
Ultrafine NiFe clusters anchored on N-doped carbon as bifunctional electrocatalysts for efficient water and urea oxidation†
 *b
*b
Abstract
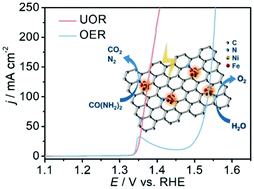
Hydrogen production through electrocatalysis is crucial in renewable energy technologies but significantly impeded by sluggish anodic reactions. Developing bifunctional anode noble-metal-free electrocatalysts towards oxygen evolution reaction (OER) and urea oxidation reaction (UOR) to boost cathodic hydrogen evolution reaction (HER) is promising but challenging to meet different reaction media and multiple applications for simultaneous clean energy production and pollution treatment. Herein, a facile one-pot thermal treatment strategy is presented to anchor NiFe nanoclusters (with a size of about 2 nm) on N-doped carbon as bifunctional electrocatalysts for both OER and UOR. Such an electrocatalyst can deliver a current density of 20 mA cm−2 with a low overpotential of 260 mV and a small Tafel slope of 42 mV dec−1 for OER, superior to the state-of-the-art Ru-based materials. Besides, this electrocatalyst also shows excellent activity for UOR with the need for just 1.37 V (vs. RHE) to attain a current density of 100 mA cm−2. In a two-electrode electrolyzer for both cathodic HER and anodic UOR, only a cell voltage of 1.50 V is required to drive a current density of 10 mA cm−2, which is 140 mV lower than that of overall water splitting electrolysis (1.64 V). The excellent electrooxidative performance can be attributed to the improved conductivity, abundant active sites and fast charge transfer and transport benefiting from the ultrafine structure of NiFe clusters and their synergistic effect with N-doped carbon.
 Please wait while we load your content...
Please wait while we load your content...

