRA-400, affording a white crystalline solid of tris-(2-skatyl)methylphosphonium chloride (7).
Deprotonation of tris-indole 7 with either KHMDS or KH gives the TSMPK2 salt (1). A highly pure material (>99% according to NMR) was isolated by crystallization from acetonitrile/diethyl ether. X-ray crystal structure determination confirmed the expected molecular structure with a K:P ratio of 2:1 (Fig. 1).
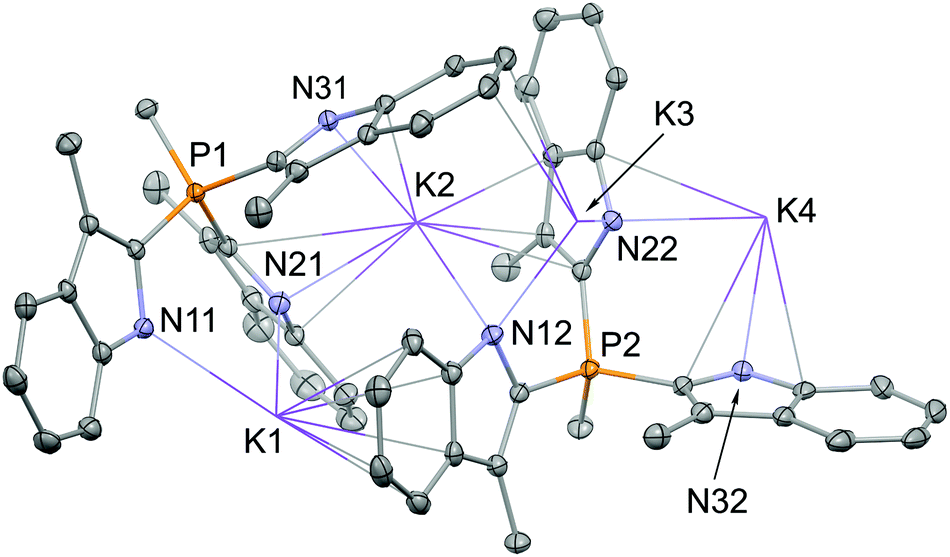 | ||
| Fig. 1 Asymmetric unit of the TSMPK2 (1) crystal. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms and acetonitrile solvent molecules are omitted for clarity. Potassium atoms are shown in a wireframe style. | ||
The K+ cations are involved in an extensive network of cation–N and cation–π interactions (η1 to η6). The K–N distances vary between 2.723(4) and 3.425(4) Å, and the K–C distances between 2.995(4) and 3.510(4) Å. With such a large variation of distances it is not possible to derive a clearly defined coordination number. Overall, K+ coordination leads to the formation of one-dimensional chains in the [110] direction, which are stacked upon each other in c-direction. This stacking is interrupted by the inclusion of acetonitrile solvent molecules between every second layer (see ESI section S3†).
The existence of a free alkaline trianionic-monocationic phosphonium salt is, to our knowledge, unprecedented. Quaternary phosphonium salts are generally incompatible with basic counterions and convert either into ylides,26 phosphoranes27,28 or form charge-transfer complexes followed by complex decomposition manifolds.29 The exceptions, however, feature electronically stabilized bulky anions (e.g. diphenylamide30 and 2,4,6-trimethylphenolate31). We speculate that the stability of 1 is due to the above reasons as well: the anionic indolide nitrogen atoms lose a lot of their nucleophilicity due to being involved in an aromatic π-system, and the phosphonium center is too encumbered to accommodate a fourth indolide in its vicinity. Additionally, the latter would experience electrostatic repulsion from other anionic indolides already present in a molecule.
Coordination chemistry of TSMP
In order to study the ligation behaviour of the novel TSMP platform, we undertook a series of metal exchange reactions with salt 1. It shows rich coordination chemistry with a number of first-row transition metal salts (Scheme 2).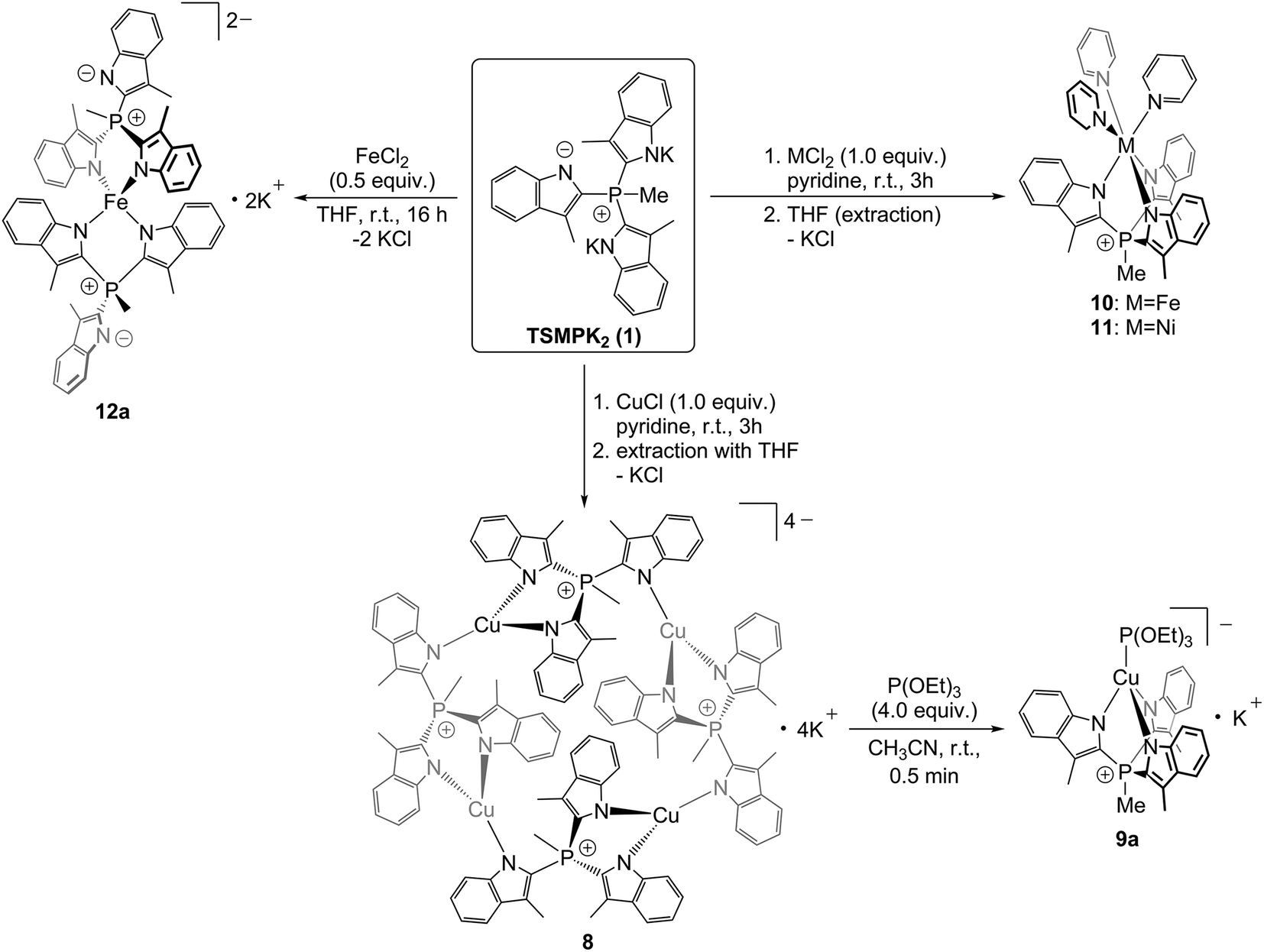 | ||
| Scheme 2 Coordination behavior of the TSMP ligand platform. | ||
Reacting equimolar amounts of TSMPK2 salt 1 with cuprous chloride in pyridine led to an orange-red solution, from which yellow crystalline compound 8 was isolated upon freeing from solvent in vacuo and extraction with THF. NMR spectroscopy in acetonitrile-d3 reveals one predominant species, albeit an oligomer. To elaborate, the 31P NMR spectrum (Fig. 2) shows a single quartet consistent with 2JP,H coupling with a methyl group. In the 1H NMR spectrum (Fig. 2), there are two singlets in a 2:1 ratio corresponding to aromatic methyl groups CH3ArA and CH3ArB. Moreover, multiplets in the aromatic region are present in the same ratio and, according to magnitude gCOSY (see ESI section S4.2†), constitute two distinct spin systems. This implies 2:1 inequivalence of indolide units in the complex. A NOESY spectrum (see ESI section S4.2†) shows a correlation between the phosphonium methyl group CH3P and two aromatic CH3ArA (mapped out in Fig. 2) but not CH3ArB, from which it follows that CH3ArB is pointing away from the phosphonium methyl group. Lastly, H4A and H7A have comparable correlation intensities with CH3ArA. Since NOE rapidly decays with distance, and H7A is clearly further away from CH3ArA than H4A, it can only be the case if there is an equivalent methyl group in the vicinity of H7A, i.e. if the complex is oligomerized. Based on this reasoning, in Fig. 2 we suggest an oligomeric structure of the complex 8 in solution.
 | ||
| Fig. 2 1H (400 MHz) and 31P NMR (162 MHz) spectra of 8 in acetonitrile-d3. Lilac arrows in the structural formula indicate diagnostic NOESY correlations discussed in the text. Only one of two equivalent CH3P–CH3ArA correlations is shown. | ||
It is worth mentioning that the NOESY spectrum also features multiple exchange peaks between indolide groups A and B, which means that these positions exchange on the mixing timescale. This could either be due to reversible dissociation of the oligomer to smaller units or to an intramolecular exchange process. Furthermore, the asterisk-labeled aromatic peaks in the spectrum show exchange with some of the assigned signals, which suggests that they may belong to a minor unassigned form of 8 in solution. However, the number and intensity of the peaks do not allow to deduce molecular connectivity.
While the degree of oligomerization in solution is unclear, our NMR assignment is consistent with the X-ray structure of a crystal grown from pyridine/hexane. Complex 8 crystallizes as a cyclic {[(TSMP)Cu]K}4 tetramer (Fig. 3) with distorted trigonal planar CuN3 centers and Cu–N distances ranging within 1.926(3)–2.007(3) Å (see ESI section S3†). The TSMP ligand adopts a μ2:κ2:κ1 coordination mode, which has been previously observed for other Cu(I) complexes with scorpionate ligands.32 The unit cell contains two independent tetrameric molecules, both being located on exact, crystallographic inversion centers. K+ ions in the proximity of indolide moieties show clear cation–π interactions. Their environment is saturated by coordinated pyridine molecules. The two independent {[(TSMP)Cu]K}4 molecules differ in the number of K+-coordinated pyridines: 14 pyridine molecules for the first tetramer, and 12 for the second. There are also significant differences between the two independent molecules in the coordination mode of K+ to the indolide moieties (see ESI section S3†). The content of the crystallographic unit cell is completed by six non-coordinated pyridine molecules.
 | ||
| Fig. 3 Molecular structure of 8, 9b, 10, 11 and 12b according to X-ray crystal structure determination. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms, some counterions and non-coordinated solvent molecules are omitted for clarity. The asymmetric unit of 8 contains two independent molecular fragments that make up two independent molecules; only one full molecule is shown. Symmetry code: ii: 1 − x, 1 − y, 1 − z. The asymmetric units of 10 and 11 contain two independent molecules, only one of which is shown. Selected bond distances and angles are provided in the ESI section S3.† | ||
The oligomeric structure of {[(TSMP)Cu]K}4 (8) in solution can be broken down upon addition of one equivalent of P(OEt)3 per equivalent of copper. The 1H NMR spectrum immediately simplifies to a single aromatic methyl peak, four aromatic multiplets, a methylphosphonium doublet and an ethyl group of triethylphosphite (see ESI section S4.3†), signifying the formation of the C3-symmetric structure [(TSMP)CuP(OEt)3]K (9a). Furthermore, NOE spectra (see ESI section S4.4†) do not show a correlation analogous to H4A–H7A in 8 (Fig. 2), which points at a monomeric structure of 9a in solution. This assignment is, again, consistent with an X-ray crystal structure determination of the 18-crown-6 adduct 9b crystallized from acetonitrile/benzene/ether (Fig. 3). With the exception of 18-crown-6 peaks, solution NMR spectra of 9a and 9b are identical. The complex features a distorted tethahedral CuN3P center (angle variance33 of 244.58 deg2) with N^Cu^N angles varying from 92.38(8) to 95.06(7)°. The Cu–P bond length is 2.1201(2) Å, as expected for Cu(I),34 and Cu–N distances lie within 2.0622(19)–2.0759(19) Å (see ESI section S3†), similarly to those in tetranuclear 8. The [(TSMP)CuP(OEt)3]− anion has approximate C3 symmetry with the P(OEt)3 ligand in an approximately staggered conformation with respect to the scorpionate [O^P^Cu^N 41.79(11)°]. The geometry of 9b closely resembles that of the neutral tris(pyrazolyl)methanide analogue {[C(3,5-Me2pz)3]CuP(OMe)3}, which features Cu–N bonds in the range 2.047(2)–2.102(2) Å and a Cu–P bond length of 2.122(2) Å.34b
Reactions of equimolar amounts of TSMPK2 (1) with either FeCl2 or NiCl2·dme adduct in pyridine give, correspondingly, bright-yellow or olive-brown solutions. Subsequent evaporation of solvent and extraction with THF yield yellow (TSMP)Fe(Py)3 (10) and green (TSMP)Ni(Py)3 (11) complexes. All attempts to form related complexes with CoCl2 under the same conditions led to intractable mixtures of products.
According to 1H NMR of 10 and 11 in pyridine-d5, these compounds are paramagnetic, and the number of lines with their integral intensity correspond to three-fold symmetric metallabicyclo[2.2.2]octane topology. Remarkably, in both spectra, the spacing between the pyridine peaks deviates from the normal values by up to 0.3 ppm with α-pyridine hydrogens being most affected. This difference is likely due to a hyperfine shift induced in pyridine hydrogens upon labile coordination to the metal centers. X-ray structure determination of crystals grown from pyridine/hexane reveals isostructural octahedral complexes that feature one TSMP and three pyridine ligands (10 and 11, respectively, in Fig. 3). Importantly, the FeN6 core in 10 has much shorter bonds with TSMP nitrogens than pyridine nitrogens, viz. 2.158(6)–2.201(6) Å vs. 2.261(7)–2.351(6) Å (see ESI section S3†). In fact, the latter are even longer on average than Fe–N distances in the [FePy6]2+ solvate: 2.22(3)–2.29(3) Å.35 This is consistent with rather weak bonding and the observed lability of pyridine ligands in solution. The NiN6 core in 11 shows a similar situation: the Ni–N distances for the TSMP nitrogen atoms are within 2.115(7)–2.148(7) Å, whereas the corresponding values for pyridine nitrogens are 2.163(8)–2.241(8) Å (see ESI section S3†). The long metal–ligand bond lengths in 10 and 11 indicate high-spin electronic states.36 This assignment is also supported by the effective solution magnetic moments measured by Evans method in pyridine-d5: 5.19μB for Fe(II) complex 10 and 2.82μB for Ni(II) complex 11, whereas the spin-only expectation values for S = 2 and S = 1 metal centers, respectively, are 4.90 and 2.82μB.
Interestingly, replacement of three pyridine ligands in 10 with another equivalent of TSMP gives a highly air-sensitive bright-yellow tetracoordinate complex [(TSMP)2Fe]K2, 12a (Scheme 2). The complex can be crystallized from acetonitrile/toluene/ether as the tetrakis(benzo-15-crown-5) adduct 12b (Fig. 3). The NMR spectra of 12a and 12b in solution are identical with the exception of benzo-15-crown-5 peaks. Compound 12b in the crystal features a pseudo-tetrahedral FeN4 core where each TSMP ligand coordinates with two arms while the third remains uncoordinated (κ2 mode). The N^Fe^N angles vary from 96.35(13)° to 119.68(13)° (see ESI section S3†). The angle variance33 of 111.29 deg2 is, consequently, rather large. The distortion is mainly caused by the chelate effect: the dihedral angle between the N11–Fe1–N21 and the N12–Fe1–N22 planes is 85.2(2)° and deviates only slightly from perfect 90°. The Fe–N distances of 2.017(3)–2.028(3) Å indicate a high-spin electronic state of the Fe(II) center. A comparison with structurally related high-spin complexes37 shows similar bonding distances of >2.0 Å. Solution effective magnetic moment measurement by Evans method in pyridine-d5 gives 5.17μB, which is close to the spin-only expectation value of 4.90μB for an S = 2 metal center. The tetrahedral geometry of 12a contrasts with the common octahedral geometry of neutral and dicationic pyrazolate-based bis(scorpionate) Fe(II) complexes38 and of the tris-pyridine complex 10. We speculate that the reduction in coordination number is due to electrostatic repulsion of the negatively-charged indolides as well as their weaker π-accepting properties as compared to pyridine or pyrazolate ligands.
Conclusions
A dianionic C3-symmetric tris-skatylmethylphosphonium (TSMP) ligand platform can be synthesized in the form of dipotassium salt TSMPK2 (1). This system is the first dianionic homoscorpionate capable of metal exchange and fills a gap in the assortment of charged scorpionates. Despite a possibility of recombination between negatively charged indolides and a positively charged phosphonium atom, salt 1 is stable both in the solid state and solution, which is likely due to a combination of electronic and steric factors.The potassium cations in TSMPK2 (1) are exchangeable for other metals, demonstrating the versatility of TSMP as a ligand for transition metals. The expected scorpionate κ3 binding mode is observed in octahedral, high spin complexes (TSMP)M(Py)3 (10: M = Fe; 11: M = Ni) as well as in the tetrahedral complex [(TSMP)CuP(OEt)3]− (9). In addition, the bridging μ2:κ2:κ1 mode is preferred with Cu(I) in the absence of a co-ligand, affording the tetrameric complex {[(TSMP)Cu]K}4 (8). Finally, the bidentate κ2 mode is observed in the tetracoordinate 2:1 Fe(II) complex [(TSMP)2Fe]2−, which displays a high-spin ground state.
The rich coordination chemistry of the dianionic homoscorpionate ligand invites further investigations. Amongst other, it has a potential to electronically stabilize high-valent metal states due to its electron-rich character, but additional derivatization might be required for kinetic stabilization. Studies in these directions are currently ongoing in our laboratories.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the NoNoMeCat Marie Skłodowska-Curie training network funded by the European Union under the Horizon2020 Program (675020-MSCA-ITN-2015-ETN). Support with NMR spectroscopic analysis by Dr J. T. B. H. Jastrzebski is gratefully acknowledged. The X-ray diffractometer has been financed by the Netherlands Organization for Scientific Research (NWO).Notes and references
- The following review and references therein: S. Trofimenko, Chem. Rev., 1993, 93, 943–980 Search PubMed.
- (a) S. Trofimenko, Scorpionates, Imperial College Press, London, 1999; pp. 1–25 Search PubMed; (b) C. Pettinari and C. Santini, Comprehensive Coordination Chemistry II: From Biology to Nanotechnology. Volume 1: Fundamentals: Ligands, Complexes, Synthesis, Purification, and Structure, ed. A. B. P. Lever and J. A. McCleverty, T. J.Meyer, Elsevier Pergamon, Amsterdam, Boston, 2004, pp. 159–210 Search PubMed; (c) D. M. Tellers, S. J. Skoog, R. G. Bergman, T. B. Gunnoe and W. D. Harman, Organometallics, 2000, 19, 2428–2432 Search PubMed.
- C. Janiak, Coord. Chem. Rev., 1997, 163, 107–216 Search PubMed.
- The following reviews and references therein: (a) C. Santini, M. Pellei, G. G. Lobbia and G. Papini, Mini-Rev. Org. Chem., 2010, 7, 84–124 Search PubMed; (b) M. Pellei and C. Santini, Biomimetic Based Applications, ed. A. George, InTech, 2011 Search PubMed.
- (a) The following review and references therein: C. Slugovc, R. Schmid and K. Kirchner, Coord. Chem. Rev., 1999, 185–186, 109–126 Search PubMed; (b) M. M. Díaz-Requejo, T. R. Belderraín, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2001, 123, 3167–3168 Search PubMed; (c) M. M. Díaz-Requejo, T. R. Belderraín and P. J. Pérez, Chem. Commun., 2000, 19, 1853–1854 Search PubMed; (d) M. E. Morilla, M. M. Díaz-Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Pérez, Chem. Commun., 2002, 24, 2998–2999 Search PubMed; (e) S. Milione, C. Montefusco, T. Cuenca and A. Grassi, Chem. Commun., 2003, 10, 1176–1177 Search PubMed; (f) H. V. R. Dias and J. Wu, Angew. Chem., Int. Ed., 2007, 46, 7814–7816 Search PubMed.
- For example: (a) R. M. Silva, C. Gwengo, S. V. Lindeman, M. D. Smith and J. R. Gardinier, Inorg. Chem., 2006, 45, 10998–11007 Search PubMed; (b) B. Zhai, W.-Z. Shen, X.-Y. Chen, H.-B. Song, W. Shi and P. Cheng, Inorg. Chem. Commun., 2006, 9, 1293–1296 Search PubMed; (c) J. A. McCleverty and M. D. Ward, Acc. Chem. Res., 1998, 31, 842–851 Search PubMed; (d) C. Janiak, T. G. Scharmann, P. Albrecht, F. Marlow and R. Macdonald, J. Am. Chem. Soc., 1996, 118, 6307–6308 Search PubMed.
- The following review and references therein: P. Martini, M. Pasquali, A. Boschi, L. Uccelli, M. Giganti and A. Duatti, Molecules, 2018, 23, 2039 Search PubMed.
- For example: (a) C. G. J. Tazelaar, E. Nicolas, T. van Dijk, D. L. J. Broere, M. Cardol, M. Lutz, D. Gudat, J. C. Slootweg and K. Lammertsma, Dalton Trans., 2016, 45, 2237–2249 Search PubMed; (b) M. M. Bittner, S. V. Lindeman, C. V. Popescu and A. T. Fiedler, Inorg. Chem., 2014, 53, 4047–4061 Search PubMed; (c) A. A. Fischer, J. R. Miller, R. J. Jodts, D. M. Ekanayake, S. V. Lindeman, T. C. Brunold and A. T. Fiedler, Inorg. Chem., 2019, 58, 16487–16499 Search PubMed; (d) A. A. Fischer, N. Stracey, S. V. Lindeman, T. C. Brunold and A. T. Fiedler, Inorg. Chem., 2016, 55, 11839–11853 Search PubMed.
- For example: (a) L. F. Szczepura, L. M. Witham and K. J. Takeuchi, Coord. Chem. Rev., 1998, 174, 5–32 Search PubMed; (b) T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 8506–8520 Search PubMed.
- The following review and references therein: E. T. Papish, N. A. Dixon and M. Kumar, Molecular Design in Inorganic Biochemistry. Structure and Bonding, ed. D. Rabinovich, Springer, Berlin, Heidelberg, 2013, vol. 160, pp. 115–150 Search PubMed.
- For example: (a) G. T. Sazama and T. A. Betley, Inorg. Chem., 2010, 49, 2512–2524 Search PubMed; (b) T. S. Barnard and M. R. Mason, Organometallics, 2001, 20, 206–214 Search PubMed.
- For example: (a) M. Garner, J. Reglinski, I. Cassidy, M. D. Spicer and A. R. Kennedy, Chem. Commun., 1996, 355, 1975–1976 Search PubMed; (b) J. Reglinski, M. Garner, I. D. Cassidy, P. A. Slavin, M. D. Spicer and D. R. Armstrong, J. Chem. Soc., Dalton Trans., 1999, 13, 2119–2126 Search PubMed.
- For example: J. F. Dunne, J. Su, A. Ellern and A. D. Sadow, Organometallics, 2008, 27, 2399–2401 Search PubMed.
- For example: X. Hu, Y. Tang, P. Gantzel and K. Meyer, Organometallics, 2003, 22, 612–614 Search PubMed.
- For example: (a) A. L. Sargent, E. P. Titus, C. G. Riordan, A. L. Rheingold and P. Ge, Inorg. Chem., 1996, 35, 7095–7101 Search PubMed; (b) I. R. Shapiro, D. M. Jenkins, J. C. Thomas, M. W. Day and J. C. Peters, Chem. Commun., 2001, 2152–2153 Search PubMed.
- For example: (a) E. E. Pullen, A. L. Rheingold and D. Rabinovich, Inorg. Chem. Commun., 1999, 2, 194–196 Search PubMed; (b) W. C. Blackwell III, D. Bunich, T. E. Concolino, A. L. Rheingold and D. Rabinovich, Inorg. Chem. Commun., 2000, 3, 325–327 Search PubMed; (c) F. Neumeyer, M. I. Lipschutz and T. D. Tilley, Eur. J. Inorg. Chem., 2013, 2013, 6075–6078 Search PubMed.
- T. N. Sorrell, W. E. Allen and P. S. White, Inorg. Chem., 1995, 34, 952–960 Search PubMed.
- W.-J. Su and L.-C. Liang, Inorg. Chem., 2018, 57, 553–556 Search PubMed.
- V. S. Joshi, V. K. Kale, K. M. Sathe, A. Sarkar, S. S. Tavale and C. G. Suresh, Organometallics, 1991, 10, 2898–2902 Search PubMed.
- The following review and references therein: H. R. Simmonds and D. S. Wright, Chem. Commun., 2012, 48, 8617–8624 Search PubMed.
- A. Frazer, B. Piggott, M. Harman, M. Mazid and M. B. Hursthouse, Polyhedron, 1992, 11, 3013–3017 Search PubMed.
- The following review and references therein: I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 35, 5836–5865 Search PubMed.
- F. Armbruster, I. Fernández and F. Breher, Dalton Trans., 2009, 29, 5612–5626 Search PubMed.
- A. Steiner and D. Stalke, Inorg. Chem., 1995, 34, 4846–4853 Search PubMed.
- E. Martin-Mothes, E. Puig, L. Vendier, C. Bijani, M. Grellier and S. Bontemps, Dalton Trans., 2018, 47, 10139–10146 Search PubMed.
- G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318–1330 Search PubMed.
- G. Wittig and M. Rieber, Justus Liebigs Ann. Chem., 1949, 562, 187–192 Search PubMed.
- (a) H. Schmidbaur and H. Stühler, Angew. Chem., Int. Ed. Engl., 1972, 11, 145–146 Search PubMed; (b) H. Schmidbaur, W. Buchner and F. H. Koehler, J. Am. Chem. Soc., 1974, 96, 6208–6210 Search PubMed.
- N. A. A. Nesmeyanov, O. A. Rebrova, V. V. Mikul'shina, P. V. Petrovsky, V. I. Robas and O. A. Reutov, J. Organomet. Chem., 1976, 110, 49–57 Search PubMed.
- M. G. Davidson and S. Lamb, Polyhedron, 1997, 16, 4393–4395 Search PubMed.
- M. G. Davidson, J. Chem. Soc., Chem. Commun., 1995, 9, 919–920 Search PubMed.
- (a) E. Haldón, M. Delgado-Rebollo, A. Prieto, E. Álvarez, C. Maya, M. C. Nicasio and P. J. Pérez, Inorg. Chem., 2014, 53, 4192–4201 Search PubMed; (b) S. M. Carrier, C. E. Ruggiero, R. P. Houser and W. B. Tolman, Inorg. Chem., 1993, 32, 4889–4899 Search PubMed.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 Search PubMed.
- (a) R. Mothes, T. Rüffer, Y. Shen, A. Jakob, B. Walfort, H. Petzold, S. E. Schulz, R. Ecke, T. Gessner and H. Lang, Dalton Trans., 2010, 39, 11235 Search PubMed; (b) I. Krummenacher, H. Rüegger and F. Breher, Dalton Trans., 2006, 1073–1081 Search PubMed.
- R. J. Doedens and L. F. Dahl, J. Am. Chem. Soc., 1966, 88, 4847–4855 Search PubMed.
- (a) J. D. Oliver, D. F. Mullica, B. B. Hutchinson and W. O. Milligan, Inorg. Chem., 1980, 19, 165–169 Search PubMed; (b) A. Michaud, F.-G. Fontaine and D. Zargarian, Inorg. Chim. Acta, 2006, 359, 2592–2598 Search PubMed; (c) R. Wanke, M. F. C. Guedes Da Silva, S. Lancianesi, T. F. S. Silva, L. M. D. R. S. Martins, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2010, 49, 7941–7952 Search PubMed.
- (a) W. D. Morris, P. T. Wolczanski, J. Sutter, K. Meyer, T. R. Cundari and E. B. Lobkovsky, Inorg. Chem., 2014, 53, 7467–7484 Search PubMed; (b) A. Panda, M. Stender, R. J. Wright, M. M. Olmstead, P. Klavins and P. P. Power, Inorg. Chem., 2002, 41, 3909–3916 Search PubMed; (c) D. M. Granum, P. J. Riedel, J. A. Crawford, T. K. Mahle, C. M. Wyss, A. K. Begej, N. Arulsamy, B. S. Pierce and M. P. Mehn, Dalton Trans., 2011, 40, 5881–5890 Search PubMed; (d) J. R. Hagadorn and J. Arnold, Inorg. Chem., 1997, 36, 132–133 Search PubMed; (e) B. Vendemiati, G. Prini, A. Meetsma, B. Hessen, J. H. Teuben and O. Traverso, Eur. J. Inorg. Chem., 2001, 2001, 707–711 Search PubMed; (f) C. A. Nijhuis, E. Jellema, T. J. J. Sciarone, A. Meetsma, P. H. M. Budzelaar and B. Hessen, Eur. J. Inorg. Chem., 2005, 2005, 2089–2099 Search PubMed; (g) L. Zhang, L. Xiang, Y. Yu and L. Deng, Inorg. Chem., 2013, 52, 5906–5913 Search PubMed; (h) F. Liu, X. Qiao, M. Wang, M. Zhou, H. Tong, D. Guo and D. Liu, Polyhedron, 2013, 52, 639–644 Search PubMed.
- (a) J. P. Jesson, S. Trofimenko and D. R. Eaton, J. Am. Chem. Soc., 1967, 89, 3148–3158 Search PubMed; (b) C. Janiak, S. Temizdemir, S. Dechert, W. Deck, F. Girgsdies, J. Heinze, M. J. Kolm, T. G. Scharmann and O. M. Zipffel, Eur. J. Inorg. Chem., 2000, 1229–1241 Search PubMed; (c) D. L. Reger, J. R. Gardinier, W. R. Gemmill, M. D. Smith, A. M. Shahin, G. J. Long, L. Rebbouh and F. Grandjean, J. Am. Chem. Soc., 2005, 127, 2303–2316 Search PubMed; (d) B. Moubaraki, B. A. Leita, G. J. Halder, S. R. Batten, P. Jensen, J. P. Smith, J. D. Cashion, C. J. Kepert, J.-F. Létard and K. S. Murray, Dalton Trans., 2007, 4413–4426 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and characterization data for all new compounds, additional figures. CCDC 2010902–2010907. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02601h |
| This journal is © The Royal Society of Chemistry 2020 |