
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogenation/dehydrogenation of N-heterocycles catalyzed by ruthenium complexes based on multimodal proton-responsive CNN(H) pincer ligands†
Práxedes
Sánchez
 a,
Martín
Hernández-Juárez
b,
Nuria
Rendón
*a,
Joaquín
López-Serrano
a,
Laura L.
Santos
a,
Eleuterio
Álvarez
a,
Margarita
Paneque
a and
Andrés
Suárez
*a
a,
Martín
Hernández-Juárez
b,
Nuria
Rendón
*a,
Joaquín
López-Serrano
a,
Laura L.
Santos
a,
Eleuterio
Álvarez
a,
Margarita
Paneque
a and
Andrés
Suárez
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA). CSIC and Universidad de Sevilla. Avda. Américo Vespucio 49, 41092, Sevilla, Spain. E-mail: nuria@iiq.csic.es; andres.suarez@iiq.csic.es
bCentro de Investigaciones Químicas. Universidad Autónoma del Estado de Hidalgo. Km. 14.5 Carretera Pachuca-Tulancingo. C.P. 42184, Mineral de la Reforma, Hidalgo, Mexico
First published on 7th July 2020
Abstract
Ru complexes based on lutidine-derived pincer CNN(H) ligands having secondary amine side donors are efficient precatalysts in the hydrogenation and dehydrogenation of N-heterocycles. Reaction of a Ru-CNN(H) complex with an excess of base produces the formation of a Ru(0) derivative, which is observed under catalytic conditions.
Following pioneering work by Noyori et al. on the use of metal complexes based on ligands bearing primary or secondary amine donors capable of getting involved in reversible metal-amine/metal-amido interconversion,1 a considerable diversity of ligands containing Brønsted acid/base functionalities have been developed.2 Among others, lutidine-derived metal complexes, which are readily deprotonated at the pincer methylene arms with concomitant dearomatization of the pyridine central moiety, have received significant attention.3 Both lutidine- and NH-containing pincer complexes have provided highly active and selective catalysts for a broad variety of hydrogenation4 and dehydrogenation reactions.5
Recently, the Milstein group has reported novel Ru complexes incorporating lutidine-based PNN(H) ligands containing secondary amines as side donors that are efficient catalysts in the (de)hydrogenation of polar substrates.6,7 These complexes are active ester and amide hydrogenation catalysts under very mild conditions, and catalyze the dehydrogenative coupling of alcohols to esters at low temperatures. Interestingly, reaction of a Ru-PNN(H) complex with 2 equiv. of base produced the formation of an enamino anionic Ru(II) species,6 which according to DFT calculations catalyzes the dehydrogenative coupling of alcohols solely through amine-metal/amido-metal interconversion.8
On the other hand, hydrogenation and acceptorless dehydrogenation are low environmental impact processes for the reduction of aromatic N-heterocycles to their corresponding saturated derivatives and the oxidation of the latter products to the parent N-heteroarenes, respectively.9 Although the principle of microscopic reversibility dictates that species able to catalyze the hydrogenation of N-heterocycles should also be active in the reverse dehydrogenation process, the number of catalytic systems that are able to perform both transformations is scarce, being these mainly based on M-PNHP (M = Fe, Co) complexes10 or costly CpIr derivatives.11
Pincer complexes based on N-heterocyclic carbenes (NHCs) have received an increased attention as catalysts in hydrogenation and dehydrogenation reactions.12 An interesting modification of the structure of lutidine-derived PNP and PNN ligands consists on the substitution of the P-donors by NHC groups.13 Herein, we report a series of Ru complexes stabilized with lutidine-derived CNN(H) pincer ligands incorporating secondary amino groups that are suitable catalytic precursors in both the hydrogenation and dehydrogenation of N-heterocycles. Because of the presence of two acidic functionalities in the ligands, these complexes might exhibit metal–ligand cooperation based on pyridine aromatization/dearomatization or amine-metal/amido-metal interconversion. However, preliminary NMR spectroscopic data revealed the unexpected formation of a zero-valent Ru complex under catalytic conditions.
Aiming to synthesize Ru-CNN(H) complexes, the imidazolium salts 1a–c were made react with Ag2O in CH2Cl2 to yield the silver complexes 2a–c (Scheme 1). Subsequent reactions of 2a–c with RuHCl(CO)(PPh3)3 in THF, followed by treatment with NaBF4 and PPh3 in CH3CN, allowed the isolation of the Ru-CNN(H) complexes 3a–c in moderate to good yields (33–85%). In the 1H NMR spectrum, the hydrido ligand of 3a gives rise to a doublet resonance at −7.79 ppm with a large 2JHP of 114.0 Hz, evincing the trans arrangement of the hydrido and PPh3 ligands. Similar NMR features are observed for complexes 3b and 3c. This structural arrangement was further confirmed in the solid state by an X-ray diffraction study of 3a and 3b (ESI†).
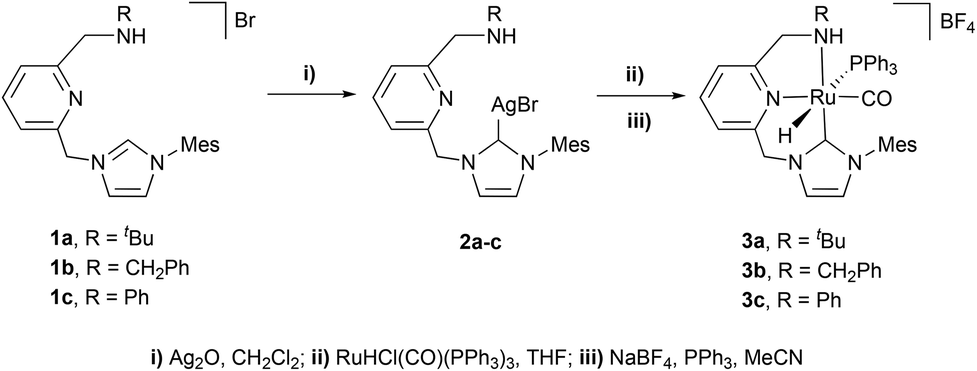 | ||
| Scheme 1 Synthesis of Ru–CNN(H) complexes. | ||
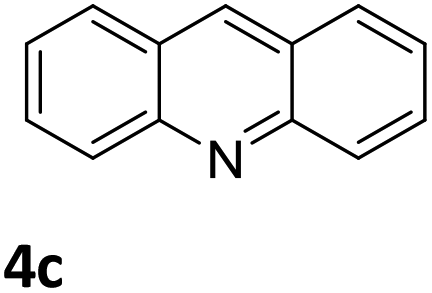
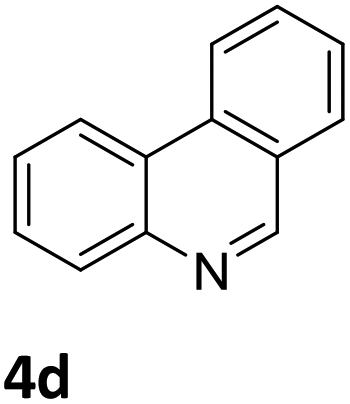
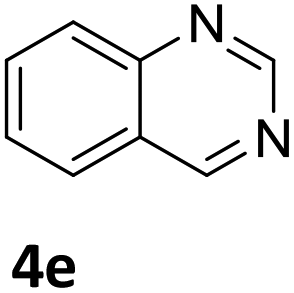
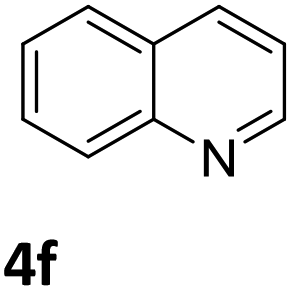
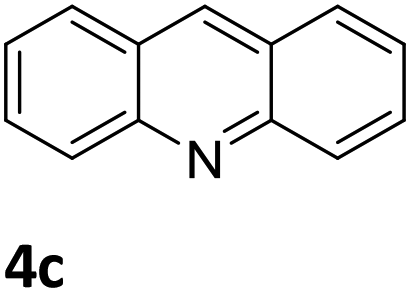
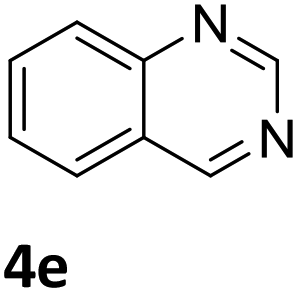
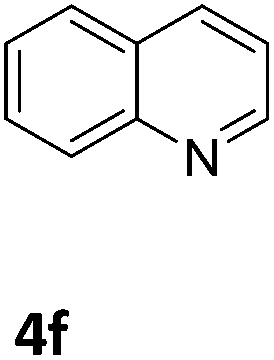
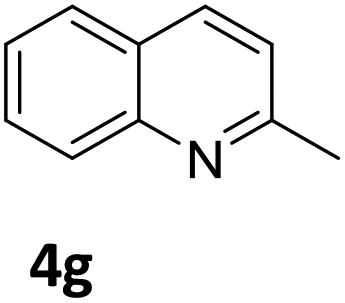
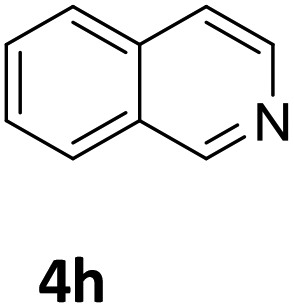
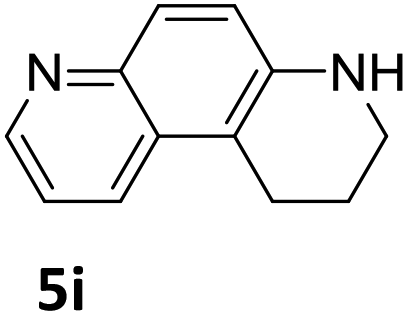
Next, the catalytic behavior of 3a–c in the hydrogenation of N-heterocycles was examined (Table 1). In the presence of KOtBu (5 mol%), complex 3a (0.5 mol%) catalyzed the hydrogenation of quinoxaline (4a) under 4 bar of H2 at 80 °C leading to full conversion in 6 h (entry 1). However, under the same conditions, complexes 3b and 3c were found to be less active than 3a (entries 2 and 3). Based on these results, complex 3a was tested in the hydrogenation of a series of N-heterocycles. For example, 2-methylquinoxaline (4b) was reduced under the above conditions with high conversions in 24 h (entry 4). Similarly, using 0.5 mol% of 3a, acridine (4c) and phenanthridine (4d) were hydrogenated with elevated conversions in 19 h (entries 5 and 6). However, the hydrogenation of quinazoline (4e), quinoline (4f) and quinaldine (4g) required harsher reaction conditions (1.0 mol% 3a, 95 °C, 48 h) to proceed to higher than 95% conv (entries 7–9). Under the later conditions, an increase of the H2 pressure to 10 bar was used for the hydrogenation of isoquinoline (4h) (entry 10). Finally, under 4 bar of H2, selective reduction of one of the N-containing rings of 4,7-phenanthroline (4i) was achieved, while the hydrogenation of both N-heterocycles was accomplished at 10 bar of H2 (entries 11 and 12).
| Entry | Substrate | Product | Cat. | Yield (%) |
|---|---|---|---|---|
| Reaction conditions: Unless otherwise noted: 4 bar H2, 80 °C, 2-methyltetrahydrofuran, 0.5 mol% Ru–CNN(H), 5 mol% KOtBu; [S] = 0.24 M. Yields were determined by 1H NMR spectroscopy using mesitylene as internal standard.a 95 °C.b 1.0 mol% 3a.c 10 bar H2. | ||||
| 1 |

|
|
3a | >99 (6 h) |
| 2 | 3b | 23 (6 h) | ||
| 3 | 3c | 22 (6 h) | ||
| 4 |
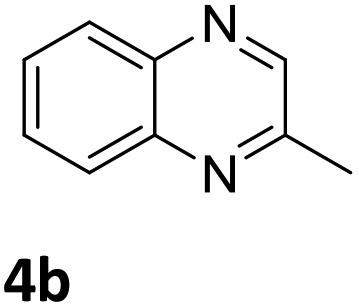
|
|
3a | >99 (24 h) |
| 5 |
|

|
3a | 98 (19 h) |
| 6 |
|

|
3a | 92 (19 h) |
| 7a,b |
|

|
3a | 98 (48 h) |
| 8a,b |
|
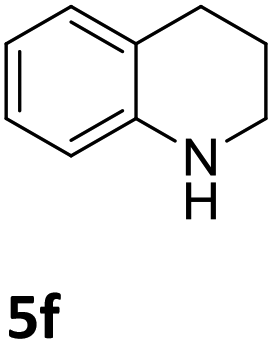
|
3a | >99 (48 h) |
| 9a,b |
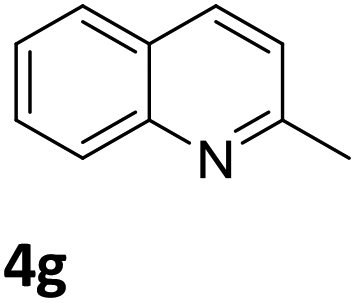
|
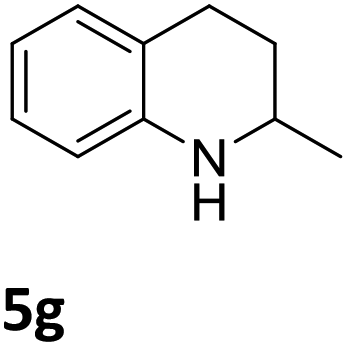
|
3a | 95 (48 h) |
| 10a,b,c |
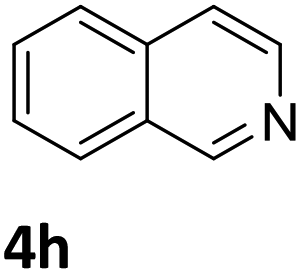
|
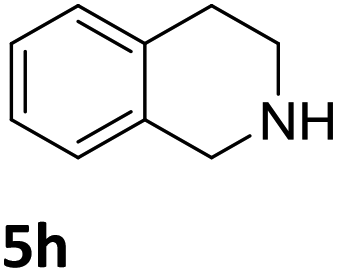
|
3a | 74 (24 h) |
| 11a,b |

|
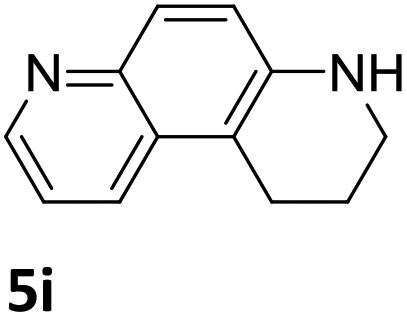
|
3a | >99 (24 h) |
| 12a,b,c |
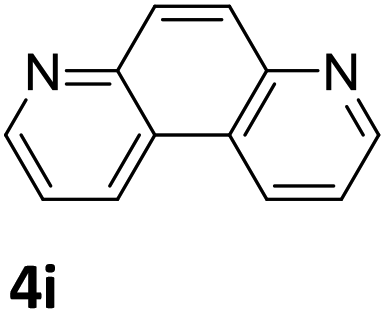
|
|
3a | 93 (72 h) (+7% 5i) |
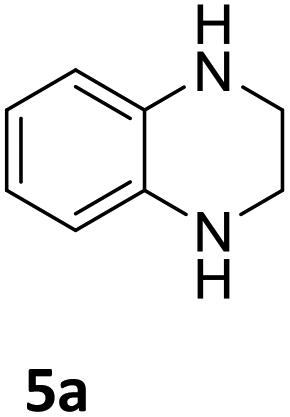
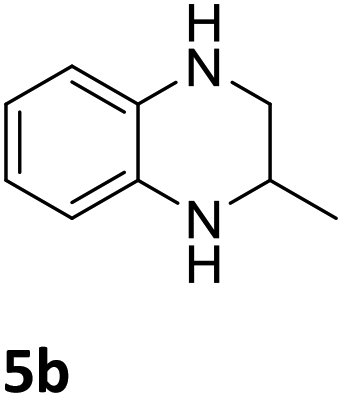
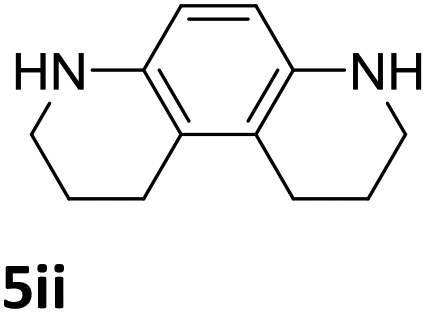
The catalytic performance of 3a in the dehydrogenation of the N-heterocycles 5 was also investigated (Table 2). Initially, dehydrogenation of 1,2,3,4-tetrahydroquinoxaline (5a) was examined using 4.0 mol% of 3a and 60 mol% of KOtBu in 2-methyltetrahydrofuran at 85 °C, leading to complete formation of 4a after 24 h (entry 1). However, a higher temperature was required for the reaction of 5b (160 °C, o-xylene) (entry 2). In addition, the oxidation of other N-heterocyclic substrates was examined. 9,10-Dihydroacridine (5c) was dehydrogenated in refluxing o-xylene with moderate catalytic activity (entry 3). Meanwhile, under the same conditions, hydrogen release from 5d and 5e took place with higher than 94% conv. (entries 4 and 5). The dehydrogenation of 5f, 5g and 5h proceeded with only low to moderate conversions after 48 h (entries 6–8); whereas 5i yielded 4,7-phenanthroline in 74% NMR yield (entry 9). To the best of our knowledge, complex 3a is the first example of a Ru derivative that catalyzes both the hydrogenation and dehydrogenation of a series of N-heterocycles. In addition, the catalytic activity of 3a in the reduction of N-heteroarenes lies in the range of most CpIr systems, although it is less efficient in the dehydrogenation reactions (0.1–5 mol% Ir, 74–160 °C).11 Moreover, the performance of 3a is superior to that of the Fe-PNHP catalyst in the hydrogenation of N-heterocycles (3 mol% Fe, 80 °C, 5 bar H2), while it is slightly poorer in the dehydrogenation reactions (3 mol% Fe, xylene reflux).10
| Entry | Substrate | Product | Yield (%) |
|---|---|---|---|
| Reaction conditions, unless otherwise noted: 160 °C, o-xylene, 4.0 mol% 3a, 60 mol% KOtBu. [S] = 0.12 M. Yields were determined by 1H NMR spectroscopy using mesitylene as internal standard.a 85 °C, 2-methyltetrahydrofuran. [S] = 0.06 M. | |||
| 1a |
|
|
>99 (24 h) |
| 2 |
|
|
85 (24 h) |
| 3 |
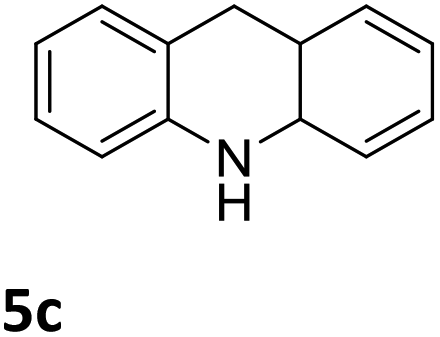
|
|
76 (48 h) |
| 4 |

|

|
94 (24 h) |
| 5 |
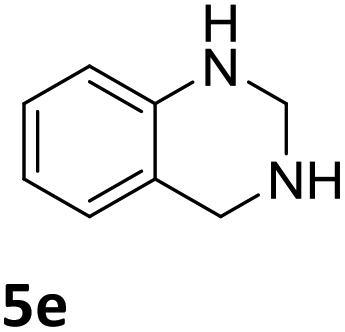
|
|
>99 (24 h) |
| 6 |

|
|
50 (48 h) |
| 7 |

|
|
27 (48 h) |
| 8 |

|
|
27 (48 h) |
| 9 |
|
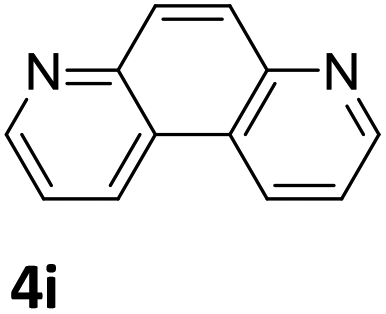
|
74 (48 h) |
To investigate the likely Ru species formed under catalytic conditions, complex 3a was treated with KOtBu (1.3 equiv.) in THF-d8 producing the instantaneous dark red coloring of the initially clear solution. Formation of the deprotonated complex 6 was ascertained by 1H NMR spectroscopy (Scheme 2), which shows the hydrido ligand of 6 giving rise to a broad doublet at −7.64 ppm with a large 2JHP of 140.2 Hz, indicative of its trans coordination to PPh3. Selective deprotonation of the CH2-NHC arm of 3a was evident from the observation of a singlet signal at 4.38 ppm (integrating to 1H), due to the methyne ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–NHC bridge, and two doublets of doublets at 3.43 (2JHH = 12.0 Hz, 3JHH = 12.0 Hz) and 3.09 (2JHH = 12.0 Hz, 3JHH = 1.9 Hz) ppm, attributable to the CH2-NH moiety. Subsequent treatment of the same THF-d8 solution of complex 6 with an excess of KOtBu (10 equiv.) or KHMDS (3 equiv.) produced a dark violet coloring of the sample (Scheme 2). The NMR spectra of the reaction mixture pointed out to the formation of the anionic complex 7, which is structurally related to an enamino Ru(II) complex previously reported by Milstein et al.6 The hydride and the enamino CH–NtBu hydrogens of the pincer ligand resonate as singlets at −17.09 and 6.69 ppm, respectively, in the 1H NMR experiment, while the inequivalent methylene protons of the CH2–NHC bridge appear as mutually coupled doublets at 4.83 and 4.56 ppm (2JHH = 12.7 Hz). Also of note, the hydrogens of the pincer dearomatized central ring produce upfield shifted resonances in the range between 5.33 and 6.54 ppm.
CH–NHC bridge, and two doublets of doublets at 3.43 (2JHH = 12.0 Hz, 3JHH = 12.0 Hz) and 3.09 (2JHH = 12.0 Hz, 3JHH = 1.9 Hz) ppm, attributable to the CH2-NH moiety. Subsequent treatment of the same THF-d8 solution of complex 6 with an excess of KOtBu (10 equiv.) or KHMDS (3 equiv.) produced a dark violet coloring of the sample (Scheme 2). The NMR spectra of the reaction mixture pointed out to the formation of the anionic complex 7, which is structurally related to an enamino Ru(II) complex previously reported by Milstein et al.6 The hydride and the enamino CH–NtBu hydrogens of the pincer ligand resonate as singlets at −17.09 and 6.69 ppm, respectively, in the 1H NMR experiment, while the inequivalent methylene protons of the CH2–NHC bridge appear as mutually coupled doublets at 4.83 and 4.56 ppm (2JHH = 12.7 Hz). Also of note, the hydrogens of the pincer dearomatized central ring produce upfield shifted resonances in the range between 5.33 and 6.54 ppm.
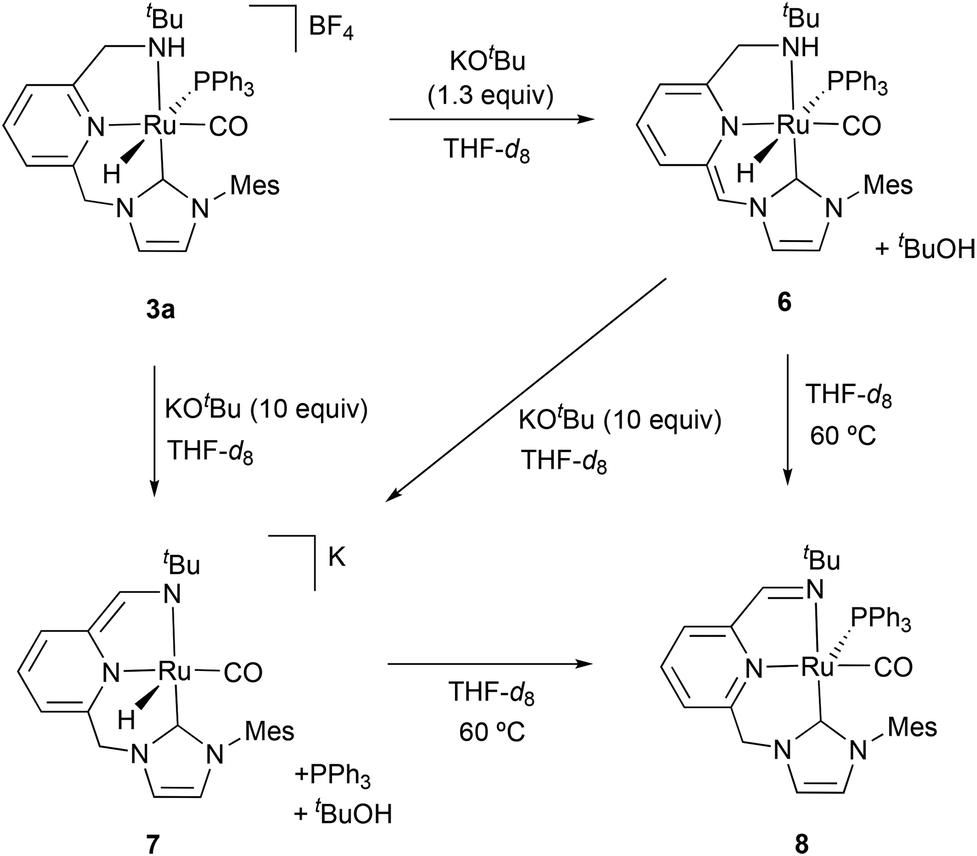 | ||
| Scheme 2 Generation of Ru complexes 6–8. | ||
THF-d8 solutions pressurized with H2 (3 bar) of the in situ generated complexes 6 and 7 were analyzed by NMR spectroscopy in order to determine the potential of these species to perform H2 activation. However, noticeable changes in their 1H NMR experiments were not observed. Alternatively, pressurization of solutions of complexes 6 and 7 with D2 (3 bar) produced the H/D exchange of the hydride ligands and the protons of the methylene and methyne bridges (ESI†), evincing the ability of complexes 6 (after PPh3 decoordination) and 7 to produce the reversible activation of H2 in a ligand-assisted process or through the participation of an external base.
Unexpectedly, heating to 60 °C solutions of the in situ formed species 6 or 7 produces the clean generation of a new species, which has been spectroscopically characterized as the Ru(0) imine complex 8 (Scheme 2).14 The 1H NMR spectrum of the resulting dark blue solutions indicates the presence of the imine moiety by the appearance of a doublet at 7.92 ppm (4JHP = 3.7 Hz). The 13C{1H} NMR spectrum presents the resonances corresponding to the CO ligand and the C2–NHC carbon as doublets at 216.1 (JCP = 10 Hz) and 191.3 (JCP = 7 Hz) ppm, respectively. Finally, the coordination of PPh3 is manifested in the 31P{1H} NMR experiment by the appearance of a singlet at 50.5 ppm. Complex 8 can be regarded as derived from 6 by the formal loss of a H2 molecule.
It is worth noting that imine Ru(0) complexes structurally related to 8 have been recently reported by Keith, Chianese et al. (Scheme 3).14 These derivatives (such as A), which were shown to be highly active ester hydrogenation catalysts, were isolated from the reactions with base of Ru–PNN and –CNN complexes bearing dialkylamino side donors. Moreover, hydrogen activation by A led to the formation of a Ru dihydride complex (B) in which the imine ligand fragment was hydrogenated to amine. Interestingly, solutions of the Ru(0) derivative 8 were active in the hydrogenation of N-heterocycles.14,15 Thus, THF-d8 solutions containing this complex were able to hydrogenate (3 bar H2) 2-methylquinoxaline (10 equiv.) at 60 °C, being 8 the only detectable metal species during the catalytic reaction and suggesting that this is the catalyst resting state (ESI†). Similarly, addition of 2-methylquinoxaline (10 equiv.) to a THF-d8 solution of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 6 and 7 produced the instantaneous formation of 8. Subsequent pressurization with H2 (3 bar) and heating to 65 °C the resulting solution brought about the hydrogenation of the N-heteroarene (ESI†).
:1 mixture of 6 and 7 produced the instantaneous formation of 8. Subsequent pressurization with H2 (3 bar) and heating to 65 °C the resulting solution brought about the hydrogenation of the N-heteroarene (ESI†).
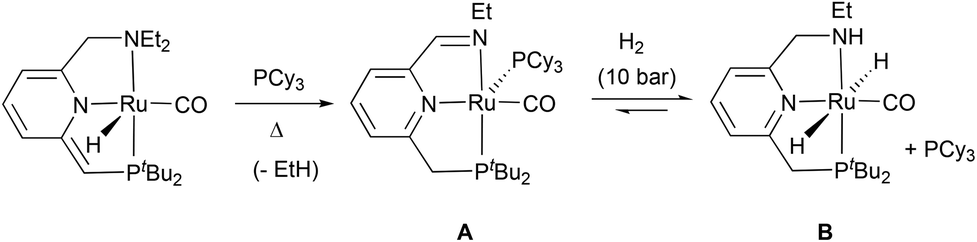 | ||
| Scheme 3 Complexes A and B reported by Keith, Chianese et al. | ||
In an attempt to determine the likely formation of dihydride species similar to B, a THF-d8 solution of complex 8 was pressurized with H2 (4 bar) and analyzed by NMR spectroscopy. Contrary to previously observed in the reaction of A with H2,14 the 1H NMR spectrum of this solution did not reveal changes in the temperature range between −80 and 55 °C. DFT calculations (B3LYP-D3, 6-31 g(d,p)/SDD) showed that hydrogen activation by 8 to yield a dihydride Ru complex analogous to B is endergonic by 9.5 kcal mol−1 (ESI†). Moreover, H/D exchange upon exposure to D2 (3 bar) of an in situ generated solution of 8 did not occur even after prolonged (72 h) heating to 65 °C. However, in the latter experiment, formation of HD was observed, what can be ascribed to the generation of deuteride species (Ru-D) under the NMR detection limit that react with tBuOH resulting from the deprotonation of 3a with KOtBu (ESI†).
In conclusion, ruthenium complexes 3 incorporating CNN(H) pincer ligands are efficient catalyst precursors in the hydrogenation of N-heteroarenes and in the acceptorless dehydrogenation of N-heterocycles. Although complexes 3 contain two potential sites for metal–ligand cooperation, reaction of 3a with base ultimately furnishes the Ru(0) complex 8, whose formation is observed under catalytic conditions. Further studies to determine the nature of the metal species participating in the catalytic cycle are being carried out.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support (FEDER contribution) from the Spanish Ministry of Science and Innovation (CTQ2016-80814-R) and Junta de Andalucía (PY18-3208), and the use of computational facilities of the Supercomputing Center of Galicia (CESGA) are gratefully acknowledged. We thank support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed.
- D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 CrossRef PubMed.
- A. Suárez, Phys. Sci. Rev., 2018, 3, 20170028 Search PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228 CrossRef CAS PubMed.
- E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem. – Eur. J., 2014, 20, 15727 CrossRef CAS PubMed; S. Kar, M. Rauch, A. Kumar, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2020, 10, 5511 CrossRef PubMed.
- P. Hu, E. Fogler, Y. Diskin-Posner, M. A. Iron and D. Milstein, Nat. Commun., 2015, 6, 6859 CrossRef CAS PubMed; A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, J. Am. Chem. Soc., 2018, 140, 7453 CrossRef PubMed.
- C. Hou, J. Jiang, Y. Li, C. Zhao and Z. Ke, ACS Catal., 2017, 7, 786 CrossRef CAS.
- Z. X. Giustra, J. S. Ishibashi and S. Y. Liu, Coord. Chem. Rev., 2016, 314, 134 CrossRef CAS; R. H. Crabtree, ACS Sustainable Chem. Eng., 2017, 5, 4491 CrossRef.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564 CrossRef CAS PubMed; R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5, 6350 CrossRef.
- R. Yamaguchi, C. Ikeda, Y. Takahashi and K. Fujita, J. Am. Chem. Soc., 2009, 131, 8410 CrossRef CAS PubMed; K. Fujita, Y. Tanaka, M. Kobayashi and R. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 4829 CrossRef PubMed; K. Fujita, T. Wada and T. Shiraishi, Angew. Chem., Int. Ed., 2017, 56, 10886 CrossRef PubMed; J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 6983 CrossRef PubMed; á. Vivancos, M. Beller and M. Albrecht, ACS Catal., 2018, 8, 17 CrossRef; B. Maji and J. Choudhury, Chem. Commun., 2019, 55, 4574 RSC.
- D. A. Hey, R. M. Reich, W. Baratta and F. E. Kühn, Coord. Chem. Rev., 2018, 374, 114 CrossRef CAS.
- C. del Pozo, M. Iglesias and F. Sánchez, Organometallics, 2011, 30, 2180 CrossRef CAS; E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826 CrossRef; Y. Sun, C. Koehler, R. Tan, V. T. Annibale and D. Song, Chem. Commun., 2011, 47, 8349 RSC; G. A. Filonenko, D. Smykowski, B. M. Szyja, G. Li, J. Szczygiel, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 1145 CrossRef; M. Hernández-Juárez, J. López-Serrano, P. Lara, J. P. Morales-Cerón, M. Vaquero, E. Álvarez, V. Salazar and A. Suárez, Chem. – Eur. J., 2015, 21, 7540 CrossRef PubMed; X. Wu, L. Ji, Y. Ji, E. H. M. Elageed and G. Gao, Catal. Commun., 2016, 85, 57 CrossRef.
- T. He, J. C. Buttner, E. F. Reynolds, J. Pham, J. C. Malek, J. M. Keith and A. R. Chianese, J. Am. Chem. Soc., 2019, 141, 17404 CrossRef CAS PubMed.
- A. Anaby, M. Schelwies, J. Schwaben, F. Rominger, A. S. K. Hashmi and T. Schaub, Organometallics, 2018, 37, 2193 CrossRef CAS; A. Eizawa, S. Nishimura, K. Arashiba, K. Nakajima and Y. Nishibayashi, Organometallics, 2018, 37, 3086 CrossRef; D. J. Tindall, M. Menche, M. Schelwies, R. A. Paciello, A. Schäfer, P. Comba, F. Rominger, A. S. K. Hashmi and T. Schaub, Inorg. Chem., 2020, 59, 5099 CrossRef PubMed; N. Govindarajan, V. Sinha, M. Trincado, H. Grützmacher, E. J. Meijer and B. de Bruin, ChemCatChem, 2020, 12, 2610 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; X-ray diffraction analysis; and DFT calculations details. CCDC 1968868 [3a·CH2Cl2] and 1968869 [3b·2CH3OH]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02326d |
| This journal is © The Royal Society of Chemistry 2020 |