
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[4 + 2]-Cycloadditions of a thiazol-based tricyclic 1,4-diphosphinine and a new easy 1,4-diphosphinine protection deprotection strategy†
Imtiaz
Begum‡
a,
Tim
Kalisch‡
a,
Gregor
Schnakenburg
a,
Zsolt
Kelemen
b,
László
Nyulászi
 *b and
Rainer
Streubel
*a
*b and
Rainer
Streubel
*a
aInstitut für Anorganische Chemie der Rheinischen Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Straße 1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn
bDepartment of Inorganic and Analytical Chemistry and MTA-BME Computation Driven Chemistry Research Group, Budapest University of Technology and Economics, Szt Gellert ter 4, 1111 Budapest, Hungary. E-mail: nyulaszi@mail.bme.hu
First published on 1st September 2020
Abstract
Diels–Alder-reactions of a thiazol-2-thione-based, tricyclic 1,4-di-phosphinine were investigated, showing that the central aromatic π-system can react with various dienophiles. The reaction with 4-phenyl-1,2,4-triazoline-3,5-dione was special as the product revealed a remarkable sensitivity towards light, thus enabling the photochemical deprotection of the tricyclic 1,4-diphosphinine.
Introduction
An interesting aspect of phosphinine chemistry is to enable access to phosphabarrelenes using the somewhat dienic nature of the phosphinine π-system.1 The first synthesis of phosphabarrelenes was achieved by Märkl in 1968 using reactions of a phosphinine with different alkynes2 and, later on, also with arynes.3 Phosphabarrelenes are considered important ligands and, besides several other applications,4 they were used in the hydrogenation4a of alkenes and rhodium-catalyzed hydroformylation.5 An interesting addition to this field, was the one-pot tandem reaction sequence under hydroformylation conditions using phosphabarrelene rhodium complexes, reported by Müller and co-workers.6Interestingly, Krespan reported the first 1,4-diphosphabarrelene I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 (Fig. 1) even before the phosphabarrelene. Compound I was obtained via thermolysis of hexafluoro-2-butyne in the presence of red phosphorus, using iodine as catalyst. This methodology needs a long reaction time at elevated temperatures (200 °C) under autogenous pressure, and yet the maximum yield was only 43%. A more efficient synthetic protocol to diphosphabarrelene would be from 1,4-diphosphinines using reactions with alkynes. However, until very recently only one example of a 1,4-diphosphinine was known. Kobayashi and co-workers synthesized the same compound (I) via reaction of tetrakis(trifluoromethyl)-1,4-diphosphabenzene with hexafluoro-2-butyne.8 The aforementioned 1,4-diphosphinine of Kobayashi appeared to be a rather unstable compound and its synthesis is somewhat laborious, thus hampering further studies on complexes of I.
7 (Fig. 1) even before the phosphabarrelene. Compound I was obtained via thermolysis of hexafluoro-2-butyne in the presence of red phosphorus, using iodine as catalyst. This methodology needs a long reaction time at elevated temperatures (200 °C) under autogenous pressure, and yet the maximum yield was only 43%. A more efficient synthetic protocol to diphosphabarrelene would be from 1,4-diphosphinines using reactions with alkynes. However, until very recently only one example of a 1,4-diphosphinine was known. Kobayashi and co-workers synthesized the same compound (I) via reaction of tetrakis(trifluoromethyl)-1,4-diphosphabenzene with hexafluoro-2-butyne.8 The aforementioned 1,4-diphosphinine of Kobayashi appeared to be a rather unstable compound and its synthesis is somewhat laborious, thus hampering further studies on complexes of I.
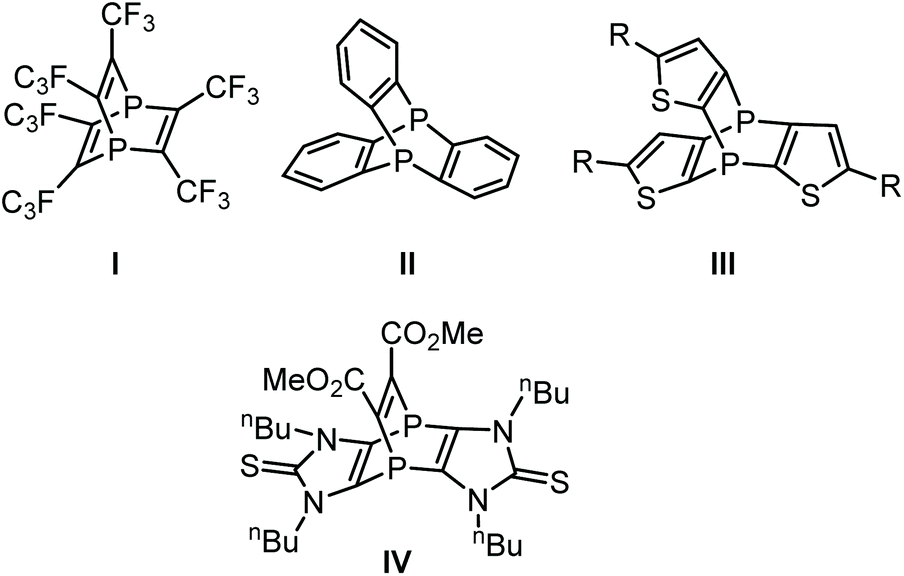 | ||
| Fig. 1 Examples of 1,4-diphosphabarrelenes I–IV. | ||
Other synthetic approaches to 1,4-diphosphabarrelenes (Fig. 1) have been reported. Weinberg and co-workers described the synthesis of II in 1971 via thermolysis of ortho-dichlorobenzene and white phosphorus in the presence of catalytic amounts of FeCl3 (20% yield).9 Further attempts by Yosuke et al.10 to optimize the synthetic protocol were unsuccessful, i.e., the yields were even lower (9%). Synthesis of 4,8-diphospha[3,4-a:7,8-a]thiophenotriptycene III was reported by Nakayama and co-workers using the reaction of tris(thiophen-2-yl)phosphane P-oxide with tris(phenoxy)phosphane using nBuLi as base;11 but again the yield was low (11%).
Very recently, we investigated the reactivity of an imidazole-based tricyclic 1,4-diphosphinine12 towards some C,C-based dienophiles in [4 + 2]-cycloaddition reactions. The corresponding 1,4-diphosphabarrelenes such as IV were obtained in very good yields (up to 95%).13a The first examples of [4 + 1]-cycloadditions were also reported recently using, somewhat exotic, group 13 element(I) compounds (Al, Ga);13b these products showed a thermal retro-reaction.
As facile access to a thiazole-based tricyclic 1,4-diphosphinine was recently described14 revealing differences in product stabilities, i.e. it allowed for the isolation of a stable mono anion,14 whereas in case of the imidazole-based 1,4-diphosphinine only intermediates were obtained,15 we became interested to investigate [4 + 2]-cycloaddition reactions and the stability of the products obtained.
Results and discussion
The dienic nature of tricyclic 1,4-diphosphinine 114 was first probed with dimethyl acetylenedicarboxylate (DMAD) (Scheme 1). At 50 °C in toluene the red color of the 1,4-diphosphinine 1 started to disappear and a yellow-orange turbid solution was formed, from which compound 2 was isolated in 73% yield. The 31P{1H} NMR spectrum of 2 (CD2Cl2) showed one signal at −75.4 ppm, comparable to literature-known compounds (−87.1 and −93.4 ppm for III11 and −87.3 ppm for IV13) and, therefore, was assigned to the 1,4-diphosphabarrelene 2. The 1H NMR spectrum of compound 2 showed a characteristic signal at 3.82 ppm for the protons of the two OCH3 groups.
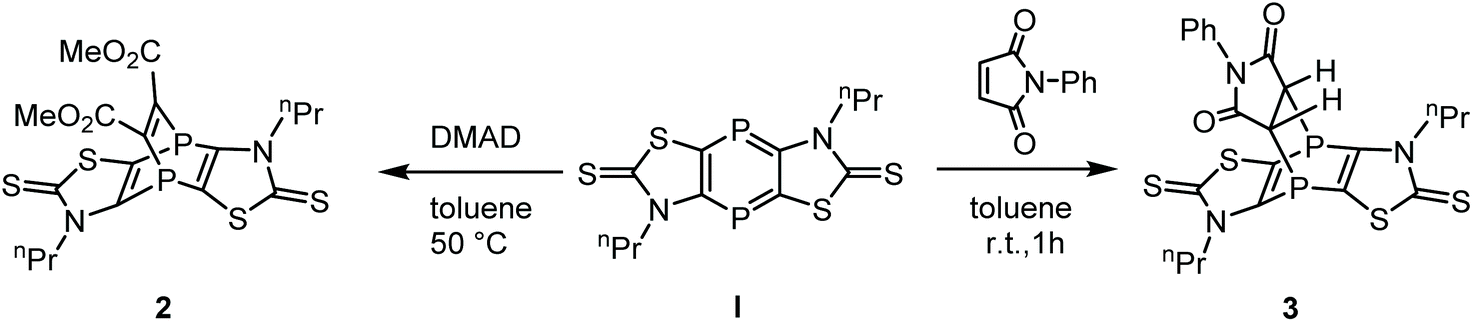 | ||
| Scheme 1 [4 + 2]-Cycloaddition reactions of 1,4-diphosphinine 1 to afford 2 and 3. | ||
The carbon nuclei of the latter were assigned to the signal at 53.4 ppm in the 13C{1H} NMR spectrum, and the carbon resonance at 157.6 ppm (dd, 1JP,C = 15.1 Hz, 2JP,C = 14.0 Hz) to the sp2-hybridized carbon atoms of the C2 bridge. The EI-MS spectrum showed the molecular ion peak of 2 (m/z 518.0), but also one fragment revealing the loss of the alkyne moiety to give the cation of 1 (m/z 376.0). According to our B3LYP-D3/6-311+G** calculations the fragmentation of 2˙+ to 1˙+ and the alkyne moiety is endothermic by 40.0 kcal mol−1, but this energy is easily provided by the ionizing electron, giving the explanation for the observation of both 2˙+ and 1˙+. The single crystal X-ray diffraction analysis confirmed the molecular structure of 2 (Fig. 2); selected bond lengths and angles are given in the figure caption. The 1.336(4) Å C13–C14 bond distance is in the expected range (cf. with the 1.329(9) Å for the related molecule IV).13 This also holds for the distance P1–C1 of 1.846(3) Å being within the range of phosphabarrelenes (1.87 to 1.82 Å), e.g., the value in IV is 1.820(6) Å.13
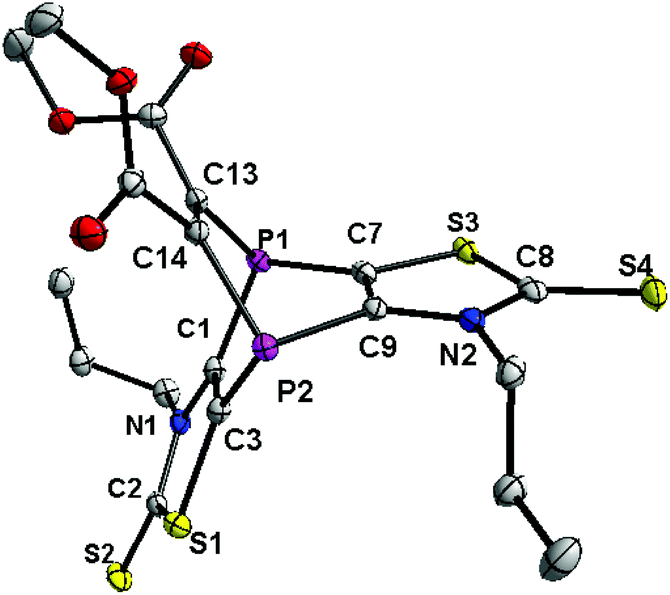 | ||
| Fig. 2 Diamond plot of molecular structure of 2 in solid state. Thermal ellipsoids are drawn at a 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) P1–C1 1.846(3), P1–C7 1.818(3), C1–C3 1.348(4), P1–C13 1.870(3), C13–C14 1.336(4), C1–P1–C7 95.63(13). | ||
To have more insight into the formation of 2, B3LYP-D3/6-311+G** level DFT calculations were performed (Gibbs free energies were calculated at 25 °C and atmospheric pressure—for details see ESI†). The reaction Gibbs free energy is −13.7 kcal mol−1 and the corresponding barrier amounts to 17.0 kcal mol−1, in accordance with the relative mild reaction conditions.
To test a heterocyclic electron-deficient 2π system, the reaction of 1 with 1-phenyl-pyrole-2,5-dione, having a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, was investigated. A very selective reaction occurred at ambient temperature forming 3 as the single product (Scheme 1) as the 31P{1H} NMR spectrum of the reaction solution revealed, and two doublets at −74.3 ppm and −74.7 ppm (3JP,P = 28.8 Hz) of 3 were observed. In the 31P NMR spectrum (Fig. 3) the signals show a further splitting derived from the 2J and 3JP,H couplings. This spectrum was simulated as an ABXY spin system (A, B = 31P, X, Y = 1H) using the program gNMR.16,17 Compound 3 was isolated as white powder in excellent yields (94%) and fully characterized including MS and elemental analysis.
C bond, was investigated. A very selective reaction occurred at ambient temperature forming 3 as the single product (Scheme 1) as the 31P{1H} NMR spectrum of the reaction solution revealed, and two doublets at −74.3 ppm and −74.7 ppm (3JP,P = 28.8 Hz) of 3 were observed. In the 31P NMR spectrum (Fig. 3) the signals show a further splitting derived from the 2J and 3JP,H couplings. This spectrum was simulated as an ABXY spin system (A, B = 31P, X, Y = 1H) using the program gNMR.16,17 Compound 3 was isolated as white powder in excellent yields (94%) and fully characterized including MS and elemental analysis.
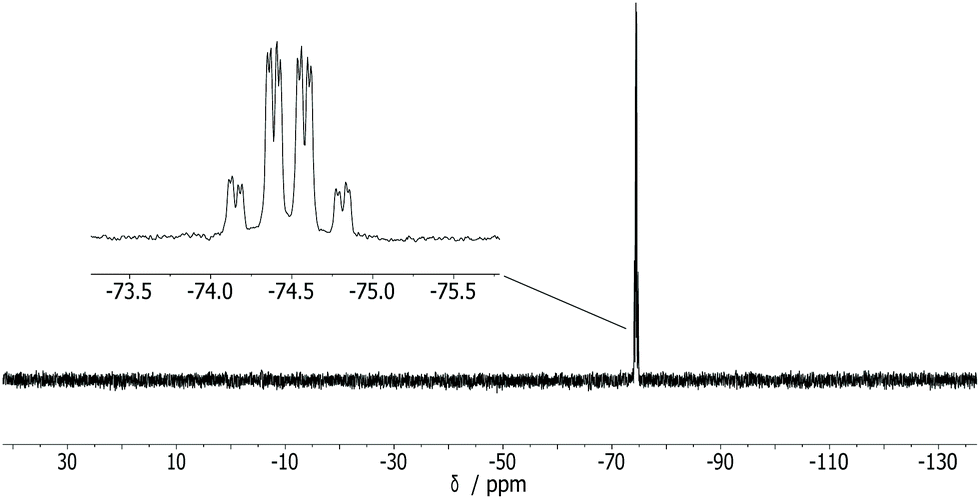 | ||
| Fig. 3 31P NMR spectrum of 3 in CD2Cl2. | ||
The 1H and 13C{1H} NMR spectra of 3 showed two sets of signals for each group of the different structural subunits, confirming the lower symmetry in 3 compared to 2. The constitution of 3 was unequivocally confirmed by single crystal X-ray diffraction analysis, but the quality wasn't sufficient to allow for a detailed discussion.
According to our calculations, the formation of 3 is an exergonic reaction (ΔG = −2.6 kcal mol−1 under ambient conditions) and the reaction barrier 15.6 kcal mol−1, being somewhat lower than in case of 2, thus being also in agreement with a product formation at lower temperature.
To examine an even more reactive electron-deficient 2π system 4-phenyl-1,2,4-triazoline-3,5-dione (in the following: triazoline), having a NN bond, was used. Indeed, the reaction with 1 in toluene started already at −20 °C (Scheme 2), but 90 °C was needed to achieve full conversion as both reactants as well as the product have rather low solubility. Upon cooling to ambient temperature, the product 4 precipitated and was purified by filtration and washing with toluene and n-pentane (yield 67%). The 31P{1H} NMR spectrum of 4 showed one signal at −45.8 ppm, indicating a rapid inversion at the N-centers. The constitution of 4 was further confirmed by 1H and 13C NMR as well as IR spectra. The HRMS spectrum of 4 showed the molecular ion peak (m/z 550.9893), being in good agreement with the calculated value of 550.9897.
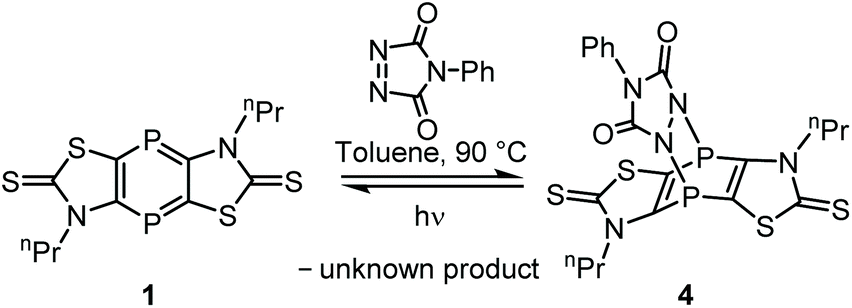 | ||
| Scheme 2 [4 + 2]-Cycloaddition of 1,4-diphosphinine 1 to afford 4. | ||
In accordance with the reaction at low temperature the activation barrier of the cycloaddition is very low (ΔG# = 5.6 kcal mol−1), and the B3LYP-D3/6-311+G**reaction Gibbs free energy of –15.6 kcal mol−1 indicates an exergonic reaction. (Further calculations at other levels predicted even larger reaction energies, see ESI.†)
A remarkable instability of 4 (in solution) towards light was observed as 1 was formed again (after it had been fully consumed) if a sample stood in plain daylight. A photochemical reaction was confirmed via irradiation of a solution of 4 in dichloromethane with UV light for 1.5 h (Hg lamp) as the 31P{1H} NMR monitoring revealed the formation of 1 (48% by NMR integration), while some residual 4 was still present (52% by NMR integration).
However, we observed that – after the photochemical reaction – 4 was not formed, again. This suggested that instead of a clean [4 + 2]-cycloreversion a (subsequent?) decomposition of the triazoline took place under these conditions. Hence, UV/vis spectroscopic measurements of the starting materials – 1,4-diphosphinine 1 and the triazoline – as well as of the [4 + 2]-cycloaddition product 4 and the reaction mixture after irradiation were performed (Fig. 4).18,19 A comparison of the spectrum of pure triazoline,20 showing two absorption bands at 335 and 545 nm, with the spectrum of the product mixture (after irradiation) in dichloromethane showed no absorption band at 545 nm. This was clear evidence that the triazoline had not been formed from 4 and/or had subsequently reacted to give a new unknown compound (Scheme 2). Independent support was obtained by the following experiment, monitored by 31P{1H} NMR spectroscopy. After irradiation, the reaction mixture was subjected to the same conditions used before for the formation of 4 (toluene, 90 °C), now using an additional amount of triazoline or having no additional reagent. The NMR spectroscopic monitoring confirmed our conclusion made beforehand as the addition of triazoline led to a conversion of the 1,4-diphosphinine (to 4), while without addition no [4 + 2]-cycloaddition took place, and the 1,4-diphosphinine 1 remained in the mixture.
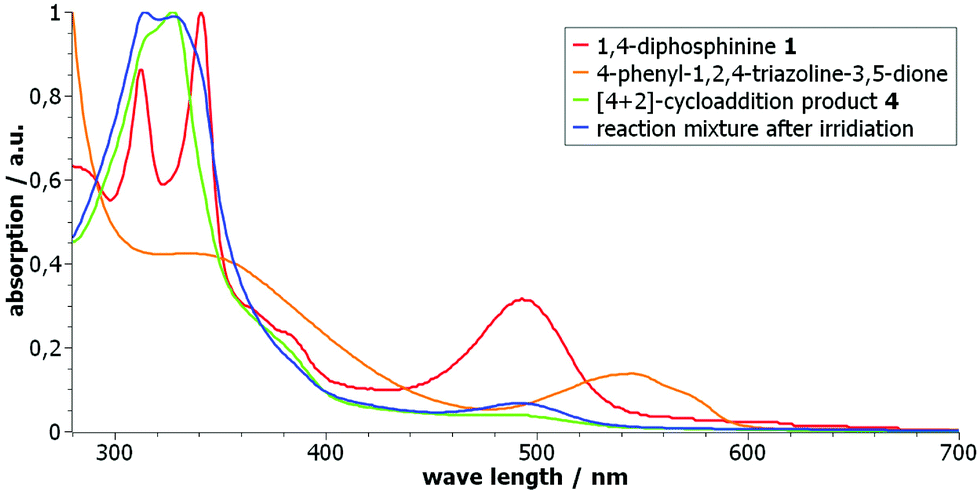 | ||
| Fig. 4 UV/vis studies in CH2Cl2 of 1, 4-phenyl-1,2,4-triazole-3,5-dione, 4 and the reaction mixture after irradiation. The curves were individually normalized to their highest value. | ||
We have attempted to optimize the excited state of 4 by B3LYP-D3/6-311+G** TD DFT calculations (which describe well the spectrum of 1,14 and 4) to see if the excited state shows any sign of decomposition. Unfortunately, none of the structure of the lowest four excited states had significantly different geometry from the ground state of 4 itself, thus we were not able to obtain any hint from these calculations on the mechanism of the photochemical decomposition of 4.
Conclusions
A facile, high-yield access to new polycyclic 1,4-diphosphabarrelenes and related bicyclic heterocycles via [4 + 2]-cycloaddition reaction is presented.It was shown that reactions with double bond systems possess lower reaction energy barriers compared to an alkyne as supported by DFT calculations, being remarkably low for 4-phenyl-1,2,4-triazoline-3,5-dione. Preliminary results from photochemical studies showed that a “retro”-[4 + 2]-cycloaddition is possible, thus yielding the tricyclic 1,4-diphosphine 1. The latter finding represents a new, easier photoremoval strategy of protection and deprotection of the 1,4-diphosphinine ring.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R.S. is grateful to the University of Bonn and the EU (Cost Action CM 1302 “Smart Inorganic Polymers”) for support of this work. L. N. is grateful for BME Nanotechnology and Materials Science TKP2020 IE grant of NKFIH Hungary (BME IE-NAT TKP2020) as well as for the Alexander von Humboldt Stiftung for re-invitation.Notes and references
- N. Mézailles and P. Le Floch, Curr. Org. Chem., 2006, 10, 3–25 CrossRef.
- G. Märkl and F. Lieb, Angew. Chem., Int. Ed. Engl., 1968, 7, 733–733 CrossRef.
- G. Märkl, F. Lieb and C. Martin, Tetrahedron Lett., 1971, 12, 1249–1252 CrossRef.
- (a) B. Breit and E. Fuchs, Synthesis, 2006, 2121–2128 CrossRef CAS; (b) O. Piechaczyk, M. Doux, L. Ricard and P. le Floch, Organometallics, 2005, 24, 1204–1213 CrossRef CAS; (c) M. Blug, C. Guibert, X.-F. Le Goff, N. Mezailles and P. Le Floch, Chem. Commun., 2008, 203 Search PubMed.
- (a) B. Breit and E. Fuchs, Chem. Commun., 2004, 694–695 RSC; (b) E. Fuchs, M. Keller and B. Breit, Chem. – Eur. J., 2006, 12, 6930–6939 CrossRef CAS.
- P. S. Bauerlein, I. A. Gonzalez, J. J. M. Weemers, M. Lutz, A. L. Spek, D. Vogt and C. Müller, Chem. Commun., 2009, 4944–4946 RSC.
- C. G. Krespan, J. Am. Chem. Soc., 1961, 83, 3432–3433 CrossRef CAS.
- (a) Y. Kobayashi, J. Kumadaki, A. Ohsawa and W. Hamana, Tetrahedron Lett., 1976, 41, 3715–3716 CrossRef; (b) Y. Kobayashi, S. Fujino and J. Kumadaki, J. Am. Chem. Soc., 1981, 103, 2465–2466 CrossRef CAS; (c) Y. Kobayashi, S. Fujino, H. Hamana, Y. Hanzawa, S. Morita and I. Kumadaki, J. Org. Chem., 1980, 45, 4683–4685 CrossRef CAS.
- K. G. Weinberg and E. B. Whipple, J. Am. Chem. Soc., 1971, 93, 1801–1802 CrossRef CAS.
- U. Yosuke, S. Jun, S. Munenori, Y. Gaku and M. Yasuhiro, Bull. Chem. Soc. Jpn., 2009, 82, 819–828 CrossRef.
- A. Ishii, R. Yoshioka, J. Nakayama and M. Hoshino, Tetrahedron Lett., 1993, 34, 8259–8262 CrossRef CAS.
- A. Koner, G. Pfeifer, Z. Kelemen, G. Schnakenburg, L. Nyulászi, T. Sasamori and R. Streubel, Angew. Chem., Int. Ed., 2017, 56, 9231–9235 CrossRef CAS.
- (a) A. Koner, Z. Kelemen, G. Schnakenburg, L. Nyulászi and R. Streubel, Chem. Commun., 2018, 54, 1182–1184 RSC; (b) A. Koner, B. M. Gabidullin, Z. Kelemen, L. Nyulászi, G. Nikonov and R. Streubel, Dalton Trans., 2019, 48, 8248–8253 RSC.
- I. Begum, G. Schnakenburg, Z. Kelemen, L. Nyulászi, R. T. Boeré and R. Streubel, Chem. Commun., 2018, 54, 13555–13558 RSC.
- A. Koner, PhD Thesis, University of Bonn, 2017.
- P. H. Budzeelar, gNMR Version 5.0.6.0, IvorySoft, 2006 Search PubMed.
- The following chemical shifts and coupling constants were extracted from the simulated spectrum: δ/ppm = −74.67 (ddd, 3JP,P = 28.51 Hz, 2JP,H = 8.69 Hz, 3JP,H = 2.66 Hz), −74.32 (ddd, 3JP,P = 28.51 Hz, 2JP,H = 7.78 Hz, 3JP,H = 4.08 Hz).
- It should be noted that this is the first investigation of the photochemistry of diphosphabarrelene-type compounds including a 1,4-diphosphinine. For comparison, phosphinines and phosphabarrelenes have been studied in more detail. For a recent example (and references), see: (a) M. Rigo, M. Weber and C. Müller, Chem. Commun., 2016, 52, 7090–7093 RSC; (b) M. Bruce, M. Papke, A. W. Ehlers, M. Weber, D. Lentz, N. Mézailles, J. C. Slootweg and C. Müller, Chem. – Eur. J., 2019, 25, 14332–14340 CrossRef CAS.
- Compounds 2 and 3 don't show any reaction under these photochemical conditions.
- D. I. Schuster, L. Wang and J. M. van der Veen, J. Am. Chem. Soc., 1985, 107, 7045–7053 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols, NMR spectra and details of the X-ray diffraction analyses as well as the DFT calculations. CCDC 1941597 and 1941598. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02529a |
| ‡ The first two authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |