
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMnI complex redox potential tunability by remote lewis acid interaction†
Anandi
Srinivasan
a,
Jesús
Campos
 b,
Nicolas
Giraud
c,
Marc
Robert
ad and
Orestes
Rivada-Wheelaghan
*a
b,
Nicolas
Giraud
c,
Marc
Robert
ad and
Orestes
Rivada-Wheelaghan
*a
aUniversité de Paris, Laboratoire d'Electrochimie Moléculaire, CNRS, F-75006 Paris, France. E-mail: orestes.rivada@u-paris.fr
bInstituto de Investigaciones Químicas (IIQ), Consejo Superior de Investigaciones Científicas (CSIC) and University of Sevilla, Avenida Américo Vespucio 49, 41092 Sevilla, Spain
cUniversité de Paris, Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, CNRS, F-75006 Paris, France
dInstitut Universitaire de France (IUF), F-75005 Paris, France
First published on 24th July 2020
Abstract
In this work we provide direct experimental evidence on the correlation of remote interactions between a newly synthesized MnI-complex (1) and different alkali cations with redox potential tuning. Furthermore we report the electrochemical behavior of 1 towards carbon dioxide, including the effects of added alkali salts using cyclic voltammetry.
To Make our planet great again,1 measures to minimize carbon dioxide (CO2) concentration includes its use as an abundant C1 source for renewable fuels.2,3 In this regard, homogeneous and heterogeneous electrocatalysis of the CO2 reduction reaction (CO2RR) has attracted wide attention.4 The physical operating conditions of homogeneous molecular catalysis prevents obtaining the same performances as with heterogeneous systems.5 Yet, working in homogeneous conditions provides means of understanding the structure–activity relationship in molecular electrocatalysis. In particular, secondary-sphere coordination effects in metal complexes have shown to markedly impact the catalytic activity.6–9 However, the development of ligand sets and their corresponding metal complexes can be synthetically time consuming. Thus, alternatively, a single metal complex bearing a chemically switchable ligand could be used.10 Hereof, several groups have explored this approach in coordination chemistry and homogeneous catalysis commonly involving chemical modification or Lewis (or Brønsted) acid/base interaction at remote binding sites of the ligand platform leading to the modification of metal center electronics.11–13
Along these lines, we report the synthesis and complexation of an NHC-ligand platform (NHC = N-heterocyclic carbene) to a MnI center, bearing a 2-pyridone ring in which the O-atom is directed at the second coordination sphere of the Mn-center (Scheme 1). We examined the effects of potassium cation on the MnI-complex through 1H NMR studies. It allowed identifying interactions of the cation with the O-atom of the pyridone complex and probing the effects on the electronic properties of the MnI-center by cyclic voltammetry (CV). These studies demonstrate electronic tuning at the metal center through ligand interaction with alkali salts. Moreover, we evaluated their impact on the electrochemical performance of the Mn-complex under CO2.14,15
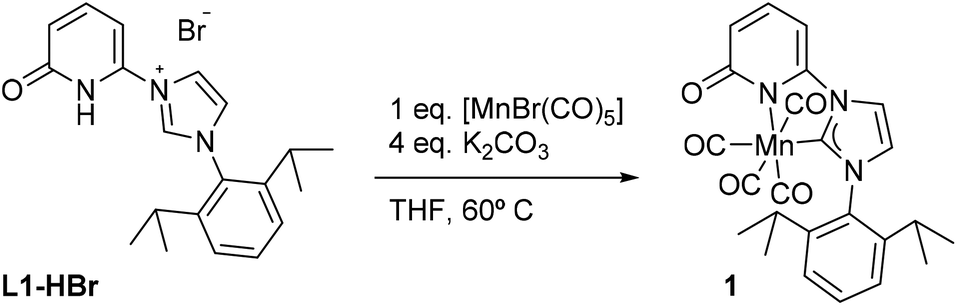 | ||
| Scheme 1 Formation of complex 1. | ||
The ligand precursor 1-(2,6-diisopropylphenyl)-3-(6-pyridonyl) imidazolium bromide (L1-HBr) was synthesized following the procedure reported elsewhere.16 The 1H NMR spectrum of L1-HBr exhibits the characteristic imidazolium peak at 10.33 ppm in CDCl3, coupled to two signals at chemical shifts, δ = 9.12 and 7.40 ppm, corresponding to the CH at the backbone of the imidazolium ring (Fig. S1, ESI†). The identity of the ligand was further verified by ESI-HRMS. Complex 1 was synthesized by deprotonating L1-HBr with 4 equivalents of K2CO3 in the presence of [MnBr(CO)5] in freshly distilled THF at 60 °C for 18 h (Scheme 1). Work up of the reaction gave complex 1 as a yellow solid in good yields (>80%). Complex 1 was unstable in MeCN or CH2Cl2, showing the broadening of NMR signals in DCM-d2 after several hours at RT. However, it remained stable in THF-d8 and/or DMF-d7 at RT, exhibiting no change in their spectra after long periods of time (>2 weeks). The 1H NMR spectrum (Fig. S6 and S7, ESI†) of 1 in THF-d8 exhibits only one septet signal for the –CH– fragment of the isopropyl groups, indicating the presence of a complex with structural symmetry. Moreover, the signals appearing at δ = 7.23(m), 6.52(d), and 6.09(d) ppm indicate the dearomatization of the pyridone group. The 13C{1H} NMR spectrum shows a peak at δ = 193.6 ppm which was attributed to the carbene carbon attached to the MnI center (CNHC–Mn) while the peaks at δ = 216.0(2C), 211.9(1C), and 210.5(1C) ppm have been assigned to the carbonyl ligands. In addition, the IR spectrum shows four carbonyl bands at ϑCO = 2013, 1994, 1924, and 1900 cm−1 (Fig. S10, ESI†) in agreement with the structure proposed in Scheme 1, furthermore, complex 1 was crystallized from a concentrated solution of diethyl ether at −30 °C and its structure was determined by X-ray diffraction analysis. Complex 1 crystallizes in the tetragonal P41212 space group and the metal center adopts a six-coordinate arrangement bearing four CO and one L1 ligand coordinated in a bidentate fashion. The pyridone ring is deprotonated as it is deduced from the localization of the double bonds at the ring and the C–O bond distance of 1.272(10) Å, with the Mn–C–O angle comprising the CO ligand opposite to the carbene carbon deviate slightly from planarity (MnI–C24–O5 = 171.4(8)°) (Fig. 1, Table S6, ESI†). With 1 characterized and its structure confirmed, we investigated possible interactions of alkali cations (perchlorate salts of Li+, Na+, K+, and Mg2+) with the O-atom of the pyridone in DMF and its effects on the electronic properties of the coordinatively saturated MnI-complex.
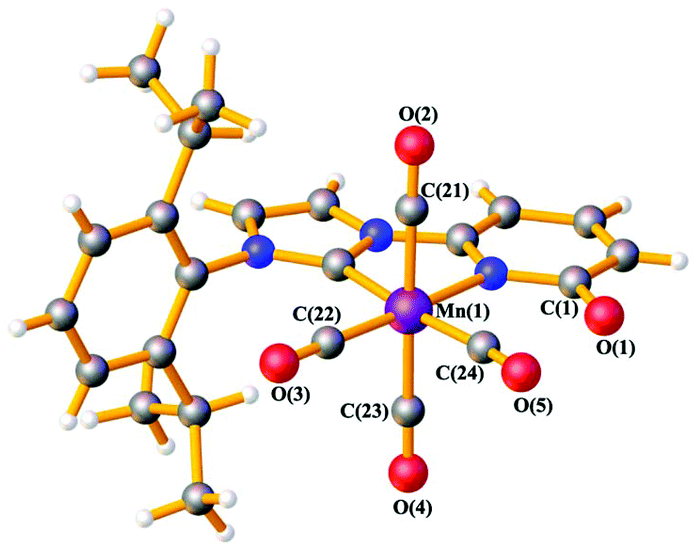 | ||
| Fig. 1 Ball and stick representation of 1·Et2O at 193 K obtained from X-ray diffraction data. Solvent molecule is omitted. Mn: violet, O: red, N: blue, C: grey, H: white. | ||
In situ IR spectroscopic studies under Ar atmosphere, in anhydrous DMF solution of 1 (1 mM) containing different concentrations of the various alkali salts did not exhibit any shift in the CO stretching bands due to 1⋯[M]+ interactions (Fig. S17–S20, ESI†). The use of DMF in IR studies did not permit to detect the interactions of the 2-pyridone carbonyl band and its interaction with the alkali cations, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯[M]+, because of overlapping with the solvent bands. Attempts to detect these possible interactions were performed by NMR titration of 1 (6 mM) under Ar with [K][B(ArF)4] (B(ArF)4 = tetrakis[3,5-bis(trifluoromethyl)phenyl] borate) (ESI, Fig. S14 and S15†). The non-coordinating [B(ArF)4] counterion was used to increase the solubility of the alkali salt in DMF-d7. The addition of [K][B(ArF)4] induced higher chemical shift variation (Δδ) for protons located at the 2-pyridone ring (a, b, Fig. 2) than for those located at the backbone of the NHC (d, e, Fig. 2) or at the diip-group (f and g, Fig. 2; diip = C6H3-2,6-iPr2). Since complex 1 is coordinatively saturated, this distinct chemical shift trend observed is presumably assigned to the weak interaction of K+⋯O-atom at the 2-pyridone ring and is consistent with the bare shift observed for diip-group signals (affinity constant of the equilibrium Kd ∼ 0.4 M, Table S1,† page S16). The different responses of the two H-signals from the NHC-backbone can be explained by the inductive effect generated from [M]+⋯O-atom interaction at the pyridone-ring (larger shift observed for d than for e, Fig. 2).
O⋯[M]+, because of overlapping with the solvent bands. Attempts to detect these possible interactions were performed by NMR titration of 1 (6 mM) under Ar with [K][B(ArF)4] (B(ArF)4 = tetrakis[3,5-bis(trifluoromethyl)phenyl] borate) (ESI, Fig. S14 and S15†). The non-coordinating [B(ArF)4] counterion was used to increase the solubility of the alkali salt in DMF-d7. The addition of [K][B(ArF)4] induced higher chemical shift variation (Δδ) for protons located at the 2-pyridone ring (a, b, Fig. 2) than for those located at the backbone of the NHC (d, e, Fig. 2) or at the diip-group (f and g, Fig. 2; diip = C6H3-2,6-iPr2). Since complex 1 is coordinatively saturated, this distinct chemical shift trend observed is presumably assigned to the weak interaction of K+⋯O-atom at the 2-pyridone ring and is consistent with the bare shift observed for diip-group signals (affinity constant of the equilibrium Kd ∼ 0.4 M, Table S1,† page S16). The different responses of the two H-signals from the NHC-backbone can be explained by the inductive effect generated from [M]+⋯O-atom interaction at the pyridone-ring (larger shift observed for d than for e, Fig. 2).
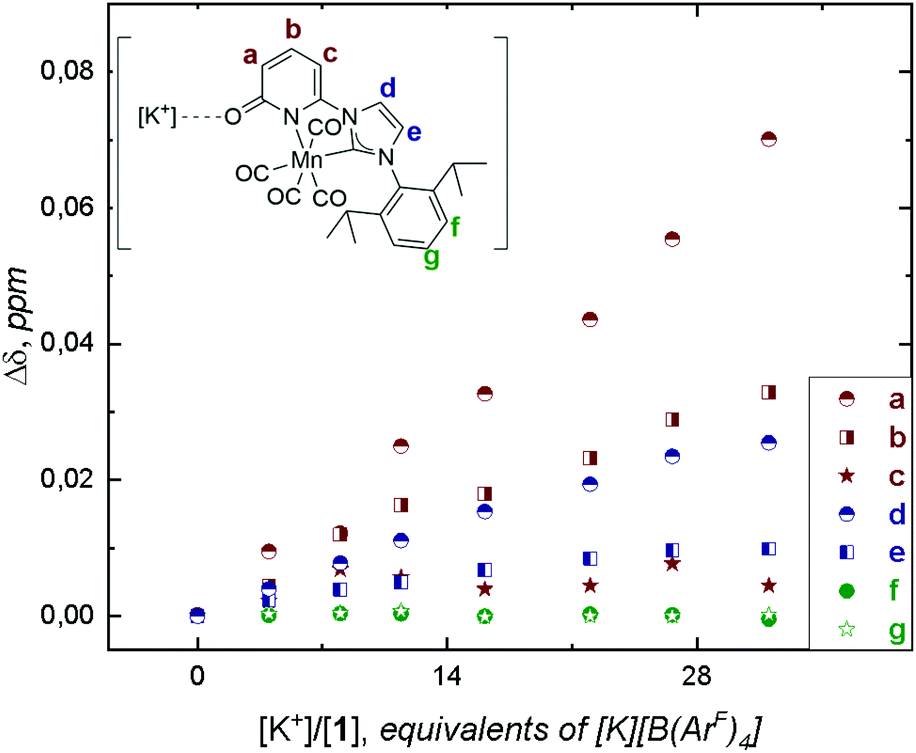 | ||
| Fig. 2 Evolution of 1H chemical shifts of 6 mM of 1 in 0.5 mL DMF-d7 upon titration with 0.1 M TBAH and [K][B(ArF)4] at 293 K. Proton labelling is shown on the molecular structure of 1 (top left). | ||
To obtain insights into the electronic perturbation at the Mn center caused by the interaction with Lewis acids, we then performed electrochemical measurements on 1. The electrochemistry of 1 was examined in dry DMF using a glassy carbon working electrode, a Pt counter electrode, and a Saturated Calomel Electrode (SCE) as a reference electrode (T = 293 K). CV of 1 (1 mM) in dry DMF with 0.1 M TBAH under Ar exhibited a single partially irreversible one electron reduction curve. Fast scan rate analysis led to estimate 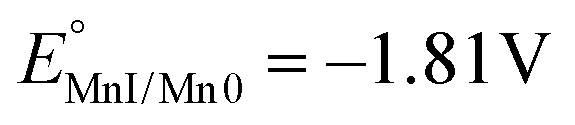 vs. SCE (black dashed line, Fig. 3A–D). Complex 1 has more negative potential compared to Mn-bipyridine systems,17 reflecting the strong electron donating properties of NHC and formally negative charge of the N-atom of the pyridone ring that acts as a strong π-donor.18,19 Progressive addition of different alkali salts under Ar revealed a positive shift in the peak potential, as the alkali salts concentration increase, with the largest shift obtained upon Mg(ClO4)2 addition (Fig. 3A–D).
vs. SCE (black dashed line, Fig. 3A–D). Complex 1 has more negative potential compared to Mn-bipyridine systems,17 reflecting the strong electron donating properties of NHC and formally negative charge of the N-atom of the pyridone ring that acts as a strong π-donor.18,19 Progressive addition of different alkali salts under Ar revealed a positive shift in the peak potential, as the alkali salts concentration increase, with the largest shift obtained upon Mg(ClO4)2 addition (Fig. 3A–D).
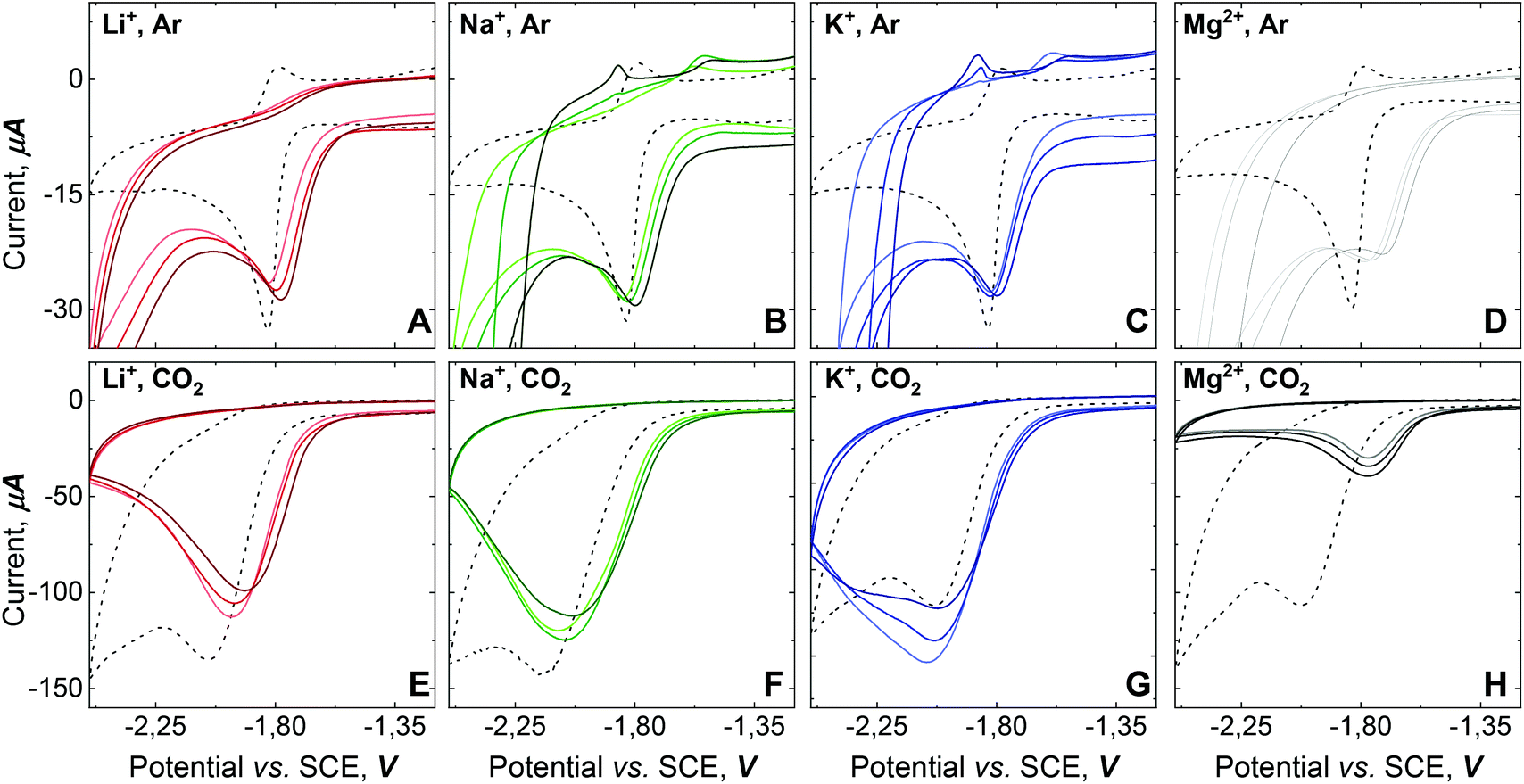 | ||
| Fig. 3 CVs of 1 (1 mM) under Ar (A–F) and CO2 (E–H), at 100 mV·s−1 in DMF, 0.1 M TBAH, at 293 K. No salt added (dashed line), LiClO4 (red), NaClO4 (green), KClO4 (blue), Mg(ClO4)2 (grey). Concentrations from lighter to darker colour: 45, 90 and 180 mM. | ||
Moreover, when the half peak potential (Ep/2) is plotted vs. the log[Mn+], a linear trend is observed suggesting a Nernstian response upon addition of the different alkali salts (ESI, Fig. S33†).20 Linear fitting gives a slope value close to the theoretical, for T = 293 K in agreement with one electron process and a stoichiometry of one [M]n+ interacting with complex 1. Therefore, along with our IR and NMR studies, these results confirm that the remote interaction of the alkali cations with the O-atom at the L1 platform induce redox tuning at 1.
Consequently, we investigated the electrochemical behavior of 1 under CO2 atmosphere. CVs of 1 (1 mM) under saturated CO2 atmosphere exhibit a four-fold current enhancement, consistent with CO2 electrocatalytic reduction.17 However, the curve shape indicates either a diffusion limited process or modification of the electrode surface by reaction product or decomposition of the catalyst in the reaction layer (dashed line, (Fig. 3E–H).4,21
The addition of alkali salts to molecular complexes have been shown to improve the efficiency of electrocatalytic CO2 reduction due to their capacity to assist C–O bond cleavage.14,15,22,23 However, the addition of alkali cations to 1 has a contrary effect; with current inhibition when Mg(ClO4)2 is present, current decrease for Li+ or Na+-perchlorate salt additions and slight current enhancement observed for KClO4 additions (Fig. 3E–H). Besides the distinct response over 1 that each alkali cation in solution generates under CO2 atmosphere, the shift of the reduction potential towards more positive values is indicative of cation assistance for the CO2 reduction. Moreover, the wave shape response after adding alkali salts strongly points toward a modification of the electrode surface, due to catalyst decomposition (vide infra).4,21
To delineate the origin of the cathodic current enhancement, bulk electrolysis studies were performed using a glassy carbon plate as a working electrode. The gaseous headspace of the sealed electrolysis cell was analyzed after the end of each experiment. Results are summarized in Table S4, ESI,† showing that CO was the only gas produced. Controlled potential electrolysis (CPE) at −1.8 V of 1 (5 mM) in 0.1 M TBAH DMF solution produced more CO (8.2 μmol) under CO2 than under Ar atmosphere (2.7 μmol) under the same experimental conditions. During the 1 hour of CPE, the current response dropped within the first 10 minutes under Ar and CO2 atm, due to the passivation of the electrode surface under such reduction conditions (ESI, Fig. S38 and S39†). Visual inspection of the electrode surface after CPE revealed a yellowish thin film deposited on the surface of the glassy carbon electrode. Ex situ X-ray photoelectron spectroscopy (XPS) analysis suggests that a de-metalation process occurred during CPE leading to the deposition of Mn on the electrode surface. The peaks at 653 and 642 eV (Mn 2p) and at 89 and 84 eV (Mn 3s) indicate the presence of Mn oxides (MnOn) on the electrode surface. The MnOn was presumably formed at the end of the electrolysis when the glassy carbon electrode was removed from the cell and expose to air. Bulk electrolysis performed in the presence of KClO4 generated a similar amount of CO under CO2 or Ar atmosphere (2.6 μmol and 2.7 μmol, respectively), with a simultaneous decrease in cathodic current and passivation of the electrode surface (ESI, Fig. S40 and S41†). These results are consistent with cyclic voltammetry studies in the presence of alkali salts, during which modification of the electrode surface occurs. A comparison of XPS analysis after bulk electrolysis with and without KClO4 show similar MnOn deposition on the surface of the electrodes (ESI, Fig. S42 and S43†). Additionally, a high concentration of 1 was detected in solution by IR analysis after bulk electrolysis (Fig. S23 and S24, ESI†). No carbonate bands were detected either by IR or {1H}13C NMR analysis of post electrolysis solution. Therefore, these results are indicative of partial decomposition of complex 1, which passivates the glassy carbon electrode surface during CPE and prevents further production of CO.
In conclusion, we report a new NHC-MnI complex, 1, and demonstrate the interaction of various alkali cations with pending O-atom of the 2-pyridone ring and its effects on the redox potential of the Mn-center, demonstrating electronic tunability of the complex with alkali cation concentration variation in DMF solutions. Furthermore, we performed CV studies under CO2 atmosphere and observed electrocatalytic activity for complex 1 (CO production) as well as the effects caused by the different alkali salts present in solution on the reduction potential. Despite the instability of the complex in electrocatalytic conditions, this report shows that pyridonate-platforms can be used to induce electrochemical tuneability in the presence of Lewis acids. Currently, we are targeting analog derivatized ligand platforms to exploit cooperative effects for the electrochemical CO2RR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge the financial support from the Investissement l'Avenir, specifically through the MOPGA call no. ANR-18-MPGA-0012. Dr Phillippe Decorse (Université de Paris) is warmly thanked for providing XPS analysis and Dr F. Mavré, Dr K. Torbensen and Dr E. Anxolabéhère-Mallart are acknowledged for their helpful discussions.Notes and references
- Make our planet great again, is an initiative of the President of the Republic of France Emmanuel Macron, Launched on 1st June 2017 following the decision of the United States to leave the Paris Agreement on the climate. https://makeourplanetgreatagain.fr/.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- F. Marques Mota and D. H. Kim, Chem. Soc. Rev., 2019, 48, 205–259 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367 CrossRef CAS PubMed.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 42–49 CrossRef PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- M. H. H. Rønne, D. Cho, M. R. Madsen, J. B. Jakobsen, S. Eom, É. Escoudé, H. C. D. Hammershøj, D. U. Nielsen, S. U. Pedersen, M.-H. Baik, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2020, 142, 4265–4275 CrossRef PubMed.
- A. M. Allgeier and C. A. Mirkin, Angew. Chem., Int. Ed., 1998, 37, 894–908 CrossRef PubMed.
- K. T. Horak, D. G. VanderVelde and T. Agapie, Organometallics, 2015, 34, 4753–4765 CrossRef CAS.
- A. J. M. Miller, Dalton Trans., 2017, 46, 11987–12000 RSC.
- H. F. Cheng, A. I. d'Aquino, J. Barroso-Flores and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 14590–14594 CrossRef CAS PubMed.
- A. Zhanaidarova, H. Steger, M. H. Reineke and C. P. Kubiak, Dalton Trans., 2017, 46, 12413–12416 RSC.
- M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 1386–1393 CrossRef CAS PubMed.
- J. Messelberger, A. Grünwald, P. Stegner, L. Senft, W. F. Heinemann and D. Munz, Inorganics, 2019, 7, 65 CrossRef CAS.
- M. Stanbury, J.-D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 361, 120–137 CrossRef CAS.
- F. Franco, M. F. Pinto, B. Royo and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2018, 57, 4603–4606 CrossRef CAS PubMed.
- T. H. T. Myren, A. Alherz, J. R. Thurston, T. A. Stinson, C. G. Huntzinger, C. B. Musgrave and O. R. Luca, ACS Catal., 2020, 1961–1968, DOI:10.1021/acscatal.9b04773.
- A. J. Bard and L. J. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2nd edn, 2001 Search PubMed.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, Chem. Soc. Rev., 2019, 48, 2927–2945 RSC.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Phys. Chem., 1996, 100, 19981–19985 CrossRef CAS.
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, syntheses, NMR, IR, CVs, CPE and XPS characterization. CCDC 2001353. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02467h |
| This journal is © The Royal Society of Chemistry 2020 |