
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomers in chlorido and alkoxido-substituted oxidorhenium(V) complexes: effects on catalytic epoxidation activity†
Jörg A.
Schachner
 *,
Ferdinand
Belaj
and
Nadia C.
Mösch-Zanetti
*
*,
Ferdinand
Belaj
and
Nadia C.
Mösch-Zanetti
*
Institute of Chemistry, Inorganic Chemistry, University of Graz, Schubertstr. 1, 8010 Graz, Austria. E-mail: joerg.schachner@uni-graz.at
First published on 3rd August 2020
Abstract
The syntheses and characterizations of oxidorhenium(V) complexes trans-dichlorido [ReOCl2(PPh3)(L1a)] (trans-2a), cis-dichlorido [ReOCl2(PPh3)(L1b)] (cis-2b) and ethoxido-complex [ReO(OEt)(L1b)2] (4b), ligated with the dimethyloxazoline-phenol ligands HL1a and HL1b are described. The bidentate ligand HL1a (2-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-phenol) is unsubstituted on the phenol ring; ligand HL1b (2-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-4-nitrophenol) contains a nitro group in para-position to the hydroxy group. In the reaction of precursor complex [ReOCl3(PPh3)2] and HL1a the two stereoisomers cis/trans-2a, with respect to chlorido ligands, are formed. The solid state structures of both isomers cis- and trans-2a were determined by single crystal X-ray diffraction analysis. In contrast, with ligand HL1b, only the cis-isomer cis-2b was obtained. Ethoxido-complex 4b is exclusively obtained when precursor [ReOCl3(OPPh3)(SMe2)] is reacted with 2 equiv. of HL1b in ethanol in the presence of the base 2,6-dimethylpyridine (lutidine). If no lutidine is added, chlorido-complex [ReOCl(L1b)2] (3b) is obtained. Complexes [ReOCl2(PPh3)(L1a)] (cis/trans-2a), [ReOCl2(PPh3)(L1b)] (cis-2b), [ReO(OMe)(L1a)2] (4a) and [ReO(OEt)(L1b)2] (4b) were tested as homogeneous catalysts in the benchmark reaction of cyclooctene epoxidation. The influence of isomerism and effects of ligand substitutions on catalytic activity was investigated. Based on the time-conversion plots it can be concluded that cis/trans-isomerism does not influence catalytic activity, but electron-withdrawing substituents, as in cis-2b, 3b and 4b, show a beneficial effect.
Introduction
With Herrmann's seminal papers on the convenient synthesis1 and the superior catalytic activity of methyltrioxorhenium (MTO) in olefin epoxidation and oxidation,2 a surge of research in high-valent oxidorhenium chemistry was initiated.3 In olefin epoxidation catalysis, MTO remains the most investigated and active rhenium-based catalyst to this day, reaching turnover numbers (TONs) of >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.4–6 However, MTO requires H2O2 as oxidant, which leads to formation of stoichiometric amounts of H2O in epoxidation reactions. This limits the use of MTO for epoxides which are prone to hydrolytic ring opening under acidic or aqueous reaction conditions.7 Complexes containing the oxidorhenium(V) [ReO]3+ core were identified as a possible solution to the ring-opening problems of MTO, as such complexes are stable in water,8 whereas MTO acts as an acid in the presence of water.9 The first two examples of such oxidorhenium(V) complexes were disclosed in 1996 and 1997, again by the group of Herrmann, employing tetra- and bidentate Schiff-base ligands.10,11 Over the next two decades several research groups around the world, including ours, contributed to oxidorhenium(V) chemistry and its application in homogeneous catalytic epoxidation of cyclooctene.4,8,12–15 We have been exploring the chemistry of oxidorhenium(V) complexes with different ON-bidentate ligands of the type [ReOCl2(PPh3)(ON)] (referred to as mono-ligated) and [ReOCl(ON)2] (referred to as bis-ligated) complexes.
000.4–6 However, MTO requires H2O2 as oxidant, which leads to formation of stoichiometric amounts of H2O in epoxidation reactions. This limits the use of MTO for epoxides which are prone to hydrolytic ring opening under acidic or aqueous reaction conditions.7 Complexes containing the oxidorhenium(V) [ReO]3+ core were identified as a possible solution to the ring-opening problems of MTO, as such complexes are stable in water,8 whereas MTO acts as an acid in the presence of water.9 The first two examples of such oxidorhenium(V) complexes were disclosed in 1996 and 1997, again by the group of Herrmann, employing tetra- and bidentate Schiff-base ligands.10,11 Over the next two decades several research groups around the world, including ours, contributed to oxidorhenium(V) chemistry and its application in homogeneous catalytic epoxidation of cyclooctene.4,8,12–15 We have been exploring the chemistry of oxidorhenium(V) complexes with different ON-bidentate ligands of the type [ReOCl2(PPh3)(ON)] (referred to as mono-ligated) and [ReOCl(ON)2] (referred to as bis-ligated) complexes.
The rich coordination chemistry and structural features of oxidorhenium(V) complexes have been extensively reviewed by Sergienko and Machura over the last few years.8,16,17 In principle there are seven stereoisomers possible for mono-ligated complexes of type [ReOX2L(ON)] (a–g, Fig. 1) and six for bis-ligated complexes of type [ReOX(ON)2] (A–F, Fig. 1).18 Except for isomers b, f and A, these complexes also show octahedral chirality.
 | ||
| Fig. 1 Top row: possible stereoisomers a–g for mono-ligated complexes; bottom row: possible stereoisomers A–F for bis-ligated complexes; isomers in brackets have not been isolated yet. | ||
For isomers a and b, the cis/trans label refers to the orientation of the two X ligands. For bis-ligated isomers A–D, the N,N-cis/trans label was established in literature, referring to the position of the nitrogen atoms. Isomers c–g and E–F have not been observed yet. In our research, focus was placed on two classes of ON-bidentate ligands, namely pyrazole-phenol (HpyzR) as well as oxazoline (Hoz) and dimethyloxazoline-phenol ligands (HdmozR), with different electron-withdrawing and -donating substituents R on the phenol moiety (Fig. 2).
 | ||
| Fig. 2 Pyrazole- and oxazoline-phenol based ON-bidentate ligands investigated in oxidorhenium(V) chemistry. | ||
The Hoz ligand led to the rare case where both N,N-trans and N,N-cis isomers of the complex [ReOCl(oz)2] were formed in equal amounts and were successfully isolated.19,20 In contrast, complexes containing a ligand from the dimethyloxazoline HL1 family have exclusively yielded N,N-trans complexes so far.19–21,22 Their application in two benchmark homogeneous catalytic reactions, namely cyclooctene epoxidation and perchlorate reduction was studied in a systematic manner.12,13,19,21,23–25 Whereas both mono- and bis-ligated complexes show activity in epoxidation catalysis, only the bis-ligated complexes are active in perchlorate reduction. In addition we found that N,N-trans [ReOCl(oz)2] shows superior activity in perchlorate reduction over N,N-cis [ReOCl(oz)2].19 Therefore, we were interested to also test both N,N-cis/trans [ReOCl(oz)2] isomers in epoxidation catalysis, but in contrast to perchlorate reduction it was found that both stereoisomers displayed the same, mediocre activity in cyclooctene epoxidation (TON = 30 and 40, resp.).21 With an electron-withdrawing NO2 substituent on the ligand, N,N-trans complex [ReOCl(L1b)2] (3b) displayed much higher epoxidation activity, giving more than twice as many turnovers as unsubstituted complex [ReOCl(L1a)2] (3a) (3a, TON = 37; 3b, TON = 80).21 A similar observation was made for nitro-substituted complex [ReOCl(pyzNO2)2].25 Thus, complexes equipped with electron-withdrawing substituents are one key feature for active epoxidation catalysts.
Within this manuscript we present further studies on the influence of stereoisomers and substituent effects with regards to catalytic epoxidation activity. We were able to isolate and fully characterize the two oxidorhenium(V) coordination isomers cis- and trans-2a [ReOCl2(PPh3)(L1a)]. The observation of two isomers is in contrast to cis-2b [ReOCl2(PPh3)(L1b)], where only the cis-isomer was obtained. The stereochemistry of all three complexes was confirmed by single crystal X-ray diffraction. Bis-ligated complexes [ReOCl(L1a)2] (3a) and [ReOCl(L1b)2] (3b) react in an alcoholic solvent in the presence of a suitable base to the alcoholato complexes [ReO(OMe)(L1a)2] (4a)19 and [ReO(OEt)(L1b)2] (4b) respectively (Scheme 3). The series of complexes were used as catalysts in the epoxidation of cyclooctene allowing the investigation of the influence of stereoisomeric as well as electrophilic properties, and were compared to previously published complexes 3a, 3b and 4a.19,21 To the best of our knowledge, complexes cis/trans-2a is the first pair such stereoisomers that is investigated in epoxidation catalysis.
Results and discussion
Synthetic procedures
A general overview of the synthesis of mono- and bis-ligated complexes with ligands HL1 is shown in Scheme 1. Precursor [ReOCl3(PPh3)2] exclusively yields mono-ligated complexes of type [ReOCl2(PPh3)(L1)], precursor [ReOCl3(OPPh3)(SMe2)] exclusively bis-ligated complexes of type [ReOCl(L1)2], independent of the chosen metal to ligand stoichiometry. | ||
| Scheme 1 General scheme of formation of complexes and their isomers with ligand HL1; abbreviations used to indicate stereoisomers are given (n.ob. = not observed; B = Brønsted base, e.g. 2,6-dimethylpyridine, NaH). | ||
The synthesis of mono-ligated complex [ReOCl2(PPh3)(L1a)] (cis-2a) was reported in 2014.19 During a later synthesis, after isolation of product cis-2a as previously described,19 upon further cooling of the remaining supernatant to 8 °C, a small amount of a green crystalline material was obtained, giving a different 1H NMR spectrum than cis-2a. This material proved to be the stereoisomer trans-2a (Scheme 2). In contrast, with nitro-substituted ligand HL1b, under otherwise identical conditions, only stereoisomer cis-2b could be isolated.
 | ||
| Scheme 2 Syntheses of complex cis-2a,19trans-2a and cis-2b (n. ob. = not observed). | ||
The 1H NMR spectrum of trans-2a shows a time-averaged Cs-symmetric molecule in solution, with both methyl groups and the methylene protons of the oxazoline moiety appearing as singlets (Fig. S1†). The coordinated PPh3 ligand appears as two well-separated multiplets centered at 7.88 and 7.50 ppm (CDCl3, Fig. S1†). In the 31P NMR spectrum, a shift of −5.35 ppm is observed (Fig. S3†), which is almost identical to the shift of free PPh3 (−5.47 ppm). Single crystals suitable for X-ray diffraction analysis of cis- and trans-2a were obtained confirming their isomeric structures (see below). trans-2a shows a limited stability in CDCl3 solution under ambient atmosphere. After several days, signals for OPPh3 are appearing.
The 1H NMR spectrum of complex cis-2b gave the expected pattern of a coordinated L1b ligand moiety and a molecule of PPh3 (Fig. S4 and S5†). The coordinated PPh3 ligand shows a peak at −17.35 ppm (CDCl3) in the 31P NMR spectrum (Fig. S6†). Complex cis-2b also shows a limited stability in CDCl3 solution. After several days decomposition occurs, indicated by a color change from initial green to brown, and the appearance of several new signals in the NMR spectra, which could not be assigned to a new complex.
The synthesis of chlorido-complex N,N-trans [ReOCl(L1b)2] (3b) in acetonitrile solution has been previously published (Scheme 3).21 However, when EtOH was the reaction solvent and lutidine was added, the reaction solution turned dark-brown under reflux conditions. Upon cooling of the reaction mixture, a brown micro-crystalline material could be isolated in high yields. In the supernatant, the by-product lutidine hydrochloride could be identified. As single crystals of high enough quality of this material could not be grown, the assignment of the structure of 4b as a symmetric ethoxido-complex (Scheme 3) is based on the following analytical data: in the 1H NMR spectrum, a diagnostic quartet of doublets at 3.50 ppm (2H) and a corresponding triplet at 0.81 ppm (3H) could be observed (Fig. S8†), indicating the presence of an ethoxido ligand. Furthermore, only one set of L1b ligand signals was observed, consistent with the formation of a symmetric complex (B, Fig. 1). By integration, the ratio of the ethoxido to L1b ligand moieties is 1:2, confirming the stoichiometry as [ReO(OEt)(L1b)2] (Fig. S7†). The proposed structure of 4b (Scheme 3) is of C2-symmetry, consistent with the observed 1H NMR spectrum (3b has C1-symmetry21). In the IR spectrum, the Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency is found at 912 cm−1, whereas the related band of the cis-[ReOCl]3+ core of 3b is located at 962 cm−1.21 Such a significant bathocromic shift would be expected for a trans-[ReO(OR)]3+ core, due to competition of the ethoxido ligand for π bonding with the metal dπ orbitals.12,13,26 A mass spectrum of 4b showed a molecular ion peak with a correct isotope pattern matching for [ReO(OEt)(L1b)2]. The same spectroscopic and structural features were also observed for previously published methoxido-coordinated complex [ReO(OMe)(L1a)2] (4a), whose solid state structure was confirmed by X-ray crystallography.19 Complex 4a was obtained in a similar fashion from a reaction in MeOH with residual amounts of the base NaH still present. In case of 4b, the added lutidine played a similar role, probably by deprotonating EtOH and inducing chlorido abstraction. In absence of lutidine, expected chlorido-complex 3b is obtained from synthesis in EtOH (Scheme 3).
O stretching frequency is found at 912 cm−1, whereas the related band of the cis-[ReOCl]3+ core of 3b is located at 962 cm−1.21 Such a significant bathocromic shift would be expected for a trans-[ReO(OR)]3+ core, due to competition of the ethoxido ligand for π bonding with the metal dπ orbitals.12,13,26 A mass spectrum of 4b showed a molecular ion peak with a correct isotope pattern matching for [ReO(OEt)(L1b)2]. The same spectroscopic and structural features were also observed for previously published methoxido-coordinated complex [ReO(OMe)(L1a)2] (4a), whose solid state structure was confirmed by X-ray crystallography.19 Complex 4a was obtained in a similar fashion from a reaction in MeOH with residual amounts of the base NaH still present. In case of 4b, the added lutidine played a similar role, probably by deprotonating EtOH and inducing chlorido abstraction. In absence of lutidine, expected chlorido-complex 3b is obtained from synthesis in EtOH (Scheme 3).
 | ||
| Scheme 3 Formation of 3b in absence of lutidine; formation of 4b in presence of lutidine in EtOH. | ||
Crystallography
Single crystal X-ray diffraction analyses confirmed the stereochemistry of cis-2a, trans-2a and cis-2b. Molecular views of cis/trans-2a are given in Fig. 3, of cis-2b in Fig. 4. Selected bond lengths and angles are shown in Table 1. Details on crystallographic refinements can be found in the ESI.†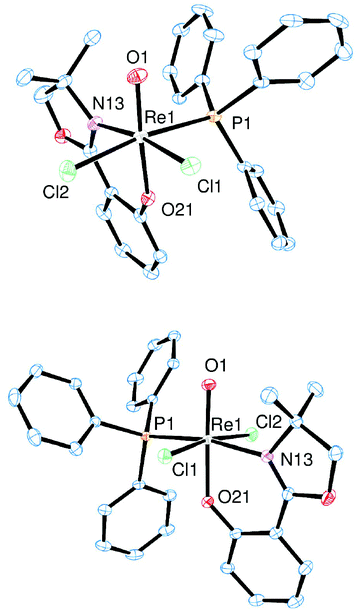 | ||
| Fig. 3 Molecular views (50% level) of complexes cis-[ReOCl2(PPh3)(L1a)] (cis-2a, top) and trans-[ReOCl2(PPh3)(L1a)] (trans-2a, bottom) (H atoms and solvent molecules omitted for clarity). | ||
 | ||
| Fig. 4 Molecular view (50% level) of complex cis-[ReOCl2(PPh3)(L1b)] (cis-2b) (H atoms omitted for clarity). | ||
| [Å] | Re1O1 |
Re1–O21 | Re1–P1 | Re1–Cl1 | Re1–Cl2 | Re1–N13 |
|---|---|---|---|---|---|---|
| cis-2a | 1.684(3) | 1.947(3) | 2.4961(11) | 2.3747(11) | 2.4019(12) | 2.166(3) |
| trans-2a | 1.6917(13) | 1.9481(13) | 2.4723(5) | 2.3981(4) | 2.4026(5) | 2.1423(16) |
| cis-2b | 1.6775(19) | 1.9969(18) | 2.4749(7) | 2.3632(7) | 2.3766(7) | 2.155(2) |
| [°] | O1–Re1–O21 | Cl1–Re1–Cl2 | N13–Re1–P1 |
|---|---|---|---|
| cis-2a | 168.16(13) | 86.39(4) | 94.54(10) |
| trans-2a | 177.71(6) | 171.181(16) | 168.60(4) |
| cis-2b | 167.51(8) | 86.27(3) | 168.96(6) |
Single crystals for cis-2a were obtained by slow evaporation of a saturated dichloromethane solution, for trans-2a of a saturated acetonitrile solution. Both complexes show a distorted octahedral coordination of the rhenium center, with the trans arrangement of the phenolate oxygen O21 to the oxido ligand O1 (Fig. 3). For trans-2a, the two chlorido ligands are in trans orientation to each other, in contrast to most previously described mono-ligated oxidorhenium(V) complexes of that type.12,17,19,25,27 Only few examples were disclosed, where both the cis and trans isomers of the same ligand set were isolated and characterized.28 For cis-2a and trans-2a, the respective Re1O1 and Re1–O21 bond distances are the same within experimental error. Single crystals of cis-2b were obtained by slow evaporation of a concentrated solution in EtOAc. Complex cis-2b shows the same isomeric arrangement as cis-2a (isomer a, Fig. 1). The electron-withdrawing influence of the L1b ligand moiety is reflected by the slightly shortened Re1O1 and elongated Re1–O21 bond, in comparison to cis-2a.
Epoxidation
The catalytic epoxidation of cyclooctene (Scheme 4) of complexes trans-2a, cis-2b and 4b together with the two previously published complexes cis-2a and 4a was investigated. The results of the two alkoxido-complexes 4a and 4b are compared to previously obtained epoxidation data of the respective chlorido-complexes 3a and 3b.21 Of particular interest was the influence on catalytic activity of the cis/trans-isomerism of 2a and the effect chlorido/alkoxido substitution in complexes 3a/4a and 3b/4b. Finally, the influence of electron-withdrawing substituents was studied by comparison of catalytic activity between cis-2a and cis-2b. Accordingly, all complexes were tested using 3 equiv. of tert-butylhydroperoxide (TBHP) as oxidant (Scheme 4) with a 1 mol% catalyst loading in CHCl3 at 50 °C. Reaction progress was monitored by withdrawing aliquots and analysis with gas chromatography mass spectrometry. | ||
| Scheme 4 Catalytic epoxidation of cyclooctene using tert-butylhydroperoxide (TBHP). | ||
Time-conversion plots for cis/trans-2a and cis-2b are given in Fig. 5, for complexes 3a/b and 4a/b in Fig. 6. A summary of turnover numbers (TONs) and turnover frequencies (TOFs) can be found in Table 2. In general all tested complexes are catalytically active without induction period. The highest activities were displayed by complexes equipped with the electron-withdrawing ligand HL1b. Mono-ligated complex [ReOCl2(PPh3)(L1b)] (cis-2b) showed the highest TON of 95 after only 12 h of reaction time. Similarly, complex [ReOCl(L1b)2] (3b) reached a TON of 82, complex [ReO(OEt)(L1b)2] (4b) the essentially same TON of 84. Complexes equipped with ligand HL1a showed in general lower activities. Mono-ligated complexes [ReOCl2(PPh3)(L1a)] (cis/trans-2a) showed average activities (TON = 69 and 65, resp.). The lowest overall activities were displayed by chlorido-complex [ReOCl(L1a)2] (3a, TON = 37)21 and methoxido-complex [ReO(OMe)(L1a)2] (4a, TON = 39).
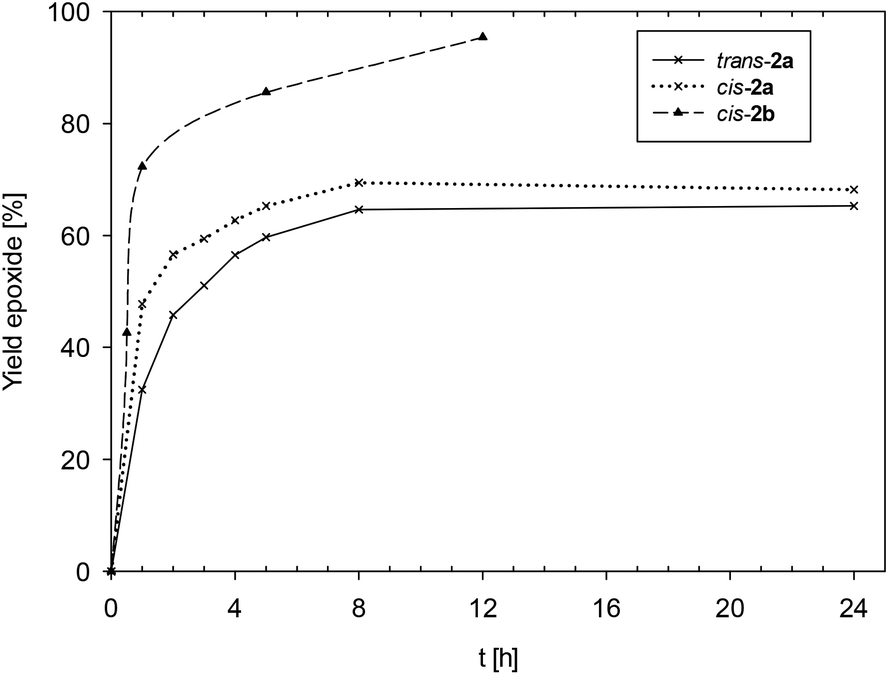 | ||
| Fig. 5 Comparison of epoxide yields for isomeric complexes cis/trans-2a and cis-2b. | ||
 | ||
| Fig. 6 Comparison of epoxide yields of complexes 3a/4a and 3b/4b; data for 3a–b was previously published.21 | ||
| cis-2a | trans-2a | cis-2b | 3a | 4a | 3b | 4b | |
|---|---|---|---|---|---|---|---|
| General conditions: 1 mol% catalyst loading, 3 equiv. TBHP (5.5 M in decane), CHCl3, 50 °C; data for 3a and 3b from ref. 21.a TOF calculated after maximum conversion.b TOF calculated after 1 h. | |||||||
| TON (TOF h−1) | 69 (8.6) | 65 (8.1) | 95 (7.9) | 37 (1.5) | 39 (4.8) | 82 (3.4) | 84 (9.9) |
| TOFa [h−1] | 8.6 | 8.1 | 7.9 | 1.5 | 4.8 | 3.4 | 9.9 |
| TOFb1 h [h−1] | 47 | 32 | 72 | 6.4 | 11 | 22 | 59 |
Data presented in Fig. 5 displays that isomeric complexes cis/trans-2a both reach a maximum yield of epoxide after 8 h reaction time (65 and 69%), after which no conversion of cyclooctene occurs. Also, the yield of epoxide remains constant, indicating that complexes cis/trans-2a to do not further react with the formed epoxide. Based on the displayed similar activity (within experimental error), it seems that epoxidation catalysis is rather insensitive to stereoisomers. The same observation was made for the two N,N-cis/trans stereoisomers of [ReOCl(oz)2] (TON trans-[ReOCl(oz)2] = 30; TON cis-[ReOCl(oz)2] = 40).21 The positive influence of the electron-withdrawing nitro group in cis-2b is easily visible from the data displayed in Fig. 5. Complex cis-2b reaches >95% conversion to epoxide after only 12 h. In any given time interval it shows a higher activity than unsubstituted complex cis-2a. For the chlorido/alkoxido complexes [ReOX(L1a)2] (3a/4a) and [ReOX(L1b)2] (3b/4b) the data presented in Fig. 6 shows that both complexes equipped with the NO2-substituted ligand L1b have a higher catalytic activity than with un-substituted ligand L1a. Ethoxido-complex 4b proved to be the most active catalyst of the four (TON = 84), also showing the highest initial rate, reaching a TOF1 h of 59 h−1 in the first hour (Table 2). In comparison, the respective chlorido-complex 3b shows a smaller TOF1 h of 22 h−1 in the first hour, but a similar activity after 8 h (TON = 82) (Table 2).21 Both complexes 3b and 4b reach a maximum yield of epoxide after 8 h. After that time, the amount of epoxide remains constant in case of 3b.
In contrast to chlorido-complex 3b, the formed epoxide gets consumed again by ethoxido-complex 4b at longer reaction times, dropping from 84% after 8 h to 66% after 24 h. This behavior is often caused by over-oxidation of the epoxide to cyclooctanone or hydrolytic ring-opening to the respective diol. Complexes 3a and 4a, equipped with the unsubstituted ligand L1a, produced a similar yield of epoxide after 24 h, albeit with a quite different activity profile. Similar to ethoxido complex 4b, the symmetric methoxido-complex 4a showed a higher initial reaction rate (TOF1 h = 11 h−1) compared to asymmetric chlorido-complex 3a (TOF1 h = 6.4 h−1). Complex 4a reached a maximum productivity after 8 h, after which the yield of epoxide remained unchanged, similar to 4b. Complex 3a showed a slow but steady increase in epoxide over the 24 h reaction time without reaching a plateau (Fig. 6).
An overview of calculated turnover numbers (TONs) and turnover frequencies (TOFs) is given in Table 2. With the data at hand, a trend can be observed that both symmetric OMe/OEt-ligated complexes 4a and 4b are more active in epoxidation catalysis compared to their asymmetric Cl-ligated analogues 3a and 3b. In literature, there are only few other published examples of such Cl/OR substituted pairs of complexes, that were tested in cyclooctene epoxidation.12,13 Two examples are complexes [ReOX(icq)2]12 (X = Cl or OMe, icq = isoquinoline-1-carboxylic acid) and [ReOX(pic)2]13 (X = Cl or OMe, pic = picolinic acid). These complexes differ in their chemistry from [ReO(X)(L1)2] complexes quite significantly. Both chlorido complexes adopt the asymmetric N,N-cis configuration in the solid state, which is also retained in both methoxido ligands. In cyclooctene epoxidation, the observed data does not allow to deduce a simple trend for chlorido substitution with an alkoxido ligand. Whereas for [ReOX(pic)2] both complexes (X = Cl or OMe) have essentially the same catalytic activities,13 [ReO(OMe)(icq)2] showed higher activities compared to [ReOCl(icq)2].12 In case of both pairs of chlorido/alkoxido complexes 3a/4a and 3b/4b, the substitution of the chlorido with an alkoxido ligand results in a more active catalyst, especially in the first 4 h of reaction time. If this higher activity is a result of the alkoxido ligand or the isomerization to a symmetric isomer cannot be answered with the data at hand. A substantial increase of catalyst activity is effected by the electron-withdrawing ligand L1b.
Mechanistic investigations in rhenium epoxidation chemistry are dominated by the chemistry of MTO, where both a peroxido and a bis-peroxido complex could be identified as catalytically active intermediates.5,15,29 Considering the electrophilic nature of the oxygen atom transfer from such a peroxide moiety to the olefin, a beneficial effect of electron-withdrawing ligands is expected, as such ligands withdraw electron density form the rhenium center and thereby increase the electrophilicity of a coordinated peroxide ligand. Exactly this behaviour, higher activity in epoxidation catalysis, was observed for ligand HL1b over HL1a. It also explains the lesser influence of stereoisomerism, as observed for cis/trans-2a. The mechanism of epoxidation for oxidorhenium(V) complexes is much less investigated, and an analogous peroxide complex could so far not be observed. There is however some evidence that a sequential oxidation of the starting mono-oxido to a bis-oxido and finally also a tris-oxido rhenium(VII) species is occurring on the way to the catalytic active species. Such a tris-oxido complex could then enter a catalytic cycle very similar to MTO.10,11,17,30
Conclusions
Within this manuscript we present a further example of stereoisomeric cis/trans-complexes in oxidorhenium(V) chemistry. Complexes cis-2a and trans-2a could be isolated, and their stereochemistry characterized by single crystal X-ray diffraction analysis. They are the first example of a pair of such stereoisomers that were tested in epoxidation catalysis. The influence of electron-withdrawing substituents was probed by complex cis-2b. Furthermore, another example of a reaction of an oxidorhenium(V) complex with an alcoholic solvent is presented, namely the formation of ethoxido complex 4b in EtOH in presence of the base lutidine. A similar reaction had been observed for methoxido complex 4a, obtained in MeOH in the presence of NaH. The complexes cis/trans-2a, cis-2b, 4a and 4b where then tested in catalytic epoxidation of cyclooctene and compared to their respective counterparts 3a and 3b. The activities between complexes did vary significantly, with yields of epoxide found between 37 and 95%. The two stereoisomers cis and trans-2a have a very similar catalytic activity profile, not influenced by the position of the ligands around the rhenium center. For the chlorido/alkoxido-complexes 3a/3b and 4a/4b, a twice as high catalytic activity of complexes with the nitro-substituted ligand L1b (3b and 4b) was observed compared to complexes containing the unsubstituted ligand L1a (3a and 4a). The highest activity was displayed by complex cis-2b, again showing the positive effect of nitro-substituted ligand L1b. We had previously observed a similar beneficial effect of electron-withdrawing substituents on pyrazole-phenol ligands in epoxidation activity.24,25 Selectivities towards epoxide were high for all tested complexes. Only in case of ethoxido-complex 4b, epoxide was further consumed to other products at longer reaction times. The main side product observed was ring-opened cyclooctane diol. To summarize, the electron-withdrawing nature of ligand HL1b is a more important contribution to catalyst activity than stereoisomerism (cis/trans) or nature of monodentate ligand X (Cl vs. OMe/OEt).Materials and methods
General
The rhenium precursors [ReOCl3(PPh3)2]31 and [ReOCl3(OPPh3)(SMe2)]32 were prepared according to previously published methods. The ligands HL1a33 and HL1b34 were synthesized by published routes. Syntheses of complexes cis-2a, 4a,193a and 3b21 were previously published. Additional unpublished analytical data (IR and UV-Vis data of 4a) has been included here. Chemicals were purchased from commercial sources and were used without further purification. NMR spectra were recorded with a Bruker (300 MHz) instrument. Chemical shifts are given in ppm and are referenced to residual protons in the solvent. Signals are described as s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublet), t (triplet), qd (quartet of doublets), m (multiplet) and coupling constants (J) are given in Hertz (Hz). Mass spectra were recorded with an Agilent 5973 MSD – Direct Probe using the EI ionization technique. Samples for infrared spectroscopy were measured on a Bruker Optics ALPHA FT-IR Spectrometer equipped with an ATR diamond probe head. GC-MS measurements were performed on an Agilent 7890 A with an Agilent 19091J–433 column coupled to a mass spectrometer type Agilent 5975 C. Elemental analyses were carried out using a Heraeus Vario Elementar automatic analyzer at the Graz University of Technology. Crystallographic data (excluding structure factors) for cis-2a, trans-2a and cis-2b were deposited with the Cambridge Crystallographic Data Center as supplementary publication no. CCDC 1854788, 1854789 and 2011198.†Epoxidation of olefin
In a typical experiment, 2–3 mg of catalyst (1 mol%) were dissolved in 0.5 mL CHCl3 and mixed with cyclooctene (1 equiv.), 50 μL of mesitylene (internal standard) and were heated to the reaction temperature. A Heidolph Parallel Synthesizer 1 was used for all epoxidation experiments. Then the oxidant (3. equiv.) was added. Aliquots for GC–MS (20 μL) were withdrawn at given time intervals, quenched with MnO2 and diluted with HPLC grade EtOAc. The reaction products were analysed by GC-MS (Agilent 7890 A with an Agilent 19091J–433 column coupled to a mass spectrometer type Agilent 5975 C), and the epoxide produced from each reaction mixture was quantified vs. mesitylene as the internal standard.N), 1590 (m, CN), 1448 (m), 1434 (m), 1266 (m), 1087 (m), 968 (s, ReO), 885 (m), 750 (s), 685 (s), 530 (s), 510 (s), 489 (m), 391 (w); EI-MS (m/z): 725.1 (M+); UV (CH2Cl2) λmax, nm (ε): 635 (69); elemental analysis calculated for C29H27Cl2NO3PRe·CH3CN (725.6 g mol−1): C 48.56, H 3.94, N 3.65; found C 49.38, H 3.81, N 3.80.
N), 1314 (s, -NO2), 1285 (m, -NO2), 988 (s, ReO), 847 (m), 744 (s), 710 (s), 525 (s), 507 (s), 491 (m); EI-MS (m/z): 262.1 (PPh3) (no M+ visible); UV (CH2Cl2) λmax, nm (ε): 649 (82); elemental analysis calculated for C29H26Cl2N2O5PRe (770.6 g mol−1): C 45.20, H 3.40, N 3.64; found C 45.81, H 3.51, N 3.64.
N), 1467 (m), 1245 (m), 1108 (m), 1076 (m), 927 (s, ReO), 851 (m), 760 (s), 697 (m), 664 (m), 577 (m), 482 (s); UV (CH2Cl2) λmax, nm (ε): 580 (116).
N), 81.54 (–CH2–), 74.61 (–C(CH3)2–), 66.07 (–OCH2CH3), 27.16 (–CH3), 27.10 (–CH3), 17.84 (–OCH2CH3). ATR-IR (cm−1): 2969 (w), 2854 (w), 1608 (s, CN), 1556 (w), 1468 (m, –NO2), 1409 (w), 1305 (s, –NO2), 1275 (m), 1112 (m), 1067 (m), 947 (m), 912 (m, ReO), 833 (m), 748 (w), 704 (m), 666 (m), 532 (m), 408 (m); EI-MS (m/z): 718.3 (M+), 673.3 (M+ − OEt); UV (CH2Cl2) λmax, nm (ε): 544 (238); elemental analysis calculated for C24H27N4O10Re (717.7 g mol−1): C 40.16, H 3.79, N 7.81; found C 40.13, H 3.79, N 7.57.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from NAWI Graz is gratefully acknowledged.Notes and references
- W. A. Herrmann, J. G. Kuchler, J. K. Felixberger, E. Herdtweck and W. Wagner, Angew. Chem., Int. Ed., 1988, 27, 394–396 CrossRef.
- (a) W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed., 1991, 30, 1638–1641 CrossRef; (b) W. A. Herrmann and M. Wang, Angew. Chem., Int. Ed., 1991, 30, 1641–1643 CrossRef.
- (a) C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef PubMed; (b) A. J. L. Pombeiro, M. F. C. G. da Silva and R. H. Crabtree, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Inc, 2011 Search PubMed; (c) R. G. Harms, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2015, 296, 1–23 CrossRef CAS.
- J. W. Kück, R. M. Reich and F. E. Kühn, Chem. Rec., 2016, 16, 349–364 CrossRef PubMed.
- S. Huber, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2014, 751, 25–32 CrossRef CAS.
- S. Yamazaki, Org. Biomol. Chem., 2007, 5, 2109–2113 RSC.
- (a) W. Adam and C. M. Mitchell, Angew. Chem., Int. Ed., 1996, 35, 533–535 CrossRef CAS; (b) T. R. Boehlow and C. D. Spilling, Tetrahedron Lett., 1996, 37, 2717–2720 CrossRef CAS; (c) P. Altmann, M. Cokoja and F. E. Kühn, Eur. J. Inorg. Chem., 2012, 3235–3239 CrossRef CAS.
- B. Machura, M. Wolff and I. Gryca, Coord. Chem. Rev., 2014, 275, 154–164 CrossRef CAS.
- W. A. Herrmann, J. Organomet. Chem., 1990, 382, 1–18 CrossRef CAS.
- W. A. Herrmann, M. U. Rauch and G. R. J. Artus, Inorg. Chem., 1996, 35, 1988–1991 CrossRef CAS.
- F. E. Kühn, M. U. Rauch, G. M. Lobmaier, G. R. J. Artus and W. A. Herrmann, Chem. Ber., 1997, 130, 1427–1431 CrossRef.
- B. Machura, M. Wolff, E. Benoist, J. A. Schachner and N. C. Mösch-Zanetti, Dalton Trans., 2013, 42, 8827–8837 RSC.
- B. Machura, M. Wolff, D. Tabak, J. A. Schachner and N. C. Mösch-Zanetti, Eur. J. Inorg. Chem., 2012, 3764–3773 CrossRef CAS.
- S. Dinda, M. G. B. Drew and R. Bhattacharyya, Catal. Commun., 2009, 10, 720–724 CrossRef CAS.
- F. E. Kühn, A. M. Santos and W. A. Herrmann, Dalton Trans., 2005, 34, 2483–2491 RSC.
- (a) V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2016, 61, 1708–1726 CrossRef CAS; (b) V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2017, 62, 1327–1342 CrossRef CAS; (c) V. S. Sergienko, Russ. J. Inorg. Chem., 2017, 62, 751–759 CrossRef CAS; (d) V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2018, 63, 631–641 CrossRef CAS; (e) B. Machura, Coord. Chem. Rev., 2005, 249, 591–612 CrossRef CAS; (f) B. Machura, Coord. Chem. Rev., 2005, 249, 2277–2307 CrossRef CAS.
- M. Wolff and B. Machura, Rev. Inorg. Chem., 2020, 40, 47–73 Search PubMed.
- B. Machura, M. Wolff, E. Benoist, J. A. Schachner, N. C. Mösch-Zanetti, K. Takao and Y. Ikeda, Polyhedron, 2014, 69, 205–218 CrossRef CAS.
- J. A. Schachner, B. Terfassa, L. M. Peschel, N. Zwettler, F. Belaj, P. Cias, G. Gescheidt and N. C. Mösch-Zanetti, Inorg. Chem., 2014, 53, 12918–12928 CrossRef CAS PubMed.
- J. Liu, D. Wu, X. Su, M. Han, S. Y. Kimura, D. L. Gray, J. R. Shapley, M. M. Abu-Omar, C. J. Werth and T. J. Strathmann, Inorg. Chem., 2016, 55, 2597–2611 CrossRef CAS PubMed.
- J. A. Schachner, B. Berner, F. Belaj and N. C. Mösch-Zanetti, Dalton Trans., 2019, 48, 8106–8115 RSC.
- (a) J. Liu, X. Su, M. Han, D. Wu, D. L. Gray, J. R. Shapley, C. J. Werth and T. J. Strathmann, Inorg. Chem., 2017, 56, 1757–1769 CrossRef CAS PubMed; (b) B. Terfassa, J. A. Schachner, P. Traar, F. Belaj and N. C. Mösch-Zanetti, Polyhedron, 2014, 75, 141–145 CrossRef CAS.
- (a) B. Terfassa, P. Traar, M. Volpe, N. C. Mösch-Zanetti, V. J. T. Raju, N. Megersa and N. Retta, Eur. J. Inorg. Chem., 2011, 4434–4440 CrossRef CAS; (b) N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2016, 55, 5973–5982 CrossRef CAS PubMed.
- N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2014, 53, 12832–12840 CrossRef CAS PubMed.
- P. Traar, J. A. Schachner, L. Steiner, A. Sachse, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 1983–1990 CrossRef CAS PubMed.
- B. Machura, M. Wolff, I. Gryca and R. Kruszynski, Polyhedron, 2012, 40, 93–104 CrossRef CAS.
- (a) B. Machura, R. Kruszynski and J. Kusz, Polyhedron, 2007, 26, 3455–3464 CrossRef CAS; (b) B. Machura and J. Kusz, Polyhedron, 2008, 27, 923–932 CrossRef CAS; (c) B. Machura, M. Wolff, W. Cieślik and R. Musioł, Polyhedron, 2013, 51, 263–274 CrossRef CAS.
- (a) B. Machura, R. Kruszynski and J. Kusz, Polyhedron, 2008, 27, 1679–1689 CrossRef CAS; (b) W. K. Rybak, A. Skarżyńska, L. Szterenberg, Z. Ciunik and T. Głowiak, Eur. J. Inorg. Chem., 2005, 4964–4975 CrossRef CAS; (c) B. Machura, M. Wolff, J. Kusz and R. Kruszynski, Polyhedron, 2009, 28, 2949–2964 CrossRef CAS.
- (a) I. V. Yudanov, J. Struct. Chem., 2007, 48, S111–S124 CrossRef CAS; (b) F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149–4164 CrossRef; (c) J. H. Espenson, Chem. Commun., 1999, 479–488 RSC.
- (a) R. Sarkar, A. Hens and K. K. Rajak, RSC Adv., 2015, 5, 15084–15095 RSC; (b) K. R. Grünwald, G. Saischek, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 7162–7171 CrossRef PubMed.
- J. Chatt and G. A. Rowe, J. Chem. Soc., 1962, 4019 RSC.
- B. D. Sherry, R. N. Loy and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4510–4511 CrossRef CAS PubMed.
- P. G. Cozzi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1995, 34, 2921–2930 CrossRef CAS.
- H. C. Aspinall, O. Beckingham, M. D. Farrar, N. Greeves and C. D. Thomas, Tetrahedron Lett., 2011, 52, 5120–5123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for trans-2a, cis-2b and 4b, zoom in of 1H NMR spectrum of complex 4b of the ethoxido region (Fig. S1–9); data on cyclovoltammetry; crystallographic details on data acquisition, bond lengths and angles and CCDC numbers for trans-2a (1854788), cis-2a (1854789) and cis-2b (2011198). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02352c |
| This journal is © The Royal Society of Chemistry 2020 |