
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis-cyclometallated Ir(III) complexes containing 2-(1H-pyrazol-3-yl)pyridine ligands; influence of substituents and cyclometallating ligands on response to changes in pH†
Noreldin S. Y.
Abdolla‡
 ,
David L.
Davies
*,
Mark P.
Lowe
and
Kuldip
Singh
,
David L.
Davies
*,
Mark P.
Lowe
and
Kuldip
Singh
School of Chemistry, University of Leicester, Leicester LE1 7RH, Libya. E-mail: dld3@le.ac.uk
First published on 13th August 2020
Abstract
Bis-cyclometallated Ir(III) complexes containing 2-(1H-pyrazol-3-yl)pyridine ligands have been synthesised. Their absorption is almost unchanged with changes in pH however the emission intensities vary by a factor of up to three and the complexes have emission pKas in the range 8.0 to 10.0. Substituents on the pyrazole have only a minor effect on the emission pKa. Surprisingly the complexes with phenylpyrazole cyclometallated ligands 3aL1–3 showed an intensity decrease with increasing pH (switch off) whilst the corresponding phenylpyridine ones 3cL1–3 showed an increase in emission intensity with increasing pH. Putting electron-withdrawing CF3 substituents on the cyclometallating phenyls reduced the pKa of the complexes to 6.8–7.8, thereby extending the useful pKa range; however, in general it tended to reduce the magnitude of the change in emission intensity. Surprisingly the CF3-substituted complexes also showed a complete reversal in the direction of the intensity change when compared to their respective unsubstituted congeners.
Introduction
Luminescent pH sensors have attracted significant interest for measuring pH in biological environments due to their excellent sensitivity, minimal damage to living samples, specificity, the availability of a wide range of indicator dyes, high signal-to-noise ratios and the ability to continuously monitor rapid pH changes. Furthermore, fluorescence microscopic imaging allows mapping of the spatial and temporal distribution of H+ within living cells.1 To date, pH sensors have primarily focussed on organic molecules; however, transition metal and rare earth metal complexes have also been studied.2 Transition metal complexes are attractive as possible pH sensors due to significant Stokes shifts for easy separation of excitation and emission, tuneable emission wavelength, and relatively long emission lifetimes compared to organic molecules allowing for possible luminescent lifetime imaging of pH.3 Several luminescent complexes of the platinum group metal ions have been used as pH sensors, notably Ru(II),4 Re(I),5 and Pt(II).1a,6Iridium complexes have also been used for pH sensing. An early example by Licini and Williams used Ir bis-terpyridine complexes, (Fig. 1) containing either pyridyl or phenol substituents as the pH responsive groups which exhibit changes in lifetime and intensity with changing pH.7 The pKas of the complexes ranged from 4.1 (cf., pyridinium pKa = 5.25) to 8.1 (cf., phenol pKa = 10.0), in both cases the cationic electron-withdrawing metal terpyridyl unit reduces the basicity of the pendant group, pyridine or phenol, respectively.
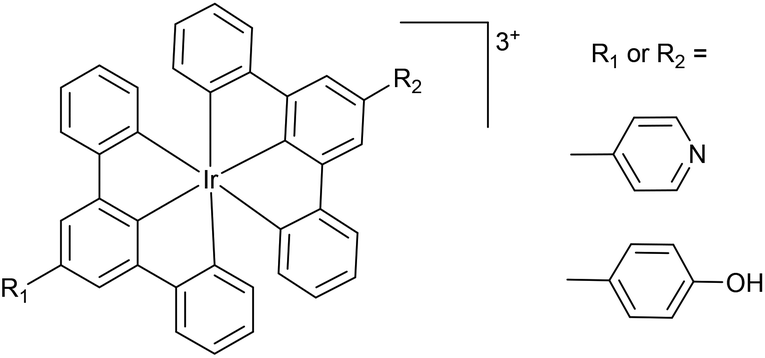 | ||
| Fig. 1 Early examples of pH sensitive luminescent Ir(III) complexes.7 | ||
The coordination of a functionalised ligand to a metal tends to lead to a reduction in electron density due to electron donation to the metal and hence should lead to a lowering of the pKa for the ligand in the ground state. However, it should be noted that it is harder to predict the outcome for an excited state.
In the last 20 years there has been huge interest in the use of cyclometallated Ir(III) complexes of general formula [Ir(C^N)2(X^Y)]n+ (C^N = a cyclometallated ligand, X^Y = a neutral or anionic bidentate ligand n = 0,1; Fig. 2 structure A) which have interesting photophysical properties and have found applications particularly in OLEDS, sensors and as labels for biological imaging.8 A comparatively small number show pH responsive emission, changing wavelength and/or intensity of emission. A number of different pH responsive ligands have been used, and some of these are shown in Fig. 2. The pH responsive group can be on the cyclometallating (C^N) ligands or on the ancillary (X^Y) ligand. For example, tris-cyclometallated complexes with protonatable pyridyl9 or amine10 substituents (B or CFig. 2) have been reported. Bis-cyclometallated complexes with ligands D were non-emissive at low pH 2–5 but gave red emission at higher pH which was ascribed to deprotonation of the carboxylic acid groups.11 The pKa value was approximately 7.0, suggesting that the probe may useful for monitoring pH in biological systems.
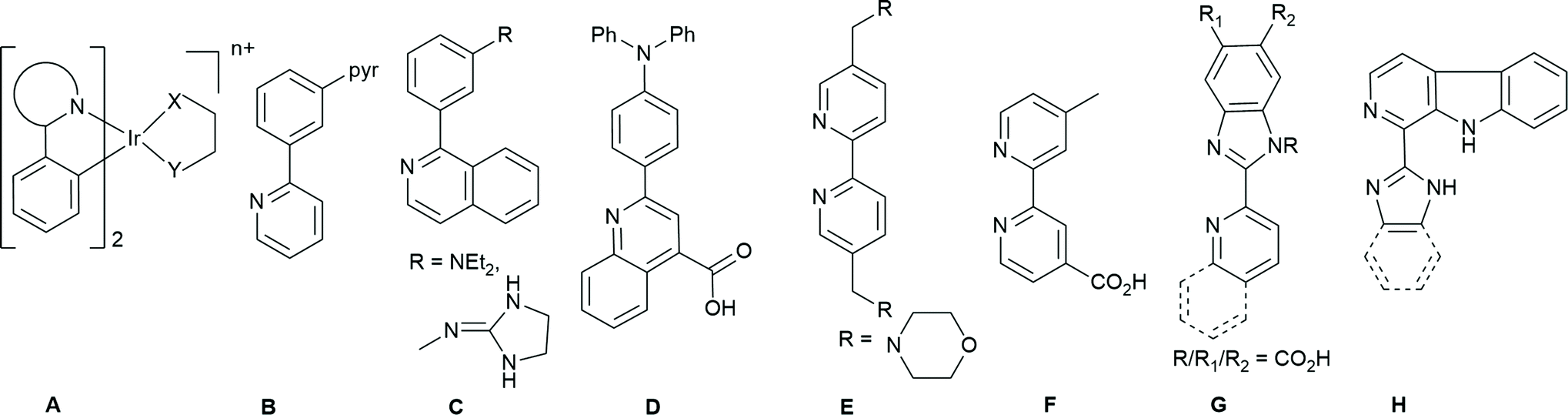 | ||
| Fig. 2 Examples of pH responsive cyclometallating ligands (B to D) or ancillary ligands ligands (E to H) used in bis- or tris-cyclometallated Ir complexes. | ||
Several proton responsive N^N ligands have been used as the ancillary X^Y ligand in bis-cyclometallated complexes (Fig. 2E–H). For example, functionalised bipyridine ligands, with morpholine E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 or carboxylic acid substituents F.13 Several groups have investigated complexes with imidazole or benzimidazole containing ligands e.g.G.14 Similar carboline-containing ligands H gave pH-sensitive iridium(III) complexes used in lysosome targeted photodynamic therapy (PDT).15 The complexes were more emissive at low pH with pKa values between 3.6 and 4.4.
12 or carboxylic acid substituents F.13 Several groups have investigated complexes with imidazole or benzimidazole containing ligands e.g.G.14 Similar carboline-containing ligands H gave pH-sensitive iridium(III) complexes used in lysosome targeted photodynamic therapy (PDT).15 The complexes were more emissive at low pH with pKa values between 3.6 and 4.4.
Although, imidazole-based ligands have been investigated, pyrazole ligands with an NH moiety have been much less studied. Lam et al. studied a Re-carbonyl complex containing a bidentate pyrazolyl–pyridine.5b Cyclometallated Pt complexes with tridentate pyrazolyl-containing ligands have also been studied which show pH dependent emission some of which have been in used in cell imaging.6b,16 Hence, we have investigated Ir(III) bis-cyclometallated complexes with 2-(1H-pyrazol-3-yl)pyridine ligands. 2-(1H-pyrazol-3-yl)pyridine itself has a pKa of 11.6.17 We have investigated the effect of the nature of the cyclometallated ligands, phenylpyrazole and phenylpyridine respectively and ones with an electron-withdrawing CF3 substituent, and of substituents on the pyrazole.
Results and discussion
Synthesis
The bis-cyclometallated dimers 1a–d (Chart 1) were prepared by a literature method18 and the data are consistent with those published.18,19 The syntheses of cationic complexes 2aHL1–3 are outlined in Scheme 1. The dimer 1a was reacted with the relevant 2-(1H-pyrazol-3-yl)pyridine (HL1–3) in the presence of KPF6 in methanol at 60 °C under microwave irradiation for 20–40 minutes (see Experimental section). After work up, complexes 2aHL1–3 were formed in good to excellent yields. | ||
| Chart 1 Iridium dimers used. | ||
 | ||
| Scheme 1 Preparation of cationic bis-cyclometallated phenylpyrazole complexes. | ||
1H and 13C NMR spectra for all the complexes were assigned using two-dimensional NMR experiments such as TOCSY, COSY, NOESY, and HMQC. The coordination of the N^N ligand removes the C2-symmetry of the dimers, causing the two C^N ligands to become inequivalent and therefore doubling the number of peaks for the cyclometallated ligands. The 1H and 13C NMR spectra of 2aHL1–3 are similar to each other therefore, only the assignment of complex 2aHL1 is discussed in detail.
The 1H NMR spectrum of complex 2aHL1 contains twenty-one aromatic environments. There are two doublets of doublets at δ 6.30 and 6.32, characteristic of cyclometallated phenyl protons which are shifted to high field due to ring current effects.20 This then allows assignment of the signals for the phenyl and pyrazole rings of the C^N ligands using the TOCSY and NOESY spectra (Fig. S1 and 2†). Likewise, the protons of the pyrazole of the N^N ligand are easily identified in the TOCSY spectrum as doublets at δ 7.83 and δ 7.20 slightly shifted downfield compared to the free ligand (by ca. δ 0.4 and 0.2, respectively) presumably due to the coordination of the (N^N) ligand to the metal. The doublet at δ 7.20 shows an NOE to a doublet of triplets at ca. δ 8.20 which is therefore assigned as the closest pyridine proton (He see ESI† for NMR labelling) and the other pyridine protons Hf–h are assigned from the TOCSY and COSY spectra. The protons He–g are shifted slightly downfield compared to the free ligand (ca. δ 0.2 to 0.4) as might be expected on coordination to the metal, however, the proton next to the N atom (Hh) is observed at δ 8.01 about δ 0.7 upfield compared to the free ligand (δ 8.68) due to ring currents from the neighbouring phenyl ring (A), as noted previously, e.g. for [Ir(R-ppz)2(bipy)]PF6.18 Proton Hh also shows an NOE to a pyrazole proton multiplet at ca. δ 7.07 which is therefore assigned as H7B. This distinguishes the two cyclometallating ligands and hence allows for assignment of all the other protons. The NH proton was not observed, probably due to exchange with D2O in the solvent (CD3CN). The 13C NMR spectrum showed the expected number of signals, and were assigned using the HSQC and HMBC spectra. The high resolution mass spectrum (ASAP) shows a molecular ion with characteristic Ir isotopes pattern for the cation at m/z 624.1497 (624.1488 calculated for C26H21193IrN7).
The 1H NMR spectra of 2aHL2 and 2aHL3 are similar to that of 2aHL1 except for the substituent on the N^N pyrazole, a singlet due to a tBu at δ 1.26 for 2aHL2 and extra signals in the aromatic region for 2aHL3. For 2aHL3 a downfield singlet at δ 12.01 was assigned to the NH proton. The 13C NMR spectra showed the expected number of signals and both complexes showed an ion corresponding to the cationic complex in their high resolution (ASAP) mass spectra.
The neutral complexes 3aL1–3 corresponding to loss of a proton from 2aHL1–3 were synthesised in good yields by reaction of dimer 1a with ligands HL1–HL3 and NaOMe in a mixture of DCM/methanol (2:1) (Scheme 2). Additionally, a CF3 substituted dimer 1b was also used to investigate the effect of an electron withdrawing substituent on the cyclometallating ligand, providing complexes 3bL1–3.
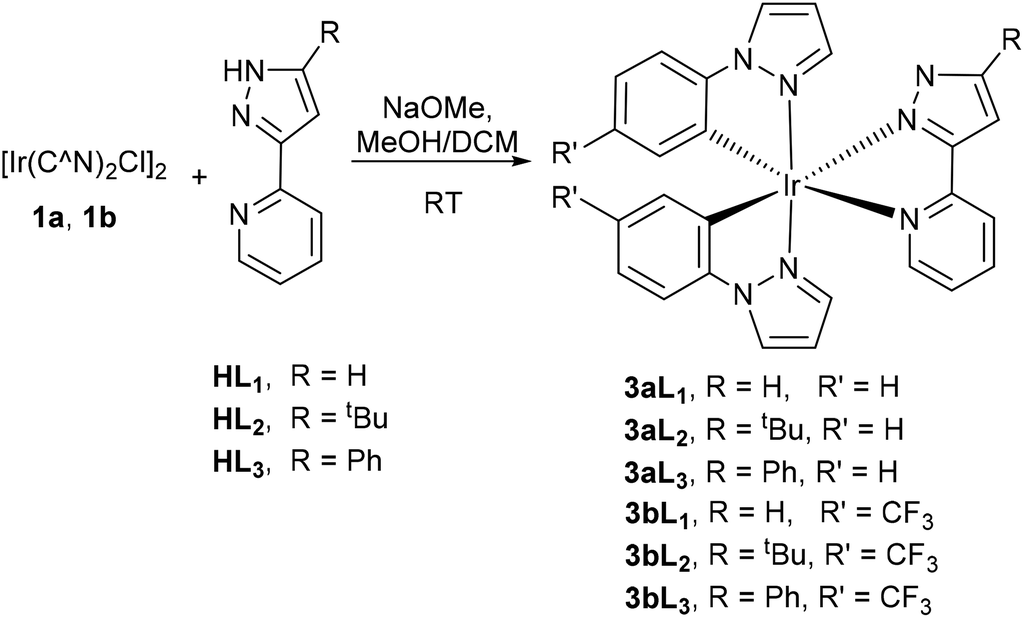 | ||
| Scheme 2 Preparation of neutral bis-cyclometallated complexes of phenyl pyrazole. | ||
The 1H and 13C NMR spectra of 3aL1–3, are similar to those for 2aHL1–3, respectively, and assignments have been made on this basis. As expected, in the majority of cases the deprotonation of the ancillary ligand causes an upfield shift of the protons of the N^N ligand in the 1H NMR spectra compared to their corresponding cationic complexes. For example, for 3aL1 the pyrazole protons Ha,b are observed as mutually coupled doublets at δ 7.47 and 6.82, respectively, upfield compared to the corresponding signals in the cationic complex 2aHL1 (δ 7.83 and 7.20 respectively) consistent with deprotonation of the pyrazole. Unusually, for 3aL1 the most upfield resonance in the aromatic region is a pyrazole proton not a cyclometallated phenyl. The spectra of the CF3-substituted complexes 3bL1–3 are similar to those of the unsubstituted complex but with two fewer resonances in the aromatic region. The 19F NMR spectra of 3bL1–3 exhibit two singlets at about −60 ppm corresponding to two different CF3 groups. All of the neutral complexes show ions due to [M + H]+ in their high-resolution mass spectra.
The corresponding complexes containing cyclometallated phenylpyridine ligands in place of phenylpyrazole were prepared by analogous procedures to give cationic complexes 2cHL1–3 and neutral complexes 3cL1–3 and CF3-substituted complex 3dL2 respectively (see Chart 2). These were fully characterised by 1H and 13C NMR spectroscopy and high-resolution mass spectra.
 | ||
| Chart 2 Labelling of bis-cyclometallated complexes of phenylpyridine. | ||
The 1H NMR spectra of 2cHL1–3 show the cyclometallated phenyl protons as the most upfield aromatic signals, between δ 6.23, and 6.35. The pyridine proton next to nitrogen for both the cyclometallating ligands and the N^N ligand are all influenced by ring currents from the other ligands and hence are shifted upfield compared to the free ligands, being observed between δ 7.9 and 7.5. The 1H NMR spectra for neutral complexes 3cL1–3 show the cyclometallated phenyl protons between δ 6.23 and 6.39 within 0.1 ppm of the related cationic complexes. As for 3aL1–3 deprotonation of the pyrazole NH proton 3cL1–3 leads to an approximately 0.5 ppm upfield shift for pyrazole proton Hb. All the other protons are within 0.2–0.3 ppm of the signals in the corresponding cationic complexes 2cHL1–3. The 13C NMR spectra show the expected number of signals for 2cHL1–3. The 1H and 13C NMR spectra of 3dL2 are similar to 3cL2, except the signals for one proton on each the phenyl rings of the C^N ligands have been replaced by a CF3 group, as confirmed by 19F NMR spectroscopy. The high-resolution mass spectra (ASAP) each show a molecular ion for the cations 2cHL1–3 and the protonated molecular ions for 3cL1–3 and 3dL1–3.
X-ray crystallography
The cationic complexes were relatively easy to crystallise as their PF6 salts, hence complexes 2aHL1–3 and 2cHL1–3 were characterised by X-ray crystallography. In addition, two of the neutral molecules 3aL2 and 3cL3 were also characterised by X-ray crystallography. As examples the structures of two cationic/neutral pairs 2aHL2 and 3aL2, and 2cHL3 and 3cL3 are shown in Fig. 3 and 4 respectively, the remaining structures are in Fig. S3 and S4† with selected bond lengths and angles in Tables S1 and S2.† | ||
| Fig. 3 Left, structure of the cation of 2aHL2 and right the structure of 3aL2 showing 50% ellipsoids. All hydrogen atoms (except NH) have been omitted for clarity. | ||
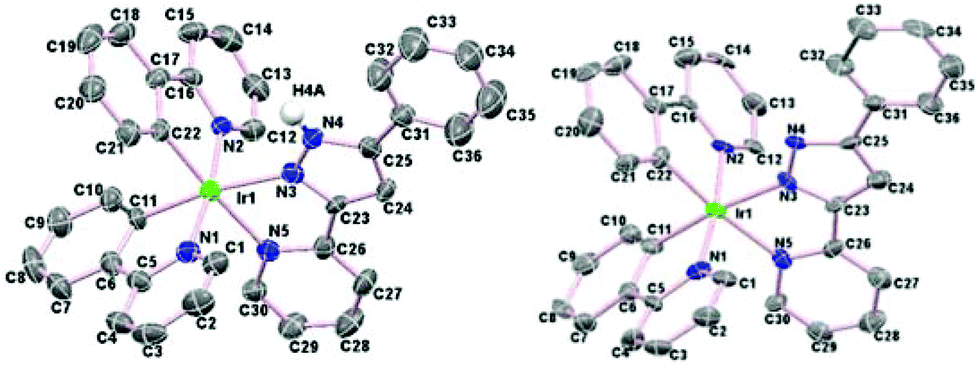 | ||
| Fig. 4 Left, structure of the cation of 2cHL3 and right the structure of 3cL3 showing 50% ellipsoids. All hydrogen atoms (except NH) have been omitted for clarity. | ||
All the crystal structures show the expected distorted octahedral coordination geometry with cis metallated carbons and trans nitrogen atoms. The chelate bite angles for the cyclometallated ligands are all about 80°, and about 75° for the N^N ligands. The Ir–N bond lengths to the N^N ligand are longer than those to the cyclometallating ligands, due to the former being trans to the C atoms, as observed previously.21 For example, for complex 2aHL1, [Ir(1)–N(5), 2.116(7) and Ir(1)–N(7), 2.148(6) Å] are longer than [Ir(1)–N(1), 2.036(7), and Ir(1)–N(3), 2.031(6) Å] similar trends are seen for complexes 2aHL2–3. The same trend in Ir–N bond lengths is also seen for the neutral complexes. The Ir–N bond distance to the anionic pyrazole of the N^N ligand [2.122(6) Å in 3aL2 and 2.088(7) Å in 3cL3] is shorter than that in the cationic analogue [2.175(8) Å in 2aHL2, and 2.149(4) Å in 3cL3] though the other Ir–N and Ir–C bonds and the chelate bite angles are statistically almost the same in both pairs of complexes. The structures of 2aHL1–3 and 2cHL1 and 2cHL3 show the presence of waters of crystallisation and most of the structures show hydrogen bonding from the NH of the protonated pyrazole to the PF6 anion or to a water molecule.
Photophysical properties
The UV-vis absorption spectra of all 16 complexes are shown in Fig. S5–S8† and the data are collated in Table S3.† Cationic complexes 2aHL1–3, and their neutral counterparts 3aL1–3 are shown in Fig. S5† whilst the CF3-substituted complexes 3bL1–3 are compared with their unsubstituted analogues, 3aL1–3 in Fig. S6.† Similarly, spectra for phenylpyridine complexes 2cHL1–3, and 3cL1–3 are in Fig. S7† and 3cL2, is compared with 3dL2 in Fig. S8.† All the complexes show strong bands between 225 and 300 nm due to π → π* transitions in addition the phenylpyridine complexes 2cHL1–3, 3cL1–3 and 3dL2 show weak bands between 380–405 nm (possibly having contributions from 1MLCT transitions). Adding a CF3 substituent to the C^N ligand causes only minor changes, and these are in the short wavelength region. The substituent on the N^N ligand also has very little effect on the λmax. For both the phenylpyrazole and phenylpyridine complexes there are only minor differences in uv-vis spectra between the cationic and neutral species and these are in the uv region, a slight red shift (10 to 30 nm) in λmax occurring on deprotonation. Since there is no significant change (<20%) in absorbance at any wavelength, these complexes would not be expected to be good pH sensors in their ground states.Emission spectra of the complexes were run in MeCN in aerated solutions. Each pair of cationic and neutral complexes (e.g., 2aHL1 and 3aL1) were irradiated at the same excitation wavelength (see Fig. S9 and 10† for excitation spectra). The emission spectra of phenylpyrazole cationic complexes 2aHL1–3 and their neutral analogues 3aL1–3 are shown in Fig. 5a whilst those of 3aL1–3 are compared with their CF3-substituted analogues 3bL1–3 in Fig. 5b and all the associated data are reported in Table S4.†
 | ||
| Fig. 5 Emission spectra of (a) cationic and neutral complexes 2aHL1–3 (—) and 3aL1–3 (– – –), (b) normalised emission spectra of neutral complexes 3aL1–3 (– – –) and 3bL1–3 (….), all in MeCN at 0.02 mM at room temperature in air; excitation at 320–325 nm. | ||
All three cationic complexes 2aHL1–3 show a broad emission band, each with a similar λmax at 502, 495, and 508 nm, respectively. Hence, the substituent on the pyrazole has only a small effect on the emission wavelength. The corresponding neutral complexes 3aL1–3 show similar spectra with a slight blue shift in λmax of 27 nm (1053 cm−1) for 3aL1, 2 nm (82 cm−1) for 3aL2 and 12 nm (476 cm−1) for 3aL3. The emission intensity for the neutral complexes 3aL1–3 are all reduced by about 50% compared with the cationic complexes 2aHL1–3. Hence, these complexes have potential as luminescent pH sensors. Neutral complexes 3bL1–3 with an electron-withdrawing substituent (CF3) on the phenyl ring each show a broad emission band which is blue-shifted compared to the corresponding unsubstituted complexes 3aL1–3, with 3bL2 showing the largest shift of 35 nm (1550 cm−1) (Fig. 5b and Table S4†), respectively. A blue shift in emission by addition of electron-withdrawing groups to the C^N cyclometallating ligands is a well-known phenomenon that has been ascribed to the electron-withdrawing group stabilising the HOMO.18,22
The emission spectra of the corresponding phenylpyridine complexes 2cHL1–3, and 3cL1–3 are shown in Fig. 6 and their data are reported in Table S4.† Complex 2cHL1 shows a broad emission band (λmax 505 nm); however, complexes 2cHL2–3 both show some structure in the emission band. As for the phenylpyrazole complexes, changing the R2 group on the N^N ligand has only a small effect on the emission wavelength. The neutral complexes 3cL1–3 show similar spectra to the cationic complexes, with a broad emission band with shoulders. Surprisingly there is no significant change in emission intensity between the cationic and neutral complexes, though the neutral complexes are slightly more emissive than the cationic ones which is the opposite case to the phenylpyrazole complexes. Hence, these are not expected to be good pH sensors in their excited states (see pH titrations below for further discussion). The neutral CF3-substituted complex 3dL2 showed a λmax at 502 nm, a small blue shift (ca. 4 nm) compared to the corresponding unsubstituted complex 3cL2, and also shows a distinct shoulder at longer wavelength ca. 533 nm (Fig. S10†).
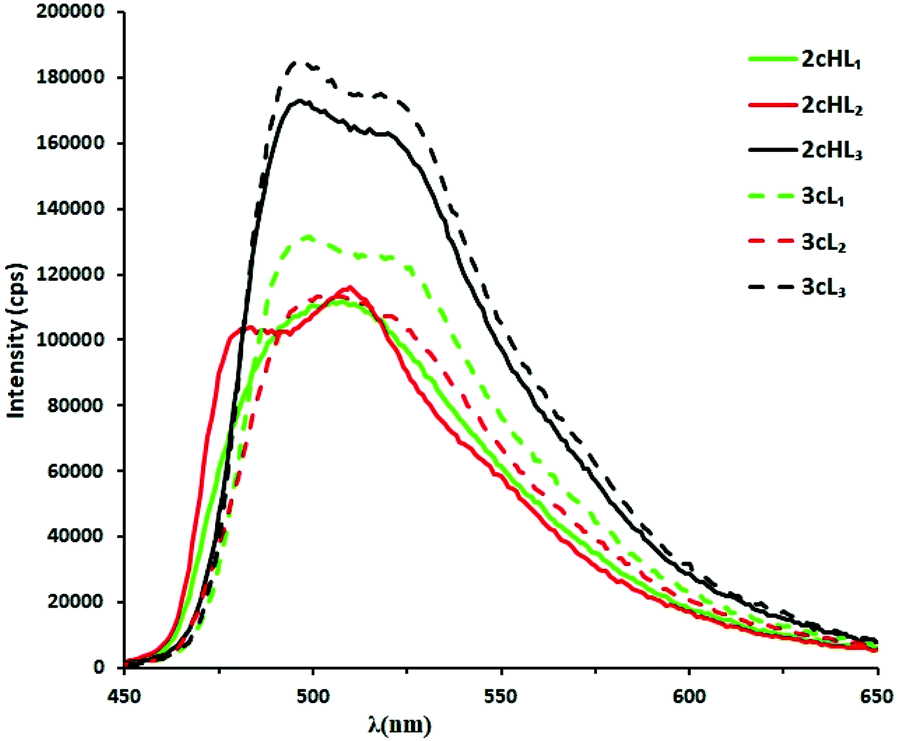 | ||
| Fig. 6 Emission spectra of cationic and neutral complexes 2cHL1–3 (—) and 3cL1–3 (– – –), in MeCN at 0.02 mM at room temperature in air; excitation at 320–325 nm. | ||
In conclusion, the UV-vis absorption spectra of all the complexes show strong bands between 200 and 300 nm due to π → π* transitions, with the phenylpyridine complexes also showing weak bands between 380–405 nm. All the complexes are emissive at room temperature in solution in air. There is no significant variation in emission wavelength upon changing the substituent on the ancillary N^N ligands, and there is very little change in λmax on deprotonation, except for complex 3aL1 which shows a blue shift of approximately 27 nm. Substituting H with an electron-withdrawing CF3 group on the C^N ligand causes a blue shift in the emission spectrum, consistent with other bis-cyclometallated Ir complexes.18,22 The effect of changing the cyclometallated ligand from phenylpyrazole to phenylpyridine is not uniform, giving a red shift in some cases and a blue shift in others. This is further complicated by the presence of shoulders on some bands.
pH titrations
To investigate their potential application as pH sensors, pH titrations of the complexes were carried out in MeCN/H2O (1:9) (see Experimental for details). As expected from the studies of the isolated neutral and cationic complexes there is very little change in the uv-vis spectra with changing pH, hence absorption spectra cannot not be used to determine ground state pKa values. The emission pKa values23 for the complexes were determined by the change in emission intensity with pH over the range ca. 3 to 12.5.
As an example, the emission pH titration data and pKa values of complex 3aL1 are illustrated in Fig. 7. The emission intensity of 3aL1 at 500 nm was relatively unchanged between pH 7.5 and pH 3.7, but above pH 7.5, there was a gradual decrease in emission intensity by about a factor of two to around pH 10, above which the intensity remained essentially constant until pH 12.3. Over the full pH range studied, there is a small blue shift (ca. 6 nm) in λmax. The emission pKa value was determined to be about 9.9 at the equivalent point, which is more acidic than the free 2-(1H-pyrazol-3-yl)pyridine ligand itself (pKa 11.6).17
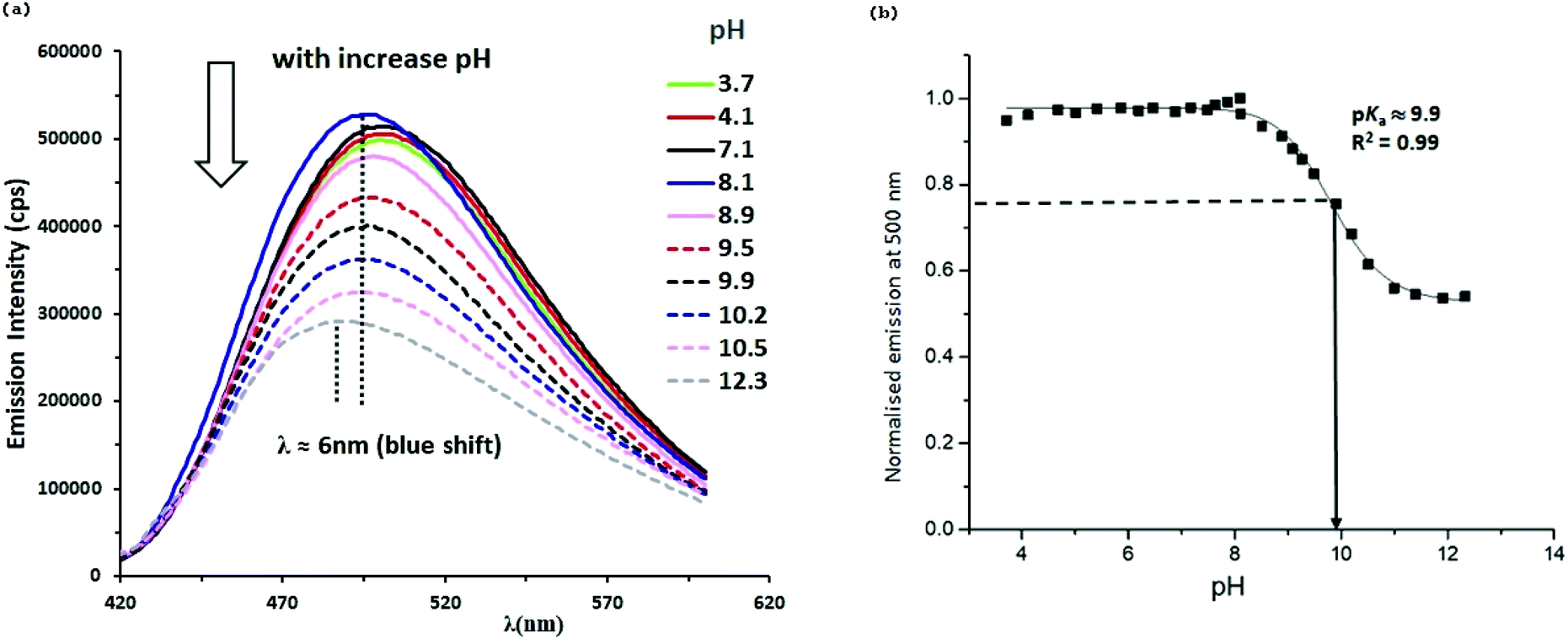 | ||
| Fig. 7 (a) Selected emission spectra of complex 3aL1 (0.02 mM) at various pH values in MeCN/H2O (1:9), in air with excitation at 324 nm. (b) Plot of normalised emission intensity of complex 3aL1 against pH. | ||
The emission pH titration spectra of complexes 3aL2–3 (Fig. S12 and 13†) were similar to that of 3aL1. In both complexes the emission intensity was relatively constant over the pH range ca. 4.9–7.5 but then decreased at higher pH by a factor of ca. 2 for 3aL2 and ca. 1.3 for 3aL3. The emission pKa values of complexes 3aL2 and 3aL3 were determined to be ca. 10.0 and 8.9, respectively (Fig. S10 and 11†), which are more acidic than the corresponding free ligands at 12.3 and 11.6, respectively.17 Hence, all three complexes 3aL1–3 can function as pH sensors. However, even though the pKa of the complexes is between 1.7 and 2.7 units lower than for the free ligands, none of them are low enough to be of use as biological pH sensors. To lower the pKa further the complexes need to be made more acidic. Hence, complexes 3bL1–3 with electron-withdrawing CF3 groups on the cyclometallating phenyls were examined.
The emission pH titration of complexes 3bL1–3 (Fig. S14–16†) did show changes with pH, however, for all three complexes replacing H with the electron-withdrawing CF3 group on the C^N ligand led to a complete reversal of the shape of the pH titration curve. Thus, at low pH, the complexes show low emission and at high pH the emission intensity is higher, which is the exact opposite of complexes 3aL1–3. From the emission intensity changes, a pKa value of 6.8 was determined for 3bL1 which is much smaller than the pKa (9.9) for the unsubstituted complex 3aL1. The emission pH titration spectra of 3bL2 and 3bL3 were similar to that of 3bL1. Showing a relatively small (1.2–1.3 fold) increase in intensity with increasing pH. The pKa values of 3bL2 and 3bL3 were calculated to be 7.8 and 7.5, respectively, both lower than the pKa of the unsubstituted analogues 3aL2 and 3aL3 of 10.0 and 8.9, respectively. Hence, addition of the CF3 substituents has, as desired, lowered the pKa of the complexes by between 1.4 and 3.1 units. As noted above, the emission intensity of the CF3-substituted complexes 3bL1 increases with increasing pH whilst the intensity of the unsubstituted analogues 3aL1 decreases with increasing pH. The reason for this difference is not known. In addition, the complexes, particularly 3bL2 and 3bL3 showed only a small change in emission intensity per unit change in pH and hence will clearly not be particularly good sensors. The smaller change in intensity could be due to reduced quantum yields or may just be increased triplet character and hence more quenching by oxygen.
As mentioned above the emission spectra for cationic and neutral phenylpyridine complexes 2cHL1–3 and 3cL1–3 were rather similar however full pH titrations were still carried out. The results for 3cL1 are shown in Fig. 8 and those for 3cL2–3 are in Fig. S17 and 18.†
 | ||
| Fig. 8 (a) Selected emission spectra of complex 3cL1 (0.02 mM) at various pH values in MeCN/H2O (1:9), in air with excitation at 324 nm. (b) Plot of normalised emission intensity of complex 3cL1 against pH. | ||
Complex 3cL1 shows two emission maxima at 479 and 505 nm. Above pH 8.0, these bands start to shift to longer wavelength (λmax 492 nm) and increase in intensity up to about pH 10.9, above which the intensity remains essentially constant. The overall increase in intensity at 492 nm is by a factor of ca. 1.6, and the pKa was 9.0, smaller than that, 11.6, of the free ligand HL1.17 Spectral changes for 3cL2–3 as a function of pH are displayed in Fig. S17 and 18.† The emission intensity and wavelength of both complexes were relatively unaffected over the pH range from ca. 4 to 7.5. Further increases in pH led to a gradual increase in intensity by a factor of ca. 1.8 and 1.7 for 3cL2 and 3cL3, respectively. The pKa values of complexes 3cL2–3 are ca. 8.0 and 9.0, respectively, again, smaller than the ground state pKa of the free ligands (12.3 for HL2 and 11.6 for HL3).17
Overall, the phenylpyridine complexes 3cL1–3 show an increase in emission intensity with increasing pH, which is the opposite trend to that observed for the phenylpyrazole complexes 3aL1–3 described above. This suggests that different molecular orbitals are involved in the emission from the two series of complexes. The pKa values for the phenylpyridine complexes are lower than those for the phenylpyrazole complexes for L1 and L2 (by approximately 1 and 2 units respectively) whilst that for L3 is about the same. However, the pKa values are still rather high for use a biological pH sensors hence the CF3 substituted complex 3dL2 was examined and the results are shown in Fig. S19.†
For 3dL2 the emission intensity decreases at high pH whilst the opposite is true for the unsubstituted complex 3cL2. Hence, as found for the phenylpyrazole complexes addition of a CF3 substituent to the C^N ligand has the effect of reversing the slope of the pH curve. From the intensity changes a pKa value of 7.8 was determined (Fig. S19†), which is surprisingly similar to that of the unsubstituted complex (pKa 8.0 for 3cL2).
Conclusions
In conclusion, for all the complexes studied changes in pH gave only very minor changes in the associated UV-vis spectra over the full pH range studied. Hence, determining the ground state pKa was not feasible. In contrast, the emission intensity of the complexes was significantly modulated (by a factor of 1.2 to 2.9) by altering the pH. Surprisingly the phenylpyrazole complexes 3aL1–3 showed an intensity decrease with increasing pH (switch off) whilst the phenylpyridine complexes 3cL1–3 showed an increase in emission intensity with increasing pH. Changing the R group on the N^N ligand had an effect of up to 1 pH unit on the subsequent pKa values. However, the trends were not the same for the different series of complexes. More surprisingly, putting CF3 groups on the cyclometallated phenyls, i.e., complexes 3bL1–3 and 3dL2, resulted in a complete reversal in the direction of the intensity change when compared to their respective unsubstituted congeners. The unsubstituted complexes had pKa values ranging from 8.0 to 10.0 and as anticipated, adding the electron-withdrawing group reduced the pKa of the complexes to 6.8–7.8, thereby extending to useful pKa range.However, whilst adding CF3 substituents had the desired effect on the pKa, in general it tended to reduce the proportional change in emission intensity per unit change in pH. The reason(s) for this difference are not currently known. It is hoped that these studies will provide useful information for the design of other pH sensitive cyclometallated iridium complexes.
Experimental
All reactions were carried out under an inert atmosphere of nitrogen unless otherwise mentioned though all work-up was carried out in air. Microwave reactions were carried out in a CEM Explorer hybrid 12 microwave synthesiser, solutions were degassed by bubbling nitrogen through the solution for 3 minutes before sealing the tube with a plastic cap. NMR spectra were recorded on a Bruker DRX400 spectrometer operating at 400.13 (1H), 376.50 (19F), 100.61 MHz (13C) and or a Bruker DRX500 spectrometer at 500 (1H) and 125 MHz (13C) respectively at ambient temperature; chemical shifts (ppm) are referred to the residual protic solvent peaks and coupling constants are expressed in Hertz (Hz). Assignments of 1H and 13C NMR signals were made where possible, using COSY, NOESY, TOCSY, HMQC, HMBC, DEPT and APT spectra. Electrospray (ES) and ASAP mass spectra were obtained using a micromass Qtra LC spectrometer in HPLC grade MeCN. The UV/Vis spectra measurements were carried out on a Shimadzu UV-1600 series spectrometer in 1 cm quartz cuvette at room temperature. Luminescence studies were performed on a Jobin Yvon Horiba Fluoromax–P spectrofluorimeter, in either reagent grade DCM or MeOH, HPLC grade MeCN or spectroscopy grade DMF (N,N-Dimethylformamide). Complexes were excited at a wavelength between 320–405 nm using a filter of 370, 399 or 450 nm. The pH values were determined using a Jenway 3510 pH meter, calibrated prior to use with standard buffer solutions of pH 4, 6 and 10 at room temperature. The pKa values for all complexes were determined by the change of emission intensity upon the pH changing from about ca. 2.5 to 12.5.2-(1H-pyrazol-3-yl)pyridine ligands HL1–3 were prepared by literature methods24 as were 1-(4-(trifluoromethyl)phenyl)-1H-pyrazole25 and 2-(4-(trifluoromethyl)phenyl)pyridine26 and the bis-cyclometallated dimers 1a–d18 and the data are consistent with those published.18,19
General procedure for the synthesis of cationic complexes [Ir(C^N)2(HN^N)]PF6
The relevant dimer 1a, c, ligand (HL1/HL2/HL3) (2.2–2.4 equiv.) and KPF6 (2.4 equiv.) were placed in a microwave vial and methanol (3 ml) was added. After degassing with nitrogen the tube was heated under microwave irradiation at 60 °C for 20–40 min. After this time, the solvent was removed in vacuo leaving behind a solid which was dissolved in DCM (15 ml) and passed through Celite. The filtrate was reduced in volume and hexane was added slowly to induce precipitation. The precipitate was isolated, washed with hexane and dried in vacuo. Samples could be recrystallised from DCM/hexane.General procedure for the synthesis of neutral complexes [Ir(C^N)2(N^N)]
A mixture of the ligand (HL1/HL2/HL3) (2.2–2.4 equiv.) and an equimolar amount of NaOMe in MeOH (3 ml) was warmed gently at 40 °C for 15 min. A solution of the appropriate dimer 1a–d (1 equiv.) in DCM (6 ml) was added and the mixture was stirred for 2–4 h at room temperature. After this time, the solvent was removed in vacuo and the residue was dissolved in DCM (15 ml) and passed through Celite. The filtrate was reduced in volume and hexane was added slowly to induce precipitation. The precipitate was isolated, washed with hexane and dried in vacuo. Samples could be recrystallised from DCM/hexane.For all complexes the crude yields after washing but before recrystallisation are reported.
:1) to give a grey yellowish solid (68 mg, 82%). 1H NMR (500 MHz, CD3CN, 298 K): δ 8.41 (1H, d, J = 2.7 Hz, H5B), 8.36 (1H, d, J = 2.7 Hz, H5A), 8.22 (1H, br. d, J = 7.8 Hz, He), 8.10 (1H, td, J = 7.8, 1.1 Hz, Hf), 7.99 (1H, br. d, J = 5.3 Hz, Hh), 7.74 (2H, br. d, J = 6.9 Hz, Hj,k), 7.55–7.46 (6H, m, Hb,l,m,n,4A,4B), 7.38 (1H, br. t, J = 6.2 Hz, Hg), 7.21 (1H, d, J = 2.1 Hz, H7A), 7.10–7.04 (3H, m, H3A,3B,7B), 6.89 (1H, t J = 7.3 Hz, H2A/2B), 6.86 (1H, t J = 7.3 Hz, H2A/2B), 6.63 (1H, t, J = 2.5 Hz, H6A), 6.61 (1H, t, J = 2.5 Hz, H6B), 6.32 (1H, d, J = 7.3 Hz, H1B), 6.28 (1H, d, J = 7.3 Hz, H1A). 13C NMR: (125 MHz, CD3CN, 298 K): δ 155.0 (Cq), 153.2 (Cq), 151.6 (Ch), 149.0 (Cq), 145.0 (Cq), 144.6 (Cq), 140.7 (Cf), 140.3 (C7A), 139.7 (C7B), 135.3 (C1A/1B), 134.0 (C1A/1B), 132.2 (Cq), 130.8 (Cn), 130.2 (2Cl,m), 129.0 (Cq), 128.8 (C5A), 128.7 (C5B), 128.4 (Cq), 127.7 (2Cj,k), 127.5 (C2A/2B), 127.2 (C2A/2B), 127.0 (Cg), 124.4 (C3A/3B), 124.1 (C3A/3B), 124.0 (Ce), 113.0 (C4A/4B), 112.6 (C4A/4B), 109.3 (C6B), 109.1 (C6A), 104.0 (Cb). HRMS (ASAP): m/z 700.1821 [700.1801 calculated for C32H25193IrN7].
:1), respectively. 3aL1 was isolated as a grey yellow solid (45 mg, 76%). 1H NMR: (500 MHz, CD3CN, 298 K): δ 8.29 (1H, d, J = 2.7 Hz, H5B), 8.26 (1H, d, J = 2.7 Hz, H5A), 7.90–7.86 (2H, m Hz, Hh,e), 7.83 (1H, td, J = 7.7, 1.5 Hz, Hf), 7.47 (1H, d, J = 2.1 Hz, Ha), 7.39 (1H, d, J = 7.6 Hz, H4A), 7.35 (1H, d, J = 7.8 Hz, H4B), 7.06 (1H, ddd, J = 7.6, 5.6, 1.2 Hz, Hg), 7.00–6.95 (2H, m, H7B, H3A), 6.91 (1H, td, J = 7.7, 1.1 Hz, H3B), 6.82–6.80 (2H, m, Hb, H2A), 6.72–6.69 (2H, m, H2B,7A), 6.53 (1H, t, J = 2.5 Hz, H6B), 6.50 (1H, t, J = 2.5 Hz, H6A), 6.34 (1H, dd, J = 7.3, 1.0 Hz, H1A), 6.22 (1H, dd, J = 7.4, 1.0 Hz, H1B). 13C NMR (125 MHz, MeOD, 298 K): δ 154.4 (Cc), 154.1 (Cd), 151.7 (Ch), 145.2 (C8A/8B), 145.1 (C8A/8B), 140.9 (Cf), 140.0 (C7A/7B), 139.5 (C7A/7B), 135.2 (C1B), 134.9 (Ca), 134.4 (C1A), 132.7 (C9A/9B), 129.2 (C9A/9B), 128.9 (C5A), 128.8 (C5B), 127.7 (C2A/2B), 127.3 (C2A/2B), 127.1 (Cg), 124.5 (C3A/3B), 124.3 (C3A/3B), 124.0 (Ce), 113.0 (C4A/4B), 112.8 (C4A/4B), 109.5 (C6A/6B), 109.3 (C6A/6B), 106.4 (Cb). HRMS (ASAP): [M + H]+, m/z 624.1489 [624.1488 calculated for C26H21193IrN7].
:6 ml). After work up, the compound was isolated as yellow solid (72 mg, 73%). For the 1H NMR spectrum assignments are given where possible but there are many overlapping peaks. 1H NMR (500 MHz, CDCl3, 300 K): δ 7.83–7.80 (3H, m), 7.74 (1H, td, J = 7.7, 1.3 Hz, Hf), 7.66–7.58 (6H, m), 7.51 (1H, m), 6.99–6.93 (4H, m, 3 × Ph, Hg), 6.87–6.84 (3H, m), 6.61 (1H, s, Hb), 6.35 (1H, dd, J = 7.5, 0.5 Hz, H1B), 6.27 (1H, dd, J = 7.5, 0.8 Hz, H1A), 1.29 (9H, s, HtBu). 13C NMR (100 MHz, CDCl3, 300 K): δ 168.4 (Cq), 167.8 (Cq), 150.4 (CH), 149.7 (Cq), 149.5 (CH), 148.3 (CH), 144.5 (Cq), 144.0 (Cq), 138.0 (CH), 137.0 (CH), 136.7 (CH), 132.3 (CH), 131.9 (CH), 130.2 (CH), 129.8 (CH), 124.4 (CH), 124.3 (CH), 123.0 (CH), 122.9 (CH 122.1 (CH), 121.8 (CH), 121.6 (CH), 120.5 (CH), 119.2 (CH), 118.7 (CH), 100.3 (Cb), 30.7 (CtBu). The quaternary carbon of the tBu was not observed. HRMS (ASAP): [M + H]+, m/z 702.2236 [702.2209 calculated for C34H31193IrN5].
:6 ml). After work up, the product was isolated as yellow solid (50 mg, 87%). The 1H NMR spectrum for this complex is very complicated with a lot of overlapping signals hence very few assignments are given. 1H NMR (500 MHz, CDCl3, 298 K): δ 7.83 (1H, bd, J = 5.5 Hz), 7.80–7.68 (5H, m), 7.66–7.52 (6H m), 7.45 (1H, td, J = 7.2, 1.1 Hz,), 7.24 (2H, t, J = 7.6 Hz), 7.11 (1H, br. t, J = 7.4 Hz), 7.01 (1H, s, Hb), 6.95 6.78 (6H, m), 6.75 (1H, td, J = 6.1, 0.9 Hz), 6.39 (1H, bd, J = 7.3 Hz, H1B), 6.33 (1H, bd, J = 7.2 Hz, H1A). Due to overlap no assignments are made 13C NMR (126 MHz, CDCl3, 298 K): δ ppm 168.6 (Cq), 168.1 (Cq), 155.7 (Cq), 155.1 (Cq), 153.4 (Cq), 151.0 (Cq), 150.6 (Cq), 150.4 (CH), 149.4 (CH), 148.2 (CH), 144.6 (Cq), 143.7 (Cq), 137.5 (CH) 136.7 (CH) 136.3 (CH) 134.6 (Cq), 132.1 (CH), 132.0 (CH), 130.1 (CH), 129.5 (CH), 128.3 (2 × CH), 126.2 (CH), 125.4 (2 × CH), 124.2 (CH), 124.1 (CH), 122.6 (CH), 122.0 (CH), 121.8 (CH), 121.4 (CH), 120.9 (CH), 119.6 (CH), 119.0 (CH), 118.4 (CH), 100.9 (CH). HRMS (ASAP): [M + H]+, m/z 722.1862 [722.1896 calculated for C36H27193IrN5].
Conflicts of interest
There are no conflicts to declare.Acknowledgements
One of us (NSYA) is grateful to the Omar Al-Mukhtar University Tobruk Libya and the Libyan Embassy in the UK for financial support and to the Libya Ministry of Education Programme for a scholarship.Notes and references
- (a) C. Y. S. Chung, S. P. Y. Li, M. W. Louie, K. K. W. Lo and V. W. W. Yam, Chem. Sci., 2013, 4, 2453–2462 RSC; (b) B. Tang, X. Liu, K. Xu, H. Huang, G. Yang and L. An, Chem. Commun., 2007, 3726–3728 RSC; (c) H. R. Kermis, Y. Kostov, P. Harms and G. Rao, Biotechnol. Prog., 2002, 18, 1047–1053 CrossRef CAS PubMed; (d) L. Cao, X. Li, S. Wang, S. Li, Y. Li and G. Yang, Chem. Commun., 2014, 50, 8787–8790 RSC; (e) M. Y. Wu, K. Li, Y. H. Liu, K. K. Yu, Y. M. Xie, X. D. Zhou and X. Q. Yu, Biomaterials, 2015, 53, 669–678 CrossRef CAS PubMed; (f) Y. Ge, A. Liu, J. Dong, G. Duan, X. Cao and F. Li, Sens. Actuators, B, 2017, 247, 46–52 CrossRef CAS.
- (a) K. Qiu, Y. Chen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2019, 378, 66–86 CrossRef CAS; (b) S. Das, D. Saha, S. Karmakar and S. Baitalik, J. Phys. Chem. A, 2012, 116, 5216–5226 CrossRef CAS PubMed; (c) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC; (d) B. V. Harbuzaru, A. Corma, F. Rey, J. L. Jordá, D. Ananias, L. D. Carlos and J. Rocha, Angew. Chem., Int. Ed., 2009, 48, 6476–6479 CrossRef CAS PubMed; (e) T.-T. Meng, H. Wang, Z.-B. Zheng and K.-Z. Wang, Inorg. Chem., 2017, 56, 4775–4779 CrossRef CAS PubMed; (f) V. W. W. Yam, V. K. M. Au and S. Y. L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (g) M. Bar, D. Maity, S. Deb, S. Das and S. Baitalik, Dalton Trans., 2017, 46, 12950–12963 RSC.
- (a) J. N. Demas and B. A. DeGraff, Anal. Chem., 1991, 63, 829A–837A CrossRef CAS; (b) J. N. Demas and B. A. DeGraff, J. Chem. Educ., 1997, 74, 690–695 CrossRef CAS.
- (a) B. Higgins, B. A. DeGraff and J. N. Demas, Inorg. Chem., 2005, 44, 6662–6669 CrossRef CAS PubMed; (b) H. J. Kim, Y.-C. Jeong, J. Heo, J. I. Rhee and K.-J. Hwang, Bull. Korean Chem. Soc., 2009, 30, 539–540 CrossRef CAS; (c) S. San Tan, S. Yanagisawa, K. Inagaki, Y. Morikawa and M. B. Kassim, Phys. Chem. Chem. Phys., 2017, 19, 25734–25745 RSC; (d) H. J. Yu, Z. F. Hao, H. L. Peng, R. H. Rao, M. Sun, R. Alana, W. C. Ran, H. Chao and L. Yu, Sens. Actuators, B, 2017, 252, 313–321 CrossRef CAS; (e) F. Yu, C. Shen, T. Zheng, W. K. Chu, J. Xiang, Y. Luo, C. C. Ko, Z. Q. Guo and T. C. Lau, Eur. J. Inorg. Chem., 2016, 2016, 3641–3648 CrossRef CAS.
- (a) T. Mizuno, T. Fukumatsu, M. Takeuchi and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 2000, 407–413 RSC; (b) M. H. W. Lam, D. Y. K. Lee, K. W. Man and C. S. W. Lau, J. Mater. Chem., 2000, 10, 1825–1828 RSC; (c) M. Cattaneo, F. Fagalde, C. D. Borsarelli and N. E. Katz, Inorg. Chem., 2009, 48, 3012–3017 CrossRef CAS PubMed; (d) T. W. Tseng, S. Mendiratta, T. T. Luo, T. W. Chen and Y. P. Lee, Inorg. Chim. Acta, 2018, 477, 312–317 CrossRef CAS.
- (a) K. M. C. Wong, W. S. Tang, X. X. Lu, N. Zhu and V. W. W. Yam, Inorg. Chem., 2005, 44, 1492–1498 CrossRef CAS PubMed; (b) J. L. L. Tsai, T. Zou, J. Liu, T. Chen, A. O. Y. Chan, C. Yang, C. N. Lok and C. M. Che, Chem. Sci., 2015, 6, 3823–3830 RSC.
- M. Licini and J. A. G. Williams, Chem. Commun., 1999, 1943–1944 RSC.
- (a) M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS; (b) M. K. Nazeeruddin, C. Klein, M. Grätzel, L. Zuppiroli and D. Berner, in Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 363–390 Search PubMed; (c) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS; (d) E. Longhi and L. De Cola, in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons Ltd, 2017, pp. 205–274 Search PubMed; (e) D. L. Ma, S. Lin, W. Wang, C. Yang and C. H. Leung, Chem. Sci., 2017, 8, 878–889 RSC; (f) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC; (g) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS; (h) Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC; (i) K. K. S. Tso and K. K. W. Lo, in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons Ltd, 2017, pp. 415–477 Search PubMed.
- A. Nakagawa, Y. Hisamatsu, S. Moromizato, M. Kohno and S. Aoki, Inorg. Chem., 2014, 53, 409–422 CrossRef CAS PubMed.
- A. Kando, Y. Hisamatsu, H. Ohwada, T. Itoh, S. Moromizato, M. Kohno and S. Aoki, Inorg. Chem., 2015, 54, 5342–5357 CrossRef CAS PubMed.
- J. Weng, Q. Mei, W. Jiang, Q. Fan, B. Tong, Q. Ling and W. Huang, Analyst, 2013, 138, 1689–1699 RSC.
- K. Qiu, L. Ke, X. Zhang, Y. Liu, T. W. Rees, L. Ji, J. Diao and H. Chao, Chem. Commun., 2018, 54, 2421–2424 RSC.
- M. Qian, Z. Wang, H. Qi and H. Qi, Inorg. Chem. Commun., 2019, 106, 95–98 CrossRef CAS.
- (a) D.-K. Cao, J.-S. Hu, M.-Q. Li, D.-P. Gong, X.-X. Li and M. D. Ward, Dalton Trans., 2015, 44, 21008–21015 RSC; (b) A. J. Hallett, B. D. Ward, B. M. Kariuki and S. J. A. Pope, J. Organomet. Chem., 2010, 695, 2401–2409 CrossRef CAS; (c) A. F. Henwood, Y. Hu, M. T. Sajjad, G. K. V. V. Thalluri, S. S. Ghosh, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel, N. Robertson and E. Zysman-Colman, Chem. – Eur. J., 2015, 21, 19128–19135 CrossRef CAS PubMed; (d) L. Murphy, A. Congreve, L. O. Palsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743–8745 RSC; (e) S. A. Rommel, D. Sorsche, N. Rockstroh, F. W. Heinemann, J. Kübel, M. Wächtler, B. Dietzek and S. Rau, Eur. J. Inorg. Chem., 2015, 2015, 3730–3739 CrossRef CAS.
- L. He, Y. Li, C. P. Tan, R. R. Ye, M. H. Chen, J. J. Cao, L. N. Ji and Z. W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- (a) C.-K. Koo, Y.-M. Ho, C.-F. Chow, M. H.-W. Lam, T.-C. Lau and W.-Y. Wong, Inorg. Chem., 2007, 46, 3603–3612 CrossRef CAS PubMed; (b) C. K. Koo, K. L. Wong, C. W. Y. Man, Y. W. Lam, L. K. Y. So, H. L. Tam, S. W. Tsao, K. W. Cheah, K. C. Lau, Y. Y. Yang, J. C. Chen and M. H. W. Lam, Inorg. Chem., 2009, 48, 872–878 CrossRef CAS PubMed; (c) C.-K. Koo, K.-L. Wong, C. W.-Y. Man, H.-L. Tam, S.-W. Tsao, K.-W. Cheah and M. H.-W. Lam, Inorg. Chem., 2009, 48, 7501–7503 CrossRef CAS PubMed.
- T. Y. Lin, K. C. Tang, S. H. Yang, J. Y. Shen, Y. M. Cheng, H. A. Pan, Y. Chi and P. T. Chou, J. Phys. Chem. A, 2012, 116, 4438–4444 CrossRef CAS PubMed.
- D. L. Davies, M. P. Lowe, K. S. Ryder, K. Singh and S. Singh, Dalton Trans., 2011, 40, 1028–1030 RSC.
- N. M. Shavaleev, F. Monti, R. Scopelliti, N. Armaroli, M. Grätzel and M. K. Nazeeruddin, Organometallics, 2012, 31, 6288–6296 CrossRef CAS.
- P. J. Spellane, R. J. Watts and C. J. Curtis, Inorg. Chem., 1983, 22, 4060–4062 CrossRef CAS.
- (a) W. Jiang, Y. Gao, Y. Sun, F. Ding, Y. Xu, Z. Bian, F. Li, J. Bian and C. Huang, Inorg. Chem., 2010, 49, 3252–3260 CrossRef CAS PubMed; (b) F. Neve, M. La Deda, A. Crispini, A. Bellusci, F. Puntoriero and S. Campagna, Organometallics, 2004, 23, 5856–5863 CrossRef CAS; (c) E. Orselli, G. S. Kottas, A. E. Konradsson, P. Coppo, R. Fröhlich, L. De Cola, A. van Dijken, M. Büchel and H. Börner, Inorg. Chem., 2007, 46, 11082–11093 CrossRef CAS PubMed; (d) Q. Zhao, S. Liu, M. Shi, C. Wang, M. Yu, L. Li, F. Li, T. Yi and C. Huang, Inorg. Chem., 2006, 45, 6152–6160 CrossRef CAS PubMed.
- (a) M. K. Nazeeruddin, R. T. Wegh, Z. Zhou, C. Klein, Q. Wang, F. De Angelis, S. Fantacci and M. Grätzel, Inorg. Chem., 2006, 45, 9245–9250 CrossRef CAS PubMed; (b) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- We have called the pKa determined from the fluorescence titration the emission pKa. The difference between this and the excited state pKa is explained in J. G. Vos, Polyhedron, 1992, 11, 2285–2299 CrossRef CAS.
- (a) W. S. Yu, C. C. Cheng, Y. M. Cheng, P. C. Wu, Y. H. Song, Y. Chi and P. T. Chou, J. Am. Chem. Soc., 2003, 125, 10800–10801 CrossRef CAS PubMed; (b) X. Y. Yu, L. Deng, B. Zheng, B. R. Zeng, P. Yi and X. Xu, Dalton Trans., 2014, 43, 1524–1533 RSC.
- M. Pichette Drapeau, T. Ollevier and M. Taillefer, Chem. – Eur. J., 2014, 20, 5231–5236 CrossRef CAS PubMed.
- C. Liu, X. Lv, Y. Xing and J. Qiu, J. Mater. Chem. C, 2015, 3, 8010–8017 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data (CIF), extra molecular structures, absorption and emission spectra and pH titrations, NMR spectra. CCDC 2014697–2014704. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02434a |
| ‡ Current address, Chemistry Department, Faculty of Science, Omar Al-Mukhtar University, Torbruk, Libya. |
| This journal is © The Royal Society of Chemistry 2020 |