
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of alkaline-earth stannanide complexes by hydride-mediated distannane metathesis and organostannane dehydrogenation†
Louis J.
Morris
a,
Nasir A.
Rajabi
 a,
Mary F.
Mahon
a,
Ian
Manners
b,
Claire L.
McMullin
*a and
Michael S.
Hill
*a
a,
Mary F.
Mahon
a,
Ian
Manners
b,
Claire L.
McMullin
*a and
Michael S.
Hill
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
bDepartment of Chemistry, University of Victoria, Victoria BC V8P 5C2, Canada
First published on 9th July 2020
Abstract
The synthesis of heteroleptic complexes with calcium– and magnesium–tin bonds is described. The dimeric β-diketiminato calcium hydride complex, [(BDI)Ca(μ-H)]2 (ICa) reacts with Ph3Sn–SnPh3 to provide the previously reported μ2-H bridged calcium stannanide dimer, [(BDI)2Ca2(SnPh3)(μ-H)] (3). Computational assessment of this reaction supports a mechanism involving a hypervalent stannate intermediate formed by nucleophilic attack of hydride on the distannane. Monomeric calcium stannanides, [(BDI)Ca(SnPh3)·OPPh3] (8·OPPh3) and [(BDI)Ca(SnPh3)·TMTHF] (8·TMTHF, TMTHF = 2,2,5,5-tetramethyltetrahydrofuran) were obtained from ICa and Ph3Sn–SnPh3, after addition OPPh3 or TMTHF. Both complexes were also synthesised by deprotonation of Ph3SnH by ICa in the presence of the Lewis base. The calcium and magnesium THF adducts, [(BDI)Ca(SnPh3)·THF2] (8·THF2) and [(BDI)Mg(SnPh3)·THF] (9·THF), were similarly prepared from [(BDI)Ca(μ-H)·(THF)]2 (ICa·THF2) or [(BDI)Mg(μ-H)]2 (IMg) and Ph3SnH. An excess of THF or TMTHF was essential in order to obtain 8·TMTHF, 8·THF2 and 9·THF in high yields whilst avoiding redistribution of the phenyl-tin ligand. The resulting Ae–Sn complexes were used as a source of [Ph3Sn]− in salt metathesis, to provide the known tristannane Ph3Sn–Sn(t-Bu)2–SnPh3 (11). Nucleophilic addition or insertion with N,N′-di-iso-propylcarbodiimide provided the stannyl-amidinate complexes, [(BDI)Mg{(iPrN)2CSnPh3}] (12) and [(BDI)Ca{(iPrN)2CSnPh3}·L] (13·TMTHF, 13·THF, L = TMTHF, THF). The reactions and products were monitored and characterised by multinuclear NMR spectroscopy, whilst for compounds 8, 9, 12, and 13·THF, the X-ray crystal structures are presented and discussed.
Introduction
Although Grignard's ubiquitous organomagnesium compounds have been widely used as synthetic reagents for over a century, the catalytic potential of alkaline-earth (Ae) reagents was largely overlooked until the past two decades.1,2 By analogy to well-established lanthanide(III)-based catalysis,3 Ae2+ centres participate in redox-neutral catalytic cycles that are assembled from fundamental steps such as polarised 2σ–2π insertion- and 2σ–2σ metathesis.2 In many cases, reactivity is better described by non-concerted processes involving attack of an Ae-bound nucleophile on a substrate, such as a silane, that is capable of expanding its coordination sphere.4–7 As such, the heavier alkaline earths (Mg–Ba) are adept at mediating catalytic dehydrocoupling,5,8–15 hydrofunctionalisation,16–23 and even reductive hydrogenation reactions.24–26 We have previously reported the use of silylboranes to perform the catalytic ‘disilacoupling’ of amines and boranes; a non-dehydrogenative process thought to be dependent on Ae-mediated redox-neutral metathesis of N–H and Si–B σ-bonds (Scheme 1, top).27 A model reaction between a β-diketiminato (BDI) magnesium butyl complex and the silylborane, PhMe2Si-Bpin (Bpin = pinacolatoboryl), resulted in elimination of nBu-Bpin and isolation of the magnesium silanide complex, 1 (Scheme 1, bottom).27 Computational assessment has suggested that this reaction is best described by nucleophilic attack of a butyl group on the boron centre to provide a borate intermediate from which the silyl group is subsequently transferred to magnesium.28 Bis(pinacolato)diboron (B2pin2), which contains a non-polar B–B σ-bond, was shown to react in a similar way with [(BDI)MgBu] to provide an isolable diboranate complex, 2a (Scheme 1, bottom). Treatment of 2a with 4-dimethylaminopyridine (DMAP) promoted heterolysis of the B–B bond and delivered the terminal magnesium boryl species, 2b, which is a source of the nucleophilic [Bpin]− anion.29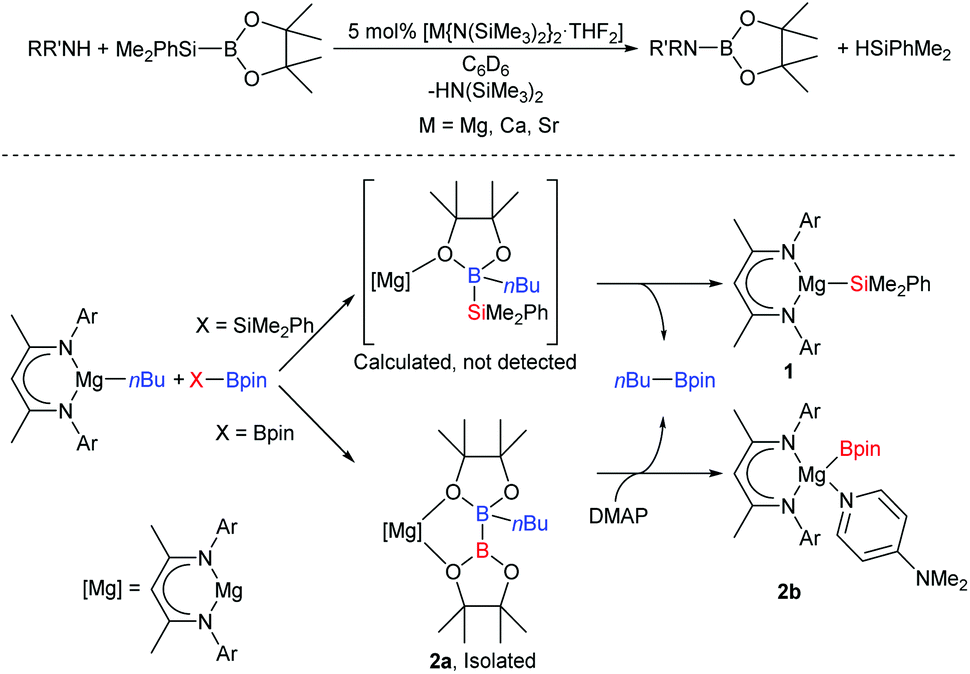 | ||
| Scheme 1 Top, alkaline-earth mediated boron–nitrogen ‘desilacoupling’; bottom, synthesis of magnesium silyl and boryl complexes from a nucleophilic alkyl-magnesium complex.27–29 Ar = Dipp = 2,6-di-isopropylphenyl, DMAP = 4-dimethylaminopyridine. | ||
The Ae-centred manipulation of boron-, silicon-, and organic substrates has, thus, received significant attention. In contrast, comparable reports of Ae-mediated reactivity suitable for the construction of catalytic cycles involving organostannanes, which could provide an attractive route towards materials such as polystannanes30,31 or act as sources of organostannane cross-coupling reagents,32,33 are lacking. The majority of published Ae-mediated organotin chemistry focusses on the irreversible, stoichiometric reaction between the group 2 element and organotin halides, distannanes and silastannanes.34–41 We recently reported that the BDI-calcium stannanide complexes 3 and 4 may be accessed through deprotonation of commercially available triphenylstannane by the soluble calcium hydride complex, ICa (Scheme 2a).42 Crystallographically characterised examples of Ae–Sn bonds were previously limited to the calcium and magnesium complexes 5 and 6, and the barium species 7 (Scheme 2b). Compound 5 was readily prepared by the oxidative-addition of hexamethyldistannane to calcium metal,43 the synthesis of 6 and 7 utilised salt metathesis routes from group 1-metallated precursors.44–46 Neither of these strategies, however, is likely to be amenable to incorporation into catalytic cycles. Since the formation of 3 and 4 is redox neutral at calcium and generates H2 instead of insoluble salts as a by-product, therefore, it holds attractive potential for the development of Ae-based catalysts for processes such as hydrostannylation or stannane dehydrocoupling.
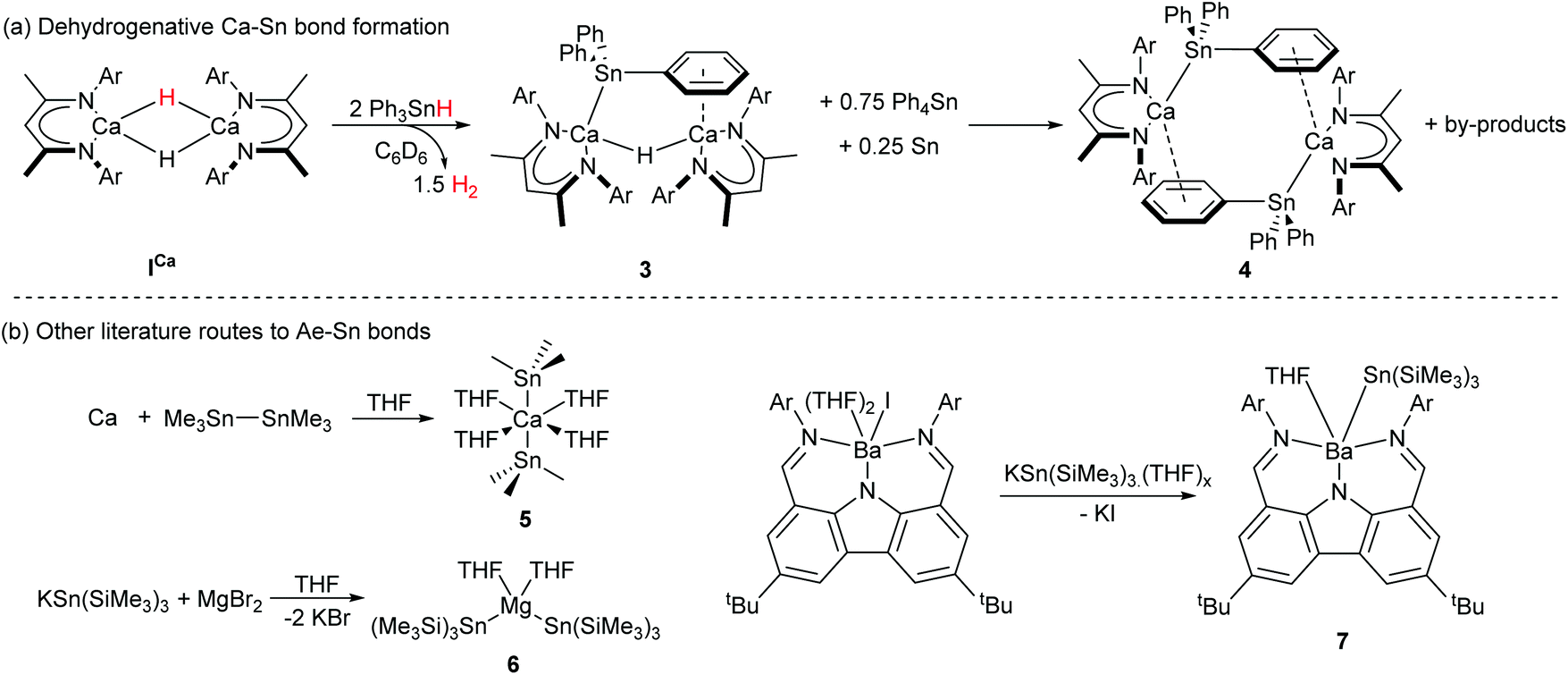 | ||
| Scheme 2 (a) Dehydrogenative synthesis of Ca–Sn bonded compounds 3 and 4.42 (b) Crystallographically characterised literature compounds containing Ae–Sn bonds prepared by salt-metathesis44–46 or oxidative addition routes.43 Ar = 2,6-di-isopropylphenyl. | ||
Distannanes are synthetically useful precursors to organotin radicals,47–53 as well as 1,2-distannylated alkanes and alkenes via transition metal-catalysed distannylation of alkenes and alkynes.54 Such organotin compounds are valuable cross-coupling reagents in organic synthesis.49,55 Although the heterogeneous reaction of distannanes with solid alkali56 and alkaline-earth41,43 metals is well-known, the manipulation of distannanes by soluble s-block complexes has not been described. By analogy to the nucleophilic substitution-like process operative in the formation of Mg-silyl and -boryl species 1 and 2a/2b, we speculated that molecular calcium hydride and alkyl derivatives may react with Ph3Sn–SnPh3, providing an alternative route to nucleophilic calcium stannanide complexes. These investigations were motivated by the limitations encountered during our previously described synthesis of 3 and 4.42 Firstly, ICa also promotes redistribution of the organotin substrate, culminating in the generation of homoleptic SnPh4 and (presumably) SnH4, the latter of which rapidly decomposes to give Sn(0) and H2. Secondly, the strongly bound dimer of 3 retains a μ2-hydride ligand and the subsequent formation of 4 is low-yielding and slow, impeding any rational assessment of the reactivity of these unusual compounds. In this contribution, therefore, we describe the facile and high yielding synthesis of well-defined, monomeric Ae-stannanide complexes and a preliminary assessment of their nucleophilic reactivity.
Results and discussion
Reaction of ICa with Ph3Sn–SnPh3 and synthesis of compound 3
When ICa was dissolved in C6D6 with an equimolar quantity of Ph3Sn–SnPh3, the reaction mixture bubbled gently and darkened from pale-yellow to orange brown over the course of six hours. The respective μ-hydride and BDI-γ-CH proton resonances of ICa at δ 4.27 and 4.83 ppm in the in situ1H NMR spectrum were replaced by two new singlets of relative intensity 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at δ 4.75 and 3.83 ppm. The latter signal displayed unresolved 117/119Sn satellites with 2J(117/119Sn–1H) = 94 Hz, while the corresponding 119Sn{1H} NMR spectrum revealed complete consumption of the distannane and the appearance of a signal at δ −139.8 ppm, which was accompanied by the generation of Ph4Sn (δ −126 ppm).57 These observations were consistent with the formation of the μ-H-bridged dimeric calcium stannanide, 3, whilst the brown colouration was assigned to formation of colloidal tin.42 Although the slow formation of compound 4 was identified by its resonance at δ −158.5 ppm in the 119Sn{1H} NMR spectrum after a further five days at room temperature, complete conversion to this product was not obtained (Scheme 3).
:1 at δ 4.75 and 3.83 ppm. The latter signal displayed unresolved 117/119Sn satellites with 2J(117/119Sn–1H) = 94 Hz, while the corresponding 119Sn{1H} NMR spectrum revealed complete consumption of the distannane and the appearance of a signal at δ −139.8 ppm, which was accompanied by the generation of Ph4Sn (δ −126 ppm).57 These observations were consistent with the formation of the μ-H-bridged dimeric calcium stannanide, 3, whilst the brown colouration was assigned to formation of colloidal tin.42 Although the slow formation of compound 4 was identified by its resonance at δ −158.5 ppm in the 119Sn{1H} NMR spectrum after a further five days at room temperature, complete conversion to this product was not obtained (Scheme 3).
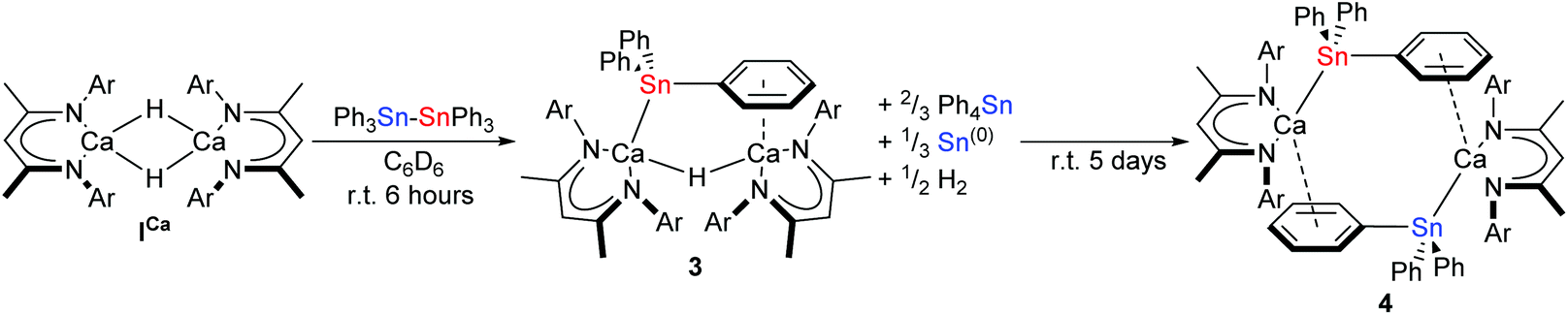 | ||
| Scheme 3 Synthesis of 3 and 4 by reaction of ICa with Ph3Sn–SnPh3 in C6D6. Ar = 2,6-di-isopropylphenyl. | ||
Computational and mechanistic investigation of ICa mediated Ph3Sn–SnPh3 activation
In order to assess the mechanism of Ph3Sn–SnPh3 activation, the reaction between ICa and Ph3Sn–SnPh3 was investigated by density functional theory (DFT, Fig. 1a, BP86 optimised, see ESI† for full details of computational methodology). Although we cannot, at this juncture, discount the operation of competitive single electron-based processes, consistent with the reported reactivity of compound ICa thus far,25 these calculations are suggestive of a metathesis-based reactivity. Following the initial formation of a van der Waals encounter complex (A, ΔG = +8.7 kcal mol−1), the distannane is subjected to nucleophilic attack by one of the μ2-hydride ligands (Ha) via transition state TSAB (Fig. 1b, ΔG‡ = +12.7 kcal mol−1), at which the Sna–Snb bond is marginally elongated from 2.85 Å (calculated for Ph3Sn–SnPh3) to 2.87 Å. Inspection of the Caa–Ha and Ha–Sna bond lengths (2.30 Å and 2.14 Å, respectively) in the subsequent intermediate, B (Fig. 1c, ΔG = +5.2 kcal mol−1), is suggestive of the transfer of Ha to Sna and the formation of a hypervalent stannate anion with a Sna–Snb distance of 2.95 Å. The Sna–Snb distance elongates to 3.55 Å in the transition state TSBC (ΔG‡ = +8.2 kcal mol−1), facilitating cleavage of the stannate anion and concerted formation of a Caa–Snb bond (distance in TSBC = 2.24 Å) to give intermediate C (ΔG = +2.5 kcal mol−1). Subsequent dissociation of Ph3SnaHa provides 3, at ΔG = −5.2 kcal mol−1. Whilst the overall process is only moderately exergonic, the modest kinetic barrier is consistent with the room temperature reaction conditions. Meanwhile, rapid consumption of the resultant molecule of Ph3SnH provides a thermodynamic driving force, yielding H2 and a second molecule of 3.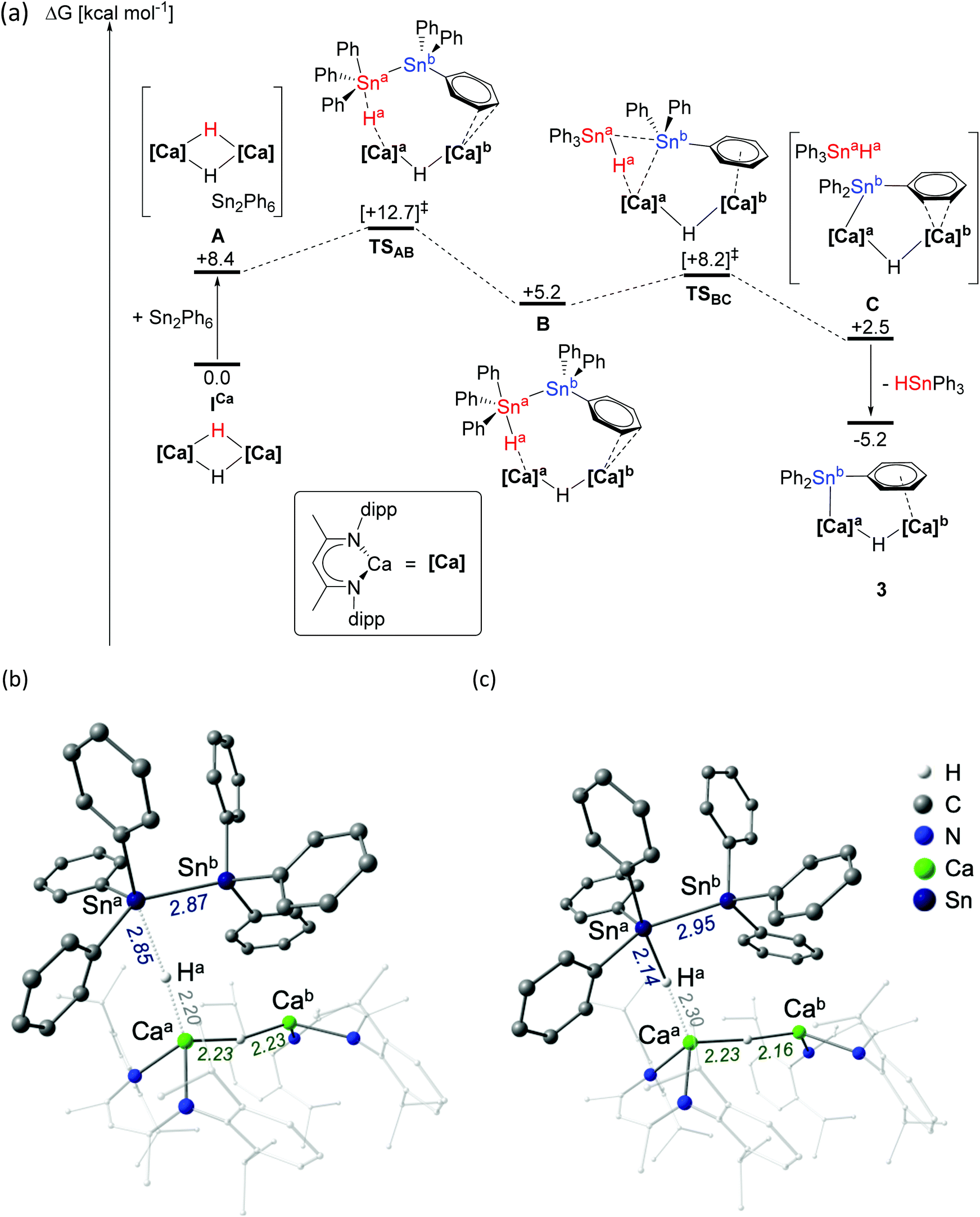 | ||
| Fig. 1 (a) DFT calculated free energy surface (BP86-D3(BJ)-benzene/BS2//BP86/BS1, kcal mol−1) for the reaction of ICa with (Ph3Sn)2; structures of (b) transition state TSAB and (c) intermediate B. See ESI† for full details. | ||
Experimental evidence in support of this mechanism was obtained by carrying out the analogous reaction between Ph3Sn–SnPh3 and the n-hexyl-calcium complex [(BDI)Ca(Hex)]2 (II). The relatively poor solubility of both substrates in C6D6 and the greater steric demand of the hexyl ligand compared to the hydride of ICa resulted in sluggish reaction kinetics. Nevertheless, after gentle heating to 40 °C for 48 hours, the characteristic triplet corresponding to the α-CH2 protons of II at δ −0.71 ppm was all but absent from the 1H NMR spectrum. Although this observation was accompanied by almost complete redistribution to [(BDI)2Ca]58 as the only soluble BDI-containing product, a resonance at δ −98.2 ppm in the corresponding 119Sn{1H} NMR spectrum revealed Ph3Sn(Hex) as the predominant tin-containing species.59 The absence of any unambiguously identifiable alkyl or stannyl-calcium species, such as a n-hexyl-containing analogue of 3, may be attributed to the likely low thermal stability of such intermediates. Formation of Ph3Sn(Hex), however, can be rationalised by attack of a calcium-bound n-hexyl-nucleophile on the distannane, with subsequent transfer of [Ph3Sn]− to calcium and elimination of Ph3Sn(Hex) (Scheme 4). Whereas the tetraorganostannane is inert towards further reactivity under these conditions, a similar reaction with ICa would yield Ph3SnH, which is rapidly deprotonated by a second molecule of ICa to provide 3.
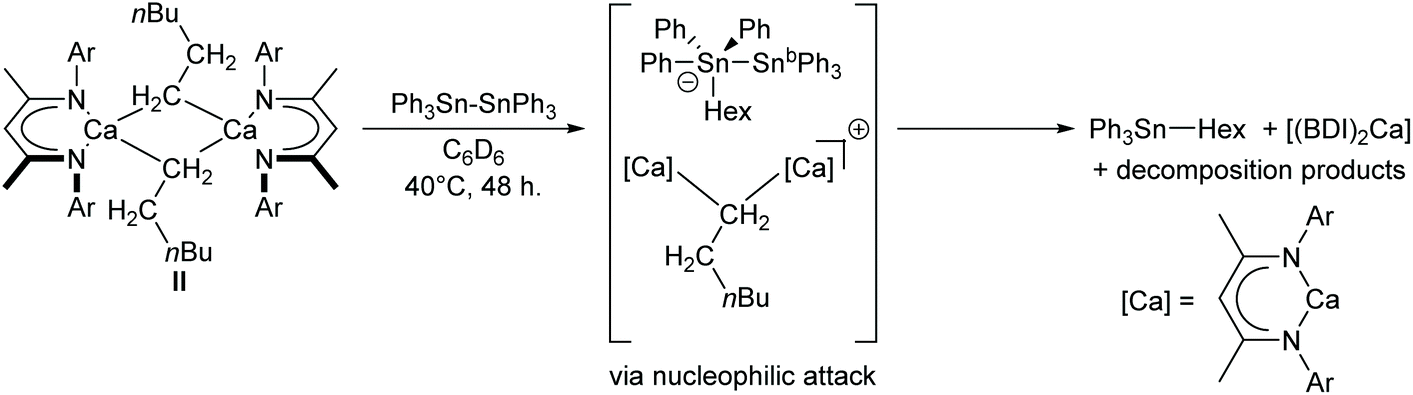 | ||
| Scheme 4 Postulated mechanism for the reaction between II and Ph3Sn–SnPh3. Ar = 2,6-di-isopropylphenyl. | ||
Synthesis and NMR characterisation of monomeric alkaline-earth stannanides 8 and 9·THF
Compound 4 could only be accessed in low yields following fractional crystallisation of the crude products obtained from reaction of ICa with Ph3SnH42 or Ph3Sn–SnPh3. It was also anticipated that the μ2-hydride of 3 would provide a likely complication in subsequent efforts to assess the reactivity of the Ca–Sn bond. With this in mind, we speculated that addition of a Lewis base would encourage fragmentation of the dimer, result in reaction of both hydride ligands, and provide a high-yielding route towards a well-defined monomeric calcium stannanide. Similar strategies have previously been applied successfully to achieve, for example, the isolation of monomeric magnesium complexes comprising terminal hydride and boryl ligands.29,60,61To this end, the reaction between ICa and Ph3Sn–SnPh3 was repeated and, after quantitative conversion of ICa was ascertained by 1H NMR spectroscopy, an equimolar equivalent of Ph3PO was added to the in situ generated solution of 3 (Scheme 5). Upon standing at room temperature for 24 hours, the reaction mixture took on an opaque dark-brown appearance and, in addition to several minor species, a major new BDI-γ-CH resonance was observed to have emerged at δ 5.23 ppm. The 119Sn{1H} NMR spectrum displayed a doublet at δ −146 ppm, whose coupling constant of 10 Hz is consistent with the sparse number of 3J(31P–117/119Sn) coupling constants that have been reported.62,63 Notwithstanding some minor peaks at δ 88–89 ppm and 71 ppm, consistent with this observation, the 31P{1H} NMR spectrum was free of evidence for any unligated phosphine oxide. The spectrum also comprised a major resonance at δ 36.4 ppm, which displayed unresolved 117/119Sn satellites with an approximate coupling constant consistent with that observed in the 119Sn{1H} NMR spectrum. Recrystallisation of the crude product mixture from toluene/hexane provided single-crystals of the monomeric Ph3PO-adduct 8·OPPh3 in low yield, from which the molecular structure was determined by X-ray diffraction analysis (Fig. 2a). Crystals of the known compound [(BDI)Ca(OPPh2)]2 were also obtained from the same sample and identified from the unit cell-parameters determined by X-ray diffraction.64 This observation is consistent with the calcium hydride-mediated reduction chemistry previously reported for phosphine oxides,64 and helps to account for the low yield and poor selectivity of this reaction. Compound 8·OPPh3 was, however, obtained cleanly from the single-step reaction of ICa with two equivalents each of Ph3SnH and Ph3PO in C6D6. Although high solubility of the crystalline product obtained from this reaction provided a low, unoptimised isolated yield, it displayed identical NMR resonances to those described above.
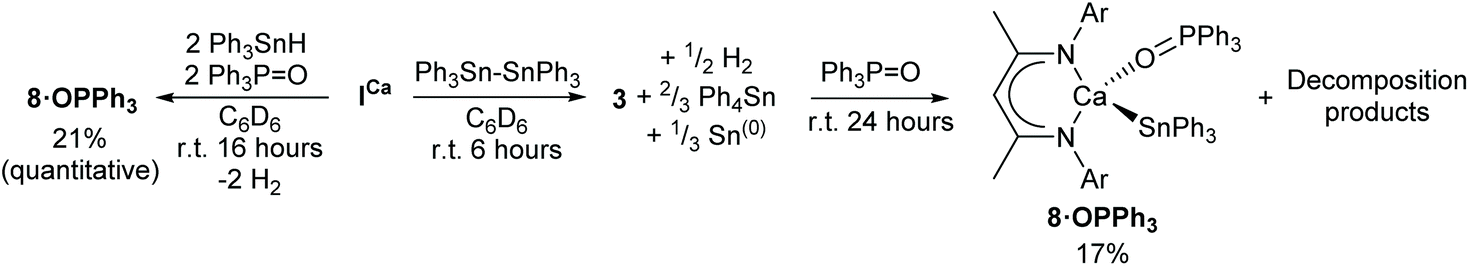 | ||
| Scheme 5 Synthesis of the Ph3PO-adduct, 8·OPPh3. Yields refer to unoptimised crystalline yield, those in parentheses refer to % spectroscopic conversion determined by in situ1H NMR spectroscopy. Ar = 2,6-di-isopropylphenyl. | ||
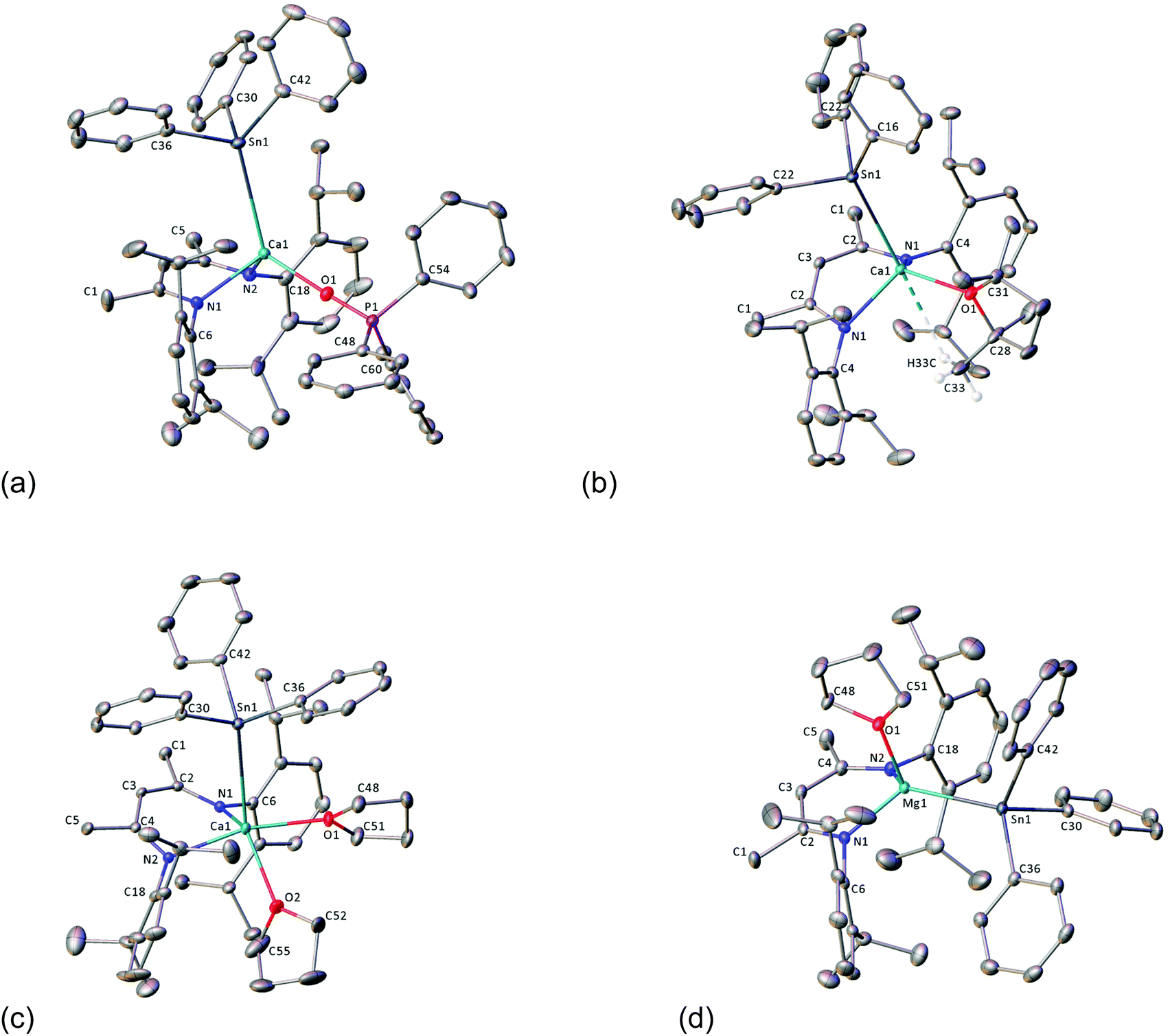 | ||
| Fig. 2 X-ray crystal structures of (a) 8·OPPh3, (b) 8·TMTHF, (c) 8·THF2, (d) 9·THF. Ellipsoids are shown at the 30% probability level and hydrogen atoms are omitted for clarity except for those bound to C33 in 8·TMTHF. Where disorder is present only the major component is shown. | ||
Mindful of phosphine oxide reactivity towards reductive and/or nucleophilic alkaline-earth complexes,61,64 it was decided that 2,2,5,5-tetramethyltetrahydrofuran (TMTHF) would be a better choice of Lewis-base. Westerhausen and co-workers have recently reported the use of TMTHF to prepare monomeric amide complexes [Ae{N(SiMe3)2}2·TMTHF] (Ae = Mg, Ca, Sr, Ba), in which the TMTHF ligand is highly labile in solution.65 We reasoned that, whilst coordination of TMTHF would encourage monomerisation, its relatively labile binding compared to more common bases such as THF or DMAP, might enhance the reactivity of the resultant calcium stannanide complex. Hence, ICa was dissolved in C6D6 with two equivalents each of Ph3Sn–SnPh3 and TMTHF (Scheme 6). Analysis of the crude reaction mixture by 1H NMR spectroscopy showed complete conversion of the starting materials after two days at room temperature. A new product, 8·TMTHF, was characterised by a broadened resonance at δ 5.21 ppm corresponding to the γ-CH of the BDI ligand backbone. The 119Sn{1H} NMR spectrum comprised a resonance at δ −170.6 ppm in addition to a signal which was readily assigned as Ph4Sn at δ −126 ppm. Colourless block-like single crystals deposited from the reaction mixture overnight and were shown to be the monomeric TMTHF-solvated calcium stannanide, compound 8·TMTHF, by X-ray diffraction analysis (Fig. 2b). Compound 8·TMTHF could also be obtained by reacting ICa with two equivalents of Ph3SnH in toluene (Scheme 6). Organostannane redistribution to Ph4Sn was completely circumvented by use of a ten-fold excess of TMTHF, and 8·TMTHF was deposited as colourless crystals on standing at room temperature overnight in 68% yield.
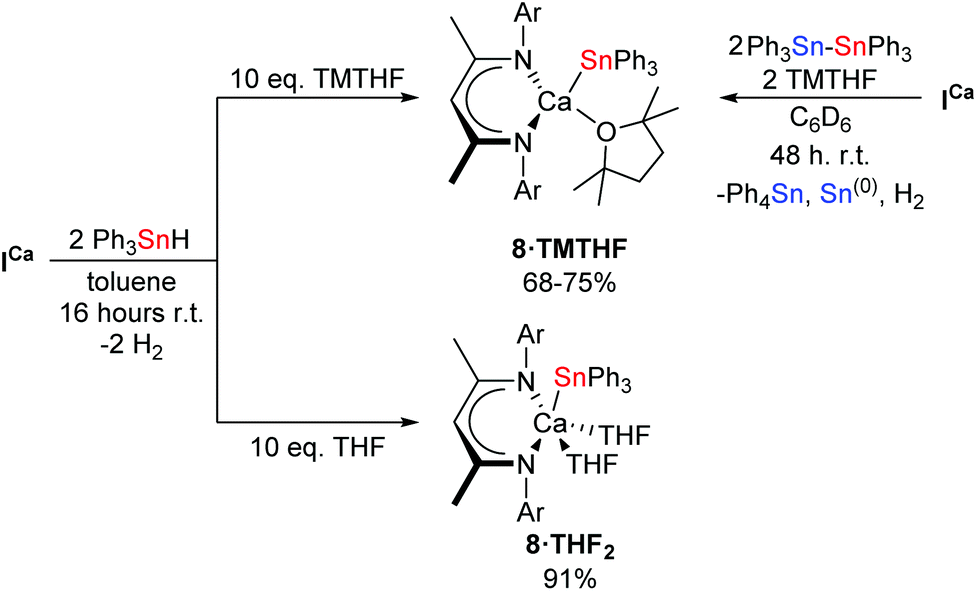 | ||
| Scheme 6 Synthesis of 8·TMTHF and 8·THF2. Ar = 2,6-di-isopropylphenyl. | ||
Once crystallised, 8·TMTHF is sparingly soluble in aromatic solvents but is readily soluble in THF. The 1H NMR spectrum in d8-THF displayed a single, well-defined BDI environment, while resonances observed at δ 1.80 and 1.16 ppm suggested displacement of TMTHF from the calcium centre by the NMR solvent. The resultant 119Sn{1H} chemical shift was also substantially perturbed with a single resonance appearing at δ −137.3 ppm. Despite poor solubility, attempts to obtain NMR spectra of isolated and vacuum-dried crystals of 8·TMTHF in C6D6 were successful, albeit the resonances were weak and broadened. Nevertheless, we were interested to find that two species were clearly discernible by 1H NMR spectroscopy. Although both species were identifiable as 8·TMTHF by the BDI γ-CH resonance at δ 5.21 ppm and the Lewis base-free compound, 4 (δ 5.02 ppm for γ-CH of the BDI backbone),42 their contrasting solubility in aromatic solvents prevented any confident, quantitative analysis of their relative abundance in solution. The apparent lability of TMTHF under vacuum was, however, further supported by the low relative intensity of its associated 1H resonances when vacuum-dried samples were re-dissolved in d8-THF. In order to investigate the viability of 8·TMTHF as a convenient precursor to 4, therefore, isolated crystals were stirred in the solid state under vacuum at 80 °C for sixteen hours. The resultant pale-yellow powder was only partially soluble in d8-toluene and, although the relative ratio of the two species determined by integration of the 1H NMR spectrum was increased in favour of 4, substantial quantities of 8·TMTHF remained. Both species could be clearly discerned in the resulting 119Sn{1H} NMR spectrum, which comprised two resonances at δ −160.5 (4) and −170.7 ppm (8·TMTHF).
The solution-state behaviour of 8·TMTHF was also investigated by variable temperature 1H NMR in d8-toluene. Whilst separate environments for 4 and 8·TMTHF could be discerned at 298 K, the γ-CH signals coalesced to a single broad resonance at δ 5.11 ppm above 318 K. Similarly, resonances assigned to free TMTHF experienced a pronounced and simultaneous upfield shift with increasing temperature. Although no more quantitative information could be extracted from these experiments, both of these observations suggest the establishment of a coordination–decoordination equilibrium when isolated 8·TMTHF samples are dissolved in arene solvents, facilitated by the lability of coordinated TMTHF.
The THF-solvated calcium hydride, [(BDI)CaH·THF]2 (ICa·THF2) was also reacted with two equivalents of Ph3SnH under a ten-fold excess of THF in toluene. After stirring overnight at room temperature, volatiles were removed under vacuum to provide the bis-THF adduct, 8·THF2, as a pale cream-coloured powder in high yield (Scheme 6). Its molecular structure (Fig. 2c) was determined by X-ray diffraction analysis performed on single crystals obtained by slow evaporation of a saturated toluene/THF solution. Compound 8·THF2 is readily soluble in aromatic solvents and displays a well-defined 1H NMR spectrum in C6D6 or d8-toluene. The single 119Sn environment resonates at δ −138.4 ppm in C6D6. When dissolved in d8-THF, the 1H and 119Sn{1H} NMR spectra of 8·THF2 were identical to that of 8·TMTHF in the same solvent, supporting the hypothesis that TMTHF is readily displaced from the calcium centre in THF-solution.
Whilst ICa reacts rapidly with two equivalents of Ph3SnH to provide 3, the magnesium congener, [(BDI)MgH]2 (IMg) reacts much more slowly. Although, approximately 50% of the initial Ph3SnH was observed to have redistributed to Ph4Sn after five days at room temperature (Scheme 7), the 1H NMR spectrum showed no net consumption of IMg. In addition, no other significant BDI- or Sn-containing products could be detected by either 1H or 119Sn{1H} NMR spectroscopy. Repetition of the reaction in toluene with a 10-fold excess of THF, however, not only suppressed organostannane redistribution, but also accelerated consumption of Ph3SnH. The monomeric magnesium stannanide complex, 9·THF, was, thus, obtained in near quantitative yield as a colourless powder after stirring for 16 hours at room temperature and removal of volatiles under vacuum (Scheme 7). Single-crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of hexane vapour into a THF solution at −30 °C, providing confirmation of the solid-state structure (Fig. 2d). Compound 9·THF is readily soluble in aromatic solvents and THF and, albeit the 1H resonances associated with the iso-propyl resonances were substantially broadened in C6D6 at 25 °C, both the 1H and 13C{1H} NMR spectra were indicative of a single BDI-environment. Similarly, the 119Sn{1H} NMR spectrum displayed a single resonance at δ −155.4 ppm in C6D6.
 | ||
| Scheme 7 Synthesis of 9·THF. Ar = 2,6-di-isopropylphenyl. | ||
X-ray diffraction analysis of 8·OPPh3, 8·THF2, 8·TMTHF and 9·THF
Compounds 8·OPPh3, 8·THF2 and 9·THF each crystallise in the monoclinic space group, P21/c, whilst the crystal structure of 8·TMTHF adopts the P21/m space group (Fig. 2a–d; selected bond distances and angles are presented in Table 1). Whilst the geometries of the four-coordinate calcium centres in 8·OPPh3 and 8·TMTHF are best described as distorted tetrahedra, 9·THF adopts a near trigonal-pyramidal geometry, with the magnesium centre situated 0.557(1) Å above an equatorial plane defined by the nitrogen and tin atoms (Σangles = 342°). The geometry of the five-coordinate calcium centre in 8·THF2 can be considered as a heavily distorted trigonal bipyramid, with the [Ph3Sn]− and one THF ligand in the axial positions and the BDI ligand and the second THF molecule occupying the equatorial sites. Compound 8·TMTHF is bisected through C3, the C16–C21 phenyl ring, and the furan ring by a mirror plane that is intrinsic to the space group, such that half a molecule is present per asymmetric unit. The methyl groups of the TMTHF ligand were disordered across the crystallographic mirror and a weak anagostic interaction was observed between one methyl group and the calcium centre. This is manifested by a H33C–Ca1 distance of 2.85(6) Å, and the consequently contrasting Ca–O–C angles: Ca1–O1–C28 = 113.88(16)°, Ca1–O1–C31 = 135.75(16)°. The Ca–Sn bond lengths of compounds 8·OPPh3, 8·TMTHF and 8·THF2 all lie within a narrow range established for compounds 3 (3.2137(4) Å) and 4 (3.3221(6) Å) and are comparable to the analogous distance in Westerhausen's calcium trimethylstannanide derivative (5, 3.2721(3) Å).43 The phosphine-oxide adduct, 8·OPPh3, displays an apparently more compressed coordination sphere, with shorter Ca–Sn, –N, and –O bonds than the furan-coordinated analogues. As a likely result of the steric congestion imposed by the bulky BDI ligand on the relatively weakly Lewis-basic triphenylstannanide anion, the Mg–Sn bond of 9·THF is longer (2.8340(6) Å) than that of the [Sn(SiMe3)3]− based complex, 6 (2.817(1) Å), the only other crystallographically characterised example of this type of bond in the literature.45 In the calcium complexes, the metal centres project by 1.266(2) Å (8·THF2), 1.449(2) Å (8·OPPh3) and 1.575(2) Å (8·TMTHF) from the mean plane of the BDI ligand backbone, and the [Ph3Sn]− moiety is located above the BDI-ligand backbone. In contrast, the smaller ionic radius of magnesium results in a 0.742(2) Å displacement of the metal centre from the BDI-plane in 9·THF, forcing the stannanide ligand away from the iso-propyl groups, and into the ‘pocket’ defined by the flanking Dipp groups of the BDI-ligand. The calcium complexes all display slightly compressed C–Sn–C angles, thus distorting the geometry of the otherwise tetrahedral tin centres. The Ca1–O1–P1 angle of 8·OPPh3 is approaching linearity (173.47(10)°), whilst the Ca–O and O–P distances are similar to those of the alkyl-calcium Ph3PO adduct, [(Ph3PO)2Ca{CH(SiMe3)2}2] previously reported by Hill et al.64| Compound | 8·TMTHF | 8·OPPh3 | 8·THF2 | 9·THF | |
|---|---|---|---|---|---|
| Sn1–Ca1 | 3.2470(5) | Sn1–Ae1 | 3.2304(4) | 3.2951(5) | 2.8340(6) |
| Sn1–C16 | 2.197(3) | Sn1–C30 | 2.195(2) | 2.200(2) | 2.184(2) |
| Sn1–C22 | 2.203(2) | Sn1–C36 | 2.199(2) | 2.204(3) | 2.176(2) |
| Sn1–C42 | 2.192(2) | 2.205(3) | 2.183(2) | ||
| Ca1–O1 | 2.3768(19) | Ae1–O1 | 2.2020(15) | 2.3723(18) | 2.0513(15) |
| Ae1–O2 | 2.406(2) | ||||
| Ca1–N1 | 2.3349(15) | Ae1–N1 | 2.3014(17) | 2.336(2) | 2.0485(17) |
| Ae1–N2 | 2.3189(16) | 2.348(2) | 2.0449(18) | ||
| P1–O1 | 1.5044(15) | ||||
| P1–C48 | 1.797(2) | ||||
| P1–C54 | 1.800(2) | ||||
| P1–C60 | 1.799(2) | ||||
| C22–Sn1–Ca1 | 112.60(5) | C30–Sn1–Ae1 | 105.87(5) | 116.46(7) | 117.77(6) |
| C36–Sn1–Ae1 | 118.23(6) | 115.74(7) | 120.00(6) | ||
| C22–Sn1–C22′ | 99.37(11) | C30–Sn1–C36 | 99.65(8) | 102.12(10) | 100.80(8) |
| C16–Sn1–C22 | 97.62(7) | C42–Sn1–C36 | 98.60(8) | 102.74(10) | 101.57(8) |
| C16–Sn1–Ca1 | 131.74(8) | C42–Sn1–Ae1 | 128.90(6) | 121.44(7) | 113.02(5) |
| C42–Sn1–C30 | 100.93(8) | 94.76(9) | 100.76(8) | ||
| O1–Ca1–Sn1 | 109.60(5) | O1–Ae1–Sn1 | 122.09(4) | 89.97(5) | 105.21(5) |
| O1–Ae1–O2 | 75.66(8) | ||||
| O2–Ae1–Sn1 | 142.83(7) | ||||
| N1–Ca1–O1 | 118.71(5) | O1–Ae1–N1 | 105.76(6) | 102.09(7) | 104.24(7) |
| N1–Ca1–O2 | 118.41(9) | ||||
| O1–Ae1–N2 | 112.13(6) | 165.72(7) | 102.25(7) | ||
| N2–Ae1–O2 | 90.51(8) | ||||
| N1–Ca1–Sn1 | 112.64(4) | N1–Ae1–Sn1 | 109.38(4) | 97.89(5) | 122.92(5) |
| N1–Ca1–N1′ | 82.33(8) | N1–Ae1–N2 | 81.07(6) | 81.14(7) | 93.52(7) |
| N2–Ae1–Sn1 | 117.58(4) | 103.45(5) | 125.61(5) | ||
| C28–O1–Ca1 | 113.88(16) | O1–P1–C48 | 110.55(9) | ||
| C31–O1–Ca1 | 135.75(16) | O1–P1–C54 | 110.49(9) | ||
| O1–P1–C60 | 111.32(9) | ||||
| C48–P1–C54 | 108.26(9) | ||||
| C48–P1–C60 | 110.27(9) | ||||
| C60–P1–C54 | 105.81(10) | ||||
| P1–O1–Ca1 | 173.47(10) |
Salt metathesis reactions with 8·TMTHF and 9·THF
With a series of well-defined monomeric Ae-stannanide derivatives in hand, we undertook an initial exploration of their reactivity. The highly ionic nature of the Ae–Sn bond suggests that they can be considered as hydrocarbon-soluble salts of the Ph3Sn− anion. As such, compounds 8·TMTHF and 9·THF were reacted with 0.5 equivalents of t-Bu2SnCl2 in C6D6 (Scheme 8). Both reactions provided a relatively clean 1H NMR spectrum indicative of the formation of a single major BDI-containing product. A pair of resonances at δ −76.9 and −137.20 ppm in the 119Sn{1H} NMR spectrum was consistent with formation of the alternating tristannane, Ph3Sn–Sn(t-Bu)2–SnPh3 (11). The identity of this compound was confirmed by X-ray diffraction and NMR spectroscopic analysis performed on single crystals isolated by fractional recrystallisation of the crude product mixture from hexane/toluene. Unfortunately, a satisfactory sample of the calcium-containing by-product (10Ca) could not be isolated from the reaction involving 8·TMTHF. When 9·THF was used, however, colourless crystalline blocks were deposited from the reaction mixture and identified as the known chloride complex, [(BDI)Mg(μ-Cl)]210Mg, by comparison to the published unit cell parameters and NMR spectra.66 | ||
| Scheme 8 Synthesis of alternating tristannane 11 by salt metathesis of Ae-stannanides and t-Bu2SnCl2. Ar = 2,6-diisopropylphenyl, L = THF, TMTHF. | ||
Compound 11 was first isolated in 33% yield (versus 78% in the current work) by Adams and Dräger in 1987 and synthesised by salt metathesis of the lithiated precursor, Ph3SnLi, with t-Bu2SnI2 in THF and/or toluene.67 Notably, although selectivity could be improved by variation of reaction stoichiometry, solvent polarity and concentration, this earlier approach yielded a mixture of Ph3Sn-capped tetra-, penta-, and hexa-stannanes such that the published crystal structure of 11 was as a component of a co-crystal with the tetrastannane, Ph3Sn–Sn(t-Bu)2–Sn(t-Bu)2–SnPh3. For completeness, therefore, the crystal structure of the pure tristannane, 11, is included in the ESI (Fig. S1†). This reaction presents 8·TMTHF and 9·THF as promising alternatives to group 1 metallated organostannanes in salt metathesis reactions.68–71
Insertion/nucleophilic addition of Ae–Sn bonds to N,N′-di-iso-propylcarbodiimide
As an initial assay of the potential utility of BDI-Ae stannanides to engage in catalytically relevant insertion reactions with unsaturated small molecules, 8·TMTHF, 8·THF2 and 9·THF were reacted with one equivalent of N,N′-di-iso-propylcarbodiimide (DIC) in C6D6 (Scheme 9). To the best of our knowledge, the resultant compounds provide the first reported C-organostannyl analogues of the ubiquitous amidinate class of N,N-donor anions. Compound 9·THF required 48 hours to cleanly convert DIC into the stannyl-amidinate complex, 12, at room temperature. Compound 12 was characterised by an upfield-shifted resonance at δ −186.9 ppm in the 119Sn{1H} NMR spectrum and a characteristic resonance at δ 181.9 ppm in the 13C{1H} NMR spectrum, corresponding to the central carbon atom of the Mg-ligated stannyl-aminidate ligand. The 1H NMR spectrum was indicative of a single, symmetrical BDI environment, with equivalent N-iso-propyl environments and characteristic SnPh3 resonances with 119/117Sn satellites. THF was absent from the isolated product, which was obtained as a colourless powder by removal of volatiles under vacuum and which could be crystallised from methylcyclohexane at −30 °C. The resultant colourless blocks were subjected to single-crystal X-ray diffraction analysis to provide the molecular structure of compound 12 (Fig. 3). | ||
| Scheme 9 Synthesis of stannylamidinates 12, 13·THF and 13·TMTHF. Ar = 2,6-di-isopropylphenyl, L = THF, TMTHF. Yields refer to unoptimised isolated yield, with quantitative spectroscopic conversion determined by 1H NMR spectroscopy. Ar = 2,6-di-isopropylphenyl. | ||
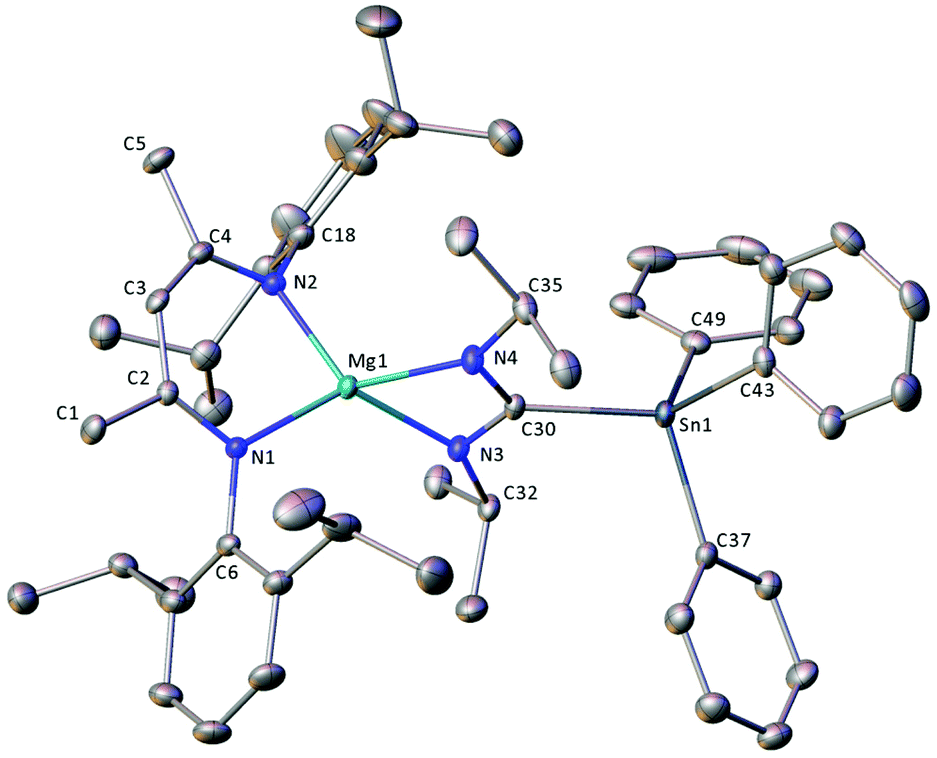 | ||
| Fig. 3 X-ray crystal structure of compound 12. Ellipsoids are shown at the 30% probability level and hydrogen atoms are omitted for clarity. The crystal structure contains two methylcyclohexane molecules per unit cell, which were heavily disordered across inversion centres and treated using a solvent mask algorithm. Selected bond lengths (Å) and angles (°): Sn1–C30 2.2030(14), Sn1–C37 2.1420(16), Sn1–C43 2.1524, Sn1–C49 2.1409(16), Mg1–N1 2.0328(13), Mg1–N2 2.0446(13), Mg1–N3 2.0456(13), Mg1–N4 2.0763(13), N3–C30 1.3353(19), N4–C30 1.3311(19), C38–Sn1–C30 109.38(6), C37–Sn1–C43 100.64(6), C43–Sn1–C30 123.56(6), C49–Sn1–C30 107.77(6), C49–Sn1–C37 110.98(6), C49–Sn1–C43 104.12(6), N1–Mg1–N2 94.98(5), N1–Mg1–N3 128.51(5), N1–Mg1–N4 125.70(5), N2–Mg1–N3 122.53(6), N2–Mg1–N4 121.01(6), N3–Mg1–N4 66.12(5), N3–C30–Sn1 119.36(10), N4–C30–Sn1 125.54(11), N4–C30–N3 114.99(13). | ||
The calcium complexes were more reactive towards DIC compared to 9·THF. Compound 8·TMTHF provided a clear, colourless solution of the calcium stannyl-amidinate, 13·TMTHF, after 60 minutes of sonication at room temperature. A further reaction at room temperature for 16 hours also provided quantitative spectroscopic conversion to 13·TMTHF, and 13·THF was obtained in a similar manner from 8·THF2 (Scheme 9). Compounds 13·TMTHF and 13·THF were isolated as colourless powders after removing volatiles from the reaction mixture and displayed similar 1H, 13C{1H}, and 119Sn{1H} NMR spectra to 12. Compared to 12, the 119Sn{1H} resonances of 13·TMTHF and 13·THF exhibited slightly upfield shifts to δ −193.8 and −196.1 ppm, respectively, whilst the stannyl-amidinate ‘backbone’ carbon nuclei resonated at δ 179.1 and 177.1 ppm in the corresponding 13C{1H} NMR spectra. The 119Sn and 117Sn satellites could also be clearly discerned for the tin-bonded amidinate 13C resonance of 13·TMTHF to provide coupling constants of 1J(119Sn) = 360.4 Hz, 1J(117Sn) = 344.7 Hz. The BDI and stannyl-amidinate ligands of both complexes display a set of resonances indicative of high symmetry and, in contrast to 12, the presence of a single coordinated TMTHF or THF was clearly discerned by 1H NMR spectroscopy. Although attempts to acquire single crystals of 13·TMTHF were unsuccessful, colourless plate-like single crystals suitable for X-ray analysis of 13·THF were obtained by cooling a hexane/methylcyclohexane solution to −30 °C.
Compound 12 crystallises in the monoclinic space group, P21/c, with one molecule of the magnesium complex and one disordered solvent region, equating to two methylcylohexane molecules, per unit cell. The solid state structure of 12 (Fig. 3) consists of a four-coordinate distorted tetrahedral magnesium centre, bonded to a BDI ligand via N1 and N2, and to a stannyl-amidinate ligand via N3 and N4. Although the Mg–N bond distances are all of a similar length, the magnesium centre is co-planar to the latter ligand but projects out of the mean N1–C2–C3–C4–N2 plane of the BDI ligand by 0.7483(16) Å. The two bidentate ligands are effectively perpendicular, such that the angle between the mean planes defined by N1–Mg1–N2 and N3–Mg1–N4–C30, is 90.47(6)°. Although no directly analogous stannyl-amidinate ligands have been reported previously, the C30–Sn1 bond length is unremarkable (2.2030(14) Å). The C30–N3 and C30–N4 bond lengths (1.3353(19), 1.3311(19) Å) are slightly longer, and the N3–C30–N4 angle (114.1(11)°) is slightly more acute, than those previously reported in homo- and heteroleptic N,N′-di-iso-propylformamidinate calcium complexes (ca. 1.28(3)–1.328(6) Å, 118.6(4)–121.3(2)°).72,73
Compound 13·THF crystallised in the monoclinic Cc space group and, unusually, contains four crystallographically independent molecules per unit cell (Fig. S2†). Because of this, and a fall-off in diffraction intensity at higher Bragg angles arising from the thin plate-like morphology of the crystal, a detailed discussion of the structure is unwarranted. The gross features of the compound are, however, unambiguous and the four molecules display only minor structural differences. The X-ray crystal structure of the Ca1/Sn1-containing molecule is shown in Fig. 4. The BDI, stannyl-amidinate and THF ligands are arranged about the five-coordinate calcium centre such that N1, N2, and N3 lie in an approximate equatorial plane, with O1 located axially. The two chelating ligands are arranged in a similar way to those in 12, with an average twist angle of approximately 93° between the mean planes defined by NBDI–Ca–NBDI, and Nam–Mg1–Nam–Cam, respectively. Significant variations in the structural metrics pertaining to the stannyl-amidinate ligands of 12 and 13·THF were not unambiguously discernible, but the larger ionic radius and higher coordination number of calcium results in displacement of the metal centre by approximately 2.5 Å from the mean plane of the BDI ligand backbone.
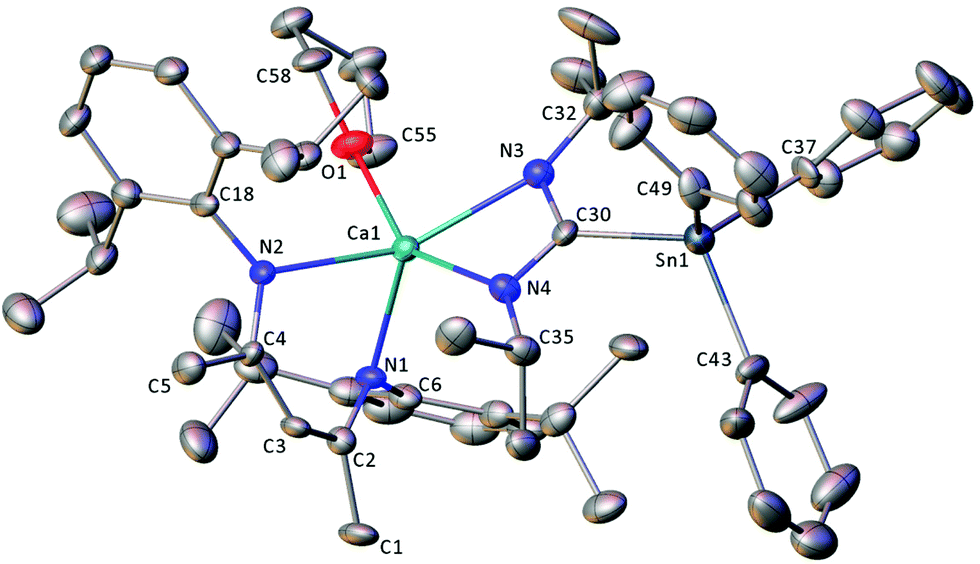 | ||
| Fig. 4 X-ray crystal structure of compound 13·THF. Ellipsoids are shown at the 30% probability level and hydrogen atoms are omitted for clarity. Only one of the four crystallographically independent molecules present in the unit cell is shown. Selected bond lengths (Å) and angles (°): Sn1–C30 2.214(12), Sn1–C37 2.165(10), Sn1–C43 2.167(13), Sn1–C49 2.094(13), Ca1–O1 2.377(10), Ca1–N1 2.382(10), Ca1–N2 2.341(10), Ca1–N3 2.373(11), Ca1–N4 2.377(11), N3–C30 1.380(17), N4–C30 1.311(16), C37–Sn1–C30 120.5(5), C37–Sn1–C43 100.1(5), C43–Sn1–C30 113.7(5), C49–Sn1–C30 107.4(5), C49–Sn1–C43 107.7(6), O1–Ca1–N1 97.9(4), N2–Ca1–O1 89.8(4), N2–Ca1–N1 80.2(4), N2–Ca1–N3 146.0(4), N2–Ca1–N4 108.9(4), N3–Ca1–O1 150.4(4), N4–Ca1–N1 107.6(4), N3–C30–Sn1 123.7(13), N4–C30–Sn1 121.2(9), N4–C30–N3 114.1(11). | ||
Conclusions
In conclusion, dimeric calcium and magnesium hydrides ICa, ICa·THF2, and IMg deprotonate triphenylstannane in the presence of an excess of coordinating Lewis base to provide clean access to well-defined monomeric Ae-stannanide complexes in good yield. Calcium stannanide complexes are also accessible through distannane heterolysis by nucleophilic attack of a calcium hydride. A preliminary exploration of the reactivity arising from the resultant compounds demonstrates their potential as well-defined, soluble sources of the [Ph3Sn]− anion in salt metathesis and nucleophilic addition reactions. Further work continues to explore the nature and reactivity of bonds between heavier p-block elements and the heavier alkaline earths.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the EPSRC through grant numbers EP/N014456/1, EP/R020752/1, and the EPSRC Centre for Doctoral Training in Catalysis (EP/L016443/1). This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath. I. M. thanks the University of Bristol for support and the Canadian Government for a Canada 150 Research Chair.Notes and references
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS PubMed.
- J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782–16785 CrossRef CAS PubMed.
- C. Bellini, J.-F. Carpentier, S. Tobisch and Y. Sarazin, Angew. Chem., Int. Ed., 2015, 54, 7679–7683 CrossRef CAS PubMed.
- C. Bellini, V. Dorcet, J. F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 4564–4583 CrossRef CAS PubMed.
- E. Le Coz, Z. Zhang, T. Roisnel, L. Cavallo, L. Falivene, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2020, 26, 3535–3544 CrossRef CAS PubMed.
- A. C. A. Ried, L. J. Taylor, A. M. Geer, H. E. L. Williams, W. Lewis, A. J. Blake and D. L. Kays, Chem. – Eur. J., 2019, 25, 6840–6846 CrossRef CAS PubMed.
- F. Buch and S. Harder, Organometallics, 2007, 26, 5132–5135 CrossRef CAS.
- M. S. Hill, D. J. Liptrot, D. J. MacDougall, M. F. Mahon and T. P. Robinson, Chem. Sci., 2013, 4, 4212–4222 RSC.
- C. Bellini, V. Dorcet, J. F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 4564–4583 CrossRef CAS PubMed.
- A. Baishya, T. Peddarao and S. Nembenna, Dalton Trans., 2017, 46, 5880–5887 RSC.
- E. Le Coz, V. Dorcet, T. Roisnel, S. Tobisch, J. F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2018, 57, 11747–11751 CrossRef CAS PubMed.
- E. Le Coz, S. Kahlal, J.-Y. Saillard, T. Roisnel, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2019, 25, 13509–13513 CrossRef CAS PubMed.
- L. J. Morris, M. S. Hill, M. F. Mahon, I. Manners, F. S. McMenamy and G. R. Whittell, Chem. – Eur. J., 2020, 26, 2954–2966 CrossRef CAS PubMed.
- M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042–2043 CrossRef CAS PubMed.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 2953–2956 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497–499 CrossRef CAS.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC.
- F. Buch, H. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741–2745 CrossRef CAS PubMed.
- V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2014, 50, 2311–2314 RSC.
- L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Chem. Sci., 2019, 10, 8108–8118 RSC.
- D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., 2020, 59, 310–314 CrossRef CAS PubMed.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed.
- (a) A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed; (b) M. S. Hill, M. F. Mahon, A. S. S. Wilson, C. Dinoi, L. Maron and E. Richards, Chem. Commun., 2019, 55, 5732–5735 RSC; (c) A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon, L. Maron and E. Richards, Angew. Chem., 2020, 59, 1232–1237 CrossRef CAS PubMed.
- H. Bauer, M. Alonso, C. Fischer, B. Rosch, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 15177–15182 CrossRef CAS PubMed.
- D. J. Liptrot, M. Arrowsmith, A. L. Colebatch, T. J. Hadlington, M. S. Hill, G. Kociok-Köhn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 15280–15283 CrossRef CAS PubMed.
- N. A. Rajabi, C. L. McMullin and M. S. Hill, unpublished results.
- A.-F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef PubMed.
- W. Caseri, Chem. Soc. Rev., 2016, 45, 5187–5199 RSC.
- M. Trummer, F. Choffat, P. Smith and W. Caseri, Macromol. Rapid Commun., 2012, 33, 448–460 CrossRef CAS PubMed.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS.
- M. M. Heravi and L. Mohammadkhani, J. Organomet. Chem., 2018, 869, 106–200 CrossRef CAS.
- C. Tamborski and E. J. Soloski, J. Am. Chem. Soc., 1961, 83, 3734–3734 CrossRef CAS.
- H. M. J. C. Creemers, J. G. Noltes and G. J. M. Vanderke, J. Organomet. Chem., 1968, 14, 217–221 CrossRef CAS.
- J. C. Lahournere and J. Valade, J. Organomet. Chem., 1970, 22, C3–C4 CrossRef CAS.
- M. Westerhausen and T. Hildenbrand, J. Organomet. Chem., 1991, 411, 1–17 CrossRef CAS.
- R. Hummeltenberg, K. Jurkschat and F. Uhlig, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 123, 255–261 CrossRef CAS.
- C. Kayser, R. Klassen, M. Schurmann and F. Uhlig, J. Organomet. Chem., 1998, 556, 165–167 CrossRef CAS.
- U. Hermann, M. Schurmann and F. Uhlig, J. Organomet. Chem., 1999, 585, 211–214 CrossRef CAS.
- U. Englich, K. Ruhlandt-Senge and F. Uhlig, J. Organomet. Chem., 2000, 613, 139–147 CrossRef CAS.
- L. J. Morris, M. S. Hill, I. Manners, C. L. McMullin, M. F. Mahon and N. A. Rajabi, Chem. Commun., 2019, 55, 12964–12967 RSC.
- M. Westerhausen, Angew. Chem., Int. Ed. Engl., 1994, 33, 1493–1495 CrossRef.
- R. Fischer, J. Baumgartner, C. Marschner and F. Uhlig, Inorg. Chim. Acta, 2005, 358, 3174–3182 CrossRef CAS.
- D. Matioszek, N. Katir, S. Ladeira and A. Castel, Organometallics, 2011, 30, 2230–2235 CrossRef CAS.
- P. M. Chapple, S. Kahlal, J. Cartron, T. Roisnel, V. Dorcet, M. Cordier, J.-Y. Saillard, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2020, 59, 9120–9126 CrossRef CAS PubMed.
- S. K. Sur and J. P. Colpa, Organometallics, 1989, 8, 2749–2751 CrossRef CAS.
- W. P. Neumann, H. Hillgartner, K. M. Baines, R. Dicke, K. Vorspohl, U. Kobs and U. Nussbeutel, Tetrahedron, 1989, 45, 951–960 CrossRef CAS.
- T. N. Mitchell, K. Kwetkat, D. Rutschow and U. Schneider, Tetrahedron, 1989, 45, 969–978 CrossRef CAS.
- T. N. Mitchell, J. Organomet. Chem., 1986, 304, 1–16 CrossRef CAS.
- T. N. Mitchell, A. Amamria, H. Killing and D. Rutschow, J. Organomet. Chem., 1986, 304, 257–265 CrossRef CAS.
- A. Khan, A. J. Lough, R. A. Gossage and D. A. Foucher, Dalton Trans., 2013, 42, 2469–2476 RSC.
- T. N. Mitchell, A. Amamria, H. Killing and D. Rutschow, J. Organomet. Chem., 1983, 241, C45–C47 CrossRef CAS.
- H. Yoshida, A. Shinke and K. Takaki, Chem. Commun., 2013, 49, 11671–11673 RSC.
- J. S. Stehouwer, N. Jarkas, F. Zeng, R. J. Voll, L. Williams, V. M. Camp, E. J. Malveaux, J. R. Votaw, L. Howell, M. J. Owens and M. M. Goodman, J. Med. Chem., 2008, 51, 7788–7799 CrossRef CAS PubMed.
- M. S. Alnajjar and H. G. Kuivila, J. Am. Chem. Soc., 1985, 107, 416–423 CrossRef CAS.
- B. Wrackmeyer, Annu. Rep. NMR Spectrosc., 1999, 38, 203–264 CrossRef CAS.
- S. Harder, Organometallics, 2002, 21, 3782–3787 CrossRef CAS.
- G. M. Allan, R. A. Howie, J. N. Low, J. M. S. Skakle, J. L. Wardell and S. Wardell, Polyhedron, 2006, 25, 695–701 CrossRef CAS.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- A.-F. Pécharman, N. A. Rajabi, M. S. Hill, C. L. McMullin and M. F. Mahon, Chem. Commun., 2019, 55, 9035–9038 RSC.
- A. C. T. Kuate, R. A. Lalancette, T. Bannenberg and F. Jäkle, Angew. Chem., Int. Ed., 2018, 57, 6552–6557 CrossRef PubMed.
- J. W. Chen, D. A. M. Parra, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 10202–10205 CrossRef CAS PubMed.
- M. S. Hill, M. F. Mahon and T. P. Robinson, Chem. Commun., 2010, 46, 2498–2500 RSC.
- S. Krieck, P. Schuler, H. Gorls and M. Westerhausen, Inorg. Chem., 2018, 57, 13937–13943 CrossRef CAS PubMed.
- A. P. Dove, V. C. Gibson, P. Hormnirun, E. L. Marshall, J. A. Segal, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 3088–3097 RSC.
- S. Adams and M. Dräger, Angew. Chem., Int. Ed. Engl., 1987, 26, 1255–1256 CrossRef.
- S. Adams and M. Dräger, J. Organomet. Chem., 1987, 323, 11–20 CrossRef CAS.
- D. Reed, D. Stalke and D. S. Wright, Angew. Chem., Int. Ed. Engl., 1991, 30, 1459–1460 CrossRef.
- P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1993, 554–555 RSC.
- T. Schollmeier, U. Englich, R. Fischer, I. Prass, K. Ruhlandt, M. Schurmann and F. Uhlig, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1462–1470 CAS.
- S. B. Kim, C. X. Yang, T. Powers, L. M. Davis, X. B. Lou and R. G. Gordon, Angew. Chem., Int. Ed., 2016, 55, 10228–10233 CrossRef CAS PubMed.
- J. Dyall, M. S. Hill, M. F. Mahon, L. Teh and A. S. S. Wilson, Dalton Trans., 2019, 48, 4248–4254 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray diffraction analysis of compounds 8·OPPh3, 8·TMTHF, 8·THF2, 9·THF, 11, 12, 13·THF, details for the computational analysis and atomic coordinates of computed structures. CCDC 2003513–2003519. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02406f |
| This journal is © The Royal Society of Chemistry 2020 |